

Abstract
Dairy cattle of different ages experience different living conditions and varied frequency of antibiotic administration that likely influence the distribution of microbiome and resistome in ways that reflect different risks of microbial transmission. To assess the degree of variance in these distributions, fecal and soil samples were collected from six distinct housing areas on commercial dairy farms (n = 7) in Washington State. 16S rRNA gene sequencing indicated that the microbiota differed between different on-farm locations in feces and soil, and in both cases, the microbiota of dairy calves was often distinct from others (P < 0.05). Thirty-two specific antibiotic resistance genes (ARGs) were widely distributed on dairies, of which several clinically relevant ARGs (including cfr, cfrB, and optrA) were identified for the first time at U.S. dairies. Overall, ARGs were observed more frequently in feces and soil from dairy calves and heifers than from hospital, fresh, lactation and dry pens. Droplet-digital PCR demonstrated that the absolute abundance of floR varied greatly across housing areas and this gene was enriched the most in calves and heifers. Furthermore, in an extended analysis with 14 dairies, environmental soils in calf pens had the most antibiotic-resistant Escherichia coli followed by heifer and hospital pens. All soil E. coli isolates (n = 1,905) are resistant to at least 4 different antibiotics, and the PFGE analysis indicated that florfenicol-resistant E. coli is probably shared across geographically-separated farms. This study identified a discrete but predictable distribution of antibiotic resistance genes and organisms, which is important for designing mitigation for higher risk areas on dairy farms.
Keywords: Antibiotic resistance, housing areas, dairy farm, soil, calf, discrete distribution
1. Introduction
Antimicrobial resistance poses a significant threat to public health (Blair et al., 2015; Laxminarayan et al., 2013), and enteric bacteria from livestock are an important source of antibiotic-resistance genes (ARGs) (Aarestrup, 2015; Liu et al., 2016b; Tian et al., 2017; Van Boeckel et al., 2015). While much is known about the gut microbiota and antibiotic resistome from animals, knowledge about the proximate environment of these animals is limited (Cheng et al., 2013; Hu et al., 2017). On-farm soil, which is frequently mixed with bedding and receives repeated influx of feces and urine from resident animals, may be critical to long-term persistence of antimicrobial-resistant bacteria on farms (Burgos et al., 2005; Liu et al., 2016a). When animals are treated with antibiotics, fecal shedding of enriched populations of ARGs together with excreted antibiotics can drive “blooms” of antibiotic-resistant bacteria by several orders-of-magnitude in farm soil (Call et al., 2013; Liu et al., 2016a). Elevated populations of soil-borne resistant bacteria increases the probability of transmission to and colonization of naïve animals (Davis et al., 2005; Liu et al., 2016a; Rasmussen and Casey, 2001). If reservoirs are formed in soil after exposure to antibiotic-treated animals, then at the farm scale, differential use of antibiotics will likely contribute to a heterogeneous distribution of environmental reservoirs. Identifying predictable “hotspots” on farms could help to identify targeted strategies for limiting the persistence of antibiotic-resistant bacteria in food-animal environments.
Food-producing animals are housed differently according to their age and production phase (Mandel et al., 2016). Although every farm is unique with respect to layout and operation, most U.S. dairies include areas for pre-weaned calves (< 7–8 weeks; calf pens), post-weaned heifers (< 13–15 months; heifer pens), postpartum cows (fresh pens), lactating cows (lactation pen), non-lactating cows (dry pens) and cows that are isolated for various therapeutic treatments (hospital pens). Dairy cattle experience age-dependent development of gut microbiota from birth to adulthood (Dill-McFarland et al., 2017), and the composition of the microbial community is closely related to the antibiotic resistome (Forsberg et al., 2014). In particular, dairy calves typically have a higher prevalence of antibiotic-resistant bacteria (79.2% resistance to tetracycline in pre-weaned calves) than older animals (14.2% resistance to tetracycline in lactating cow) even in the absence of antibiotic effects (Khachatryan et al., 2006; Khachatryan et al., 2004). Consequently, farm soil will receive fecal loading with differed microbiota and varied levels of antibiotic-resistant bacteria depending on the life-stage of resident animals.
Moreover, the inherent differences between life stages are further differentiated by exposure to different therapeutic regimens. For example, the veterinary analog to chloramphenicol (florfenicol) is primarily used in young dairy calves to treat respiratory disease (Davis et al., 2015), and excreted florfenicol residues remain functionally available in soil (Subbiah et al., 2011). A third-generation cephalosporin (ceftiofur) is used systemically to treat respiratory disease, metritis and foot rot, but it is most commonly infused into the udder to treat mastitis (Davis et al., 2015). Besides exposure to different groups of animals, each distinct farm area is often exposed to different degrees of sunlight, temperature changes, and moisture, while potentially having different beddings materials (Miller, 1979). Collectively, these factors will likely contribute to distinct microbial communities and resistomes in soil across housing areas.
We hypothesized that the microbiota and prevalence of ARGs in both animals (i.e. feces) and soil differ in a predictable manner across different housing areas on dairy farms. Fresh fecal and soil samples were collected from commercial dairy farms in Washington State, and 16S rRNA gene sequencing was applied to examine the microbiota. In addition, we focused on assessing the prevalence of 32 ARGs that confer resistance to actively used antibiotics (e.g., ceftiofur and florfenicol) according to a recent survey of 30 commercial farms (Davis et al., 2015). Ultimately, in an effort to quantify the risk of antibiotic resistance, we mapped the distribution and density of antibiotic-resistant E. coli in environmental soil across different housing areas. Our results indicate that there is a discrete distribution of antibiotic resistant bacteria across housing areas where ARGs and antibiotic-resistant bacteria are more frequently observed in feces and soil from dairy calves and heifers. These findings present a unique opportunity to design targeted and cost-effective strategies to limit the extent and persistence of antibiotic resistance in dairy production.
2. Materials and methods
2.1. Sampling farms, sample and isolate collection
Sampling occurred between July and November 2015. Fecal and soil samples were collected at seven non-adjacent commercial dairy farms (40 to 150 km apart) in central Washington State based on willingness to allow sampling. For each dairy farm, we sampled six distinct housing areas (pre-weaned calf pens, post-weaned heifer pens, lactating pens, fresh pens, dry lot and hospital pens). Pre-weaned calves (< 6–8 weeks) were housed in individual hutches (pre-weaned calf pens). Un-bred, post-weaned heifers (< 13–15 months) were group housed (post-weaned heifer pens). Post-partum cows were usually housed in a fresh pen (typically for 2 to 3 weeks after calving), and lactating cow were housed in lactating pens. Cows between the end of lactation and subsequent calving were considered “dry cows” and these are housed in a “dry lot”. Sick cows were often moved to “hospital pens” for treatment. Specifically, one fecal sample and two soil samples were collected from each housing area resulting in 42 fecal samples and 84 soil samples. Fresh feces (~2 g) were obtained directly from the rectum of dairy calves and heifers. Adult fecal samples were collected from the ground immediately after they were passed by the animals. Animals were chosen for sampling based on a randomization of ear tag numbers per group. Topsoil samples (~5 g) were collected at the gate entrance as well as the center of housing pens from the first 1–2 cm depth within a 4-cm2 area following published protocol (Liu et al., 2016a). All samples were immediately stored on ice and transported to the lab for DNA extractions.
Between April and May 2016, we extended sampling at 14 dairies, which included previously sampled farms (n = 7), for isolation of antibiotic-resistant E. coli. Specifically, ten soil samples (~5 g) were collected from each housing area for a total of 840 samples (14 farms × 60 samples farm−1 = 840 samples). For rectangular-shaped pens holding multiple animals, soil samples were systematically collected approximately equidistant along two diagonal transects. For irregular-shaped pens, samples were collected to maximize distance between sample points. Single, centrally located samples were collected from separate pre-weaned calf hutches and all sampled dairies managed their calves using individual housing. Soil samples were immediately stored on ice and transported to lab to quantify the abundance of presumptive E. coli. Briefly, we performed 1:10 serial dilutions for both soil samples and typically plated the 10−3 dilution to obtain countable colonies. Soil slurries were kept at 4°C for at least one additional day if different dilutions were needed for accurate counts. MacConkey agar plates were prepared with ceftiofur (8 μg ml−1; Sigma-Aldrich, St. Louis, MO, USA) or florfenicol (50 μg ml−1; Sigma-Aldrich) following published concentrations (Liu et al., 2016a) to enumerate presumptive antibiotic-resistant E. coli. We archived 1–2 presumptive E. coli isolates from each plate resulting in 649 ceftiofur-resistant E. coli and 1,256 florfenicol-resistant E. coli. All isolates were stored in 96-well plates as glycerol stocks at −80°C for further antibiotic susceptibility testing.
2.2. DNA extraction, 16S rRNA gene sequencing and data analysis
Fecal DNA was extracted using a QIAmp® DNA Stool Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. DNA was extracted from soil samples using a Power Soil Kit (MoBio, Carlsbad, CA, USA) following published modifications (Cadillo-Quiroz et al., 2006). Quality and concentrations of DNA extractions were estimated by spectrophotometer analysis (NanoDrop ND-2000, Thermo Fisher Scientific, Waltham, MA, USA). 16S rDNA libraries were prepared by amplifying the V4 region of 16S rRNA gene using the primers 520F(5-AYTGGGYDTAAAGNG-3) and 802R(5-TACNVGGGTATCTAATCC-3). PCR products were checked by agarose-gel electrophoresis (2%) and purified using a Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries were generated using a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following manufacturer’s recommendations and index codes were added. The quality of the library preparations was assessed using Qubit 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. Libraries were sequenced using an Illumina MiSeq (250-bp PE) platform. Amplicon sequencing data generated in this study have been deposited with the NCBI SRA (PRJNA480049) and are publicly available.
