

ABSTRACT
The extracellular ATP/adenosine axis in the tumor microenvironment (TME) has emerged as an important immune-regulatory pathway. Nucleoside triphosphate diphosphohydrolase-1 (NTPDase1), otherwise known as CD39, is highly expressed in the TME, both on infiltrating immune cells and tumor cells across a broad set of cancer indications. CD39 processes pro-inflammatory extracellular ATP to ADP and AMP, which is then processed by Ecto-5ʹ-nucleotidase/CD73 to immunosuppressive adenosine. Directly inhibiting the enzymatic function of CD39 via an antibody has the potential to unleash an immune-mediated anti-tumor response via two mechanisms: 1) increasing the availability of immunostimulatory extracellular ATP released by damaged and/or dying cells, and 2) reducing the generation and accumulation of suppressive adenosine within the TME. Tizona Therapeutics has engineered a novel first-in-class fully human anti-CD39 antibody, TTX-030, that directly inhibits CD39 ATPase enzymatic function with sub-nanomolar potency. Further characterization of the mechanism of inhibition by TTX-030 using CD39+ human melanoma cell line SK-MEL-28 revealed an uncompetitive allosteric mechanism (α < 1). The uncompetitive mechanism of action enables TTX-030 to inhibit CD39 at the elevated ATP concentrations reported in the TME. Maximal inhibition of cellular CD39 ATPase velocity was 85%, which compares favorably to results reported for antibody inhibitors to other enzyme targets. The allosteric mechanism of TTX-030 was confirmed via mapping the epitope to a region of CD39 distant from its active site, which suggests possible models for how potent inhibition is achieved. In summary, TTX-030 is a potent allosteric inhibitor of CD39 ATPase activity that is currently being evaluated in clinical trials for cancer therapy.
KEYWORDS: CD39, ATP, enzyme inhibitor, enzyme kinetics, enzyme mechanism, allosteric regulation, monoclonal antibody, cancer, cancer therapy
Introduction
CD39, encoded by the gene ectonucleoside triphosphate diphosphohydrolase-1 (ENTPD1), is an integral membrane protein that metabolizes extracellular adenosine triphosphate (ATP) and adenosine diphosphate (ADP) to adenosine monophosphate (AMP); AMP is then further converted to adenosine via another ecto-enzyme, ecto-5ʹ-nucleotidase/CD73.1,2 Extracellular adenosine suppresses the immune system via signaling through four adenosine (P1) receptors expressed on immune cells.3 In contrast, extracellular ATP is pro-inflammatory, regulating innate and adaptive immune responses through agonizing various P2X and P2Y receptors ubiquitously expressed on immune cells.4–7 While there are other potential sources of extracellular adenosine,8–10 CD39 appears to be the major extracellular enzyme catabolizing ATP in the tumor microenvironment (TME).11,12
CD39 is constitutively expressed on immune cell populations, such as B cells, natural killer cells, dendritic cells, Langerhans cells, monocytes, macrophages, mesangial cells, neutrophils, activated T cells, and regulatory T cells (Tregs), as well as endothelial cells.13,14 During chronic viral infection in humans and in a mouse model, CD39 is a CD8+ T cell exhaustion marker.15 In an oncology setting, CD39 is highly expressed by tumor-infiltrating lymphocytes, especially Tregs, exhausted CD8+ T cells, and myeloid-derived cells such as myeloid-derived suppressor cells.16 CD39 is the rate-limiting enzymatic step in the production of immunosuppressive adenosine, and should therefore be viewed as an immunological switch that shifts ATP-driven pro-inflammatory immune cell activity toward an anti-inflammatory, immunosuppressed state mediated by adenosine. Indeed, murine tumor models demonstrate that significant anti-tumor activity achieved by CD39 blockade required activation of proinflammatory ATP signaling pathways in addition to reduction of accumulated adenosine.17 Thus, blockade of CD39 enzymatic activity represents a unique therapeutic target for oncology indications.18–20
An inhibitor of CD39 should be exquisitely specific, lacking reactivity to other ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) family members that regulate ATP.21 For example, NTPDase2 (CD39L1) activates platelets in the vasculature,21 and NTPDase8 is the major ectonucleotidase active in the liver.22 A disruption of either nucleotide hydrolase’s activity function by a promiscuous NTPDase inhibitor may cause undesired toxicities. Outside of the enzymes of NTPDase family, extracellular nucleotide pyrophosphatase/phosphodiesterase-1 (NPP1) and tissue-nonspecific alkaline phosphatase (TNAP) also hydrolyze ATP. However, both enzymes have reduced catalytic activity at neutral pH and are not expected to significantly contribute to ATP catabolism in the slightly acidic TME.2 The published small molecule inhibitors of CD39, such as sodium polyoxotungstate (POM-1), 6-N, N-diethyl-D-β,γ-dibromomethylene-ATP-trisodium salt (ARL-67156), adenylyl-imidophosphate (AMP-PNP), and sodium azide, are neither CD39-specific nor have favorable drug-like properties.23–29 A monoclonal antibody (mAb) inhibitor of CD39 ATPase activity would be anticipated to meet the specificity requirements for a therapeutic.
Inhibition of CD39 activity could be achieved mechanistically through downregulation of cell surface CD39 by an antibody that did not block enzyme activity. However, an inhibitor of CD39 enzymatic activity that directly prevents ATP binding and/or hydrolysis is preferable because antibodies that rely on downregulation are likely to have greater target-mediated drug disposition leading to poor pharmacokinetic characteristics and rapid clearance.30 In the TME and at sites of metastases, extracellular ATP levels can reach 100–500 µM (compared to nanomolar range in healthy tissues), but can be rapidly hydrolyzed by CD39.7,11 In this environment, an allosteric antibody inhibitor of CD39 (noncompetitive or uncompetitive) is the optimal mechanism to sustain inhibition across a broad range of ATP concentrations. A potent noncompetitive or uncompetitive allosteric inhibitor would be insensitive to substrate levels and maintain its maximal inhibition even at high concentrations of ATP or ADP. In contrast, a competitive inhibitor, like the small molecule inhibitors POM-1, ARL, and AMP-PNP, could exhibit diminished inhibition at the high ATP levels found in the TME due to displacement by substrate.31
Advanced clinical-stage efforts to target the adenosine axis in the TME have focused on A2A and A2B receptors and CD73.32–36 Three anti-CD39 antibodies (TTX-030, SRF617, IPH5201) have recently entered clinical studies, but the mechanism of enzymatic inhibition was not delineated.17,36–38 Here, we describe TTX-030, a novel CD39-specific inhibitor that offers a dual mechanism for enhancing tumor immunity by reducing adenosine accumulation while concurrently maintaining proinflammatory extracellular ATP. We characterize the steady-state kinetics of CD39 activity and show that TTX-030 inhibits NTPDase activity in human cells and tissues via uncompetitive allosteric mechanisms. Furthermore, TTX-030 maintains robust inhibition of CD39 cellular ATPase activity in vitro under therapeutically relevant conditions such as wash-out of free antibody, continuous addition of ATP, and at TME-like acidic pH.
Results
Characterization of enzymatic properties of cellular CD39
CD39 is reported to have broad NTPase and NDPase specificities,39,40 so the rate of purine and pyrimidine nucleotide substrate hydrolysis was compared using a melanoma cell line (SK-MEL-28) that endogenously expresses CD39. Inorganic phosphate, Pi, a product of the CD39-mediated hydrolysis reaction converting ATP to ADP and AMP, was kinetically monitored. Figure 1a shows the initial velocity of Pi release plotted against substrate concentration with the data fit to a Michaelis-Menten model (Equation 1). NTP and ADP substrates were hydrolyzed with a similar maximal velocity (Vmax), and the Michaelis constant (Km) fell within a range of approximately 30 to 70 µM (Table S1). The Vmax and Km for AMP, which is hydrolyzed by endogenously expressed CD73 on SK-MEL-28 cells, were greater than that of the CD39-hydrolyzed substrates. When higher substrate concentrations are tested, nucleotide hydrolysis follows a substrate inhibition model41 (Figure S1). Kinetic enzymatic assessment of other CD39+ primary cells and cell lines confirmed Km for ATP catabolism ranges from approximately 10–200 µM2 (Table S2), and that Vmax correlated (Pearson r = 0.93, p < .001) with CD39 expression levels (Figure S2).
Figure 1.
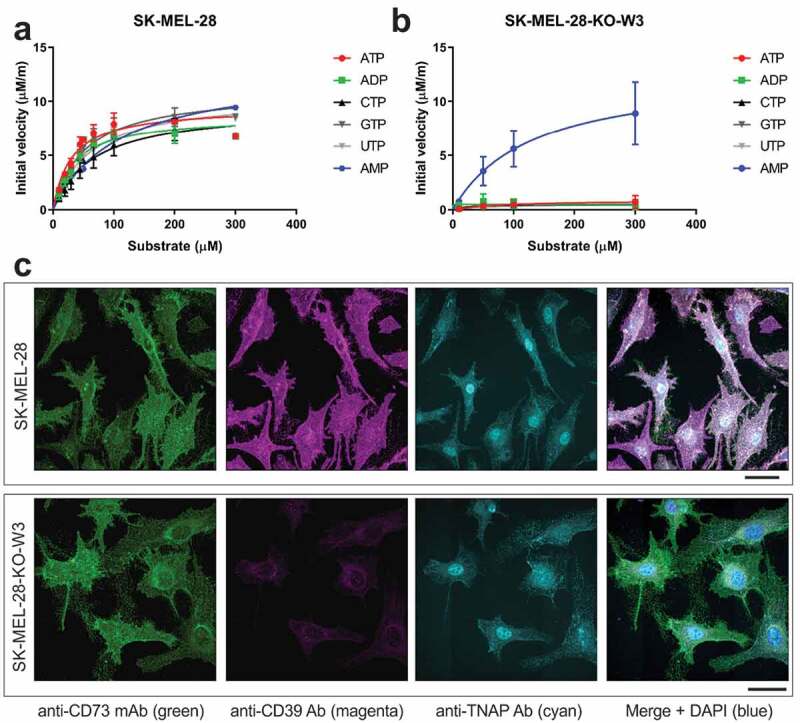
Deletion of CD39 in SK-MEL-28 cells is accompanied by near-complete loss of NTP/ADPase but not AMPase activities. A, The initial velocities of wildtype SK-MEL-28 CD39-mediated Pi release were plotted against a titration of substrate concentrations and fit to the Michaelis-Menten model. Error bars, S.D., n= 2. B, A CD39 KO derivative of the SK-MEL-28 cell line (SK-MEL-28-KO-W3) showed a near-complete reduction of Pi release for all substrates other than AMP, confirming the deletion of CD39. AMP hydrolysis is mediated by CD73. Error bars, S.D., n= 2. C, Confocal microscopy analysis of CD73, CD39, and TNAP expression in wildtype SK-MEL-28 and SK-MEL-28-KO-W3 cells. Scale bars, 40 µm
A CD39 knockout (KO) derivative of the SK-MEL-28 cell line (SK-MEL-28-KO-W3) enabled the determination of the specific contribution of CD39-mediated hydrolysis, compared to other NTPDases (Figure 1b). The Vmax for ATP was reduced from 9.5 µM/min in wild type (WT) cells to 0.9 µM/min upon deletion of CD39 (Table S1). The residual ATPase activity observed in the KO cells may be attributed to minor contribution of other ecto-nucleotidases, including TNAP (Figure 1c).2 The Vmax for other NTPs and ADP was similarly diminished upon knocking out CD39. CD73-associated AMP hydrolysis was unaffected by knocking out CD39 (Figure 1b and Table S1). Immunofluorescence staining of SK-MEL-28 and SK-MEL-28-KO-W3 cells confirmed deletion of CD39 in KO cells, and further demonstrated comparable staining patterns for other purine-converting ectoenzymes, CD73 and TNAP (Figure 1c).
