

Abstract
Infecting millions of people, the SARS‐CoV‐2 is evolving at an unprecedented rate, demanding advanced and specified analytic pipeline to capture the mutational spectra. In order to explore mutations and deletions in the spike (S) protein — the most‐discussed protein of SARS‐CoV‐2 — we comprehensively analyzed 35,750 complete S protein‐coding sequences through a custom Python‐based pipeline. This GISAID‐collected dataset of until 24 June 2020 covered six continents and five major climate zones. We identified 27,801 (77.77% sequences) mutated strains compared to reference Wuhan‐Hu‐1 wherein 84.40% of these strains mutated by only a single amino acid (aa). An outlier strain (EPI_ISL_463893) from Bosnia and Herzegovina possessed six aa substitutions. We also identified 11 residues with high aa mutation frequency, and each contains four types of aa variations. The infamous D614G variant has spread worldwide with ever‐rising dominance and across regions with different climatic conditions alongside L5F and D936Y mutants, which have been documented throughout all regions and climate zones, respectively. We also found 988 unique aa substitutions spanned across 660 residues, which differed significantly among different continents (p = .003) and climatic zones (p = .021) as inferred with the Kruskal–Wallis test. Besides, 17 in‐frame deletions at four sites adjacent to receptor‐binding‐domain were determined that may have a possible impact on attenuation. This study provides a fast and accurate pipeline for identifying mutations and deletions from the large dataset for coding and also non‐coding sequences as evidenced by the representative analysis on existing S protein data. By using separate multi‐sequence alignment, removing ambiguous sequences and in‐frame stop codons, and utilizing pairwise alignment, this method can derive both synonymous and non‐synonymous mutations (strain_ID reference aa:mutation position:strain aa). We suggest that the pipeline will aid in the evolutionary surveillance of any SARS‐CoV‐2 encoded proteins and will prove to be crucial in tracking the ever‐increasing variation of many other divergent RNA viruses in the future. The code is available at https://github.com/SShaminur/Mutation-Analysis.
Keywords: Climate, Geography, Mutations, SARS‐CoV‐2, Spike (S) protein | COVID‐19
1. INTRODUCTION
Mutations in the viral genomes serve as the building blocks of viral evolution and remain the main reason for the novelty in evolution (Baer, 2008; Duffy, 2018). However, mutations in the viral genomes are not restricted to their replication since they can also result from spontaneous nucleic acid damage over time in different host populations or from editing of the genetic materials. Thus, a large portion of mutations, either at nucleotides (nt) and/or change in amino acids (aa) levels, are harmful (Loewe & Hill, 2010). RNA viruses like SARS‐CoV‐2 generally have higher mutation rates; however, a few of these mutations are correlated with differential virulence, evolving ability, and traits considered beneficial for viruses (Duffy, 2018; Islam et al., 2020). Inherent high mutation rate of SARS‐CoV‐2 has already produced many descendants from the original Wuhan strain, which complicates its genotyping. The ability of the structural proteins especially spike protein, in different strains of the SARS‐CoV‐2 to undergo rapid changes have enabled their genomes to emerge in novel hosts, escape vaccine‐induced immunity and evolve in diverse geo‐climatic conditions (Duffy, 2018; Islam et al., 2020; Loewe & Hill, 2010). Moreover, spontaneous mutation is a key parameter in modelling the genetic structure and evolution of populations (Drake & Holland, 1999). Therefore, investigation of the increased rate of non‐synonymous mutations in the SARS‐CoV‐2 genomes could be an important tool in assessing the genetic health of the populations.
SARS‐CoV‐2 comprises of four major structural proteins—specifically spike (S) glycoproteins, envelope (E) proteins, membrane (M) proteins and nucleocapsid (N) proteins (Ahmed et al., 2020; Rahman et al., 2020; Wu et al., 2020). The entry of SARS‐CoV‐2 into the host cells is mediated by the transmembrane S protein which consists of two functional subunits responsible for binding to the host cell receptor (S1 subunit), and for fusing the viral and cellular membranes (S2 subunit) (Walls et al., 2020). The higher antigenic and surface exposure properties of the S protein facilitate the attachment and entry of viral particles into the host cells through the host angiotensin‐converting enzyme 2 (ACE2) receptor (Grant et al., 2020; Shang et al., 2020; Zhou et al., 2019). Therefore, the spike contains highest variations and determines, to some extent, the viral host range (Coutard et al., 2020; Wu et al., 2020). Furthermore, the S protein is the main target of neutralizing antibodies (Abs) upon infection and is thus one of the most important structures for therapeutics and vaccine design (Rahman et al., 2020; Walls et al., 2020).
The continuing rapid transmission and global spread of COVID‐19 have raised intriguing questions regarding the evolution and adaptation of SARS‐CoV‐2 in diverse geographic and climatic conditions driven by non‐synonymous mutations, deletions and/or replacements (Bal et al., 2020; Islam et al., 2020; Pachetti et al., 2020). The capability of the different strains of SARS‐CoV‐2 strains for swiftly adapting to diverse environments could be linked with their geographic distributions. Though not yet well studied, evidence suggests that the transmission of SARS‐CoV‐2 infections and per day mortality rate from this infection is positively associated with weather conditions, and the diurnal temperature range (DTR) (Su et al., 2016; Islam et al., 2020). However, the exact role of geo‐climatic conditions on SARS‐CoV‐2 is unknown, but it would be worth keeping in mind that this novel disease originated from wildlife before spreading to humans (Harvey, 2020). Therefore, genomic mutation analysis of SARS‐CoV‐2 strains, integrated with geographic and climatic data, would provide a fuller understanding of the origin, dispersal and dynamics of the evolving SARS‐CoV‐2 virus. Although several reports predicted possible adaptations at the nucleotide and aa level, along with structural heterogeneity in viral proteins, especially in the S protein (Armijos‐Jaramillo et al., 2020; Islam et al., 2020; Phan, 2020; Sardar et al., 2020), most of these studies were carried out few complete representative genomes from a limited geographic area. As the genome number is increasing day by day, regular in‐house monitoring of the crucial components such as the S protein is urgently necessary to understand the genomic basis and evolution of the diagnostic RT‐PCR primer. There are a few pipelines (Yin, 2020) and websites (https://mendel.bii.astar.edu.sg/METHODS/corona/beta/MUTATIONS/hCoV19_Human_2019_WuhanWIV04/hCoV‐19_Spike_new_mutations_table.html) in GSAID where aa change or substitution can be observed. In order to provide an alternative tool with a wider range of functions, we present an easy rapid pipeline that will assist in the alignment of large volumes of viral genomes, remove low‐quality sequences and in‐frame stop codons and provide in‐house non‐synonymous mutation analysis of large volumes of sequences while requiring minimal knowledge of the command line. This tool can perform this analysis for any other proteins as required. This study aimed to investigate the mutational spectra of aa utilizing this novel methodology in the S proteins in 35,750 complete genome sequences of the SARS‐CoV‐2 belonging to 135 countries and/regions, and five climatic zones around the world, retrieved from the global initiative on sharing all influenza data (GISAID) (https://www.gisaid.org/) up to 24 June 2020 (Data S1).
2. MATERIALS AND METHODS
2.1. Genomic data collection and processing
To decipher the genetic variations of the S glycoprotein, we retrieved 53,981 complete (or near‐complete) genome sequences of SARS‐CoV‐2, available at the global initiative on sharing all influenza data (GISAID) (https://www.gisaid.org/) up to 24 June 2020. These sequences belonged to infected patients from 135 countries and/or regions from across six continents (Data S1). Using pyfasta (https://github.com/brentp/pyfasta), we split the total genome into 6 separate files having around 8,900 sequences in each. We aligned each file through the MAFFT (maximum limit 10,000 sequences) online server (https://mafft.cbrc.jp/alignment/server/add_fragments.html?frommanual) using default parameters (Katoh et al., 2002). The complete genome sequence of SARS‐CoV‐2 Wuhan‐Hu‐1 strain (Accession NC_045512, Version NC_045512.2) was used as a reference genome.
