

Abstract
The recently realized biochemical phenomenon of energy conservation through electron bifurcation provides biology with an elegant means to maximize utilization of metabolic energy. The mechanism of coordinated coupling of exergonic and endergonic oxidation-reduction reactions by a single enzyme complex has been elucidated through optical and paramagnetic spectroscopic studies revealing unprecedented features. Pairs of electrons are bifurcated over more than 1 volt of electrochemical potential by generating a low-potential, highly energetic, unstable flavin semiquinone and directing electron flow to an iron-sulfur cluster with a highly negative potential to overcome the barrier of the endergonic half reaction. The unprecedented range of thermodynamic driving force that is generated by flavin-based electron bifurcation accounts for unique chemical reactions that are catalyzed by these enzymes.
Flavin-based electron bifurcation has recently gained acceptance as a fundamental mechanism of biological energy conservation, in addition to substrate-level phosphorylation and electron-transport-linked phosphorylation1–3. Bifurcation is essential to the biochemistry that drives microbial life at the thermodynamic limits observed for global anaerobic processes, including methanogenesis, acetogenesis and hydrogen metabolism4–6. A key feature common to all bifurcating enzymes is the ability to use free energy generated by an exergonic oxidation-reduction reaction to drive a coupled endergonic reaction. Although general aspects of the mechanism have been inferred, the specific details of how electron bifurcation mechanistically occurs remain unknown3,7–12. This study demonstrates how the flavin-based bifurcating enzyme NADH-dependent ferredoxin-NADP+ oxidoreductase I (Nfn) catalyzes the formation of an energy-rich product, reduced ferredoxin (Fd), from the less energetic donor, NADPH, by coupling this reaction to the thermodynamically favorable reduction of NAD+ by NADPH:
Electron bifurcation is involved in modulating the redox state of the pyridine nucleotide pools to balance catabolic (NADH-dependent) and anabolic (NADPH-dependent) reactions during anaerobic metabolism. Flux in anabolic and catabolic reactions as well as the state of the NADH/NAD+ and NADPH/NADP+ redox pools are not in thermodynamic equilibrium. The ratio of NADPH to NADP+ is generally >1, whereas NADH/NAD+ is generally <1, such that conversion of NADPH to NAD+ is exergonic. In most anaerobic organisms, a driving force from an additional cellular electron carrier, a low-potential iron-sulfur-containing protein, Fd (midpoint potential (Em) ≈ −450 mV), is coupled to the interconversion of NADPH and NADH through the activity of Nfn to result in a thermodynamically favorable NADH-dependent reduction of NADP+.
Conceptually, electron bifurcation was first conceived to explain the so-called Q cycle of the quinone-based system, catalyzed by Complex III of the aerobic respiratory chain that is dependent on the two-electron redox cofactor quinol2. Flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN) is used as a two-electron redox cofactor in flavin-based bifurcating systems that generate low-potential, high-energy compounds such as hydrogen gas13 and reduced ferredoxin. Flavin-based bifurcation is as important for energy coupling in anaerobic metabolism as the quinone-based system is in aerobic metabolism. Moreover, it is evident that electron bifurcation is widespread among all domains of life1,5,6,14, yet a fundamental mechanistic understanding that is essential for explaining the energetics of these processes remains elusive.
Here we show that the high-barrier reaction is driven by formation of an unstable flavin anionic semiquinone (ASQ) intermediate that promotes one of the electrons from the two-electron donor NADPH to a highly reduced state sufficient to reduce ferredoxin. This flavin intermediate has not been previously observed because of its high reactivity. Its characterization, along with the energetic landscape of the additional redox cofactors in Nfn, allows us to construct a mechanistic understanding of how electrons are bifurcated over more than 1 volt of electrochemical potential, a range typically associated with photo-based biology15. An understanding of how this reaction is achieved in Nfn will not only guide the design of novel catalysts with large driving forces and high efficiencies, but also lead to advances in our understanding of metabolic pathways and the design of new bioconversion processes through synthetic biology.
RESULTS
The exergonic branch of electron transfer
The previously reported structure of Nfn from Thermotoga martima (Tm)7 and the high-resolution structure of Pyrococcus furiosus (Pf NfnI) reported here (Fig. 1; Supplementary Results, Supplementary Fig. 1 and Supplementary Table 1) reveal a heterodimeric structure, composed of a large (L) subunit and a small (S) subunit, that harbors two cofactor chains that form bifurcated electron transfer pathways from a central FAD (L-FAD) in the large subunit. These structures provide a framework for probing the cofactors that are required for electron bifurcation by Nfn, the unprecedented properties of which are elucidated here through optical and electron paramagnetic spectroscopic studies. Reduction of both NAD+ and Fd by Nfn enzymes is coupled to the bifurcation of electrons from NADPH oxidation. The strict substrate requirement for activity has been assessed for Pf NfnI by electrochemical as well as solution-based assays and is comparable to that of Nfn from other organisms (Supplementary Table 2). The exergonic coupling of the NADP(H) and NAD(H) half reactions in Nfn is presumed to be mediated by NADPH oxidation at L-FAD followed by electron transfer to the proximal [2Fe-2S] cluster and subsequently the NAD(H) binding site at S-FAD (Figs. 1 and 2a). The [2Fe-2S] cluster is unusual, with a relatively high (positive) reduction potential (Em8 = +80 mV, pH 8; ref. 16) and coordination by one Asp and three Cys ligands. The transport of electrons from L-FAD along this pathway therefore transitions through this thermodynamic barrier to reduce oxidized (OX) S-FAD (two-electron OX/HQ couple Em8 = −276 mV; Supplementary Table 3).
Figure 1 |. Pf NfnI structure and orientation of cofactors.
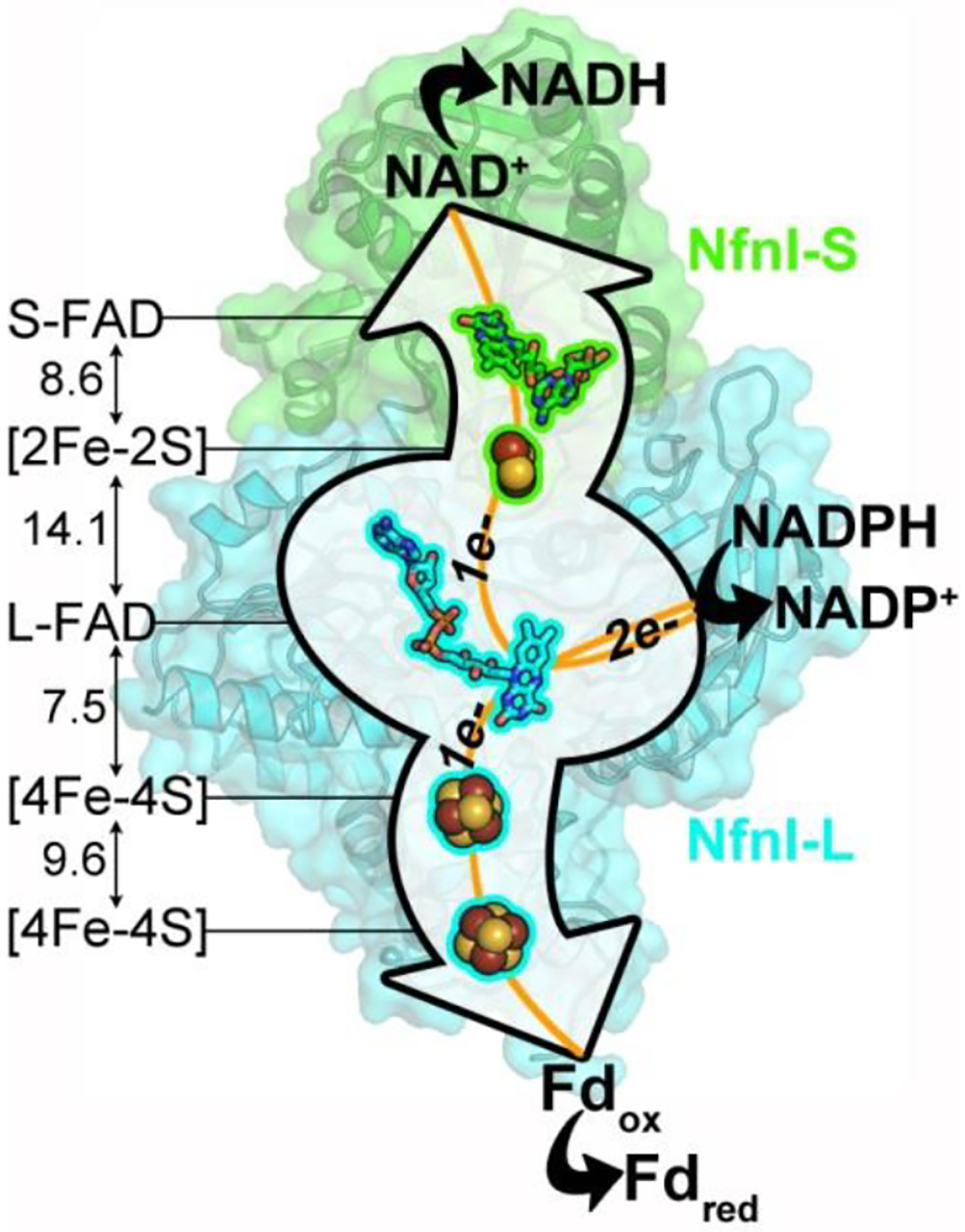
Pf NfnI is a heterodimer of 31 kDa NfnI-S and 53 kDa NfnI-L. NfnI-S contains S-FAD and a [2Fe-2S] cluster with an unusual Asp ligand. NfnI-L contains one FAD (L-FAD), which is the site of electron bifurcation, and two [4Fe-4S] clusters. The L-FAD proximal [4Fe-4S] cluster coordination includes an unusual Glu ligand. S-FAD and L-FAD bind NADH and NADPH, respectively. Ferredoxin (Fd) is proposed to bind near the distal [4Fe-4S] cluster of NfnI-L. Edge-to-edge distances between cofactors are given in Ångström (Å). Pf NfnI shows sequence and structural similarity to the flavin-based electron bifurcating enzyme from Thermotoga maritima (NfnAB, PDB 4YRY). Fdox, oxidized Fd; Fdred, reduced Fd.
