

Summary
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is a novel human respiratory viral infection that has rapidly progressed into a pandemic, causing significant morbidity and mortality. Blood clotting disorders and acute respiratory failure have surfaced as the major complications among the severe cases of coronavirus disease 2019 (COVID‐19) caused by SARS‐CoV‐2 infection. Remarkably, more than 70% of deaths related to COVID‐19 are attributed to clotting‐associated complications such as pulmonary embolism, strokes and multi‐organ failure. These vascular complications have been confirmed by autopsy. This study summarizes the current understanding and explains the possible mechanisms of the blood clotting disorder, emphasizing the role of (1) hypoxia‐related activation of coagulation factors like tissue factor, a significant player in triggering coagulation cascade, (2) cytokine storm and activation of neutrophils and the release of neutrophil extracellular traps and (3) immobility and ICU related risk factors.
Keywords: COVID‐19, cytokine storm, IL‐6, pulmonary embolism, SARS‐CoV‐2, thrombosis
Abbreviations
- ARDS
acute respiratory distress syndrome
- COVID‐19
coronavirus disease 2019
- DHF
dengue haemorrhagic fever
- DIC
disseminated intravascular coagulation
- Egr‐1
early growth response‐1
- HIF1
hypoxia inducible factor 1
- IL
Interleukin
- NET
neutrophil extracellular traps
- PE
pulmonary embolism
- PS
protein s
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- TF
tissue factor
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is a newly emerged positive sense RNA virus belonging to the family of betacoronaviruses. 1 , 2 Members of this family of coronaviruses have crossed the species barrier, adapted to humans and get transmitted effectively from person to person through the respiratory route. So far, humankind has witnessed seven different human coronaviruses with varying incubation times, degrees of transmissibility, and disease severity, which are ordered by mortality rate as MERS‐CoV > SARS‐CoV > SARS‐CoV‐2 > HKU1 ≃ NL63 ≃ OC43 ≃ 229E. 3 , 4 , 5 , 6 Among these, SARS‐CoV‐2 is unique with a relatively prolonged incubation time. 2 In addition to the acute respiratory failure associated with this virus, disturbances in haemostatic balance have emerged as a key issue in moderately and severely ill patients. These disturbances can result in hypercoagulability disseminated intravascular coagulation (DIC), which can contribute to organ failure, stroke, and heart and kidney complications. 7 , 8 , 9 , 10 Here, we briefly review and outline the current knowledge on the progression of COVID‐19, and how multifactorial pathologies and molecular responses influence the coagulation system and the fibrinolytic system during COVID‐19.
2. SARS‐CoV‐2 DISEASE PROGRESSION
Typically, SARS‐CoV‐2 infection runs a course of illness that is over by 20 days post infection. Initial infection can manifest a range of clinical symptoms, including dry cough, sore throat, fever, malaise, myalgias, gastrointestinal symptoms such as anorexia, nausea and diarrhoea. 11 , 12 Some patients also present a temporary loss of taste and smell. 13 If the infection progresses, by the end of second week and early third week, patients exhibit shortness of breath or dyspnoea, a range of haematological irregularities such as lymphopenia and neutrophilia, coagulation abnormalities such as pulmonary embolism (PE), blood thickening, and strokes and, rarely, neurological symptoms. 14 , 15
Patients with severe disease can develop a condition called “cytokine storm”, in which cytokines, released by lymphocytes, monocytes and alveolar macrophages that encounter virus, contribute to damaging inflammatory responses. Damage to the liver and other virus induced factors, indicated by an increase in D‐dimers, combined with the cytokine storm, leads to altered blood coagulation factors and DIC. The terminal stages of SARS‐CoV‐2 infection in patients that die often include acute respiratory distress syndrome (ARDS), stroke, myocardial injury, and multiorgan function damage. 15
3. HYPERCOAGULABILITY AND CLOTTING IN SARS‐CoV‐2
Patients with COVID‐19 present with evidence, most commonly in the form of elevated levels of D‐dimer, of activation of the coagulation system. More than 70% of the deaths related to COVID‐19 are associated with deregulation of the mechanisms that control blood clotting. Blood thickening and clotting are important to prevent excessive loss of blood due to injury. In infection, localized clotting or systemic clotting are part of the innate immune response to limit the spread of the pathogen. 16 However, this clotting response might be associated with harmful effects. When released into the blood stream, a blood clot or thrombus can block the arteries supplying oxygenated blood, resulting in an embolism and death of the oxygen‐starved tissue. 17 When coagulation is insufficiently controlled, DIC may evolve, resulting in a clinical syndrome that involves both widespread microvascular thrombosis (referred to as microthrombosis) from excessive clotting and enhanced bleeding from depletion of clotting factors. 18
3.1. Hypoxia‐induced coagulopathy in SARS‐CoV‐2 infection
Alveoli, small thin‐walled bulb‐like structures, are the structural and functional units of the lungs where blood gets oxygenated through a very thin interstitial space between the capillary and alveoli. The oxygenated blood returns to the heart, where it is pumped to other parts of the body. In ARDS due to SARS‐CoV‐2 infection or other lung infection or injury, the lungs become inflamed and fluid fills the interstitial space, the capillaries become leaky, and the alveoli fill with proteinaceous liquid that prevents oxygen exchange (Figure 1). The resulting hypoxic condition necessitates artificial respiration or assisted breathing. The hypoxic environment reduces the blood anti‐coagulation and activates pro‐coagulation factors that may promote hypoxia‐induced thrombosis. 19
FIGURE 1.