2.2. Mutation frequency analysis
MEGA 7 was used to differentiate the spike protein of SARS‐CoV‐2 from multiple sequence alignment (Sudhir Kumar et al., 2016). Sequence cleaner (https://github.com/metageni/Sequence‐Cleaner) with set parameters of minimum length (m = 3,822), percentage N (mn = 0), keep_all_duplicates, and remove_ambiguous was employed to remove all ambiguous, and low‐quality sequences. We utilized SeqKit toolkit (seqkit grep ‐s ‐p "‐" in.fa > out.fa) to apprehend gap containing strains for deletion analysis (Shen et al., 2016). Internal stop codon containing sequences were removed by using SEquence DAtaset builder (SEDA; https://www.sing‐group.org/seda/). Amino acid mutation analysis was done with bio‐python program using pairwise alignment (https://github.com/SShaminur/Mutation‐Analysis). The custom Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/) server was used to make the Venn diagrams, and visualize the data. Swiss‐Model, a structure homology‐modelling server (https://swissmodel.expasy.org/), was used to predict the 3D structure (template, PDB ID:6VSB) of the S protein of the reference genome, and the structure was visualized in PyMOL (DeLano, 2002; Rahman et al., 2020; Waterhouse et al., 2018). Furthermore, we divided the S glycoprotein mutation of SARS‐CoV‐2 data according to their geographic origins from six continents—Europe, Asia, North America, South America, Africa and Australia, and five related climatic zones—temperate, tropical, diverse, dry and continental (Kissler, Tedijanto, Goldstein, & Yonatan, 2020). To estimate the case fatality (mortality) rates of SARS‐CoV‐2 infections, we collected information on total infected cases and total reported deaths in these countries from the World Health Organization (WHO) COVID‐19 Reports up to 24 June 2020 (WHO Reports, 2020).
2.3. Pipeline validations
The overview of the methods is described in Figure 1. The SARS‐CoV‐2 genomes are increasing very rapidly in the Global initiative on sharing all influenza data (GISAID), but not all genomes are of high quality or complete. So, non‐synonymous mutation analysis with particular crucial part of the virus like S or other structural protein gives statistically more significant insights rather considering the complete genome of the SARS‐CoV‐2 virus. Of the total S protein sequences, sequence cleaner removed 33.77% of the low‐quality or ambiguous sequences. Of the rest cleaned sequences (66.23%), we found ten in‐frame stop codon containing sequences which were eventually removed using SEDA (https://www.sing‐group.org/seda/manual/operations). SeqKit toolkit was used to arrest gap containing sequences which identified around 453 sequences, and we also carefully checked the in‐frame deletion, and 103 strains containing in‐frame deletions. SNP‐sites is a very efficient tools for nucleotide variation detection in different format like multi‐fasta alignment, variant call format (VCF), and relaxed phylip format (Page et al., 2016) but this tool is highly dedicated for nucleotide. Snippy (Seemann, 2015) is another tool where nucleotide and protein variation can also be detected, but for large data set with ambiguous sequences will require a separate processing to entrust more accurate results. This pipeline gives the non‐synonymous mutation results in a file format (Strain_ID Reference_aa:Mutation_Position:Strain_aa) that will assist in the downstream analysis like unique mutation, unique position mutation, mutational frequency and strains having number of mutation (Figure 2). Moreover, synonymous mutations analysis for large datatset can also be applied by this tool. For deletion analysis, this pipeline helped in decreasing the size of sequences (just 453 sequences from 53,981 sequences). Details of the current pipeline and coding are deposited in the Github (https://github.com/SShaminur/Mutation‐Analysis).
Figure 1.
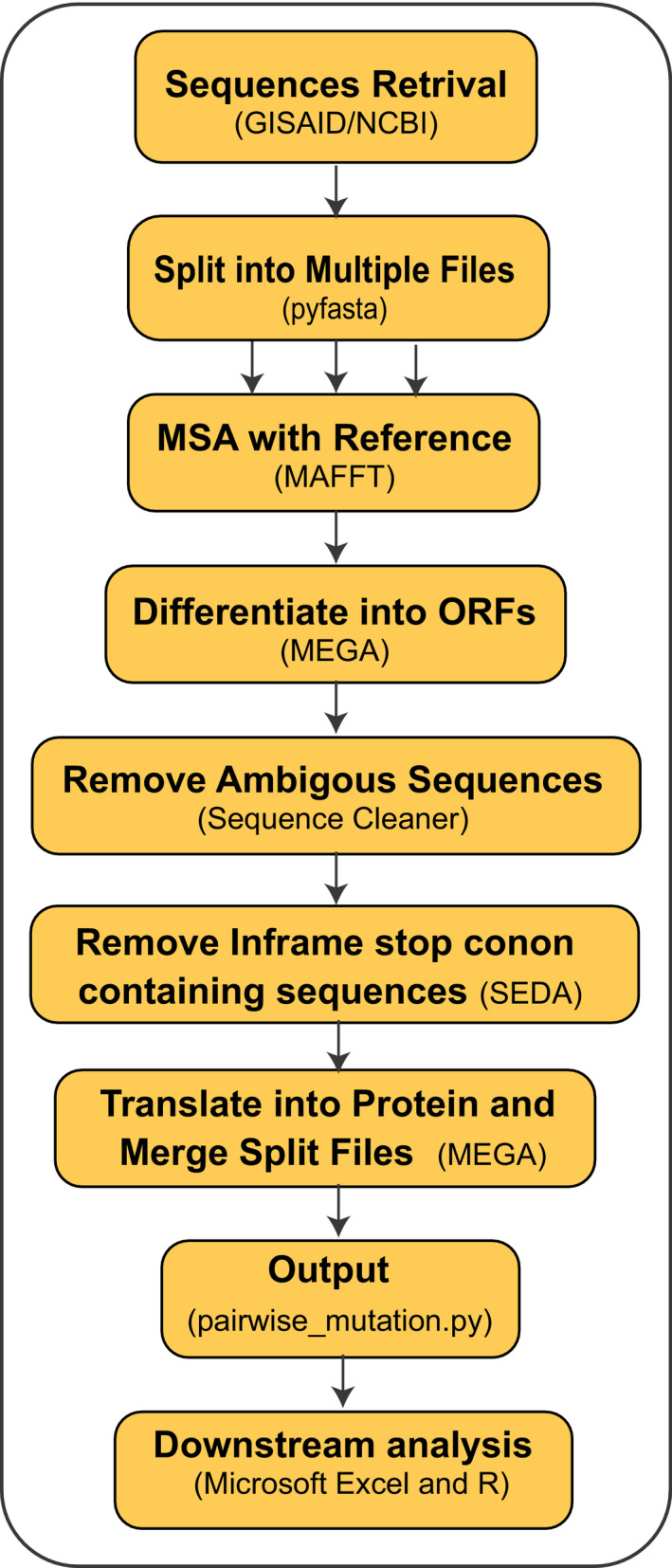
Workflow of the pipeline used for non‐synonymous mutation analyses in this study. File splitting needs if the number of sequences is more than 10,000. Through these methods, nucleotide mutations can also be calculated. Here: MSA: multiple sequence alignment, and ORFs: open reading frames [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
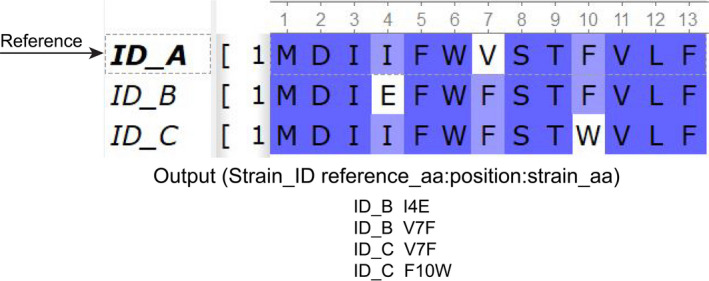
Overview of the input and results output representing changes in aa position (white background) in different strains of SARS‐CoV‐2 in regard to reference genome [Colour figure can be viewed at wileyonlinelibrary.com]
2.4. Statistical analysis
Wu–Kabat variability coefficient was employed to calculate the aa position variability in regard to evolutionary adaptation (Garcia‐Boronat et al., 2008; Kabat et al., 1977). The variability coefficient was calculated using the following formula:
N = total number of sequences in the alignment, k = number of different aa at a given position, and n = frequency of the most common aa at that position.
We used Microsoft Excel 2016 to calculate the frequency, percentages, Wu–Kabat variability coefficient calculation using the above mentioned formula and overall data management (David, 2017). Wu–Kabat variability coefficient plot was visualized in RStudio by using ggplot2 package (Wickham, 2011). Frequency lolliplot was also visualized in RStudio with the trackViewer Vignette package (https://bioconductor.org/packages/release/bioc/vignettes/trackViewer/inst/doc/trackViewer.htm) (Ou et al., 2020a,2020b). To measure the morbidity and case fatality rates, and association between the S protein mutational spectra and case fatality rates, we applied non‐parametric test Kruskal–Wallis rank sum test (Hoque et al., 2019) using IBM SPSS (SPSS, version 23.0, IBM Corp., NY USA).