Figure 2 |. Exergonic branch for electron transfer between NADP(H) and NAD(H) in Pf NfnI.
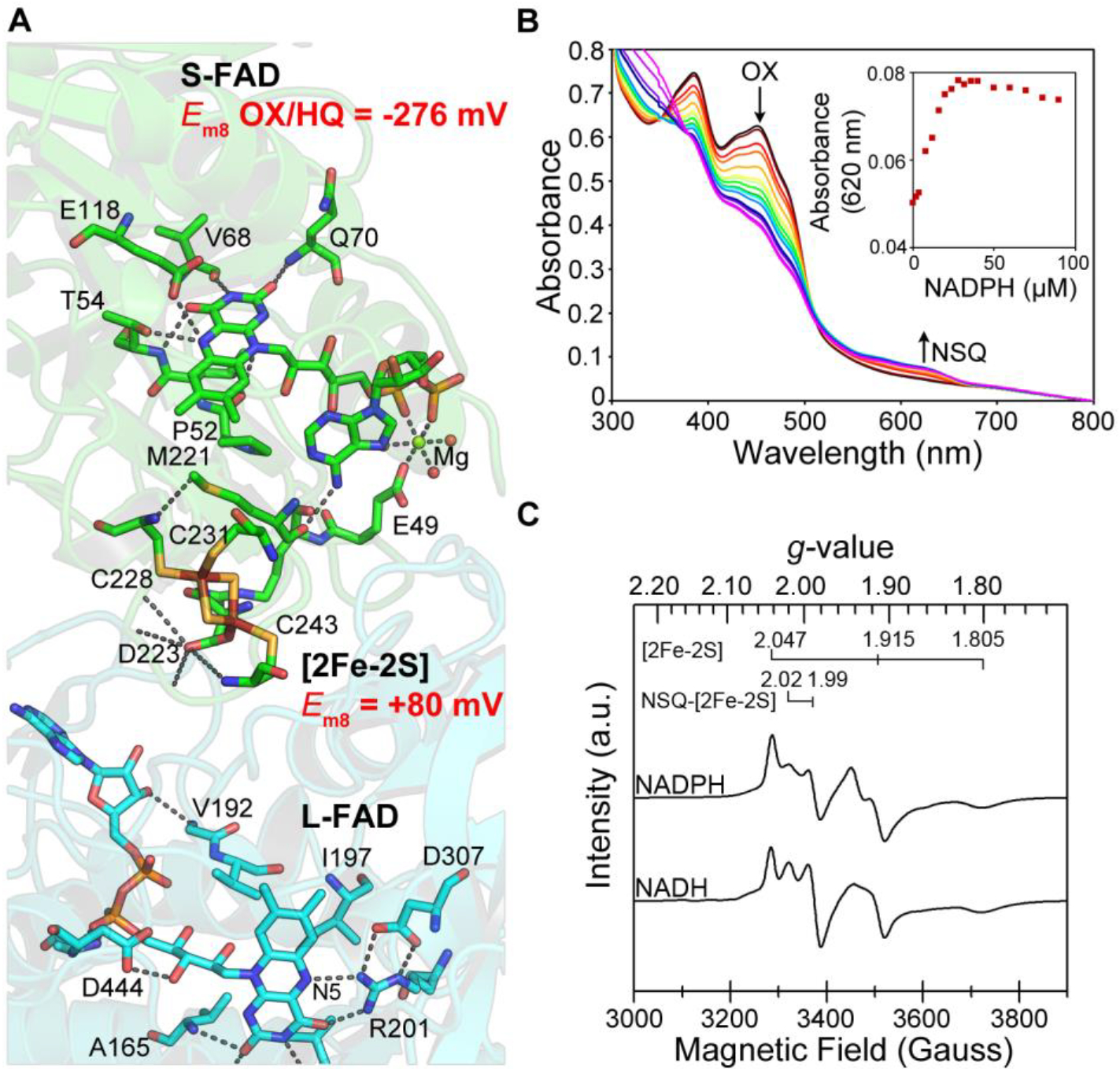
(a) Electron-transfer pathway between L-FAD and S-FAD. Em8 values are shown for [2Fe-2S] (ref. 16) and S-FAD (determined in this work). (b) A stable neutral semiquinone (NSQ) species of S-FAD is formed by reductive titration with NADPH under steady-state conditions and correlates with a decrease in OX. The increase in NSQ (A620 nm) as a function of NADPH concentration is shown in Supplementary Figure 3. (c) EPR spectra from reduction with NADH or NADPH. a.u., arbitrary units. n = 1 experiment for b and c.
EPR analysis of Pf NfnI after reduction by NAD(P)H revealed a complex spectrum with rhombic signals, consistent with reduced [2Fe-2S]+ and [4Fe-4S]+ clusters (Fig. 2c), as well as an isotropic signal (g = 2.004) resolved at a higher temperature, typical of a flavosemiquinone (Supplementary Fig. 5). An overlapping signal with near axial symmetry in the g = 2 region was also present, which displayed covariant temperature dependence with the [2Fe-2S]+ cluster, gradually shifting to a more uniform isotropic symmetry above 80 K. This signal is thought to arise from spin interaction of a flavosemiquinone species with the reduced [2Fe-2S]+ cluster. A similar spin interaction has also been observed as a g = 2 signal originating from a [2Fe-2S]+ cluster and FMN NSQ in dihydroorotate dehydrogenase B iron-sulfur flavoprotein (DHODB)17, which has homology with Pf NfnI-S. The large line width (28 gauss) for the isotropic signal resolved at higher temperatures, observed both here and for DHODB, can further be explained as arising from spin coupling between the two centers17. Thus, the reduction of Pf NfnI cofactors by the substrate NADPH shows clear evidence of strong spin interaction between [2Fe-2S]+ and S-FAD NSQ. This interaction assists in the efficient transfer of electrons along the exergonic branch during bifurcation.