Inflammation and fluid accumulation in alveoli due to severe acute respiratory syndrome coronavirus 2 infection
Animal studies showed that, under hypoxic conditions such as that caused by infection, tissue factor (TF) is produced by the endothelial and subendothelial smooth muscle cells of the vasculature and leukocytes of the lungs. 20 , 21 , 22 Early growth response‐1 (Egr‐1), a cellular mediator, stimulates the transcription of the gene encoding TF via vascular endothelial growth factor, initiating the local procoagulant response. 23 Homozygous Egr‐1–null mutant mice placed in a hypoxic environment show no change in TF abundance in the lungs. 24 TF initiates the clotting process through thrombin formation. Thrombin is a serine protease that catalyses coagulation‐related reactions and converts soluble fibrinogen into insoluble fibrin. 25
Hypoxia also reduces the abundance of protein S (PS), a natural anticoagulant produced primarily in the liver. 26 The amount of PS is inversely correlated with the amount of hypoxia inducible factor 1 (HIF1), a transcriptional regulator stabilized under hypoxic conditions. This inverse relationship establishes a molecular link between hypoxia and thrombosis. Cells respond to hypoxia through HIF1, a dimeric transcription factor composed of HIF1α and HIF1β. In the cytoplasm, HIF1α is continuously degraded in an O2‐dependent manner. 27 Patients deficient in PS, with the Factor V Leiden mutation, have reduced endogenous antithrombotic activity and are vulnerable to enhanced fibrin deposition at hypoxemic sites. 28 Thus, we hypothesize that the COVID‐19–induced hypoxia may cause a PS deficiency, which elevates thrombotic risk.
In severe cases of COVID‐19, D‐dimer, a small protein fragment that is a fibrin degradation product of the clot‐dissolving process, is significantly increased, and this protein is a reliable marker of disease severity. 29 As a sensitive marker of coagulation and fibrinolysis, D‐dimer abundance is also useful in diagnosing deep venous thrombosis or PE. 30 D‐dimer abundance also displays a positive correlation with liver dysfunction. By the third week of infection in patients with severe COVID‐19, hypercoagulability issues occur. Autopsy examinations of deceased patients revealed blood clots in the liver, kidney, heart and lungs. D‐dimer is significantly increased and positively correlates with disease severity in patients infected with various other hemorrhagic viral infections, such as Ebola, Hantavirus hemorrhagic fever with renal syndrome and Dengue Hemorrhagic Fever (DHF). 30 , 31 , 32
3.2. An exacerbated immune response and neutrophil‐derived extracellular traps in lungs during SARS‐CoV‐2 infection
Severe illness and disease complications present in the later stage of SARS‐CoV‐2 infection. In this later stage, use of an interleukin 6 (IL6) inhibitor (toculizumab) or a recombinant human IL1 receptor antagonist (anakinra) can reduce illness, indicating that collateral organ damage may result from an overactive immune response rather than from direct induction of tissue damage by the virus. 33 , 34
Pro‐ and anti‐inflammatory cytokines and chemokines, comprising IL1β, IL2, IL6, IL7, IL8, IL10, IL17, interferon γ (IFNγ), IFNγ‐inducible protein 10, monocyte chemoattractant protein 1, granulocyte‐colony stimulating factor, macrophage inflammatory protein 1α and tumour necrosis factor‐α in the plasma, are elevated during SARS‐CoV‐2 infection. 12 , 35 , 36 , 37 This increase in cytokines and chemokines could play a major role in the altered blood coagulation process and severe disease pathology.