Biochemical characterization of high affinity and selective anti-CD39 mAb
A fully human hinge-stabilized42 IgG4 mAb, TTX-030, was developed against human CD39 that binds to and inhibits both soluble and cellular CD39. TTX-030 binds to an epitope on CD39 distinct from commercial anti-hCD39 mAb A1 as shown by epitope binning using the antigen-binding fragment (Fab) of TTX-030 (Figure 2a). As determined by multiple methods, TTX-030 binds to cell-surface expressed CD39 with a range of binding affinities from approximately 100 to 500 pM (Figure 2b and Figure S3). TTX-030 demonstrated selectivity to CD39, as it did not bind to other NTPDase family members, either as soluble NTPDase2, NTPDase3 or NTPDase8 (Figure S4), nor to Chinese hamster ovary (CHO) cells overexpressing each of these related NTPDases (Figure 2b). TTX-030 cross-reacts with cynomolgus monkey CD39, but not murine CD39 overexpressed on CHO cells (Figure S5). No significant binding of TTX-030 to CD39 KO cell line SK-MEL-28-KO-W3 was observed (data not shown).
Figure 2.
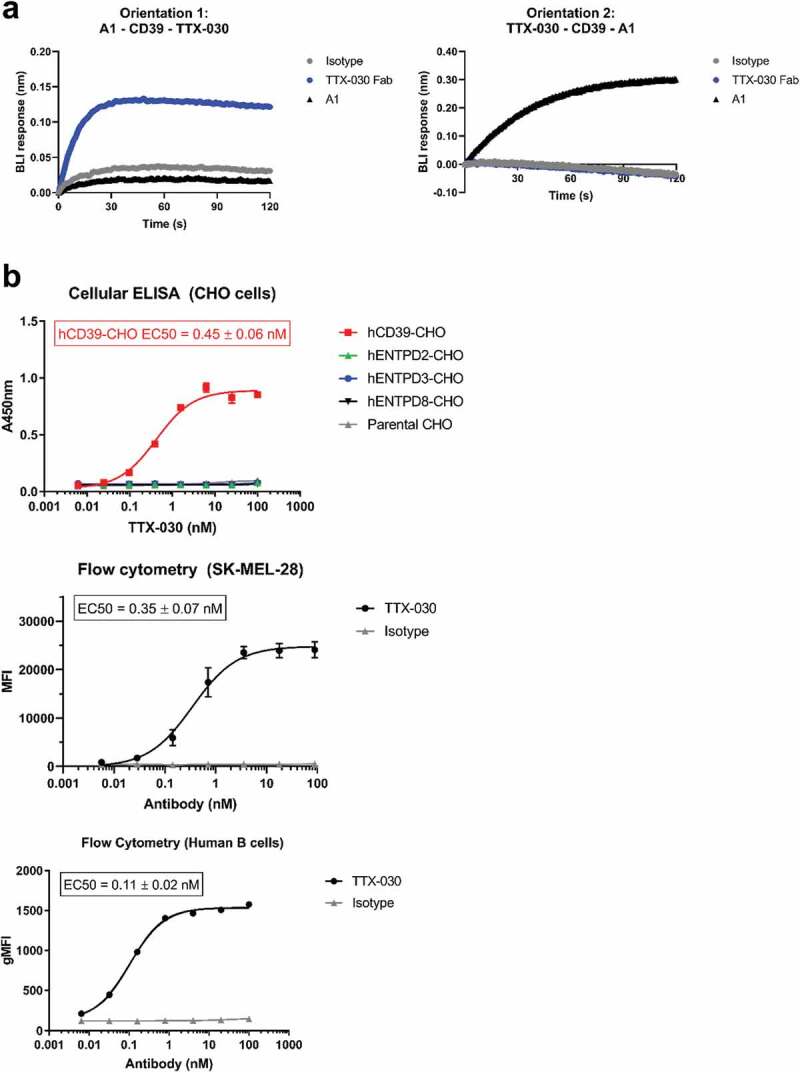
Anti-CD39 mAb, TTX-030, binds to CD39 with high affinity and specificity. A, TTX-030 Fab binds to a distinct epitope from anti-CD39 clone A1 mAb as measured by biolayer interferometry (BLI). Left panel, A1 mAb was captured to anti-murine IgG Fc biosensors and saturated with rhCD39-ECD. The competition association step sensorgram is shown with TTX-030 Fab, but not A1 or isotype control, producing a BLI binding response. Right panel, TTX-030 Fab was captured to anti-human Fab-CH1 biosensors and saturated with rhCD39-ECD. The competition association step sensorgram is shown with A1, but not TTX-030 Fab or isotype control, producing a BLI binding response. B, By cellular ELISA, TTX-030 had a subnanomolar EC50 for CHO cells overexpressing human CD39 but did not bind CHO cells overexpressing other extracellular human NTPDases. By flow cytometry, TTX-030 had a subnanomolar EC50 for SK-MEL-28 and primary human B cells. Error bars, S.D.; n= 3. MFI, mean fluorescence intensity.
TTX-030 is an uncompetitive allosteric inhibitor of cellular CD39
TTX-030 was shown to inhibit the extracellular domain (ECD) of recombinant human CD39 (rhCD39-ECD) ATPase activity using an endpoint Malachite Green-based assay that detects the release of free phosphate (Pi). TTX-030 inhibited rhCD39-ECD ATPase activity with an IC50 of 0.20 ± 0.06 nM with 55% maximal inhibition (Figure 3a). For comparison, competitive and nonspecific NTPDase inhibitor POM-1, at a threefold molar excess concentration above substrate, maximally inhibited rhCD39-ECD by 80%. Anti-CD39 clone A1 does not inhibit rhCD39-ECD ATPase activity (Figure S6). The hydrolysis of other NTPs by rhCD39-ECD was similarly inhibited by TTX-030 (Figure 3b). Importantly, TTX-030 inhibits not only soluble CD39-ECD, but membrane-bound cellular CD39 NTPase activity as well. TTX-030 maximally inhibited NTP and ADP hydrolysis on CD39+ SK-MEL-28 cells by up to 80% after a 15-min treatment with substrate (Figure 3c). TTX-030 also inhibits CD39 ATPase activity on primary human B cells (Figure 3d). We also employed lead nitrate-based enzyme histochemistry approach to evaluate the inhibitory potency of TTX-030 and POM-1 in human tonsils known to express high CD39 activity.43 Figure 3e depicts representative staining images of human tonsillar CD39-mediated ADPase activity, showing selective localization of ADP-specific brown staining in the interfollicular area. This activity was markedly reduced in the presence of POM-1 (10 μM), while TTX-030 (0.5 μg/ml) nearly completely blocked ADPase activity. Noteworthy, since POM-1 is a highly reversible CD39 inhibitor, it was necessary to maintain it in the assay medium during both the pre-treatment step and subsequent tissue incubation with the ADP substrate. By contrast, pretreatment of the human tonsils with TTX-030 and subsequent washing of the antibody prior to adding ADP exerted similar inhibitory effects on the measured ADPase activity (data not shown).
Figure 3.
