

Abstract
To cope with sudden changes in the external environment, the budding yeast Saccharomyces cerevisiae orchestrates a multi-faceted response that spans many levels of physiology. Several studies have interrogated the transcriptome response to endoplasmic reticulum (ER) stress and the role of regulators such as the Ire1 kinase and Hac1 transcription factor. However, less is known about responses to ER stress at other levels of physiology. Here, we used quantitative phosphoproteomics and computational network inference to uncover the yeast phosphoproteome response to the reducing agent DTT and the upstream signaling network that controls it. We profiled wild-type cells and mutants lacking IRE1 or MAPK kinases MKK1 and MKK2, before and at various times after DTT treatment. In addition to revealing downstream targets of these kinases, our inference approach predicted new regulators in the DTT response, including cell-cycle regulator Cdc28 and osmotic-response kinase Rck2, which we validated computationally. Our results also revealed similarities and surprising differences in responses to different stress conditions, especially in the response of Protein Kinase A (PKA) targets. These results have implications for the breadth of signaling programs that can give rise to common stress response signatures.
Keywords: yeast stress response, Unfolded Protein Response, Phosphoproteomic, signaling network
Graphical Abstract
INTRODUCTION
Free-living microbes are frequently exposed to sudden and often drastic changes to their environment. Thus, cells have evolved intricate methods to sense when there is a problem and mount the proper physiological response to cope with stressful conditions. The budding yeast Saccharomyces cerevisiae reacts to diverse stresses by mounting a multi-faceted response that includes dynamic changes to the transcriptome, proteome, phosphoproteome, and other cellular levels. These together activate defense strategies to combat the stressor, often in concert with transient reduction of growth and cell-cycle progression while cells acclimate. Many studies have profiled transcriptome changes in yeast responding to different stressors (reviewed in 1); however, much less is known about changes to the phosphoproteome, including phosphorylation events that mediate downstream effects as well as those that regulate cellular signaling to coordinate cellular responses.
An important stress encountered by yeast and other organisms is the accumulation of misfolded proteins in the lumen of the endoplasmic reticulum (ER), which occurs in response to environmental conditions and during human disease (reviewed in 2). To cope, eukaryotic cells mount a response called the unfolded protein response (UPR) 3,4. The UPR activates a transcriptional program, regulated by the yeast transcription factor Hac1, that induces genes encoding ER chaperones, lipid biosynthetic enzymes, and ER-associated protein degradation (ERAD) factors to effectively increase ER-folding capacity 5. Concurrently, ER stress leads to selective mRNA degradation and translational suppression to decrease ER protein import and thus the ER protein-folding burden 6–8. The UPR is activated by a regulatory system partially conserved from yeast to humans. Whereas humans utilize a trio of upstream regulators, including Ire1, PERK, and ATF6 3,4, yeast utilize a single pathway controlled by the ER stress-sensing kinase Ire1 9,10. Upon ER stress, misfolded proteins bind directly to the Ire1 ER luminal domain, causing Ire1 to oligomerize 11. This activates the Ire1 catalytic RNase domain 12, which excises an intron in the HAC1 transcript 13 and produces functional Hac1 to induce over 380 targets 5. Ire1 can also be activated by other related stresses including lipid bilayer stress 14. Once cells have regained ER homeostasis, Ire1 deoligomerizes, effectively shutting down the UPR 15.
ER stress induced by the reducing agent DTT is an interesting case compared to other yeast stresses. Whereas acute doses of many stresses induce rapid transcriptome responses (peaking within 15-30 minutes after treatment), DTT treatment produces a multi-phase response 16. Transcripts linked to the UPR are induced rapidly, within 15 minutes of ER stress 17,5,18. However, other transcriptomic signatures of the stress response are not observed until hours after DTT exposure, when growth rate also declines. This includes the Environmental Stress Response (ESR), a common gene expression response to diverse stresses. ESR activation includes induced expression of ~300 genes involved in stress defense and reduced expression of ~600 genes involved in ribosome biogenesis and growth 17,16. Under optimal conditions, the ESR is suppressed (reflected by high expression of ribosome-biogenesis and growth-promoting genes and low abundance of stress-defense transcripts), in part via signaling through the Protein Kinase A (PKA) and TOR pathways 19–21. During an acute stress response, PKA and TOR are thought to be suppressed to enable ESR activation by stress-activated signaling pathways 22.
While the UPR is activated directly by unfolded ER proteins, late-phase responses are likely responding to secondary effects of unresolved ER stress. Defects in secretion may inflict stress on the cell wall/membrane, explaining activation of the Hog1-mediated hyperosmotic response (HOG) and cell wall integrity (CWI) MAPK pathways 23–27.
Hog1 is not required for UPR-mediated gene expression 27, but is thought to influence autophagy and metabolism during unresolved ER stress and is important for acclimation 26–29. The CWI pathway is triggered in response to long-term ER stress and is necessary for stimulation of the Cch1-Mid1 Ca2+ channel, which leads to intracellular Ca2+ accumulation and subsequent activation of the phosphatase calcineurin. Defects in this response have little impact on UPR activation but are important for viability during long-term ER stress 23. Despite the implication of these other pathways, a prior study by Pincus et al. showed that the transcriptional response to 5 mM DTT can be recapitulated simply by over-expressing functional Hac1 while inactivating PKA 18, suggesting that PKA suppression is the prime contributor to the late-phase DTT transcriptional response.
To better understand the signaling response to DTT, we measured dynamic changes in protein phosphorylation during ER stress by performing quantitative phosphoproteomics on wild-type and mutant S. cerevisiae responding to DTT. Whereas the early response showed a modest number of phosphorylation changes, the late response was much broader and included proteins involved in diverse functions. Network inference implicated potential regulators in both the early and late responses, and identified responses dependent on Ire1 and the CWI pathway. Surprisingly, the response of PKA targets was strikingly different from that seen in other conditions including the immediate response to sodium chloride (NaCl) stress, where PKA targets are globally reduced in phosphorylation level. Our results reveal commonalities and distinctions in the phosphoproteome responses to different stressors.
EXPERIMENTAL PROCEDURES AND METHODS
Strains and growth conditions for phosphoproteomics
All strains were of the BY4741 background. The ire1Δ::KANMX strain was obtained from Thermo Scientific, Waltham, MA 30. Deletion strains were verified by diagnostic PCR. The mkk1Δ::KANMX mkk2Δ::KANMX strain was generated by mating BY4741 mkk1Δ::KANMX (AGY0047) and BY4742 mkk2Δ::KANMX (AGY0062). Following sporulation, tetrads were dissected, and cells were subjected to diagnostic PCR to verify the original deletions. Subsequently, a halo mating type assay was performed, and MATa variants were selected and used for all experiments 31.
To assess how DTT affects the phosphoproteome, a single replicate time series was completed by growing BY4741 for at least 7 generations in YPD at 30°C to early log phase (OD600 ~ 0.3-0.4). An aliquot was collected before and 5, 15, 60, and 120 min after treatment with 2.5 mM final concentration DTT (Ultrapure, Invitrogen). Immediately following the 60 min collection, an equal volume of pre-warmed YPD containing 2.5 mM DTT was added to the culture to ensure cells remained in log phase for the final collection. This was important for the longer time course to ensure that cells remained in log phase at the final time point, far from the diauxic shift in which thousands of transcripts and peptides changes due to glucose depletion. The ire1Δ::KANMX and mkk1Δ::KANMX mkk2Δ::KANMX mutants, along with a paired wild type, were collected before, 15, and 120 min after DTT treatment in biological duplicate, diluted at 60 min identically to the wild-type timecourse. Together, this produced biological triplicates for 0, 15, and 120 min wild-type samples and duplicate measurements for each mutant. Cells were collected by centrifugation and immediately flash frozen until proteomic analysis.
Mass Spectrometry Workflow
Tryptic digest, TMT labelling, phosphopeptide enrichment, and fractionation
Lysate preparation was as previously described 32. Briefly, cell pellets were thawed on ice, washed, resuspended in lysis buffer (8 M urea, 50 mM Tris pH 8.0, and protease and phosphatase inhibitor cocktail table, Roche, Indianapolis, IN), and lysed by glass bead milling (Retsch, Newton, PA). Proteins were reduced by incubation for 45 min at 55 °C with 5 mM dithiothreitol, cooled to room temperature, and alkylated by addition of 15mM iodoacetamide in the dark for 45 min before quenching with 5 mM dithiothreitol. Lysates were digested with trypsin in diluted urea buffer (~ 1.5 M) at 1:50 trypsin:protein (Promega, Madison, WI) over night, then acidified by the addition of 10% TFA, desalted over a Sep-Pak (Waters, Milford, MA), and lyophilized to dryness in a SpeedVac (Thermo Fisher, Waltham, MA). Peptides were labelled with tandem mass tags (Pierce TMT, Rockford, IL), according to the manufacturer’s instruction. The first 5-plex experiment explored the wild-type BY4741 response to DTT (0, 5, 15, 60, 120 min post DTT addition in channels 126, 127C, 128C, 129C, and 130C, respectively). The subsequent 10-plex TMT experiments measured the ire1Δ, mkk12Δ, and wild-type BY4741 response to DTT at 0, 15, 120 min, labeled in successive TMT channels in that numerical order. Labelled peptides were then mixed in 1:1 ratio, and the resulting mixture was desalted over a Sep-Pak. ~2.5 mg of the labelled peptide mixture were used to enrich for phosphopeptides via immobilized metal affinity chromatography (IMAC) using magnetic beads (Qiagen, Valencia, CA), according to the published method 33. Flow through unmodified peptides were lyophilized to dryness and ~500 μg was fractionated, as described below.
High-pH reverse phase liquid chromatography was used to fractionate enriched phosphopeptides and unmodified flow-through peptides.. Mobile phase A consisted of 20 mM ammonium formate pH 10.0; mobile phase B contained 80% acetonitrile in 20 mM ammonium formate pH 10.0. Phosphopeptides and unmodified peptides were fractionated into 16 factions, concatenated into 8 combined fractions, and lyophilized to dryness. Each fraction of phosphopeptides was resuspended in 15 μl 0.2% formic acid for LC-MS/MS analysis. Two injection replicas of 5 μl were analyzed via LC-MS/MS analyses. ~2 μg of each fraction of unmodified peptides were injected for the analysis.
