

Abstract
Embryonic development is a complex process that is unamenable to direct observation. In this study, we implanted a window to the mouse uterus to visualize the developing embryo from embryonic day 9.5 to birth. This removable intravital window allowed manipulation and high-resolution imaging. In live mouse embryos, we observed transient neurotransmission and early vascularization of neural crest cell (NCC)–derived perivascular cells in the brain, autophagy in the retina, viral gene delivery, and chemical diffusion through the placenta. We combined the imaging window with in utero electroporation to label and track cell division and movement within embryos and observed that clusters of mouse NCC-derived cells expanded in interspecies chimeras, whereas adjacent human donor NCC-derived cells shrank. This technique can be combined with various tissue manipulation and microscopy methods to study the processes of development at unprecedented spatiotemporal resolution.
Different methods are used to study embryonic development in mice, a common mammalian research model (1). However, each has its limitations. Histology on fixed embryos does not capture developmental dynamics. Ultrasound or magnetic resonance imaging has limited resolution and does not take advantage of transgenic strains with fluorescent reporters (2, 3). The peri-implantation to early organogenesis stages can be imaged by means of optical coherence microscopy or light sheet fluorescence microscopy after removal of the mouse embryo from the uterus and culture in medium for 24 to 48 hours (4-6). However, despite efforts to mimic the intrauterine environment (7), culturing embryos past embryonic day 9 (E9), when various organs form, remains difficult because of the inability to recapitulate the feto-placental nutritional exchange (8-10).
Intravital imaging has been used to observe organs at high resolution but not whole embryos (11-16). We developed an implantable window for imaging the mouse embryo from E9.5 to birth. The window is circular, with a 10-mm inner diameter and 1.5-mm depth, and is covered with a glass coverslip that can be removed for manipulating the embryo (Fig. 1, A and B). The embryo becomes easily identifiable under a dissection microscope around E9.5, when the allantois fuses with the chorionic plate to form a labyrinthine layer that separates fetal and maternal blood vessels and provides a large surface area for gas and nutrient exchange (17). We developed a surgical procedure (supplementary materials, materials and methods) to implant the window and strip the decidua and uterine muscle, which impedes the view of the embryo from E9.5 to E12.5 (Fig. 1, C to E), without interfering with embryo survival as indicated by a beating heart (movie S1). To minimize motion from the dam’s breathing, we sutured the uterine wall to the dam’s abdominal muscle and the abdominal muscle to the window and designed a 3D-printed clip to stabilize the window (fig. S1A). If an embryo was to be tracked beyond E13.5 (when it tended to retract somewhat into the mother’s abdominal cavity), its position was adjusted with a new window (fig. S1, B and C). The survival rate of embryos was 65.6% (21 of 32) after window implantation.
Fig. 1. Intravital imaging of mouse embryos.

(A and B) Schematic illustration of (A) side view and (B) top view of the embryonic window. (C) Illustration of a mouse embryo at E9.5. UW, uterine muscle; De, decidua; PYS, parietal yolk sac; VYS, visceral yolk sac; Am, amnion. (D) Overview of the stripping surgical protocol. (E) Embryo growth from E9.5 to E10.0. (F) Embryo growth from E12.5 to E13.5. (G and H) An elliptical window used for imaging later stages. (G) Schematic illustration of the elliptical window. (H) View of the elliptical window at E16.5. (I to K) Embryo weights at (I) E14.5, 3 days after stripping surgery; (J) E15.5; and (K) E18.5, 3 days after nonstripping surgery. Data are mean ± SD; n = 3 dams, **P < 0.01. (L) Timeline of embryo development and imaging procedures.
After E12.5, the decidua diminished, and the uterine wall became transparent (fig. S1D); therefore, there was no need to strip the decidua and uterine muscle (Fig. 1F). The window allowed observation of the embryo until birth; this view was reduced to partial after the embryo outgrew the window (typically by E15.5) (fig. S2A). We 3D-printed a larger, elliptical-shaped window to image the entire embryo at older stages, from E12.5 until birth (Fig. 1, G and H, and fig. S2B). The survival rates of embryos underneath the circular and elliptical windows were 81.9% (59 of 72) and 84.1% (37 of 44), respectively.