The response of immune cells may also contribute to thrombotic risk. 17 , 18 In infected lung tissue, alveolar macrophages and resident immune cells attract neutrophils and other immunomodulatory cells. Neutrophils are an important part of the innate immune response to heal damaged tissues and resolve infections. These cells clear pathogens by producing a burst of reactive oxygen species (referred to as a respiratory burst) by phagocytosis, and by the formation of neutrophil extracellular traps (NETs), which are web‐like structures of DNA and proteins expelled from the neutrophils (Figure 2). NETs trap pathogens. 38 , 39 , 40 , 41 , 42 Autopsy results indicate the formation of NETs in the patients with severe COVID‐19 disease. 43
FIGURE 2.
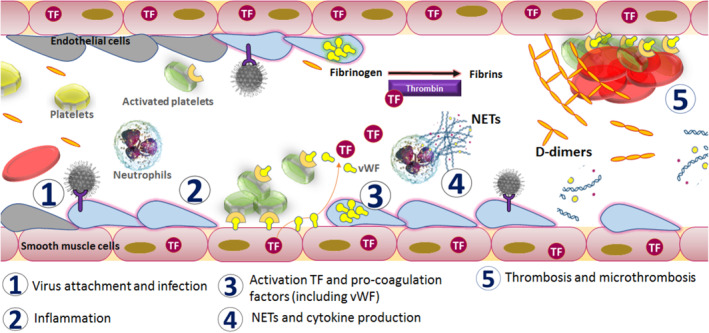
Mechanism of activation of coagulopathy in severe acute respiratory syndrome coronavirus 2 infection
Activation of neutrophils by the cytokines results in the release of chemoattractants and an increase in the recruitment of neutrophils to the site of inflammation. NETs produced by neutrophils induce macrophages to secrete IL1β, which in turn enhances NET formation, creating a self‐amplifying loop. 44 Electrostatic interactions between the NET histones and platelet phospholipids activate the blood coagulation pathway. 45 Neutrophil elastase (a serine protease) is one of the key enzymes that contributes to NET formation. 46 We hypothesize that this enzyme also promotes the coagulation process by digesting inhibitors of coagulation, such as antithrombin III and TF pathway inhibitor. Excessive NET formation can trigger a cascade of inflammatory reactions that can destroy surrounding tissues, facilitate microthrombosis, and result in permanent organ damage, including the vital pulmonary, cardiovascular and renal systems. Elevated levels of NETs accelerate thrombosis in arteries and veins. 47 , 48 , 49 NETs are also detected in a severe form of dengue fever, DHF, which also involves multiorgan failure and is associated with fatal outcomes. 50
The non‐pharmacological measures to sustain severe COVID‐19 patients may also contribute to hypercoagulopathy. Immobilization, mechanical ventilation and central catheters in the intensive care units are risk factors for developing issues related to blood clotting. 51
4. CONCLUSION
We are in the midst of an unprecedented pandemic situation due to a new, rapidly emerging coronavirus. With time and experience, we are learning more about the clinical features and disease pathogenesis of this novel pathogen. In recent weeks, blood hypercoagulability has emerged as a major clinical symptom in severe COVID‐19 patients. Current SARS‐CoV‐2 literature is rapidly evolving on a daily basis. With the current understanding, we propose a possible mechanism that is driving the process of coagulopathy in COVID‐19. Upon entry and binding to its receptor, the virus establishes itself in the upper respiratory tract and the lung tissues. The cells of the endothelial lining and the innate immune surveillance cells, such as alveolar macrophages and neutrophils, are initially infected by the virus. The infection‐mediated inflammation and hypoxic condition result in the release of TF, IL6, and other chemokines, which attract more neutrophils and lymphocytes into the lungs. Depending on the cell type, activated neutrophils and endothelial cells release NETs or pro‐coagulation factors. Reactive immune molecules intended for infected cells leak into the blood circulation and trigger the clotting process by activating platelets and thrombin, resulting in the induction of a microthrombosis process that leads to PE, DIC and multiorgan failure due to nutrient‐ and oxygen‐starvation (Figure 2). In some rare cases, over utilization of platelets may result in bleeding from orifices.