3. RESULTS AND DISCUSSIONS
3.1. Geo‐climatic distribution of strains
Trimming of the low‐quality, ambiguous and non‐human host RNA sequences resulted in 35,750 (66.23%) cleaned and full‐length S protein sequences (Data S1). These sequences belonged to 135 countries and/or regions from six continents (Europe, Asia, North America, South America, Africa and Australia) and five major climatic zones (temperate, tropical, diverse, dry and continental) around the world (Data S1). European countries and/or regions had the highest percentage (58.90%) of S protein sequences, followed by North American (25.78%), Asian (9.34%), Australian (3.61%), South American (1.21%) and African (1.18%) countries or regions. On the other hand, the temperate climatic zone covered the majority of these S protein sequences (60.18%), followed by diverse (33.08%), continental (3.25%), tropical (2.81%) and dry (0.69%) climatic conditions (Data S1). We selected the complete genome sequence SARS‐CoV‐2 Wuhan‐Hu‐1 strain (Accession NC_045512, Version NC_045512.2) as a reference genome. Through non‐synonymous mutations analysis, we found 27,801 (77.77%) mutated strains of the SARS‐CoV‐2 in the cleaned sequences (n = 35,750). Furthermore, country or region‐specific aa change patterns revealed the highest number of mutated SARS‐CoV‐2 strains in England (7,067) followed by USA (6,501), Wales (3,002), Scotland (1,463), Netherlands (1,194), Australia (681), Belgium (596) and Denmark (582) (Data S1).
3.2. Evolutionary footprint in spike
Our mutational analyses revealed a total of 988 unique amino acid (aa) substitutions distributed across 660 positions of SARS‐CoV‐2 S protein. Among these positions, 250 showed two or more aa variations in a certain position (Figure 3a, Table 1, Data S2). The primary structure of the S protein is 1,274 aa; of them, 51.81% aa positions (n = 660) undergo aa‐level evolution worldwide. The positions‐specific aa variability of S protein was visualized in Wu–Kabat protein variability plot (Figure 3b). The current variability analysis identified 19 positions showing Wu–Kabat variability coefficient ˃4 indicating high variability of these positions. However, 614 (48.19%) positions had coefficient 1 indicating invariability of the positions compared to the reference strain of SARS‐CoV‐2 (Figure 3b). Remarkably, we found eleven highly variable sites (position: 32, 142, 146, 215, 261, 477, 529, 570, 622, 778, 791, 1,146, 1,162) showing four types of aa variations in a single position (Table 1). We also found that positions 52, 185 and 410 in the S glycoprotein underwent to 3, 2 and 1 aa substitutions, respectively (Table 1, Data S2). Notably, position 614 showed two variants, substitution D614G (Aspartic acid ˃ Glycine) found in ⁓74.82% (n = 26,749) of the cleaned sequences (⁓96.22% of the mutated sequences), and another variant D614N (Aspartic acid ˃ Asparagine) was observed only in four strains from England and Wales (EPI_ISL_439400, EPI_ISL_443658 and EPI_ISL_445498, EPI_ISL_472913). The variant D614G in the S protein has overcome the wild‐type variant from China since its first appearance in Germany on 28 January 2020 (Comandatore et al., 2020; Eaaswarkhanth et al., 2020; Kim et al., 2020; Korber et al., 2020; Trucchi et al., 2020). Moreover, variant frequencies of a recurrent pattern of G614 increase at multiple geographic levels: national, regional and municipal has also been reported through dynamic tracking. The shift might occur even in local epidemics where the original D614 form was well established prior to introduction of the G614 variant (Korber et al., 2020).
Figure 3.
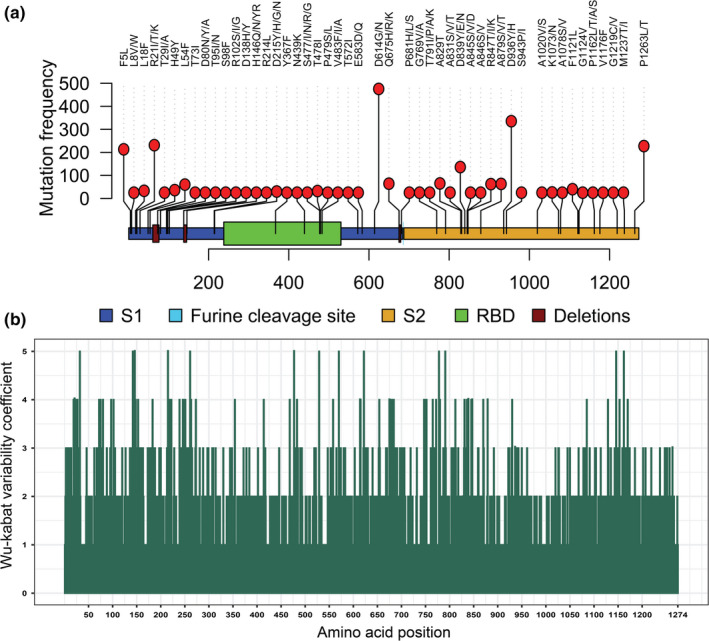
Mutational mapping and Wu–Kabat variability analysis of SARS‐CoV‐2 S protein. (a) Mapping and frequency distribution of recurrently occurred mutations in the S protein of ≥25 SARS‐CoV‐2 strains. Deletion sites of the S protein were also visualized in the Lolliplot graph. (b) Wu–Kabat protein variability coefficient plot of SARS‐CoV‐2 S protein. Here, variability coefficient 1 indicates the conservancy, whereas coefficients ˃1 indicate relative variability of the respective position. The more the coefficient value the more the variability or diversity [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
Amino acid variations in the S protein of the SARS‐CoV‐2 according to their position
| Position in S | Number of variations | Name of amino acid |
|---|---|---|
| 19 | 3 | T19P, T19I, T19S |
| 21 | 3 | R21I, R21T, R21K |
| 22 | 3 | T22N, T22I, T22A |
| 26 | 3 | P26L, P26S, P26R |
| 27 | 3 | A27V, A27T, A27S |
| 32 | 4 | F32L, F32Y, F32I, F32V |
| 72 | 3 | G72E, G72W, G72R |
| 75 | 3 | G75D, G75V, G75R |
| 80 | 3 | D80N, D80Y, D80A |
| 97 | 3 | K97E, K97N, K97R |
| 102 | 3 | R102S, R102I, R102G |
| 142 | 4 | G142D, G142A, G142V, G142S |
| 146 | 4 | H146Q, H146N, H146Y, H146R |
| 148 | 3 | N148Y, N148K, N148S |
| 153 | 3 | M153T, M153I, M153V |
| 183 | 3 | Q183H, Q183R, Q183L |
| 215 | 4 | D215Y, D215H, D215G, D215N |
| 218 | 3 | Q218R, Q218E, Q218L |
| 222 | 3 | A222V, A222S, A222P |
| 239 | 3 | Q239K, Q239R, Q239H |
| 246 | 3 | R246I, R246K, R246S |
| 247 | 3 | S247R, S247I, S247N |
| 251 | 3 | P251S, P251H, P251L |
| 261 | 4 | G261V, G261S, G261D, G261R |
| 263 | 3 | A263T, A263S, A263V |
| 273 | 3 | R273M, R273K, R273S |
| 354 | 3 | N354D, N354K, N354S |
| 414 | 3 | Q414R, Q414K, Q414P |
| 468 | 3 | I468F, I468T, I468V |
| 477 | 4 | S477I, S477N, S477R, S477G |
| 483 | 3 | V483F, V483I, V483A |
| 529 | 4 | K529M, K529N, K529R, K529E |
| 558 | 3 | K558N, K558Q, K558R |
| 570 | 4 | A570S, A570V, A570D, A570T |
| 615 | 3 | V615I, V615F, V615L |
| 622 | 4 | V622F, V622L, V622I, V622A, A623V |
| 654 | 3 | E654D, E654Q, E654K |
| 675 | 3 | Q675H, Q675R, Q675K |
| 677 | 3 | Q677H, Q677R, Q677Y |
| 681 | 3 | P681H, P681L, P681S |
| 684 | 3 | A684V, A684T, A684S |
| 747 | 3 | T747A, T747I, T747N |
| 750 | 3 | S750N, S750R, S750I |
| 752 | 3 | L752I, L752R, L752F |
| 765 | 3 | R765L, R765H, R765C |
| 772 | 3 | V772L, V772I, V772A |
| 778 | 4 | T778S, T778A, T778N, T778I |
| 780 | 3 | E780D, E780Q, E780V |
| 791 | 4 | T791I, T791A, T791K, T791P |
| 812 | 3 | P812S, P812T, P812L |
| 831 | 3 | A831S, A831V, A831T |
| 836 | 3 | Q836H, Q836P, Q836L |
| 838 | 3 | G838S, G838V, G838D |
| 839 | 3 | D839Y, D839E, D839N |
| 845 | 3 | A845S, A845V, A845D |
| 847 | 3 | R847T, R847I, R847K |
| 870 | 3 | I870S, I870T, I870V |
| 879 | 3 | A879S, A879V, A879T |
| 930 | 3 | A930S, A930V, A930T |
| 1,085 | 3 | G1085R, G1085E, G1085L |
| 1,129 | 3 | V1129L, V1129A, V1129I |
| 1,146 | 4 | D1146Y, D1146H, D1146E, D1146N |
| 1,153 | 3 | D1153A, D1153H, D1153Y |
| 1,162 | 4 | P1162L, P1162T, P1162A, P1162S |
| 1,170 | 3 | S1170T, S1170Y, S1170P |
Here, the position(s) where more than 2 variations occurred are represented.
