

Abstract
Tafazzin, which is encoded by the TAZ gene, catalyzes transacylation to form mature cardiolipin and shows preference for the transfer of a linoleic acid (LA) group from phosphatidylcholine (PC) to monolysocardiolipin (MLCL) with influence from mitochondrial membrane curvature. The protein contains domains and motifs involved in targeting, anchoring, and an active site for transacylase activity. Tafazzin activity affects many aspects of mitochondrial structure and function, including that of the electron transport chain, fission-fusion, as well as apoptotic signaling. TAZ mutations are implicated in Barth syndrome, an underdiagnosed and devastating disease that primarily affects male pediatric patients with a broad spectrum of disease pathologies that impact the cardiovascular, neuromuscular, metabolic, and hematologic systems.
Keywords: mitochondrial biology, rare mitochondrial diseases, apoptosis, clinical case reports
1. Introduction
TAZ, also known as G4.5, is a 10kb gene located at position 28 on the q arm of chromosome X within a gene-rich, 450kb cluster of 13-16 small genes with CpG islands initially identified by Bione et al. as potential candidates for involvement in neuromuscular and cardiovascular disorders (Bione et al., 1993; Bione et al., 1996). TAZ encodes the transacylase protein tafazzin (Table 1), so named by Bione et al. based on a masochistic comic character named Tafazzi from an Italian sports show, apparently due to the difficulty they encountered in the original identification and characterization of this protein (Bione et al., 1996). Tafazzin, located in the inner and outer mitochondrial membranes (IMM and OMM), acts as an acyl-specific transacylase that is essential to lipid metabolism through cardiolipin (CL) remodeling. CL remodeling, in turn, is essential for mitochondrial respiratory chain homeostasis, and disruptions to this process as a result of TAZ mutations have been shown to be a major cause of the complex, multi-system Barth syndrome. Mutations in the TAZ gene are associated with severe cardiovascular defects observed in Barth syndrome (BTHS), including endocardial fibroelastosis (EFE), X-linked dilated cardiomyopathy 3A (CMD3A, and left ventricular noncompaction (LVNC). As such, the gene is known by several aliases, including BTHS, EFE and EFE2, CMD3A, and LVNCX (Table 1). Due to unfortunate naming conventions, the TAZ gene and its tafazzin protein product have been confused in the literature with the TAZ protein, and WWTR1 gene, which are associated with various cancers (NCBI, 2019). TAZ and tafazzin are not known to have any role in cancer. To date, there is no cure for Barth syndrome, and treatments for tafazzin deficiencies have focused on symptom-based management. Deeper investigation of TAZ, tafazzin, and cardiolipin is necessary to increase our collective understanding of mitochondrial biology and may help find treatments for Barth syndrome and mitochondrial myopathies.
Table 1. Essential properties and identifiers of TAZ and tafazzin.
The table contains a summary of the essential properties, identifiers, and names of the TAZ gene and the encoded tafazzin protein in Homo sapiens (human) and Mus musculus (mouse) (NCBI, 2019).
| Species | Homo sapiens (human) | Mus musculus (mouse) |
| Gene | TAZ | TAZ |
| Synonyms | BTHS, EFE, EFE2, CMDA3, LVNCX, G4.5 | 5031411C02Rik, 9130012G04Rik, AW107266, AW552613, G4.5 |
| NCBI Gene ID | 6901 | 66826 |
| Chromosomal location | chrXq28:154,411,524-154,421,726 | chrX:74,282,697-74,290,151 |
| Length (nt) | 10,171 | 7,454 |
| Exons/Introns | 11/10 | 10/9 |
| NCBI Gene ID | 6901 | 66826 |
| UniProt ID | Q16635 | I7HJS2 |
| Ensembl ID | ENSG00000102125 | ENSMUST00000069722.12 |
| Length (aa) | 292 | 263 |
| Molecular weight (Da) | 33,459 | 30,433 |
2. Structure
Tafazzin is part of a superfamily of acyltransferases based on conserved regions and motifs identified through sequence alignment with other acyltransferase proteins involved in phospholipid biosynthesis (Neuwald, 1997; Acehan et al., 2007). The crystal structure of tafazzin has not been determined by x-ray crystallography; the closest homologous protein is plant glycerol-3-phosphate acyltransferase (G3PAT) from Cucurbita moscata and Spinacea oleracea (PDB ID: 1IUQ) (Tamada et al., 2004), which has just 18.1% sequence identity with human tafazzin (Waterhouse et al., 2018). The half-life of tafazzin in mammalian cells is much shorter than that of many other mitochondrial proteins at only 3-6 hours (Xu et al., 2015); the median half-life for mouse mitochondrial proteins is 17.2 days in the heart and 4.26 days in the liver (Kim et al., 2012). This rapid turnover rate has likely contributed to the difficulty in elucidating the structure of tafazzin or acquiring a detailed understanding of its post-translational modifications (PTMs) from mass spectrometry data.
2.1. Tafazzin active site.
The putative phospholipid-binding site of human tafazzin is a 57 amino acid cleft with two open ends and a stretch of conserved, positively charged residues, based on homology modeling using ALAdeGAP for improved amino acid sequence alignment (Hijikata et al., 2011; Hijikata et al., 2015). Tafazzin and related hydrolases contain a conserved histidine residue required for their enzymatic action (Neuwald, 1997). In human tafazzin, His-69 (His-77 in yeast) and Asp-74 form part of a conserved HX4D motif seen in acyltransferases composed of a histidine (His) and aspartic acid (Asp) separated by any 4 amino acids (X4) (Heath and Rock, 1998; Xu et al., 2006; Abe et al., 2016). The HX4D motif facilitates the Asp-His dyad mechanism seen commonly in serine proteases, whereby Asp raises the pKa of His and aids the deprotonation of a hydroxyl group (Tang et al., 2018).
2.2. Mitochondrial localization and membrane anchoring.
Mitochondrial localization and membrane anchoring domains in tafazzin are of critical importance to its role in cardiolipin remodeling. In the H9c2 rat cardiomyocyte cell line, TAZ encodes two peptides external to the active site of the tafazzin protein that act independently to direct it to mitochondria (Dinca et al., 2018). The first of these sequences, encoded in exon 3 and spanning residues 84-95 on the tafazzin protein, targets exclusively to mitochondria, while the second, encoded in exon 7/8 and forming residues 185-220, also targets other cytosolic compartments (Dinca et al., 2018). The yeast TAZ1 orthologue is homologous to human TAZ and has been used extensively to study the structure of tafazzin, its function, and in modeling Barth syndrome (Vaz et al., 2003; Gu et al., 2004; Ma et al., 2004; Claypool et al., 2006). In yeast, tafazzin has been shown to localize to membrane leaflets facing the intermembrane space (IMS) between the IMM and OMM, where it associates peripherally due to its lack of a transmembrane domain (Gawrisch, 2012; Abe et al., 2016). A hydrophobic sequence from residues 215-232 in yeast tafazzin confers its characteristic interfacial anchoring behavior in both the IMM and OMM (Herndon et al., 2013). Together, the translocase of the outer membrane (TOM) and the translocase of the inner membrane (TIM) facilitate tafazzin’s movement across and insertion into the outer membrane, as well as its anchoring to inner membrane regions of intermediate density (Herndon et al., 2013).
2.3. TAZ mutations and effects on tafazzin structure.
In characterizing the mutations of a family of patients with Barth syndrome, many unique forms of tafazzin were identified based on differential splicing events, ranging in length from 129 to 292 amino acids and affecting regions throughout the protein (Bione et al., 1996). Many of the shorter forms of the protein lack a 30-residue hydrophobic N-terminus thought to contain a localization signal sequence, as well as modifications to the hydrophilic center of the protein in a 71 amino acid domain profuse with glycine and glutamic acid (Bione et al., 1996). Mutations within the localization region result in mistargeting that directs the protein into inner membrane leaflets facing the matrix, rather than facing the IMS (Herndon et al., 2013). Whited et al. categorized TAZ mutations into 7 functional classes based on the pathogenic loss-of-function mechanisms of each mutation (Whited et al., 2013). The largest class of mutations, Class 1, contains frameshift and splice-site mutations along the length of the gene. Class 2 and 3 mutations are both found in the membrane anchor domain: Class 2 mutations, including V224R, V223D, and I226P variants, represent pleiotropic biochemical defects and often result in mitochondrial mistargeting, while Class 3 mutations (G230R) affect tafazzin macromolecular assembly. Class 4 mutations are composed primarily of missense mutations resulting in catalytically inactive tafazzin and Class 5 mutations, including L90P and N109V, encode hypomorphic alleles which retain transacylase activity. Class 6 mutations, including A88R and L148H, have folding and assembly defects, and Class 7 mutations result in temperature sensitive proteins that undergo activity loss before degradation. There is limited knowledge regarding a link between the different classes of TAZ mutations and disease severity. Whether tafazzin is rendered catalytically inactive, mistargeted, or incapable of membrane anchoring, there does not appear to be a clear distinction between phenotypic presentations of patients with different mutations. The diverse nature of TAZ mutations is clearly demonstrated in the expansive database maintained and regularly updated by the Barth Syndrome Foundation, which actively collects new data from healthcare professionals on both pathogenic and benign variants (https://barthsyndrome.org/research/tazdatabase.html). Mutations along the length of the TAZ gene, their frequency, and their pathogenicity (benign, pathogenic, or unknown effect) are depicted in Figure 1. Exonic, pathogenic variants along the length of the tafazzin protein, their frequency, and the type of genetic mutation from which they arise (deletion, frame shift, point mutation, or stop codon) are represented in Figure 2, along with the primary protein domains extracted from the literature.
Figure 1. TAZ genetic mutations, frequency and pathogenicity.

The frequency of intronic and exonic mutations across the length of the TAZ gene are shown here, categorized by pathogenicity and mapped to the nucleotide (nt) position on the gene, with exons represented by thick grey bars below the x-axis. Patient mutation data was acquired from the Barth Syndrome Foundation TAZ database (https://barthsyndrome.org/research/tazdatabase.html).
Figure 2. Tafazzin domains, mutation frequency, and mutation type.

The frequency of pathogenic, exonic mutations across the length of the tafazzin protein are shown here, categorized by mutation type, with key functional domains of the tafazzin protein displayed below the x-axis. Tafazzin contains a transmembrane helix (TM helix) and a membrane anchor at positions 15-34 and 215-232, respectively. The acyltransferase active site spans 176 amino acids (aa) from residue 41-217, with His77 forming part of the His-Asp motif (HX4D). Mitochondrial targeting domains are encoded in exon 3 as well as exon 7/8, spanning protein residues 84-95 and 185-220, respectively. Patient mutation data was acquired from the Barth Syndrome Foundation TAZ database (https://www.barthsyndrome.org/research/tazdatabase.html).
