

Abstract
Severe cases of COVID‐19 are associated with extensive lung damage and the presence of infected multinucleated syncytial pneumocytes. The viral and cellular mechanisms regulating the formation of these syncytia are not well understood. Here, we show that SARS‐CoV‐2‐infected cells express the Spike protein (S) at their surface and fuse with ACE2‐positive neighboring cells. Expression of S without any other viral proteins triggers syncytia formation. Interferon‐induced transmembrane proteins (IFITMs), a family of restriction factors that block the entry of many viruses, inhibit S‐mediated fusion, with IFITM1 being more active than IFITM2 and IFITM3. On the contrary, the TMPRSS2 serine protease, which is known to enhance infectivity of cell‐free virions, processes both S and ACE2 and increases syncytia formation by accelerating the fusion process. TMPRSS2 thwarts the antiviral effect of IFITMs. Our results show that SARS‐CoV‐2 pathological effects are modulated by cellular proteins that either inhibit or facilitate syncytia formation.
Keywords: fusion, interferon, SARS‐CoV‐2, syncytia
Subject Categories: Immunology
Cells infected with SARS‐CoV‐2 can fuse with neighbouring cells in a process accelerated by infectivity‐enhancing host factor TMPRSS2 and restricted by antiviral IFITM proteins.

Introduction
COVID‐19 consists of a spectrum of syndromes from a mild, flu‐like illness to severe pneumonia. Disease severity is linked to lung epithelial destruction, resulting from both immune‐mediated damages and viral cytopathic effects. SARS‐CoV‐2 infection of respiratory epithelial cells likely activates monocytes, macrophages, and dendritic cells, resulting in secretion of proinflammatory cytokines (Huang et al, 2020; Ong et al., 2020; Zhou et al, 2020). Excessive systemic cytokine production may lead to thrombosis, hypotension, acute respiratory distress syndrome (ARDS), and fatal multi‐organ failure. The innate type‐I and type‐III interferon (IFN) response, which normally controls viral replication is also reduced in severe cases (Blanco‐Melo et al, 2020; preprint: Hadjadj et al, 2020; Park & Iwasaki, 2020). However, prolonged IFN‐production aggravates disease by impairing lung epithelial regeneration (Broggi et al, 2020; Major et al, 2020). In the lung, SARS‐CoV‐2 infects ciliated cells in the airway, alveolar type 2 pneumocytes, and epithelial progenitors among others (Bost et al, 2020; Hou et al, 2020; Subbarao & Mahanty, 2020). SARS‐CoV‐2 and other coronaviruses are cytopathic (Freundt et al, 2010; preprint: Gorshkov et al, 2020; Ogando et al, 2020; Ren et al, 2020; Tang et al, 2020). The death of infected cells is also a trigger of immune activation.
SARS‐CoV‐2 entry into cell is initiated by interactions between the spike glycoprotein (S) and its receptor, angiotensin‐converting enzyme 2 (ACE2), followed by S cleavage and priming by the cellular protease TMPRSS2 or other surface and endosomal proteases (Letko et al, 2020; Matsuyama et al, 2020; Hoffmann et al, 2020b). The structure of S in complex with ACE2 has been elucidated (Lan et al, 2020; Walls et al, 2020; Wang et al, 2020). S consists of three S1‐S2 dimers, displaying conformational changes upon virus entry leading to fusion. Besides fusion mediated by virions, S proteins present at the plasma membrane can trigger receptor‐dependent syncytia formation. These syncytia have been observed in cell cultures and in tissues from individuals infected with SARS‐CoV‐1, MERS‐CoV, or SARS‐CoV‐2 (Franks et al, 2003; Matsuyama et al, 2010; Chan et al, 2013; Qian et al, 2013; preprint: Giacca et al, 2020; Hoffmann et al, 2020a; Tian et al, 2020; Xu et al, 2020), but they were not precisely characterized. It has been proposed that they may originate from direct infection of target cells or from the indirect immune‐mediated fusion of myeloid cells. Fused pneumocytes expressing SARS‐CoV‐2 RNA and S proteins were observed in post‐mortem lung tissues of 20 out of 41 COVID‐19‐infected patients, indicating that productive infection leads to syncytia formation, at least in critical cases (preprint: Giacca et al, 2020).
SARS‐CoV‐2 replication is in part controlled by the innate host response, through mechanisms that are currently being unveiled. Interferon‐stimulated genes (ISGs) inhibit discrete steps of the viral life cycle. At the entry level, the interferon (IFN)‐Induced Transmembrane proteins (IFITM1, IFITM2, or IFITM3) block many viruses by inhibiting virus–cell fusion at hemifusion or pore formation stages (Shi et al, 2017). IFITMs act by modifying the rigidity and/or curvature of the membranes in which they reside (Abdel Motal et al, 1993; Compton Alex et al, 2014; Shi et al, 2017; Zani & Yount, 2018). Due to different sorting motifs, IFITM1 is mostly found at the plasma membrane, whereas IFITM2/3 accumulates in the endo‐lysosomal compartment after transiting through the surface. IFITMs inhibit SARS‐CoV, 229E, and MERS‐CoV entry, but promote infection by HCoV‐OC43, a coronavirus that causes the common cold (Huang et al, 2011; Bertram et al, 2013; Warren et al, 2014; Wrensch et al, 2014; Zhao et al, 2014; Zhao et al, 2018). IFITMs, as well as other ISGs, including LY6E and Cholesterol 25‐hydrolase (CH25H), impair SARS‐CoV‐2 replication by blocking the fusion of virions (Pfaender et al, 2020; preprint: Zang et al, 2020; Zhao et al, 2020). Most of the experiments regarding these ISGs have been performed with single‐cycle viral pseudotypes. Little is known about the impact of IFITMs on SARS‐CoV‐2‐induced syncytia formation.
Here, we characterized the mechanisms of SARS‐CoV‐2‐induced cell–cell fusion and examined how syncytia formation is impacted by IFITMs and TMPRSS2.
Results
Syncytia formation by SARS‐CoV‐2‐infected cells
We first examined whether SARS‐CoV‐2‐infected cells may form syncytia. To this aim, we derived U2OS cells stably expressing ACE2. We selected this cell line because its flat shape facilitates imaging. We generated U2OS‐ACE2 cells carrying a GFP–Split complementation system (Buchrieser et al, 2019), in which two cells separately produce half of the reporter protein, producing GFP only upon fusion (Fig 1A). These U2OS‐ACE2‐derived cells, that we termed “S‐Fuse” cells, were exposed to various doses of SARS‐CoV‐2. Video‐microscopy analysis showed that syncytia appeared rapidly, starting at 6 h post‐infection and grew in size, as bystander cells are incorporated in fused cells (Fig 1B and C and Movie EV1). Most of the syncytia end up dying, as assessed by the acquisition of propidium iodide (PI) (Fig 1B and C and Movie EV1). The extent of fusion was then quantified by measuring the GFP+ area with a high‐content imager. The total area of syncytia within each well correlated with the viral inoculum, indicating that the assay provides a quantitative assessment of viral infection (Fig 2A). S was expressed by the syncytia, but also by single infected cells that have not yet fused, as assessed by immunofluorescence (Fig 1D). Flow cytometry on unpermeabilized infected cells further showed that S was present at the surface (Fig 1E).
Figure 1. SARS‐CoV‐2 induced syncytia formation.
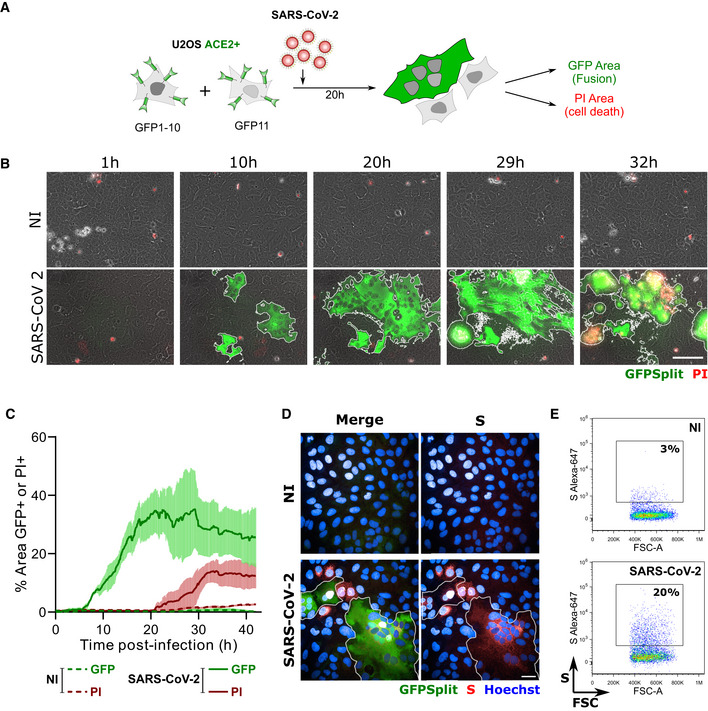
-
AGFP‐Split U2OS‐ACE2 were co‐cultured at a 1:1 ratio and infected with SARS‐CoV‐2. Syncytia formation and cell death was monitored by video microscopy or at endpoint using confocal microscopy and high‐content imaging.