Symptomatic treatment along with close monitoring and measurement of platelet counts and levels of D‐dimer and fibrinogen might be beneficial for early diagnosis of PE in patients with COVID‐19. Because the immune overreactions are not exclusive to SARS‐CoV‐2, existing anti‐inflammatory therapeutics, such as tocilizumab or anakinra, to improve the ARDS and reduce the impact of cytokine storm, could help in minimizing COVID‐19 severity and morbidity. Also, integrating antiviral treatment with promising antiviral candidates, such as hydroxychloroquine and remdesivir, that target SARS‐CoV‐2 entry and viral replication, could speed recovery times and perhaps even reduce death rates. Future observations and experimental studies will play a vital role in portraying a clear picture in understanding the physiological, molecular and cellular signalling pathways that contribute to SARS‐CoV‐2–related thrombus formation.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Sujit Pujhari conceived the idea. Sujit Pujhari and Sanjeeta Paul did the literature search, drew the figures and wrote the manuscript. Sujit Pujhari, Sanjeeta Paul, Jasmina Ahluwalia and Jason L. Rasgon edited the manuscript. All the authors discussed and approved the manuscript.
References
REFERENCES
- 1. Gorbalenya AE, Baker SC, Baric RS, et al. The species severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5:536‐544. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2020;17:181‐192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Wit E, van Doremalen N, Falzarano D, et al. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523‐534. 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dijkman R, Jebbink MF, El Idrissi NB, et al. Human coronavirus NL63 and 229E seroconversion in children. J Clin Microbiol. 2008;46:2368‐2373. 10.1128/JCM.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forni D, Cagliani R, Clerici M, Sironi M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2020;25:35‐48. 10.1016/j.tim.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bikdeli B, Madhavan MV, Jimenez D, et al. COVID‐19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow‐up. J Am Coll Cardiol. 2020;75(23):2950–2973. 10.1016/j.jacc.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and antiphospholipid antibodies in patients with covid‐19. N Engl J Med. 2020;382:e38. 10.1056/NEJMc2007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wichmann D, Sperhake JP, Lutgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID‐19: a prospective cohort study. Ann Intern Med. 2020;M20–2003. 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemostasis. 2020;18:844‐847. 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gandhi RT, Lynch JB, Del Rio C. Mild or moderate covid‐19. N Engl J Med. 2020. 10.1056/NEJMcp2009249. [DOI] [PubMed] [Google Scholar]
- 12. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497‐506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spinato G, Fabbris C, Polesel J, et al. Alterations in smell or taste in mildly symptomatic outpatients with SARS‐CoV‐2 infection. J Am Med Assoc. 2020;323(20):2089–2209. 10.1001/jama.2020.6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carod‐Artal FJ. Neurological complications of coronavirus and COVID‐19. Rev Neurol. 2020;70:311‐322. 10.33588/rn.7009.2020179. [DOI] [PubMed] [Google Scholar]
- 15. Helms J, Tacquard C, Severac F, et al. High risk of thrombosis in patients with severe SARS‐CoV‐2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020. 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Esmon CT. Interactions between the innate immune and blood coagulation systems. Trends Immunol. 2004;25:536‐542. 10.1016/j.it.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemostasis. 2011;9(Suppl 1):182‐188. 10.1111/j.1538-7836.2011.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kitchens CS. Thrombocytopenia and thrombosis in disseminated intravascular coagulation (DIC). Hematology Am Soc Hematol Educ Program 2009:240‐246. 10.1182/asheducation-2009.1.240. [DOI] [PubMed] [Google Scholar]
- 19. Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res. 2019;181:77‐83. 10.1016/j.thromres.2019.07.013. [DOI] [PubMed] [Google Scholar]
- 20. Lawson CA, Yan SD, Yan SF, et al. Monocytes and tissue factor promote thrombosis in a murine model of oxygen deprivation. J Clin Invest. 1997;99:1729‐1738. 10.1172/JCI119337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Monteiro RQ, Lima LG, Goncalves NP, et al. Hypoxia regulates the expression of tissue factor pathway signaling elements in a rat glioma model. Oncol Lett. 2016;12:315‐322. 10.3892/ol.2016.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. White TA, Witt TA, Pan S, et al. Tissue factor pathway inhibitor overexpression inhibits hypoxia‐induced pulmonary hypertension. Am J Respir Cell Mol Biol. 2010;43:35‐45. 10.1165/rcmb.2009-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lucerna M, Mechtcheriakova D, Kadl A, et al. NAB2, a corepressor of EGR‐1, inhibits vascular endothelial growth factor‐mediated gene induction and angiogenic responses of endothelial cells. J Biol Chem. 2003;278:11433‐11440. 10.1074/jbc.M204937200. [DOI] [PubMed] [Google Scholar]