A strain from Bosnia_and_Herzegovina (EPI_ISL_463893) had the highest number of aa changes/substitutions (n = 6) at six positions (R246I, L276I, T430A, D614G, S750N, L922V) of S protein. Also, we found that 84.8% (n = 23,576) of the mutated sequences carried just a single aa mutation throughout the S proteins. The remaining 13.44%, 1.63%, 0.11% and 0.01% of the mutated sequences contained 2, 3, 4 and 5 aa changes, respectively (Data S2). Moreover, we did not find any of such non‐synonymous aa mutation in the full‐length S protein of 18 countries and/or regions including Anhui, Brunei, Cambodia, Changzhou, Chongqing, Foshan, Ganzhou, Guam, Hefei, Jiangxi, Jingzhou, Jiujiang, Lishui, Nepal, Philippines, Qatar, Yingtan and Yunnan. This indicates S protein homogeneity of these countries/regions with the reference sequence from Wuhan, China (Data S1).
The RBD region (Watanabe et al., 2020) (aa position: 338–530) showed non‐synonymous mutations at 82 different positions in 516 strains, whereas in the S1 site and S2 site, there were 362 and 297 positional recurrent mutations, respectively. Moreover, in the furin cleavage site (R685 and S686), we also observed a non‐synonymous mutation (S686G) in a single strain (Russia/Krasnodar‐63401/2020|EPI_ISL_428867|2020‐03‐11) (Data S2). We also found aa substitutions at six positions within the RBD region that are directly involved in binding with ACE‐2 receptor (Wang et al., 2020; Yuan et al., 2020) including N439K (Scotland, Romania), L455F (England), A475V (USA, Australia), and F456L, Q493L and N501Y (USA) (Data S2). All these mutations were found between March and April at a lower frequency (N439K with maximum frequency in 41 Scottish strains and one Romanian strain), except Q493L found in two USA strains reported in May. Q493R position showed variation in an English strain (EPI_ISL_470150) found in April. Furthermore, 18 substitutions at fourteen positions, previously reported to interact with anti‐SARS‐CoV‐2 antibody (Yuan et al., 2020), were found in the strains from Bangladesh, England, Portugal, Wales, Shanghai, France, USA, Scotland, Russia, Latvia, Netherlands, South Africa, Bosnia and Herzegovina, Belgium, Bosnia and Australia (Data S2) during the time frame March to May. Discontinuation of the mutants globally may be linked to reduction of virus pathogenicity and virulence fitness affecting transmission dynamics. However, the unavailability of these variants may result due to rejection of the variants with a lower ratio when generating the final consensus sequences and insufficient sequences reporting from unusual asymptomatic patients. Moreover, eight glycosylated sites of S protein underwent aa conversions including three substitutions in the NTD region (N17K, N74K and N149H), five substitutions at four sites in the S1 region (N17K, N74K, N149H, N603S and N603K) and four mutations in the S2 region (N717T, N1074D, N1158S and N1194S) (Watanabe et al., 2020). Furthermore, a total of 50 aa substitutions within the S protein were observed that incorporated asparagine (N) in S protein of SARS‐CoV‐2 including seven within the RBD region (S359N, K378N, K417N, K458N, S477N, T523N and K529N) (Data S2). These substitutions alter glycosylation sites and it nature, though it needs further investigations. Overall, the aa substitutions related to asparagine in the RBD (ACE binding domain) and/or in S1/2 domains nearer to the glycosylated sites may affect the glycosylation shield, folding of S protein, host–pathogen interactions, viral entry and finally immune modulation, thus affecting antibody recognition and viral pathogenicity (Ou et al., 2020a,2020b; Watanabe et al., 2020). Overall, these variability profiles may have notable implications in therapeutic and/or prophylactic interventions targeting the S protein of SARS‐CoV‐2.
3.3. In‐frame deletions resided adjacent S glycoprotein
Besides site‐specific mutations, our analysis revealed 17 in‐frame deletions of ranged nucleotides across the SARS‐CoV‐2 S protein sequences originating from different countries worldwide (Table 2, Data S2). Notably, we considered the deletions that occurred in at least two strains at a certain position as deletions. All of the identified deletions distributed throughout the nucleotide sequence 179–2035 fall into four major regions of S protein, that is nt position ranges 179–226 (61–76 aa: NVTWFHAIHVSGTNGT), 413–433 (138–144 aa: DPFFLGVY), 724–732 (241–244: LLAL) and 2021–2035 (675–679 aa: QTQTN). Amino acid deletions at positions 61–76, 138–144, and 241–244 are near the RBD region. Among them, deletions of positions 61–76 and 141–144 are surface exposed, but 241–244 are situated at the inner surface of the predicted S protein (Figure 4). Also, deleted aa at positions 675–679 are located in the C‐terminal transmembrane domain of S protein. Surface exposed deletions near the RBD region may have significant impact on host–pathogen interaction and immune modulation.
Table 2.
Deletion sites observed across the S glycoprotein
| Nucleotide position | Amino acid position | Deleted amino acid | Countries | Number of strains |
|---|---|---|---|---|
| 179–217 | 61–73 | NVTWFHAIHVSGT | England | 1 |
| 200–226 | 68–76 | IHVSGTNGT | Taiwan, Malaysia | 2 |
| 201–224 | 68–75 | IHVSGTNG | Thailand | 1 |
| 203–208 | 69–70 | HV | Sweden, England, Australia | 3 |
| 413–421 | 138–140 | DPF | Sweden | 1 |
| 418–420 | 140 | F | England, Sichuan | 3 |
| 420–431 | 141–144 | LGVY | England, Iceland, USA, Scotland, Kenya | 16 |
| 420–422 | 141 | L | England | 1 |
| 422–430 | 141–143 | LGV | Portugal, England, Iceland, Scotland | 4 |
| 423–431 | 142–144 | GVY | England, Netherlands | 3 |
| 428–430 | 143 | V | USA, Belgium | 4 |
| 428–433 | 143–144 | VY | England | 2 |
| 429–431 | 145 | Y | England, Canada, Slovenia, Jordan, Netherlands, Saudi_Arabia, Scotland, USA, Spain, Wales, India, Australia | 48 |
| 724–732 | 241–243 | LLA | China, England, Belgium, Scotland, Netherlands | 6 |
| 724–726 | 241 | L | USA | 2 |
| 727–732 | 243–244 | AL | England, Wales, Spain, Sichuan | 6 |
| 2021–2035 | 675–679 | QTQTN | Taiwan, Malaysia | 2 |
Countries represent the origin of strains where the deletions found. We considered the deletions that occurred in at least two strains in a certain position.
Figure 4.