LC-MS/MS
All capillary columns were prepared in house, as previously described 34. All used solvents were LC-MS grade and purchased from Thermo Fisher Scientific (Waltham, MA). Mobile phase buffer A consisted of 0.2% formic acid water; mobile phase B consisted of 70% acetonitrile, 0.2% formic acid in water, and 5% DMSO. Columns were heated to 60-65°C inside an in-house made heater. Peptides were loaded onto a column in 0% B and separated at a flow rate of 300-400 nl/min over a 90 min gradient for phosphopeptides. Eluting peptides were analyzed on a quadrupole-ion trap-Orbitrap hybrid Fusion® (Thermo Scientific, San Jose, CA). Orbitrap survey scans were performed at a resolving power of 60,000 at 200 m/z with an AGC target of 1x106 ions and maximum injection time set to 100 ms. The instrument was operated in the Top Speed mode with 2 s cycles and monoisotopic precursor selection turned on. All tandem MS/MS scans were acquired on precursors with charge states of 2-6 with ion count target was set to 5x104. For TMT-labelled samples tandem scans were collected in the Orbitrap at a resolving power of 60,000 at 200 m/z using HCD fragmentation with normalized collision energy of 35, dynamic exclusion of 25 s, and the maximum injection time of 350 ms (for enriched phosphopeptides).
Data analysis
The raw data corresponding to TMT-labelled peptides were searched against Saccharomyces genome database (SGD) of yeast protein isoforms (Version 64-1-1) (downloaded 12.02.2014) 35 and processed using the COMPASS software suite 36. Carbamidomethylation (+57.0513 Da) of cysteine residues and TMT 10plex (+229.1629 Da) on N-termini of proteins and lysine residues were included as fixed modifications. Oxidation of methionine (+15.999 Da) and TMT 10plex on tyrosine (+229.1629 Da) were included as variable modifications. Average mass tolerances of 125 ppm and 0.015 Da were allowed for MS1 precursor searches and MS2 fragment searches, respectively. Up to three missed cleavages on tryptic peptides following the proline rule were allowed. 1% false discovery rate (FDR) correction was performed on all identified peptides and proteins. TMT reporter region quantification was performed using an in-house software TagQuant, as previously described 37. Briefly, the raw reporter ion intensity in each TMT channel was corrected for isotope impurities, as specified by the manufacturer for the used product lot, and normalized for mixing differences by equalizing the total signal in each channel. In cases where no signal was detected in a channel, the missing value was assigned with the noise level in the original spectrum (i.e. noise-band capping of missing channels), and the resultant intensity was not corrected for impurities or normalized for uneven mixing. All proteomic data is available from the PRIDE database under accession number PXD017396.
Criteria used to define phosphopeptides reproducibly changing in response to DTT
Since protein levels vary late in the timecourse, we normalized the wild type 120 min phosphopeptide log2(fold changes) to the 120 min log2(fold-change) in protein abundance. For phosphopeptides whose corresponding proteins were missing values from that replicate, the average log2(fold change) in protein abundance from the other replicates was used. Phosphopeptides responding to DTT but lacking protein measurements in all replicates were omitted from the analysis. We did not normalize phosphopeptides to protein levels at the 15 min time point, as the measured protein abundances at 15 min versus 0 min were very similar (R2 = 0.99 and slope ~1.0) and only 5 proteins displayed a statistically significant abundance change wild-type cells at an FDR < 0.05.
We scored significant phosphopeptide changes in two ways. First, we scored peptide changes and protein changes separately using linear models in edgeR 38; peptides measured in at least two replicates whose significant (FDR < 0.05) log2(fold-change) could not be explained by the underlying protein log2(fold-change) can be considered significant. However, to better account for underlying protein changes, we also calculated the log2 (fold-change) in protein abundance-normalized phosphopeptides, across timepoints and across strains. Because edgeR cannot accommodate the normalized, ratioed data, here we identified all phosphopeptides exhibiting ≥1.5-fold normalized change at 120 or 15 min versus 0 min in 2 of 3 replicates, and with the same directionality of change, as reproducibly changing. Because some clearly reproducible normalized-peptide values were missed by edgeR, we focused our analysis on normalized fold-change calls but cite FDRs in the manuscript in specific cases. All FDR values and fold-change calls are available in Dataset S1. Hierarchical clustering the log2 ratios for post-DTT vs. pre-stress peptide abundances was performed using Cluster 3.0 39.
Identification of recurrent phosphorylation motifs using motif-X
To identify enriched motifs in the DTT phosphorylation data, we ran motif-X40, independently on pooled lists of peptides, defined as those significantly increased at 15 min, reproducibly induced at 120 min, or reproducibly repressed at 120 min after DTT. A custom script was used to map peptides from the motif-X analysis against the yeast proteome to recover gene and phospho-site identifiers. We ran motif-X using the following parameters: extend from: SGD yeast proteome; central character: s* or t*; width: 13; occurrences: 10; significance: 1x10−6. This approach revealed 8 enriched motifs that capture 67% of all peptides 120 min post-DTT. Groups of phosphopeptides with similar phosphorylation changes in response to DTT and similar motifs were grouped together.
Defining mutant dependencies and module groupings
As with wild-type samples, phosphopeptide abundance at the 120 min timepoint was normalized to underlying protein changes, so as to score changes in protein phosphorylation. Phosphopeptides that changed significantly in wild-type were scored as having a dependency on Ire1 or Mkk1/Mkk2 if they showed a 30% or more difference in abundance compared to the paired wild-type samples, in both replicates at either 15 min or 120 min post-DTT treatment, and with the same directionality of change in the two replicates. Linear models were also applied in edgeR to identify significant changes (FDR < 0.05) in phosphopeptides or proteins, as described above. Phosphopeptides with larger DTT-dependent induction or repression in the mutant compared to WT were termed amplified responses, whereas those with smaller induction or repression were termed defective responses, respectively. Phosphopeptides that changed in abundance in the wild type but failed to meet these criteria in the mutant comparison were classified as no-phenotype. These classifications were used to further partition peptide groups above into modules, such that module peptides share the same directionality of phosphorylation change at a given timepoint, the same phospho-motif as identified by motif-X, and the same mutant dependencies or lack thereof. Peptides affected by both mutants were represented multiple times according to each mutant dependency.
Identification of shared interactors
We compiled a background dataset of protein interactions as follows: physical interactions from Biogrid 41 were retained for low-throughput measurements and high-throughput measurements if observed in at least two studies; protein-RNA interactions were excluded. We added to this kinase-substrate and phosphatase-substrate interactions from the KID database 42 as previously described 43. To identify putative module regulators, we identified proteins within the background network that interact with more of the proteins whose peptides are represented in each module than expected by chance using the hypergeometric test comparing to all proteins in the background dataset, with Benjamini-Hochberg p-value correction < 0.05 taken as significant. These are termed shared interactors (SIs) 44.
Functional enrichment analysis
Gene Ontology (GO) enrichments were calculated by the hypergeometric test using the website FunSpec 45. P-values < 1x10−4 were taken as significant. Enriched categories are available in Supporting Material S3. Enrichment of proteins interacting with PKA was done using the list of proteins interacting with Tpk1, Tpk2, or Tpk3 as downloaded from Biogrid 41.
In vitro PKA activity
Cells were grown in YPD to an OD600 ~0.5 before 2.5 mM DTT was added to the culture. 20 mL of culture was harvested by centrifugation and flash frozen to fix cells, immediately before and at 15, 60, and 120 min after DTT addition. Cells were diluted at 60 min into YPD + 2.5 mM DTT as described above to maintain the protocol used for proteomic sampling. Cell pellets were resuspended in 1 mL SB buffer (1 M sorbitol, 20 mM Tris HCl, pH 7.4) with 300 units of zymolyase (Amsbio) and 10 μL of protease inhibitor cocktail IV (Millipore), and incubated at 30°C for 10 minutes. Spheroplasts were collected by centrifuging, washed in SB buffer, then resuspended in 750 μL HLB buffer (10 mM Tris HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.3% (vol/vol) NP-40, 10% (vol/vol) glycerol) with 10 μL of protease inhibitor cocktail IV and incubated for 10 minutes on ice. Cells were lysed through ten rounds in a Dounce homogenizer. Total protein abundance was calculated by a Bradford assay. The PKA Kinase Activity Assay Kit (Abcam, ab139435) was performed following the manufacturer’s protocol. The reaction was detected in a TECAN Infinite 200 Pro at a wavelength of 450 nm. Background-subtracted PKA signal was normalized to protein abundance for relative PKA activity. Significant differences in relative PKA activity among samples were calculated with a paired T-test from three biological replicates.
RESULTS
We designed an experimental workflow to characterize the effect of DTT treatment on the yeast phosphoproteome at varying times before and after treatment. We used quantitative mass spectrometry and isobaric tagging to quantify phosphopeptides before and at 5, 15, 60, and 120 min after treatment with 2.5 mM DTT, triplicated at 15 and 120 min (see Methods). This dose of DTT provides enough stress to reduce cell growth but enables acclimation at a new growth rate. The timecourse captured the early response regulated as part of the UPR as well as the later phase when DTT-mediated transcriptional changes peak and growth is reduced (Fig 1A, 17). To ensure that cells did not enter the yeast diuaxic shift during the longer time course (when hundreds of transcripts and proteins change in abundance), cells were diluted immediately after sampling at 60 min to maintain them in logarithmic phase at the time the final 120 min sample was collected. The dilution did not affect the growth rate (Fig 1A).
Figure 1. Phosphoproteomic response to DTT treatment.