-
BStill images of GFP (syncytia) and propidium iodide (PI) (cell death) at different time points. Scale bar: 100 µm.
-
CQuantification of U2OS‐ACE2 fusion and death by time‐lapse microscopy. Results are mean ± SD from three fields per condition.
-
DS staining of infected U2OS‐ACE2 cells analyzed by immunofluorescence. The Hoechst dye stains the nuclei. Scale bar: 40 µm.
-
ESurface S staining of infected U2OS‐ACE2 cells analyzed by flow cytometry. Results are representative of at least three independent experiments.
Figure 2. Impact of TMPRSS2 and IFITMs on syncytia formation by U2OS‐ACE2‐infected cells.
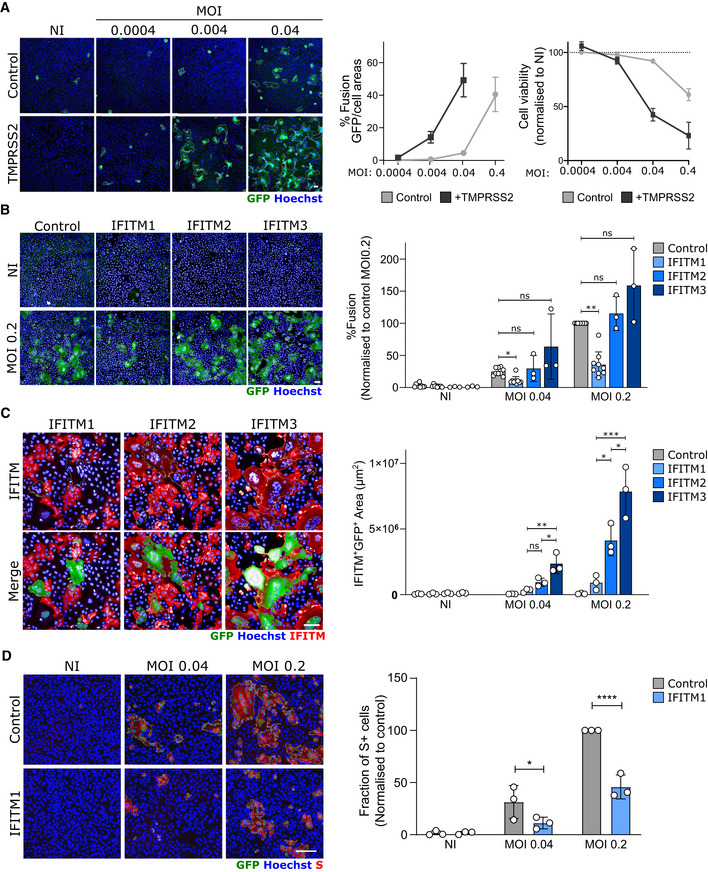
-
ATMPRSS2 increases fusion and cell mortality. Right panel: Areas of GFP+ cells and nuclei count normalized to NI.
-
BIFITM1, but not IFITM2 and 3, inhibits SARS‐CoV‐2‐induced syncytia formation. Right panels: Area of GFP+ cells
-
CThe fusion of IFITM1+ cells with U2OS‐ACE2 syncytia is drastically reduced. Right panel: Area of IFITM+ GFP+ cells
-
DIFITM1 decreases the number of infected cells. Right panel: Fraction of cells positive for S, normalized to control cells (transduced with pQCXIP‐empty vector).
Data information: Left panels: one representative experiment is shown. Scale bars: 100 µm. Right panels: Data are mean ± SD of 3–9 independent experiments. Statistical analysis: B–C: One‐way ANOVA, D: Two‐way ANOVA. ns: non‐significant, *P < 0. 05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Impact of IFITMs and TMPRSS2 on syncytia formation
We then asked whether TMPRSS2 and IFITM1, IFITM2, and IFITM3 impact syncytia formation. We generated S‐Fuse cells stably producing each of the four proteins. Their expression was verified by flow cytometry or Western blotting (Fig EV1). We then compared their sensitivity to SARS‐CoV‐2 infection. The presence of TMPRSS2 increased the appearance of fused cells by 5‐ to 10‐fold (Fig 2A). On the contrary, IFITM1 significantly inhibited syncytia formation (Fig 2B and Movie EV1). Therefore, TMPRSS2 and IFITM1 exert opposite effects on syncytia formation. IFITM2 and 3 were poorly active (Fig 2B), probably because they mostly accumulate within the endosomal compartment, which limits their ability to alter fusion events occurring at the plasma membrane. Since only 40% of each cell population expressed high levels of IFITM, as assessed by flow cytometry (Fig EV1) and immunofluorescence (Fig 2C), we asked whether IFITM+ cells were present in the syncytia. A co‐staining with anti‐IFITM antibodies indicated that syncytia did not incorporate IFITM1+ cells present in the culture, whereas this was not the case for IFITM2 and 3 (Figs 2C and EV3). Moreover, the inhibitory effect of IFITM1 on syncytia was associated a decreased number of S+ cells (Fig 2D).
Figure EV1. ACE2 and IFITM expression in U2OS‐ACE2 cell derivatives.
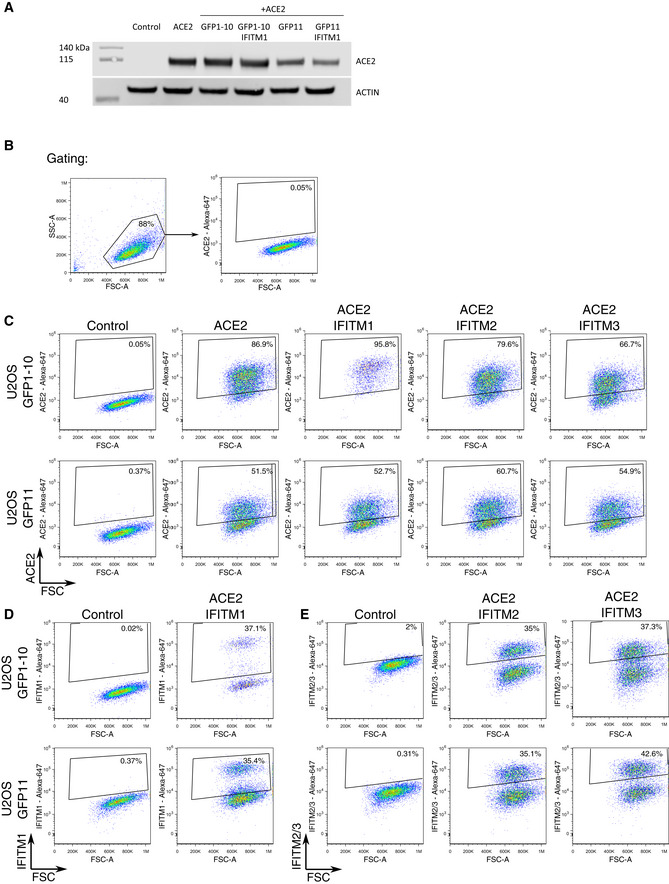
-
AACE2 expression assessed by Western blot in U2OS‐ACE2 GFP‐split cells expressing or not IFITM1. Cell lysates were analyzed by for ACE2 and actin as a loading control.
-
B–EAnalysis of ACE2 and IFITMs levels by flow cytometry B. Gating strategy. Cells were gated by size and granularity. Positive and negative gates were then set on control U2OS cells lacking the protein of interest. C. ACE2 levels on parental U2OS (control) and various IFITM derivatives. D. IFITM1 levels on parental U2OS (control) and U2OS‐ACE2 IFITM1 cells. E. IFITM2 and IFITM3 levels on parental U2OS (control) and U2OS‐ACE2 IFITM2 and U2OS‐ACE2 IFITM3 cells.
Figure EV3. Expression of IFITM1, IFITM2, and IFITM3 in SARS‐CoV‐2‐infected syncytia.