Sequencing reads were de-multiplexed by using Sabre software (https://github.com/najoshi/sabre) and were then loaded into QIIME 2 (qiime tools import --type ‘SampleData[PairedEndSequencesWithQuality]’ --source-format PairedEndFastqManifestPhred33) (Caporaso et al., 2010). The sequence quality control and feature table construction was performed using DADA 2 (qiime dada2 denoise-paired --p-trim-left-f 21 --p-trim-left-r 23 --p-trunc-len-f 242 --p-trunc-len-r 250) (Callahan et al., 2016). Alpha diversity as measured by Shannon index, which considers both the richness and evenness of a sample, was calculated based on a rarefied feature table (qiime diversity core-metrics-phylogenetic; > 2000 reads). Beta diversity was examined based on Bray-Curtis distance matrices using non-metric multidimensional scaling (NMDS) in R (Team, 2018). A 2D plot was used if stress was < 0.2, where stress reflects how well the ordination summarizes the observed distances among the samples. The number of dimensions (of the plot) was increased until a plot with stress < 0.2 was produced. Ellipses shapes/paths were calculated with the veganCovEllipse function from the vegan package in R. The same method was used to construct ellipses for the NMDS ordination analysis of PCR data. Taxonomy was assigned using QIIME2 (qiime feature-classifier classify-sklearn) against the Greengenes Database. LEfSe (Segata et al., 2011) was used to identify bacterial families which are differentially abundant between fecal and soil samples.
2.3. The prevalence and abundance of targeted ARG
DNA used for microbiota analysis were further subjected to ARG profiling. Specifically, the presence of a panel of 32 ARGs that confer resistance to nine classes of antibiotics (aminoglycoside, beta-lactams, fluoroquinolones, fosfomycin, macrolide-lincosamide-streptogramin (MLS), oxazolidinones, amphenicols, tetracyclines, and glycopeptides) was characterized for each tested DNA sample. All conventional PCR assays were performed using the Thermo Scientific DreamTaq™ Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) per manufacturer’s instructions, with a final concentration of 2 μM of each primer for assessed ARGs. Previously validated primer sequences were used in this study (Table S1), and we followed published studies for optimal PCR conditions. PCR products were analyzed by agarose-gel electrophoresis (1%) in 1X TAE buffer, and PCR-positive amplicons were purified and sequenced to further rule out potential mismatches. Negative controls were included in all PCR assays to detect potential contamination. PCR results were recorded as presence or absence of ARGs, and the percent positive samples (detection rate) was calculated as the ratio of PCR-positive samples divided by total samples. Ordination analysis (NMDS) was performed with Jaccard distance matrices to explore the ARG profiles at the community level.
In addition to the prevalence analysis using conventional PCR, Droplet Digital PCR (ddPCR) was applied to assess the absolute abundance of ARG. Specifically, the quantity of floR was assessed using ddPCR with a QX100 Droplet Digital PCR Platform (Bio-Rad, Hercules, CA, USA) as previously described (Liu et al., submitted). Briefly, reactions occurred in a total of 20 μL volume including 900 nM each primer (Forward: 5’-GGAGCAGCTTGGTCTTCAAC and Reverse: 5’-CGTAGATGACGACACCCTCA), 250 nM floR Probe (5’-FAM-XXX-BHQ1–3’),1X PCR master mix and 2 μL DNA samples. Raw data were initially analyzed QuantaSoft software (Bio-Rad) and then imported into R (version 3.4.4) for downstream analysis. E. coli strain H4H (Call et al., 2010) and sterile water were used as positive and negative controls, respectively.
2.4. Antibiotic susceptibility testing
Frozen archives of E. coli isolates were resuscitated in LB broth at 37°C (without antibiotic; Luria-Bertani medium; Fisher Biotech, Fair Lawn, NJ) by overnight incubation one day prior to antibiotic-resistance testing. A 96-pin replicator (flamed twice between plates; Boekel Scientific, Feasterville-Trevose, PA, USA) was used to transfer isolates from 96-well plates onto MacConkey agar plates (150 × 15 mm) that contained one antibiotic (Caudell et al., 2018). A panel of ten antibiotics including ampicillin, amoxicillin/clavulanic acid, ceftiofur, chloramphenicol (a substitute for florfenicol), tetracycline, ciprofloxacin, streptomycin, kanamycin, sulfamethoxazole and trimethoprim were used in this assay (Table S2). Positive (E. coli strain H4H) and negative (E. coli strain K-12) controls, with or without specific antibiotic resistance phenotypes, were added manually to each plate. Isolates that grew on antibiotic containing media (“1”) were considered resistant to that drug, while failure to grow (“0”) denoted susceptibility. A non-antibiotic containing plate was used to confirm growth from every well.
2.5. Filter-mating conjugation assay
To evaluate the possibility of plasmid-mediated transmission of ARGs, a filter-conjugation assay (Subbiah et al., 2011) was used to determine if the dominant multidrug resistant phenotypes were associated with conjugative plasmids. Donor strains (n = 20) were randomly selected from the archived collection of ceftiofur-resistant (demonstrating resistance to ampicillin, ceftiofur, chloramphenicol, kanamycin, streptomycin, sulfamethoxazole, tetracycline and trimethoprim; 10 isolates) and florfenicol-resistant (demonstrating resistance to ampicillin, chloramphenicol, streptomycin, sulfamethoxazole and tetracycline; 10 isolates) E. coli. We used E. coli GeneHogs® (rifampicin resistant) as the recipient cell for ceftiofur-resistant E. coli and E. coli K12 (nalidixic acid resistant) as recipient cell for florfenicol-resistant E. coli. Briefly, fresh overnight cultures of donor and recipient strains (OD600 = 0.8), were mixed (100-μl total volume) and added to a nitrocellulose membrane overlaid on LB agar plates without antibiotics. After overnight incubation (37°C), the mixed culture was diluted with 1 mL LB broth and spread (100 μL) onto LB agar plates. For ceftiofur-resistant E. coli conjugation experiments, the plates contained rifampicin (32 μg ml−1; Sigma-Aldrich) and ceftiofur (8 μg ml−1). For florfenicol-resistant E. coli conjugation experiments, the plates contained florfenicol (50 μg ml−1) and nalidixic acid (30 μg ml−1; Sigma-Aldrich). Original cultures and the number of transconjugants were enumerated and the conjugation efficiency was calculated by dividing the number of CFU of transconjugants by the number of original donors.
2.6. Pulsed-field gel electrophoresis (PFGE)
Seventy florfenicol-resistant E. coli were randomly selected from 14 farms (5 strains each farm) and high-molecular-weight DNA was extracted from each isolate following a published protocol (CDC, 2013). The restriction enzyme XbaI (New England Biolabs, Beverly, MA, USA) was used for in-gel digestion at 37°C for 2.5 h. Restriction fragments were resolved with a contour-clamped homogeneous electric field CHEF-DR III pulsed-field system (Bio-Rad, Hercules, CA, USA) at 6 Vcm-1 for 18 h and with two liters of standard 0.5 X TBE running buffer maintained at 14°C. Gels were stained in distilled water with ethidium bromide (1 μg ml-1) for 20 min and then de-stained for 30 min. The resulting PFGE gels were visualized with a ChemiDoc™ Imaging System (Bio-Rad), and the sizes of the PFGE fragments were estimated by comparison with the PFGE marker (Salmonella enterica, serotype Braenderup H9812; 4 markers gel-1) ranging from 20.5 to 1135 kb. Macro-restriction digest data was summarized as Dice coefficients as implemented by BioNumerics v6.6 (Applied Maths, NV). An unpaired group means algorithm (UPGMA) was used to construct a dendrogram by using iTOL v4 (Letunic and Bork, 2019).
2.7. Statistical analysis
For microbiota and ARG profiling, the statistical significance of the percentage (detection rate), Shannon index and ddPCR data were analyzed by using a Kruskal-Wallis test, and the pairwise Mann-Whitney U-tests were used for multiple comparisons when needed [false discovery rate (FDR) adjustments were made to control for false positives]. Differences in beta-diversity, as visualized by NMDS, were tested using adonis2 in the vegan package (version 2.4–4) after checking for differences in dispersion using betadisper (Anderson and Walsh, 2013). Pairwise permutation MANOVAs (post hoc test for multiple comparisons) was performed in R with the RVAideMemire package (version 0.9-69-3) (Harvé, 2015), and significant differences were evaluated after FDR adjustment for α = 0.05.
For E. coli isolates, all bacterial CFU counts were transformed to log10 CFU prior to statistical analyses. Zero values were adjusted to the analytical detection limit of our assays (60 and 100 CFU g−1 of sample for soil and feces, respectively) by following the formula “substitution = log (RAND()*(detection limit))” in Excel (Microsoft Corp., Redmond, WA) prior to analysis. General linear models were developed to partition variance for log CFU between farms (n = 14; random factor), and housing areas (n = 6; fixed factor). A Tukey-Kramer multiple-comparison test was used to assess differences between least-squares means for housing areas (α = 0.05). NCSS 2007 software (LLC. Kaysville, UT) was used to assess the general linear models.
3. Results
3.1. The fecal and soil microbiota of dairy cattle varies across housing areas
To assess the variance in microbiota across housing areas, samples collected from seven commercial dairy farms were subjected to 16S rRNA gene sequencing. Quality-filtered 16S rRNA gene sequences (1.21 million) were obtained for 42 fecal samples (11,889 ± 5,225 reads per sample) (mean ± s.d.) and 84 soil samples (8,464 ± 1,546 reads per sample). In feces, the Shannon index varied greatly between different animal groups (P = 0.01), of which dairy cattle from hospital pens possessed the least diverse microbiota (P < 0.05) (Fig. 1a). Similarly, beta diversity varied significantly for cattle housed in different locations (P = 0.001) (Fig. 1b). In particular, calves were relatively distinct from cattle housed in hospital, lactation and dry pens, while the fecal microbiota of lactation and dry cattle were also different from each other (P < 0.05) (Fig. 1b). Ruminococcaceae was detected most often (~12.7% relative abundance) followed by Pseudomonadaceae (~11.8%), Moraxellaceae (~11.3), Lachnospiraceae (~6.0%) and Bacteroidaceae (~5.5%) (Fig. 1c).