The endergonic branch of electron transfer
The coupled endergonic half reaction between NADP(H) and Fd is mediated by electron transfer through two [4Fe-4S] clusters: a proximal cluster located near L-FAD and a distal cluster close to the protein surface where Fd is proposed to bind (Figs. 1 and 3a). Square wave voltammetry (SWV) measurements yielded pH-independent oxidation-reduction peaks at Em8 = −513 mV and −718 mV (Fig. 3b and Supplementary Fig. 6), which we assigned to each of the two [4Fe-4S] clusters. Thus, the thermodynamics of this branch are poised to rapidly transfer electrons to Fd (Em8 ≈ −368 mV)18. In agreement with these observations, EPR analysis of reduced Pf NfnI revealed two overlapping [4Fe-4S] signals that change in relative intensity under varying reduction potentials (Fig. 3c and Supplementary Fig. 7). When Pf NfnI was poised at a lower reduction potential (−510 mV compared to −488 mV), the EPR signal intensity increased and adopted a more axial symmetry as a result of an increased contribution from one of the [4Fe-4S]+ cluster signals, presumably the one with a relatively lower Em value. Accordingly, this signal can be assigned to the L-FAD proximal [4Fe-4S]+ cluster with an Em8 = −718 mV from SWV (Fig. 3b). Reduction with titanium citrate also appeared to cause an increased reduction of the low-potential [4Fe-4S]+ cluster signal, although overlap of the cluster and titanium citrate signals in the g = 1.94 region complicated the analysis19. Evidence of spin coupling between the two [4Fe-4S]+ clusters upon reduction at lower potentials was given by the overall EPR signal adopting greater intensity in the middle of the spectrum near gav (average g values) = 1.96–1.98 with accompanying loss in signal rhombicity20. This coupling of the clusters establishes that they form a functional conduit for rapid electron exchange between L-FAD and Fd.
Figure 3 |. Endergonic electron transfer between NADP(H) and Fd in Pf NfnI.
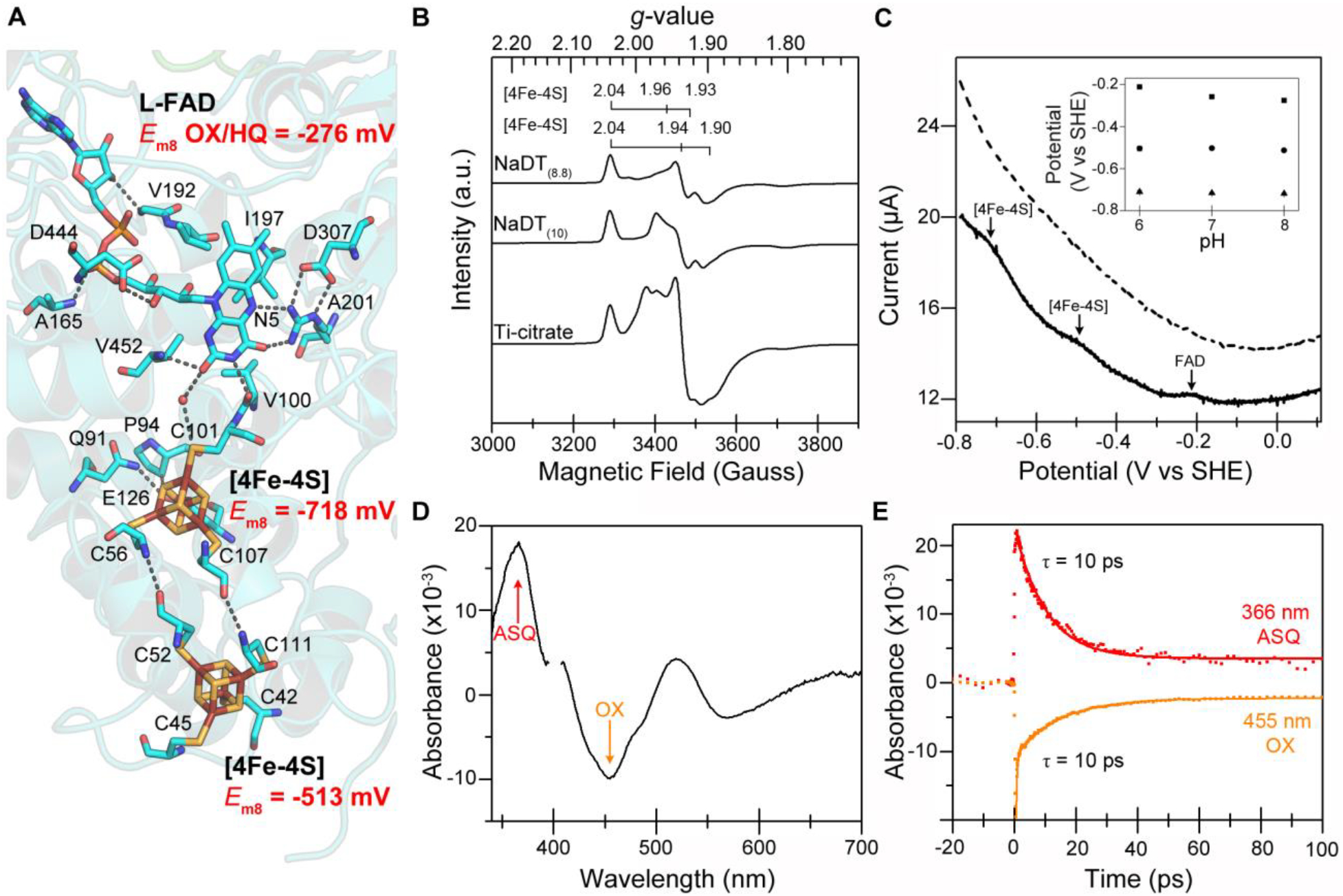
(a) Key residues that act to orient and control flavin reactivity at the bifurcating flavin site. (b) Square wave voltammograms from Pf NfnI adsorbed to an electrode (top) and a protein-free electrode (bottom). The FAD signal arises from both flavins (n = 3 experiments). SHE, standard hydrogen electrode. The pH dependency of the signals is shown in Supplementary Figure 6. (c) Reduction with sodium dithionite (pH 8.8 = −488 mV; pH 10 = −510 mV) or Ti-citrate (scaled by 0.5) was used to assign the two [4Fe-4S] cluster signals. More negative potentials led to relative increases in intensity of the uniquely coordinated proximal [4Fe-4S] cluster signal (g = 2.04, 1.96 and 1.93). (d) TAS difference spectra of Pf NfnI treated with NADPH at a time delay of 3 ps. The L-FAD ASQ absorption (366 nm) and OX bleach (455 nm) are shown. Spectral contributions from the pump at 400 nm have been removed for clarity. (e) TAS kinetic traces (data, points; fit, solid lines) of L-FAD ASQ decay (top) and OX bleach recovery (bottom) are correlated in time (i.e., ASQ absorption arises from OX bleach) with half-lives of 10 ps (ASQ = 10.2 ± 0.2 ps, OX = 10.0 ± 0.7 ps). n = 1 experiment for c; n = 2 experiments for d and e.
The substantial difference between the reduction potentials of the two L-FAD-proximal iron-sulfur clusters (+80 mV versus −718 mV) requires a considerable separation of electron energies during the oxidation of NADPH to accomplish energy conservation. There are two mechanisms by which this might be accomplished: via a low-potential (≤ −718 mV) redox couple of L-FAD SQ/hydroquinone (HQ) or by a low-potential OX/SQ couple, where SQ is a NSQ or ASQ species. A SQ/HQ poised at ≤ −718 mV would present a substantial thermodynamic barrier to reduction of L-FAD by NADPH (Em8 NADP+/NADPH = −380 mV). However, there is precedent for low-potential SQ intermediates for both free flavin21 and the high-energy quinol intermediates in the cytochrome bc1 complex Q cycle22. A low-potential L-FAD SQ would also be expected to be a highly reactive intermediate with a very short lifetime, and our steady-state experiments of Pf NfnI reveal no short-lived intermediates (spectroelectrochemical titration; Supplementary Fig. 8) and only a long-lived stable S-FAD NSQ when reduced with NADPH (Fig. 2b).