- 24. Yan SF, Zou YS, Gao Y, et al. Tissue factor transcription driven by Egr‐1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxia. Proc Natl Acad Sci USA. 1998;95:8298‐8303. 10.1073/pnas.95.14.8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938‐949. 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 26. Pilli VS, Datta A, Afreen S, Catalano D, Szabo G, Majumder R. Hypoxia downregulates protein S expression. Blood. 2018;132:452‐455. 10.1182/blood-2018-04-841585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Semenza GL. Hypoxia‐inducible factor 1 (HIF‐1) pathway. Sci STKE 2007;2007:cm8. 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 28. Koeleman BP, van Rumpt D, Hamulyak K, Reitsma PH, Bertina RM. Factor V Leiden: an additional risk factor for thrombosis in protein S deficient families? Thromb Haemost. 1995;74:580‐583. [PubMed] [Google Scholar]
- 29. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054‐1062. 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rollin PE, Bausch DG, Sanchez A. Blood chemistry measurements and D‐Dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J Infect Dis. 2007;196 Suppl 2:S364‐S371. 10.1086/520613. [DOI] [PubMed] [Google Scholar]
- 31. Korva M, Rus KR, Pavletic M, et al. Characterization of biomarker levels in Crimean‐Congo hemorrhagic fever and Hantavirus fever with renal syndrome. Viruses 2019;8(11):686. 10.3390/v11080686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sridhar A, Sunil Kumar BM, Rau A, Rau ATK. A correlation of the platelet count with D‐dimer levels as an indicator for component therapy in children with dengue hemorrhagic fever. Indian J Hematol Blood Transfus. 2017;33:222‐227. 10.1007/s12288-016-0686-7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu X, Han M, Li T, et al. Effective treatment of severe COVID‐19 patients with tocilizumab. Proc Natl Acad Sci USA. 2020. 10.1073/pnas.2005615117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aouba A, Baldolli A, Geffray L, et al. Targeting the inflammatory cascade with anakinra in moderate to severe COVID‐19 pneumonia: case series. Ann Rheum Dis. 2020. 10.1136/annrheumdis-2020-217706. [DOI] [PubMed] [Google Scholar]
- 35. Wu D, Yang XO. TH17 responses in cytokine storm of COVID‐19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. 2020. 10.1016/j.jmii.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020. 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033‐1034. 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880‐15885. 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1777‐1783. 10.1161/ATVBAHA.111.242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279‐287. 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 41. Kaplan MJ, Radic M. Neutrophil extracellular traps: double‐edged swords of innate immunity. J Immunol. 2012;189:2689‐2695. 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laridan E, Martinod K, De Meyer SF. Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost. 2019;45:86‐93. 10.1055/s-0038-1677040. [DOI] [PubMed] [Google Scholar]
- 43. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, et al. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652. 10.1084/jem.20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meher AK, Spinosa M, Davis JP, et al. Novel role of IL (Interleukin)‐1beta in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2018;38:843‐853. 10.1161/ATVBAHA.117.309897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oehmcke S, Morgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun. 2009;1:225‐230. 10.1159/000203700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887‐896. 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 47. Gollomp K, Kim M, Johnston I, et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight. 2018;3(18):e99445. 10.1172/jci.insight.99445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lachowicz‐Scroggins ME, Dunican EM, Charbit AR, et al. Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma. Am J Respir Crit Care Med. 2019;199:1076‐1085. 10.1164/rccm.201810-1869OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Teague HL, Varghese NJ, Tsoi LC, et al. Neutrophil subsets, platelets, and vascular disease in Psoriasis. JACC Basic Transl Sci. 2019;4:1‐14. 10.1016/j.jacbts.2018.10.008S2452-302X(18)30279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Opasawatchai A, Amornsupawat P, Jiravejchakul N, et al. Neutrophil activation and early features of NET formation are associated with dengue virus infection in human. Front Immunol. 2019;9:3007. 10.3389/fimmu.2018.03007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klok FA, Kruip M, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19. Thromb Res. 2020. 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]