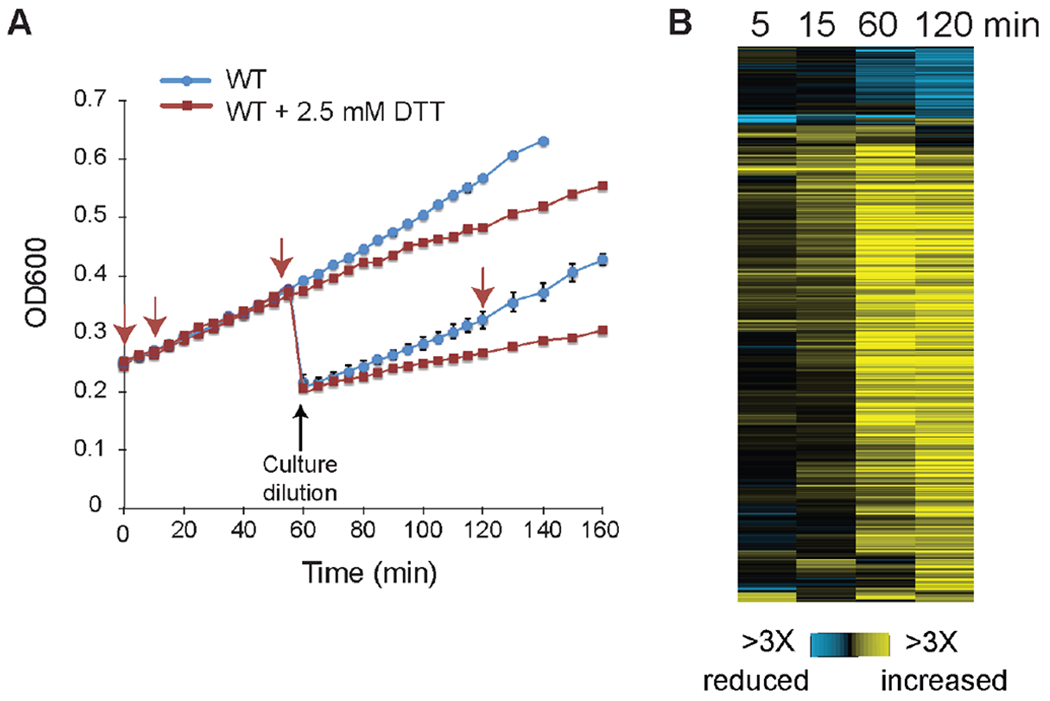
A) Growth of a wild-type culture with (red curve) and without (blue curve) 2.5 mM DTT treatment added at 0 min. Data represent the average and standard deviation of biological duplicates, for cultures with and without dilution at 60 min (which was required to maintain cells in logarithmic phase for the longer time proteomic timecourse). Arrows indicate the timing of sample collection. B) Timecourse of 502 unique phosphopeptides (rows) identified as having a reproducible change in phosphorylation at either 15 or 120 min after DTT treatment and measured in at least three timepoints, see text for details. Data represent the log2(fold change) in phosphopeptide abundance according to the key; 15 and 120 min samples show the average of biological triplicates, and 120 min sample data was normalized to underlying changes in protein abundance.
Of the 7,814 phosphopeptides identified (mapping to 1,983 proteins), we selected those with reproducible changes in phosphopeptide abundance during the early (15 min) or late (120 min) phase of the DTT response (FDR < 0.05 or reproducible 1.5X change, see Methods) (Fig 1B). Given the likelihood of long-term protein changes, phosphopeptide abundance at 120 min was normalized to underlying protein abundance to measure changes in phosphorylation status. The early response constituted only 107 peptides with reproducible 1.5X changes, from 90 proteins, and compared to all proteins in the proteome was enriched for kinases (including Ire1, FDR = 0.015), proteins localized to the bud and/or cell periphery, and proteins involved in cell-cycle regulation (P < 1x10−4, hypergeometric test). Almost all (98%) of these peptides showed increased phosphorylation, including on serine 840 (S840) and S841 of Ire1 that are autophosphorylated in trans during ER stress, consistent with UPR activation 46.
In contrast, the late response to DTT was substantially larger, consisting of 771 peptides that changed by >1.5X, mapping to 522 proteins. The response was also more diverse, since 19% of phosphopeptides showed phosphorylation decrease in addition to the 81% with phosphorylation increases. Late-induced phosphoproteins (Fig 1B) were heavily enriched for kinases, including members of the CWI pathway (Pkc1 and Bck1, FDR <0.07), several in the HOG pathway (Sln1, Ssk2, Pbs2, Hog1, Rck2, FDR < 0.05), Yck1/2 and their upstream activators Pkh1/2 that respond to membrane stress (FDR < 0.05), calmodulin-dependent kinase Cmk2, cell cycle regulators (Cdc28, Swe1, Cbk1, Cdc14, FDR < 0.05) and other stress-regulated kinases (Rim15, Yak1, Sak1, FDR < 0.05). Proteins with increasing phosphorylation were also enriched for those involved in budding, the actin cytoskeleton, endocytosis, cytokinesis, stress granule formation, proteins localized to the membrane/cell wall, and proteins whose mRNAs are induced with the environmental stress response 17(P<1x10−4, hypergeometric test). De-phosphorylated proteins (Fig 1B) were enriched for ribosomal proteins, consistent with regulated translation after DTT stress 6,47, and other proteins localized to the nucleolus, cell wall, and plasma membrane (P < 1x10−4).
We next profiled the DTT response in cells lacking IRE1 or the CWI MAPKKs MKK1 and MKK2, both known to respond to DTT stress, in biological duplicate at two timepoints (see Methods). Ire1 autophosphorylates upon ER stress but is not known to phosphorylate other targets 4. We identified several proteins with amplified phosphorylation in the ire1Δ mutant compared to wild type, likely representing secondary responses at both 15 and 120 min post stress (Dataset S1). Only two phosphopeptides displayed mildly reduced phosphorylation at 15 min (although they did not meet the FDR selection), mapping to kinase Kkq8 and hydrophillin Stf2 that influences removal of reactive oxygen species and is transcriptionally induced as part of the UPR 5,48,49. In contrast, we identified 68 peptides that changed in wild-type but showed defects at 120 min in the ire1Δ mutant; two thirds of these showed smaller changes in the mutant compared to wild-type (Dataset S1). The group of peptides that failed to get fully phosphorylated was enriched for proteins involved in ER functions (including ER chaperone Kar2, and Sfb2 and Sec31 important for ER to Golgi secretion) and included several kinases (including Kkq8 and poorly characterized Tda1 and Iks1).
In contrast to the minimal effects of IRE1 deletion, deletion of MKK1 and MKK2 affected a larger number of peptides, including 14 at 15 min and 217 at 120 min, the vast majority (84%) of which showed defects in DTT-induced phosphorylation. In total, 42% of phosphorylation events scored in wild-type were affected by Mkk1/2, confirming previous findings that the CWI pathway plays a role in the adaptive response to unresolved ER stress 25. Proteins with defects in DTT-induced phosphorylation were enriched for kinases, proteins localized to the bud neck, cell periphery, and actin cytoskeleton, and proteins linked to endocytosis, carbohydrate metabolism, and osmotic stress response. Interestingly, proteins that failed to get dephosphorylated in response to DTT were enriched for ribosomal proteins (P < 1x10−4, hypergeometric test, Supporting Material S3). These results capture known functions of the CWI pathway 50 but also raise the possibility that CWI may influence translational regulation after DTT stress.
Inferring the kinases that regulate DTT-dependent phosphorylation changes
While our results confirm the involvement of Ire1 and Mkk1/Mkk2 in the DTT response, most of the phosphorylation changes seen in wild-type cells were independent of these kinases, pointing to other regulators. To implicate kinases mediating these changes, we used a computational pipeline developed previously in our lab for kinase inference 32. The method first identifies subsets or ‘modules’ of phosphopeptides that are likely to be co-regulated, since they share the same pattern of phosphorylation change, same mutant dependencies (or lack thereof), and harbor similar sequences around the phosphorylation site (known as “phospho-motifs”). Because many kinases physically interact with their targets, the method next identifies kinases that interact with more of a module’s constituent proteins than expected by chance, based on a background network of published protein-protein and kinase-substrate interactions (FDR < 0.05, see Methods). These “shared interactors” are implicated as potential regulators of the module. Kinases are further elevated as potential regulators if their known specificity 51 matches the phospho-motif identified for that module. In this way, the method identifies modules of potentially coregulated phosphopeptides and suggests kinases and other regulators that may control them. We note that some shared interactor proteins are not regulators but represent other relationships, e.g. members of the same multi-subunit protein families as module constituents.
We partitioned DTT-responsive phosphopeptides into modules of potentially coregulated phosphopeptides, including 3 main modules identified in the early response and 30 identified in the late response, which together captured 44% of the reproducibly changing phosphopeptides (others that lack a clear phospho-motif are not represented in modules). Twenty-seven captured peptides showed a phosphorylation difference in the ire1Δ response, 10 modules (106 peptides) showed dependency on Mkk1/Mkk2, and 10 modules (235 peptides) were scored as independent of both regulators (Supporting Material S1).
We next identified kinases and phosphatases that interacted with more of a module’s constituents than expected by chance (FDR < 0.05, see Methods). Although this approach has some limitations (see Discussion), it identified many kinases as shared interactors of specific modules, implicating them as candidate regulators (Supporting Material S2). Several of these cases involved Ire1 and CWI components (Fig 2). For example, one module represented seven proteins that were phosphorylated at 120 min, shared an Arginine-X-X-Serine (RxxS) motif at their phosphorylation site, and showed reduced induction in the ire1Δ mutant (Fig 2A). Ire1 was identified as a shared interactor of this module (FDR < 0.02), based on its known physical interactions with kinase Kkq8 and ERAD protein Add37 52. Although Ire1 may not phosphorylate the proteins directly, that several other proteins are also linked to ER functions or induced with the UPR suggests a functional connection. Another module of peptides displayed defective phosphorylation in the mkk1Δ/mkk2Δ mutant responding to DTT and was connected to the Mkk1/2 regulated MAPK Slt2 as a shared interactor (FDR < 0.02); this module was enriched for proteins linked to CWI processes, including budding, cell polarity, endocytosis, and the actin cytoskeleton (P < 1e-4, hypergeometric test), consistent with the notion that Mkk1/2-dependent phosphorylation is mediated by Slt2. Thus, the method can successfully implicate likely regulators and phosphoproteome changes they may help to coordinate (see Discussion).