Unique properties of the bifurcating flavin site
To probe for L-FAD SQ, we used transient UV-vis absorption spectroscopy (TAS), a high-sensitivity method for observing chemical species that exist on very short time scales (picoseconds to nanoseconds). Upon illumination of NADPH-reduced Pf NfnI, we observed a very short-lived ASQ (absorption peak at 366 nm, τ = 10 ps) that was assigned to the one-electron reduced, bifurcating L-FAD (Fig. 3d,e). This lifetime is a function of electron transfer between L-FAD ASQ and the proximal [4Fe-4S] cluster. Using the driving force and distance dependence of electron transfer23 and the measured ASQ lifetime of 10 ps (Fig. 3e), the reduction potential values for L-FAD can be calculated as EOX/ASQ = −911 mV and EASQ/HQ = +359 mV (pH 8, ΔE = 1.2 V; Supplementary Table 3). The formation of a low-potential ASQ is facilitated by the structure around the N5 position of L-FAD (Supplementary Fig. 4b); the conserved Arg (R201) that is within hydrogen bonding distance of N5 controls the protonation state of L-FAD. We hypothesize that the hydrogen bond between R201 and N5 obstructs protonation of the L-FAD semiquinone, preventing formation of a lower-energy NSQ. In the physiological reaction, the two-electron reduction of L-FAD by NADPH involves a hydride transfer to N5 to form HQ. Formation of an ASQ in the physiological mechanism results from an initial electron and proton transfer event to convert L-FAD HQ to ASQ.
Protein dynamics
Solution-phase analysis of Pf NfnI, with and without NADP(H), by hydrogen-deuterium exchange mass spectrometry (HDX-MS) revealed isotopic exchange in the protein amide backbone, including the region around the L-FAD N5 hydrogen bonding network as well as the surface between the large and small subunits (Fig. 4a). Binding of NADP(H) also resulted in a subtle conformational change that affected the location of the proposed Fd binding site (Supplementary Fig. 9). The solvent-exposed face of L-FAD that is occupied by NADP(H) becomes desolvated, particularly around the N5 R201 (Fig. 4b,c). R201 is part of an extensive hydrogen bonding network that extends toward the protein surface. Thus, the hydrogen bonding and solvation networks at L-FAD N5 are highly controlled during binding and catalytic turnover of NADP(H) to promote the coupling of proton and electron transfer events at L-FAD with binding of substrates NAD(H) and NADP(H) and of Fd.
Figure 4 |. NADP(H) binding affects both short and long range interactions in Pf NfnI.
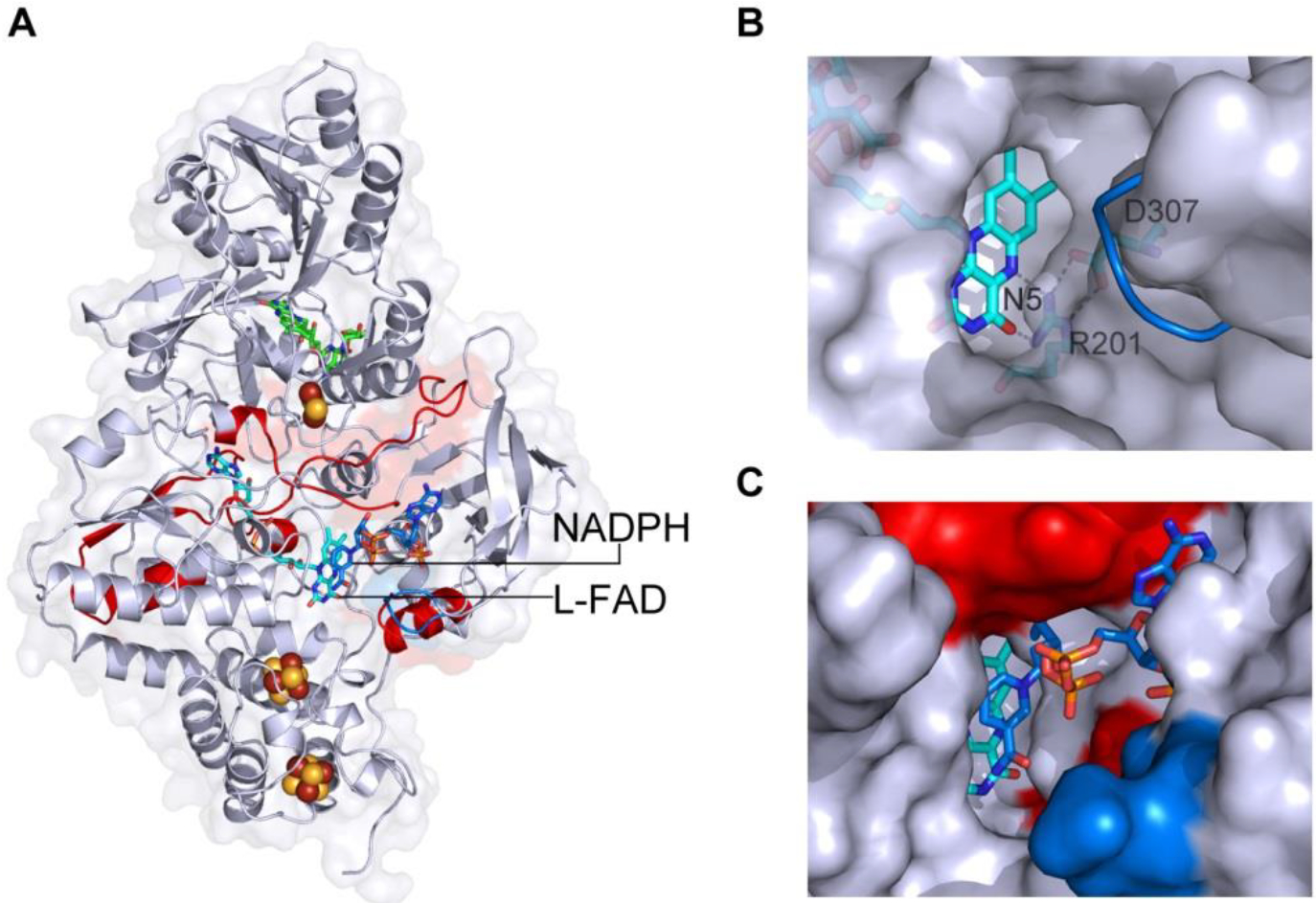
HDX-MS shows proton exchange in backbone residues after NADPH binding (red, more change; blue, less change). Increased exchange (red) with NADP+ occurred at L-FAD toward the protein surface and along the interface between NfnI-S and NfnI-L. (b) Unbound Pf NfnI showing NADP(H) binding cavity with L-FAD (cyan) and N5 interaction region. NADP(H) binding causes a loop rearrangement (blue) as visualized by superimposition of secondary structure of the NADP(H) bound Pf NfnI. (c) NADP(H) binding cavity with bound NADPH (blue sticks) along with HDX changes (red) and structural rearrangements (blue).
DISCUSSION
Both the exergonic and endergonic branches of Pf NfnI contain cofactors that are coupled to establish efficient electron transferring pathways for the reduction of NAD+ and Fd. Additionally, these cofactors operate within a thermodynamic landscape set forth by a unique bifurcating flavin. The reduction potentials of the L-FAD OX/ASQ and ASQ/HQ couples (calculated at −911 mV and +359 mV, respectively) (Supplementary Table 3) are highly crossed24, resulting in a thermodynamically unstable ASQ. This property of ASQ was predicted to be a requirement for flavin-based electron bifurcation24; however, previous experimental evidence for an ASQ in flavin-based bifurcating enzymes has been lacking because of the difficulty in detecting this short-lived, unstable species. Pf NfnI employs the properties of a crossed-potential flavin not only to perform two-electron chemistry, but also to take advantage of a highly negative one-electron couple of L-FAD OX/ASQ to drive the reduction of the low-potential acceptor, ferredoxin. Owing to the higher reduction potential of the EASQ/HQ couple of a crossed-potential L-FAD, the first oxidation of fully reduced L-FAD is directed to the exergonic branch (Supplementary Table 4). The second oxidation, that of the unstable ASQ, is then directed to the endergonic branch, providing enough power to reduce Fd (Supplementary Table 4).