Fig. 1.
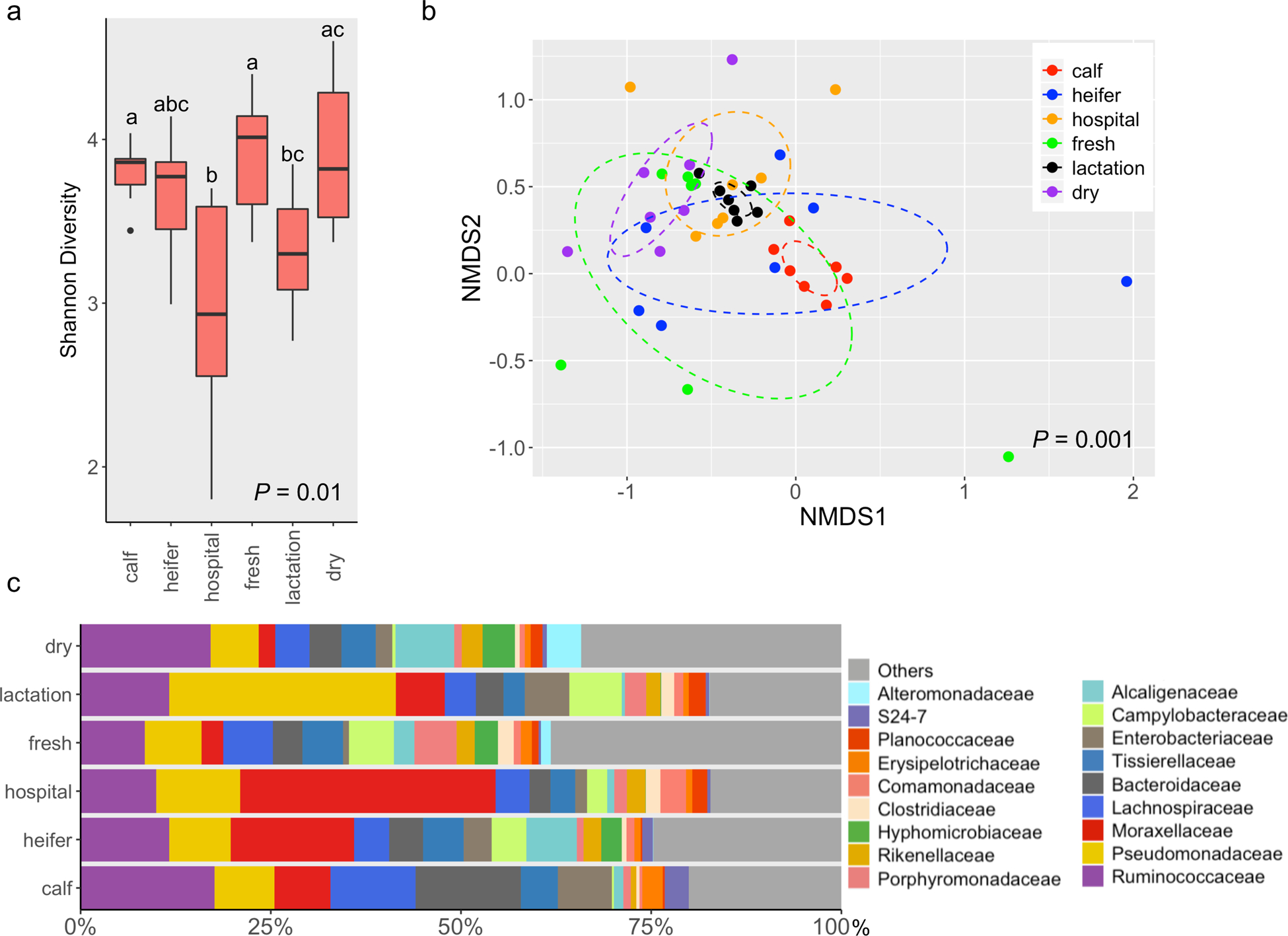
The fecal microbiota of dairy cattle differs across housing areas. (a) Boxplots of alpha diversity as measured by Shannon diversity index. The Shannon index values were presented as the median (central black horizontal line); the lower and upper hinges correspond to the 25th and 75th percentiles. Outliers are displayed as small black circles. This design applies to all the following boxplots in this study. The data were analyzed by a Kruskal-Wallis test followed by the pairwise Mann-Whitney U-tests for multiple comparisons. Different letters indicate statistically significant groups. (b) 3-D NMDS of dairy fecal samples based on Bray-Curtis matrices (stress = 0.16). The centroid of each ellipse represents the group mean, and the shape was defined by the covariance within each group; this design applies to all the following NMDS plots in this study. The data were analyzed by PERMANOVA followed by pairwise permutation MANOVAs for multiple comparisons. (c) Bar plot depicting the relative abundance of bacterial families over time; bacterial families which has a relative abundance less than 1% were grouped into “Others”.
In contrast, the alpha diversity (Shannon index) from soil was not significantly different among housing areas (P = 0.74) (Fig. 2a), but the soil community composition as measured by beta-diversity demonstrated overall difference (P = 0.002) (Fig. 2b). Specifically, the soil collected from dairy calf pen exhibited a distinct microbiota from other housing areas, while heifer soil microbiota was also significantly different from other groups except soil from dry pens (P < 0.05) (Fig. 2b). Corynebacteriaceae (~7.5%) together with Staphylococcaceae (~7.4%), Planococcaceae (~6.5%), Enterobacteriaceae (~4.7%) are the most abundant microbial families in soil, collectively representing approximately 26% of the sequence reads (Fig. 2c).
Fig. 2.
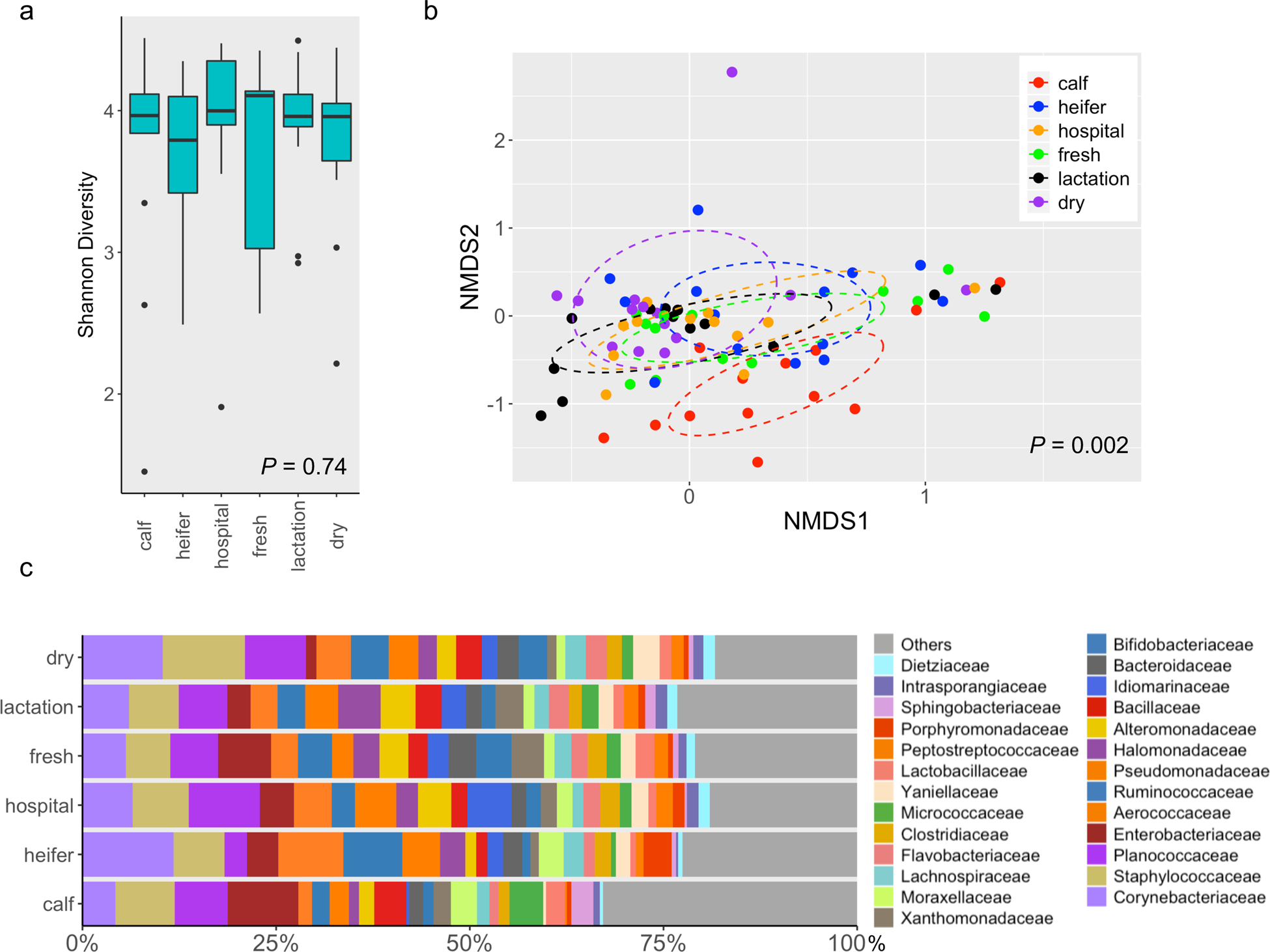
The soil microbiota differs across housing areas on dairy farms. (a) Boxplots of alpha diversity as measured by Shannon diversity index. (b) 3-D NMDS of dairy soil samples based on Bray-Curtis matrices (stress = 0.14). (c) Bar plot depicting the relative abundance of bacterial families over time; bacterial families which has a relative abundance less than 1% were grouped into “Others”.