Figure 2. The network implicates known regulators in the 120 min response to DTT.
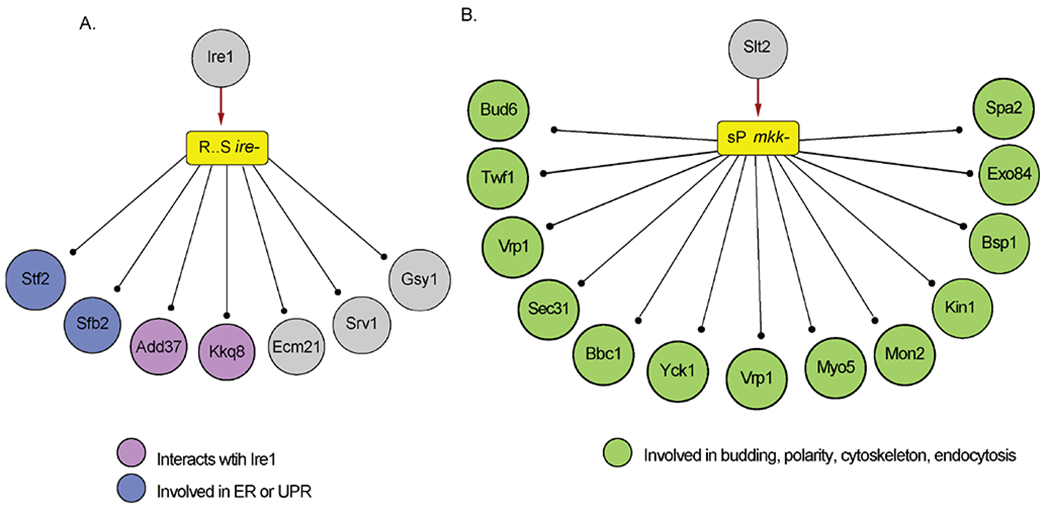
Modules of peptides are represented by a colored rectangle, with yellow indicating increased phosphorylation. Text in the module represents the consensus sequence around the phospho-motif and the mutant dependency. Circles represent proteins, ball and stick lines indicate module constituent proteins, and red arrows indicate connections between a shared interactor and the module. (A) A module of seven proteins with defective phosphorylation at 120 min in the ire1Δ mutant (ire−). (B) A module of 28 proteins with defective phosphorylation at 120 min in the mkk1Δ/mkk2Δ mutant (mkk−), showing the subset of constituents associated with enriched processes.
Cdc28 is implicated in the early response to DTT
In response to stress, cells often transiently arrest cell-cycle progression, allowing them to reallocate resources towards defense. The proline-directed, cyclin-dependent kinase Cdc28 regulates the cell cycle at different phases by phosphorylating distinct targets depending on available cyclins 53. We observed increased phosphorylation of Cdc28 on tyrosine 19 (Y19) during the early DTT response, independent of Ire1 and Mkk1/2. Phosphorylation of this residue by the kinase Swe1 coincides with cell-cycle delay after heat shock 54, osmostress 55, UV damage 56, and growth on non-preferred carbon sources 57, whereas a non-phosphorylatable allele (cdc28-Y19F) leads to defective arrest after heat shock 54. Swe1 is known to mediate G2/M arrest during ER stress 23, suggesting that it may phosphorylate Cdc28 early after DTT treatment. Cdc28 Y19 phosphorylation was observed as early as 5 min after DTT treatment but peaked at 120 min, similar to the timing of reduced culture growth rate (see Fig 1).
In addition to becoming phosphorylated in response to DTT, Cdc28 was also implicated as a shared interactor with proteins in one of the three early-response modules (Fig 3), as well as two late-response modules whose phospho-motifs matched Cdc28 specificity (Supporting Material S2). Although the modules also linked to additional kinases as shared interactors (Supporting Material S2), the early-response module encompassed phosphorylation sites matching Cdc28 specificity 51, included several known Cdc28 targets (such as START regulator Whi5), and was enriched for proteins localizing to the bud neck during division (P < 1x10−4) 45. To test computationally if these sites may be targets of Cdc28, we compared to a previously published dataset of phosphopeptides dependent on Cdc28 activity. The module was enriched for phosphorylated sites that are reduced upon Cdc28 inhibition (P = 0.005, hypergeometric test) 59,54. In contrast, the only other shared interactor whose specificity matched the phospho-motif, Hog1, was not phosphorylated at this early time point as it was at 120 min, and there was no enrichment among module constituents for sites phosphorylated by Hog1 during NaCl stress 32. Together, these results suggest that Cdc28 is active during the early DTT response, perhaps regulated directly in response to ER stress (see Discussion).
Figure 3. Cdc28 is identified as a shared interactor 15 min after DTT treatment.

A module of peptides with induced phosphorylation 15 min after DTT treatment and harboring a serine-Proline (sP) consensus phosphorylation motif, as shown in Figure 2. Green circles highlight known Cdc28 targets 58 or proteins that display defective phosphorylation upon Cdc28 inhibition 59,54. Specific phosphorylated residues that overlapped between our analysis and previous studies of Cdc28 are annotated. Several other proteins (including Cyk3 and Rph1) were also identified as affected by Cdc28 inhibition by 59, but the Cdc28-dependent defect fell below the threshold of target identification.
Rck2 is implicated in the late ER stress response
Hog1 is activated in response to unresolved ER stress 26–28; however, its precise contribution to ER homeostasis has not been elucidated. An important Hog1 target during osmostress is the kinase Rck2, whose phosphorylation by Hog1 reduces translational elongation 60,61.
We discovered that Rck2 is phosphorylated during the late DTT response and implicated as a shared interactor of several phosphopeptide modules. DTT treatment induced Rck2 phosphorylation on several sites (T44, S46, S187), including S46 that is phosphorylated during osmotic stress in a Hog1-dependent manner 32. Rck2 was also identified as a shared interactor for multiple late-responding modules, three of whose constituents (collectively spanning 79 sites) harbor phospho-sites that match Rck2’s basophilic R/KxxS specificity. One module constituent mapped back to Rck2, raising the possibility of auto-phosphorylation during DTT stress. A second module was dependent on Ire1 and represents the same module as in Fig 2A, and the third module was also connected to PKA subunits (see more below).
To test the prediction that many of these phosphopeptides are targets of Rck2, we compared our data to a phosphoproteomic study of an rck2 mutant subjected to NaCl stress 62. The Rck2-connected modules were enriched for phospho-sites (P = 0.0018) and proteins (P = 1e-9) whose phosphorylation is dependent on Rck2 during NaCl stress 62. These mapped to proteins involved in processes that contribute to the adaptive response to ER stress, including lipid metabolism (Swh1, Pik1), ubiquitination (Bul1, Ubx7), and vesicular transport (Imh1, Sfb2)5,4. Swh1 promotes production of the membrane sterol ergosterol 63, which at higher levels is correlated with increased heat and ethanol stress resistance 64. Taken together, we hypothesize that Rck2 contributes to ER stress acclimation, perhaps in a manner regulated by DTT-responsive Hog1.
Increased phosphorylation of PKA targets 120 min after DTT treatment.
PKA signaling promotes growth-related processes including ribosome biogenesis while suppressing the stress response via direct phosphorylation of stress-responsive regulators 16. Thus, it is presumed that PKA signaling must be suppressed to enable the stress response including activation of the ESR 19–21,65,66. Indeed, our prior network analysis of the NaCl-responsive phosphoproteome revealed widespread decrease in phosphorylation of known and predicted PKA sites 32. Furthermore, Pincus et al showed that drug-induced PKA inhibition, concurrent with artificial Hac1 activation, could recapitulate the late-acting DTT-responsive transcriptome, including ESR activation, in the absence of any added stress 18. Thus, we expected signatures of global PKA suppression after DTT treatment, including decreased phosphorylation of known PKA phospho-sites.
Surprisingly, we did not see the same signature of globally reduced PKA signaling seen during acute NaCl treatment. Unlike the NaCl stress response 32, in which PKA subunits were shared interactors with many de-phosphorylated peptides, after DTT treatment PKA subunits Tpk1 and Tpk2 associated with late-responding modules (scored as either dependent or independent of Mkk1/2) and one early-response module, all containing peptides with increased phosphorylation (Fig 4A). Seventy-three of the late-responding phosphopeptides harbored basophilic PKA phospho-motifs [RxS, RxxS], although other kinases also recognize such motifs 67,51,68 including Rck2 that was a shared interactor of one module (Fig 4A). Nonetheless, nearly a third of the proteins in these modules interact with one or more PKA catalytic subunits, above what is expected by chance (P = 8e-6, hypergeometric test)41, and several sites are known PKA substrates, including S22 on pyruvate kinase Pyk1 69 and S83 on the neutral trehalase Nth1 70 (FDR < 0.05, Fig 4A). PKA signaling is regulated by feedback mechanisms, since PKA phosphorylates upstream regulators influencing its activation 71. We observed phosphorylation increases on several upstream PKA regulators, including sites on Ras2 (S207, FDR = 0.09), Ras GEF Cdc25 (S629, FDR = 0.03), and adenylate cyclase-associated Srv2 (S343, FDR = 1e-5). In contrast to modules with increased phosphorylation, PKA subunits were not implicated as shared interactors of de-phosphorylated modules. None of the de-phosphorylated modules with basophilic phospho-motifs was enriched for PKA-interacting proteins (P = 0.99), and only one of their phosphopeptides is implicated as a PKA target 72.
Figure 4. Peptide constituents of PKA-associated modules.