To test the effect of the window on the growth of the embryos, we implanted circular or elliptical windows at E11.5, E12.5, and E15.5 and weighed the embryos 3 days after implantation. Embryo weight was reduced by 28.2, 11.7, and 12.0%, respectively, relative to littermates that were not placed under a window (Fig. 1, I to K). Hematoxylin and eosin staining showed no structural abnormalities in the embryos underneath the window (fig. S2, C to E), and there were no differences in terms of complete blood components (fig. S2, F to H). Dams showed no notable signs of inflammation or anemia after window implantation (fig. S2I). All eight dams implanted with the circular windows gave birth vaginally to all pups (including the ones underneath the window), whereas only 2 of 7 dams with elliptical windows gave birth naturally, owing to the lack of abdominal contractions (fig. S2J and movie S2). The dam fed the pups normally (movie S3), and pups imaged embryonically underneath the windows were indistinguishable from their littermates and grew without noticeable abnormalities.
Transgenic mice with cell lineage–specific expression of fluorescent reporter proteins are commonly used to study development in live organisms (4). We used two types of microscopy—stereoscopic microscopy and two-photon microscopy—to observe embryos in Wnt1-Cre-tdTomato mice. In this transgenic strain, Wnt1-Cre is expressed in the dorsal neuroepithelium from E8.5, which leads to lineage labeling of neural crest cells (NCCs) and their descendants as well as the dorsal central nervous system (18-20). We crossed Wnt1-Cre mice with ROSA26-CAG-tdTomato mice to create the transgenic strain and placed the optical windows on dams at E11.5. The midbrain of F1 embryos was clearly visible through the window and was imaged continuously for 6 hours with a stereoscopic microscope (Fig. 2, A and B, and movie S4).
Fig. 2. Tracking live embryo development in transgenic mice.
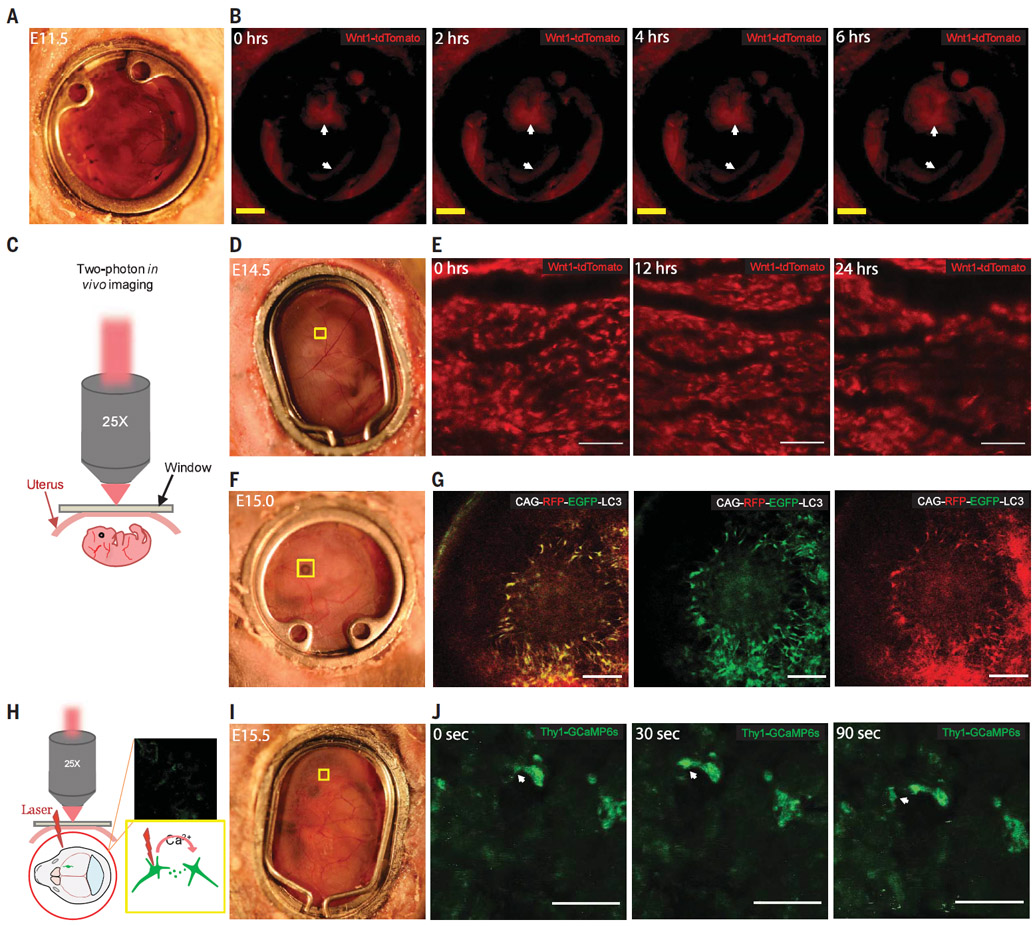
(A and B) Light-field view and 6-hour continuous fluorescence imaging, respectively, of a Wnt1-cre:tdTomato E11.5 mouse embryo. Neurons in the midbrain are labeled with tdTomato, as indicated with the arrows. Scale bar, 2 mm. (C) Schematic of in vivo two-photon microscopy. (D and E) Light-field view and 24-hour imaging of the marked brain region, respectively, of an E14.5 Wnt1-cre:tdTomato mouse embryo. Scale bar, 100 μm. (F and G) Light-field view and fluorescent imaging, respectively, of the marked eye region of a CAG-RFP-EGFP-LC3 mouse embryo (RFP, right; eGFP, middle; overlay, left). Scale bar, 100 μm. (H) Schematic illustration of the imaging setup. (I and J) Light field view and GCaMP6s signal, respectively, in the marked brain region in a Thy1-GCaMP6s embryo at E15.5. White arrows indicate neurotransmission. Scale bar, 25 μm.