Fecal and soil samples had distinct community composition (P = 0.001) (Fig. S1a), and in particular, soil possessed a more diverse microbiota than feces as measured by Shannon index (P = 0.006) (Fig. S1b). Differential abundance analysis indicated that the bacteria belonging to the families Corynebacteriaceae, Staphylococcaceae, Planococcaceae etc. were significantly more abundant in soil samples. In contrast, bacteria within families such as Ruminococcaceae, Lachnospiraceae and Tissierellaceae were more abundant in feces (Fig. S2).
3.2. The prevalence of fecal and soil ARGs varies across housing areas on dairy farms
Microbial composition is thought to be closely correlated with the prevalence of ARGs (Forsberg et al., 2014). In feces, the presence of 32 ARGs was not significantly divergent among housing areas (P = 0.08), but feces from dairy calves were more likely to test positive (Fig. 3a). Fecal ARG diversity, as measured by ordination analysis, exhibited different patterns across groups of animals (P = 0.004) (Fig. 3b) with diversity in calves being significantly different from animals housed in all other areas but heifers (P < 0.05) (Fig. 3b). Among all 32 ARGs, beta-lactam resistance genes including blaCMY-2 (~38.1% detection rate on average for six housing areas) and blaTEM (~40.5%), one MLS resistance gene mefA/E (~92.9%), two phenicol resistance genes fexB (~52.4%) and floR (~83.3%), and one tetracycline resistance gen tet(B) (~69%) were observed from cattle feces across all six housing areas (Fig. 3c). ARGs including aacA-aphD (aminoglycoside), ermA (MLS), cfr (oxazolidinones), cfrB (oxazolidinones), and tet(M) (tetracyclines) were solely detected in feces of dairy calves; ermC (MLS) and vanB (vancomycin) were only observed in heifer; npmA (aminoglycoside), ermB (MLS) and optrA (oxazolidinones) were only found in feces from calf and heifer (Fig. 3c). Notably, this is the first study demonstrated the presence of cfr, cfrB, and optrA of dairy origin in the United States.
Fig. 3.
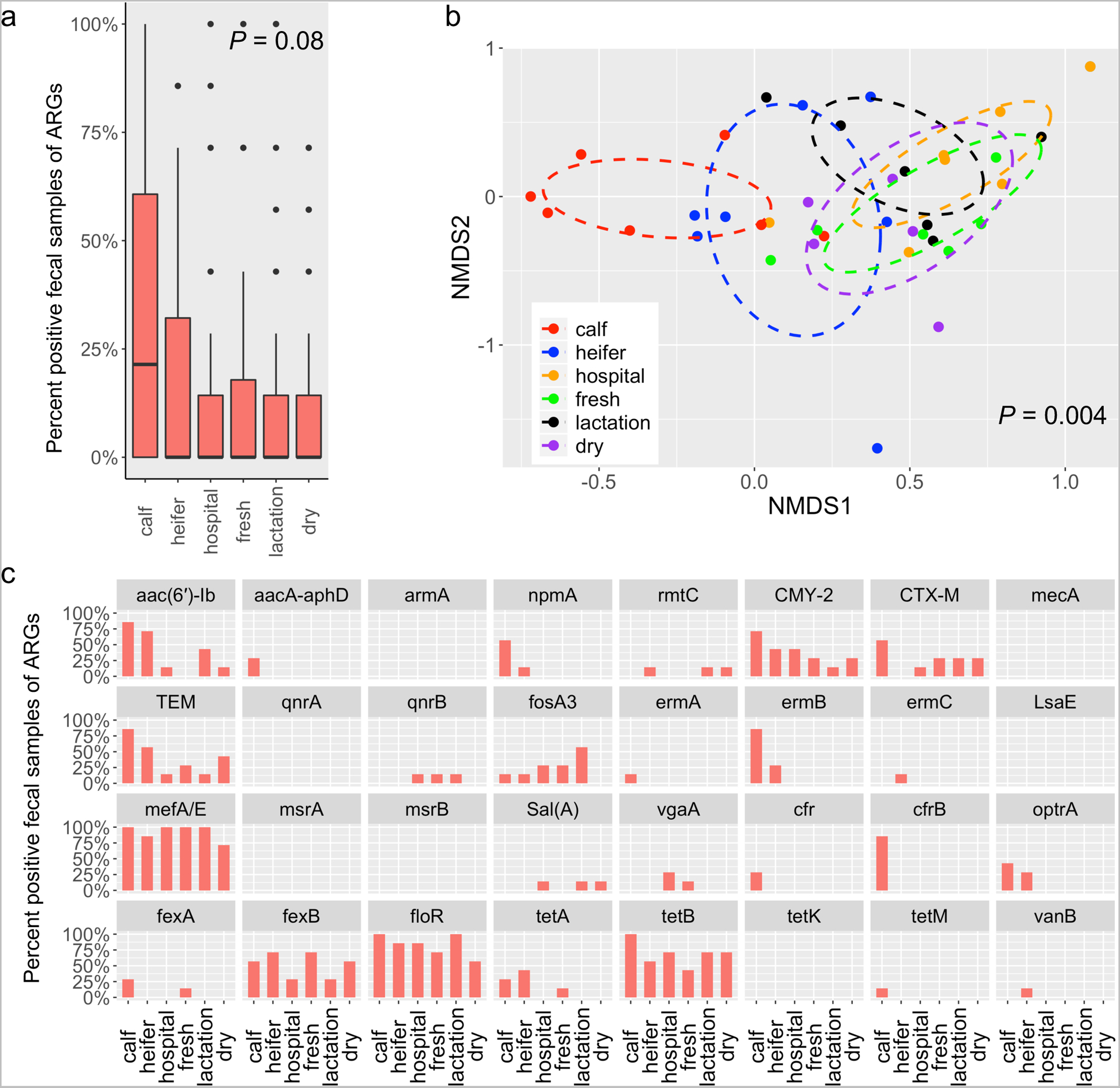
The prevalence of targeted ARGs in fecal samples vary across housing areas. (a) Boxplots of detection rate of ARGs grouped by housing regions. The detection rate was calculated as the ratio of PCR-positive samples divided by total samples. This was applied to all the following analyses in this study. (b) 3-D NMDS ordination of fecal samples based on Jaccard matrices (stress = 0.13). (c) Bar plot depicting the detection rate of individual ARGs across housing areas.
Consistent with observations in feces, the prevalence of ARGs was significantly higher from calf pens compared to hospital, lactation and dry pens (P < 0.05) (Fig. 4a). Ordination analysis indicated that the ARG diversity was significantly different among housing types in soil (P = 0.007) (Fig. 4b). The ARG structure in dairy calf hutches was significantly different from hospital and dry pens, while heifer pens possessed different ARG profiles from hospital and lactation pens (P < 0.05) (Fig. 4b). Of 32 ARGs, 12 genes including aac(6’)-ib (~66.7%; aminoglycoside), blaCMY-2 (~21.4%; beta-lactam), blaTEM (~54.8%; beta-lactam),ermA (~70.2%; MLS), ermC (~40%; MLS), mefA/E (~85.7%; MLS), sal(A) (~13.1%; MLS), fexA (~41.7%; phenicol), fexB (~67.9%; phenicol), floR (~75%;phenicol), tet(A) (~23.8%;tetracycline), and tet(B) (~65.5%;tetracycline) were present in all housing areas (Fig. 4c). One aminoglycoside resistance gene, rmtC, and one fosfomycin resistance gene, fosA3, were detected in soil of dairy calf hutches (Fig. 4c).
Fig. 4.
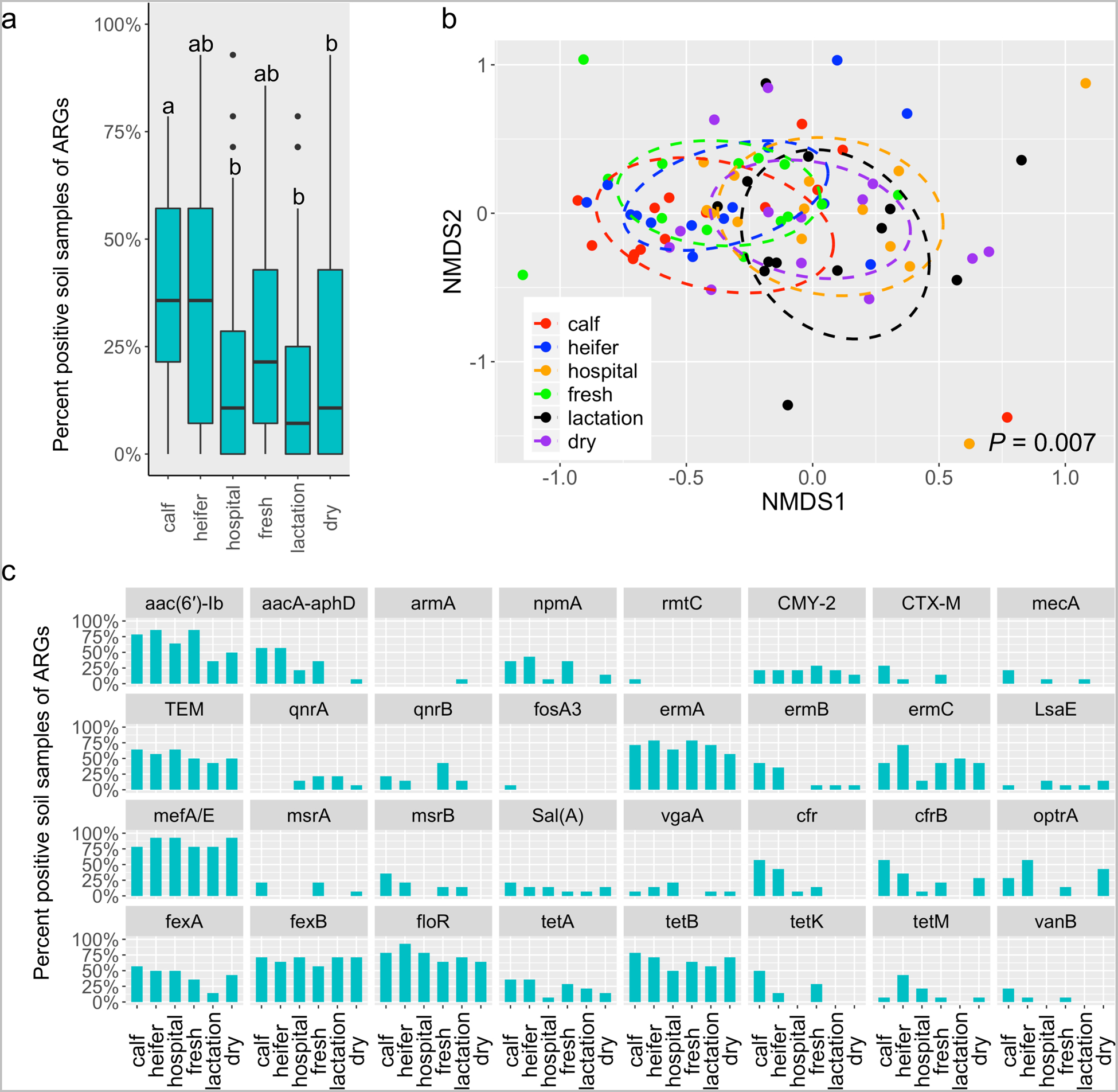
The prevalence of targeted ARGs in soil samples vary across housing areas. (a) Boxplots of detection rate of ARGs grouped by housing regions. Different letters indicate statistically significant groups. (b) 3-D NMDS ordination of soil samples based on Jaccard matrices (stress = 0.14). (c) Bar plot depicting the detection rate of individual ARGs across housing areas.