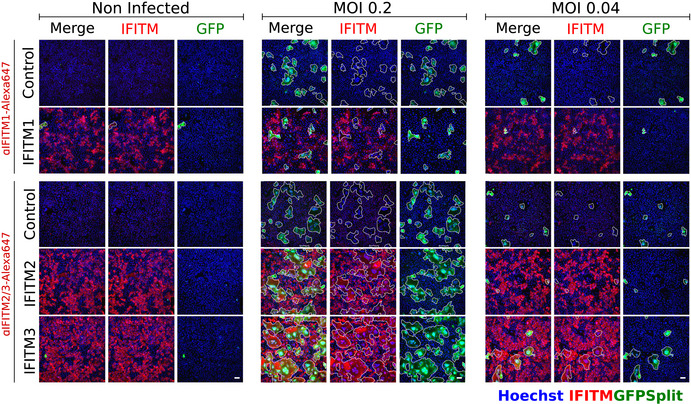
SARS‐CoV‐2‐induced syncytia in U2OS cells are IFITM1 dim or negative, but IFITM2/3 positive. Left: Non‐infected U2OS‐ACE2 cells. Middle and right: Cells infected with SARS‐CoV‐2 at the indicated MOI. The GFP area is delimited in white. With IFITM1, the fused GFP area does not overlap with the red IFITM1 area. For IFITM2 and IFITM3, an overlap with the GFP is visible. Scale bars: 100 µm. Quantifications are presented Fig 2C.
Figure EV2. Image quantification methodology.
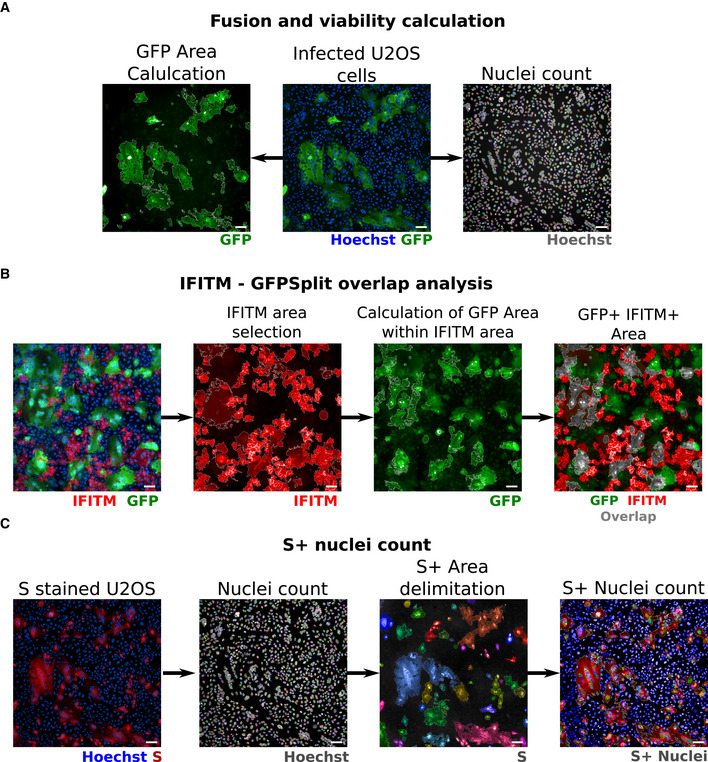
-
AQuantification of fusion and viability. To measure the extent of cell–cell fusion, the GFP area was automatically delimited, measured, and then divided by the total cell area. For cell viability, nuclei were automatically counted, and the total number of nuclei per well was normalized to that of non‐infected control cells.
-
BQuantification of syncytia expressing IFITMs. For IFITM‐GFP overlap quantification, the IFITM+ area was first selected on the 647 nm channel (image 2) and the GFP‐positive area was quantified within the IFITM+ area (image 3). Image 4 shows the overlap area in gray for simpler visualization.
-
CQuantification of infected cells expressing the S protein. The cells were stained with anti‐S antibodies, and the nuclei were detected with Hoechst. The S+ area was delimited (each selected object is pseudo‐colored). Nuclei present within the S+ area were scored and divided by the total number of nuclei, to calculate the number of infected cells per well.
Data information: Scale bars: 100 µm. The same field was used in this example for A and C, Hoechst images are therefore identical in A and C.
We next assessed whether other cell types form syncytia upon SARS‐CoV‐2 infection. To this aim, we used 293T cells transiently transfected with ACE2 and also generated stable A549‐ACE2 cells. The two cell lines readily formed syncytia upon infection (Fig EV4A and B). In order to rule out the possibility that syncytia formation is solely dependent on ACE2 over‐expression, we investigated the naturally permissible Vero cells with the GFP‐split system. We did not detect fused infected Vero cells (Fig EV5A); thus, we used as donors U20S‐ACE2‐infected cells that we co‐cultivated with uninfected Vero cells. Numerous heterocellular syncytia were formed in a short period of time (8 h) (Fig EV5D). The ability of Vero cells to fuse was again confirmed when donor 293T cells were transfected with S and co‐cultivated with Vero E6 acceptor cells (Fig EV5C). Additionally, Vero cells are also capable of forming syncytia upon transfection of only the S protein (Fig EV5B). Of note, Caco2 cells did not fuse upon SARS‐CoV‐2 infection (Fig EV4C). Taken together, our data strongly suggest that the ability to form syncytia upon SARS‐CoV‐2 infection is dependent on cell type as well as on the surface levels of S and ACE2. Fusion is detected in Vero cells with endogenous levels of ACE2.
Figure EV4. Characterization of SARS‐CoV‐2‐induced syncytia in different cell lines.
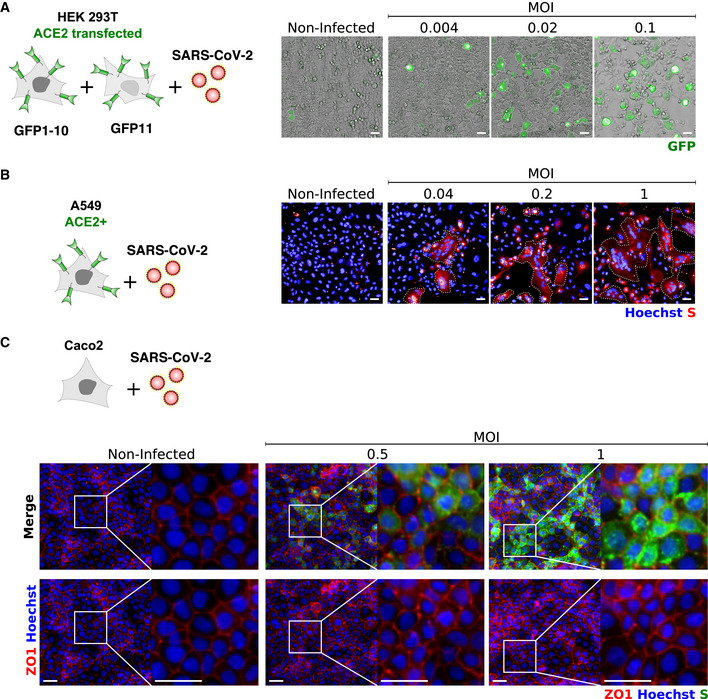
-
A293T‐GFP1‐10 and ‐GFP11 (1:1 ratio) were transfected with ACE2 and infected with SARS‐CoV‐2 at the indicated MOIs. Cell fusion was visualized by the GFP+ area 18 h post‐infection. One representative image per condition is shown, and GFP area is delimited in white.
-
BA549‐ACE2 cells were infected with SARS‐CoV‐2 at the indicated MOIs for 24 h and stained with anti‐S antibodies and Hoechst. One representative image per condition is shown, and syncytia are manually delimited in white.
-
CCaco2 cells were infected with SARS‐CoV‐2 at the indicated MOIs for 24 h and stained for ZO1 tight junction marker, Hoechst and S. No clear syncytia formation was observed at any of the MOIs tested.
Data information: Scale bar: 50 µm. Data are representative of three independent experiments.
Figure EV5. Characterization of syncytia formation in Vero cells.
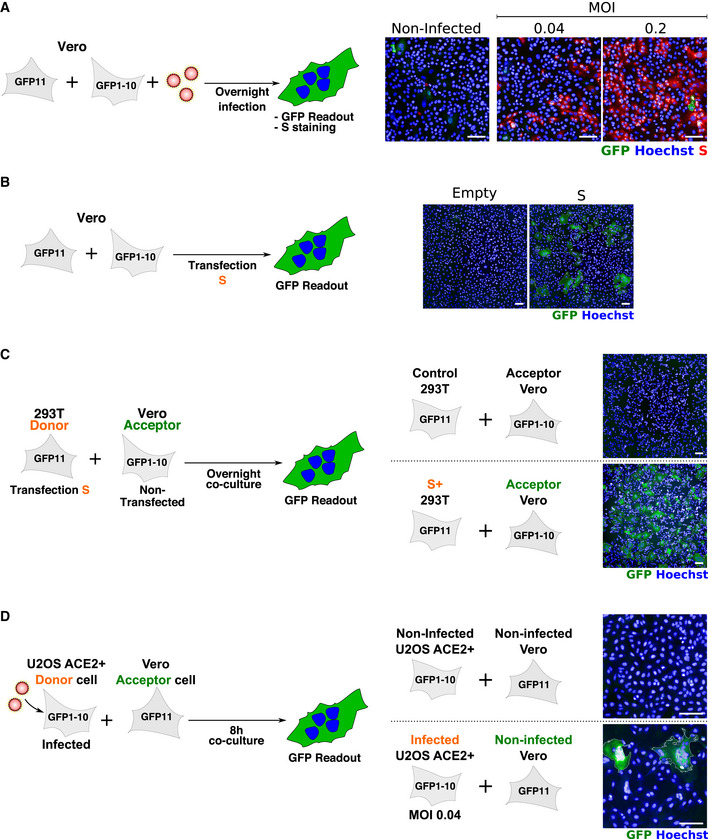
-
AVero‐GFP1‐10 and ‐GFP11 (1:1 ratio) were infected with SARS‐CoV‐2 for 24 h and stained with anti‐S antibodies and Hoechst. Cell fusion was visualized by the GFP+ area. No syncytia were observed in infected Vero cells.