Two-photon microscopy was then used to image the tdTomato-labeled cells in the midbrain from E10.5 to E11.0, with the decidua and uterine muscle stripped (Fig. 2C and fig. s3A to E). We next implanted the window at E13.5. Using the vasculature on the surface of the uterus and in the embryonic brain as a roadmap, we were able to track the tdTomato+ cells in the mouse embryonic brain for 24 hours (Fig. 2, D and E, and fig. S4, A to C). We further observed tdTomato+ cells in a mesh-like, 200- to 300-μm-deep fluorescent layer in the E13.5 embryo (fig. S4D). To identify these cells, we fixed the whole embryos and stained for various markers and discovered that these meshlike cells overlapped with blood vessels marked by CD31+ endothelial cells and had differentiated into platelet-derived growth factor receptor–β+ (PDGFR-β+) perivascular cells (fig. S4, E and F). Because previous reports showed that NCCs differentiate into perivascular cells in the thymus by E13.5 in mice (21), this observation indicated that NCCs also differentiate into perivascular cells to form vasculature in the brain as early as E13.5.
We then imaged embryos in transgenic mice that harbor the autophagy reporter CAG-RFP-eGFP-LC3, in which a CAG promoter drives expression of red fluorescent protein (RFP), enhanced green fluorescent protein (eGFP), and the phagosome marker LC3. Because RFP (pKa 4.5, where Ka is the acid dissociation constant) is more stable than eGFP (pKa 5.9) at low pH, this strain reports intracellular pH and can be used to distinguish different phagocytic cellular compartments. Both eGFP and RFP are expressed in phagosomes and autophagosomes, whereas eGFP is quenched in autolysosomes (which are more acidic) (22). The presence of overlapping RFP and eGFP signals indicates autophagosomes in the retina of E15.0 embryos (Fig. 2, F and G), which was confirmed ex vivo by fixed embryos (fig. S5A) (23, 24). We also imaged Lgr5+ stem cells in the developing intestine using the LGR5-DTR-EGFP strain (fig. S5B) (25).
Genetically encoded calcium indicators (GECIs) such as GCaMP have been used to monitor neuronal activity (26, 27). We used GCaMP6 transgenic mice to image neuronal activity in the developing brain (Fig. 2H). We implanted the optical window into GP4.3 (Thy1-GCaMP6s) mice at E15.5 and observed neurotransmission among three cells, as shown in movie S5 (Fig. 2, I and J).
The placenta is a barrier that impedes passage of chemicals such as drugs into the embryo, but the diffusion rates for molecules are difficult to assess (28, 29). As a proof of concept, we assessed the permeability of the placenta to fluorescein by injecting fluorescein through the dam’s orbital vein and monitoring its passage through the umbilical cord and subsequent diffusion into the embryo. Fluorescein passed through the umbilical cord 1.5 min after injection and diffused into the whole embryo 30 min after injection (Fig. 3, A and B, and movie S6).