Consistent with observed differences in microbiota, ordination analysis demonstrated that the ARG profile in soil was significantly different from that in feces (P = 0.001) (Fig. S3a). Nevertheless, the prevalence of ARGs in soil samples positively correlates with that in fecal samples (Spearman correlation rho = 0.53, P < 0.001) (Fig. S3b). Moreover, ARGs were more frequently detected in soil compared to feces (P < 0.001) (Fig. S3c). Specifically, all assessed ARGs (n=32) were detected in soil samples from at least one housing area, while ARGs including armA (aminoglycoside), mecA (beta-lactam), qnrA (fluoroquinolones), LsaE (MLS), msrA (MLS), msrB (MLS), and tet(K) (tetracyclines) were under the detection limit in feces (Fig. S3d). In summary, our results indicate that both feces and soil are important sources of ARGs and, in both cases, dairy calves possess the highest prevalence of ARGs among animals from all housing areas.
3.3. The absolute abundance of floR varies across housing areas on dairy farms
Conventional PCR evaluated the presence or absence (prevalence) of ARGs, and absolute quantification (abundance) with ddPCR can further assess the risk of observed ARGs. We next determined the abundance of floR, which is the most prevalent florfenicol resistance gene in Gram-negative bacteria (Blickwede and Schwarz, 2004; Cloeckaert et al., 2000). In our dataset, this gene was present in both fecal (~83.3% detection rate) and soil (~75%) samples across all housing areas. The abundance of floR varied significantly among six measured areas in both feces (P = 0.0004) and soil (P = 0.001) (Fig. 5). In feces, calf and heifer animals possessed the highest abundance of floR, and in particular, the quantity of floR in dairy calves was significantly higher than that from hospital, fresh, lactation and dry animals. The floR gene was more abundant in feces from heifer animals than that from hospital and lactation cow (Mann-Whitney U-tests, FDR adjusted P < 0.05) (Fig. 5). In soil, floR was more abundant in calf and heifer pens than fresh and lactation pens, while floR in fresh pens was significantly more abundant than in soil from dry lots (P < 0.05) (Fig. 5). floR showed similar overall abundance between fecal (~2.16-log) and soil (~1.88-log) samples (P = 0.23). The distribution of floR abundance across housing areas is positively correlated with the observed overall ARG detection rate in both fecal (r = 0.85, P = 0.03) (Fig. S4a) and soil samples (r = 0.82, P = 0.04) (Fig. S4b), making floR a promising indicator to predict the general prevalence of antibiotic resistance (AMR) on dairy farms.
Fig. 5.
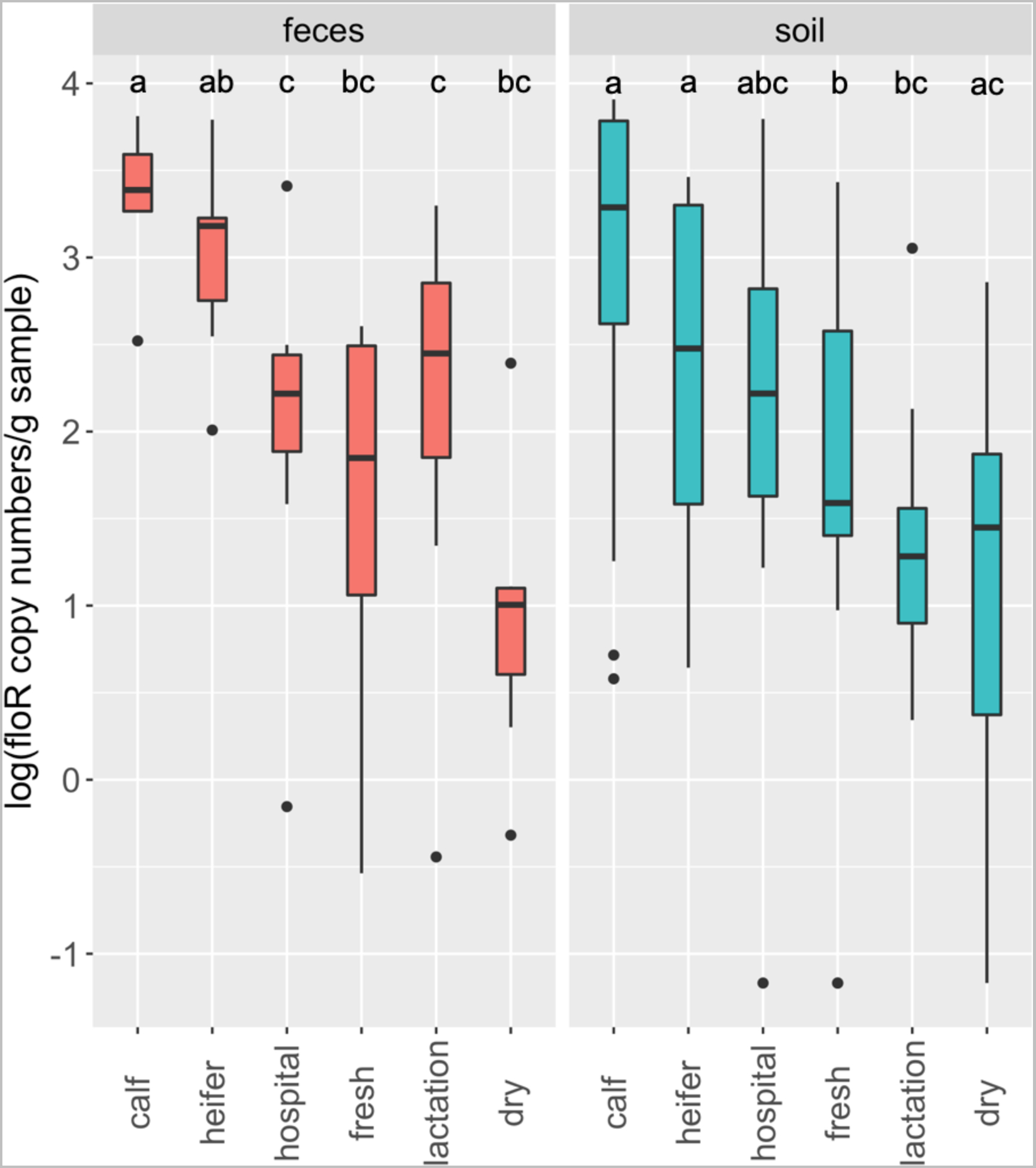
The absolute abundance of floR across housing areas in both fecal and soil samples. The data were analyzed by a Kruskal-Wallis test followed by the pairwise Mann-Whitney U-tests for multiple comparisons. Different letters indicate statistically significant groups.
3.4. Abundance of ceftiofur- and florfenicol-resistant E. coli in soil differs by housing areas
We further quantified the population of antibiotic-resistant bacteria in environmental soil with extended sampling on 14 dairy farms, with 840 soil samples from six housing areas included in this analysis. Least-squares adjusted means for ceftiofur-resistant E. coli at the farm level (n = 14 farms) ranged between 1.53 and 2.77-log CFU g−1. Counts for housing areas ranged between 1.46 (lactation pens) to 2.9-log CFU g−1 (calf pens) with three statistically distinct groupings (Fig. 6a). Least-squares adjusted means for florfenicol-resistant E. coli counts at the farm level varied between 1.73 and 2.9-log CFU g−1, while housing type ranged from 1.57 (lactation pens) to 3.3-log CFU g−1 (calf pens) (Fig. 6b).
Fig. 6.