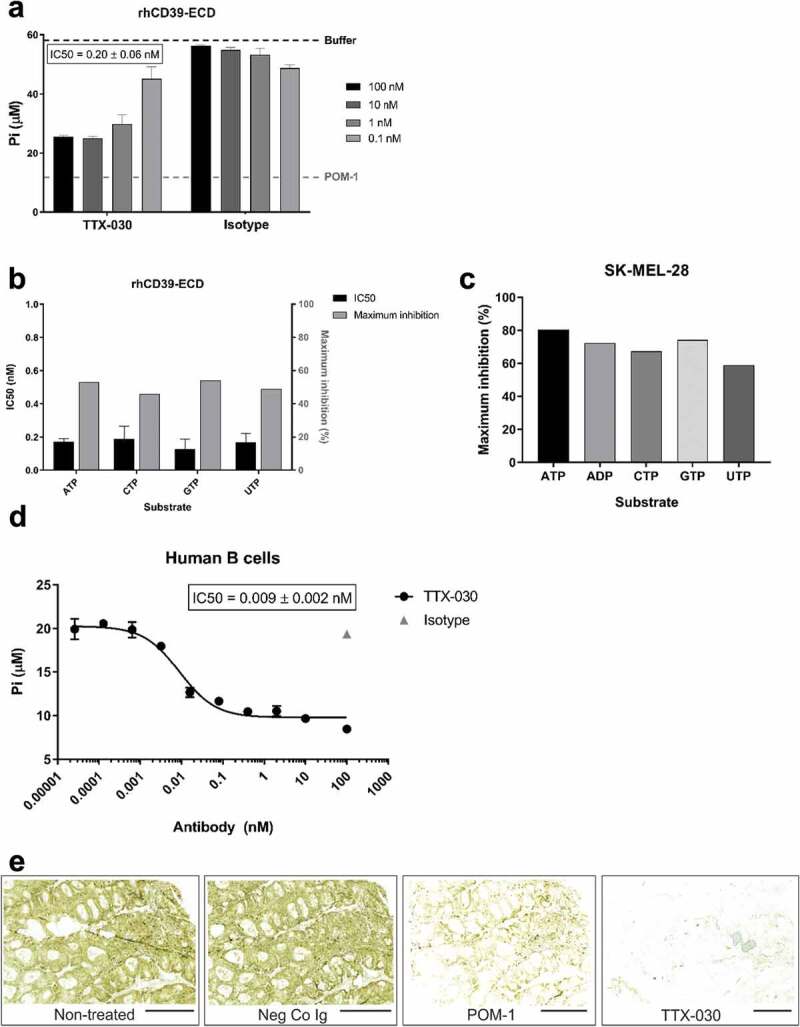
TTX-030 inhibits soluble and cell-surface expressed CD39-mediated NTP and ADP hydrolysis. A, A titration of TTX-030 or isotype control (hIgG4) was pre-incubated with rhCD39-ECD for 1 h prior to the addition of ATP substrate. The amount of free Pi was quantified after a 30 min incubation with ATP. TTX-030 inhibited rhCD39-ECD ATPase activity with an IC50 of 0.20 ± 0.06 nM with 55% maximal inhibition. Nonselective inhibitor POM-1 at 150 µM inhibited rhCD39-ECD activity by approximately 80%. Error bars, S.D.; n= 2. B, The assay in A using rhCD39-ECD was repeated with other NTP substrates. TTX-030 has similar potency (IC50) and maximal inhibition of CD39 NTPase activity. Error bars, S.D.; n= 2. C, TTX-030 maximally inhibits SK-MEL-28 cellular CD39 NTPase and ADPase activity to a greater extent than rhCD39-ECD. Maximal percent inhibitions are reported here without correcting for residual non-CD39-mediated NTP/NDPase activity. D, TTX-030 inhibits CD39-mediated Pi release of primary human B cells. E, TTX-030 and POM-1 inhibit tonsillar CD39-mediated ADPase activity in situ. Enzyme histochemistry images were captured using slide scanner. Nearly complete inhibition of brown staining in the presence of TTX-030 precludes proper comparative quantification of staining intensities in control versus treated sections. Scale bars, 3 mm
The mechanism of inhibition of TTX-030 was determined by enzyme kinetics studies under initial velocity conditions. These assays reveal TTX-030 to be an uncompetitive inhibitor (α < 1) of SK-MEL-28 CD39-mediated ATP hydrolysis (Figure 4a). Global fitting across multiple concentrations of ATP and TTX-030 to the partial, mixed inhibition equation (Equation 2) yielded α = 0.42 ± 0.03 and β = 0.23 ± 0.02. The maximal inhibition by TTX-030 was estimated to be 85% (β = 0.15) when corrected for the residual, non-CD39-related ATPase velocity of SK-MEL-29-KO-W3 cells (Figure 1b). TTX-030 similarly inhibited HUVEC and OAW42 CD39-mediated ATP hydrolysis via an uncompetitive mechanism (Table S3). Monovalent TTX-030 Fab similarly inhibits SK-MEL-28 ATP hydrolysis with α = 0.37 ± 0.06 and β = 0.20 ± 0.01. An α < 1 indicates preferential binding of TTX-030 to ATP-bound CD39 over free (i.e., non-substrate-bound) CD39.41 However, as binding studies in the absence of ATP revealed (Figure 2b), TTX-030 differs from the classical uncompetitive inhibitor definition in that it also tightly binds substrate-free enzyme. In fact, a highly related precursor to TTX-030 bound to rhCD39-ECD in the presence of non-hydrolyzable ATP analog, AMP-PNP, with increased monovalent affinity compared to free rhCD39-ECD (Figure 4b). Non-CD39-specific small molecule inhibitor ARL 67156 was confirmed to inhibit SK-MEL-28 ATPase activity by a competitive mechanism (α ≫ 1) (Figure S7), similar to previously published data with competitive inhibitor POM-1.12,44,45
Figure 4.
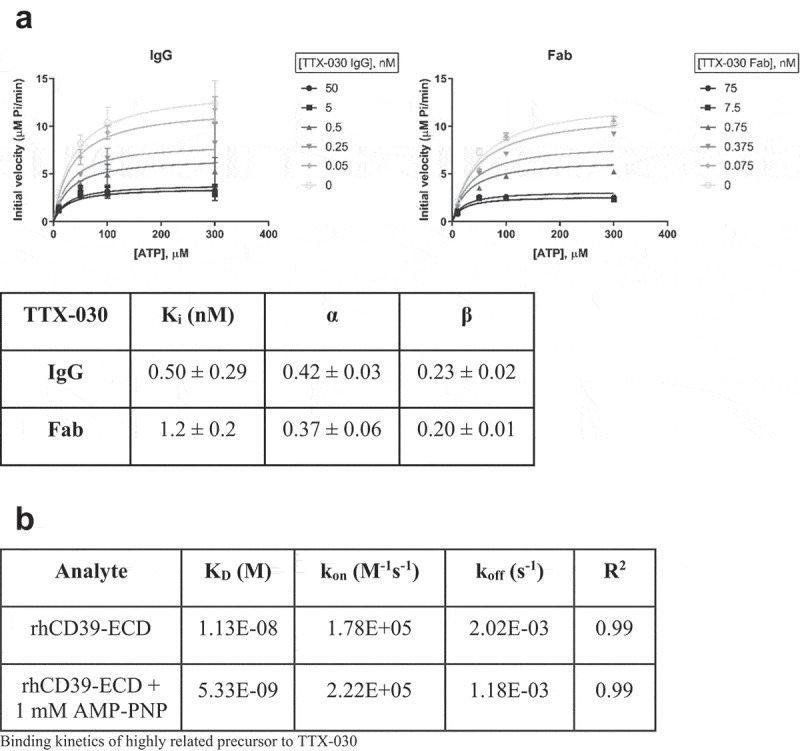
TTX-030 is an uncompetitive inhibitor of CD39 ATPase activity. A, SK-MEL-28 CD39-mediated ATPase initial velocities of Pi release as a function of ATP concentration were plotted for a titration of TTX-030 IgG (left panel) or Fab (right panel) and globally fit to the partial, mixed inhibition equation to obtain the inhibitor constant, Ki, α, and β. α is a parameter that determines the mechanism of inhibition where α < 1 indicates an uncompetitive mechanism; β is a parameter that determines the extend of partial inhibition where zero indicates complete inhibition and 1.0 indicates no inhibition. IgG, Error bars, S.D., n= 4. Fab, Error bars, S.E., n= 1. B, Binding kinetics of a highly related precursor to TTX-030 to recombinant extracellular domain of human CD39, rhCD39-ECD, in the presence or absence of non-hydrolyzable ATP analog, AMP-PNP. Binding to rhCD39-ECD was approximately 2-fold stronger in the presence of 1 mM AMP-PNP, supporting an uncompetitive mechanism. Data were globally fit to 1:1 binding model. KD, dissociation constant; kon, association (or on) rate; koff, dissociation (or off) rate
The activity of CD39 was also measured chromatographically as the rate of 3HATP conversion into dephosphorylated 3H-metabolites, determined as pooled ADP, AMP, and nucleoside fractions. Consistent with the above colorimetric Pi-liberating assay data, radio-thin-layer chromatograph (TLC) analysis of3HATP hydrolysis by SK-MEL-28 cells provides an independent line of evidence of uncompetitive inhibition mechanism of CD39 by TTX-030 (Figure 5a). Additional time-course analysis of product formation revealed that incubation of SK-MEL-28 cells with 100 μM 3HATP caused its progressive hydrolysis, with a respective rise in ADP and AMP as intermediate metabolites, and the formation of the ultimate reaction product, adenosine. Pretreatment of the cells with POM-1 or a suboptimal concentration of TTX-030 significantly inhibited the whole pattern of sequential3HATP hydrolysis to3Hadenosine (Figure 5b).
Figure 5.
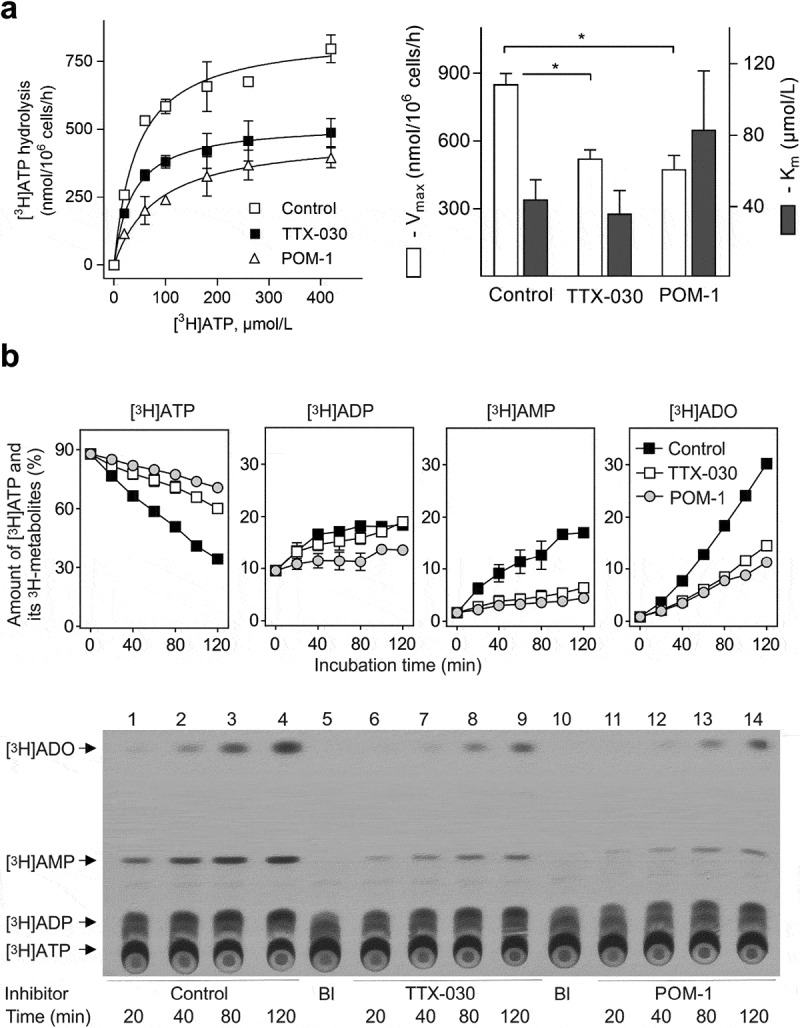
Effect of TTX-030 and POM-1 on the pattern of3HATP metabolism by SK-MEL-28 cells. A, Radio-TLC assays tracked the inhibition of CD39-mediated hydrolysis of3H-labeled ATP of SK-MEL-28 cells by POM-1 (10 µM) and a suboptimal treatment of TTX-030 (200 ng/ml). The kinetic parameters (Vmax and Km) for all catalytic activities were calculated from the presented curves and summarized in the right panel. Error bars, S.E., n= 3. *P < .05 as compared with control. B, Cultured SK-MEL-28 cells were incubated with 100 μM3HATP and subsamples of the medium were collected at timed intervals, followed by TLC separation and quantification of the amount of3HATP and its dephosphorylated3H-metabolites, ADP, AMP and adenosine (ADO). The ordinate shows the relative content of3H-labeled nucleotides and ADO expressed as percentage of total radioactivity added. Error bars, S.E., n= 2. Lower panel depicts a representative autoradiographic image of an overall pattern of metabolism of3HATP by control and treated cells. The blank (Bl) shows the radiochemical purity of3HATP in the absence of cells. Arrows indicate the positions of nucleotide and ADO standards
Epitope mapping confirms allosteric mechanism of inhibition
As described above (Figure S5), TTX-030 does not cross-react with murine CD39. Human-murine CD39 chimeras were therefore interrogated to ascertain the general binding site on cellular CD39. TTX-030 loses binding to hCD39 when residues between E142-Y159 are replaced with murine CD39 residues Q143-S158 (Table 1 and Figure S8). In contrast, the non-inhibitory anti-CD39 mAb A1 retained binding to this same human-mouse CD39 chimera and lost the ability to bind a human-mouse CD39 chimera in which residues S274-L278 are replaced by their murine counterparts. The TTX-030 binding site on CD39, E142-Y159, is distal to the putative ATP binding site, which includes, but is not limited to, essential residues E174 and S218.27,46 Taken together, these data demonstrate that TTX-030 is not binding to the active site region of the molecule, supporting an allosteric mechanism of action (MOA).