3. Function
Tafazzin plays a critical role in cardiolipin remodeling, limits the structural diversity of CL molecular species, and restricts CL composition to two fatty acids, typically linoleic and oleic acids (Schlame, 2008). Tafazzin displays a preference for the transfer of linoleic acid (LA) from phosphatidylcholine (PC) to monolysocardiolipin (MLCL) and may be affected by and contribute to the negative curvature of the IMM and OMM (Schlame et al., 2017). Through its effects on CL, tafazzin impacts many aspects of mitochondrial structure-function, including inner membrane curvature, oxidative phosphorylation (OXPHOS), supercomplex formation, oxidative stress repair, apoptosis, and fission and fusion (Haines and Dencher, 2002; Schug and Gottlieb, 2009; Baile et al., 2014; Vernon et al., 2014; Frohman, 2015).
3.1. Transacylase activity.
Tafazzin is an acyl-specific transferase that catalyzes reversible acyl transfer reactions between phospholipids and lysophospholipids in a CoA-independent manner, playing a critical role in the deacylation-reacylation cycle of cardiolipin (Xu et al., 2006; Epand et al., 2015; Schlame et al., 2017). Generally, transacylases exhibit phospholipase activity and catalyze acylation and deacylation through a free enzyme acyl intermediate. Tafazzin, on the other hand, does not exhibit phospholipase activity, nor does it utilize the free enzyme acyl intermediate mechanism; it acylates and re-acylates, but deacylation occurs independently of tafazzin (Xu et al., 2006). After de novo synthesis of CL from phosphatidylglycerol by CL synthase (Crd1 in yeast, hCLS1 in humans) (Chen et al., 2006; Ye et al., 2016), the remodeling process is initiated with cardiolipin deacylation to form MLCL by the cardiolipin specific phospholipase Cld1 in yeast (Beranek et al., 2009) or the calcium-independent phospholipase A2 (iPLA2) in humans (Mancuso et al., 2007a; Mancuso et al., 2007b; Malhotra et al., 2009; Yoda et al., 2010; Hsu et al., 2013). In order for MLCL produced by Cld1 to be exposed to tafazzin in the IMS, it must be transported through a different and unknown remodeling step (Baile et al., 2013). In mammals, tafazzin functions along with other enzymes to achieve CL remodeling, including MLCL acyltransferase (MLCLAT), acyl-CoA:lysocardiolipin acyltransferase (ALCAT), and phospholipase (Ye et al., 2016). Remodeling by tafazzin adds an acyl residue to immature CL, most frequently in the form of a linoleoyl residue in humans (Houtkooper et al., 2009b; Minkler and Hoppel, 2010). Tafazzin reacylates MLCL in a single-step acyl group transfer reaction (Figure 3) from a variety of phospholipids (PL), including CL, PC, phosphatidylethanolamine (PE), and phosphatidic acid (PA). Thus, tafazzin effectively acts as a shuttle for specific acyl groups between different phospholipids (Xu et al., 2006; Schlame, 2013).
Figure 3. Mechanism of acyltransferase activity by tafazzin.
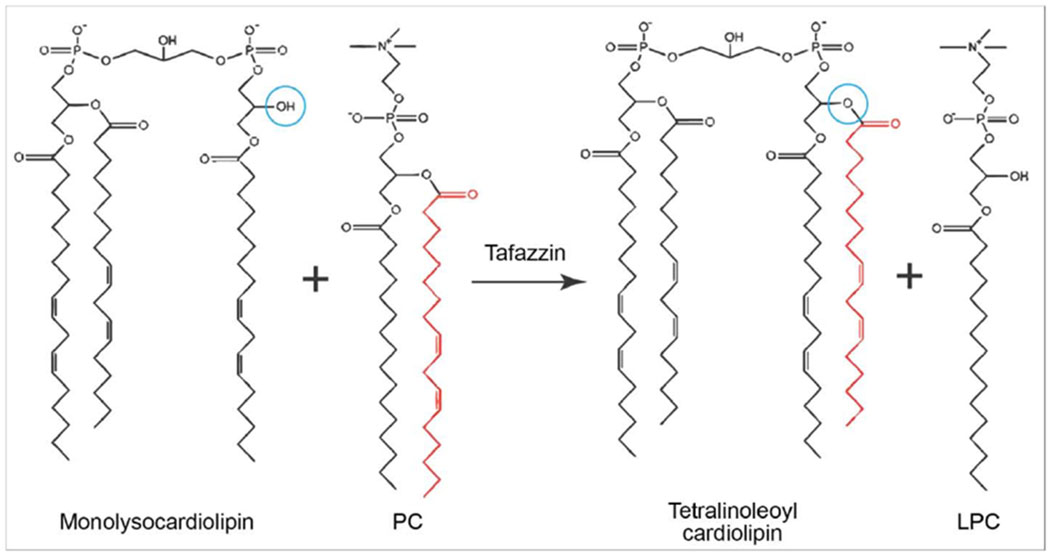
Tafazzin acts as a shuttle for specific acyl groups between different phospholipids to generate mature cardiolipin, Tafazzin transfers an acyl side chain from a phospholipid such as phosphatidylcholine (PC) to reacylate monolysocardiolipin (MLCL) in a single-step acyl group transfer reaction, resulting in the formation of lysophosphatidylcholine (LPC) and the mature tetralinoleoyl form of cardiolipin. The red acyl side chains indicate the acyl group that is transferred by tafazzin, and the blue circles indicate the location on MLCL where the new acyl chain is added to form the mature tetralinoleoyl cardiolipin.
3.2. Acyl specificity and sensing curvature.
Tafazzin shows a clear preference for the transfer of an LA group from PC to MLCL to form mature CL (Schlame et al., 2017). This remodeling process converts cardiolipin into a mature composition that contains a predominance of tetralinoleoyl moieties. This results in an enrichment of tetralinoleoyl-cardiolipin (CL4) in the IMM (Houtkooper et al., 2009b). Indeed, Xu et al. report that, in Drosophila melanogaster, tafazzin can catalyze acyl transfer using multiple substrates, yet has a preference for the transfer of linoleoyl groups from PC to MLCL at a rate 10 times greater than that of oleoyl groups and twenty times greater than that of arachidonoyl groups, indicating a clear predilection for CL and PC substrates (Xu et al., 2006). Conflicting explanations for this preference have been proposed, namely that tafazzin has an inherent enzymatic preference for specific acyl residues, or that it acts on the basis of energy minimization and is influenced by the surrounding mitochondrial microenvironment. Abe et al. propose that tafazzin exhibits acyl specificity for the PC to MLCL reaction, and that its function is predominately centered on the transacylation of unsaturated acyl PC to MLCL under any conditions (Abe et al., 2016). Schlame et al., on the other hand, propose the ‘thermodynamic remodeling’ hypothesis, whereby a perturbation of the lipid bilayer state and the physical properties of the lipid membrane determines tafazzin’s preference for specific acyl groups (Schlame et al., 2012; Epand et al., 2015). Schlame et al. propose that alternative phospholipases and acyltransferases (MLCAT and ALCAT), as well as the thermodynamic properties of lipids, provide the acyl specificity in CL remodeling and that tafazzin itself has no kinetic properties that suggest any sort of acyl-specificity. This mechanism proposes that since CL formation by tafazzin is reversible and has a minimal overall free energy, tafazzin’s role is to non-specifically transfer acyl groups among phospholipids to achieve optimal lipid composition and reduce the impact of membrane constraints (Schlame et al., 2012; Schlame et al., 2017).
According to the model proposed by Schlame et al. tafazzin specificity ultimately depends on the physical characteristics and packing properties of the lipid domain, including structural order and state. In D. melanogaster, tafazzin requires phospholipids that have a propensity to form non-bilayer phases such as HII phase, which is characterized by its negative curvature, disorganized acyl chains, and low packing order (Schlame et al., 2012; Schlame et al., 2017). Stable lipid bilayers were found to have the lowest rate of reaction by tafazzin, while lipids in the hexagonal or micellar phases, which were characterized by packing order changes due to positive or negative curvature, had significantly higher reaction rates. In addition to the rate of reaction, curvature was also shown to determine the specificity of acyl transfer (Epand et al., 2015). The cristae of the IMM have negatively curved lipid monolayers and a predominance of phospholipids with small polar head groups, such as CL, and asymmetric, unsaturated hydrocarbon chains, such as linoleic acid. CL and linoleic acid specificity may thus be driven by curvature segregation of phospholipids based on physical properties of the lipid domains, which causes tafazzin to transacylate phospholipids that are located in negatively curved monolayers (Gawrisch, 2012; Schlame et al., 2012). In Saccharomyces cerevisiae, however, Abe et al. determined that tafazzin can efficiently catalyze a transacylation reaction even in a highly ordered lipid bilayer domain. Further, they posit that tafazzin has a unique acyl chain specificity for the PC to MLCL reaction in which tafazzin acts selectively to transfer PC’s sn-2 acyl chain to MLCL’s sn-1 and sn-2 positions. They determined that these reactions can occur in any environment, regardless of packing order and thermodynamic considerations (Abe et al., 2016). These studies illustrate the propensity of tafazzin to transfer a linoleoyl group from PC to MLCL; however, more research into the specific mechanisms is required to fully understand the process and specificity of tafazzin’s actions. Further experiments with NMR analysis by groups such as Epand et al. may aid in elucidating these mechanisms due to its ability to probe curvature properties of lipid assemblies and observe structures with minor isotropic resonance (Epand et al., 2015).
3.3. Tafazzin and cardiolipin in mitochondrial structure and function.
Cardiolipin, modified by tafazzin, constitutes 13 - 20% of the total phospholipid mass and exhibits a cone-shaped structure that facilitates its distribution into mitochondrial cristae (Daum and Vance, 1997; Vernon et al., 2014). In the tafazzin-impaired fibroblasts of Barth syndrome patients, a greater proportion of saturated acyl chain substitution compromises this cone-shaped structure, and CL is heavily depleted with a concurrent accumulation of MLCL species (Oemer et al., 2018). CL assists in various aspects of OXPHOS, supporting the stability and function of the mitochondrial respiratory chain complexes through linkages between acyl chains (Houtkooper et al., 2009b). CL binds selectively to the c-rings of ATP synthase, which is required for the function and assembly of the ATP synthase. It also smooths the rotation (Mehdipour and Hummer, 2016) and facilitates dimerization for efficient ATP synthesis (Acehan et al., 2011). The structural properties of CL and its pK above 8 facilitate trapping proteins in the IMS. This is thought to achieve proton localization for ATP synthase function and minimization of pH fluctuations (Haines and Dencher, 2002). CL also interacts with other proteins such as the ATP/ADP translocase, pyruvate carrier, and carnitine carrier, assisted by glycerol bridges which enable flexibility for interaction with diverse surface shapes.