Fig. 3. Imaging chemical diffusion, embryonic AAV transduction, and cell movements after in utero electroporation.
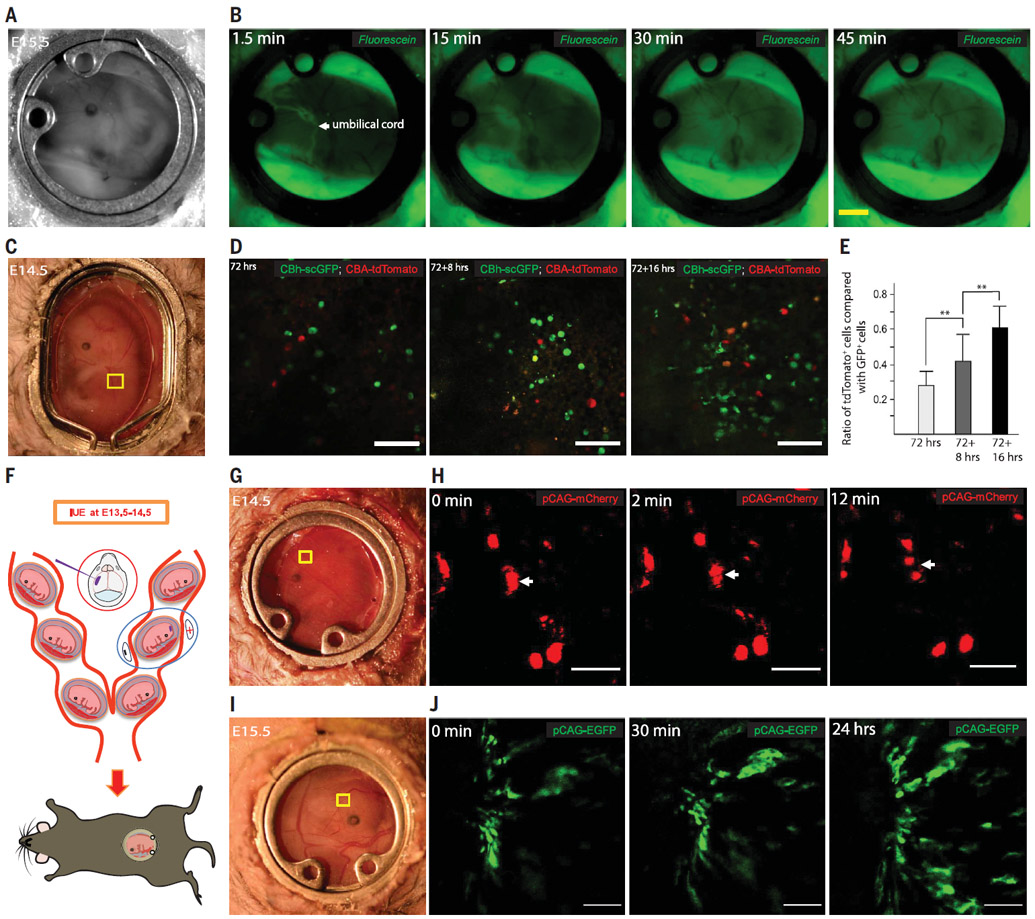
(A) View of an embryo at E15.5. (B) Diffusion of fluorescein into an embryo after dam retro-orbital injection. Scale bar 2 mm (C) Light-field view of an E14.5 embryo, 72 hours after AAV8g9-pTR-CBh-scGFP and AAV8g9-pTR-CBA-tdTomato co-injection. (D) GFP and tdTomato expression in the marked region in (C). Scale bar, 100 μm. (E) Ratio of tdTomato+ and GFP+ cells. Data are mean ± SD; n = 3 dams, **P < 0.01. (F) Schematic of in utero electroporation. (G) Light-field view of an E14.5 embryo, 1 day after electroporation of pCAG-mCherry. (H) Movement of cells in the region marked in (G). Scale bar, 50 μm. (I) Light-field view of an E15.5 embryo, 1 day after electroporation of pCAG-EGFP. (J) Twenty four–hour imaging of the brain region marked in (I) showing cell migration. Scale bar, 100 μm.