Table 1.
Allosteric mechanism of inhibition confirmed via mapping epitope to a region of CD39 distal to active site
| Chimera # | FASTA residue # from species of origin in chimera |
Mouse amino acid sequence | mAb binding to human-mouse CD39-CHO chimeras |
|||
|---|---|---|---|---|---|---|
| Human | Mouse | Human | A1 | TTX-030 | ||
| 1 | M1-E110 | L111-Q123 | T124-V510 | L111STELIPTSKHHQ | Yes | Yes |
| 2 | M1-E142 | Q143-S158 | Y159-V510 | Q143SADEVLAAVSTSLKS | Yes | No |
| 3 | M1-G187 | R188-K204 | Q206-V510 | R188FTQEQSWLSLISDSQK | Yes | Yes |
| 4 | M1-S274 | G274-V276 | L278-V510 | G274GV | No | Yes |
Robust inhibition of SK-MEL-28 ATP hydrolysis by TTX-030
The in vitro inhibition of SK-MEL-28 CD39 ATPase activity by TTX-030 was tested under conditions designed to stress antibody function and/or simulate the TME, including extensive washing post-antibody incubation, repeated addition of ATP, and at TME-like acidic pH. Previous assays included a 1 h cell pretreatment step with TTX-030 before ATP was added without washing. Here, SK-MEL-28 cells were treated with TTX-030 titrated into media, then unbound antibody was washed off, and ATP subsequently was added. Both TTX-030 Fab and full IgG formats were tested to distinguish between affinity and avidity effects of washing. Figure 6a shows that both TTX-030 Fab and full IgG formats sustain 80% maximal inhibition (β ≈ 0.20) of SK-MEL-28 ATPase activity despite the wash step. The potency of inhibition (as measured by Ki and IC50) of bivalent TTX-030 was only weakened by 2- to threefold with washing whereas the monovalent Fab version of TTX-030 was 20- to 50-fold less potent following washing. Extending the antibody preincubation step from 1 to 25 h before washing recovered the potency of bivalent TTX-030 to similar levels as without washing. Longer antibody incubation steps may induce receptor internalization47 and offer an alternative mechanism for inhibition of cell-surface receptor function, but we did not detect significant levels of CD39 internalization on SK-MEL-28 cells even with overnight incubations of TTX-030, while an anti-CD73 antibody does internalize (Figure S9). For TTX-030 Fab, extension of the preincubation step overnight improved potencies compared to the shorter 1 h preincubation, but Ki and IC50 were still approximately 10-fold less potent with a post-Fab incubation wash step. Collectively, these results suggest that the avidity of bivalent TTX-030 drives its slower off-rate and more potent inhibition (IC50 and Ki) compared to monovalent TTX-030 Fab. However, the preserved maximal inhibition (β) of TTX-030 Fab suggests any loss in potency during stressed conditions can be overcome by adding excess TTX-030 Fab.
Figure 6.
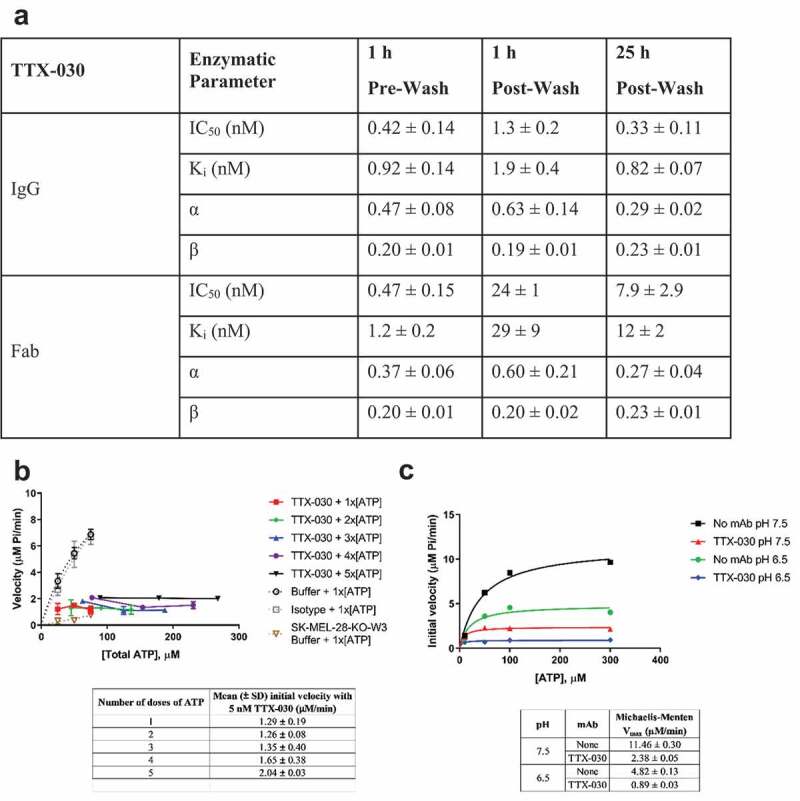
TTX-030 robustly inhibits CD39 under stressed and/or TME-like conditions. A, Washing unbound TTX-030 prior to adding ATP and extending the TTX-030 pre-incubation step from 1 h to 25 h had no effect on maximal inhibition, (1-β). However, the potency (Ki and IC50) was reduced, especially for TTX-030 Fab. SK-MEL-28 CD39-mediated ATPase initial velocities of Pi release as a function of ATP concentration were determined for a titration of TTX-030 (full length and Fab) and globally fit to the partial, mixed inhibition equation to obtain inhibitor constant, Ki, α, and β. α is a parameter that determines the mechanism of inhibition where α < 1 indicates an uncompetitive mechanism; β is a parameter that determines the extend of partial inhibition where zero indicates complete inhibition and 1.0 indicates no inhibition. Error, S.E., n= 1. IC50 was calculated from plotting initial velocities as a function of TTX-030 concentration for each ATP concentration and fitting to a three-parameter nonlinear regression model. Error, S.D., n= 3–4 across [ATP]. B, TTX-030 continued to inhibit cellular CD39 ATPase activity despite a continuous replenishment of an ATP reservoir. A dose titration of ATP was added to the reaction every 3 min and then the Pi release was determined over time. SK-MEL-28 CD39-mediated ATPase initial velocities as a function of ATP concentration were plotted for a titration of TTX-030. Only the first dose of ATP is shown for buffer only and isotype controls because the release of Pi was no longer at steady state at subsequent doses. Error bars, S.E., n= 1. C, TTX-030 (10 nM) maintained equal maximal inhibition of SK-MEL-28 CD39-mediated ATPase activity at pH 7.5 and TME-like pH 6.5. The Pi release velocity in the absence of TTX-030 was reduced at lower pH. Error bars, S.E., n= 1
TTX-030 continually inhibited SK-MEL-28 CD39 ATP hydrolysis velocity when a dose titration of ATP was added to the same reaction every 3 min (Figure 6b). After 5 ATP spikes, TTX-030 maintained 80% inhibition using an estimated Michaelis-Menten Vmax of 10 µM/min without inhibitor. These results confirm TTX-030 can maintain robust inhibition of CD39 enzymatic activity despite continuous replenishment of an ATP reservoir as would be expected from dying tumor cells.48
All the characterization of CD39 ATPase activity described above were performed at neutral pH. Since the TME is known to be slightly acidic,49 the potency and affinity of TTX-030 was tested under acidic conditions. At pH 6.5, TTX-030 maintained its maximal inhibition of SK-MEL-28 cells (Figure 6c). In the absence of TTX-030, the ATP hydrolysis velocity at pH 6.5 was 60% slower compared to pH 7.5 otherwise using the same assay conditions. TTX-030 also equivalently binds to recombinant CD39-ECD at pH 7.4 and pH 6.5 with a monovalent affinity, KD, of approximately 1.5 nM under each condition (Figure S10).
Discussion
For the first time, we describe and fully characterize a specific, allosteric antibody inhibitor of human CD39, TTX-030. We demonstrate TTX-030 is an uncompetitive inhibitor via steady-state enzyme kinetic analysis and confirm the allosteric inhibition MOA by demonstrating that TTX-030 binds to CD39 on a region distal to the hydrolysis/enzymatic active site. This feature of allostery allows TTX-030 to potently inhibit CD39 NTPase activity at the elevated ATP conditions found in the TME. Furthermore, robust inhibition was maintained in slightly acidic conditions and during repeated ATP pulsing.