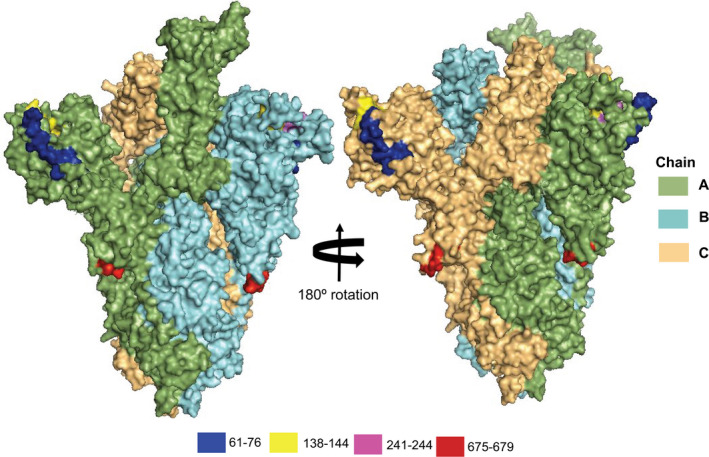
Structural visualization of S protein deletion sites. The four aa deleted positions (61–76, 138–144, 241–244, and 675–679) in the S protein of the reference genome, SARS‐CoV‐2 Wuhan‐Hu‐1 strain (Accession NC_045512, Version NC_045512.2). The positions are visualized in the tertiary (3D) structure of S protein using PyMOl. The smudge, cyan and light orange colours represent the A, B and C chains of SARS‐CoV‐2 spike protein, respectively. Blue, yellow, magenta and red colours represent the aa deletion position of 61–76, 138–144, 241–244, and 675–679, respectively [Colour figure can be viewed at wileyonlinelibrary.com]
Among the deletions, nucleotide deletion positioned at 418–433 (aa position 140–144) faced frequent overlapped deletions among strains of multiple countries (Table 2). Notably, a single aa in‐frame deletion of nucleotides positioned 429–431 (aa position 145) with the highest frequency in 48 strains from multiple countries and/or regions including Australia, England, Canada, Slovenia, Jordan, Netherlands, Saudi_Arabia, Scotland, USA, Spain, Wales and India. A strain from Taiwan (EPI_ISL_444275) showed two coevolving deletions at nt positions 200–226 (68–76 aa:IHVSGTNGT) and nt positions 2021–2035 (675–679 aa:QTQTN). Moreover, two deletions at nt positions 418–420 (140 aa:F) and 727–732 (243–244 aa:AL) were coevolved in a Sichuan strain (EPI_ISL_451369). No other strain had such coevolving deletions, thereby indirectly indicating the negative impact of the deletions on virus fitness and human‐to‐human transmissibility. Noteworthy, a 5‐aa deletion (675–679 aa: QTQTN) at the upstream of the polybasic cleavage site of S1–S2 and a 21‐nt deletion 23596–23617 (aa‐NSPRRAR) including the polybasic cleavage site in clinical samples and cell‐isolated virus strain likely benefit the SARS‐CoV‐2 replication or infection in vitro and under strong purification selection in vivo (Liu et al., 2020). Moreover, attenuated SARS‐CoV‐2 variants with 15–30‐bp deletions (Del‐mut) at the S1/S2 junction were reported to show less virulence in an animal model (Lau et al., 2020).
These deletions may affect viral adaptations to human, virus–host interactions for infections, attenuation, pathogenicity and immune modulations by potentially influencing the tertiary structures and functions of the associated proteins (Phan, 2020). However, further studies are required for the mechanistic clarification and functional implication of these deletions in the SARS‐CoV‐2 S glycoprotein. The deletion mutations identified in this study should be also considered for current vaccine development.
3.4. Geo‐climatic scenario of amino acid heterogeneity in the spike protein of SARS‐CoV‐2 and associated disease severity
Considering geo‐climatic impacts on aa changes in the S protein of the SARS‐CoV‐2, we sought to determine the possible residue positions, and total number of mutations in the S protein sequences from 135 countries and/or territories, and five climatic zones worldwide. Nine hundred and eighty‐eight (988) unique aa replacements across 660 positions along the S protein were identified which differed significantly among different continents (p = .003, Kruskal–Wallis test) and climatic zones (p = .021, Kruskal–Wallis test). We found that the frequency of aa changes in the S protein remained substantially higher in the SARS‐CoV‐2 genome sequences of Europe (62.02%), followed by North America (25.50%), Asia (6.83%), Australia (2.89%), South America (1.41%) and Africa (1.35%) (Figure 5a, Data S1). Among these replacements, aa residues at position 5 (L5F) and 614 (D614G) were found to be the common in Asia, Europe, North America, South America, Africa and Australia (Figure 5b). Moreover, 408, 127, 139, 17, 10 and 8 unique aa replacements (mutation that found only once in a sequence) were identified in the S protein sequences of Europe, Asia, North America, Australia, South America and Africa, respectively (Figure 5b, Data S3). In addition to unique aa mutations, 244, 146, 194, 61, 19 and 23 accessory aa replacements (mutations shared with at least two continents) were also found in the S protein of SARS‐CoV‐2 genomes sequenced from Europe, Asia, North America, Australia, South America and Africa, respectively (Figure 5b, Data S3). Significantly higher unique mutations in European (p = .0121, Kruskal–Wallis test), Asian (p = .0177, Kruskal–Wallis test) and American (p = .0391, Kruskal–Wallis test) sequences point out the geographic clustering pre‐disposition of the virus. However, further phylogenic study targeting those unique and accessory mutations may lead to a better understanding of global phylodynamics, and thereby guiding the regional control strategy for the COVID‐19 pandemic.
Figure 5.
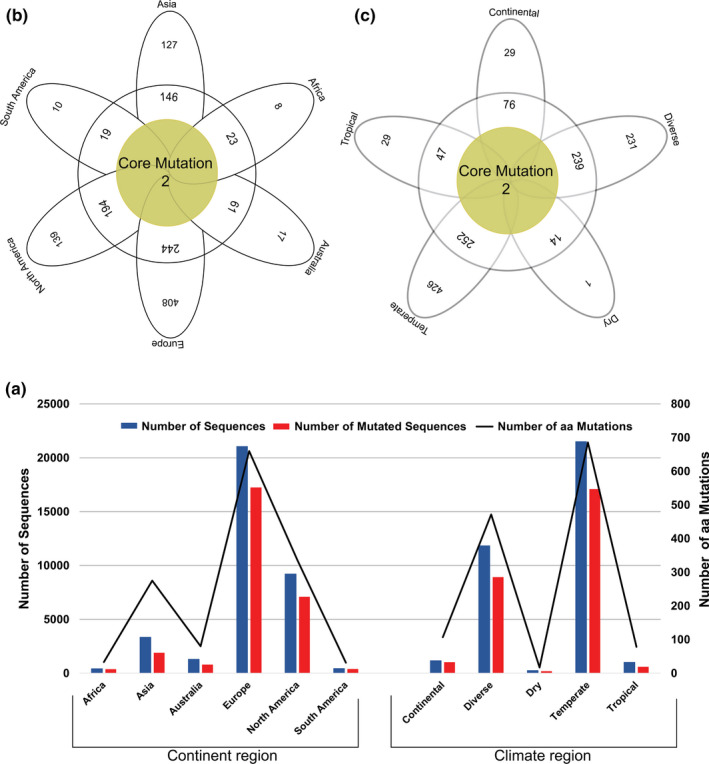
The frequency spectra of aa mutations in the S protein of the SARS‐CoV‐2. (a) Number of sequences, number of mutated sequences and number of aa mutations with respective to continent and climate region. Number of aa mutation in Africa, Asia, Australia, Europe, North America and South America were 33, 275, 80, 660, 335 and 31, respectively, and those were in continental, diverse, dry, temperate and tropical climatic conditions 107, 472, 17, 686 and 78, respectively. The aa mutations are represented according to (b) geographic areas and (c) different climate zones. We found two core shared aa mutations at residue position 5 (L5F) and 614 (D614G) in Asia, Europe, Africa, Australia, North America and South America, and two core shared mutations at residue positions of 614 (D614G) and 936 (D936Y) in continental, diverse, dry, tropical and temperate climatic conditions. In both cases (b and c), the middle brown circles represent frequency of aa substitutions shared by all variables, and the frequency of aa substitutions shared by at least two continents/climate zones are shown in white circle. The white coloured outer ribbons represent unique aa mutations in each individual region and climate zone [Colour figure can be viewed at wileyonlinelibrary.com]
This study also explores the non‐synonymous mutations in the S protein of the SARS‐CoV‐2 genomes across five different climatic conditions worldwide. We found significant (p = .021, Kruskal–Wallis test) variations in aa mutation patterns in different climatic conditions keeping the highest (p = .017, Kruskal–Wallis test) frequency of unique aa mutations in the temperate region. Our analysis showed that only two core aa substitutions at positions 614 (D614G) and 936 (D936Y) were shared across all the climatic zones (Figure 5c). Furthermore, 426, 231, 29, 29 and 1 unique aa replacement were found in the S protein sequences of the temperate, diverse, tropical, continental and dry climatic conditions, respectively. In addition, we also identified 252, 239, 47, 76 and 14 shared aa replacements in temperate, diverse, tropical, continental and dry climatic conditions, respectively, where non‐synonymous mutations occurred in at least two climatic zones (Figure 5c, Data S3). RNA viruses like SARS‐CoV‐2 might have remarkable capabilities to adapt to new environments and confront different selective pressures they encounter (Watanabe et al., 2020). The mutational evolution geographic and climatic patterns of mutational evolution of SARS‐CoV‐2 S protein were visualized in Figure 6.