Adeno-associated viruses (AAVs) are used as vehicles for gene therapy (30, 31), but the kinetics by which AAVs permeate the placenta and express exogenous genes in embryos have not been studied. We injected AAV serotypes 8 and 9 (AAV8 and AAV9) carrying a GFP reporter into the tail vein of dams at E11.5 and observed GFP expression in embryos at E14.5, 72 hours after injection (fig. S6, A and B). We confirmed similar GFP expression in the embryos from injected dams without the window (fig. S6C) and GFP expression in the placenta (fig. S6D). To assess the speed of viral transduction into the embryo, we injected a single-stranded AAV vector (ssAAV8-eGFP) and a self-complementary AAV vector (scAAV8-tdTomato) (32) into the tail vain of dams at E11.5. We started imaging the embryos at E14.5, 72 hours after injection. We initially detected more GFP signal than tdTomato signal, although the difference in expression diminished within 16 hours (Fig. 3, C to E). This experiment demonstrates the use of intravital imaging of AAV gene delivery and suggests that different AAV vectors have different transduction dynamics, an important consideration when targeting a specific embryonic stage.
In utero electroporation (IUE) can be used to introduce DNA into embryos to study development and disease. This technique allows the fluorescent labeling of a sparse population of neural cells in the cerebral cortex (33). We combined our window surgery with IUE to track electroporated cells. We introduced CAG.mCherry plasmid DNA at E13.5 in utero in the ventricle and electroporated the embryos to sparsely label cells in the cerebral cortex (Fig. 3F). One day after electroporation, we implanted the embryonic window on top of the electroporated embryo and observed two cells separating after a recent division (Fig. 3, G and H, and movie S7). We also used CAG.eGFP to tracked cell migration (Fig. 3, I and J, and movie S8) and used CAG.mCherry. CAAX to label neuronal membrane (fig. S7).
We used blastocyst injection to introduce foreign cells into an embryo to produce blastocyst chimeras. We injected exogenous eGFP-labeled C57Bl/6;Col1a1(GFP) mouse embryonic stem cells (mESCs) into mouse blastocysts at E2.5 and implanted the embryos into timed pregnant surrogate mice, and we implanted optical windows in the dams at E14.5 (Fig. 4A). Using stereoscopic microscopy, we observed the distribution of labeled donor cells in the entire embryo (Fig. 4B) and observed the heartbeat, uterine contraction, and overall embryonic development continuously for 12 hours (fig. S8A and movie S9). The embryo died after 12 hours, although the mother survived. We tracked specific embryonic regions for 24 hours by using skin blood vessels as position reference markers (Fig. 4C).
Fig. 4. Imaging chimeric embryos.

(A) Schematic of imaging a chimeric embryo grown from blastocysts microinjected with labeled mESCs. (B) A blastocyst chimeric embryo, injected with GFP+ cells, at E14.5 and E15.5. Scale bar, 2 mm. (C) Imaging from E14.5 to E15.5 (24 hours) of the region marked in (B). Scale bar, 100 μm. (D) Chimeric embryo are co-injected with PSC-derived hNCCs (GFP+) and mNCCs (tdTlomato+) at E8.5 and imaged at E14.5. (E to H) Position of hNCC (eGFP) and mNCC (tdTomato) in an E14.5 embryo, 6 days after injection. (E) Bright field. (F) Overlay. (G) tdTomato. (H) eGFP. Scale bar, 2 mm. (I) Twenty four–hour imaging of the area marked in (E) to (H). Scale bar, 100 μm.
Interspecies chimeras provide a tool for studying human development in vivo (34, 35). Integration of exogenous pluripotent stem cell (PSC)–derived human NCCs (hNCCs) and mouse NCCs (mNCCs) into E8.5 mouse embryos showed varying efficiency (36). To directly compare integration of hNCCs and mNCCs in a single host embryo, we co-injected eGFP-labeled hNCCs and tdTomato-labeled mNCCs into E8.5 mouse embryos, which we then imaged starting at E14.5 (Fig. 4D). Using a stereoscopic microscope, we identified embryonic areas consisting of tdTomato-labeled cells, the progeny of injected mNCCs (Fig. 4, E to G). However, the stereoscopic microscope did not have sufficient resolution to detect areas of eGFP-labeled cells (Fig. 4H).