Under steady-state conditions, TTX-030 maximally inhibits ATP hydrolysis by CD39+ SK-MEL-28 cells by 85%. The extent of inhibition by TTX-030 compares favorably to reported antibody inhibitors to other enzyme targets: 70% inhibition of CD39 by IPH5201,36 74% or 80% inhibition of CD73 by MEDI-9447 or IPH5301, respectively,32,36 56% inhibition of full-length MMP9 by GS-5745,50 50% inhibition of LOXL2 by AB0023,51 80% inhibition of CD38 by SAR650984,52 and 20% inhibition of CD38 by daratumumab.53 An important caveat is that many antibody inhibitors reported in the literature were only characterized against recombinant, soluble proteins rather than the cellular protein, which can dramatically change its potency. Here, we selected TTX-030 based on its inhibition of cellular CD39 and in situ tissue enzyme histochemistry. We demonstrated that TTX-030 had enhanced maximal inhibition of cellular CD39 ATPase activity compared to recombinant CD39 (85% versus 55%, respectively). TTX-030 allosterically inhibits cellular CD39 enzymatic activity without inducing internalization, which offers another point of differentiation compared to anti-CD73 inhibitors.54 Moreover, most published antibody inhibitors were not fully characterized using steady-state conditions. Rather, the percent inhibition of enzyme activity was reported for a single substrate concentration in an endpoint assay. When considering the translatability of in vitro enzymatic inhibition to in vivo models or a clinical setting, the optimal enzyme inhibitor will sustain inhibition at the range of physiological substrate concentrations, whereas an inhibitor with a suboptimal MOA will not. To illustrate, the nonselective competitive CD39 inhibitor, ARL (Figure S7), inhibits ATPase activity to significantly different extents depending on the concentrations of substrate and inhibitor. At 50 µM ATP, 10 µM ARL inhibits CD39 activity by 50%. At a TME-like 300 µM ATP, 10 µM ARL inhibition drops to 25%. In contrast, the novel uncompetitive inhibitor described here demonstrates a trend of enhanced inhibition of ATP hydrolysis by CD39 at elevated substrate concentrations (Figure 4a). This feature could be crucial for preserving the high local ATP concentration in the tumor environment to levels sufficient to elicit ATP-mediated activation of purinergic signaling, including P2X7 driven proinflammatory inflammasome responses. The eATP-P2X7-inflammasome axis yielded significant antitumor activity in preclinical models by both TTX-030 and an inhibitory anti-murine CD39 mAb.17,55 For example, the in vitro impact of TTX-030 on human immune cells in the presence of ATP includes enhanced T-cell function, dendritic cell maturation and function, and inflammasome activation (data not shown).55
In the absence of a published hCD39 crystal structure, we can try to model how TTX-030 allosterically inhibits CD39 by turning to structures of other related NTPDases, for example, LpNTPDase1 (27% sequence identity), rat NTPDase1 (74%), and rat NTPDase2 (42%).12,27,40,56,57 Structural data from these other NTPDases indicate that substrate hydrolysis is dependent on domain closure motions. One possibility is that the allosteric binding site of TTX-030 on CD39 could disrupt the conformational change required for catalysis. The crystal structure of LpNTPDase1 with non-hydrolyzable substrate (competitive inhibitor AMP-PNP) and transition-state analogs revealed that nucleotides bind the open form of LpNTPDase1 and are hydrolyzed following a 15° domain closure motion that allows the active site to achieve suitable geometry for catalysis.40 This domain closure motion is significantly less than the reported 114° conformational switch that CD73 undergoes during AMP hydrolysis.58,59 The restriction of hydrolytic domain motion has also been proposed as the mechanism of inhibition of small molecule inhibitors (akin to the nonselective POM-1 inhibitor of human CD39) of the ECD of rat NTPDase1.12 The authors hypothesized that the polyoxometallates inhibit NTPase activity via restricting the enzyme’s flexibility to adopt the necessary closed domain, but the transmembrane (TM) helices of NTPDase1 were removed to obtain a crystal structure. The TM domains not only anchor NTPDase1 to the membrane, but are also important for enzymatic function.60,61 Removal of one or both TM domains from native CD39 results in a 90% reduction in enzymatic activity. Therefore, caution should be taken interpreting conformational flexibility of the ECD-only molecule compared to membrane-bound CD39.
Thus, disruption of local domain motions plays a critical role in the regulation of the family of NTPDases, to which CD39 belongs. We mapped (Table 1) the TTX-030 CD39 epitope to a region between E142 and Y159, at the N-terminal lobe of the enzyme, and distal to the active site of CD39. The novel, projected TTX-030 binding site is distinct from the putative binding sites of published polyoxometallates inhibitors to rat ENTPDase1. We propose that TTX-030 binds CD39 such that it does not affect substrate binding, as supported by the ability of a TTX-030 precursor to bind in the presence of AMP-PNP (Figure 4b) and the steady-state enzyme kinetic data (Figure 4a). Rather, TTX-030 may prevent or disturb the subsequent domain closure motion required for efficient nucleotide hydrolysis. Since TTX-030 inhibits both CD39-ECD (no TM domains) and surface-bound CD39 (TM domains intact), the mechanism of TTX-030 inhibition goes beyond solely affecting the TM domains.
Aside from crystal structures with small molecule inhibitors, a published ecto-nucleotidase inhibitory mAb, hN3-B3S, was mapped to an epitope on the C-terminal lobe of human NTPDase3, a related human ecto-nucleotidase family member (37% sequence identity with hNTPDase1).25 The rat NTPDase2 crystal structure57 was used as a template to model human NTPDase3 in silico.62 The authors originally proposed an allosteric mechanism of inhibition of NTPDase3 activity caused by either deformation of the active site or restriction of enzyme flexibility.25 However, more precise mapping of hN3-B3S to NTPDase3 suggested the antibody-bound proximal to the active site and inhibited via steric hindrance (i.e., a competitive enzymatic inhibitor).62 Therefore, the inhibition of hNTPDase3 ATPase activity would be expected to be negatively impacted at high ATP concentrations.
The prevention or abatement of CD39 oligomerization is another potential mechanism by which TTX-030 allosterically inhibits CD39 ATPase activity. Oligomeric forms of membrane-expressed NTPDases have increased catalytic activity compared to monomers.21 A TTX-030-mediated disruption of CD39 oligomerization could also explain why TTX-030 has enhanced inhibition of cellular CD39 compared to rhCD39-ECD. Recombinant soluble ECD lacks TM domains and, while it can form dimers, oligomers of TM-truncated constructs do not have the same structure as cellular CD39 oligomers and have low ATPase activity.12,60 Intriguingly, we also identified distinct antibodies with greater rhCD39-ECD inhibition than TTX-030, yet lacked the ability to inhibit cellular hCD39 ATPase activity (data not shown). We hypothesize that these antibodies may bind epitopes or conformations presented by soluble rhCD39-ECD that are not physiologically presented by cellular CD39. This disconnect between soluble ECD and cellular CD39 biology informed our antibody discovery campaign to select for (and thoroughly characterize) therapeutic leads based on cellular CD39 inhibition rather than ECD inhibition. Our observation of substrate inhibition behavior at high ATP concentrations in the absence of TTX-030 (Figure S1)41 could also be explained by an obstruction of CD39 oligomerization. The nonhyperbolic dependence of ATPase velocity on substrate concentration suggests that, at sufficiently high ATP concentrations, a second molecule of ATP may bind to the CD39-ATP complex, rendering it less active. The second ATP molecule bound to CD39 may be preventing further oligomerization. Indeed, rat NTPDase1 monomers pre-complexed with AMP-PNP (ATP analog) could not dimerize because of the obstruction of the nucleotide-binding site.12
Extensive characterization of the steady-state kinetics of cellular CD39 NTPase activity revealed TTX-030 has an uncompetitive mechanism of inhibition. This allosteric mechanism allows TTX-030 to maintain its inhibition of CD39 despite the elevated extracellular ATP conditions of the TME. By potently and specifically inhibiting the enzymatic function of CD39, TTX-030 dually increases the concentration of immunostimulatory extracellular ATP and reduces the accumulation of immunosuppressive extracellular adenosine. Therefore, in human cancers, TTX-030 may help the immune system’s ability to detect and attack tumors. TTX-030 is currently being evaluated in Phase 1 clinical trials for cancer therapy (NCT03884556 and NCT04306900).
Materials and methods
Reagents and materials
Small molecule NTPDase inhibitors ARL 67156 trisodium salt and sodium metatungstate monohydrate (POM-1) were purchased from Tocris and Alfa Aesar, respectively. Due to residual phosphate in the POM-1 formulation, POM-1 was desalted using Zeba spin columns (Thermo Scientific) prior to use in inhibition assays. The following antibodies were used in this study: a mouse anti-CD39 (clone A1, eBioscience, Cat: 14–0399-82), guinea pig anti-human CD39 (hN1-1 C, provided by Prof. Jean Sevigny; http.//ectonucleotidases-ab.com), mouse anti-CD73 (clone 4G4, generated in-house),63 and polyclonal rabbit anti-ALPL (TNAP) antibody (MyBioSource, Cat: MBS003198). Secondary Alexa Fluor® 488-conjugated goat anti-mouse (Cat: A-11001) and Alexa Fluor® 633-conjugated goat anti-rabbit (Cat: A-21070) antibodies, and ProLong® Gold Antifade reagent with 4,6-diamidino-2-phenylindole (DAPI) were from Invitrogen™ (ThermoFisher Life Technologies). Cy™3-conjugated donkey anti-guinea pig IgG was from Jackson ImmunoResearch Laboratories (Cat: 706–165-148). Recombinant ectodomain of human and mouse CD39 (ectonucleoside triphosphate diphosphohydrolase-1, NTPDase-1) and human NTPDase2, 3, and 8, were obtained from R&D Systems. ATP, ADP, and AMP were purchased from Acros Organics. Other nucleotides were from Thermo Fisher. The Malachite Green Phosphate Detection Kit was purchased from R&D Systems, and the EnzChek Phosphate Assay Kit was purchased from Invitrogen. [2,8–3H]ATP was from Perkin Elmer. SpectraMax i3x (Molecular Devices) or Cytation 5 (BioTek Instruments) were used for plate-based assays. BD LSRFortessa™ X-20 (BD Biosciences) was used for flow cytometry assays. Isotype control was an in-house preparation of palivizumab hIgG4, hinge stabilized (S228P) unless otherwise indicated.
Cell lines and primary cells
Cell lines expressing CD39 were purchased from ATCC and grown according to the ATCC recommended protocol. Cells were detached from flasks using TrypLE™ (ThermoFisher). To generate the CD39 KO cell line, SK-MEL-28-KO-W3, CRISPR-Cas9 editing was used to remove the CD39 gene from the adherent human melanoma skin cell line, SK-MEL-28, as previously described,64 followed by limiting dilution to obtain a stable subclone. CD39 receptor number was determined via BD Quantibrite™ beads.65 Pearson correlation coefficient was calculated using built-in function within GraphPad Prism (GraphPad Software).
Human B cells were isolated from ¼ of a full-size leukopak (Stemcell Technologies) using the EasySep™ Human B cell Isolation Kit (Stemcell Technologies) and following the manufacturer’s recommended protocol. B cells were isolated from peripheral blood mononuclear cells by immunomagnetic negative selection.
Generation of inhibitory antibodies against human CD39
Anti-CD39 mAbs were selected from a synthetic human antibody library that displays full-length IgG on the surface of yeast cells as generally described previously.66,67 TTX-030 was selected based on its ability to inhibit and bind CD39 with high affinity.
Biolayer interferometry binding kinetics and binning
All binding kinetics and epitope binding assays were performed using established procedures for the Octet Red96 (ForteBio).68,69 Briefly, for selectivity assays, anti-human IgG Fc capture (AHC) biosensors were used to capture TTX-030 or hIgG4 isotype control. The biosensors were dipped for 300 sec into wells containing 100 nM rhCD39-ECD or each human NTPDase followed by a 600 sec dissociation step. For sandwich epitope binning assay, the anti-hCD39 mAb A1 was loaded onto anti-murine IgG Fc capture (AMC) biosensors and 50 nM rhCD39-ECD was added to saturation. Next, 1.25 µg/ml TTX-030 Fab, isotype control, or A1 were added. For assays determining the effect of non-hydrolyzable ATP analog, AMP-PNP, a highly related precursor of TTX-030 was immobilized onto AHC biosensors and binding kinetics to a titration of rhCD39-ECD were determined in the presence or absence of 1 mM AMP-PNP. Data were globally fit to the 1:1 binding model. All steps were performed at 30°C and 1000 rpm. All Octet data analysis was performed using ForteBio Data Analysis v10.0.