The lipid-to-protein mass ratio of OXPHOS complexes located in the mitochondrial cristae is 22:78 (Lotan and Nicolson, 1981), meaning that each complex is surrounded by just 40-400 lipid molecules (Xu et al., 2019). The molecular packing of lipids in a bilayer with such a high protein density causes elastic stress on the curvature of the membrane (Brown, 2017). Tafazzin remodeling is triggered by OXPHOS complex assembly so as to mitigate this stress and stabilize the membrane by generating CL species with reduced free energy (Xu et al., 2019). The OXPHOS complexes I, III, and IV also form supercomplexes within the mitochondrial cristae such as the I1III2IVn=1–4 “respirasome” (Schagger and Pfeiffer, 2000; Schagger, 2001; Stuart, 2008; Gu et al., 2016). CL is directly involved in the formation and maintenance of supercomplexes, providing structural support for trimer- and tetramerization (Zhang et al., 2002; Mileykovskaya and Dowhan, 2014). Furthermore, supercomplexes are disrupted and destabilized in Barth syndrome patients due to the loss of mature CL from the IMM (McKenzie et al., 2006).
Cardiolipin’s intimate association with the electron transport chain brings it into close proximity with reactive oxygen species (ROS) generated by OXPHOS complexes and which have been shown to target CL. The proximity and its enrichment in long-chain polyunsaturated fatty acid (PUFA) chains make CL susceptible to the attack. In the process of lipid peroxidation, highly reactive oxygen free radicals oxidize the fatty acid chains of CL to form lipid peroxides (Paradies et al., 2002). Oxidative damage leads to a loss of functional CL, a basis for mitochondrial dysfunction (Lesnefsky and Hoppel, 2008; Shi, 2010). CL remodeling removes and replaces acyl chains damaged by oxidative stress and is thought to play a key role in oxidative stress repair mechanisms (Baile et al., 2014) and the recovery of the normal oxidative functions of mitochondria (Baile et al., 2013). Aside from its damaging effects, ROS is also critical in mitochondrial and intracellular signaling, particularly in the context of cardioprotection from ischemia-reperfusion injury (Garlid, 2000). Phospholipids in the bilayer, such as cardiolipin, can be oxidized to form hydroperoxy fatty acids, which are hypothesized to carry the cardioprotective signal from mitochondria (Garlid et al., 2013).
CL forms membrane domains localized to negatively curved regions and induced by mitochondrial creatine kinase (mtCK) and cytochrome c that play critical roles in energy transfer, apoptosis, and functional recovery from ischemic insult (Laclau et al., 2001; Epand et al., 2007; Renner and Weibel, 2011). The microdomains occur at contact sites where the IMS narrows such that the IMM and OMM are positioned in close proximity to one another (Epand et al., 2007; Renner and Weibel, 2011; Pennington et al., 2018). The IMS at these contact sites is replete with mtCK, which induces their formation, recruits CL, and provides stabilization (Speer et al., 2005; Epand et al., 2007). Mature tetralinoleoyl-CL species generated by tafazzin remodeling are required for the formation of these domains and CL4 depletion disrupts their formation, which may explain the mitochondrial impairment observed in Barth syndrome and cardiac ischemia-reperfusion injury (Sparagna et al., 2007; Paradies et al., 2015; Pennington et al., 2018). Further, mtCK is functionally coupled to adenine nucleotide translocase (ANT) in the IMM to facilitate efficient energy transfer by shuttling high-energy phosphates from the mitochondria to the cytosol through the voltage-dependent anion channel (VDAC) of the outer membrane (Laclau et al., 2001; Saks et al., 2006). During ischemia, for example, this IMS structure-function is disrupted, reducing the functional coupling of mtCK and ANT and increasing the permeability of the OMM to ADP, thereby limiting energy transfer processes and exacerbating damage from an ischemic event such as a heart attack (Laclau et al., 2001). The cardioprotective ischemic preconditioning (IPC) protocol opens the mitochondrial ATP-sensitive K+-channel (mitoKATP), which causes matrix swelling and results in preservation of IMS volume, contact sites, and tight coupling between mtCK and ANT (Garlid, 2000; Laclau et al., 2001; Costa and Garlid, 2009). CL clustering at these contact sites is dependent on the octameric structure of mtCK, which readily binds to anionic phospholipids and may mediate intermembrane contact by binding to VDAC on the OMM (Epand et al., 2007).
Cardiolipin mediates apoptosis through its interactions with members of the Bcl-2 family, caspases, Bid, Bax, and Bak, with a direct impact on the apoptotic signaling cascade (Schug and Gottlieb, 2009). The total level of CL as well as the oxidative state of its acyl side chains directly impacts apoptosis by regulating cytochrome c mobilization; decreased CL content or oxidation of the normally unsaturated acyl side chains releases cytochrome c from the membrane (Schug and Gottlieb, 2009). This can be prevented with antioxidants (Petrosillo et al., 2003; Tyurina et al., 2006) and by the presence of mitochondrial redox proteins (Ran et al., 2004; Enoksson et al., 2005). Therefore, CL remodeling by tafazzin restores cytochrome c affinity for CL and its localization in the membrane by replacing oxidized fatty acids with non-oxidized acyl groups (Ye et al., 2016). Mobilized cytochrome c released from the mitochondrial membrane activates caspases 8 and 9, which cleave Bid to produce its truncated and active form, tBid (Kantari and Walczak, 2011). Once recruited by mitochondrial carrier homologue 2 (MTCH2), tBid activates apoptosis via Bax and Bak activation (Katz et al., 2012). Caspases 8 and 9, activate caspase 3, which drives apoptosis and inhibits ROS production (Brentnall et al., 2013). The apoptotic pathway is further amplified by this ROS inhibition, as it is a ROS signal that is responsible for protective mitoKATP opening that blocks the mitochondrial permeability transition (MPT) and prevents apoptosis (Costa and Garlid, 2008; Garlid et al., 2013).
Additionally, through interactions with both inner and outer mitochondrial membranes and proteins such as the GTPase Opa1, CL plays an important role in mitochondrial fission, fusion, and mitophagy (Frohman, 2015; Hsu et al., 2015). TAZ deficiency reduces the generation of mitophagosomes and prevents initiation of mitophagy, further exacerbating the already reduced function of mitochondrial populations in TAZ-deficient organisms (Hsu et al., 2015). Under mitochondria-stress conditions, CL has been shown to promote mitochondrial fusion and membrane tethering with L-Opa1 and trans-Opa1, respectively, further illustrating its multifunctional importance in mitochondrial form and function (Ban et al., 2017).
4. Clinical Significance: Barth syndrome
Considering its critical role in the construction and maintenance of the IMM, it is of little surprise that TAZ has been implicated in a broad spectrum of disease pathologies that impact the cardiovascular, neuromuscular, metabolic, and hematologic systems. TAZ mutations are specifically associated with the multi-faceted Barth syndrome. An X-linked autosomal recessive disorder also known as 3-Methylglutaconic Aciduria Type II (3MGA2) (Barth et al., 1983; Aprikyan and Khuchua, 2013; Clarke et al., 2013). Characterized initially by Barth et al. in 1983 (Barth et al., 1983) as a uniformly lethal disease that affects only males, it has now been found that the age distribution ranges between 0 to 49 years, and symptoms peak around puberty (Barth et al., 2004). At least one female patient with BTHS has been identified (Cosson et al., 2012). The Barth Syndrome Foundation reports that 151 living Barth patients have been identified up to 2012 and 10 new patients are diagnosed each year in the United States with no apparent racial or ethnic predilections. BTHS is estimated to appear in 1 out of every 300,000 to 400,000 live births, although predictions have suggested that the prevalence is actually closer to 1 out of every 140,000 live births as a result of the generally accepted notion that the disease is underdiagnosed (Cantlay et al., 1999; Barth Syndrome Foundation, 2019). In an effort to impose structure on otherwise unstructured clinical language contained in clinical case reports on BTHS (among other diseases), Caufield et al. extracted metadata from the reports and characterized patient symptomology using codes from the International Statistical Classification of Diseases and Related Health Problems (ICD-10) (Caufield et al., 2018b; Caufield et al., 2018a). The MitoCases platform (http://mitocases.org/) houses data on mitochondrial diseases, including Barth syndrome. Figure 4 displays the distribution of 997 instances of 206 unique ICD-10 codes represented across 54 clinical case reports covering 133 patients with BTHS, including top symptoms overall as well as top cardiovascular symptoms. Classifying Barth syndrome symptomology using ICD-10 codes has the potential to facilitate a greater understanding of the disease, its phenotypes, as well as to aid in diagnosis and treatment.
Figure 4. Complex symptomology of Barth syndrome codified by ICD-10.

Symptom occurrence codified using the 10th edition of the International Statistical Classification of Diseases and Related Health Problems (ICD-10) from 133 Barth syndrome patients described in 54 clinical case reports (please see References Cited: Clinical Case Reports). Panel (A) presents the full collection of 997 instances of 206 unique ICD-10 codes across all patients in these reports, grouped by disease category (Caufield et al., 2018b; Caufield et al., 2018a). Cardiovascular diseases and symptoms are the most highly represented among all ICD-10 categories (n = 272). Panel (B) highlights the top 10 symptoms from all ICD-10 categories. Panel (C) depicts the top 10 cardiovascular symptoms. All data is housed on the MitoCases platform (http://mitocases.org/) along with detailed metadata on the medical information contained in the text of each CCR.
BTHS diagnosis and treatment is complicated and frequently delayed due to the complexity and variation of disease presentation. Early cardiomyopathy and hypertrophy combined with neutropenia (a low neutrophil count in the blood) is a hallmark of the disease, but confirmation of the diagnosis typically relies on genetic analysis of TAZ. Over 160 mutations or errors in the TAZ gene have been identified in BTHS patients, with a wide variety of onset, progression, and severity (Aprikyan and Khuchua, 2013). 3-methylglutaconic acid (3-MGA) and CL content levels are often used to identify BTHS, but they are not always a reliable indicator, which led some to propose using an HPLC-tandem mass spectrometry blood spot assay to measure the ratio of MLCL to CL4. Although indirect, this highly specific biochemical measure of tafazzin function has the potential to provide a clinically valid method for BTHS diagnosis (Kulik et al., 2008; Houtkooper et al., 2009a; Bowron et al., 2015; Thompson et al., 2016). Combining biochemical analyses with physical tests, such as the 6-minute walk test (6MWT), may allow clinicians to determine the extent of the musculoskeletal impact and cardiac function in patients who survive infancy and those with unknown mitochondrial deficiencies. A combination of these procedures may help to improve diagnostic abilities and shape patient-specific treatment plans (Thompson et al., 2016).