Figure 6.
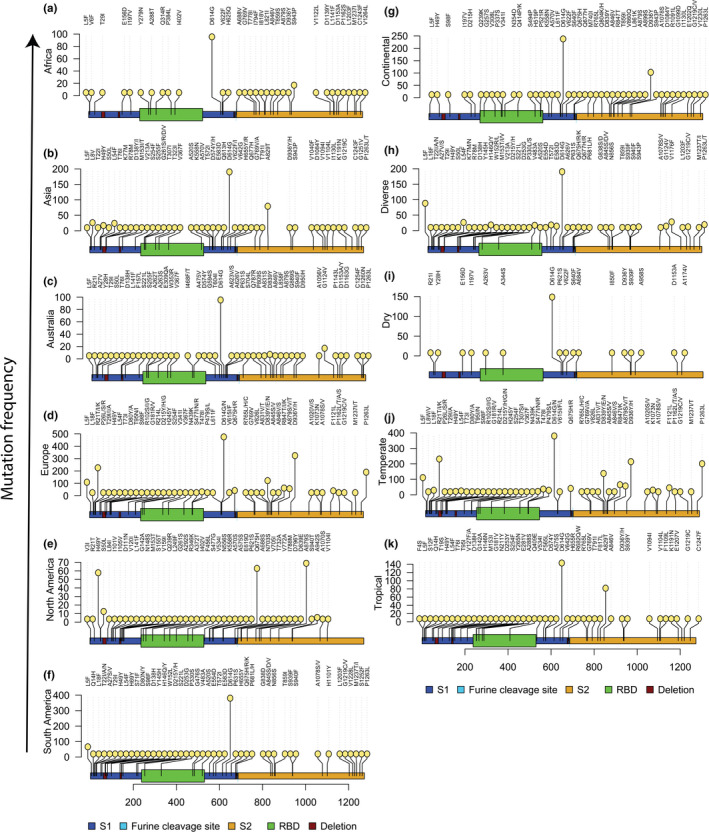
Variations in aa mutations across the S protein sequences of the SARS‐CoV‐2 according to different geographic regions (continental) (a‐f) and climatic conditions (g‐k). Top 45 high‐frequency mutations in each domain (continent or climatic zone) were considered for Lolliplot visualization. We found 614 positions where mutations occurred in 1,481, 377, 743, 16,842, 6,880 and 428 S protein sequences of Asia, Africa, Australia, Europe, North America and South America, respectively, and 959, 8,654, 156, 16,531 and 435 sequences in continental, diverse, dry, temperate and tropical condition, respectively. We normalized the graphs keeping position 614 as the highest mutational frequency in each graph [Colour figure can be viewed at wileyonlinelibrary.com]
The genomic variability of SARS‐CoV‐2 strains manifested by mutations in the spike protein scattered across the globe underlay geographically specific aetiological effects. One important effect of mapping mutations is the development of antiviral therapies targeting specific regions, for example the spike region of the SARS‐CoV‐2 genomes (Callaway, 2020). Our current findings corroborate the study completed by Deshwal (2020), who reported the highest SARS‐CoV‐2 infections and case fatality rates in European countries. In another study, Pachetti et al. (2020) reported two non‐synonymous mutations (R203K and L3606F) that were shared across ORFs of the SARS‐CoV‐2 genomes of six continents, and recurrent mutations were also common in different countries along with unique mutations. Nevertheless, mutations in the structural proteins of the SARS‐CoV‐2, especially in the spike proteins, are driven by the geographic locations that diverged differently, possibly due to the environment, demography and the low fidelity of reverse transcriptase (Brassey et al., 2020; Pachetti et al., 2020; Su et al., 2016).
In this study, we found 14.16%, 11.72%, 10.05%, 9.31%, 3.30%, 3.00%, 2.30%, 2.07%, 1.65% and 1.63% case fatality rates in United Kingdom Italy, France, Spain, Belgium, Germany, Russia, Netherlands, Sweden and Turkey, respectively (Data S3). Among the tropical Asian countries, higher mortality rates from SARS‐CoV‐2 infections were estimated in Iran (4.76%), India (4.72%), China (2.56%), Pakistan (1.38%) and Indonesia (1.11%), and rest of the countries had less than 1.0% case fatality rates. Moreover, in the diverse climatic conditions of the American countries or territories (both North and South Americans), the United States (5.67%) and Brazil (5.14%) had relatively higher mortality rates from SARS‐CoV‐2 pandemics, and rest of the countries in these continents had substantially lower disease severity rates (<1.0%). Case fatality or mortality rates from SARS‐CoV‐2 infections in rest of the two continents (Africa and Australia) remained much lower, and only 2.19%, 1.40% and 1.26% death rates were found in South Africa, Australia and Algeria, respectively. The rest of the countries and/or territories of these two continents had less than 1.0% mortality rates (Data S3).
The predominantly higher mortality rates and unique aa mutations in the S protein sequences of the many European temperate countries might be associated with higher number of SARS‐CoV‐2 genome sequences deposited to the global databases like GISAID during that time compared to other continents. However, the correlation of such higher aa mutation frequency with viral pathogenesis needs to be ascertained. Moreover, it is worth noting that reported disease severity (may not represent the actual severity) might be affected by several other factors like healthcare facilities, average age group and genetic context of the population and control strategies adopted by the countries. Irrespective of the significance of geography for emerging infectious disease epidemiology, the effects of global mobility upon the genetic diversity, and molecular evolution of SARS‐CoV‐2 are under‐appreciated and only beginning to be understood. The recent monograph on the spatial epidemiology of COVID‐19 makes no reference to the genetic disparity of SARS‐CoV‐2 (Brassey et al., 2020; Harvey, 2020; Pachetti et al., 2020; Su et al., 2020).
3.5. Mutational comparison of the S proteins of SARS‐CoV‐2, SARS‐CoV and BatCoV
We compared the S protein mutations of the SARS‐CoV‐2 with the SARS‐CoV reference strain (NCBI accession no. NC_004718) and Bat coronavirus RaTG13 strain (NCBI accession no. MN996532). The identity, similarity and gap of the S protein between the Wuhan strain of the SARS‐CoV‐2 and RaTG13 were 97.3%, 98.3% and 0.4%, respectively, and those between the Wuhan strain SARS‐CoV‐2 and SARS‐CoV were 76.2%, 86.9% and 2.1%, respectively (Table S1). These findings are in line with many of the previously published reports (Swatantra Kumar et al., 2020; Tang et al., 2020; Wrapp et al., 2020). We found mutations in the variable regions between SARS‐CoV‐2 and RaTG13, and these recurrent mutations (S50L, T76I, A372T, N439K) are supposed to be converted to RaTG13 from SARS‐CoV‐2 (Table S1). Furthermore, we also found 45 mutation sites in the variable regions between the SARS‐CoV‐2 and SARS‐CoV which resulted in the conversion of SARS‐CoV‐2 to SARS‐CoV (Figure 7).
Figure 7.
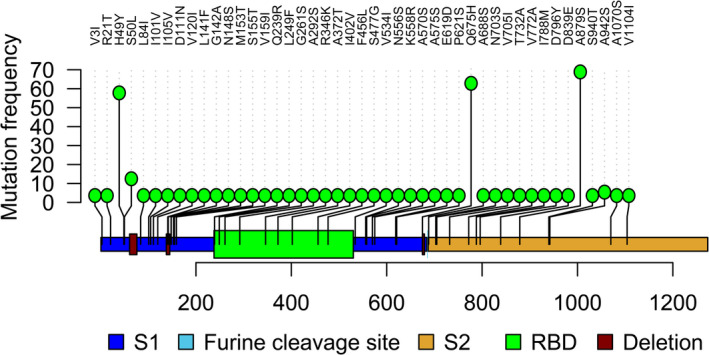
Lolliplot mapping of mutational conversion from SARS‐CoV‐2 to SARS‐CoV with their frequency. We identified 45 sites in the SARS‐CoV‐2 S protein with substitutions resulting in aa homogeneity with the S protein of SARS‐CoV [Colour figure can be viewed at wileyonlinelibrary.com]
The RaTG13 genome possessed a deletion site (681–684 aa) in respect to the SARS‐CoV‐2 genome, and we also found deletions at a very close position (675–679 aa) in two strains of SARS‐CoV‐2 (Table 2). The SARS‐CoV also possessed deletions in respect to the Wuhan reference strain of the SARS‐CoV‐2 at aa positions (72–78, 144–147, 243–247, 256–257, 679–682). In this study, we also found deletion at different aa positions (61–76, 138–145, 241–244, 675–679) in different strains of SARS‐CoV‐2. Therefore, these types of deletions suggest that different strains of the SARS‐CoV‐2 are acquiring the traits of SARS‐CoV. Moreover, a recent study reported that the S1 protein of the Pangolin‐CoV is much more closely related to SARS‐CoV‐2 than to RaTG13 (Uddin et al., 2020). However, this phenomenon of evolving mutations and/or recurrent mutations should be interpreted using a larger dataset from different host populations and geo‐climatic conditions.