The data indicate that the first electron transfer occurs along the exergonic branch from the L-FAD ASQ/HQ (Em = +359 mV) to the [2Fe-2S] cluster (Em = +80 mV). Although this is an apparent thermodynamic barrier, individual electron transfer steps that have positive changes in potential are possible in an overall thermodynamically favorable reaction (i.e., NADPH to NAD+ has a negative overall ΔG)25,26. This phenomenon is rationalized by the application of Marcus theory to biological electron transfer summarized in the Page-Moser-Dutton equation23. The potential difference between redox cofactors in electron transfer pathways strongly influences the overall rate in oxidation-reduction reactions, which may be an important feature of flavin-based electron bifurcation to gate the transfer of electrons. Strict control of electron transfer is required for efficient bifurcation and for preventing short-circuit reactions that lead to nonproductive reaction pathways owing to the highly reactive bifurcating flavin intermediate.
Our biophysical analysis of Pf NfnI provides the first direct insight into the mechanism of flavin-based electron bifurcation and the requisite structural features, some of which are unprecedented in redox enzyme biochemistry. Energy-conserving bifurcating enzymes must couple microscopic electron-transfer reactions that span a wide potential range far from the equilibrium values of the biochemical half reactions. The separation of electron pairs by L-FAD over a free-energy gap of ~1.2 V, coupled to redox cofactors that span ~0.8 V in reduction potential, are very rare in biological systems. Other enzymes that operate over such a broad potential range are typically light driven, for example, photosynthetic reaction centers15 and electron transfer complexes22. Highly unusual structural features of Nfn are required, in part, to kinetically control the highly reactive, low-potential flavin ASQ in a manner that conserves electrochemical potential. The kinetic, thermodynamic and structural principles revealed in this work provide a fundamental basis for understanding how bifurcating enzymes have evolved to merge unique cofactors with catalytic sites to accomplish a diverse array of biochemical reactions that underpin this third mechanism of metabolic energy conservation. This knowledge also presents an opportunity for the design of synthetic enzymes able to utilize the high-energy, low-potential flavin-based intermediate to perform thermodynamically unfavorable catalysis.
METHODS
PCR product and strain constructions.
List of primers used for strain construction:
DN.039, 5’-CTAGGTCTATCTTCCTCCCTT C-3’
DN.040, 5’-GGTGTTCCTCAAACATTTTCAAGTATGCACATCACCCTACAA G-3’
DN. 041, 5’-ATCCATCGGGCAATTCATGG-3’
DN.060, 5’-GATTATTGGGAGGTGGAGAAAAATGCATCACCACCATCACCACCACCATCACGGTTTCAAAATTTTAAGAAAAGAGAGGC-3’
SP2.088, 5’-CTTGAAAATGTTTGAGGAACACC-3’
SP2.055, 5’-TTTTCTCCACCTCCCAATAATC-3’
A linear PCR construct was designed to insert an NfnI overexpression cassette into the Pf genome at the native nfnI gene location. The nfnI expression cassette, consisting of the 182 bp slp promoter27, the 9× His-tag sequence (targeting insertion at the N-terminal of nfnI-S gene PF1328), and the pyrF pop-out marker cassette containing 65 bp identical flanking regions for marker removal via homologous recombination27, was sandwiched between ~0.5 kb homologous flanking regions of the nfnI-S ATG start codon. Each PCR product was obtained separately, and the full linear ~2.5 kb PCR construct was assembled via splicing by overlap extension and PCR28.
The NfnI overexpression PCR construct was transformed into Pf COM1, as previously described29. The transformants were cultured and purified thrice on liquid and solid defined cellobiose medium29. Genomic DNA was isolated using the ZymoBead Genomic DNA Kit (Zymo Research) and isolates were screened by PCR, using primers that target outside of the homologous flanking regions. A Pf strain containing the NfnI overexpression construct was sequence verified and designated as MW367.
Purification of His-tagged NfnI expressed in Pyrococcus furiosus
Recombinant strains overexpressing Pf NfnI were grown on a 15 L scale as previously described30. Cells were collected by centrifugation at 7,000 × g for 10 min and stored at −20 °C until use. Cells (~40–50 g) were lysed under strict anaerobic conditions by resuspension in 25 mM phosphate buffer pH 7 in a Coy anaerobic chamber. Cell-free extracts were prepared by centrifuging at 100,000 × g for 1 h to remove particles and membrane fragments. The resulting supernatant was loaded onto a 5 ml HisTrap FF crude (GE Healthcare Bio-Sciences) and eluted with a gradient of imidazole in phosphate buffer pH 7 according to manufacturer instructions. Fractions containing Pf NfnI activity were pooled, concentrated by Amicon Ultra-4 ultrafiltration centrifugal filters (Merck Millipore). About 2 ml of Pf NfnI concentrate was loaded onto a HiLoad Superdex 200 prep grade XK 16/60 column (GE Healthcare Bio-Sciences) equilibrated and developed with 25 mM Tris, 500 mM NaCl, pH 8. Active fractions judged pure by gel electrophoresis were pooled and stored at −80 °C until use. The typical yield obtained for Pf overexpressed NfnI was 0.3–0.5 mg g−1 wet cell paste. Anaerobic conditions were maintained throughout the purification and 1 mM dithiothreitol was used to maintain reducing conditions.
Pf NfnI activity assay
Dye-linked activity assays were performed in 50 mM MOPS pH 7.5 with NADH and NADPH using benzyl viologen (BV; monitored at 600 nm, ε = 7.4 mM−1cm−1) as the electron acceptor. Specific activity (U mg−1) is defined as 1 μmol BV (min/mg protein)−1. For the bifurcating assay, 2NADPH + 2Fdox + NAD+ → 2NADP+ + H+ + NADH + 2Fdred, the reduction of Fd at 425 nm (ε = 13 mM−1 cm−1)31 was followed using 0.5 mM NADPH, 1 mM NAD+ and 25 μM Pf Fd. The confurcating reaction was measured by following the oxidation of Fd in which the Fd was first reduced with equal molar amount Ti-citrate before adding 1 mM NADP+, 1 mM NADH and enzyme. Specific bifurcating activity (U mg−1) is defined as 1 μmol Fd (min/mg protein)−1. For the pH-dependent activities of Pf NfnI the following buffers were used: 50 mM sodium acetate, pH 5; 50 mM MES, pH 6; 50 mM MOPS, pH 7; 50 mM EPPS, pH 8; 50 mM CHES, pH 9; and 50 mM CAPS, pH 10.
Electrochemistry
Electrochemical experiments were performed in a glove box filled with a nitrogen atmosphere in a temperature-controlled, water-jacketed, all glass electrochemical cell with a pyrolytic graphite edge working electrode (0.2 cm2 surface area), a 3 M Ag/AgCl reference electrode, and a Pt wire counter electrode. The reference electrode was calibrated using Fe(CN)63−/4− as a standard, and all potentials have been converted to SHE using this calibration. The mixed buffer contains 15 mM each of 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES), 2-[N’-cyclohexylamino]ethanesulfonic acid (CHES), 2-[N’-morpholino]ethanesulfonic acid (MES), N’-tris[hydroxymethyl]methyl-3-amino-propanesulfonic acid (TAPS), and sodium acetate with 0.1 M NaCl as supporting electrolyte in purified water (resistivity = 18.2 MΩ cm−1). Solutions were titrated to the required pH with HCl or NaOH.
Protein films were prepared by abrading the graphite electrode surface with sandpaper, polishing with an aqueous slurry of 1 μm alumina (Buehler), and thorough sonication and rinsing. A 5 μL aliquot of protein solution was dried onto the clean electrode surface.