4.1. Cardiovascular pathology.
Cardiomyopathy is a major characteristic of BTHS resulting from TAZ mutations. TAZ mutations lead to altered acyl chain composition and lipid peroxidation, and this can result in a failure of the sarcomeric action required to generate a sufficient power stroke. Disruption of the uniform contraction of sarcomeres can severely weaken the tissue, enlarge the left ventricular chamber, result in partial or incomplete contraction, and lead to decreased ejection volume. This results in the gradual thinning of the ventricular wall, stretching and dilation of cardiac chambers, and a cardiomyopathic phenotype of Barth syndrome, characterized by a weakened heart and diminished contractility (Barth et al., 2004; Ikon and Ryan, 2017). Among all patients, about 95% exhibited a history of cardiomyopathy, with 41.5% of all diagnosed cardiomyopathies occurring from birth to one month of age. Furthermore, statistical analysis revealed that cardiac function of patients declines over time (Roberts et al., 2012). Contrary to ATP depletion, Wang et al. suggest ROS as the main cause of cardiomyocyte dysfunction and cardiovascular impairments such as defective sarcomere assembly and contractile stress (Wang et al., 2014). The typical approach to treating the cardiovascular symptoms of Barth syndrome is to follow the treatment paradigm for heart failure. This includes: 1) diuretics for fluid retention (e.g., spironolactone or furosemide), 2) angiotensin-converting enzyme (ACE) inhibitors for vasodilation to reduce afterload (e.g., captopril), 3) positive inotropes to increase contractility and as an antiarrhythmic (e.g., digoxin), and 4) beta blockers to reduce heart rate (e.g., propranolol, carvedilol). Regular echocardiography is used to monitor cardiovascular function and ejection fraction (McCanta et al., 2008; Masarone et al., 2017). Severe forms of cardiac symptoms in BTHS patients necessitate heart transplantation. Spencer et al. reported nine out of 73 (12%) patients referred to the BTHS Registry (https://barthsyndromeregistry.patientcrossroads.org) who have undergone cardiac transplantation are alive at the last update (Roberts et al., 2012; Clarke et al., 2013). Transplantation is generally successful (Clarke et al., 2013); among four BTHS patients described in Mangat et al., one developed a severe infection but they did not show an increased rate of rejection and rated their quality of life as good (Ronghe et al., 2001; Mangat et al., 2007).
Cardiomyopathy in BTHS includes dilated cardiomyopathy (DCM) and left ventricular noncompaction (LVNC) (Barth et al., 2004). DCM is a specific type of cardiomyopathy characterized by an enlarged heart that is limited in function due to its inability to contract and pump blood efficiently (Araco et al., 2017; Soares et al., 2017). A patient with BTHS resulting from a c.83T>A mutation in tafazzin exhibited DCM with an ejection fraction of 30%, providing a direct association between the gene and DCM (Zapala et al., 2015). LVNC is a condition that exhibits prominent trabeculations and deep intertrabecular recesses in the left ventricle that resemble a spongy structure on the ventricular wall (Shemisa et al., 2013). One such case involves a family of 6 affected members that presented with LVNC with BTHS due to TAZ mutations (Bleyl et al., 1997). Isolated noncompaction of the ventricular myocardium (INVM) has also been found to affect the right ventricle and the interventricular septum (Bleyl et al., 1997; Barth et al., 2004). Despite the general occurrence of cardiomyopathy, there have been instances of BTHS caused by TAZ mutations with mild or late-onset cardiac involvement, as seen in Woiewodski et al. and again in Rigaud et al. Each discuss a cohort of BTHS patients exhibiting varying levels of cardiomyopathy, including two infantile patients who did not present with cardiomyopathy at the time of diagnosis (Woiewodski et al., 2017), another infantile patient whose autopsy revealed no cardiomyopathy, and one 12-year-old patient with no manifestation of cardiomyopathy (Rigaud et al., 2013). There is no clear structural or functional reason for the relatively mild presentations of certain patients, nor a direct mechanistic link between different mutations and disease presentations, representing an intriguing area of research necessary to glean a greater understanding of tafazzin and its role in disease.
4.2. Musculoskeletal pathology.
Although skeletal myopathy is often a typical characteristic of patients with disease-causing TAZ mutations, it manifests itself in a wide range of symptoms from nonexistent to severe. One of the most common musculoskeletal symptoms in BTHS patients is general and localized weakness. This includes overt muscle weakness and increased exertional fatigue due to skeletal myopathy and exaggerated by the cardiovascular complications associated with Barth syndrome (Spencer et al., 2011). Hypotonia, fatigue, and weakness can present early in life, persist, and may result in delayed motor development; most patients can walk unassisted by 2 years of age. Common phenotypes include short stature and facial dysmorphia and can extend to rarer phenotypes such as clubfoot (bilateral talipes) (Ades et al., 1993). Christodoulou et al. describes 6 cases of BTHS from four families with dysmorphic features, all of which exhibited persistent short stature. Four of the patients had also been found to exhibit similar myopathic facial appearances in conjunction with neuromuscular, cardiovascular and infectious symptoms (Christodoulou et al., 1994). A growth curve generated by examining 73 BTHS patients in Roberts et al. revealed a common down-shift in weight, length, and height relative to the normal population. Developmental delays are prevalent in BTHS patients with motor skills being the most affected, as indicated by a 65% prevalence of a delay in sitting up and a 71.6% delay in walking (Roberts et al., 2012).
Developmental delay has been treated with some success using cornstarch supplementation. This alternate source of glucose production ameliorates muscle wasting due to overnight fasting (Clarke et al., 2013). Other treatments, including oral arginine and carnitine supplementation, have centered on treating metabolic deficiencies, which improves cardiac function and muscle weakness in some patients (Ferreira et al., 1993; Rigaud et al., 2013; Vernon et al., 2014). However, while carnitine supplementation was initially offered as a treatment paradigm for all cases of BTHS (Ino et al., 1988), its effect has subsequently been called into question, and no formal assessment of the utility of arginine supplementation has been published. Thus far, both carnitine and arginine have demonstrated efficacy only in patients with those specific deficiencies (Ferreira et al., 1993; Rigaud et al., 2013; Vernon et al., 2014).
4.3. Neurological pathology.
Neurological complications tend to manifest as mild cognitive impairments in BTHS patients with TAZ mutations. While these patients were found to have a higher incidence of cognitive impairment (Mazzocco et al., 2007) and mild learning and speech difficulties (Roberts et al., 2012), many patients were found to have normal cognitive development, including a three-generation family with no cognitive impairment despite BTHS diagnosis (Ades et al., 1993). The limited neurologic involvement of BTHS is interesting given that tafazzin has been shown to play an important role in brain mitochondrial respiration and normal cognitive function (Cole et al., 2018). One postulate contends that the brain’s reliance on glucose, over tissues in the heart and liver that require high mitochondrial activity, allows the brain to have a more diverse and less tetralinoleoyl-dependent CL composition. Reducing the need for highly symmetric remodeled tetralinoleoyl-CL to achieve sufficient mitochondrial function may allow the brain to mitigate or avoid the detrimental effects of a tafazzin deficiency (Houtkooper et al., 2009b). Indeed, CL in the brain has higher amounts of arachidonic (AA) and docosahexaenoic (DHA) acids, distinct from the preference for tetralinoleoyl-CL seen in other tissues (Houtkooper et al., 2009b). Over 80% of CL in the liver and heart take the 18:2n-6 form, whereas the brain demonstrates less of a preference and has a higher concentration of saturated acyl chains, with only 48% polyunsaturated fatty acids and just 20% of its CL in the 18:2n-6 form (Corazzi and Roberti, 2009). It has also been proposed that the higher ROS scavenging capability of the brain, which is about 100 times higher than the rate of ROS generation (Starkov et al., 2014), may allow the brain to avoid the harmful effects more effectively than in other tissues even though it generates higher total levels of ROS (Cole et al., 2018).
4.4. Metabolic disorder.
3-methylglutaconic aciduria (3-MGA) is a major indicator of a variety of syndromes including BTHS, and is the result of mutations, including those in TAZ, that are linked to mitochondrial dysfunction (Su and Ryan, 2014). 3-MGA refers to increased levels of the organic acids 3-methylglutaconic acid, 3-methylglutaric acid, and 2-ethyl-hydracrylic acid in urine (Wortmann et al., 2012). BTHS patients typically demonstrate a large and consistent increase in the excretion of 3-MGA (Wortmann et al., 2013). A diagnosis of 3-MGA type II is synonymous with BTHS. While most BTHS patients exhibit varying severities of 3-MGA, a case report by Schmidt et al. describes a 15 year-old-boy with typical BTHS symptoms, such as dilated cardiomyopathy, but normal levels of organic acids, amino acids, and mucopolysaccharides in urine. Thus, there was no diagnosis of 3-MGA, despite a TAZ missense mutation in nucleotide 877 at exon 8 (Schmidt et al., 2004). Therapeutics such as riboflavin or coenzyme Q10 have been reported to show substantial improvement in some patients with 3-MGA (Wortmann et al., 2012). Overall, however, metabolic treatments vary between patients and are largely designed to target symptomatic deficiencies rather than the underlying cause of the disease (Clarke et al., 2013; Reynolds, 2015).
4.5. Hematologic pathology.
Neutropenia is one of the most frequent characteristics of BTHS caused by TAZ mutations, characterized by a decline in total number of circulating neutrophils accompanied by an increase in monocytes and eosinophils with no fluctuations in lymphocyte numbers (Clarke et al., 2013). Makaryan et al. found that neutropenia in BTHS is caused by a disruption of mitochondrial membrane potential as well as caspase-3 activation resulting in an increased rate of apoptosis of myeloid progenitor cells (Makaryan et al., 2009; Makaryan et al., 2012). Neutropenia is a particularly variant symptom, and can present itself in many different forms, from severe to mild, cyclical to non-cyclical, and intermittent to chronic (Steward et al., 2019). Severe chronic neutropenia (SCN), defined by an absolute neutrophil count of < 500/μL, is the most detrimental phenotype (Dale et al., 2006).