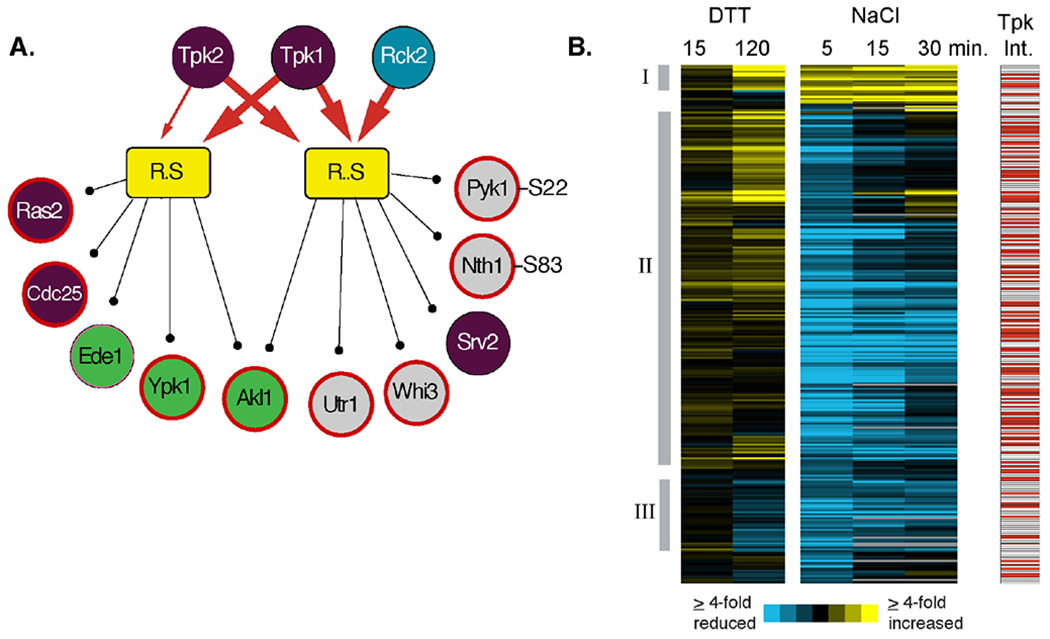
(A) Two modules of late-responding phosphopeptides with increased phosphorylation after DTT treatment and for which PKA subunits were identified as shared interactors are shown as in Figure 2. Note, in each case, we combined one module that was independent and one dependent on Mkk1/2 but for which shared interactors and phospho-motifs were otherwise identical. Heavy red arrows connect shared interactors identified at an FDR of 0.02, and light red arrows reflect shared interactors identified at FDR of 0.05. Only a subset of module constituents are shown, including regulators in the RAS/PKA pathway (purple circles), proteins involved in endocytosis (green circles), and known PKA protein targets 72 and phosphorylation sites (bold red outline) 70. (B) Hierarchical clustering of 268 peptides contained within PKA-connected modules in response to either 2.5 mM DTT or 0.7M NaCl treatment at the denoted times. Each row represents a different peptide, each column represents a different time point, and colorized data represent the log2(fold change) in phosphorylation state according to the key. The response to DTT and 5 min post NaCl treatment represent the average of biological triplicates. Red boxes to the right represent proteins that physically interact with one or more PKA catalytic subunit. Manually identified clusters discussed in the text are indicated.
To further investigate, we directly compared phosphopeptides that were measured in response to both NaCl and DTT and constituents of PKA-associated modules from one or both studies. This included 73 peptides identified in the late DTT response and 303 from the acute NaCl response 32, with 268 phosphopeptides measured in both studies. This group was heavily enriched for proteins that physically interact with one or more Tpk subunit 41 (P = 1.3e-26, hypergeometric test). Clustering of the phosphorylation changes confirmed the differences in responses but also identified some nuances (Fig 4B). One group of 13 peptides increased in phosphorylation after both treatments (Cluster I), and these included several kinases (Akl1, Ptk2, Ark1, Pik1), proteins involved in mRNA related processes (Xrn1, Pab1, Tif4631, Ssb2, Kem1), and several proteins linked to secretion or endocytosis (Sly1, Smy2, Akl1). Another group of 43 peptides (Cluster III) showed reduced phosphorylation after both NaCl and DTT treatment (but did not necessarily meet the threshold for DTT), and this group was enriched for proteins involved in cell polarity, phosphate metabolism, and Rho signaling. However, the vast majority of phosphopeptides exhibited decreased phosphorylation after NaCl treatment, with maximally reduced phosphorylation 5 min after NaCl, but exhibited either increased phosphorylation or little change 15 or 120 min after DTT treatment (Cluster II, Fig 4B). This group was enriched for kinases, proteins involved in cell polarity and cytoskeletal organization, proteins localizing to the bud tip, and stress-response factors (including trehalose metabolism enzymes and the transcription factor Msn2) (P <1e-4, hypergeometric test), functions associated with PKA activity. These results raised the possibility that PKA is not globally suppressed at this timing and dose of DTT treatment.
PKA activity in cellular lysates changes after DTT treatment
To test the activity of PKA over the course of the DTT response, we used an in vitro peptide reporter to measure PKA activity in cellular lysates, before and after cells were treated with 2.5 mM DTT. In vitro phosphorylation of the reporter peptide decreased 15 and 60 min after DTT treatment but partially recovered 120 min after treatment, corresponding to the late-phase proteomic time point (Fig 5A). Activity measured at this time point was consistently more variable even with additional replicates, perhaps due to variable recovery times. We note the challenges with this assay, in that it reports on a single peptide target in a homogenized lysate that loses cellular compartmentalization. Nonetheless, the results indicate that by this measure, PKA activity in the homogenate is decreased after DTT treatment. Comparing the timing of PKA homogenate activity and other responses revealed interesting correlations. The trough of in vitro measured PKA activity coincided with the onset of reduced cellular growth rate, although the reduced growth rate persisted even after PKA recovery at 120 min (Fig 5B). Furthermore, the peak ESR activation as measured in a previous study 17 occurred well after the drop in in vitro tested PKA activity and peaked when PKA activity was scored as resuming at 120 min (Fig 5C). We propose several models to explain these data in the Discussion.
Figure 5. PKA activity relative to other aspects of the cellular response.

(A) Average and standard deviation of in vitro PKA activity from cell lysate at various times after DTT treatment (n = 3). Timepoints statistically different from the unstressed cells are indicated with an asterisk (p<0.001). (B) Overlay of the relative PKA activity measured in (A) (blue graph) versus change in optical density (OD600) shown in Figure 1 (black, purple). (C) Comparison to the average expression change of genes induced in the ESR (iESR) or repressed as part of the ribosomal protein (RP) or ribosome biogenesis (RiBi) gene groups 73, taken from 17. Corresponding timepoints in (B) and (C) are highlighted in grey.
Shared versus stress-specific phosphorylation changes upon DTT and NaCl stresses.
We were curious to quantify other similarities and differences in the DTT versus NaCl phosphoproteome responses. Although the responses operate on different timescales, we directly compared the two datasets. There were more stress-responsive phosphorylation changes observed within 5 min of 0.7M NaCl treatment (1,249 peptides out of 8,122 measured) compared to 2.5 mM DTT exposure (771 peptides at 120 min, out of 7,814 measured), which may reflect the severity of the shock at onset. Of the phosphorylation changes observed 120 min after DTT treatment, 81% of phosphopeptides and 77% of proteins showed increased phosphorylation. In contrast, after acute NaCl stress only 37% of phosphopeptides and 40% of proteins increased in phosphorylation (Fig 6A), with much larger fraction showing phosphorylation decreases.
Figure 6. Common and condition-specific responses to DTT and NaCl.
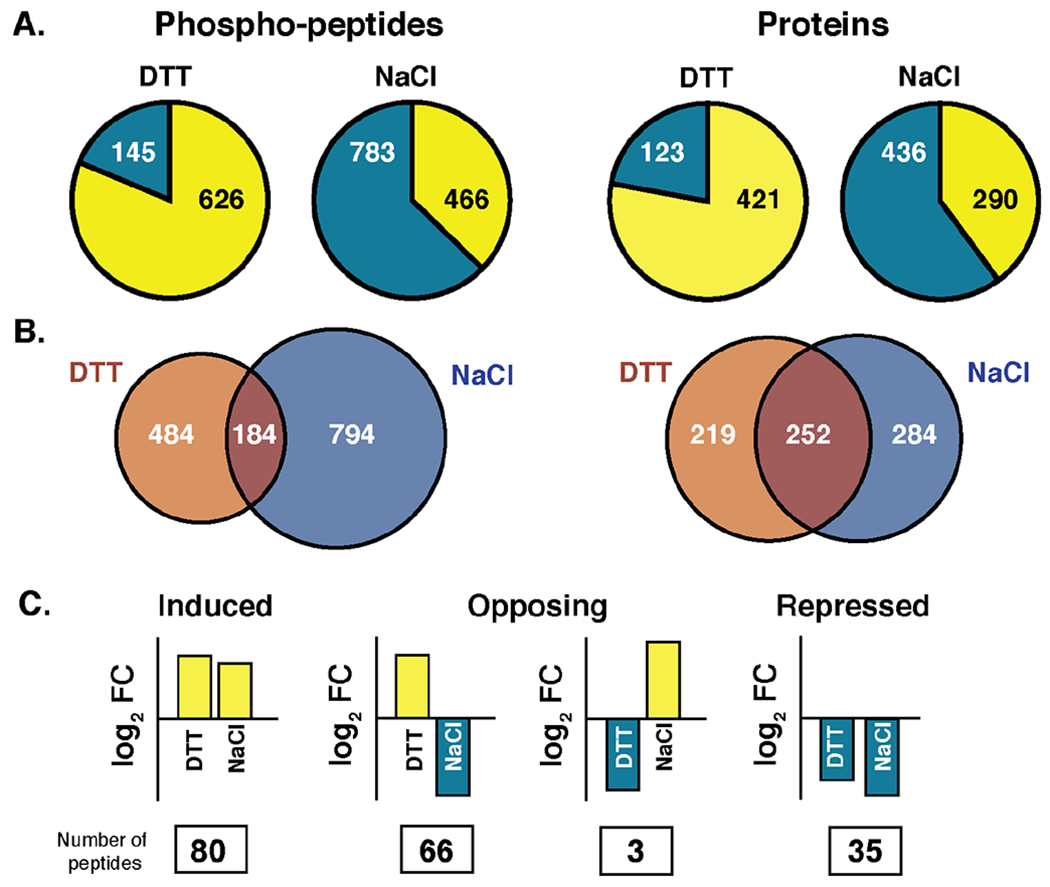
(A) Number of phosphopeptides (left) or proteins (right) with increased (yellow) or decreased (blue) phosphorylation 120 min after DTT or 5 min after NaCl. Note proteins can be counted as having both increased and decreased phosphorylation if multiple residues were affected. (B) Overlap in proteins (left) or phosphopeptides (right) that were scored as having a change in phosphorylation in response to each stress. (C) Of the 184 peptides scored as changing in both studies, the number that was induced or repressed in both studies or showed opposing phosphorylation changes according to the cartoon is listed.