-
BVero‐GFP1‐10 and ‐GFP11 (1:1 ratio) were transfected with S expression plasmid and cell fusion was visualized by the GFP+ area at 18 h post‐transfection. S expression induced extensive syncytia formation.
-
CS‐expressing 293T GFP11 (Donor cells) were co‐cultured overnight with Vero GFP1‐10 (Acceptor cells). Cell fusion was quantified by measuring the GFP+ area. S‐expressing 293Ts readily fuse with Vero cells.
-
DSARS‐CoV‐2‐infected U2OS ACE2+ GFP1‐10 (Donor cells) were co‐cultured for 8 h with Vero GFP11 (Acceptor cells). Cell fusion was quantified by measuring the GFP+ area. Infected U2OS fuse with Vero cells.
Data information: Scale bar: 50 µm. Representative images of three independent experiments.
Syncytia formation by S‐expressing cells
We further characterized the mechanisms of fusion and its regulation by IFITMs and TMPRSS2. We asked whether S alone was sufficient to trigger fusion by transfecting an expression plasmid in 293‐T cells harboring the GFP‐Split system (Fig 3A). Many large and multinucleated GFP+ cells were detected, when ACE2 was co‐expressed (Fig 3B). S‐mediated cell fusion was significantly decreased when cells were co‐transfected with flag‐tagged IFITM1, 2, or 3 plasmids, compared with a control plasmid (Fig 3B and C). IFITM1 was slightly more inhibitory than IFITM2 and 3 in this system. The transient expression of TMPRSS2 enhanced fusion by 2.5‐fold. Interestingly, when the serine protease was present, the inhibitory effect of IFITMs was no longer visible (Fig 3B and C). The anti‐fusogenic effect of IFITM1 varied with the amount of transfected IFITM1 plasmid, but TMPRSS2 counteracted IFITM1 at all doses (Fig EV6A–C). We next measured the kinetics of S‐mediated cell fusion by live video‐microscopy, monitoring in real‐time the GFP+ area. TMPRSS2 accelerated the appearance of GFP+ cells, indicating that it increases the speed of cell–cell fusion (Movie EV2 and Fig 3D). At 12 h post‐transfection, the syncytia area was already fourfold larger than in the control condition (Fig 3D). A similar kinetic analysis indicated that IFITM1 strongly inhibited fusion, whereas IFITM2 and IFITM3 were less efficient (Fig 3E). In the presence of TMPRSS2, the rapid fusion kinetics were similar with or without IFITMs (Fig 3E).
Figure 3. Impact of TMPRSS2 and IFITMs on the kinetics of fusion by S‐expressing 293T cells.
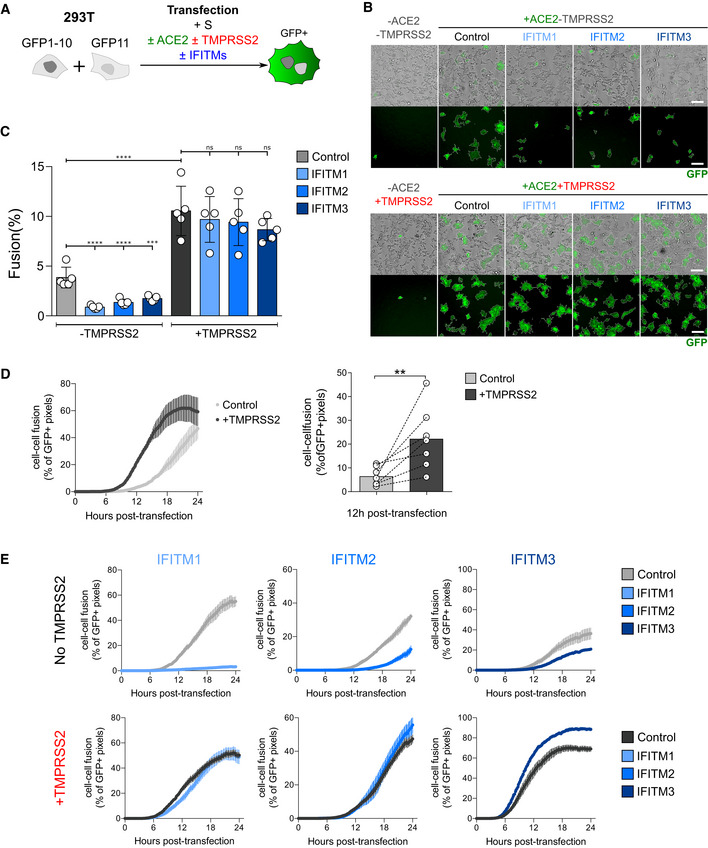
-
A293T‐GFP1‐10 and ‐GFP11 cells (1:1 ratio) were co‐transfected with S, ACE2, TMPRSS2, IFITM, or control plasmids. Cell fusion was quantified by measuring the GFP+ area by high‐content imaging after 18 h. (B and C) or analyzed over time by video microscopy (D and E).
-
BRepresentative images of cell–cell fusion. Scale bar: 100 µm.
-
CQuantification of GFP+ areas. Results are mean ± SD from five independent experiments.
-
DTMPRSS2 accelerates fusion. Cells were monitored by video microscopy, and the GFP area was quantified over time. Left panel: one representative experiment. Results are mean ± SD from three fields per condition. Right panel: Mean ± SD from seven independent experiments (at 12 h post‐transfection).
-
EImpact of TMPRSS2 and IFITMs on the kinetics of fusion by S‐expressing 293T cells. One representative out of three independent experiment is shown.
Data information: Statistical analysis: B, C: One‐way ANOVA, ns: non‐significant, ***P < 0.001, ****P < 0.0001. D: Wilcoxon matched‐pairs signed‐rank test, **P < 0.01.
Figure EV6. Dose‐response analysis of IFITM1 activity and impact of TMPRSS2 on IFITM1, 2, and 3 levels measured by Western blot.
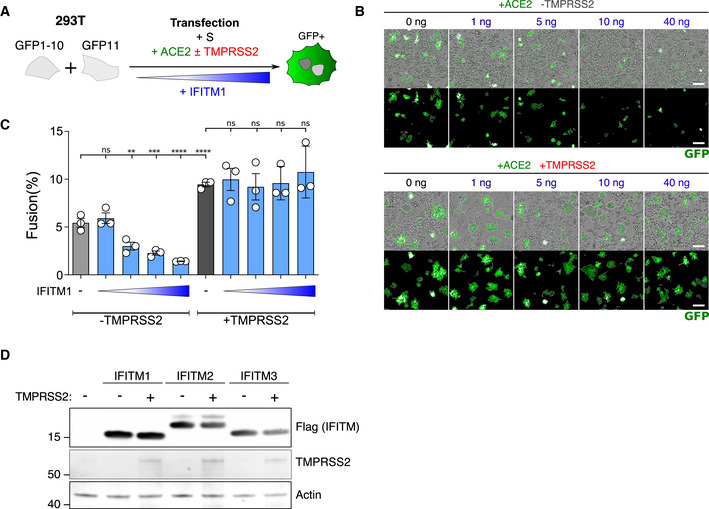
-
A293T‐GFP1‐10 and ‐GFP11 (1:1 ratio) were co‐transfected with S, ACE2, TMPRSS2, and increasing amounts of IFITM1 plasmids. Cell fusion was quantified by measuring the GFP+ area by high‐content imaging at 18 h post‐transfection
-
BRepresentative images of cell–cell fusion, with the indicated amounts of transfected plasmids. Scale bar: 100 µm.
-
CQuantification of GFP+ area. Results are mean ± SD from three independent experiments. Statistical analysis: One‐way ANOVA, ns: non‐significant, **P < 0.01, ***P < 0.001, ****P < 0.0001.
-
D293T cells were co‐transfected with TMPRSS2 and IFITM1, IFITM2, IFITM3, or control plasmids, and analyzed 18 h post‐transfection by Western blot. TMPRSS2 does not cleave or reduce the levels of IFITMs.