Cellular ELISA
The day before the assay, cells were harvested and reseeded into 96-well flat-bottom tissue culture plates (Corning No. 3595) at 30,000 cells/well in complete media. On the day of assay, cells were washed with phosphate-buffered saline (PBS) and fixed with 3.7% formaldehyde (Fisher BioReagents) for 20 min at room temperature. Fixed cells were washed again, then blocked with blocking buffer (PBS supplemented with 3% bovine serum albumin and 0.05% Tween-20) for 1 h at room temperature. TTX-030 or human IgG4 Isotype Control were serially diluted in blocking buffer for a total of eight test concentrations. Blocking buffer was removed from the cells and 50 µL of diluted antibodies were added to the cells for 1 hour at room temperature. Secondary antibodies including anti-human IgG-horseradish peroxidase (Jackson ImmunoResearch, Cat: 109–035-097) were diluted 1:10,000 in blocking buffer. Cells were then washed with PBS-T (PBS supplemented with 0.05% Tween-20) and 100 µL per well of diluted species-matched secondary detection antibodies was added for 1 hour at room temperature. Cells were washed again and 100 µL of room temperature 1-Step Ultra TMB-ELISA Substrate Solution (ThermoFisher) was added. Color developed to sub-saturation, approximately 5 min. To stop the colorimetric reaction, a 50 µL volume of 2 M sulfuric acid (Sigma Aldrich) was added to each well. The absorbance of each well was read at 450 nm on a plate reader. The A450 was plotted against antibody concentration and the EC50 was determined using GraphPad Prism.
Flow-based EC50 determination
SK-MEL-28 cells were plated at 40,000 cells per well density. Human B cells were plated at 150,000 cells per well density. Cells were washed once with fluorescence-activated cell sorting (FACS) Buffer (2% fetal bovine serum (FBS), 2 mM EDTA in PBS). TTX-030 was titrated starting from 15 µg/ml and 50 µl were added to each well. Following incubation for 30 min at 4°C, cells were washed twice with FACS buffer. Next, 50 µl of 1:200 diluted phycoerythrin-conjugated goat anti-human IgG secondary antibody (Southern Biotech, Cat: 9200–09) was added to the cells and incubated for 30 min at 4°C. The cells were washed twice and resuspended in FACS buffer supplemented with DAPI (1:1,000). Samples were acquired on a BD Fortessa X-20 using the high-throughput sampler. Data were analyzed using FlowJo software.
KinExA affinity determination
Avid cellular binding was determined using the KinExA® platform (Sapidyne Instruments) as previously described.70 Briefly, SK-MEL-28 and SK-MEL-28-KO-W3 were titrated while TTX-030 was held constant. The reactions were performed at 37°C for up to 2 hours. Following incubation, cells were centrifuged and the free fraction of TTX-030 in the supernatant was removed. Next, the supernatant containing TTX-030 was exposed to rhCD39-ECD-coated beads. Bead-bound TTX-030 was detected with a fluorescently labeled anti-human IgG detector. The equilibrium binding constant and number of CD39 molecules expressed on the cell surface were calculated.
Inhibition of recombinant ectodomain of human CD39
Endpoint (non-kinetic) rhCD39-ECD NTPase assays monitored the final inorganic phosphate (Pi) concentration using the Malachite Green Phosphate Detection kit. TTX-030 and matched isotype control were titrated from a starting final concentration of 100 nM in Tris Assay Buffer (25 mM Tris-HCl pH 7.5, 5 mM CaCl2). In parallel, rhCD39-ECD was prepared in Tris Assay Buffer to a final assay concentration of 1 nM, and desalted POM-1 was prepared to a final concentration of 150 µM. CD39 was preincubated with antibody or POM-1 for 1 h at 37°C. After preincubation, 50 µM ATP was added to the reaction mixture and the plate was returned to 37°C for another 30 min. The reaction was stopped by transferring to ice for immediate phosphate detection or to −20°C for future phosphate readout. Phosphate detection via Malachite Green was determined following the manufacturer’s instructions, and the absorbance was measured on a plate reader at 620 nm. The absorbance value was interpolated using a Pi standard curve. The IC50 and maximum inhibition were determined using GraphPad Prism.
Cellular kinetic phosphate detection assay
On the day before the assay, cells were harvested and reseeded into 96-well black wall, clear flat-bottom tissue culture plates (ThermoFisher, Catalog No. 165305) at 35,000 cells/well in 100 µL of culture medium. The plated cells were incubated overnight at 37°C, 5% CO2. On the day of the assay, the cells were typically greater than 80% confluent. Prior to treating cells with test agents, the cells were washed 2–3 times with Tris Assay Buffer to remove free phosphate and FBS. The washes consisted of adding 100–200 µL of Tris Assay Buffer to each well and discarding the supernatant via aspiration. Tris Assay Buffer (100 µL) was added to the wells of the outer borders of the plate following washing to limit any evaporation-related artifacts.
All test agents (small molecules and antibodies) were prepared in Tris Assay Buffer. To initiate treatment, the final wash was removed and 100 µL of pre-treatment test agent added to the cells. The cells were treated for 60 min at 37°C, 5% CO2 with TTX-030 starting at a dose titration of 50–200 nM. Additional controls included treatment with isotype control (hIgG4, hinge stabilized, or hFab in the same formulation buffer as TTX-030) at the same highest concentration as TTX-030, treatment with 500 µM ARL, and un-supplemented Tris Assay Buffer. POM-1 was not compatible with the reagents of the EnzChek assay. Pre-treatments of cells with ARL were shortened to 20 min. Twenty min before the completion of the pre-treatment step, PNP and MESG substrate (from EnzChek Phosphate Assay Kit) were spiked into the pre-treatment reaction to remove any residual Pi and equilibrate the reagents to 37°C prior to the addition of ATP. A Pi standard curve was prepared in Tris Assay Buffer and was also added to untreated cells 20 min prior to addition of ATP. The Pi standard curve consisted of a 6-point curve starting at 200 µM with two-fold dilutions thereof.
After the pre-treatment phase, cells were treated with a dose titration of ATP substrate (or other NTPs, ADP, or AMP, when noted) by spiking in 10 µL of ATP diluted in Assay Buffer, and the plate was immediately transferred to a SpectraMax i3x Plate Reader to monitor Absorbance at 360 nm (A360) every minute for at least 15 min. For the pH 6.5 assay, HCl was added to the Tris Assay Buffer to decrease pH from 7.5 to 6.5 and the lower pH buffer was used for all assay steps. The remainder of the assay proceeded as described above.
In situ ecto-ADPase activity assay
For localization of ADPase activities in human tonsils, a modification of the lead nitrate method was employed.43 In brief, palatine tonsils were obtained from adult patients with chronic tonsillitis undergoing routine tonsillectomy. The tonsils were washed with physiological salt solution, embedded in the cryo-mold with Tissue-Tek® O.C.T. compound (Sakura Finetek Europe B.V.), cut using a cryostat and stored at −80°C. Tonsil cryosections were pre-incubated for 45 min in Trizma-maleate sucrose buffer (TMSB; 40 mM Trizma® maleate; 0.25 M sucrose, pH 7.4) supplemented with the alkaline phosphatase inhibitor levamisole (2 mM) either in the absence (control) or presence of POM-1 (10 μM) or TTX-030 (0.5 μg/ml). The enzymatic reaction was performed then for 30 min at 37°C in a final volume of 20 ml TMSB containing 1.5 mM Pb(NO3)2, 1 mM CaCl2, 300 μM ADP, and tested CD39 inhibitors. The lead orthophosphate precipitated in the course of nucleotidase activity was visualized as a brown deposit by incubating sections in 0.5% (NH4)2S for 30 seconds followed by three washes in Trizma-maleate buffer for 5 min each. Slides were mounted with Aquatex medium (Merck, Germany). Multiple images of adjacent tissue areas were captured using Pannoramic 250 slide scanner (3DHistech Ltd.), and further stitched to a larger overview using the accompanying Pannoramic Viewer 1.15.4 software. The images of control and treated tissue were captured at identical exposure times and further acquired in parallel using Adobe Photoshop CS6 software.
Mode of inhibition analysis under steady-state conditions
All kinetic phosphate monitoring data from the plate reader were processed using Microsoft Excel and Prism version 7. First, raw A360 values were subtracted from the background A360 of wells that were not treated (Tris Assay Buffer only, no ATP, no Pi standard). Next, a second-order polynomial (quadratic equation built-in Prism) was used to fit each subtracted A360 value to the amount of Pi interpolated from the Pi standard curve. Interpolated Pi values were plotted as a function of time post-ATP addition to generate an enzyme reaction progress curve. Initial velocities were determined by calculating the slope (through built-in linear regression analysis in Prism) of the Pi vs. time curve for the first 5 to 15 min after ATP addition. Pi values that approached the upper limit of detection (typically 120 µM) as well as Pi values that were no longer increasing linearly with time were omitted from the linear regression analysis as these are no longer at steady state. The initial velocities (along the standard error of linear regression) for each treatment condition were plotted as a function of ATP concentration, [ATP]. The velocity vs substrate curves were fit using built-in nonlinear regression analysis (Michaelis-Menten (Equation 1)) within Prism.41
| (1) |
where ν = enzyme velocity, [S] = substrate concentration, Vmax = maximal reaction velocity, and Km = Michaelis-Menten constant (the substrate concentration at half maximal velocity). The Vmax and Km values obtained for untreated (no mAb) cells were used as input values for further analysis.
To determine the mechanism of CD39 inhibition (competitive, uncompetitive, or noncompetitive) by TTX-030 or ARL, a mixed model inhibition equation that allowed for partial inhibition was used (Equations 2–4).51
| (2) |
| (3) |
| (4) |
These equations were input into Prism as a user-defined equation where Vmax’ is the apparent Vmax, Km’ is the apparent Km, [I] is the inhibitor concentration, Ki is the inhibitor constant, α is a parameter that determines the mechanism of inhibition, and β is a parameter that determines the extent of partial inhibition. When α ≫ 1, the inhibitor is competitive. When α < 1, the inhibitor is uncompetitive. When α ≈ 1, the inhibitor is noncompetitive. When β = 0, there is complete inhibition and the equation becomes the standard mixed model inhibition equation.41 Vmax and Km were obtained from the Michaelis-Menten Kinetic Model using cells treated with ATP only, i.e., in the absence of inhibitor. Therefore, Vmax and Km are constants in Equation 3 and Equation 4 and are used as constraints when fitting data using the mixed model equation. The initial velocities for each condition were plotted as a function of [ATP], and the concentration of TTX-030 ([I] in nM) was used as the column title in Prism. The best-fit value and the standard error (S.E.) (i.e., the error in global fitting the equation to the data) of α, β, and Ki were reported for each condition studied. While these experiments are technically n = 1, each initial velocity value (per [I] and [S] pair) reported is calculated from at least five measurements as described above, further empowering the results. When multiple biological replicates of kinetic phosphate monitoring assays were performed, mean values of each parameter were reported and standard deviation (S.D.) was calculated with Excel.