Of the 1,462 phosphopeptides detected in both studies and changing in response to at least one stress, only 184 peptides (13%) met our threshold for change under both conditions. Interestingly, however, there was substantially higher overlap at the protein level, with 33% of changing proteins responsive to both stresses (Fig 6B). Proteins whose phosphorylation status changed in response to both stresses were enriched for kinases and proteins involved in cytoskeletal organization, endocytosis, budding and cell cycle control, P-body assembly, osmotic stress response, and proteins localized to the plasma membrane and cell wall (P < 1x10−4, hypergeometric test), suggesting shared targets and/or acclimation strategies.
We further compared the specific phospho-sites scored as responding to both stresses. The majority, 63%, showed similar directionality of change after NaCl and DTT, and this included the induced phosphorylation of stress-activated kinases (such as Cdc28, Pbs2, Hog1, Rck2, Bck1, Pkh2, and others) and proteins linked to budding and the actin cytoskeleton, and reduced phosphorylation weakly enriched for vacuolar/autophagy proteins and those involved in endocytosis and localized to the plasma membrane. In contrast, 35% of phospho-sites displayed opposite phosphorylation patterns. Many of the latter group included putative PKA targets as discussed above. We also identified a few sites with decreased phosphorylation upon DTT treatment despite phosphorylation after NaCl stress, and these included components of the eisosome complex that plays a role in endocytosis.
DISCUSSION
Here we present the phosphoproteomic response of S. cerevisiae to the ER stressor DTT. In addition to providing a glimpse of the physiological response to this model stressor, our results implicated new pathways and regulators that were not previously associated with acclimation to ER stress. They also afford an opportunity to compare the responses to disparate stresses, allowing us to begin to construct a view of how signaling networks can give rise to common responses and distinct features of environmental acclimation.
While phosphoproteomic profiling can delineate wild-type and mutant phosphorylation changes, a remaining challenge is making sense of how those responses are regulated and how they serve stress acclimation. Functional enrichments among phosphorylated peptides can implicate processes important in ER stress adaptation, and many of the enrichments we observe here correspond to known responses to prolonged ER stress. However, another approach to generate new hypotheses is through network inference to predict upstream regulators of the phosphoproteome response 74,75,32. Our approach first groups together peptides that may be coregulated, because they share the same phosphorylation dynamics, phospho-motif, and mutant dependencies, and then implicates regulators that interact physically with more of those proteins than expected by chance. This approach has been useful in identifying new regulators of the NaCl response 32 and regulators of rewired metabolism 76, and has implicated several kinases in this study for future analysis. Our method has some limitations, in that it will miss regulatory connections for kinases that lack specificity, and it can combine targets of kinase families if they share the same phospho motifs and phosphorylation patterns. For example, Rck2 and PKA subunits were both identified as shared interactors for induced phosphopeptides harboring the RxxS basophilic motif. That group was enriched for peptides phosphorylated in an Rck2-dependent manner during NaCl stress and for proteins interacting with Tpk1 – however, there was insignificant overlap at the protein level, suggesting that Rck2 and PKA target different sets of proteins in this module. The limitation of combining targets of kinases in the same families can be minimized when additional distinguishing features can split up the group, e.g. by assessing a higher number of mutants whose phenotypes can distinguish subsets. Nonetheless, the approach is a useful tool for generating new hypotheses about active regulators for further dissection. We caution that inferred connections between kinases and specific module constituents remain predictions until tested directly, e.g. by showing phosphorylation dependency on the implicated kinase, direct physical interaction between the kinase and protein, or in vitro kinase assays, as we have done for specific cases here.
Interrogating regulator mutants revealed that only a fraction of the DTT-provoked phosphorylation changes were dependent on those regulators, confirming the existence of other regulatory systems. One regulator implicated in the early response is Cdc28. Cdc28 phosphorylation was induced early during DTT treatment but peaked late during the response, when cell growth continued at a reduced rate. The simplest explanation is that Cdc28 is phosphorylated by the Swe1 kinase that controls G2/M delay during ER stress, and that Cdc28 phosphorylates known targets in its associated module and possibly other peptides (Fig 3). Swe1 activation is independent of the UPR and CWI during ER stress (this study and 23), raising question as to the upstream sensor. Interestingly, Cdc28 and the G1 cyclin Cln3 are localized to the ER during phases of the cell cycle 77 and Cdc28-dependent phosphorylation can regulate protein localization at the ER 78, raising the possibility of more direct involvement in ER stress.
Our study also revealed unexpected results at proteins and phospho-sites regulated by PKA. PKA is a known suppressor of the stress response during optimal growth 22, and thus we expected to see reduced phosphorylation of PKA targets as seen during acute NaCl stress; however, this was not the case. Several possibilities could explain this result. One possibility is that PKA is globally repressed after DTT treatment and activation of other basophilic kinases, or inhibition of responsible phosphatases, explains the observed phospho-site response. Indeed, other kinases can recognize these motifs, including Snf1 that is activated by DTT stress 28,79,29,80 and Rck2, which our study suggests; however, it seems unlikely that the vast majority of PKA-implicated sites would be similarly affected.
A second possibility is that PKA activity is locally regulated. There are many examples of localized regulation of kinase activity (e.g. 81–83), and it is perhaps likely that localized control (as opposed to global on/off states) predominates inside cells. In fact, localized regulation of PKA is well known in mammalian systems, where microenvironments of cAMP concentration can regulate PKA molecules within those environments 84. Furthermore, PKA subunits can be directed to specific substrates by PKA anchoring proteins 85, including the yeast PKA regulatory subunit Bcy1 86. This model of localized PKA activity would explain why many (but not all) known and predicted PKA phospho-sites increase after DTT treatment even though PKA activity directed against a single reporter in homogeneous lysates transiently decreases (Fig 5). It may also explain why the timing of ESR and growth rate changes do not parallel in vitro measured activity. Testing this notion will require careful delineation of PKA interactions and localized regulation in cells. Interestingly, cAMP and PKA activity are known to increase after ER stress in mammalian systems to aid in acclimation, regulating mitochondrial-ER contacts, apoptosis, and lipolysis in adipocytes 87–89, raising the possibility that a similar situation exists in yeast. So, while drug-based inhibition of PKA is sufficient to activate the ESR (albeit while completely arresting growth) 20,18, it may not reflect that all PKA-dependent phosphorylation is inhibited after ESR stress.
This study also afforded an opportunity to compare the phosphoproteome response to different stressors. Acute NaCl stress and prolonged DTT treatment are fundamentally different on several levels: 0.7M NaCl exposure has immediate and strong biophysical effects, whereas the stress of DTT may accumulate slowly over time 16,90,91. Nonetheless, there are common features to both responses, including stress on the cell wall/surface, altered cell cycle progression, reduced growth after acclimation, and activation of the ESR. It is interesting that the overlap in proteins that respond to these stresses was substantially higher than the overlap in altered phospho-sites on those proteins, especially on kinases that may be contributing to both responses. One possibility is that multiple sites are phosphorylated in response to both stresses, but that sampling limitations preclude identification in both studies. Further study will continue to illuminate how these kinases and other aspects of the shared responses produce similar versus distinct downstream effects during stress acclimation.
Supplementary Material
Dataset S1: Processed data. log2(fold change) in phosphopeptide abundance or phosphopeptide abundance normalized to changes in protein abundance, where indicated. The file also annotates peptides with significant changes in wild-type and mutant strains, according to that described in the Methods.
Supporting Material S1: Module Constituents. The file lists phosphopeptide constituents of modules identified in the paper.
Supporting Material S2: Shared-interactor kinases. A list of kinases identified as shared interactors for each module, at an FDR of 0.02 or 0.05 (where 1 = significant at that threshold, last columns). The file includes the number of module constituents that interact with a listed SI (k), the number of proteins in that module (n), the total number of proteins that interact with the SI in the background dataset (M), the total number of proteins in the background dataset (N), the p-value from the hypergeometric test, and peptides called significant at an FDR of 0.02 or 0.05.
Supporting Material S3: Functional enrichments.
Supporting Material S4: Cytoscape .sif file of modules, constituent peptides belonging to each module, and kinase/phosphatase shared interactors. Nodes are color coded for induced (yellow) or repressed (blue) modules, kinases (magenta), and phosphatases (light green).
Supporting Material S5: Source data for Figure 1B.
Supporting Material S6: Source data for Figure 4B.
Acknowledgements.
We thank Deborah Chasman for assistance with network inference methods, and members of the Gasch Lab for constructive feedback.
Funding: This work was supported by grants from the NIH (R01GM083989 to APG and R35 GM118110 to JJC). MEM was supported by T32 HG002760; ERW was supported by an NSF Graduate Research Fellowship; MP was supported by the Great Lakes Bioenergy Center funded by the U.S. Department of Energy (DE-SC0018409).