We next studied whether IFITMs and TMPRSS2 impact cell–cell fusion by acting on S‐expressing cells (“donor cells”), on ACE2‐expressing cells (“acceptor cells”) or on both. To this end, we used a co‐culture system of 293T‐GFP1‐10 donor cells with 293T‐GFP11 acceptor cells. IFITMs and TMPRSS2 were transfected into either donor or acceptor cells (Fig 4A). In the absence of TMPRSS2, IFITMs were poorly efficient when present in donor cells, but inhibited fusion in acceptor cells. As already observed, IFITM1 was more active than IFITM2 and IFITM3. When TMPRSS2 was present in donor cells, IFITMs lost the weak effect they displayed when they were also in donor cells but retained their ability to inhibit fusion when expressed in acceptor cells (Fig 4A). Finally, when TMPRSS2 was expressed in acceptor cells, inhibition of fusion by IFITM was abolished regardless of their side of expression (Fig 4A). Therefore, IFITMs and TMPRSS2 modulate the efficiency of fusion when present in the same cell as ACE2, rather than in the S‐expressing cell.
Figure 4. Effect of TMPRSS2 and IFITM on S‐ or ACE2‐expressing cells.
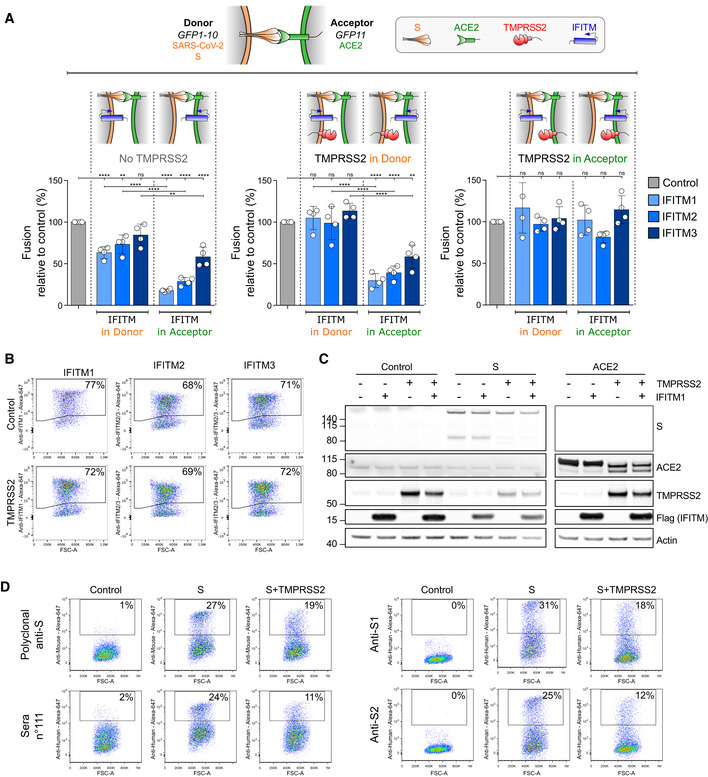
-
AS‐expressing cells (Donor cells) and ACE2‐expressing cells (Acceptor cells) were co‐transfected with TMPRSS2, IFITM, or control plasmids. Cell fusion was quantified by measuring the GFP+ area after 18 h. The indicated combinations were tested. The impact of IFITMs was measured in absence of TMPRSS2 (left panel), in the presence of TMPRSS2 in donor (middle panel) or acceptor cells (right panel). Data are mean ± SD of four independent experiments. Statistical analysis: One‐way ANOVA, ns: non‐significant, **P < 0.01, ****P < 0.0001.
-
B–DImpact of TMPRSS2 on IFITMs, ACE2, and S levels. B. TMPRSS2 does not decrease IFITM amounts measured by flow cytometry. 293T cells were transfected with the indicated IFITM plasmids, with or without TMPRSS2, and analyzed 18 h later. C. Impact of TMPRSS2 on S and ACE2, measured by Western blot. 293T cells were transfected with or without IFITM1, TMPRSS2, S, or ACE2 plasmids, and analyzed 18 h later. D. TMPRSS2 decreases S surface levels, measured by flow cytometry. 293T cells were transfected with S plasmid, with or without TMPRSS2, and analyzed 18 h later. Murine polyclonal anti‐S antibodies, one serum from a convalescent COVID‐19‐infected patient (sera n°111), and two monoclonal antibodies (anti‐S1 and anti‐S2) were tested. Data are representative of three independent experiments.
Impact of TMPRSS2 on S, IFITMs, and ACE2 levels
Our observation that TMPRSS2 counteracts the inhibitory effect of IFITMs prompted us to examine whether it degrades these antiviral proteins. Flow cytometry and Western blot showed that this is not the case, since similar levels of IFITM1, IFITM2, or IFITM3 were detected in the presence or absence of the serine protease (Figs 4B and EV6D). TMPRSS2 decreased S surface levels measured by flow cytometry with antibodies targeting the S1 and S2 domains (Fig 4D), as well as its processing, visualized by the disappearance of a 90 kDa product by Western blot (Fig 4C). In the absence of S, TMPRSS2 also triggered ACE2 processing, a phenomenon which was not inhibited by IFITM1 (Fig 4C). ACE2 cleavage by TMPRSS2 and other cellular proteases was previously reported and proposed to generate a receptor enhancing viral uptake (Haga et al, 2008; Shulla et al, 2011; Heurich et al, 2014). Altogether, our results indicate that TMPRSS2 processes both ACE2 and S, but does not degrade IFITMs.
Discussion
We report here that some SARS‐CoV‐2‐infected cells form large syncytia in culture. This phenomenon occurs in U2OS‐ACE2, 293T‐ACE2, and A549‐ACE2 cells. Naturally permissive Vero cells do not detectably fuse upon infection, but they form syncytia when they encounter infected cells or cells expressing only the Spike protein. Endogenous levels of ACE2 are thus sufficient to trigger cell fusion. Syncytia formation is thus a cell type‐dependent process and likely relies on parameters such as the amount of cellular or viral proteins involved in fusion, or intrinsic biophysical properties of the membranes. S is expressed at the surface of infected cells and is sufficient to generate fusion with neighboring cells. IFITMs inhibit syncytia formation, with IFITM1 being the most efficient. IFITM2 and IFITM3 did not inhibit fusion of infected U2OS‐ACE2 cells but were partly active in 293‐T cells expressing only S. This may be due to the intracellular location of each IFITM, or the high levels of IFITMs generated by transient transfection. Future experiments with IFITM mutants lacking sorting signals or motifs involved in palmitoylation and other post‐translational modifications will help understanding how these proteins impact SARS‐CoV‐2 fusion. Furthermore, recent reports have suggested that IFITMs may either enhance or restrict SARS‐CoV‐2 infection, depending on the experimental system and the cell type (preprint: Bozzo et al, 2020; preprint: Shi et al, 2020). Future investigations into the role endogenous IFITMs and TMPRSS2 conducted with primary cells treated or not with type‐I IFN will provide a more thorough translational understanding of viral‐induced syncytia formation.
We further show that TMPRSS2 accelerates SARS‐CoV‐2‐mediated cell–cell fusion. TMPRSS2, through its serine protease activity, is known to cleave both S and ACE2. TMPRSS2 enhances infectivity and fusogenic activity of different coronaviruses, including HCoV‐229E, MERS, and SARS‐CoV‐1 (Matsuyama et al, 2010; Glowacka et al, 2011; Shulla et al, 2011; Bertram et al, 2013; Shirato et al, 2013; Reinke et al, 2017; Kleine‐Weber et al, 2018). With SARS‐CoV‐2, a first cleavage of S by furin at the S1/S2 site is required for subsequent cleavage by TMPRSS2 at S2’ site (Hoffmann et al, 2020a). ACE2 cleavage by TMPRSS2 at a polybasic site generates a soluble fragment of the receptor (Haga et al, 2008; Shulla et al, 2011; Heurich et al, 2014). It has been proposed that this dual action on S and ACE2 facilitates virion uptake by target cells, through mechanisms that are not fully understood. The enhancement of syncytia formation described here with SARS‐CoV‐2 suggests that TMPRSS2 facilitates a step different from virion uptake. It will be worth determining which structural changes are triggered by TMPRS22 on the viral protein and its receptor, and how these changes may affect the relative affinities of the two proteins and the dynamics of the fusion process.