4. CONCLUSIONS
Our findings on non‐synonymous mutations in the spike protein of SARS‐CoV‐2 genomes suggest that the virus is continuously evolving. European, North American and Asian strains might coexist where each of them were characterized by a different mutation patterns. Moreover, the geo‐climatic distribution of the recurrent mutations in the spike deciphered a plausible link to higher mutations rates and disease severity in the European temperate countries. However, the geo‐climate effects of the observed mutations in the spike protein of SARS‐CoV‐2 on the properties of the diverse strain variants are yet to be evaluated in clinical or experimental studies. Therefore, these results need to be interpreted cautiously given the existing uncertainty about SARS‐CoV‐2 genomic data to develop potential prophylaxis and mitigation for tackling the COVID‐19 pandemic crisis. Therefore, the fast and accurate pipeline will help in an easy and accurate way to investigate the synonymous/non‐synonymous mutation, mutation frequency and deletion analysis from large number of data with a shortest possible time without having in‐depth bioinformatics knowledge.
CONFLICTS OF INTEREST
The authors of this manuscript declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
MSR, MRI, MNH, ASMRUA, MA, JA and SA conducted the overall study. MSR, MRI and MNH drafted the manuscript. MNH finally compiled the manuscript. AA, MS and MAH contributed intellectually to the interpretation and presentation of the results.
ETHICAL APPROVAL
We confirm that the ethical policies of the journal, as noted on the journal's authors guideline page, have been adhered to. No ethical approval was required since the study did not include any animal or human sample.
Supporting information
Table S1
Data S1
Data S2
Data S3
ACKNOWLEDGEMENTS
The authors sincerely appreciate the researchers worldwide who had deposited and shared the complete genomes data of SARS‐CoV‐2 and other coronaviruses to GISAID (https://www.gisaid.org/). This research utilized these precious data. The authors would also like to extend thanks to Geni Gueiros who was kind to modify his tools (Sequence cleaner) upon request from Md. Shaminur Rahman.
Rahman MS, Islam MR, Hoque MN, et al. Comprehensive annotations of the mutational spectra of SARS‐CoV‐2 spike protein: A fast and accurate pipeline. Transbound Emerg Dis. 2021;68:1625–1638. 10.1111/tbed.13834
Rahman, Islam, and Hoque equally contributed to this study.
DATA AVAILABILITY STATEMENT
This study utilized the SARS‐CoV‐2 genome sequences retrieving from the publicly available open database, GISAID. Detailed step‐by‐step methods are described in Mutation_analysis.pdf (https://github.com/SShaminur/Mutation‐Analysis).
REFERENCES
- Ahmed, S. F. , Quadeer, A. A. , & McKay, M. R. (2020). Preliminary identification of potential vaccine targets for the COVID‐19 coronavirus (SARS‐CoV‐2) based on SARS‐CoV immunological studies. Viruses, 12(3), 254. 10.3390/v12030254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armijos‐Jaramillo, V. , Yeager, J. , Muslin, C. , & Perez‐Castillo, Y. (2020). SARS‐CoV‐2, an evolutionary perspective of interaction with human ACE2 reveals undiscovered amino acids necessary for complex stability. Evolutionary Applications, 13, 2168–2178. 10.1111/eva.12980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer, C. F. (2008). Does mutation rate depend on itself. PLoS Biology, 6(2), e52. 10.1371/journal.pbio.0060052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal, A. , Destras, G. , Gaymard, A. , Bouscambert‐Duchamp, M. , Valette, M. , Escuret, V. , & Cheynet, V. (2020). Molecular characterization of SARS‐CoV‐2 in the first COVID‐19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clinical Microbiology and Infection, 26(7), 960–962. 10.1016/j.cmi.2020.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brassey, J. , Heneghan, C. , Mahtani, K. R. , & Aronson, J. K. (2020). Do weather conditions influence the transmission of the coronavirus (SARS‐CoV‐2)? Centre for Evidence‐Based Medicine, Nuffield Department of Primary Care Health Sciences, University of Oxford, March 22, 2020. https://www.cebm.net/covid‐19/do‐weather‐conditions‐influence‐the‐transmission‐of‐the‐coronavirus‐sars‐cov‐2/. [Google Scholar]
- Callaway, E. (2020). Coronavirus vaccines: Five key questions as trials begin. Nature, 579(7800), 481. 10.1038/d41586-020-00798-8 [DOI] [PubMed] [Google Scholar]
- Comandatore, F. , Chiodi, A. , Gabrieli, P. , Biffignandi, G. B. , Perini, M. , Ramazzotti, M. , & Micheli, V. (2020). Identification of variable sites in Sars‐CoV‐2 and their abundance profiles in time. BioRxiv. 10.1101/2020.04.30.071027. [DOI] [Google Scholar]
- Coutard, B. , Valle, C. , de Lamballerie, X. , Canard, B. , Seidah, N. , & Decroly, E. (2020). The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Research, 176, 104742. 10.1016/j.antiviral.2020.104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, M. (2017). Statistics for managers, using Microsoft excel: Pearson Education India. https://books.google.com.bd/books?hl=en&lr=&id=yIqqDwAAQBAJ&oi=fnd&pg=PP1&dq=Statistics+for+managers,+using+Microsoft+excel:+Pearson+Education+India&ots=flkza77qbR&sig=xTwrnFqS06zog65yAs_GeJ6EANY&redir_esc=y#v=onepage&q=Statistics%20for%20managers%2C%20using%20Microsoft%20excel%3A%20Pearson%20Education%20India&f=false. [Google Scholar]
- DeLano, W. L. (2002). The PyMOL molecular graphics system. http://www.pymol.org [Google Scholar]
- Drake, J. W. , & Holland, J. J. (1999). Mutation rates among RNA viruses. Proceedings of the National Academy of Sciences, 96(24), 13910–13913. 10.1073/pnas.96.24.13910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshwal, V. K. (2020). COVID 19: A comparative study of Asian, European, American continent. Int J Sci Res Enginee Dev, 3(2), 436–440. http://www.ijsred.com/volume3/issue2/IJSRED-V3I2P63.pdf [Google Scholar]
- Duffy, S. (2018). Why are RNA virus mutation rates so damn high? PLoS Biology, 16(8), e3000003. 10.1371/journal.pbio.3000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaaswarkhanth, M. , Al Madhoun, A. , & Al‐Mulla, F. (2020). Could the D614 G substitution in the SARS‐CoV‐2 spike (S) protein be associated with higher COVID‐19 mortality? International Journal of Infectious Diseases, 96, 459–460. 10.1016/j.ijid.2020.05.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Boronat, M. , Diez‐Rivero, C. M. , Reinherz, E. L. , & Reche, P. A. (2008). PVS: A web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Research, 36(suppl_2), W35–W41. 10.1093/nar/gkn211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, O. C. , Montgomery, D. , Ito, K. , & Woods, R. J. (2020). 3D Models of glycosylated SARS‐CoV‐2 spike protein suggest challenges and opportunities for vaccine development. BioRxiv. 10.1101/2020.04.07.030445. [DOI] [Google Scholar]
- Harvey, C. What Could Warming Mean for Pathogens like Coronavirus? E&E News, March 9, (2020). https://www.scientificamerican.com/article/what-could-warming-mean-for-pathogens-like-coronavirus/. [Google Scholar]