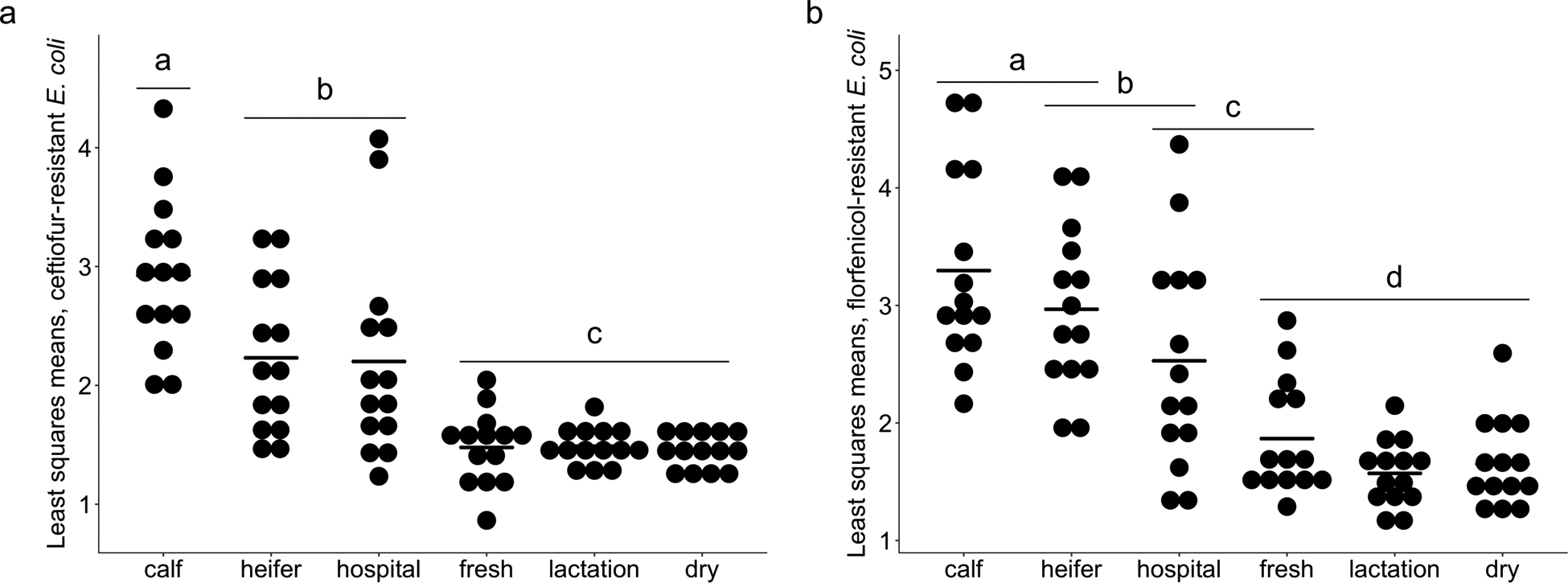
Least-squares mean Log10 CFU counts for (a) ceftiofur-resistant and (b) florfenicol-resistant E. coli. Dots represent individual farms and bolded line segments represent least-squares mean for housing type. Different letters indicate statistically significant groups based on a Tukey-Kramer multiple comparison test (P < 0.05).
For ceftiofur- and florfenicol-resistant E. coli, respectively, a high proportion of individual soil samples from calf pens (49.3% and 61.4%) and heifer pens (31.4% and 57.9%) had bacterial counts that exceeded the predicted contact-dependent ID50 (2.83-log CFU g−1) for transmission of antibiotic-resistant E. coli from soil to calves (Liu et al., 2016a). Hospital pens (26.4% and 35%) also regularly exceeded this threshold, and fresh pens were mixed (2.1% and 17.1%). Far fewer soils samples exceeded this threshold for lactating (2.1% and 4.3%) and dry lots (0.7% and 5.7%). In summary, antibiotic-resistant E. coli discretely distributed across housing areas on dairy farms with greater abundance in soil from calf pens compared to other housing areas.
3.5. Phenotypic profiling of soil-borne antibiotic-resistant E. coli isolates
A collection of 649 ceftiofur-resistant E. coli and 1,256 florfenicol-resistant E. coli isolates were obtained from pen soil samples collected in 14 dairy farms, and were further assessed for antibiotic susceptibility. All isolates were resistant to at least 4 different antibiotics with a dominant phenotype of AmpCefChlKanStrSulTetTri (41.1%) for ceftiofur-resistant E. coli, and phenotypes AmpChlStrSulTet (25.0%) and AmpCefChlKanStrSulTetTri (23.9%) for florfenicol-resistant E. coli (Table 1, Table S2). Our filter-conjugation assays further indicated that the observed multidrug resistance traits could transfer between strains. Specifically, all ceftiofur resistance phenotypes in donor cells were successfully conjugated into recipient strains with efficiencies ranging from 3.9 × 10−7 to 9.3 × 10−4. Eight of 10 florfenicol-resistant donor E. coli isolates successfully transferred the florfenicol resistance phenotype to recipients with efficiencies ranging from 2.3 × 10−7 to 4.4 × 10−2. All expected multidrug resistance phenotypes (AmpCefChlKanStrSulTetTri for ceftiofur-resistant E. coli; AmpChlStrSulTet for florfenicol-resistant E. coli) were recovered from successful transconjugants.
Table 1.
Multidrug resistance (MDR) phenotype and frequencies1.
| Ceftiofur-resistant E. coli (n=649 isolates) | ||
|---|---|---|
| MDR Phenotype2 | Positive isolates | Frequency |
| AmpCefChlKanStrSulTetTri | 267 | 41.1% |
| AmcAmpCefChlKanStrSulTetTri | 101 | 15.6% |
| AmpCefChlCipKanStrSulTetTri | 85 | 13.1% |
| AmpCefKanStrSulTetTri | 73 | 11.2% |
| AmpCefCipKanSulTetTri | 46 | 7.1% |
| AmpCefCipKanStrSulTetTri | 33 | 5.1% |
| AmcAmpCefChlCipKanStrSulTetTri | 27 | 4.2% |
| AmcAmpCefChlKanStrSulTet | 11 | 1.7% |
| AmpCefStrSulTetTri | 6 | 0.9% |
| Florfenicol-resistant E. coli (n=1,256 isolates) | ||
| MDR Phenotype | Positive isolates | Frequency |
| AmpChlStrSulTet | 314 | 25.0% |
| AmpCefChlKanStrSulTetTri | 300 | 23.9% |
| AmpCefChlCipKanStrSulTetTri | 87 | 6.9% |
| AmpChlStrSulTetTri | 75 | 6.0% |
| AmpChlKanStrSulTet | 75 | 6.0% |
| AmpChlKanStrSulTetTri | 71 | 5.7% |
| AmcAmpCefChlKanStrSulTetTri | 52 | 4.1% |
| ChlKanStrSulTet | 50 | 4.0% |
| ChlKanStrSulTetTri | 41 | 3.3% |
| AmcAmpChlKanStrSulTetTri | 27 | 2.1% |
| AmpCefChlKanStrSulTet | 25 | 2.0% |
| AmpChlStrSul | 24 | 1.9% |
| ChlStrSulTetTri | 21 | 1.7% |
| ChlStrSulTet | 14 | 1.1% |
| AmpChlCipKanStrSulTetTri | 14 | 1.1% |
| AmcAmpChlStrSulTetTri | 14 | 1.1% |
| AmpCefChlStrSulTetTri | 13 | 1.0% |
| AmcAmpChlCipKanStrSulTetTri | 12 | 1.0% |
| AmcAmpCefChlCipKanStrSulTetTri | 8 | 0.6% |
| AmpCefChlStrSulTet | 5 | 0.4% |
| AmpChlSulTetTri | 4 | 0.3% |
| AmpChlKanSulTetTri | 4 | 0.3% |
| AmpChlSulTet | 3 | 0.2% |
| AmcAmpCefChlKanStrSulTet | 3 | 0.2% |
Phenotypes occurring with a frequency <2 were not included here.
AMP (ampicillin; 32 μg/mL); AMC (amoxicillin/clavulanic acid; 32/16 μg/mL); CEF (ceftiofur; 8 μg/mL); CHL (chloramphenicol; 34 μg/mL); TET (tetracycline; 16 μg/mL); CIP (ciprofloxacin; 4 μg/mL); STR (streptomycin; 16 μg/mL); KAN (kanamycin; 64 μg/mL); SUL (sulfamethoxazole; 512 μg/mL); TRI (trimethoprim; 8 μg/mL)
Although consistent distribution patterns of antibiotic-resistant E. coli across housing areas were observed in dairy soil, there were differences between farms and phenotypes provide no information about the potential that strains of bacteria might be shared between farms. Consequently, we examined the distribution of macro-restriction digest patterns (PFGE) for florfenicol-resistant E. coli (n = 70; 5 isolates farm−1) that had the most prevalent multidrug-resistance phenotype (AmpChlStrSulTet) (Fig. S5). Florfenicol-resistant isolates are chosen for this analysis because of their overall higher prevalence compared with ceftiofur-resistant E. coli (Fig. 6), and as a result, more florfenicol-resistant E. coli isolates (n=1,256) were collected in this study than ceftiofur-resistant E. coli (n=649) (Table S2). In general, there was considerable diversity within and between farms with some sharing identical restriction-digest band patterns from independent samples (Fig. S5; farm #6 and #14). Some isolates, with the same or very similar restriction fragment patterns, were present at different farms that were geographically dispersed [Fig. S5; farm #2 and #5 (~99 km); farm #3 and #4 (~81 km); farm#1, #7 and #8 (~25–98 km); farm #12 and #13 (~73 km)]. In summary, our results indicate that all isolated soil-borne ceftiofur- and florfenicol- resistant E. coli were multidrug resistant and plasmid-mediated transmission likely explains the dissemination between strains. Moreover, antibiotic-resistant bacteria are probably shared across farms, which may contribute to long-term persistence of antibiotic resistance in dairy production.
4. Discussion
We hypothesized that dairy cattle and the corresponding soil possess distinct microbiota and distinct distributions of antibiotic-resistant organisms across housing areas. The hypothesis is supported by our results showing that younger animals, especially dairy calves, had different microbiota compared with older animals (Fig. 1b), and the soil microbiome in calf pens was also quite distinct from other housing areas (Fig. 2b). Bacterial taxonomy is closely related to the diversity of ARGs that can be found in a community (Forsberg et al., 2014). Among all the observed taxa, the bacterial family Enterobacteriaceae was present in both fecal and soil samples, and in both cases Enterobacteriaceae was represented the most in dairy calves (7.1% in feces and 9.1% in soil, respectively) (Fig. 1c&2c). This family likely harbors the most ARGs in dairy production (Liu et al., in press), and thus we speculate that the unbalanced distribution of this particular taxon contributes to a divergent prevalence of antibiotic resistance between housing areas.