Radio-TLC CD39 assays
SK-MEL-28 cells were seeded overnight onto 96-well flat bottom clear plates at a density of 5000 cells per well. CD39 activity was determined by incubating the adherent cells at 37°C in a final volume of 100 μl RPMI-1640 medium supplemented with 25 mM HEPES (pH 7.35) and containing 4 mM β-glycerophosphate and different concentrations of ATP with tracer 3HATP. In competitive assays, the cells were pretreated with POM-1 (10 µM) or TTX-030 (200 ng/ml) for 30 min prior to addition of 3HATP. Aliquots of the mixture were applied onto Alugram® SIL G/UV254 TLC sheets (Machery-Nagel), separated using appropriate solvent systems and then either developed by autoradiography using Kodak BioMax MS films (Carestream) or quantified by scintillation β-counting, as described elsewhere.63,71
Epitope mapping
Human-murine CD39 chimeras were generated to identify the binding site of TTX-030. Briefly, sequences of human and mouse CD39 were aligned with rat ENTPD1 and rat ENTPD2 crystal structures (PDB file 3zx0 and 3cj1, respectively)12,57 to identify solvent-exposed regions. These domains were then swapped, sequentially, from the human CD39 sequence to the murine CD39 sequence. Gene fragments were obtained from Genewiz and cloned into pCMV vector. CHO cells were transiently transfected with mock, WT hCD39, WT mCD39, or human-mouse chimeras. Flow cytometry assays and analysis proceeded as described above.
Alternative ATP cellular kinetic assay designs
Preincubations in media assays (“Post-wash”)
Test agents (small molecules and antibodies) were prepared in SK-MEL-28 Growth Media (DMEM (ThermoFisher) supplemented with 10% FBS (Gibco), 2 mM L-glutamine (ThermoFisher), and 100 U/mL Penicillin-Streptomycin (ThermoFisher). TTX-030 (hIgG4) was dose titrated with a starting concentration of 75 nM. TTX-030 (hFab) was titrated with a starting concentration of 200 nM. The media from the cells was removed and replaced with 100 µl media containing an indicated dose of TTX-030 for 1 or 25 h at 37°C, 5% CO2. Additional controls included treatment with isotype control mAb at the same concentration as the highest TTX-030 concentration, treatment with 500 µM ARL, or cells treated with Tris Assay Buffer (“untreated”). Pre-treatment of cells with ARL was limited to 20 min due to toxicity with longer incubation times. The remainder of the protocol and enzymatic analysis proceeded as described above.
ATP reservoir assay
Cells were pre-treated for 20 min with TTX-030 as described above. ATP (25, 50, or 75 µM) was added at time 0, 3, 9, 15, and 21 min by spiking in 10 µL of ATP diluted in Tris Assay Buffer. Cells received the same ATP treatment at each timepoint (i.e., wells that initially were spiked with 25 µM ATP continued to receive 25 µM ATP for each timepoint for a final [ATP] = 89 µM when the change in volume was considered). The assay plate was ejected from the plate reader for each ATP treatment, and the SoftMax Pro Append function was used to resume measuring A360 every min after the plate was reinserted into the reader. All incubations with test agents and ATP were carried out at 37°C, as were the kinetic measurements.
Confocal microscopy
SK-MEL-28 and CD39−/- SK-MEL-28-KO-W3 cells grown on 13-mm coverslips in 24-well plates were washed with PBS and immediately fixed for 5 min with PBS containing 4% paraformaldehyde. The adherent fixed cells were processed for staining of membrane-bound nucleotidases, as described earlier.63 The coverslips were mounted with ProLong® medium with DAPI and examined using a 3i spinning disk confocal microscope CSU-W1 with Photometrics Evolve EM-CCD camera, and Slidebook 6.0 software (Intelligent Imaging Innovations, Inc.). Z-stacks of the images were captured using C-Apochromat 40×/1.1 objective (Carl Zeiss) at identical exposure times for each antibody. Maximum intensity projection of a confocal z-stack for each channel for prepared by using Imaris 8.4 Bitplane software.
Antibody internalization assays
Receptor-mediated internalization was performed by labeling TTX-030, isotype control, and an anti-CD73 antibody (in-house preparation of CPX-006) with an amine-reactive pH-sensitive dye according to the manufacturer’s instructions (Promega, Cat: G9845).47 The dye-labeled antibodies (20 nM) were spiked into fresh medium and incubated at 37°C, 5% CO2. Following 30 min, 1 h, 2 h, 3 h, or overnight incubation, the unbound antibodies and media were washed with PBS and the fluorescence (Ex 532 nm/Em 560 nm) was measured on a plate reader. Buffer was then replaced with fresh media, the plate was returned to an incubator, and the reaction was monitored at subsequent timepoints.
Supplementary Material
Acknowledgments
We thank Dr. Jouko Sandholm at the Cell Imaging and Cytometry Unit of Turku Bioscience Centre for microscopy support and Biocenter Finland for microscopy equipment. We are also grateful to Dr. Jean Sevigny for providing the guinea pig antibody against human CD39, and to Dr. Heikki Irjala for providing the samples of human tonsils for histochemical analysis.
Abbreviations
- ADP
Adenosine diphosphate
- AHC
Anti-human IgG capture
- AMC
Anti-murine IgG capture
- AMP
Adenosine monophosphate
- AMP-PNP
Adenylyl-imidophosphate
- ATP
Adenosine triphosphate
- CHO
Chinese hamster ovary
- ECD
Extracellular domain
- ENTPD1
Ectonucleoside triphosphate diphosphohydrolase-1
- Fab
Antigen-binding fragment
- Km
Michaelis constant
- KO
Knockout
- mAb
Monoclonal antibody
- MOA
Mechanism of action
- NPP1
Nucleotide pyrophosphatase/phosphodiesterase-1
- NTPDase
Nucleoside triphosphate diphosphohydrolase
- PBS
Phosphate-buffered saline
- Pi
Inorganic phosphate
- rhCD39-ECD
Recombinant human CD39 extracellular domain
- TLC
Thin-layer chromatography
- TM
Transmembrane
- TME
Tumor microenvironment
- TMSB
Trizma-maleate sucrose buffer
- TNAP
Tissue-nonspecific alkaline phosphatase
- Tregs
Regulatory T cells
- Vmax
Maximal velocity
- WT
Wild type
Disclosure statement
Spatola, BN, Lerner, AG, Wong, C, dela Cruz, T, Welch, M, Fung, W, Kovalenko, M, Beers, C, Corbin, J, and Soros, VB are, or were, employees of Tizona Therapeutics and hold stock and/or stock options in the same. Research by Losenkova, K and Yegutkin, GG was in part sponsored by Tizona Therapeutics.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Vijayan D, Young A, Teng MW, Smyth MJ.. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709. doi: 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann H, Zebisch M, Sträter N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012;8:437–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaupel P, Multhoff G. Commentary: a metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. 2016;7:332. doi: 10.3389/fimmu.2016.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takenaka MC, Robson S, Quintana FJ. Regulation of the T cell response by CD39. Trends Immunol. 2016;37:427–39. doi: 10.1016/j.it.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bours M, Swennen E, Di Virgilio F, Cronstein B, Dagnelie P. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Jacob F, Novo CP, Bachert C, Van Crombruggen K. Purinergic signaling in inflammatory cells: P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinergic Signal. 2013;9:285–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. 2018;18:601–18. [DOI] [PubMed] [Google Scholar]
- 8.Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem Mol Biol. 2014;49:473–97. [DOI] [PubMed] [Google Scholar]
- 9.Deterre P, Gelman L, Gary-Gouy H, Arrieumerlou C, Berthelier V, Tixier J-M, Ktorza S, Goding J, Schmitt C, Bismuth G. Coordinated regulation in human T cells of nucleotide-hydrolyzing ecto-enzymatic activities, including CD38 and PC-1. Possible role in the recycling of nicotinamide adenine dinucleotide metabolites. J Immunol. 1996;157:1381–88. [PubMed] [Google Scholar]
- 10.Jackson EK, Ren J, Mi Z. Extracellular 2′, 3′-cAMP is a source of adenosine. J Biol Chem. 2009;284:33097–106. doi: 10.1074/jbc.M109.053876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36:293–303. doi: 10.1038/onc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zebisch M, Krauss M, Schäfer P, Sträter N. Crystallographic evidence for a domain motion in rat nucleoside triphosphate diphosphohydrolase (NTPDase) 1. J Mol Biol. 2012;415:288–306. doi: 10.1016/j.jmb.2011.10.050. [DOI] [PubMed] [Google Scholar]
- 13.Koziak K, Sévigny J, Robson SC, Siegel JB, Kaczmarek E. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb Haemost. 1999;82:1538–44. doi: 10.1055/s-0037-1614868. [DOI] [PubMed] [Google Scholar]
- 14.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9:1–13. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, Cosgrove C, Ledderose C, Junger WG, Robson SC, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 2015;11:e1005177. doi: 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whiteside TL. The role of regulatory T cells in cancer immunology. ImmunoTargets Therapy. 2015;4:159. doi: 10.2147/ITT.S55415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X-Y, Moesta AK, Xiao C, Nakamura K, Casey M, Zhang H, Madore J, Lepletier A, Aguilera AR, Sundarrajan A, et al. Targeting CD39 in cancer reveals an extracellular ATP-and inflammasome-driven tumor immunity. Cancer Discov. 2019;9:1754–73. doi: 10.1158/2159-8290.CD-19-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD 39 and CD 73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276:121–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boison D, Yegutkin GG. Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell. 2019;36:582–96. doi: 10.1016/j.ccell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allard D, Allard B, Stagg J. On the mechanism of anti-CD39 immune checkpoint therapy. J ImmunoTher Cancer. 2020;8:e000186. doi: 10.1136/jitc-2019-000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signal. 2006;2:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fausther M, Lecka J, Kukulski F, Lévesque SA, Pelletier J, Zimmermann H, Dranoff JA, Sévigny J. Cloning, purification, and identification of the liver canalicular ecto-ATPase as NTPDase8. Am J Physiol Gastrointest Liver Physiol. 2007;292:G785–G95. doi: 10.1152/ajpgi.00293.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. 2011;187:676–83. doi: 10.4049/jimmunol.1003884. [DOI] [PubMed] [Google Scholar]