Electrocatalytic activity was assessed using cyclic voltammetry at 20 mVs−1. One of nicotinamide adenine dinucleotide (NAD+), nicotinamide adenine dinucleotide phosphate (NADP+), nicotinamide adenine dinucleotide reduced salt (NADH), or nicotinamide adenine dinucleotide phosphate reduced salt (NADPH) was added to 1 mM final concentration. Experiments were performed at an electrode rotation rate of 2500 rpm, at pH 7, and 60 °C. Square wave voltammetry employed a step potential of 0.5 mV, an amplitude of 20 mV, and frequency of 100 Hz at 60 °C. The measured reduction potentials from SWV measurements were the average of four scans, each with different protein films on the electrode.
The experiments were controlled by a PG-STAT 128N Autolab electrochemical analyzer with GPES software. Data were analyzed with QSOAS32, free electrochemical software that can be downloaded from: http://bip.cnrs-mrs.fr/bip06/qsoas/.
Determination of kcat from electrochemical data
Equation (1) was used to calculate kcat from cyclic voltammetry data,
| (1) |
in which ilim is the limiting current, F is faraday’s constant, A is the surface area of the electrode, and Γ is the electroactive coverage of enzyme on the electrode. In single substrate reactions, ilim is defined as the maximum current measured. For the electron bifurcating reaction, ilim is the decrease in catalytic current for electrocatalytic reduction of NADP+ reduction following addition of NADH. The electroactive enzyme surface coverage was determined from the peak areas from square wave voltammetry using equation (2). The peaks at −230 and −500 mV were used in the calculation.
| (2) |
Where ν is the electrochemical potential scan rate. Since there are two molecules of FAD per-molecule of protein, the value from the −230 mV peak was divided by two. The surface coverage for Pf NfnI is 9 ± 4 pmol cm−2 averaged over five different protein films.
Determination of rates from solution assays
To compare directly catalytic rates from solution assays and electrochemistry, the rates from solution assays were converted from specific activity, in units of μmol substrate (oxidized or reduced) min−1 (mg protein)−1, to kcat in units of mol substrate (mol protein s)−1.
UV-Vis titrations with redox substrates and reagents
Titrations were performed under strict anaerobic conditions in 400 μl of 50 mM MOPS pH 7.5 using a 600 μl quartz cuvette and an Agilent Cary 100 series spectrophotometer. Data was collected between 200 and 800 nm with a 2 nm slit width and a scan rate of 600 nm min−1. Between 13 and 17 μM Pf NfnI was titrated with the addition of the following redox carriers: 7 mM TiCl3 in 200 mM sodium citrate buffer pH 7 (Ti-citrate); 1 and 5 mM NADP(H); 1 and 5 mM NAD(H), in 1–10 μl increments. Ti-citrate was prepared according to ref. 33. Pf Fd was added in equimolar amounts to the NfnI in bifurcating titrations. Fd has a strong absorption around 390 nm that partly obscures the flavin spectrum. Each titration was performed twice (n = 2) to ensure reproducibility; each figure displays a single titration (n = 1).
Structure determination and refinement
Pf NfnI crystals were obtained by the vapor diffusion method under anaerobic conditions in a Coy anaerobic chamber using 3.0 M ammonium formate as precipitant in 0.1 M HEPES buffer (pH 6.8) with 1 mM sodium dithionite and 15% v/v glycerol as a cryo-protectant. The data were collected from flash-cooled crystals with a continuous flow of liquid nitrogen at 100 K on BL12–2 (SLAC National Accelerator Laboratory). The diffraction images were indexed, integrated and scaled using HKL2000 (ref. 34). The initial structure was solved by single anomalous dispersion (SAD) using iron, tantalum or platinum as anomalous scatter (Supplementary Table 1) using phinex.autosol35 followed by phenix.autobuild36. The high-resolution structures of as-purified Pf NfnI (1.5 Å) and NADP(H) bound Pf NfnI (1.6 Å) were determined by the molecular replacement method with the model obtained from SAD experiments using phenix.phaser37. The solutions were refined and improved by phenix.refine with final R/Rfree to 16%/18% for as-purified Pf NfnI and 16%/17% NADP(H) bound structures (Supplementary Table 1). The Ramachandran plot statistics show that all residues in both structures belong to the most favorable and generously allowed regions. Model building was subsequently completed manually using COOT38. Figures were prepared using PyMol39 (http://www.pymol.org). The protein-protein docking calculations were performed using PRISM40, ClusPro41, and FRODOC42 servers. The crystal composition was confirmed by SDS-PAGE and liquid chromatography-mass spectrometry analysis. Pf NfnI structures of as-purified and NADP(H) bound forms were deposited in the PBD databank with codes 5JFC and 5JCA, respectively.
The protein-protein docking calculations were performed using PRISME40, ClusPro41 and FRODOC42 servers. The crystal composition was confirmed by SDS-PAGE and liquid chromatography-mass spectrometry analysis.
Spectroelectrochemistry
The procedures including the modified cuvette described by Massey were used with only slight changes43. An anaerobic cuvette (3 ml, 1 cm path length) was equilibrated in the glove box ([O2] ≤ 10 p.p.m.) overnight. A solution containing 13 μM Pf NfnI, 13 μM phenosafranine (Em = 311 mV vs. SHE, pH 8), 2 μM benzyl viologen (as mediator), and 400 μM xanthine as well as a solution of 6 ng of xanthine oxidase (18 μl) in 25 mM Tris, 150 mM NaCl, pH 8.0 were rendered anaerobic by 5–10 pump-purge cycles of ~30 s followed by equilibration in the glove box for 1 h. The redox dye phenosafranine was chosen because of an absence of specific interactions with Pf NfnI, as evidenced by the lack of red-shifted flavin absorbance upon addition (i.e., it did not mimic substrate or modify the flavin environment). The solution containing Pf NfnI was placed in the cuvette along with a small magnetic stir bar. Xanthine oxidase was placed in the side arm of the cuvette where it is separated from the rest of the reaction mixture. After removing the sealed cuvette from the glove box, the solution was allowed to equilibrate at 25 °C in the spectrophotometer (Varian, CARY-300 Bio) for 30 min. Initial scans were stable and showed the expected trace for fully oxidized Pf NfnI. The reaction was initiated by tipping the cuvette to mix the xanthine oxidase solution from the side arm into the Pf NfnI solution in the cuvette. The titration was performed over a period of 6 h with significant loss of oxidized flavin within the first 2 h.
Determination of FAD EOX/HQ reduction potential in Pf NfnI
SWV was used to measure the two-electron reduction potential of the two FAD cofactors, with information from SEC providing the assignment of the electrochemical signal to FAD. Due to overlapping absorbances, it was not possible to determine the contribution from the individual FAD cofactors in either of these experiments; therefore the measured reduction potential represents the contribution from both L-FAD and S-FAD. The FAD EOX/HQ reduction potential was measured by SWV at −276 ± 7 mV (mean of 4 replicates ± standard deviation; Fig. 3b) and by SEC (Supplementary Fig. 8) as −326 mV at pH 8 (n = 1). The value of EOX/HQ = −276 mV, measured by SWV is included in Supplementary Table 3 and Supplementary Table 4, which further explain how the one-electron couples are calculated for both S-FAD and L-FAD.
EPR spectroscopy
Single Pf NfnI samples (100 – 120 μM enzyme concentration, 200 ul final volume) were prepared for EPR spectroscopy by addition of either NADH (1 mM final), NADPH (1 mM final), sodium dithionite in 50 mM Tris (pH 8.8, 10 mM final), sodium dithionite in 1 M glycine (pH 10, 30 mM final), or Ti-citrate (1 mM final). The electric potentials of samples treated with sodium dithionite were measured with an ORP triode electrode (Thermo Scientific 9678BNWP) in an Mbraun glove box. The ORP triode electrode was calibrated against a standard solution (Orion 967901) and all potentials were adjusted versus SHE. Low-temperature EPR spectra were recorded on a Bruker ELEXSYS E500 CW X-band spectrometer equipped with an in-cavity cryogen-free VT system and SHQ Bruker resonator. Typical EPR parameters: microwave frequency, 9.38 GHz; sample temperature, 15.0 ± 0.5 K (4 – 100 K for temperature-dependence studies); microwave power, 1 mW; modulation frequency, 100 kHz; modulation amplitude, 10.0 G. Spin quantifications were determined by double integration of the spectra after baseline subtraction in the Origin software package and compared to copper tri-ethylamine standards at identical spectrometer conditions. Simulations were carried out in EasySpin44 using the ‘esfit’ least-squares function and were used to assist with signal assignments and g value determination.