- Hoque, M. N. , Istiaq, A. , Clement, R. A. , Sultana, M. , Crandall, K. A. , Siddiki, A. Z. , & Hossain, M. A. (2019). Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Scientific Reports, 9(1), 1–14. 10.1038/s41598-019-49468-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, M. R. , Hoque, M. N. , Rahman, M. S. , Puspo, J. A. , Akhter, M. , Akter, S. , … Hossain, M. A. (2020). Genome‐wide analysis of SARS‐CoV‐2 virus strains circulating worldwide implicates heterogeneity. Scientific reports, 10(1), 1–9. 10.1038/s41598-020-70812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat, E. , Wu, T. T. , & Bilofsky, H. (1977). Unusual distributions of amino acids in complementarity determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody‐combining sites. Journal of Biological Chemistry, 252(19), 6609–6616. [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K. I. , & Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.‐S. , Jang, J.‐H. , Kim, J.‐M. , Chung, Y.‐S. , Yoo, C.‐K. , & Han, M.‐G. (2020). Genome‐Wide Identification and Characterization of Point Mutations in the SARS‐CoV‐2 Genome. Osong Public Health and Research Perspectives, 11(3), 101. 10.24171/j.phrp.2020.11.3.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissler, S. M. , Tedijanto, C. , Goldstein, E. , Yonatan, H. G. , & Lipsitch, M. (2020). Projecting the transmission dynamics of SARS‐CoV‐2 through the postpandemic period. Science. 368(6493), 860–868. 10.1126/science.abb5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber, B. , Fischer, W. M. , Gnanakaran, S. , Yoon, H. , Theiler, J. , Abfalterer, W. , & Foley, B. (2020). Tracking changes in SARS‐CoV‐2 Spike: Evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 182(4), 812–827. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Maurya, V. K. , Prasad, A. K. , Bhatt, M. L. , & Saxena, S. K. (2020). Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019‐nCoV) and SARS coronavirus (SARS‐CoV). Virus Disease, 31(1), 1–9. 10.1007/s13337-020-00571-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S.‐Y. , Wang, P. , Mok, B.‐ W.‐Y. , Zhang, A. J. , Chu, H. , Lee, A.‐ C.‐Y. , & Song, W. (2020). Attenuated SARS‐CoV‐2 variants with deletions at the S1/S2 junction. Emerging Microbes & Infections, 9(1), 837–842. 10.1080/22221751.2020.1756700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Zheng, H. , Lin, H. , Li, L. , Yuan, R. , Peng, J. , … Wu, J. (2020). Identification of common deletions in the spike protein of SARS‐CoV‐2. Journal of virology, 94(17), 1–20. 10.1128/JVI.00790-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe, L. , & Hill, W. G. (2010). The population genetics of mutations: Good, bad and indifferent. Phil. Trans. R. Soc. B. 365, 1153–1167. 10.1098/rstb.2009.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, J. , Wang, Y.‐X. , Zhu, L. J. , Ou, M. J. , GenomicAlignments, I. , GenomicFeatures, G. , & BiocGenerics, B. (2020a). Package ‘trackViewer’. https://bioconductor.org/packages/release/bioc/manuals/trackViewer/man/trackViewer.pdf. [Google Scholar]
- Ou, X. , Liu, Y. , Lei, X. , Li, P. , Mi, D. , Ren, L. , & Hu, J. (2020b). Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nature Communications, 11(1), 1–12. 10.1038/s41467-020-15562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachetti, M. , Marini, B. , Benedetti, F. , Giudici, F. , Mauro, E. , Storici, P. , & Gallo, R. C. (2020). Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. Journal of Translational Medicine, 18, 1–9. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, A. J. , Taylor, B. , Delaney, A. J. , Soares, J. , Seemann, T. , Keane, S. J. , & Harris, S. R. (2016). SNP‐sites: rapid efficient extraction of SNPs from multi‐FASTA alignments. Microbial Genomics, 2(4), 1–5. 10.1099/mgen.0.000056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan, T. (2020). Genetic diversity and evolution of SARS‐CoV‐2. Infection, Genetics and Evolution, 81, 104260. 10.1016/j.meegid.2020.104260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, M. S. , Hoque, M. N. , Islam, M. R. , Akter, S. , Rubayet‐Ul‐Alam, A. , Siddique, M. A. , & Crandall, K. A. (2020). Epitope‐based chimeric peptide vaccine design against S, M and E proteins of SARS‐CoV‐2, the etiologic agent of COVID‐19 pandemic: An in silico approach. PeerJ, 8, e9572. 10.7717/peerj.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar, R. , Satish, D. , Birla, S. , & Gupta, D. (2020). Comparative analyses of SAR‐CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host‐virus interaction and pathogenesis. BioRxiv. 10.1101/2020.03.21.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, W. , Yang, Y. , Rao, Y. , & Rao, X. (2020). The outbreak of SARS‐CoV‐2 pneumonia calls for viral vaccines. NPJ Vaccines, 5(1), 1–3. 10.1038/s41541-020-0170-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, W. , Le, S. , Li, Y. , & Hu, F. (2016). SeqKit: A cross‐platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One, 11(10), e0163962. 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, T. (2015). Snippy: rapid haploid variant calling and core SNP phylogeny. Available, https://github.com/tseemann/snippy
- Su, S. , Wong, G. , Shi, W. , Liu, J. , Lai, A. C. , Zhou, J. , & Gao, G. F. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in Microbiology, 24(6), 490–502. 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. , Wu, C. , Li, X. , Song, Y. , Yao, X. , Wu, X. , & Qian, Z. (2020). On the origin and continuing evolution of SARS‐CoV‐2. National Science Review, 7(6), 1012–1023. 10.1093/nsr/nwaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The World Health Organization (WHO) . (2020). Novel Coronavirus (2019‐nCoV) Situation Reports (WHO, Geneva, 2020). https://apps.who.int/iris/bitstream/handle/10665/330762/nCoVsitrep23Jan2020-eng.pdf
- Trucchi, E. , Gratton, P. , Mafessoni, F. , Motta, S. , Cicconardi, F. , Bertorelle, G. , & Di Marino, D. (2020). Unveiling diffusion pattern and structural impact of the most invasive SARS‐CoV‐2 spike mutation. BioRxiv. 10.1101/2020.05.14.095620. [DOI] [Google Scholar]
- Uddin, M. , Mustafa, F. , Rizvi, T. A. , Loney, T. , Suwaidi, H. A. , Al‐Marzouqi, A. H. H. , & Stefanini, C. (2020). SARS‐CoV‐2/COVID‐19: Viral genomics, epidemiology, vaccines, and therapeutic interventions. Viruses, 12(5), 526. 10.3390/v12050526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y.‐J. , Tortorici, M. A. , Wall, A. , McGuire, A. T. , & Veesler, D. (2020). Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell, 181(2), 281–292. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Zhang, Y. , Wu, L. , Niu, S. , Song, C. , Zhang, Z. , & Yuen, K.‐Y. (2020). Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell. 181(4), 894–904. 10.1016/j.cell.2020.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, Y. , Allen, J. D. , Wrapp, D. , McLellan, J. S. , & Crispin, M. (2020). Site‐specific glycan analysis of the SARS‐CoV‐2 spike. Science, 369(6501), 330–333. 10.1126/science.abb9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, A. , Bertoni, M. , Bienert, S. , Studer, G. , Tauriello, G. , Gumienny, R. , & Bordoli, L. (2018). SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303. 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2011). ggplot2. Wiley Interdisciplinary Reviews: Computational Statistics, 3(2), 180–185. 10.1002/wics.147 [DOI] [Google Scholar]
- Wrapp, D. , Wang, N. , Corbett, K. S. , Goldsmith, J. A. , Hsieh, C.‐L. , Abiona, O. , & McLellan, J. S. (2020). Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science, 367(6483), 1260–1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. , Liu, Y. , Yang, Y. , Zhang, P. , Zhong, W. , Wang, Y. , & Li, X. (2020). Analysis of therapeutic targets for SARS‐CoV‐2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B, 10(5), 766–788. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, C. (2020). Genotyping coronavirus SARS‐CoV‐2: Methods and implications. Genomics, 112(5), 3588–3596. 10.1016/j.ygeno.2020.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, M. , Wu, N. C. , Zhu, X. , Lee, C.‐C.‐ D. , So, R. T. , Lv, H. , & Wilson, I. A. (2020). A highly conserved cryptic epitope in the receptor binding domains of SARS‐CoV‐2 and SARS‐CoV. Science, 368(6491), 630–633. 10.1126/science.abb7269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Chen, Y. , Zhang, S. , Niu, P. , Qin, K. , Jia, W. , & Zhang, L. (2019). Structural definition of a neutralization epitope on the N‐terminal domain of MERS‐CoV spike glycoprotein. Nature Communications, 10(1), 1–13. 10.1038/s41467-019-10897-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data S1
Data S2
Data S3
Data Availability Statement
This study utilized the SARS‐CoV‐2 genome sequences retrieving from the publicly available open database, GISAID. Detailed step‐by‐step methods are described in Mutation_analysis.pdf (https://github.com/SShaminur/Mutation‐Analysis).