Using two-photon microscopy, we imaged 33 embryos (in 19 dams) at E14.5 that had been injected with hNCCs and mNCCs at E8.5. We detected both tdTomato-labeled mNCC progeny and eGFP-labeled hNCC progeny cells in six embryos (18%), only tdTomato-labeled cells in 11 embryos (33%), and only eGFP-labeled cells in three embryos (9%). These rates are consistent with previously reported chimeric rates (36-38). To compare intraspecies versus interspecies contributions, we tracked embryonic regions that contained adjacent clusters of hNCC and mNCC progeny cells. Whereas tdTomato+ mNCC clusters continued to expand, GFP+ hNCC clusters shrank within 24 hours (Fig. 4, E to I, and fig. S8, B to F). Fluorescent signals from individual mNCCs and hNCCs that were separate from any cluster diminished over time (fig. S8, G to M).
We have developed an intravital imaging window for manipulating and visualizing live mouse embryos in vivo from E9.5 until birth. The capability to image embryos in utero at high resolution opens new avenues for investigation, including brain formation, peripheral nerve development, placental development, birth defects, gene editing, immune system development, environmental effects, and interspecies chimera.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Cameron (Duke University) and M. Itano (University of North Carolina) for assistance with two-photon microscopy, and the Innovation Co-Lab at Duke University. This work was supported by NSFC81670468, 2017KJXX-43, 2018SF-208, China Scholarship Council (to Q.H.); by NIH R35GM122465 and DK119795 (to X.S.); by the Emerald Foundation, the Leo Foundation, and Melanoma Research Foundation (to R.J. and M.A.C.); by NIH R37HD045022, 1R01-NS088538, and 5R01-MH104610 (to R.J.); by NIH NS083897, NS098176, NS110388 and MH119813 (to D.L.S); and by NIH R35 CA197616 (to D.G.K.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Hadjantonakis AK, Dickinson ME, Fraser SE, Papaioannou VE, Nat. Rev. Genet 4, 613–625 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP, Nat. Genet 19, 348–355 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Gregg CL, Butcher JT, Differentiation 84, 149–162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pantazis P, Supatto W, Nat. Rev. Mol. Cell Biol 15, 327–339 (2014). [DOI] [PubMed] [Google Scholar]
- 5.McDole K et al. , Cell 175, 859–876.e33 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Muñoz WA, Trainor PA, Methods Mol. Biol 1976, 107–119 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Shahbazi MN, Zernicka-Goetz M, Nat. Cell Biol 20, 878–887 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB, Int. J. Mol. Sci 15, 16153–16185 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao J et al. , Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stremmel C et al. , Nat. Commun 9, 75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holtmaat A et al. , Nat. Protoc 4, 1128–1144 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritsma L et al. , Sci. Transl. Med 4, 158ra145 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Rakhilin N et al. , Nat. Commun 7, 11800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Entenberg D et al. , Nat. Methods 15, 73–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pilz GA et al. , Science 359, 658–662 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakhilin N et al. , Nat. Commun 10, 5647 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinkenberger J, Werb Z, Nat. Genet 25, 248–250 (2000). [DOI] [PubMed] [Google Scholar]
- 18.McMahon AP, Joyner AL, Bradley A, McMahon JA, Cell 69, 581–595 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Chai Y et al. , Development 127, 1671–1679 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM, Development 127, 1607–1616 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Foster K et al. , J. Immunol 180, 3183–3189 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Lin F, Wang ZV, Hill JA, Autophagy 10, 691–693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaizuka T et al. , Mol. Cell 64, 835–849 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Gómez-Sintes R et al. , Cells 6, 37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian H et al. , Nature 478, 255–259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuryev M et al. , Front. Cell. Neurosci 9, 500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michelson NJ, Eles JR, Vazquez AL, Ludwig KA, Kozai TDY, J. Neurosci. Res 97, 620–638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leong HS et al. , Nat. Protoc 5, 1406–1417 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wick P et al. , Environ. Health Perspect 118, 432–436 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox DB, Platt RJ, Zhang F, Nat. Med 21, 121–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson CE et al. , Nat. Med 25, 427–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruenert AK et al. , PLOS ONE 11, e0152589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.dal Maschio M et al. , Nat. Commun 3, 960 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J et al. , Nature 540, 51–59 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Soldner F, Jaenisch R, Cell 175, 615–632 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen MA et al. , Proc. Natl. Acad. Sci. U.S.A 113, 1570–1575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaenisch R, Nature 318, 181–183 (1985). [DOI] [PubMed] [Google Scholar]
- 38.Cohen MA, Markoulaki S, Jaenisch R, Stem Cell Rep. 10, 1445–1452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.