Transient absorption spectroscopy (TAS)
The ultrafast (100 fs to 5.1 ns) TAS spectrometer employed in this study uses an amplified 4W Ti:sapphire laser (Libra, Coherent, 800 nm, 1 kHz, 100 fs pulse width) and the Helios spectrometer (Ultrafast Systems, LLC). A fraction of the 800 nm Libra output was frequency doubled in BBO to produce the desired pump wavelength (400 nm in the data described here) for sample excitation, which was then directed into the Helios. The pump pulses were passed through a depolarizer and chopped by a synchronized chopper to 500 Hz before reaching the sample. The pump pulse energy was 1.1 μJ per pulse at the sample. Another fraction of the 800 nm Libra output was guided directly into the Helios for generation of the probe. Within the spectrometer, a white light continuum of wavelengths including 340–800 nm was generated using a 2 mm thick CaF2 crystal. This beam was split into a probe and a reference beam. The probe beam was focused into the sample where it was overlapped with the pump beam. The transmitted probe and reference beams were then focused into optical fibers coupled to multichannel spectrometers with CMOS sensors with 1 kHz detection rates. The reference signal is used to correct the probe signal for pulse-to-pulse fluctuations in the white-light continuum. The time delay between the pump and probe pulses was controlled by a motorized delay stage. For all transient absorption measurements, the sample was made in an Mbraun glove box (N2 atmosphere), sealed in a 2 mm quartz cuvette and constantly stirred to prevent photodegradation. Pf NfnI concentrations were approximately 75–100 μM. In order to enrich for L-FAD SQ intermediates, substoichiometric amounts of NADPH were used to partially reduce Pf NfnI, leading to reduced iron-sulfur clusters (Figs. 2c and 3c; Supplementary Figs. 5 and 7) and S-FAD (NSQ) (Fig. 2b and Supplementary Fig. 2) but oxidized L-FAD. While the bifurcation reaction in Nfn is not photochemically driven, light can be used to initiate formation of ASQ in the following way: photons of sufficient energy are used to excite L-FAD OX to its excited state (FAD*). Owing to the highly oxidizing nature of FAD*, an electron is abstracted from a nearby iron-sulfur cluster to form the mechanistically relevant ASQ species that relaxes by electron transfer to reform L-FAD OX. ASQ is only observed when the iron-sulfur clusters are reduced. 82% of the photoexcited L-FAD is converted to ASQ in our TAS experiment. This value is calculated from the ΔA of the transient absorption data for ASQ (366 nm) and OX (455 nm) FAD using extinction coefficients from related species. The extinction coefficient for ASQ (15.6 mM−1 cm−1) is from glucose oxidase stabilized in the ASQ state45 and that of OX (11.3 mM−1 cm−1) from oxidized free FAD46. All experiments were conducted at room temperature. The change in absorbance signal (ΔA) was calculated from the intensities of sequential probe pulses with and without the pump pulse excitation. The data collection (350 pump shots per time point) was carried out four consecutive times and then averaged. The experiment was repeated two times. Data were corrected for spectral chirp using Surface Xplorer (Ultrafast Systems). Fitting was performed in Igor Pro using a single or double exponential fit function. ASQ and OX are fit to a constrained single exponential. OX may be fit with a double exponential function, however the extremely fast first component (fs)47 is not mechanistically relevant so has been omitted for clarity. The 550 nm peak is due to stimulated emission47.
Mass spectrometry and H/D exchange
Purified Pf NfnI complex was analyzed on a 1290 ultra high pressure (UPLC) series chromatography stack (Agilent Technologies) coupled directly to an electrospray-time of flight (ESI-TOF) mass spectrometer (Bruker Micro-TOF). Each experiment was the average of two runs and was repeated at least twice. Before infusion to ESI the Pf NfnI sample was separated on reverse-phase column (RP), PLRP-S (50 × 2.1 mm, 3 μm, 100 Å, Agilent Technologies) at 50 °C using a flow rate of 600 μl min−1 in the following conditions: 1 min, 10% B; 1.0–6.0 min, 10–70% B; 6.0–7.0 min, 10% B; solvent A = 0.1% formic acid (FA, Sigma) in water (Thermo Fisher) and solvent B = 0.1% FA in acetonitrile (ACN, Thermo Fisher). Data were acquired at rate of 2 Hz over scan range 200–3,000 m/z in positive mode with the nebulizer set to 3 bar, drying gas 7.0 l min−1, drying temperature 200 °C, capillary voltage 4.5 kV and capillary exit 100 V. Data processing and analysis was carried out in the Bruker Data Analysis software package version 4.0. Charge deconvolution was completed using a maximum entropy algorithm for H+ adducts only and 0.1 m/z data point spacing. The low-mass end was defined by the mass of the lightest component of the complex while the high-mass end was defined as 3.3× the heaviest component within the complex.
Three forms of Pf NfnI were tested using hydrogen deuterium exchange coupled with mass spectrometry (HDX-MS): as-purified Pf NfnI, NADP+ bound, and NADH bound (each form was tested in triplicate and averaged). The NADH and NADP+ were incubated with Pf NfnI at a final concentration of 1 mM. The exchange reaction was initiated by a 10-fold dilution of Pf NfnI with reaction buffer. Reaction buffer contained 100 mM Tris-HCl, 150 mM NaCl at pD 7.0 in D2O. Separate reaction buffers were also prepared containing either 1 mM NADH or 1 mM NADP+. Samples were removed and quenched to stop exchange after 1 min, 3 min, 15 min, and 60 min. At each time point, 10 uL was withdrawn and placed into quenching/digestion solution containing 1% FA (Sigma) and pepsin (final conc. 0.2 mg mL−1) at 0°C. After two min, the tube was frozen in liquid N2 and stored at −80 °C until liquid chromatography-mass spectrometry (LC-MS) analysis.
LC-MS analysis of Pf NfnI peptides was completed on a 1290 UPLC series chromatography stack (Agilent Technologies) coupled directly to a 6538 UHD Accurate-Mass Q-TOF LC/MS mass spectrometer (Agilent Technologies). Before ESI-TOF analysis, peptides were separated on a RP column (Phenomenex Onyx Monolithic C18 column, 100 × 2 mm) at 1 °C using a flow rate of 500 uL min−1 in the following conditions: 1 min, 5% B; 1.0–9.0 min, 5–45% B; 9.10–9.80 min, 95% B; 9.80–9.90 min, 5% B solvent A = 0.1% FA in water and solvent B = 0.1% FA in ACN. Data was acquired at 2 Hz over scan range 50–1700 m/z in positive mode. Electrospray settings were: nebulizer set to 3.7 bar, drying gas 8.0 L min−1, drying temperature 350 °C, capillary voltage 3.5 kV. Data dependent MS/MS with a selection window of 4 m/z was used to generate peptide sequence tags. Data processing was carried out in Agilent MassHunter Qualitative Analysis version 6.0. Peptide identification was performed using peptide analysis worksheet (PAWs, ProteoMetrics LLC.). HDX data mapped onto the structural model of NfnI using Chimera version 1.10.2 (UCSF Resource for Biocomputing, Visualization, and Informatics). Deuterium uptake was determined by monitoring shifts of the centroided peptide isotopic distribution in Agilent MassHunter Qualitative Analysis. Measured values were then used to generate a histogram of centroid peak shifts to compare relative deuterium exchange in as-purified Pf NfnI, NADH bound Pf NfnI, and NADP+ bound Pf NfnI in the following large subunit peptides: TCAADLAKMGYEVTIYEALHQPG (residues 168–190), IGTGAGTPRIYPWPGVNL (residues 245–262), TAREEEIKHAEE (residues 331–342), and IGQTPNKTFL (residues 403–412).
Statistics and general methods
Sample sizes were chosen based on precedent from relevant literature for each particular experimental technique. Samples providing significant signal to noise above baseline and compared to control experiments were determined as adequate power for detection. No data was excluded for each technique reported in this manuscript. Randomization and blind experiments were not employed.