In a cohort study, Roberts et al. describe 73 patients with BTHS and indicate that 69.1% self-reported neutropenia with varying severity (Roberts et al., 2012). Ranging from a complete lack of neutrophils to a mild decline, neutropenia may be absent at presentation and change over the course of the disease in the same patient (Clarke et al., 2013). For instance, all seven members of a family with TAZ mutations exhibited no signs of neutropenia (Ferri et al., 2015), while another case of siblings with severe BTHS both exhibited intermittent neutropenia (Bowron et al., 2015). Including persistent or intermittent forms of neutropenia, nearly 90% of BTHS patients exhibit the symptom to some degree (Clarke et al., 2013), though it is mentioned in just over 65% of available clinical case reports on Barth syndrome (Figure 4). Neutropenia is an immune system deficiency that results in diminished response to invading organisms. Therefore, decreased defense mechanisms leads to serious bacterial infections including prolonged upper respiratory tract infections, mouth ulcers (chronic aphthous stomatitis) due to Candida infections, inflamed gums and perianal dermatitis, as well as sepsis and multi-organ failure, which are frequently treated by prophylactic antibiotics (Barth et al., 2004; Dale et al., 2006; Clarke et al., 2013). Among 73 BTHS patients in Roberts et al., 65% of those with neutropenia exhibited mouth ulcers, relative to only 35% of patients without neutropenia, while 28% had a history of pneumonia and 10% had a history of blood infections (Roberts et al., 2012). Granulocyte colony-stimulating factor (G-CSF) has been identified as an effective and safe treatment for SCN (Dale et al., 2006), leading to improvement in many BTHS patients with symptoms including aphthous ulcers, bacterial infections, and lethargy (Clarke et al., 2013).
4.6. Therapies in Barth syndrome.
Although several therapeutic strategies have proved successful in select clinical presentations, treatments are focused on treating the cardiovascular, musculoskeletal, and metabolic disorders, rather than the root cause of the disease. There is currently no cure for Barth syndrome (Barth Syndrome Foundation, 2019). Based on the observation that the fatty acid environment of cells impacts CL composition, ATP synthesis, membrane potential, and ROS production, dietary fatty acids have been suggested as a therapeutic strategy to target mitochondrial lipid metabolism and ameliorate effects on bioenergetics and cardiac function in mitochondrial diseases such as BTHS (Monteiro et al., 2013). It is unclear whether these treatments have significant effects in clinical practice. Direct modulation of CL content by lipid replacement therapy using CL nanodisks has also been tested in cell and animal models of Barth syndrome. Apoptosis induced by shRNA-mediated knockdown of TAZ in cultured HL60 myeloid progenitor cells (Makaryan et al., 2012) is attenuated by incubation with CL nanodisks and confers a significant increase in cellular CL content (Ikon et al., 2015). However, translation to an in vivo setting was unsuccessful, with no alteration in the CL profile of either wildtype mice or a TAZ knockdown mouse model of Barth syndrome (Ikon et al., 2018). Another study aimed to investigate whether overexpression of an alternate CL remodeling enzyme could restore CL in TAZ-deficient cells. Lymphoblasts from Barth syndrome patients transfected with MLCLAT1 saw increased CL levels, improved mitochondrial basal respiration and proton leak, and reduced superoxide production, but only partial compensation for respiratory function and no restoration of OXPHOS supercomplex formation (Taylor et al., 2012; Mejia et al., 2018). These results show some promise, but it remains to be seen whether they can be recapitulated in a live animal model.
Elevated ROS and oxidative stress have been proposed as significant culprits in Barth syndrome and the development of cardioskeletal myopathy in these patients (Xu et al., 2005; Dudek et al., 2013; Saric et al., 2015). In vitro studies of TAZ-deficient cardiomyocytes treated with the mitochondrially-targeted Mito-Tempo antioxidant demonstrated improved contractile function, cardiac hypertrophy, and cell death (He et al., 2014; Wang et al., 2014). Mice with TAZ deficiency (TAZKD) and mitochondria-specific overexpression of catalase, however, developed cardiomyopathy and muscle weakness similar to the Barth syndrome mouse, indicating that amelioration of oxidative stress is insufficient in the in vivo setting (Johnson et al., 2018).
Peroxisome proliferator-activated receptors (PPARs) and the PPAR-gamma coactivator-1 alpha (PGC-1α) are central to energy metabolism and bioenergetics in mitochondria, presenting opportunities for treatment in a variety of mitochondrial and metabolic disorders. Bezafibrate is a fibric acid derivative pan-agonist of PPAR signaling pathways that activates oxidative metabolism genes (Huang et al., 2017). In patients with dyslipidemia or metabolic syndrome, bezafibrate reduces triglyceride levels and the incidence of myocardial infarction (Arbel et al., 2016). It also significantly decreases HbA1c in diabetic patients with dyslipidemia (Teramoto et al., 2012). Because of its role in mitochondrial bioenergetics, it has been proposed as a potential treatment for Barth syndrome as well. In a TAZKD mouse model with isoproterenol (iso) treatment to induce more significant cardiac dysfunction, bezafibrate rescued iso-induced heart failure with marked increases in left ventricular fractional shortening and ejection fraction and prevention of the development of cardiomyopathy (Huang et al., 2017). However, the treatment also caused a significant reduction in CL content and increase in MLCL/CL ratio in both wild type and TAZ knockdown mice, a common biomarker for Barth syndrome. Concurrently, mitochondrial biogenesis was amplified drastically, as indicated by a two-fold increase in mtDNA content and mitochondrial citrate synthase activity in bezafibrate-treated hearts. Additionally, the dose used in the mouse model was 60-80 times greater than is typically prescribed in dyslipidemic humans. The modification of CL content and dosage discrepancy presents significant hurdles to determine the mechanism of action, further evaluate the importance of MLCL and CL concentrations, and conduct toxicity studies before any enrollment in clinical trials (Huang et al., 2017).
Gene replacement therapy presents another avenue of exploration that has the potential to address underlying tafazzin deficiencies resulting from the TAZ mutations that cause Barth syndrome. Recombinant adeno-associated virus (rAAV) vectors provide stable and long-lasting gene transfer to the nucleus of an organism’s cells using a non-pathogenic virus with minimal immune response (Schnepp et al., 2003). The successful application of an AAV-delivered gene therapy in spinal muscular atrophy also establishes an important precedent for the safety and efficacy of this approach in a clinic setting (Mendell et al., 2017). AAV serotype 9 (AAV9) demonstrates high affinity for the heart and skeletal muscle, making it ideal for application to Barth syndrome (Bish et al., 2008). In the TAZKD mouse model of Barth syndrome, an AAV9-TAZ vector with a desmin promotor resulted in significant TAZ gene and tafazzin protein expression levels in the heart and muscle and minimal levels in the liver (Suzuki-Hatano et al., 2019a). Measures of muscular strength and fatigue as well as whole body activity (e.g., exercise and distance travelled) of the treated mice improved significantly. Increased fractional shortening and ejection fraction as well as decreased heart weight/body weight ratio indicate significant improvements in cardiac function. Mitochondrial structure and function defects were ameliorated, with improved cristae and sarcomeric organization, greater numbers and size, as well as improved mitochondrial respiration and OXPHOS complex activity (Suzuki-Hatano et al., 2019a). In further studies, multiplex tandem mass tagging-based proteomics has provided a deeper mechanistic insight into the progression of Barth syndrome and its impact on critical proteins involved in cardiac development, heart failure, transcription, translation, and carnitine biosynthesis. (Suzuki-Hatano et al., 2019b). The striking result of AAV9-TAZ gene therapy across a range of treatment ages in the mouse model of Barth syndrome paints an optimistic picture for its potential as a future clinical option for these patients.
Elamipretide (MTP-131, SS-31, Bendavia) is a novel mitochondria-targeted tetrapeptide designed to temporarily bind to CL and protect it from oxidative damage by blocking CL-mediated conversion of cytochrome c into a peroxidase, thereby preserving cristae structure, promoting OXPHOS, and maintaining mitochondrial integrity (Birk et al., 2014; Szeto, 2014). In the canine model of intracoronary microembolization-induced chronic heart failure (Sabbah et al., 1991), long-term therapy with elamipretide was demonstrated to improve left ventricular ejection fraction, normalize key plasma biomarkers including tumor necrosis factor-alpha (TNF-α) and C-reactive protein (CRP), and reverse mitochondrial deficiencies in the heart (Sabbah et al., 2016) and skeletal muscle (Sabbah et al., 2019). In explanted human cardiac ventricular tissue from patients with a wide demographic range, elamipretide improved impaired mitochondrial function in heart failure and had no effect on normal mitochondrial function in nonfailing hearts. Additionally, supercomplex function was improved, but no change was observed in the activities of OXPHOS complexes II or V (Chatfield et al., 2019). In a clinical trial for patients with heart failure with reduced ejection fraction (HFrEF), elamipretide was safe, well-tolerated, and achieved significant decreases in left ventricular end-diastolic volume (LVEDV) and end-systolic volume (LVESV) in the highest dose cohort. Ejection fraction also improved in the treatment group as compared to those administered a placebo, though the measures did not reach statistical significance (Daubert et al., 2017). Elamipretide is currently in Phase 2 clinical trials (TAZPOWER) to treat Barth syndrome specifically, and recruitment is in progress for a Phase 3 clinical trial (MMPOWER-3) in patients with primary mitochondrial myopathies (ClinicalTrials.gov [Internet]). The previous animal studies and clinical trials in heart failure bode well for successful applications of Elamipretide in the Barth syndrome and mitochondrial myopathy patient populations.
5. Conclusions
Tafazzin is a transacylase responsible for remodeling cardiolipin in the mitochondrial membrane and plays an integral role in maintaining mitochondrial structure and function. The tight bends of the cristae in the inner membrane require a specific acyl profile, afforded by the activity of tafazzin. The protein has targeting and anchoring domains that direct it to the IMM and OMM and position it to face the IMS, build mature, tetra-linoleoyl cardiolipin species, and repair damaged membranes. Mutations in TAZ produce a dysfunctional or improperly localized protein that causes Barth syndrome, a multi-factorial and devastating disease that presents in infancy and results in heart failure, neutropenia, and musculoskeletal abnormalities. Current therapeutic paradigms are wide-ranging and attempt to treat the symptoms of individual systems. No cure exists for Barth syndrome, though there are a number of different treatments in development aimed at modulating metabolic processes, reducing oxidative stress, protecting CL from degradation, as well as conducting targeted gene-replacement of TAZ. Additional studies are necessary to fully characterize and understand the unique and integral role of tafazzin in mitochondrial biology and in the manifestation of Barth syndrome.
Highlights.
TAZ encodes the tafazzin transacylase responsible for cardiolipin (CL) remodeling.
Tafazzin localizes to the mitochondrial membranes for direct access to CL.
Tafazzin generates mature CL to maintain mitochondrial structure-function.
Mutations throughout TAZ cause the rare mitochondrial disease Barth syndrome.
There is no cure for Barth syndrome, but PPAR agonists, AAV9 gene therapy, CL protection, and protection from oxidative stress are in development.