REFERENCES
- 1.Taymaz-Nikerel H, Cankorur-Cetinkaya A & Kirdar B Genome-Wide Transcriptional Response of Saccharomyces cerevisiae to Stress-Induced Perturbations. Front Bioeng Biotechnol 2016. 4, 17, doi: 10.3389/fbioe.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin JH, Walter P & Yen TS Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 2008. 3, 399–425, doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter P & Ron D The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011. 334, 1081–1086, doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 4.Gardner BM, Pincus D, Gotthardt K, Gallagher CM & Walter P Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 2013. 5, a013169, doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS & Walter P Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000. 101, 249–258, doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 6.Harding HP, Zhang Y & Ron D Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999. 397, 271–274, doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 7.Hollien J & Weissman JS Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006. 313, 104–107, doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 8.Hollien J, Lin JH, Li H, Stevens N, Walter P & Weissman JS Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 2009. 186, 323–331, doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox JS, Shamu CE & Walter P Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993. 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- 10.Mori K, Ma W, Gething MJ & Sambrook J A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993. 74, 743–756. [DOI] [PubMed] [Google Scholar]
- 11.Gardner BM & Walter P Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011. 333, 1891–1894, doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM & Walter P The unfolded protein response signals through high-order assembly of Ire1. Nature 2009. 457, 687–693, doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA & Walter P Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 2009. 457, 736–740, doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho N, Yap WS, Xu J, Wu H, Koh JH, Goh WWB, George B, Chong SC, Taubert S & Thibault G Stress sensor Ire1 deploys a divergent transcriptional program in response to lipid bilayer stress. The Journal of cell biology 2020. 219, doi: 10.1083/jcb.201909165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chawla A, Chakrabarti S, Ghosh G & Niwa M Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol 2011. 193, 41–50, doi: 10.1083/jcb.201008071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasch AP in Yeast Stress Responses Vol. 1 (eds Hohmann S & Mager P) 11–70 (Springer-Verlag; 2002). [Google Scholar]
- 17.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D & Brown PO Genomic expression programs in the response of yeast cells to environmental changes. Molecular biology of the cell 2000. 11, 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pincus D, Aranda-Diaz A, Zuleta IA, Walter P & El-Samad H Delayed Ras/PKA signaling augments the unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America 2014. 111, 14800–14805, doi: 10.1073/pnas.1409588111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boy-Marcotte E, Perrot M, Bussereau F, Boucherie H & Jacquet M Msn2p and Msn4p control a large number of genes induced at the diauxic transition which are repressed by cyclic AMP in Saccharomyces cerevisiae. J Bacteriol 1998. 180, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H & Schuller C Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes & development 1998. 12, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith A, Ward MP & Garrett S Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. The EMBO journal 1998. 17, 3556–3564, doi: 10.1093/emboj/17.13.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho YH & Gasch AP Exploiting the yeast stress-activated signaling network to inform on stress biology and disease signaling. Current genetics 2015. 61, 503–511, doi: 10.1007/s00294-015-0491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonilla M & Cunningham KW Mitogen-activated protein kinase stimulation of Ca(2+) signaling is required for survival of endoplasmic reticulum stress in yeast. Molecular biology of the cell 2003. 14, 4296–4305, doi: 10.1091/mbc.e03-02-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen TJ, Mallory MJ, Strich R & Yao TP Hos2p/Set3p deacetylase complex signals secretory stress through the Mpk1p cell integrity pathway. Eukaryot Cell 2008. 7, 1191–1199, doi: 10.1128/EC.00059-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scrimale T, Didone L, de Mesy Bentley KL & Krysan DJ The unfolded protein response is induced by the cell wall integrity mitogen-activated protein kinase signaling cascade and is required for cell wall integrity in Saccharomyces cerevisiae. Mol Biol Cell 2009. 20, 164–175, doi: 10.1091/mbc.E08-08-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bicknell AA, Tourtellotte J & Niwa M Late phase of the endoplasmic reticulum stress response pathway is regulated by Hog1 MAP kinase. The Journal of biological chemistry 2010. 285, 17545–17555, doi: 10.1074/jbc.M109.084681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres-Quiroz F, Garcia-Marques S, Coria R, Randez-Gil F & Prieto JA The activity of yeast Hog1 MAPK is required during endoplasmic reticulum stress induced by tunicamycin exposure. The Journal of biological chemistry 2010. 285, 20088–20096, doi: 10.1074/jbc.M109.063578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piao H, MacLean Freed J & Mayinger P Metabolic activation of the HOG MAP kinase pathway by Snf1/AMPK regulates lipid signaling at the Golgi. Traffic 2012. 13, 1522–1531, doi: 10.1111/j.1600-0854.2012.01406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuno T, Masuda Y & Irie K The Saccharomyces cerevisiae AMPK, Snf1, Negatively Regulates the Hog1 MAPK Pathway in ER Stress Response. PLoS genetics 2015. 11, e1005491, doi: 10.1371/journal.pgen.1005491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M & Davis RW Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science (New York, N.Y 1999. 285, 901–906. [DOI] [PubMed] [Google Scholar]
- 31.Julius D, Blair L, Brake A, Sprague G & Thorner J Yeast alpha factor is processed from a larger precursor polypeptide: the essential role of a membrane-bound dipeptidyl aminopeptidase. Cell 1983. 32, 839–852. [DOI] [PubMed] [Google Scholar]
- 32.MacGilvray ME, Shishkova E, Chasman D, Place M, Gitter A, Coon JJ & Gasch AP Network inference reveals novel connections in pathways regulating growth and defense in the yeast salt response. PLoS computational biology 2018. 13, e1006088, doi: 10.1371/journal.pcbi.1006088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose CM, Venkateshwaran M, Volkening JD, Grimsrud PA, Maeda J, Bailey DJ, Park K, Howes-Podoll M, den Os D, Yeun LH, Westphall MS, Sussman MR, Ane JM & Coon JJ Rapid phosphoproteomic and transcriptomic changes in the rhizobia-legume symbiosis. Mol Cell Proteomics 2012. 11, 724–744, doi: 10.1074/mcp.M112.019208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS & Coon JJ The one hour yeast proteome. Mol Cell Proteomics 2014. 13, 339–347, doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Karra K, Krieger CJ, Miyasato SR, Nash RS, Park J, Skrzypek MS, Simison M, Weng S & Wong ED Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic acids research 2012. 40, D700–705, doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ & Coon JJ COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 2011. 11, 1064–1074, doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee MV, Topper SE, Hubler SL, Hose J, Wenger CD, Coon JJ & Gasch AP A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol Syst Biol 2011. 7, 514, doi: 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010. 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisen MB, Spellman PT, Brown PO & Botstein D Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America 1998. 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz D & Gygi SP An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol 2005. 23, 1391–1398, doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 41.Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R, Zhang F, Dolma S, Willems A, Coulombe-Huntington J, Chatr-Aryamontri A, Dolinski K & Tyers M The BioGRID interaction database: 2019 update. Nucleic acids research 2019. 47, D529–D541, doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharifpoor S, Ba ANN, Young J-Y, van Dyk D, Friesen H, Douglas AC, Kurat CF, Chong YT, Founk K, Moses AM & Andrews BJ A quantitative literature-curated gold standard for kinase-substrate pairs. Genome biology 2011. 12, R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chasman D, Ho YH, Berry DB, Nemec CM, MacGilvray ME, Hose J, Merrill AE, Lee MV, Will JL, Coon JJ, Ansari AZ, Craven M & Gasch AP Pathway connectivity and signaling coordination in the yeast stress-activated signaling network. Molecular systems biology 2014. 10, 759, doi: 10.15252/msb.20145120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benjamini Y & Hochberg Y Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 1995. 57, 289–300. [Google Scholar]
- 45.Robinson MD, Grigull J, Mohammad N & Hughes TR FunSpec: a web-based cluster interpreter for yeast. BMC bioinformatics 2002. 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shamu CE & Walter P Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 1996. 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- 47.Iwawaki T, Hosoda A, Okuda T, Kamigori Y, Nomura-Furuwatari C, Kimata Y, Tsuru A & Kohno K Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nature cell biology 2001. 3, 158–164, doi: 10.1038/35055065. [DOI] [PubMed] [Google Scholar]
- 48.Kimata Y, Ishiwata-Kimata Y, Yamada S & Kohno K Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes to cells : devoted to molecular & cellular mechanisms 2006. 11, 59–69, doi: 10.1111/j.1365-2443.2005.00921.x. [DOI] [PubMed] [Google Scholar]
- 49.Lopez-Martinez G, Rodriguez-Porrata B, Margalef-Catala M & Cordero-Otero R The STF2p hydrophilin from Saccharomyces cerevisiae is required for dehydration stress tolerance. PLoS One 2012. 7, e33324, doi: 10.1371/journal.pone.0033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jimenez-Gutierrez E, Alegria-Carrasco E, Sellers-Moya A, Molina M & Martin H Not just the wall: the other ways to turn the yeast CWI pathway on. Int Microbiol 2019, doi: 10.1007/s10123-019-00092-2. [DOI] [PubMed] [Google Scholar]
- 51.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma JL, Sheu YJ, Sassi HE, Sopko R, Chan CS, De Virgilio C, Hollingsworth NM, Lim WA, Stern DF, Stillman B, Andrews BJ, Gerstein MB, Snyder M & Turk BE Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Science signaling 2010. 3, ra12, doi: 10.1126/scisignal.2000482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palmer EA, Kruse KB, Fewell SW, Buchanan SM, Brodsky JL & McCracken AA Differential requirements of novel A1PiZ degradation deficient (ADD) genes in ER-associated protein degradation. J Cell Sci 2003. 116, 2361–2373, doi: 10.1242/jcs.00439. [DOI] [PubMed] [Google Scholar]