Data Availability
Pf NfnI structures of the as purified and NADP(H)-bound NfnI were deposited in the PDB with codes 5JFC and 5JCA, respectively. All other data supporting the findings of this study are available within the paper and its supplementary information files.
Supplementary Material
Acknowledgements
This work and all authors were solely supported as part of the Biological and Electron Transfer and Catalysis (BETCy) EFRC, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0012518. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The Proteomics, Metabolomics, and Mass Spectrometry facility at MSU received support from the Murdock Charitable Trust and NIH 5P20RR02437 and P20GM103474. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS or the NIH. C.E.L., D.W.M., and P.W.K. were supported by the US Department of Energy under contract no. DE-AC36-08-GO28308 with the National Renewable Energy Laboratory.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Any supplementary information, chemical compound information and source data are available in the online version of the paper.
References
- 1.Buckel W & Thauer RK Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochimica et Biophysica Acta 1827, 94–113 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Mitchell P The Protonmotive Q Cycle: A General Formulation. FEBS Letters 59, 137–139 (1975). [DOI] [PubMed] [Google Scholar]
- 3.Herrmann G, Jayamani E, Mai G & Buckel W Energy Conservation via Electron-Transferring Flavoprotein in Anaerobic Bacteria. Journal of Bacteriology 190, 784–791 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters JW, Miller A-F, Jones AK, King PW & Adams MW Electron bifurcation. Current Opinions in Chemical Biology 31 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Brandt U Bifurcated ubihydroquinone oxidation in the cytochrome bc1 complex by proton-gated charge transfer. FEBS Letters 387, 1–6 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Mulkidjanian AY Activated Q-cycle as a common mechanism for cytochrome bc1 and cytochrome b6f complexes. Biochimica et Biophysica Acta 1797, 1858–1868 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Demmer JK et al. Insights into Flavin-based Electron Bifurcation via the NADH-dependent Reduced Ferredoxin:NADP Oxidoreductase Structure. Journal of Biological Chemistry 290, 21985–21995, doi: 10.1074/jbc.M115.656520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Huang H, Moll J & Thauer RK NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol 192, 5115–5123 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chowdhury NP, Klomann K, Seubert A & Buckel W Reduction of flavodoxin by electron bifurcation and sodium ion-dependent reoxidation by NAD+ catalyzed by ferredoxin-NAD+ reductase (Rnf). J. Biol. Chem 291, 11993–12002 (2016).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury NP et al. Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans. J. Biol. Chem 289, 5145–5157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F et al. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol 190, 843–850 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowdhury NP, Kahnt J & Buckel W Reduction of ferredoxin or oxygen by flavin-based electron bifurcation in Megasphaera elsdenii. FEBS J. 282, 3149–3160 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Schut GJ & Adams MWW The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol 191, 4451–4457 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandt U Energy conservation by bifurcated electron-transfer in the cytochrome-bc1 complex. Biochim. Biophys. Acta 1275, 41–46 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Ptushenko VV, Cherepanov DA, Krishtalik LI & Semenov AY Semi-continuum electrostatic calculations of redox potentials in photosystem I. Photosynth. Res 97, 55–74 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Hagen WR et al. Novel structure and redox chemistry of the prosthetic groups of the iron-sulfur flavoprotein sulfide dehydrogenase from Pyrococcus furiosus; evidence for a [2Fe-2S] cluster with Asp(Cys)3 ligands. J. Biol. Inorg. Chem 5, 527–534 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Mohsen AW, Rigby SEJ, Jensen KF, Munro AW & Scrutton NS Thermodynamic basis of electron transfer in dihydroorotate dehydrogenase B from Lactococcus lactis: analysis by potentiometry, EPR spectroscopy, and ENDOR spectroscopy. Biochemistry 43, 6498–6510 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Brereton PS, Verhagen MFJM, Zhou ZH & Adams MW Effect of iron-sulfur cluster environment in modulating the thermodynamic properties and biological function of ferredoxin from Pyrococcus furiosus. Biochemistry 37, 7351–7362 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagen WR Biomolecular EPR Spectroscopy (CRC press, 2008). [Google Scholar]
- 20.Mathews R, Charlton S, Sands RH & Palmer G On the nature of the spin coupling between the iron-sulfur clusters in the eight-iron ferredoxins. J. Biol. Chem 249, 4326–4328 (1974). [PubMed] [Google Scholar]
- 21.Anderson RF Energetics of the one-electron reduction steps of riboflavin, FMN and FAD to their fully reduced forms. Biochim. Biophys. Acta 722, 158–162 (1983). [DOI] [PubMed] [Google Scholar]
- 22.Crofts AR The cytochrome bc1 complex: function in the context of structure. Annu. Rev. Physiol 66, 689–733 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Page CC, Moser CC, Chen X & Dutton PL Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402, 47–52 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Nitschke W & Russell MJ Redox bifurcations: mechanisms and importance to life now, and at its origin: a widespread means of energy conversion in biology unfolds…. BioEssays 34, 106–109 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Crofts AR & Rose S Marcus treatment of endergonic reactions: a commentary. Biochim. Biophys. Acta 1767, 1228–1232 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Wang S, Moll J & Thauer RK Electron bifurcation involved in the energy metabolism of the acetogenic bacterium Moorella thermoacetica [DOI] [PMC free article] [PubMed]
- 27.Keller MW et al. Exploiting microbial hyperthermophilicity to produce an industrial chemical, using hydrogen and carbon dioxide. Proc. Natl. Acad. Sci. USA 110, 5840–5845 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horton RM, Hunt HD, Ho SN, Pullen JK & Pease LR Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68 (1989). [DOI] [PubMed] [Google Scholar]
- 29.Lipscomb GL et al. Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl. Environ. Microbiol 77, 2232–2238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verhagen MF, O’Rourke TW, Menon AL & Adams MW Heterologous expression and properties of the γ-subunit of the Fe-only hydrogenase from Thermotoga maritima. Biochim. Biophys. Acta 1505, 209–219 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Aono S, Bryant FO & Adams MW A novel and remarkably thermostable ferredoxin from the hyperthermophilic archaebacterium Pyrococcus furiosus. J. Bacteriol 171, 3433–3439 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fourmond V QSoas: a versatile software for data analysis. Anal. Chem 88, 5050–5052 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Zehnder AJB & Wuhrmann K Titanium (III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes. Science 194, 1165–1166 (1976). [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terwilliger TC et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr 64, 61–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 39.DeLano WL The PyMOL Molecular Graphics System (DeLano Scientific LLC, 2002). [Google Scholar]
- 40.Baspinar A, Cukuroglu E, Nussinov R, Keskin O & Gursoy A PRISM: a web server and repository for prediction of protein-protein interactions and modeling their 3D complexes. Nucleic Acids Res. 42, W285–W289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Comeau SR, Gatchell DW, Vajda S & Camacho CJ ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Garzon JI et al. FRODOCK: a new approach for fast rotational protein-protein docking. Bioinformatics 25, 2544–2551 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massey V in Flavins and Flavoproteins (eds. Curti B, Ronichi S & Zanetti G) 59–66 (Walter de Gruyter & Co., Berlin, 1991). [Google Scholar]
- 44.Stoll S & Schweiger A EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson 178, 42–55 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Johnson JL, London RE & Rajagopalan KV Covalently bound phosphate residues in bovine milk xanthine oxidase and in glucose oxidase from Aspergillus niger: a reevaluation. Proc. Natl. Acad. Sci. USA 86, 6493–6497 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aliverti A, Curti B & Vanoni MA Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol. Biol 131, 9–23 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Enescu M, Lindqvist L & Soep B Excited-state dynamics of fully reduced flavins and flavoenzymes studied at subpicosecond time resolution. Photochem. Photobiol 68, 150–156 (1998). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Pf NfnI structures of the as purified and NADP(H)-bound NfnI were deposited in the PDB with codes 5JFC and 5JCA, respectively. All other data supporting the findings of this study are available within the paper and its supplementary information files.