That TMPRSS2 counteracts the inhibitory activity of IFITMs on SARS‐CoV‐2‐mediated syncytia formation raises interesting questions. This observation is not unprecedented, as two other coronaviruses, HCoV‐229E and bat SARS‐like WIV1, employ proteolytic pathways to evade IFITM restriction (Bertram et al, 2013; Zheng et al, 2020). IFITM proteins modify the rigidity or the lipid content of cellular membranes to prevent fusion. How this biophysical constraint is overcome by TMPRSS2 will require further investigations. For instance, it will be worth assessing the motility of ACE2 and S in membranes, before and during syncytia formation, and the impact of IFITM and TMPRSS2 on these processes. Determining the role of other proteins known to inhibit virion fusion, such as Ly6E and CH25H, and other proteases such as furin will provide a global overview of the mechanisms of cell–cell fusion.
An analysis of 41 post‐mortem samples from individuals who died of COVID‐19 demonstrated extensive alveolar damage and lung vasculature thrombosis (preprint: Giacca et al, 2020). In situ hybridization showed the presence of large multinucleated pneumocytes expressing viral RNA and proteins in half of the patients (preprint: Giacca et al, 2020). Syncytia may thus be considered as a frequent feature of severe COVID‐19. It will be worth determining whether the syncytia are also generated in mild cases and whether severe or critical cases are linked to polymorphisms in IFITMs, as already reported for Flu (Shi et al, 2017; Zani & Yount, 2018) and in other IFN‐related genes. Our results pave the way for the future assessment of the role played by syncytia in viral persistence and dissemination, the destruction of alveolar architecture, and immune or inflammatory responses.
Materials and Methods
Plasmids
pQCXIP‐Empty control plasmid and pQCXIP‐IFITM1‐N‐FLAG, pQCXIP‐IFITM2‐N‐FLAG, and pQCXIP‐IFITM3‐N‐FLAG plasmids were described (Buchrieser et al, 2019). The IFITM plasmids used throughout this study, either in transfections or in the generation of stably expressing cell lines, all contain a FLAG tag on the N terminus. pQCXIP‐BSR‐GFP11 and pQCXIP‐GFP1‐10 were from Yutaka Hata (Kodaka et al, 2015; Addgene plasmid #68716; http://n2t.net/addgene:68716; RRID:Addgene_68716 and Addgene plasmid #68715; http://n2t.net/addgene:68715; RRID:Addgene_68715). pcDNA3.1‐hACE2 was from Hyeryun Choe (Li et al, 2003) (Addgene plasmid # 1786; http://n2t.net/addgene:1786; RRID:Addgene_1786). pCSDest‐TMPRSS2 was from RogerReeves (Edie et al, 2018) (Addgene plasmid # 53887; http://n2t.net/addgene:53887; RRID:Addgene_53887). pLenti6‐H2B‐mCherry was from Torsten Wittmann (Pemble et al, 2017) (Addgene plasmid # 89766; http://n2t.net/addgene:89766; RRID:Addgene_89766). pLenti6‐attB‐hACE2‐BSD was generated by cloning hACE2 from pcDNA3.1‐hACE2 into the pLenti6‐H2B‐mCherry. Briefly, hACE2 was PCR amplified adding SpeI, XhoI, and attB sites using forward primer (5’‐ TCC CTC ACT AGT ACA AGT TTG TAC AAA AAA GCA GGC TGC CAC CAT GTC AAG CTC TTC CTG GCT C ‐3’) and reverse primer (5’‐ AAA AAA CTC GAG ACC ACT TTG TAC AAG AAA GCT GGG TTT AAG CGG GCG CCA CCT ‐3’) and cloned into pLenti6‐H2B‐mCherry using XhoI and SpeI sites. phCMV‐SARS‐CoV2‐S was previously described (Grzelak et al, 2020). pLV‐EF1α‐TMPRSS2‐IRES‐Hygro was generated by gateway cloning of pCSDest‐TMPRSS2 into pLV‐EF1α‐IRES‐Hygro‐DEST.
Cells
293T cells, platinum‐E retroviral packaging cell line, A549, Caco2, and U2OS cells, and derivatives were cultured in DMEM with 10% Fetal Calf Serum (FCS) and 1% Penicillin/Streptomycin (PS). Vero cells were maintained in DMEM with 5% FCS and 1% PS. Cell lines transduced with pQCXIP pLenti6 or pLV‐IRES‐Hygro‐derived vectors were grown with 1 μg/ml puromycin, 10 μg/ml blasticidin, or 100 μg/ml hygromycin B, respectively (InvivoGen). All cells were either purchased form ATCC or gifts from members of the Institut Pasteur.
Lentiviral and Retroviral vectors
For lentiviral production, 293T cells were co‐transfected with pLenti6 or pLV‐derived vectors, packaging plasmid R8‐2, and VSV‐G plasmid as previously described (Buchrieser et al, 2019). For murine retroviral vector production, the platinum‐E (Cell Biolabs) retroviral packaging cell line was transfected with pQCXIP‐derived plasmids. Vector containing supernatants were harvested at 36, 48, and 72 h and ultracentrifuged 1 h at 4°C at 22,000 g.
Generation of stable cell lines
For lentiviral or retroviral transduction, 2 × 104 cells were resuspended in 150 µl of medium containing 5–25 μl of ultracentrifuged lentiviral or retroviral vectors. Cells were agitated 30 s every 5 min for 2 h 30 min at 37°C in a Thermomixer. For cell lines co‐expressing IFITMs and other proteins, vectors expressing the other proteins were transduced before IFITM vectors to avoid restriction of vector transduction by IFITMs. All cell lines were routinely tested for mycoplasma and found negative.
GFP‐Split fusion assay
For fusion assays with S‐expressing cells, 293T‐GFP1‐10 and 293T‐GFP11 cells (6 × 104 cells/well cells mixed at a 1:1 ratio) in 96 well plates (μClear, #655090) were transfected in suspension using Lipofectamine 2000 (Thermo) with 100 ng of DNA. 10 ng of phCMV‐SARS‐CoV2‐S, 25 ng of pCDNA3.1‐hACE2, 25 ng of pCSDest‐TMPRSS2, and 40 ng of pQCXIP‐Empty or pQCXIP‐IFITM‐N‐FLAG were used and adjusted to 100 ng DNA with pQCXIP‐Empty. 18 h post‐transfection, 21 images, covering 90% of the well surface, were acquired per well on an Opera Phenix High‐Content Screening System (PerkinElmer) and the cell confluence area and GFP area was quantified on Harmony High‐Content Imaging and Analysis Software. For “donor/acceptor” experiments, 3 × 104 293T GFP1‐10 and GFP11 expressing cells were separately transfected with 50 ng of DNA in suspension at 37°C shaking 900 rpm for 20 min using Lipofectamine 2000 (Thermo). For donor cells, 293T‐GFP1‐10 cells were transfected with 10 ng of phCMV‐SARS‐CoV2‐S, ±10 ng pCSDest‐TMPRSS2, and ±20 ng pQCXIP‐IFITM‐N‐FLAG and adjusted to 50 ng with pQCXIP‐Empty. For acceptor cells, 293T‐GFP11 cells were transfected with 10 ng of pCDNA3.1‐hACE2, ±10 ng pCSDest‐TMPRSS2, and ±20 ng pQCXIP‐IFITM‐N‐FLAG and adjusted to 50 ng with pQCXIP‐Empty. After transfection, cells were washed and resuspended in DMEM 10% FBS, mixed at a 1:1 ratio in different combinations, plated at 6 × 104 cells per well in a 96‐well plate and imaged 18 h post‐transfection. For live imaging, 2.5 × 105 GFP1‐10 and GFP11‐expressing 293T cells, mixed at a 1:1 ratio, were transfected in suspension at 37°C, with a shaking at 900 rpm for 20 min using Lipofectamine 2000 (Thermo) and 100 ng of DNA (10 ng of phCMV‐SARS‐CoV2‐S, 20 ng of pCDNA3.1‐hACE2, 20 ng of pCSDest‐TMPRSS2, and 40 ng pQCXIP‐IFITM‐N‐FLAG, adjusted to 100 ng with pQCXIP‐Empty). Cells were washed and seeded into a µ‐Dish 35 mm Quad dish (ibidi—#80416) with 2.5 × 105 cells per quadrant. Transmission and fluorescence images were taken at 37°C every 10 min up to 24 h using a Nikon BioStation IMQ, with three fields for each condition. The GFP area was quantified on ImageJ.
Antibodies
Mouse monoclonal antibodies (mAb): IFITM1 (#60074‐1‐Ig, Proteintech) 1:250 for FACS, and IF; IFITM2/3 (#66081‐1‐Ig, Proteintech) 1:250 for FACS, and IF; ACE2 (AC18F) (#AG‐20A‐0032‐C100—adipogen). Rabbit polyclonal antibodies: FLAG‐Tag DYKDDDDK (D6W5B) (#14793, Cell Signaling) 1:800 for FACS. TMPRSS2 (#HPA035787—Atlasantibodies) 1:500. ZO‐1 (#40‐2200—Thermo Fisher) 1:100. Goat polyclonal antibodies: ACE2 (AF933—R&D). SARS_Ssd3 702, SARS_Ssd3 –369, SARS_Ssd3 293, and Ascite Sso14 200705 were kindly gifted by Nicolas Escriou. SARS_Ssd3 702 antibody was used at 0.5 μg/ml for FACS and IF. Human Mab: Anti‐SARS‐CoV2 monoclonal antibodies 48 and 71 recognize two domains of the S1 protein (Planchais et al, manuscript in preparation). Secondary antibodies coupled with Alexa Fluor 488 or 647 (Invitrogen) were used at 1:500 for FACS and IF.