We surmise that the varying distribution of antibiotic resistance genes and organisms is a result of multiple practices that include different exposure to antibiotics across housing areas. While we were unable to collect the corresponding antibiotic use data for the sampled areas during this study, the presence of ARGs (Fig. 3&4) and abundance of antibiotic-resistant bacteria (Fig. 6) generally reflected the expected application of antibiotics in dairy production based on an earlier survey of 30 dairies in Washington State (Davis et al., 2015). In particular, the prevalence of beta-lactamases including blaCMY-2, blaCTX-M and blaTEM is likely associated with the commonly used beta-lactams (ceftiofur and ampicillin). In addition, the wide use of florfenicol may contribute to selection for floR and fexB. In the case of florfenicol resistance, floR was present in all pen types but was numerically more abundant for dairy calves (Fig. 5), consistent with the fact that florfenicol is primarily prescribed for treating pneumonia in calves (Davis et al., 2015). Nevertheless, other ARGs were present despite a limited probability of corresponding antibiotic selection pressure (e.g., cfr, cfrB and optrA). These ARGs have not been identified elsewhere in the United States from cattle-sourced samples, and co-selection for linked genes by other antimicrobial agents such as phenicol (e.g. florfenicol) and lincosamides (e.g. pirlimycin) (Michael et al., 2015) could explain their presence.
On a per gram basis, the prevalence of ARGs was higher in soil compared to feces. This is not unexpected because soil has a more diverse microbiome, and there are indigenous soil microbes that produce antibiotics and likely select for resistant strains (Davies and Davies, 2010). In addition, when animals are treated with antibiotics the top soil on farms is readily contaminated with new ARGs and antibiotic residues from feces and urine (Liu et al., 2016a; Zhu et al., 2013). Compared with fecal communities, our results indicate that the farm soil could be considered an important source of ARGs in dairy production.
Antibiotic-resistant E. coli in farm soil can readily colonize the host animals (Liu et al., 2016a), and subsets of resistant bacteria can favor different aged animals (Khachatryan et al., 2004). In one case, “calf-adapted” E. coli that confer resistance to streptomycin, sulfadiazine, and tetracycline (SSuT) were significantly more prevalent in calves (especially during the period when calves were on milk diet) compared to older animals (Khachatryan et al., 2004). For the present study, this specificity by age was evident for ARGs that were present across pens in the soil (Fig. 4c), but were only detected in feces of calves or heifers (i.e. aacA-aphD, npmA, ermA, ermB, ermC, cfr, cfrB, optrA, tetM, vanB) (Fig. 3c). Antibiotic-resistant bacteria favoring the enteric environment in younger animals can further contaminate the associated pen soil via fecal shedding (Liu et al., 2016a), which may in turn contribute to the relatively higher detection rate of certain ARGs in soils from calves and heifers compared to other housing areas (Fig. 4c).
In addition to the prevalence and abundance analyses of targeted ARGs, quantification of select antibiotic-resistant bacteria were consistent with a high probability that the pen soil of dairy calves serves as a reservoir of antibiotic-resistant bacteria (Fig. 6). We surmise that the differing distribution of antibiotic-resistant E. coli across housing areas is a result of mixed effects. Firstly, younger animals (especially for dairy calves) tend to have higher relative abundance of Enterobacteriaceae (Fig. 1c&2c) which is highly possible to be translated into the absolute population of overall E. coli. This community composition is likely linked to the diet in cattle (Liu et al., in press) and distinctly different gut environments between younger and older animals (e.g., abundance of volatile fatty acids). It is also possible that the characteristics of different housing pen soil environments favor the propagation of E. coli to different densities. Finally, it is possible that the observed discrete distribution of antibiotic-resistant E. coli correlated with the different use of antibiotics across animal groups. On dairy farms, younger animals tend to receive more antibiotic treatments and we know this practice leads to the bloom of antibiotic-resistant bacteria is the corresponding soil (Liu et al., 2016a). In the context of antimicrobial resistance, we consider “risk” to involve the likelihood that resistant bacteria will be transmitted between animals and potentially to people (e.g. farm workers) (Moodley and Guardabassi, 2009; Smith et al., 2013). Risk of transmission can depend on many factors (e.g., behavior), but it begins with having an abundant source of bacteria. In this regards, dairy calves serve a “hotspot” for potential transmission of antibiotic-resistant bacteria.
Of importance, the bacteria that we isolated from soil samples were all resistant to at least four of our tested antibiotics, and some of these E. coli isolates are genetically related between farms. Clearly, we need extended studies to further understand the scope of between-farm transmission events because of the implications for biosecurity for these facilities. Commercial dairies commonly use “calf raisers” in their production process and this has already been identified as a risk factor for introduction of novel antibiotic-resistant Salmonella strains onto dairy farms (Adhikari et al., 2009), and this may be the primary mechanism for disseminating antibiotic-resistant strains of other enteric bacteria as well.
In conclusion, we found that the distribution of antibiotic-resistant organisms and resistance genes for both feces and soil samples differed between housing areas, with younger animals associated with a higher prevalence of both. From a management perspective, this is important because having predictable reservoirs helps to identify where mitigation measures could be employed most effectively. Knowing that dairy calves and heifers and their associated housing areas are the most likely hotspots for ARGs, waste from these locations could be treated separately and more comprehensively (e.g., composting) to reduce or eliminate antibiotic-resistant bacteria before field application (Gou et al., 2018; Pruden et al., 2013). Similarly, farm soil in situ from these housing areas deserve more attention than others, and management strategies intended to reduce on-farm resistance should prioritize dairy calves and heifers. In contrast, “less risky” waste from other housing areas could be treated with less costly processes. The ability to partition waste according to risk offers a possibility of developing a more cost-effective mixture of management strategies, which in turn make management of risk more feasible on working farms.
Supplementary Material
Fig. S1 The direct comparison between fecal and soil microbiota on dairy farms. (a) NMDS ordination of fecal and soil samples based on Bray-Curtis matrices (3 dimensions; stress of 0.14) (b) Boxplots of alpha diversity as measured by Shannon diversity index.
Fig. S2 Differentially abundant bacterial taxa between fecal and soil samples. LDA score refers to Linear discriminant analysis score as calculated by LEfSe; scores are presented for both positive and negative values; however, absolute values of the effect size were used to interpret the scale of difference.
Fig. S3 The direct comparison of the prevalence of targeted ARGs between fecal and soil on dairy farms. (a) 3-D NMDS ordination of fecal and soil samples (Jaccard matrices, stress of 0.13). (b) The Spearman’s rank correlation of distribution of ARGs’ prevalence between fecal and soil samples. Dots represent the prevalence of individual ARGs in a specific housing area. (c) Boxplots of detection rate of ARGs in feces and soil. (d) Bar plot depicting the comparison of detection rate of individual ARGs in feces and soil.
Fig. S4 The Pearson correlation of the abundance of floR and overall detection rate of ARGs. (a) predicted correlations in fecal samples (b) predicted correlations in soil samples. The Pearson correlation was applied after checking data distribution of normality. In each type of samples, the detection rates of all assessed ARGs were averaged to represent the overall positive percentage of ARGs in a specific housing area. The absolute abundance of floR was prepared accordingly (single mean value was calculated to represent one housing area in fecal or soil samples) to perform the correlation analysis.
Fig. S5 Macro-restriction digest dendrogram for florfenicol-resistant E. coli from 14 farms. A total of 70 E. coli (five isolates randomly selected from each of 14 farms), were analyzed by macro-restriction digest and PFGE (XbaI) Dairy farms were coded 1–14 according to order of sampling. Each sample was annotated by farm ID followed by A-E. Green lines in the dendrogram represent indistinguishable fingerprints that were found in the same farm; red lines represent indistinguishable fingerprints that were observed from different farms but located in the same geographical region; and blue lines represent indistinguishable isolates were found in different farms and also different regions across the Columbia River. To the right of the dendrogram is the antibiotic resistance profile of isolates (see Table S2 for details). Filled circles represent resistance to the antibiotic and open circles represent susceptibility to the antibiotic.