Acknowledgements
The authors would like to acknowledge the support of National Institutes of Health (T32 EB16640 to Anders O. Garlid, R35 HL135772 to Peipei Ping), and the Theodore C. Laubisch endowment at UCLA to Peipei Ping. The authors are grateful to Keith Garlid and Hirsh Bhatt for their significant contributions and revisions to this manuscript, as well as Erik Lontok from the Barth Syndrome Foundation for his invaluable input.
This review and the corresponding Gene Wiki article are written as part of the Gene Wiki Review series – a series resulting from a collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Gene Wiki Initiative is supported by National Institutes of Health (R01 GM089820, U54 GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The corresponding Gene Wiki entry for this review can be found here: https://en.wikipedia.org/wiki/tafazzin/.
Abbreviations
- 3-MGA
3-methylglutaconic aciduria
- AA
arachidonic acid
- aa
amino acid
- rAAV
recombinant adeno-associated virus
- AAV9
adeno-associated virus serotype 9
- ACE
angiotensin-converting enzyme
- ALCAT
acyl-CoA:lysocardiolipin acyltransferase
- ANT
adenine nucleotide translocase
- BTHS
Barth syndrome
- CL
cardiolipin
- CL4
tetralinoleoyl-cardiolipin
- CMD3A
cardiomyopathy, dilated 3A (X-linked)
- DCM
dilated cardiomyopathy
- DHA
docosahexaenoic acid
- EFE2
endocardial fibroelastosis 2
- G3PAT
glycerol-3-phosphate acyltransferase
- G-CSF
granulocyte colony-stimulating factor
- HFrEF
heart failure with reduced ejection fraction
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- INVM
isolated noncompaction of the ventricular myocardium
- iPLA2
calcium-independent phospholipase A2
- LA
linoleic acid
- LVEDV
left ventricular end-diastolic volume
- LVESV
left ventricular end-systolic volume
- LVNC
left ventricular noncompaction
- mitoKATP
mitochondrial ATP-sensitive K+-channel
- MLCL
monolysocardiolipin
- MLCLAT
monolysocardiolipin acyltransferase
- mtCK
mitochondrial creatine kinase
- nt
nucleotide
- OMM
outer mitochondrial membrane
- OXPHOS
oxidative phosphorylation
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PL
phospholipids
- PPAR
peroxisome proliferator-activated receptor
- PGC-1α
PPAR-gamma coactivator-1alpha
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- SCN
severe chronic neutropenia
- TIM
translocase of the inner membrane
- TOM
translocase of the outer membrane
- VDAC
voltage-dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors have no competing interests to declare.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References Cited
- Abe M, Hasegawa Y, Oku M, Sawada Y, Tanaka E, Sakai Y and Miyoshi H, 2016. Mechanism for Remodeling of the Acyl Chain Composition of Cardiolipin Catalyzed by Saccharomyces cerevisiae Tafazzin. J Biol Chem 291, 15491–502. doi: 10.1074/jbc.M116.718510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acehan D, Malhotra A, Xu Y, Ren M, Stokes DL and Schlame M, 2011. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J 100, 2184–92. doi: 10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acehan D, Xu Y, Stokes DL and Schlame M, 2007. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest 87, 40–8. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ades LC, Gedeon AK, Wilson MJ, Latham M, Partington MW, Mulley JC, Nelson J, Lui K and Sillence DO, 1993. Barth syndrome: clinical features and confirmation of gene localisation to distal Xq28. Am J Med Genet 45, 327–34. doi: 10.1002/ajmg.1320450309. [DOI] [PubMed] [Google Scholar]
- Aprikyan AA and Khuchua Z, 2013. Advances in the understanding of Barth syndrome. Br J Haematol 161, 330–8. doi: 10.1111/bjh.12271. [DOI] [PubMed] [Google Scholar]
- Araco M, Merlo M, Carr-White G and Sinagra G, 2017. Genetic bases of dilated cardiomyopathy. J Cardiovasc Med (Hagerstown) 18, 123–130. doi: 10.2459/JCM.0000000000000432. [DOI] [PubMed] [Google Scholar]
- Arbel Y, Klempfner R, Erez A, Goldenberg I, Benzekry S, Shlomo N, Fisman EZ, Tenenbaum A and Group BIPS, 2016. Bezafibrate for the treatment of dyslipidemia in patients with coronary artery disease: 20-year mortality follow-up of the BIP randomized control trial. Cardiovasc Diabetol 15, 11. doi: 10.1186/s12933-016-0332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baile MG, Sathappa M, Lu YW, Pryce E, Whited K, McCaffery JM, Han X, Alder NN and Claypool SM, 2014. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J Biol Chem 289, 1768–78. doi: 10.1074/jbc.M113.525733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baile MG, Whited K and Claypool SM, 2013. Deacylation on the matrix side of the mitochondrial inner membrane regulates cardiolipin remodeling. Mol Biol Cell 24, 2008–20. doi: 10.1091/mbc.E13-03-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K and Ishihara N, 2017. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 19, 856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van ’t Veer-Korthof ET, Van der Harten JJ and Sobotka-Plojhar MA, 1983. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 62, 327–55. [DOI] [PubMed] [Google Scholar]
- Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz F.d.r.M. and Wanders RJA, 2004. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. American Journal of Medical Genetics 126A, 349–354. doi: 10.1002/ajmg.a.20660. [DOI] [PubMed] [Google Scholar]
- Barth Syndrome Foundation, 2019. What is Barth Syndrome? Barth Syndrome Foundation. [Google Scholar]
- Beranek A, Rechberger G, Knauer H, Wolinski H, Kohlwein SD and Leber R, 2009. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J Biol Chem 284, 11572–8. doi: 10.1074/jbc.M805511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA and Toniolo D, 1996. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 12, 385–9. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- Bione S, Tamanini F, Maestrini E, Tribioli C, Poustka A, Torri G, Rivella S and Toniolo D, 1993. Transcriptional organization of a 450-kb region of the human X chromosome in Xq28. Proc Natl Acad Sci U S A 90, 10977–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Bracken C, Warren JD and Szeto HH, 2014. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 171, 2017–28. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bish LT, Morine K, Sleeper MM, Sanmiguel J, Wu D, Gao G, Wilson JM and Sweeney HL, 2008. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum Gene Ther 19, 1359–68. doi: 10.1089/hum.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyl SB, Mumford BR, Brown-Harrison MC, Pagotto LT, Carey JC, Pysher TJ, Ward K and Chin TK, 1997. Xq28-linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet 72, 257–65. [PubMed] [Google Scholar]
- Bowron A, Honeychurch J, Williams M, Tsai-Goodman B, Clayton N, Jones L, Shortland GJ, Qureshi SA, Heales SJ and Steward CG, 2015. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J Inherit Metab Dis 38, 279–86. doi: 10.1007/s10545-014-9747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentnall M, Rodriguez-Menocal L, De Guevara RL, Cepero E and Boise LH, 2013. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol 14, 32. doi: 10.1186/1471-2121-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MF, 2017. Soft Matter in Lipid-Protein Interactions. Annu Rev Biophys 46, 379–410. doi: 10.1146/annurev-biophys-070816-033843. [DOI] [PubMed] [Google Scholar]
- Cantlay AM, Shokrollahi K, Allen JT, Lunt PW, Newbury-Ecob RA and Steward CG, 1999. Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 135, 311–5. [DOI] [PubMed] [Google Scholar]
- Caufield JH, Zhou Y, Garlid AO, Setty SP, Liem DA, Cao Q, Lee JM, Murali S, Spendlove S, Wang W, Zhang L, Sun Y, Bui A, Hermjakob H, Watson KE and Ping P, 2018a. Data from: A reference set of curated biomedical data and metadata from clinical case reports. Dryad Digital Repository. doi: 10.5061/dryad.r36cn90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caufield JH, Zhou Y, Garlid AO, Setty SP, Liem DA, Cao Q, Lee JM, Murali S, Spendlove S, Wang W, Zhang L, Sun Y, Bui A, Hermjakob H, Watson KE and Ping P, 2018b. A reference set of curated biomedical data and metadata from clinical case reports. Sci Data 5, 180258. doi: 10.1038/sdata.2018.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatfield KC, Sparagna GC, Chau S, Phillips EK, Ambardekar AV, Aftab M, Mitchell MB, Sucharov CC, Miyamoto SD and Stauffer BL, 2019. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl Sci 4, 147–157. doi: 10.1016/j.jacbts.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zhang XY and Shi Y, 2006. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J 398, 169–76. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou J, McInnes RR, Jay V, Wilson G, Becker LE, Lehotay DC, Platt BA, Bridge PJ, Robinson BH and Clarke JT, 1994. Barth syndrome: clinical observations and genetic linkage studies. Am J Med Genet 50, 255–64. doi: 10.1002/ajmg.1320500309. [DOI] [PubMed] [Google Scholar]
- Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI and Steward CG, 2013. Barth syndrome. Orphanet J Rare Dis 8, 23. doi: 10.1186/1750-1172-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool SM, McCaffery JM and Koehler CM, 2006. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol 174, 379–90. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov [Internet]. Identifier NCT03098797: A Trial to Evaluate Safety, Tolerability and Efficacy of Elamipretide in Subjects With Barth Syndrome (TAZPOWER). National Library of Medicine (US), Bethesda, MD. [Google Scholar]
- Cole LK, Kim JH, Amoscato AA, Tyurina YY, Bay RH, Karimi B, Siddiqui TJ, Kagan VE, Hatch GM and Kauppinen TM, 2018. Aberrant cardiolipin metabolism is associated with cognitive deficiency and hippocampal alteration in tafazzin knockdown mice. Biochim Biophys Acta Mol Basis Dis 1864, 3353–3367. doi: 10.1016/j.bbadis.2018.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazzi L and Roberti R, 2009. Lipids of Brain Mitochondria, in: Lajtha A, Tettamanti G and Goracci G (Eds.), Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids. Springer US, Boston, MA, pp. 199–221. doi: 10.1007/978-0-387-30378-9_8. [DOI] [Google Scholar]
- Cosson L, Toutain A, Simard G, Kulik W, Matyas G, Guichet A, Blasco H, Maakaroun-Vermesse Z, Vaillant MC, Le Caignec C, Chantepie A and Labarthe F, 2012. Barth syndrome in a female patient. Mol Genet Metab 106, 115–20. doi: 10.1016/j.ymgme.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Costa AD and Garlid KD, 2008. Intramitochondrial signaling: interactions among mitoKATP, PKCepsilon, ROS, and MPT. Am J Physiol Heart Circ Physiol 295, H874–82. doi: 10.1152/ajpheart.01189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AD and Garlid KD, 2009. MitoKATP activity in healthy and ischemic hearts. J Bioenerg Biomembr 41, 123–6. doi: 10.1007/s10863-009-9213-y. [DOI] [PubMed] [Google Scholar]