Flow cytometry
For intracellular staining, cells were fixed in 4% PFA for 15 to 30 min at RT and staining was performed in PBS, 1% BSA, 0.05% sodium azide, and 0.05% Saponin. Cells were incubated with primary antibodies and then with secondary antibodies for 30 min at RT. Surface staining was performed before fixation, in PBS 1% BSA. Cells were incubated with primary antibodies and then with secondary antibodies for 30 min at RT. Cells were fixed for 15 min in 4% PFA. Cells were acquired on an Attune NxT Flow Cytometer (Thermo Fisher) and data analyzed with FlowJo software.
U2OS viral‐mediated cell–cell fusion, video microscopy, and immunofluorescence
U2OS‐ACE2 GFP1‐10 or GFP11 stably expressing TMPRSS2, IFITM1, 2, 3, or control cells were mixed (1:1 ratio) and plated at 8 × 103 cells per well in a 96‐well plate (μClear, #655090), 24 h before infection. Cells were then infected with different MOI of SARS‐CoV‐2 in 150 µl of media. 18 h post‐infection, media was removed, and cells were fixed in 4% PFA for 30 min at room temperature, washed, and resuspended in PBS. For fusion and viability quantification, cells were stained for 10 min with Hoechst 33342 (1:5,000) before being imaged. For S and IFITM analysis, cells were washed twice PBS and stained with primary antibody for 45 min in PBS, 1% BSA, 0.05% sodium azide, and 0.05% Saponin in the wells. Cells were washed twice with PBS, 0.05% Saponin and stained with secondary antibody for 30 min at RT in PBS, 1% BSA, 0.05% sodium azide, 0.05% Saponin, and Hoechst 33342 (1:10,000) and washed with PBS. Cells were acquired on an Opera Phenix High Content Screening System (PerkinElmer) and analyzed on Harmony High‐Content Imaging and Analysis Software. For live imaging, U2OS‐ACE2 GFP1‐10 and GFP11 were mixed at a 1:1 ratio and plated at 4 × 104 cells per quadrant in a µ‐Dish 35 mm Quad dish (ibidi—#80416). Cells were infected the next day with SARS‐CoV‐2 (MOI 0.2) in media containing propidium iodide. Transmission and fluorescence images were taken at 37°C every 10 min, up to 48 h, using a Nikon BioStation IMQ, with three fields for each condition.
Western blot
Cells were lysed in TXNE buffer (1% Triton X‐100, 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, protease inhibitors) for 30 min on ice. Equal amounts (20–50 μg) of cell lysates were analyzed by Western blot. The following antibodies were diluted in WB‐buffer (PBS, 1% BSA, 0.05% Tween, 0.01% Na Azide): goat anti‐human ACE2 (R&D cat#AF933, 1:2,000), mouse anti‐human ACE2 (Adipogen AC18F cat #AG‐20A0032‐C100, 1:1,000), rabbit anti‐human TMPRSS2 (Atlas antibodies cat# HPA035787, 1:1,000), mouse anti‐Flag tag (Sigma cat# F1804, 1:1,000), rabbit anti‐human actin (Sigma cat#A2066, 1:2,000), and mouse ascite anti‐SARS S, (1:1,000; Siu et al, 2008). Specie‐specific secondary DyLight‐coupled antibodies were used (diluted 1:10,000 in WB‐buffer) and proteins revealed using a Licor Imager. Images were quantified and processed using Image Studio Lite software.
Virus
The strain BetaCoV/France/IDF0372/2020 was supplied by the National Reference Centre for Respiratory Viruses hosted by Institut Pasteur (Paris, France) and headed by Pr. S. van der Werf. The human sample from which the strain was isolated has been provided by Dr. X. Lescure and Pr. Y. Yazdanpanah from the Bichat Hospital, Paris, France. The viral strain was supplied through the European Virus Archive goes Global (Evag) platform, a project that has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 653316. Titration of viral stocks was performed on Vero E6, with a limiting dilution technique allowing a calculation of DCP50.
Statistical analysis
Flow cytometry data were analyzed with FlowJo v10 software (TriStar). Calculations were performed using Excel 365 (Microsoft). Figures were drawn on Prism 8 (GraphPad Software). Statistical analysis was conducted using GraphPad Prism 8. Statistical significance between different groups was calculated using the tests indicated in each figure legend.
Author contributions
Experimental strategy design, experiments: JB, JD, MH, BM, DP, MMR, FP, FGB, NC, TB, OS; Vital materials, expert advice: HM, CP, NE, SVDW; Manuscript writing: JB, OS; Manuscript editing: MMR; Manuscript reviewing, approval: All authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Acknowledgements
We thank members of the Virus and Immunity Unit for discussions and help, Mauro Giacca for discussion and sharing unpublished results, Nathalie Aulner and the UtechS Photonic BioImaging (UPBI) core facility (Institut Pasteur), a member of the France BioImaging network, for image acquisition and analysis, and Nicolas Escriou for the kind gift of anti‐SARS‐CoV‐2 antibodies. OS laboratory is funded by Institut Pasteur, ANRS, the Vaccine Research Institute (ANR‐10‐LABX‐77), Labex IBEID (ANR‐10‐LABX‐62‐IBEID), “TIMTAMDEN” ANR‐14‐CE14‐0029, “CHIKV‐Viro‐Immuno” ANR‐14‐CE14‐0015‐01 and the Gilead HIV cure program, ANR/FRM Flash Covid PROTEO‐SARS‐CoV‐2 and IDISCOVR. Work in UPBI is funded by grant ANR‐10‐INSB‐04‐01 and Région Ile‐de‐France program DIM1‐Health. HM laboratory is funded by the Institut Pasteur, the Milieu Intérieur Program (ANR‐10‐LABX‐69‐01), the INSERM, REACTing, and EU (RECOVER) grants. SVDW laboratory is funded by Institut Pasteur, CNRS, Université de Paris, Santé publique France, Labex IBEID (ANR‐10‐LABX‐62‐IBEID), REACTing, and EU (RECOVER) grant. M.M.R is supported by the Pasteur‐Paris University (PPU) International Doctoral Program. The funders of this study had no role in study design, data collection, analysis and interpretation, or writing of the article.
The EMBO Journal (2020) 39: e106267.
Data availability
This study includes no data deposited in external repositories.