Acknowledgements
We thank Lisa Jones for the guidance in samples collection and Lisa Orfe for the assistance in performing ddPCR. This study was funded in part by the Agricultural Animal Health Program, College of Veterinary Medicine, by the Washington State Agricultural Research Center, and by USDA NIFA grant 2015-68003-22998. The general program of National Natural Science Foundation of China (Grants No. 31572548) and Science and Technology Support Program of Sichuan Province, China (Grants No. 2018HH0027)
References
- Aarestrup FM, 2015. The livestock reservoir for antimicrobial resistance: a personal view on changing patterns of risks, effects of interventions and the way forward. Philos Trans R Soc Lond B Biol Sci 370, 20140085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari B, Besser TE, Gay JM, Fox LK, Davis MA, Cobbold RN, Berge AC, Hancock DD, 2009. The role of animal movement, including off-farm rearing of heifers, in the interherd transmission of multidrug-resistant Salmonella. Journal of Dairy Science 92, 4229–4238. [DOI] [PubMed] [Google Scholar]
- Anderson MJ, Walsh DCI, 2013. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecological Monographs 83, 557–574. [Google Scholar]
- Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ, 2015. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13, 42–51. [DOI] [PubMed] [Google Scholar]
- Blickwede M, Schwarz S, 2004. Molecular analysis of florfenicol-resistant Escherichia coli isolates from pigs. J Antimicrob Chemother 53, 58–64. [DOI] [PubMed] [Google Scholar]
- Burgos JM, Ellington BA, Varela MF, 2005. Presence of multidrug-resistant enteric bacteria in dairy farm topsoil. Journal of Dairy Science 88, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadillo-Quiroz H, Brauer S, Yashiro E, Sun C, Yavitt J, Zinder S, 2006. Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State, USA. Environ Microbiol 8, 1428–1440. [DOI] [PubMed] [Google Scholar]
- Call DR, Matthews L, Subbiah M, Liu JX, 2013. Do antibiotic residues in soils play a role in amplification and transmission of antibiotic resistant bacteria in cattle populations? Frontiers in Microbiology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call DR, Singer RS, Meng D, Broschat SL, Orfe LH, Anderson JM, Herndon DR, Kappmeyer LS, Daniels JB, Besser TE, 2010. blaCMY-2-positive IncA/C plasmids from Escherichia coli and Salmonella enterica are a distinct component of a larger lineage of plasmids. Antimicrob Agents Chemother 54, 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP, 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R, 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudell MA, Mair C, Subbiah M, Matthews L, Quinlan RJ, Quinlan MB, Zadoks R, Keyyu J, Call DR, 2018. Identification of risk factors associated with carriage of resistant Escherichia coli in three culturally diverse ethnic groups in Tanzania: a biological and socioeconomic analysis. Lancet Planet Health 2, e489–e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC, 2013. Standard Operating Procedure for PulseNet PFGE of Escherichia coli O157:H7, Escherichia coli non-O157 (STEC), Salmonella serotypes, Shigella sonnei and Shigella flexneri., 1–13. [Google Scholar]
- Cheng W, Chen H, Su C, Yan S, 2013. Abundance and persistence of antibiotic resistance genes in livestock farms: a comprehensive investigation in eastern China. Environ Int 61, 1–7. [DOI] [PubMed] [Google Scholar]
- Cloeckaert A, Baucheron S, Flaujac G, Schwarz S, Kehrenberg C, Martel JL, Chaslus-Dancla E, 2000. Plasmid-mediated florfenicol resistance encoded by the floR gene in Escherichia coli isolated from cattle. Antimicrob Agents Chemother 44, 2858–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Davies D, 2010. Origins and Evolution of Antibiotic Resistance. Microbiology and Molecular Biology Reviews 74, 417.-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Cloud-Hansen KA, Carpenter J, Hovde CJ, 2005. Escherichia coli O157 : H7 in environments of culture-positive cattle. Applied and Environmental Microbiology 71, 6816–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Sischo WM, Jones LP, Moore DA, Ahmed S, Short DM, Besser TE, 2015. Recent Emergence of Escherichia coli with Cephalosporin Resistance Conferred by blaCTX-M on Washington State Dairy Farms. Appl Environ Microbiol 81, 4403–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill-McFarland KA, Breaker JD, Suen G, 2017. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Scientific Reports 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N, Dantas G, 2014. Bacterial phylogeny structures soil resistomes across habitats. Nature 509, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou M, Hu HW, Zhang YJ, Wang JT, Hayden H, Tang YQ, He JZ, 2018. Aerobic composting reduces antibiotic resistance genes in cattle manure and the resistome dissemination in agricultural soils. Sci Total Environ 612, 1300–1310. [DOI] [PubMed] [Google Scholar]
- Harvé M, 2015. Package “RVAideMemoire”.
- Hu Y, Cheng H, Tao S, 2017. Environmental and human health challenges of industrial livestock and poultry farming in China and their mitigation. Environ Int 107, 111–130. [DOI] [PubMed] [Google Scholar]
- Khachatryan AR, Besser TE, Hancock DD, Call DR, 2006. Use of a nonmedicated dietary supplement correlates with increased prevalence of streptomycin-sulfa-tetracycline-resistant Escherichia coli on a dairy farm. Applied and Environmental Microbiology 72, 4583–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachatryan AR, Hancock DD, Besser TE, Call DR, 2004. Role of calf-adapted Escherichia coli in maintenance of antimicrobial drug resistance in dairy calves. Appl Environ Microbiol 70, 752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxminarayan R, Duse A, Wattal C, Zaidi AK, Wertheim HF, Sumpradit N, Vlieghe E, Hara GL, Gould IM, Goossens H, Greko C, So AD, Bigdeli M, Tomson G, Woodhouse W, Ombaka E, Peralta AQ, Qamar FN, Mir F, Kariuki S, Bhutta ZA, Coates A, Bergstrom R, Wright GD, Brown ED, Cars O, 2013. Antibiotic resistance-the need for global solutions. Lancet Infectious Diseases 13, 1057–1098. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P, 2019. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhao Z, Orfe L, Subbiah M, Call DR, 2016a. Soil-borne reservoirs of antibiotic-resistant bacteria are established following therapeutic treatment of dairy calves. Environmental Microbiology 18, 557–564. [DOI] [PubMed] [Google Scholar]
- Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, Doi Y, Tian GB, Dong BL, Huang XH, Yu LF, Gu DX, Ren HW, Chen XJ, Lv LC, He DD, Zhou HW, Liang ZS, Liu JH, Shen JZ, 2016b. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infectious Diseases 16, 161–168. [DOI] [PubMed] [Google Scholar]
- Mandel R, Whay HR, Klement E, Nicol CJ, 2016. Invited review: Environmental enrichment of dairy cows and calves in indoor housing. Journal of Dairy Science 99, 1695–1715. [DOI] [PubMed] [Google Scholar]
- Michael GB, Freitag C, Wendlandt S, Eidam C, Fessler AT, Lopes GV, Kadlec K, Schwarz S, 2015. Emerging issues in antimicrobial resistance of bacteria from food-producing animals. Future Microbiol 10, 427–443. [DOI] [PubMed] [Google Scholar]
- Miller W, 1979. Dairy cattle feeding and nutrition.
- Moodley A, Guardabassi L, 2009. Transmission of IncN plasmids carrying blaCTX-M-1 between commensal Escherichia coli in pigs and farm workers. Antimicrob Agents Chemother 53, 1709–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruden A, Larsson DG, Amezquita A, Collignon P, Brandt KK, Graham DW, Lazorchak JM, Suzuki S, Silley P, Snape JR, Topp E, Zhang T, Zhu YG, 2013. Management options for reducing the release of antibiotics and antibiotic resistance genes to the environment. Environ Health Perspect 121, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MA, Casey TA, 2001. Environmental and food safety aspects of Escherichia coli O157 : H7 infections in cattle. Critical Reviews in Microbiology 27, 57–73. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C, 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TC, Gebreyes WA, Abley MJ, Harper AL, Forshey BM, Male MJ, Martin HW, Molla BZ, Sreevatsan S, Thakur S, Thiruvengadam M, Davies PR, 2013. Methicillin-resistant Staphylococcus aureus in pigs and farm workers on conventional and antibiotic-free swine farms in the USA. PLoS One 8, e63704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah M, Mitchell SM, Ullman JL, Call DR, 2011. beta-lactams and florfenicol antibiotics remain bioactive in soils while ciprofloxacin, neomycin, and tetracycline are neutralized. Appl Environ Microbiol 77, 7255–7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C., 2018. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Tian GB, Doi YH, Shen JZ, Walsh TR, Wang Y, Zhang R, Huang X, 2017. MCR-1-producing Klebsiella pneumoniae outbreak in China. Lancet Infectious Diseases 17, 577–577. [DOI] [PubMed] [Google Scholar]
- Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, Teillant A, Laxminarayan R, 2015. Global trends in antimicrobial use in food animals. Proceedings of the National Academy of Sciences of the United States of America 112, 5649–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YG, Johnson TA, Su JQ, Qiao M, Guo GX, Stedtfeld RD, Hashsham SA, Tiedje JM, 2013. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc Natl Acad Sci U S A 110, 3435–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 The direct comparison between fecal and soil microbiota on dairy farms. (a) NMDS ordination of fecal and soil samples based on Bray-Curtis matrices (3 dimensions; stress of 0.14) (b) Boxplots of alpha diversity as measured by Shannon diversity index.
Fig. S2 Differentially abundant bacterial taxa between fecal and soil samples. LDA score refers to Linear discriminant analysis score as calculated by LEfSe; scores are presented for both positive and negative values; however, absolute values of the effect size were used to interpret the scale of difference.
Fig. S3 The direct comparison of the prevalence of targeted ARGs between fecal and soil on dairy farms. (a) 3-D NMDS ordination of fecal and soil samples (Jaccard matrices, stress of 0.13). (b) The Spearman’s rank correlation of distribution of ARGs’ prevalence between fecal and soil samples. Dots represent the prevalence of individual ARGs in a specific housing area. (c) Boxplots of detection rate of ARGs in feces and soil. (d) Bar plot depicting the comparison of detection rate of individual ARGs in feces and soil.
Fig. S4 The Pearson correlation of the abundance of floR and overall detection rate of ARGs. (a) predicted correlations in fecal samples (b) predicted correlations in soil samples. The Pearson correlation was applied after checking data distribution of normality. In each type of samples, the detection rates of all assessed ARGs were averaged to represent the overall positive percentage of ARGs in a specific housing area. The absolute abundance of floR was prepared accordingly (single mean value was calculated to represent one housing area in fecal or soil samples) to perform the correlation analysis.
Fig. S5 Macro-restriction digest dendrogram for florfenicol-resistant E. coli from 14 farms. A total of 70 E. coli (five isolates randomly selected from each of 14 farms), were analyzed by macro-restriction digest and PFGE (XbaI) Dairy farms were coded 1–14 according to order of sampling. Each sample was annotated by farm ID followed by A-E. Green lines in the dendrogram represent indistinguishable fingerprints that were found in the same farm; red lines represent indistinguishable fingerprints that were observed from different farms but located in the same geographical region; and blue lines represent indistinguishable isolates were found in different farms and also different regions across the Columbia River. To the right of the dendrogram is the antibiotic resistance profile of isolates (see Table S2 for details). Filled circles represent resistance to the antibiotic and open circles represent susceptibility to the antibiotic.