- Dale DC, Bolyard AA, Schwinzer BG, Pracht G, Bonilla MA, Boxer L, Freedman MH, Donadieu J, Kannourakis G, Alter BP, Cham BP, Winkelstein J, Kinsey SE, Zeidler C and Welte K, 2006. The Severe Chronic Neutropenia International Registry: 10-Year Follow-up Report. Support Cancer Ther 3, 220–31. doi: 10.3816/SCT.2006.n.020. [DOI] [PubMed] [Google Scholar]
- Daubert MA, Yow E, Dunn G, Marchev S, Barnhart H, Douglas PS, O’Connor C, Goldstein S, Udelson JE and Sabbah HN, 2017. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ Heart Fail 10. doi: 10.1161/CIRCHEARTFAILURE.117.004389. [DOI] [PubMed] [Google Scholar]
- Daum G and Vance JE, 1997. Import of lipids into mitochondria. Prog Lipid Res 36, 103–30. [DOI] [PubMed] [Google Scholar]
- Dinca AA, Chien WM and Chin MT, 2018. Identification of novel mitochondrial localization signals in human Tafazzin, the cause of the inherited cardiomyopathic disorder Barth syndrome. J Mol Cell Cardiol 114, 83–92. doi: 10.1016/j.yjmcc.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J, Cheng IF, Balleininger M, Vaz FM, Streckfuss-Bomeke K, Hubscher D, Vukotic M, Wanders RJ, Rehling P and Guan K, 2013. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res 11, 806–19. doi: 10.1016/j.scr.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Enoksson M, Fernandes AP, Prast S, Lillig CH, Holmgren A and Orrenius S, 2005. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem Biophys Res Commun 327, 774–9. doi: 10.1016/j.bbrc.2004.12.067. [DOI] [PubMed] [Google Scholar]
- Epand RF, Tokarska-Schlattner M, Schlattner U, Wallimann T and Epand RM, 2007. Cardiolipin clusters and membrane domain formation induced by mitochondrial proteins. J Mol Biol 365, 968–80. doi: 10.1016/j.jmb.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Epand RM, D’Souza K, Berno B and Schlame M, 2015. Membrane curvature modulation of protein activity determined by NMR. Biochim Biophys Acta 1848, 220–8. doi: 10.1016/j.bbamem.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Ferreira C, Thompson R and Vernon H, 1993. Barth Syndrome, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [Google Scholar]
- Ferri L, Donati MA, Funghini S, Cavicchi C, Pensato V, Gellera C, Natacci F, Spaccini L, Gasperini S, Vaz FM, Cooper DN, Guerrini R and Morrone A, 2015. Intra-individual plasticity of the TAZ gene leading to different heritable mutations in siblings with Barth syndrome. Eur J Hum Genet 23, 1708–12. doi: 10.1038/ejhg.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA, 2015. Role of mitochondrial lipids in guiding fission and fusion. J Mol Med (Berl) 93, 263–9. doi: 10.1007/s00109-014-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid AO, Jaburek M, Jacobs JP and Garlid KD, 2013. Mitochondrial reactive oxygen species: which ROS signals cardioprotection? Am J Physiol Heart Circ Physiol 305, H960–8. doi: 10.1152/ajpheart.00858.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid KD, 2000. Opening mitochondrial K(ATP) in the heart--what happens, and what does not happen. Basic Res Cardiol 95, 275–9. [DOI] [PubMed] [Google Scholar]
- Gawrisch K, 2012. Tafazzin senses curvature. Nat Chem Biol 8, 811–2. doi: 10.1038/nchembio.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wu M, Guo R, Yan K, Lei J, Gao N and Yang M, 2016. The architecture of the mammalian respirasome. Nature 537, 639–43. doi: 10.1038/nature19359. [DOI] [PubMed] [Google Scholar]
- Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ and Greenberg ML, 2004. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol 51, 149–58. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- Haines TH and Dencher NA, 2002. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett 528, 35–9. [DOI] [PubMed] [Google Scholar]
- He Q, Harris N, Ren J and Han X, 2014. Mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes. Oxid Med Cell Longev 2014, 654198. doi: 10.1155/2014/654198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath RJ and Rock CO, 1998. A conserved histidine is essential for glycerolipid acyltransferase catalysis. J Bacteriol 180, 1425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon JD, Claypool SM and Koehler CM, 2013. The Taz1p transacylase is imported and sorted into the outer mitochondrial membrane via a membrane anchor domain. Eukaryot Cell 12, 1600–8. doi: 10.1128/EC.00237-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata A, Yura K, Noguti T and Go M, 2011. Revisiting gap locations in amino acid sequence alignments and a proposal for a method to improve them by introducing solvent accessibility. Proteins 79, 1868–77. doi: 10.1002/prot.23011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata A, Yura K, Ohara O and Go M, 2015. Structural and functional analyses of Barth syndrome-causing mutations and alternative splicing in the tafazzin acyltransferase domain. Meta Gene 4, 92–106. doi: 10.1016/j.mgene.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Rodenburg RJ, Thiels C, van Lenthe H, Stet F, Poll-The BT, Stone JE, Steward CG, Wanders RJ, Smeitink J, Kulik W and Vaz FM, 2009a. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal Biochem 387, 230–7. doi: 10.1016/j.ab.2009.01.032. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Turkenburg M, Poll-The BT, Karall D, Perez-Cerda C, Morrone A, Malvagia S, Wanders RJ, Kulik W and Vaz FM, 2009b. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta 1788, 2003–14. doi: 10.1016/j.bbamem.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Hsu P, Liu X, Zhang J, Wang HG, Ye JM and Shi Y, 2015. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–52. doi: 10.1080/15548627.2015.1023984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YH, Dumlao DS, Cao J and Dennis EA, 2013. Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS One 8, e59267. doi: 10.1371/journal.pone.0059267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Powers C, Moore V, Schafer C, Ren M, Phoon CK, James JF, Glukhov AV, Javadov S, Vaz FM, Jefferies JL, Strauss AW and Khuchua Z, 2017. The PPAR pan-agonist bezafibrate ameliorates cardiomyopathy in a mouse model of Barth syndrome. Orphanet J Rare Dis 12, 49. doi: 10.1186/s13023-017-0605-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikon N, Hsu FF, Shearer J, Forte TM and Ryan RO, 2018. Evaluation of cardiolipin nanodisks as lipid replacement therapy for Barth syndrome. J Biomed Res 32, 107–112. doi: 10.7555/JBR.32.20170094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikon N and Ryan RO, 2017. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 52, 99–108. doi: 10.1007/s11745-016-4229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikon N, Su B, Hsu FF, Forte TM and Ryan RO, 2015. Exogenous cardiolipin localizes to mitochondria and prevents TAZ knockdown-induced apoptosis in myeloid progenitor cells. Biochem Biophys Res Commun 464, 580–5. doi: 10.1016/j.bbrc.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino T, Sherwood WG, Cutz E, Benson LN, Rose V and Freedom RM, 1988. Dilated cardiomyopathy with neutropenia, short stature, and abnormal carnitine metabolism. J Pediatr 113, 511–4. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Ferrara PJ, Verkerke ARP, Coleman CB, Wentzler EJ, Neufer PD, Kew KA, de Castro Bras LE and Funai K, 2018. Targeted overexpression of catalase to mitochondria does not prevent cardioskeletal myopathy in Barth syndrome. J Mol Cell Cardiol 121, 94–102. doi: 10.1016/j.yjmcc.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantari C and Walczak H, 2011. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta 1813, 558–63. doi: 10.1016/j.bbamcr.2011.01.026. [DOI] [PubMed] [Google Scholar]
- Katz C, Zaltsman-Amir Y, Mostizky Y, Kollet N, Gross A and Friedler A, 2012. Molecular basis of the interaction between proapoptotic truncated BID (tBID) protein and mitochondrial carrier homologue 2 (MTCH2) protein: key players in mitochondrial death pathway. J Biol Chem 287, 15016–23. doi: 10.1074/jbc.M111.328377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP and Ping P, 2012. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics 11, 1586–94. doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik W, van Lenthe H, Stet FS, Houtkooper RH, Kemp H, Stone JE, Steward CG, Wanders RJ and Vaz FM, 2008. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem 54, 371–8. doi: 10.1373/clinchem.2007.095711. [DOI] [PubMed] [Google Scholar]
- Laclau MN, Boudina S, Thambo JB, Tariosse L, Gouverneur G, Bonoron-Adele S, Saks VA, Garlid KD and Dos Santos P, 2001. Cardioprotection by ischemic preconditioning preserves mitochondrial function and functional coupling between adenine nucleotide translocase and creatine kinase. J Mol Cell Cardiol 33, 947–56. doi: 10.1006/jmcc.2001.1357. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ and Hoppel CL, 2008. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim Biophys Acta 1777, 1020–7. doi: 10.1016/j.bbabio.2008.05.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotan R and Nicolson GL, 1981. Plasma membranes of eukaryotes, Van Nostrand-Reinhold, Princeton, NJ. [Google Scholar]
- Ma L, Vaz FM, Gu Z, Wanders RJ and Greenberg ML, 2004. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem 279, 44394–9. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- Makaryan V, Dror Y and Aprikyan AA, 2009. Loss of Tafazzin (TAZ) Function and Accelerated Apoptosis of Human Bone Marrow Stem and Myeloid Progenitors in Barth Syndrome. Blood 114, 549. [Google Scholar]
- Makaryan V, Kulik W, Vaz FM, Allen C, Dror Y, Dale DC and Aprikyan AA, 2012. The cellular and molecular mechanisms for neutropenia in Barth syndrome. Eur J Haematol 88, 195–209. doi: 10.1111/j.1600-0609.2011.01725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M and Ren M, 2009. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci U S A 106, 2337–41. doi: 10.1073/pnas.0811224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso DJ, Han X, Jenkins CM, Lehman JJ, Sambandam N, Sims HF, Yang J, Yan W, Yang K, Green K, Abendschein DR, Saffitz JE and Gross RW, 2007a. Dramatic accumulation of triglycerides and precipitation of cardiac hemodynamic dysfunction during brief caloric restriction in transgenic myocardium expressing human calcium-independent phospholipase A2gamma. J Biol Chem 282, 9216–27. doi: 10.1074/jbc.M607307200. [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH and Gross RW, 2007b. Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem 282, 34611–22. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangat J, Lunnon-Wood T, Rees P, Elliott M and Burch M, 2007. Successful cardiac transplantation in Barth syndrome--single-centre experience of four patients. Pediatr Transplant 11, 327–31. PMID: 17430492. doi: 10.1111/j.1399-3046.2006.00629.x. [DOI] [PubMed] [Google Scholar]