References
- Abdel Motal UM, Zhou X, Joki A, Siddiqi AR, Srinivasa BR, Stenvall K, Dahmen J, Jondal M (1993) Major Histocompatibility Complex class I‐binding peptides are recycled to the cell surface after internalization. Eur J Immunol 23: 3224–3229 [DOI] [PubMed] [Google Scholar]
- Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann‐Winkler H et al (2013) TMPRSS2 activates the human coronavirus 229E for cathepsin‐independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87: 6150–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D et al (2020) Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell 181: 1036–1045.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, Blecher‐Gonen R, Cohen M, Medaglia C, Li H et al (2020) Host‐viral infection maps reveal signatures of severe COVID‐19 patients. Cell 181: 1475–1488 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzo C, Nchioua R, Volcic M, Wettstein L, Weil T, Krüger J, Heller S, Conzelmann C, Müller J, Gross R et al (2020) IFITM proteins promote SARS‐CoV‐2 infection of human lung cells. bioRxiv 10.1101/2020.08.18.255935 [PREPRINT] [DOI]
- Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, Clementi N, De Santis M, Mancini N, Granucci F et al (2020) Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369: 706–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J, Degrelle SA, Couderc T, Nevers Q, Disson O, Manet C, Donahue DA, Porrot F, Hillion KH, Perthame E et al (2019) IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise. Science 365: 176–180 [DOI] [PubMed] [Google Scholar]
- Chan JF, Chan KH, Choi GK, To KK, Tse H, Cai JP, Yeung ML, Cheng VC, Chen H, Che XY et al (2013) Differential cell line susceptibility to the emerging novel human betacoronavirus 2c EMC/2012: implications for disease pathogenesis and clinical manifestation. J Infect Dis 207: 1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton Alex A, Bruel T, Porrot F, Mallet A, Sachse M, Euvrard M, Liang C, Casartelli N, Schwartz O (2014) IFITM proteins incorporated into HIV‐1 virions impair viral fusion and spread. Cell Host Microbe 16: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edie S, Zaghloul NA, Leitch CC, Klinedinst DK, Lebron J, Thole JF, McCallion AS, Katsanis N, Reeves RH (2018) Survey of human chromosome 21 gene expression effects on early development in Danio rerio . G3 (Bethesda) 8: 2215–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, Reid AH, Selbs E, McEvoy CPL, Hayden CDL, Fukuoka J et al (2003) Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol 34: 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt EC, Yu L, Goldsmith CS, Welsh S, Cheng A, Yount B, Liu W, Frieman MB, Buchholz UJ, Screaton GR et al (2010) The open reading frame 3a protein of severe acute respiratory syndrome‐associated coronavirus promotes membrane rearrangement and cell death. J Virol 84: 1097–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacca M, Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, Secco I, Volpe MC, Colliva A et al(2020) Persistence of viral RNA, widespread thrombosis and abnormal cellular syncytia are hallmarks of COVID‐19 lung pathology. medRxiv 10.1101/2020.06.22.20136358 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K et al (2011) Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol 85: 4122–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorshkov K, Chen CZ, Bostwick R, Rasmussen L, Xu M, Pradhan M, Tran BN, Zhu W, Shamim K, Huang W et al(2020) The SARS‐CoV‐2 cytopathic effect is blocked with autophagy modulators. bioRxiv 10.1101/2020.05.16.091520 [PREPRINT] [DOI]
- Grzelak L, Temmam S, Planchais C, Demeret C, Tondeur L, Huon C, Guivel‐Benhassine F, Staropoli I, Chazal M, Dufloo J et al (2020) A comparison of four serological assays for detecting anti‐SARS‐CoV‐2 antibodies in human serum samples from different populations. Sci Transl Med 12: eabc3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Pere H, Charbit B, Bondet V, Chenevier‐Gobeaux C, Breillat P et al (2020) Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid‐19 patients. medRxiv 10.1101/2020.04.19.20068015 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Haga S, Yamamoto N, Nakai‐Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y (2008) Modulation of TNF‐α‐converting enzyme by the spike protein of SARS‐CoV and ACE2 induces TNF‐α production and facilitates viral entry. Proc Natl Acad Sci USA 105: 7809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurich A, Hofmann‐Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S (2014) TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88: 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Pöhlmann S (2020a) A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell 78: 779–784.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A et al (2020b) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, Kato T, Lee RE, Yount BL, Mascenik TM et al (2020) SARS‐CoV‐2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 182: 429–446.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L et al (2011) Distinct patterns of IFITM‐mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 7: e1001258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine‐Weber H, Elzayat MT, Hoffmann M, Pohlmann S (2018) Functional analysis of potential cleavage sites in the MERS‐coronavirus spike protein. Sci Rep 8: 16597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaka M, Yang Z, Nakagawa K, Maruyama J, Xu X, Sarkar A, Ichimura A, Nasu Y, Ozawa T, Iwasa H et al (2015) A new cell‐based assay to evaluate myogenesis in mouse myoblast C2C12 cells. Exp Cell Res 336: 171–181 [DOI] [PubMed] [Google Scholar]
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L et al (2020) Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature 581: 215–220 [DOI] [PubMed] [Google Scholar]
- Letko M, Marzi A, Munster V (2020) Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol 5: 562–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC et al (2003) Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, Hartmann R, Wack A (2020) Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369: 712–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F (2010) Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84: 12658–12664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, Nagata N, Sekizuka T, Katoh H, Kato F et al (2020) Enhanced isolation of SARS‐CoV‐2 by TMPRSS2‐expressing cells. Proc Natl Acad Sci USA 117: 7001–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando NS, Dalebout TJ, Zevenhoven‐Dobbe JC, Limpens RWAL, van der Meer Y, Caly L, Druce J, de Vries JJC, Kikkert M, Bárcena M et al (2020) SARS‐coronavirus‐2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology. J Gen Virol 101: 925–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong EZ, Chan YFZ, Leong WY, Lee NMY, Kalimuddin S, Haja Mohideen SM, Chan KS, Tan AT, Bertoletti A, Ooi EE et al (2020) A dynamic immune response shapes COVID‐19 progression. Cell Host Microbe 27: 879–882 e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A, Iwasaki A (2020) Type I and Type III interferons – induction, signaling, evasion, and application to combat COVID‐19. Cell Host Microbe 27: 870–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemble H, Kumar P, van Haren J, Wittmann T (2017) GSK3‐mediated CLASP2 phosphorylation modulates kinetochore dynamics. J Cell Sci 130: 1404–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaender S, Mar KB, Michailidis E, Kratzel A, Boys IN, V'Kovski P, Fan W, Kelly JN, Hirt D, Ebert N et al (2020) LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat Microbiol 5: 1330–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Dominguez SR, Holmes KV (2013) Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS‐CoV) in virus entry and syncytia formation. PLoS One 8: e76469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke LM, Spiegel M, Plegge T, Hartleib A, Nehlmeier I, Gierer S, Hoffmann M, Hofmann‐Winkler H, Winkler M, Pöhlmann S (2017) Different residues in the SARS‐CoV spike protein determine cleavage and activation by the host cell protease TMPRSS2. PLoS One 12: e0179177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, Han Y, Zhang X‐Y, Zhou W, Qiu Y et al (2020) The ORF3a protein of SARS‐CoV‐2 induces apoptosis in cells. Cell Mol Immunol 17: 881–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Kenney AD, Kudryashova E, Zhang L, Hall‐Stoodley L, Robinson RT, Kudryashov DS, Compton AA, Yount JS (2020) Opposing activities of IFITM proteins in SARS‐CoV‐2 infection. bioRxiv 10.1101/2020.08.11.246678 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Shi G, Schwartz O, Compton AA (2017) More than meets the I: the diverse antiviral and cellular functions of interferon‐induced transmembrane proteins. Retrovirology 14: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K, Kawase M, Matsuyama S (2013) Middle east respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87: 12552–12561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulla A, Heald‐Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T (2011) A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol 85: 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu YL, Teoh KT, Lo J, Chan CM, Kien F, Escriou N, Tsao SW, Nicholls JM, Altmeyer R, Peiris JSM et al (2008) The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus‐like particles. J Virol 82: 11318–11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao K, Mahanty S (2020) Respiratory virus infections: understanding COVID‐19. Immunity 52: 905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Comish P, Kang R (2020) The hallmarks of COVID‐19 disease. PLoS Pathog 16: e1008536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY (2020) Pulmonary pathology of early‐phase 2019 novel coronavirus (COVID‐19) pneumonia in two patients with lung cancer. J Thorac Oncol 15: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181: 281–292.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen K‐Y et al (2020) Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell 181: 894–904.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Griffin LM, Little AS, Huang IC, Farzan M, Pyeon D (2014) The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS One 9: e96579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrensch F, Winkler M, Pohlmann S (2014) IFITM proteins inhibit entry driven by the MERS‐coronavirus spike protein: evidence for cholesterol‐independent mechanisms. Viruses 6: 3683–3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L et al (2020) Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med 8: 420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang R, Case JB, Gomez Castro MF, Liu Z, Zeng Q, Zhao H, Son J, Rothlauf PW, Hou G, Bose S, Wang X, Vahey MD, Kirchhausen T, Fremont DH, Diamond MS, Whelan SPJ, Ding S (2020) Cholesterol 25‐hydroxylase suppresses SARS‐CoV‐2 replication by blocking membrane fusion. bioRxiv 10.1101/2020.06.08.141077 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Zani A, Yount JS (2018) Antiviral protection by IFITM3 in vivo. Curr Clin Microbiol Rep 5: 229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, Guo JT (2014) Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci USA 111: 6756–6761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sehgal M, Hou Z, Cheng J, Shu S, Wu S, Guo F, Le Marchand SJ, Lin H, Chang J et al (2018) Identification of residues controlling restriction versus enhancing activities of IFITM proteins on entry of human coronaviruses. J Virol 92: e01535‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zheng S, Chen D, Zheng M, Li X, Li G, Lin H, Chang J, Zeng H, Guo JT (2020) LY6E restricts entry of human coronaviruses, including currently pandemic SARS‐CoV‐2. J Virol 94: e00562‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Zhao X, Zheng S, Chen D, Du P, Li X, Jiang D, Guo J‐T, Zeng H, Lin H (2020) Bat SARS‐Like WIV1 coronavirus uses the ACE2 of multiple animal species as receptor and evades IFITM3 restriction via TMPRSS2 activation of membrane fusion. Emerg Microbes Infect 9: 1567–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, Guo L, Yang J, Wang C, Jiang S et al (2020) Heightened Innate Immune Responses in the Respiratory Tract of COVID‐19 Patients. Cell Host Microbe 27: 883–890 e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Data Availability Statement
This study includes no data deposited in external repositories.