- 24.Levesque S, Lavoie ÉG, Lecka J, Bigonnesse F, Sévigny J. Specificity of the ecto‐ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol. 2007;152:141–50. doi: 10.1038/sj.bjp.0707361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munkonda MN, Pelletier J, Ivanenkov VV, Fausther M, Tremblay A, Künzli B, Kirley TL, Sévigny J. Characterization of a monoclonal antibody as the first specific inhibitor of human NTP diphosphohydrolase‐3: partial characterization of the inhibitory epitope and potential applications. Febs J. 2009;276:479–96. doi: 10.1111/j.1742-4658.2008.06797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goueli SA, Hsiao K, Beavis PA. Monitoring and characterizing soluble and membrane-bound ectonucleotidases CD73 and CD39. PloS One. 2019;14:e0220094. doi: 10.1371/journal.pone.0220094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iqbal J, Shah SJA. Molecular dynamic simulations reveal structural insights into substrate and inhibitor binding modes and functionality of ecto-nucleoside triphosphate diphosphohydrolases. Sci Rep. 2018;8:1–11. doi: 10.1038/s41598-018-20971-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar M, Lowery R, Kumar V. High-throughput screening assays for cancer immunotherapy targets: ectonucleotidases CD39 and CD73. SLAS DISCOVERY: Advanc Sci Drug Discovery. 2020;25:320–26. [DOI] [PubMed] [Google Scholar]
- 29.Lee S-Y, Fiene A, Li W, Hanck T, Brylev KA, Fedorov VE, Lecka J, Haider A, Pietzsch H-J, Zimmermann H, et al. Polyoxometalates—potent and selective ecto-nucleotidase inhibitors. Biochem Pharmacol. 2015;93:171–81. doi: 10.1016/j.bcp.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Levy G. Mechanism‐based pharmacodynamic modeling. Clin Pharmacol Ther. 1994;56:356–58. doi: 10.1038/clpt.1994.149. [DOI] [PubMed] [Google Scholar]
- 31.Robinson PK. Enzymes: principles and biotechnological applications. Essays Biochem. 2015;59:1–41. doi: 10.1042/bse0590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geoghegan JC, Diedrich G, Lu X, Rosenthal K, Sachsenmeier KF, Wu H, Dall’Acqua WF, Damschroder MM. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs. 2016;8:454–67. doi: 10.1080/19420862.2016.1143182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allard D, Chrobak P, Allard B, Messaoudi N, Stagg J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett. 2019;205:31–39. doi: 10.1016/j.imlet.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J ImmunoTher Cancer. 2018;6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, George S, Hughes BGM, Hellmann MD, Shepard DR, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 2020;10:40–53. doi: 10.1158/2159-8290.CD-19-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrot I, Michaud H-A, Giraudon-Paoli M, Augier S, Docquier A, Gros L, Courtois R, Déjou C, Jecko D, Becquart O, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27:2411–25. e9. doi: 10.1016/j.celrep.2019.04.091. [DOI] [PubMed] [Google Scholar]
- 37.Holland P Targeting the adeonsine axis to treat cancer. Brisbane Immunotherapy Conference; Brisbane, Australia; 2019. [Google Scholar]
- 38.Yan J, Li X-Y, Aguilera AR, Xiao C, Jacoberger-Foisac C, Nowlan B, Robson SC, Beers C, Moesta AK, Geetha N, et al. Control of metastases via myeloid CD39 and NK cell effector function. Cancer Immunol Res. 2020;8:356–67. doi: 10.1158/2326-6066.CIR-19-0749. [DOI] [PubMed] [Google Scholar]
- 39.Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn-Schmiedeberg’s arch pharmacol. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
- 40.Zebisch M, Krauss M, Schäfer P, Lauble P, Sträter N. Crystallographic snapshots along the reaction pathway of nucleoside triphosphate diphosphohydrolases. Structure. 2013;21:1460–75. doi: 10.1016/j.str.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 41.Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. New York (NY): John Wiley & Sons; 2004. [Google Scholar]
- 42.Tam SH, McCarthy SG, Armstrong AA, Somani S, Wu S-J, Liu X, Gervais A, Ernst R, Saro D, Decker R. Functional, biophysical, and structural characterization of human IgG1 and IgG4 Fc variants with ablated immune functionality. Antibodies. 2017;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Losenkova K, Paul M, Irjala H, Jalkanen S, Yegutkin GG. Histochemical approach for simultaneous detection of ectonucleotidase and alkaline phosphatase activities in tissues. In: Purinergic signaling. New York (NY): Springer; 2020. p. 107–16. [DOI] [PubMed] [Google Scholar]
- 44.Müller CE, Iqbal J, Baqi Y, Zimmermann H, Röllich A, Stephan H. Polyoxometalates—a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioorg Med Chem Lett. 2006;16:5943–47. [DOI] [PubMed] [Google Scholar]
- 45.Köhler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Müller CE, Eltzschig HK, et al. CD39/Ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116:1784–94. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 46.Drosopoulos JH, Broekman MJ, Islam N, Maliszewski CR, Gayle RB, Marcus AJ. Site-directed mutagenesis of human endothelial cell ecto-ADPase/soluble CD39: requirement of glutamate 174 and serine 218 for enzyme activity and inhibition of platelet recruitment. Biochemistry. 2000;39:6936–43. doi: 10.1021/bi992581e. [DOI] [PubMed] [Google Scholar]
- 47.Nath N, Godat B, Zimprich C, Dwight SJ, Corona C, McDougall M, Urh M. Homogeneous plate based antibody internalization assay using pH sensor fluorescent dye. J Immunol Methods. 2016;431:11–21. doi: 10.1016/j.jim.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini J-L, Zitvogel L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8:3723–28. doi: 10.4161/cc.8.22.10026. [DOI] [PubMed] [Google Scholar]
- 49.Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Appleby TC, Greenstein AE, Hung M, Liclican A, Velasquez M, Villaseñor AG, Wang R, Wong MH, Liu X, Papalia GA, et al. Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J Biol Chem. 2017;292:6810–20. doi: 10.1074/jbc.M116.760579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodriguez HM, Vaysberg M, Mikels A, McCauley S, Velayo AC, Garcia C, Smith V. Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J Biol Chem. 2010;285:20964–74. doi: 10.1074/jbc.M109.094136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallee F, Zhou-Liu Q, Ferrari P, Pouzieux S, Lahoute C, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20:4574–83. doi: 10.1158/1078-0432.CCR-14-0695. [DOI] [PubMed] [Google Scholar]
- 53.van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, Lokhorst HM, Parren PW. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. 2016;270:95–112. doi: 10.1111/imr.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, Hammond SA, Rothstein R, Rios-Doria J, Poon E, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. 2016;5:e1208875. doi: 10.1080/2162402X.2016.1208875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lerner AG, Kovalenko M, Welch M, Cruz T, Jones J, Wong C, Spatola B, Eberhardt M, Wong A, Fung W, et al. Abstract 5012: targeting CD39 with a first-in-class inhibitory antibody prevents ATP processing and increases T-cell activation. Cancer Res. 2019;79:5012. [Google Scholar]
- 56.Zebisch M, Baqi Y, Schäfer P, Müller CE, Sträter N. Crystal structure of NTPDase2 in complex with the sulfoanthraquinone inhibitor PSB-071. J Struct Biol. 2014;185:336–41. doi: 10.1016/j.jsb.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 57.Zebisch M, Strater N. Structural insight into signal conversion and inactivation by NTPDase2 in purinergic signaling. Proceedings of the National Academy of Sciences of the United States of America 2008; 105:6882–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knapp K, Zebisch M, Pippel J, El-Tayeb A, Muller CE, Strater N. Crystal structure of the human ecto-5ʹ-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure. 2012;20:2161–73. doi: 10.1016/j.str.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Rahimova R, Fontanel S, Lionne C, Jordheim LP, Peyrottes S, Chaloin L. Identification of allosteric inhibitors of the ecto-5ʹ-nucleotidase (CD73) targeting the dimer interface. PLoS Comput Biol. 2018;14:e1005943. doi: 10.1371/journal.pcbi.1005943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grinthal A, Guidotti G. Dynamic motions of CD39 transmembrane domains regulate and are regulated by the enzymatic active site. Biochemistry. 2004;43:13849–58. doi: 10.1021/bi048644x. [DOI] [PubMed] [Google Scholar]
- 61.Grinthal A, Guidotti G. CD39, NTPDase 1, is attached to the plasma membrane by two transmembrane domains. Why? Purinergic Signal. 2006;2:391–98. doi: 10.1007/s11302-005-5907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ivanenkov VV, Crawford PA, Toyama A, Sevigny J, Kirley TL. Epitope mapping in cell surface proteins by site-directed masking: defining the structural elements of NTPDase3 inhibition by a monoclonal antibody. Protein Eng Des Sel. 2010;23:579–88. doi: 10.1093/protein/gzq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Losenkova K, Zuccarini M, Helenius M, Jacquemet G, Gerasimovskaya E, Tallgren C, Jalkanen S, Yegutkin GG. Endothelial cells cope with hypoxia-induced depletion of ATP via activation of cellular purine turnover and phosphotransfer networks. Biochimica Et Biophysica Acta (BBA) Mol Basis Dis. 2018;1864:1804–15. doi: 10.1016/j.bbadis.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 64.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 65.Pannu KK, Joe ET, Iyer SB. Performance evaluation of QuantiBRITE phycoerythrin beads. Cytometry: J Int Soc Anal Cytol. 2001;45:250–58. [DOI] [PubMed] [Google Scholar]
- 66.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–73. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vasquez M, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel. 2013;26:663–70. doi: 10.1093/protein/gzt047. [DOI] [PubMed] [Google Scholar]
- 68.Estep P, Reid F, Nauman C, Liu Y, Sun T, Sun J, Xu Y. High throughput solution-based measurement of antibody-antigen affinity and epitope binning; MAbs: Taylor & Francis:2013. 270–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abdiche YN, Miles A, Eckman J, Foletti D, Van Blarcom TJ, Yeung YA, Pons J, Rajpal A. High-throughput epitope binning assays on label-free array-based biosensors can yield exquisite epitope discrimination that facilitates the selection of monoclonal antibodies with functional activity. PLoS One. 2014;9:e92451. doi: 10.1371/journal.pone.0092451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie L, Mark Jones R, Glass TR, Navoa R, Wang Y, Grace MJ. Measurement of the functional affinity constant of a monoclonal antibody for cell surface receptors using kinetic exclusion fluorescence immunoassay. J Immunol Methods. 2005;304:1–14. doi: 10.1016/j.jim.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 71.Yegutkin GG, Henttinen T, Jalkanen S. Extracellular ATP formation on vascular endothelial cells is mediated by ecto-nucleotide kinase activities via phosphotransfer reactions. Faseb J. 2001;15:251–60. doi: 10.1096/fj.00-0268com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.