- 53.Enserink JM & Kolodner RD An overview of Cdk1-controlled targets and processes. Cell Div 2010. 5, 11, doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanshin E, Kubiniok P, Thattikota Y, D’Amours D & Thibault P Phosphoproteome dynamics of Saccharomyces cerevisiae under heat shock and cold stress. Mol Syst Biol 2015. 11, 813, doi: 10.15252/msb.20156170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clotet J, Escote X, Adrover MA, Yaakov G, Gari E, Aldea M, de Nadal E & Posas F Phosphorylation of Hsl1 by Hog1 leads to a G2 arrest essential for cell survival at high osmolarity. EMBO J 2006. 25, 2338–2346, doi: 10.1038/sj.emboj.7601095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Germain D, Hendley J & Futcher B DNA damage inhibits proteolysis of the B-type cyclin Clb5 in S. cerevisiae. J Cell Sci 1997. 110 ( Pt 15), 1813–1820. [DOI] [PubMed] [Google Scholar]
- 57.Keaton MA, Bardes ES, Marquitz AR, Freel CD, Zyla TR, Rudolph J & Lew DJ Differential susceptibility of yeast S and M phase CDK complexes to inhibitory tyrosine phosphorylation. Curr Biol 2007. 17, 1181–1189, doi: 10.1016/j.cub.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A & Tyers M BioGRID: a general repository for interaction datasets. Nucleic acids research 2006. 34, D535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP & Morgan DO Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science (New York, N.Y 2009. 325, 1682–1686, doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bilsland-Marchesan E, Arino J, Saito H, Sunnerhagen P & Posas F Rck2 kinase is a substrate for the osmotic stress-activated mitogen-activated protein kinase Hog1. Molecular and cellular biology 2000. 20, 3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teige M, Scheikl E, Reiser V, Ruis H & Ammerer G Rck2, a member of the calmodulin-protein kinase family, links protein synthesis to high osmolarity MAP kinase signaling in budding yeast. Proceedings of the National Academy of Sciences of the United States of America 2001. 98, 5625–5630, doi: 10.1073/pnas.091610798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Romanov N, Hollenstein DM, Janschitz M, Ammerer G, Anrather D & Reiter W Identifying protein kinase-specific effectors of the osmostress response in yeast. Sci Signal 2017. 10, doi: 10.1126/scisignal.aag2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang B, Brown JL, Sheraton J, Fortin N & Bussey H A new family of yeast genes implicated in ergosterol synthesis is related to the human oxysterol binding protein. Yeast 1994. 10, 341–353, doi: 10.1002/yea.320100307. [DOI] [PubMed] [Google Scholar]
- 64.Swan TM & Watson K Stress tolerance in a yeast sterol auxotroph: role of ergosterol, heat shock proteins and trehalose. FEMS Microbiol Lett 1998. 169, 191–197. [DOI] [PubMed] [Google Scholar]
- 65.Gasch AP Yeast genomic expression studies using DNA microarrays. Methods in enzymology 2002. 350, 393–414. [DOI] [PubMed] [Google Scholar]
- 66.Gorner W, Durchschlag E, Wolf J, Brown EL, Ammerer G, Ruis H & Schuller C Acute glucose starvation activates the nuclear localization signal of a stress-specific yeast transcription factor. The EMBO journal 2002. 21, 135–144, doi: 10.1093/emboj/21.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, Guo H, Jona G, Breitkreutz A, Sopko R, McCartney RR, Schmidt MC, Rachidi N, Lee SJ, Mah AS, Meng L, Stark MJ, Stern DF, De Virgilio C, Tyers M, Andrews B, Gerstein M, Schweitzer B, Predki PF & Snyder M Global analysis of protein phosphorylation in yeast. Nature 2005. 438, 679–684, doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 68.Smith FD, Samelson BK & Scott JD Discovery of cellular substrates for protein kinase A using a peptide array screening protocol. Biochem J 2011. 438, 103–110, doi: 10.1042/BJ20110720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Portela P, Moreno S & Rossi S Characterization of yeast pyruvate kinase 1 as a protein kinase A substrate, and specificity of the phosphorylation site sequence in the whole protein. Biochem J 2006. 396, 117–126, doi: 10.1042/BJ20051642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schepers W, Van Zeebroeck G, Pinkse M, Verhaert P & Thevelein JM In vivo phosphorylation of Ser21 and Ser83 during nutrient-induced activation of the yeast protein kinase A (PKA) target trehalase. J Biol Chem 2012. 287, 44130–44142, doi: 10.1074/jbc.M112.421503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vandamme J, Castermans D & Thevelein JM Molecular mechanisms of feedback inhibition of protein kinase A on intracellular cAMP accumulation. Cell Signal 2012. 24, 1610–1618, doi: 10.1016/j.cellsig.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 72.Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O’Donnell L, Reguly T, Nixon J, Ramage L, Winter A, Sellam A, Chang C, Hirschman J, Theesfeld C, Rust J, Livstone MS, Dolinski K & Tyers M The BioGRID interaction database: 2015 update. Nucleic Acids Res 2015. 43, D470–478, doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ho YH, Shishkova E, Hose J, Coon JJ & Gasch AP Decoupling Yeast Cell Division and Stress Defense Implicates mRNA Repression in Translational Reallocation during Stress. Current biology : CB 2018. 28, 2673–2680 e2674, doi: 10.1016/j.cub.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Senbabaoglu Y, Sumer SO, Sanchez-Vega F, Bemis D, Ciriello G, Schultz N & Sander C A Multi-Method Approach for Proteomic Network Inference in 11 Human Cancers. PLoS computational biology 2016. 12, e1004765, doi: 10.1371/journal.pcbi.1004765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Invergo BM & Beltrao P Reconstructing phosphorylation signalling networks from quantitative phosphoproteomic data. Essays Biochem 2018. 62, 525–534, doi: 10.1042/EBC20180019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Myers KS, Riley NM, MacGilvray ME, Sato TK, McGee M, Heilberger J, Coon JJ & Gasch AP Rewired cellular signaling coordinates sugar and hypoxic responses for anaerobic xylose fermentation in yeast. PLoS genetics 2019. 15, e1008037, doi: 10.1371/journal.pgen.1008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Verges E, Colomina N, Gari E, Gallego C & Aldea M Cyclin Cln3 is retained at the ER and released by the J chaperone Ydj 1 in late G1 to trigger cell cycle entry. Molecular cell 2007. 26, 649–662, doi: 10.1016/j.molcel.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 78.Yahya G, Parisi E, Flores A, Gallego C & Aldea M A Whi7-anchored loop controls the G1 Cdk-cyclin complex at start. Molecular cell 2014. 53, 115–126, doi: 10.1016/j.molcel.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 79.Ferrer-Dalmau J, Randez-Gil F, Marquina M, Prieto JA & Casamayor A Protein kinase Snf1 is involved in the proper regulation of the unfolded protein response in Saccharomyces cerevisiae. The Biochemical journal 2015. 468, 33–47, doi: 10.1042/BJ20140734. [DOI] [PubMed] [Google Scholar]
- 80.Kimura Y, Irie K & Mizuno T Expression control of the AMPK regulatory subunit and its functional significance in yeast ER stress response. Scientific reports 2017. 7, 46713, doi: 10.1038/srep46713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Denis V & Cyert MS Molecular analysis reveals localization of Saccharomyces cerevisiae protein kinase C to sites of polarized growth and Pkc1p targeting to the nucleus and mitotic spindle. Eukaryotic cell 2005. 4, 36–45, doi: 10.1128/EC.4.1.36-45.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rubenstein EM & Schmidt MC Mechanisms regulating the protein kinases of Saccharomyces cerevisiae. Eukaryotic cell 2007. 6, 571–583, doi: 10.1128/EC.00026-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takahashi S & Pryciak PM Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Current biology : CB 2008. 18, 1184–1191, doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnstone TB, Agarwal SR, Harvey RD & Ostrom RS cAMP Signaling Compartmentation: Adenylyl Cyclases as Anchors of Dynamic Signaling Complexes. Mol Pharmacol 2018. 93, 270–276, doi: 10.1124/mol.117.110825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Torres-Quesada O, Mayrhofer JE & Stefan E The many faces of compartmentalized PKA signalosomes. Cell Signal 2017. 37, 1–11, doi: 10.1016/j.cellsig.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 86.Galello F, Moreno S & Rossi S Interacting proteins of protein kinase A regulatory subunit in Saccharomyces cerevisiae. J Proteomics 2014. 109, 261–275, doi: 10.1016/j.jprot.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 87.Deng J, Liu S, Zou L, Xu C, Geng B & Xu G Lipolysis response to endoplasmic reticulum stress in adipose cells. The Journal of biological chemistry 2012. 287, 6240–6249, doi: 10.1074/jbc.M111.299115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aguileta MA, Rojas-Rivera D, Goossens V, Estornes Y, Van Isterdael G, Vandenabeele P & Bertrand MJ A siRNA screen reveals the prosurvival effect of protein kinase A activation in conditions of unresolved endoplasmic reticulum stress. Cell Death Differ 2016. 23, 1670–1680, doi: 10.1038/cdd.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Song YF, Hogstrand C, Wei CC, Wu K, Pan YX & Luo Z Endoplasmic reticulum (ER) stress and cAMP/PKA pathway mediated Zn-induced hepatic lipolysis. Environ Pollut 2017. 228, 256–264, doi: 10.1016/j.envpol.2017.05.046. [DOI] [PubMed] [Google Scholar]
- 90.Proft M & Struhl K MAP kinase-mediated stress relief that precedes and regulates the timing of transcriptional induction. Cell 2004. 118, 351–361, doi: 10.1016/j.cell.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 91.Ke R, Ingram PJ & Haynes K An integrative model of ion regulation in yeast. PLoS computational biology 2013. 9, e1002879, doi: 10.1371/journal.pcbi.1002879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1: Processed data. log2(fold change) in phosphopeptide abundance or phosphopeptide abundance normalized to changes in protein abundance, where indicated. The file also annotates peptides with significant changes in wild-type and mutant strains, according to that described in the Methods.
Supporting Material S1: Module Constituents. The file lists phosphopeptide constituents of modules identified in the paper.
Supporting Material S2: Shared-interactor kinases. A list of kinases identified as shared interactors for each module, at an FDR of 0.02 or 0.05 (where 1 = significant at that threshold, last columns). The file includes the number of module constituents that interact with a listed SI (k), the number of proteins in that module (n), the total number of proteins that interact with the SI in the background dataset (M), the total number of proteins in the background dataset (N), the p-value from the hypergeometric test, and peptides called significant at an FDR of 0.02 or 0.05.
Supporting Material S3: Functional enrichments.
Supporting Material S4: Cytoscape .sif file of modules, constituent peptides belonging to each module, and kinase/phosphatase shared interactors. Nodes are color coded for induced (yellow) or repressed (blue) modules, kinases (magenta), and phosphatases (light green).
Supporting Material S5: Source data for Figure 1B.
Supporting Material S6: Source data for Figure 4B.