- Masarone D, Valente F, Rubino M, Vastarella R, Gravino R, Rea A, Russo MG, Pacileo G and Limongelli G, 2017. Pediatric Heart Failure: A Practical Guide to Diagnosis and Management. Pediatr Neonatol 58, 303–312. doi: 10.1016/j.pedneo.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Mazzocco MM, Henry AE and Kelly RI, 2007. Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr 28, 22–30. doi: 10.1097/01.DBP.0000257519.79803.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCanta AC, Chang AC and Weiner K, 2008. Cardiomyopathy in a child with neutropenia and motor delay. Curr Opin Pediatr 20, 605–7. doi: 10.1097/MOP.0b013e32830a990a. [DOI] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR and Ryan MT, 2006. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol 361, 462–9. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Mehdipour AR and Hummer G, 2016. Cardiolipin puts the seal on ATP synthase. Proc Natl Acad Sci U S A 113, 8568–70. doi: 10.1073/pnas.1609806113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia EM, Zegallai H, Bouchard ED, Banerji V, Ravandi A and Hatch GM, 2018. Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J Biol Chem 293, 7564–7577. doi: 10.1074/jbc.RA117.001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, Kissel JT, Nagendran S, L’Italien J, Sproule DM, Wells C, Cardenas JA, Heitzer MD, Kaspar A, Corcoran S, Braun L, Likhite S, Miranda C, Meyer K, Foust KD, Burghes AHM and Kaspar BK, 2017. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377, 1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E and Dowhan W, 2014. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem Phys Lipids 179, 42–8. doi: 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkler PE and Hoppel CL, 2010. Separation and characterization of cardiolipin molecular species by reverse-phase ion pair high-performance liquid chromatography-mass spectrometry. J Lipid Res 51, 856–65. doi: 10.1194/jlr.D002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro JP, Oliveira PJ and Jurado AS, 2013. Mitochondrial membrane lipid remodeling in pathophysiology: a new target for diet and therapeutic interventions. Prog Lipid Res 52, 513–28. doi: 10.1016/j.plipres.2013.06.002. [DOI] [PubMed] [Google Scholar]
- NCBI, 2019. TAZ tafazzin [ Homo sapiens (human) ] Gene ID: 6901 National Center for Biotechnology Information. [Google Scholar]
- Neuwald AF, 1997. Barth syndrome may be due to an acyltransferase deficiency. Curr Biol 7, R465–6. [DOI] [PubMed] [Google Scholar]
- Oemer G, Lackner K, Muigg K, Krumschnabel G, Watschinger K, Sailer S, Lindner H, Gnaiger E, Wortmann SB, Werner ER, Zschocke J and Keller MA, 2018. Molecular structural diversity of mitochondrial cardiolipins. Proc Natl Acad Sci U S A 115, 4158–4163. doi: 10.1073/pnas.1719407115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Paradies V, Ruggiero FM and Petrosillo G, 2015. Cardiolipin alterations and mitochondrial dysfunction in heart ischemia/reperfusion injury. Clinical Lipidology 10, 415–429. [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M and Ruggiero FM, 2002. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 286, 135–41. [DOI] [PubMed] [Google Scholar]
- Pennington ER, Sullivan EM, Fix A, Dadoo S, Zeczycki TN, DeSantis A, Schlattner U, Coleman RA, Chicco AJ, Brown DA and Shaikh SR, 2018. Proteolipid domains form in biomimetic and cardiac mitochondrial vesicles and are regulated by cardiolipin concentration but not monolyso-cardiolipin. J Biol Chem 293, 15933–15946. doi: 10.1074/jbc.RA118.004948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM and Paradies G, 2003. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J 17, 2202–8. doi: 10.1096/fj.03-0012com. [DOI] [PubMed] [Google Scholar]
- Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts LJ 2nd, Herman B, Richardson A and Van Remmen H, 2004. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem 279, 55137–46. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- Renner LD and Weibel DB, 2011. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc Natl Acad Sci U S A 108, 6264–9. doi: 10.1073/pnas.1015757108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds S, 2015. Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach. J Multidiscip Healthc 8, 345–58. doi: 10.2147/JMDH.S54802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud C, Lebre AS, Touraine R, Beaupain B, Ottolenghi C, Chabli A, Ansquer H, Ozsahin H, Di Filippo S, De Lonlay P, Borm B, Rivier F, Vaillant MC, Mathieu-Dramard M, Goldenberg A, Viot G, Charron P, Rio M, Bonnet D and Donadieu J, 2013. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis 8, 70. doi: 10.1186/1750-1172-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AE, Nixon C, Steward CG, Gauvreau K, Maisenbacher M, Fletcher M, Geva J, Byrne BJ and Spencer CT, 2012. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A 158A, 2726–32. doi: 10.1002/ajmg.a.35609. [DOI] [PubMed] [Google Scholar]
- Ronghe MD, Foot AB, Martin R, Ashworth M and Steward CG, 2001. Non-Epstein-Barr virusassociated T-cell lymphoma following cardiac transplantation for Barth syndrome. Acta Paediatr 90, 584–6. [PubMed] [Google Scholar]
- Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S and Zhang K, 2016. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ Heart Fail 9, e002206. doi: 10.1161/CIRCHEARTFAILURE.115.002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Gupta RC, Singh-Gupta V and Zhang K, 2019. Effects of elamipretide on skeletal muscle in dogs with experimentally induced heart failure. ESC Heart Fail 6, 328–335. doi: 10.1002/ehf2.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET and Goldstein S, 1991. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol 260, H1379–84. doi: 10.1152/ajpheart.1991.260.4.H1379. [DOI] [PubMed] [Google Scholar]
- Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A and Wallimann T, 2006. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol 571, 253–73. doi: 10.1113/jphysiol.2005.101444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saric A, Andreau K, Armand AS, Moller IM and Petit PX, 2015. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front Genet 6, 359. doi: 10.3389/fgene.2015.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, 2001. Respiratory chain supercomplexes. IUBMB Life 52, 119–28. doi: 10.1080/15216540152845911. [DOI] [PubMed] [Google Scholar]
- Schagger H and Pfeiffer K, 2000. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19, 1777–83. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, 2008. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res 49, 1607–20. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, 2013. Cardiolipin remodeling and the function of tafazzin. Biochim Biophys Acta 1831, 582–8. doi: 10.1016/j.bbalip.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Schlame M, Acehan D, Berno B, Xu Y, Valvo S, Ren M, Stokes DL and Epand RM, 2012. The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat Chem Biol 8, 862–9. doi: 10.1038/nchembio.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Xu Y and Ren M, 2017. The Basis for Acyl Specificity in the Tafazzin Reaction. J Biol Chem 292, 5499–5506. doi: 10.1074/jbc.M116.769182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MR, Birkebaek N, Gonzalez I and Sunde L, 2004. Barth syndrome without 3-methylglutaconic aciduria. Acta Paediatr 93, 419–21. [DOI] [PubMed] [Google Scholar]
- Schnepp BC, Clark KR, Klemanski DL, Pacak CA and Johnson PR, 2003. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol 77, 3495–504. doi: 10.1128/jvi.77.6.3495-3504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT and Gottlieb E, 2009. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta 1788, 2022–31. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Shemisa K, Li J, Tam M and Barcena J, 2013. Left ventricular noncompaction cardiomyopathy. Cardiovasc Diagn Ther 3, 170–5. doi: 10.3978/j.issn.2223-3652.2013.05.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, 2010. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J Biomed Res 24, 6–15. doi: 10.1016/S1674-8301(10)60003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P, Rocha G, Pissarra S, Soares H, Flor-de-Lima F, Costa S, Moura C, Doria S and Guimaraes H, 2017. Neonatal dilated cardiomyopathy. Rev Port Cardiol 36, 201–214. doi: 10.1016/j.repc.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA and Moore RL, 2007. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res 48, 1559–70. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- Speer O, Back N, Buerklen T, Brdiczka D, Koretsky A, Wallimann T and Eriksson O, 2005. Octameric mitochondrial creatine kinase induces and stabilizes contact sites between the inner and outer membrane. Biochem J 385, 445–50. doi: 10.1042/BJ20040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CT, Byrne BJ, Bryant RM, Margossian R, Maisenbacher M, Breitenger P, Benni PB, Redfearn S, Marcus E and Cade WT, 2011. Impaired cardiac reserve and severely diminished skeletal muscle O(2) utilization mediate exercise intolerance in Barth syndrome. Am J Physiol Heart Circ Physiol 301, H2122–9. doi: 10.1152/ajpheart.00479.2010. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Andreyev AY, Zhang SF, Starkova NN, Korneeva M, Syromyatnikov M and Popov VN, 2014. Scavenging of H2O2 by mouse brain mitochondria. J Bioenerg Biomembr 46, 471–7. doi: 10.1007/s10863-014-9581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward CG, Groves SJ, Taylor CT, Maisenbacher MK, Versluys B, Newbury-Ecob RA, Ozsahin H, Damin MK, Bowen VM, McCurdy KR, Mackey MC, Bolyard AA and Dale DC, 2019. Neutropenia in Barth syndrome: characteristics, risks, and management. Curr Opin Hematol 26, 6–15. doi: 10.1097/MOH.0000000000000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart RA, 2008. Supercomplex organization of the oxidative phosphorylation enzymes in yeast mitochondria. J Bioenerg Biomembr 40, 411–7. doi: 10.1007/s10863-008-9168-4. [DOI] [PubMed] [Google Scholar]
- Su B and Ryan RO, 2014. Metabolic biology of 3-methylglutaconic acid-uria: a new perspective. J Inherit Metab Dis 37, 359–68. doi: 10.1007/s10545-013-9669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki-Hatano S, Saha M, Rizzo SA, Witko RL, Gosiker BJ, Ramanathan M, Soustek MS, Jones MD, Kang PB, Byrne BJ, Cade WT and Pacak CA, 2019a. AAV-Mediated TAZ Gene Replacement Restores Mitochondrial and Cardioskeletal Function in Barth Syndrome. Hum Gene Ther 30, 139–154. doi: 10.1089/hum.2018.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki-Hatano S, Saha M, Soustek MS, Kang PB, Byrne BJ, Cade WT and Pacak CA, 2019b. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Mol Ther Methods Clin Dev 13, 167–179. doi: 10.1016/j.omtm.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, 2014. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171, 2029–50. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada T, Feese MD, Ferri SR, Kato Y, Yajima R, Toguri T and Kuroki R, 2004. Substrate recognition and selectivity of plant glycerol-3-phosphate acyltransferases (GPATs) from Cucurbita moscata and Spinacea oleracea. Acta Crystallogr D Biol Crystallogr 60, 13–21. doi: 10.1107/s0907444903020778. [DOI] [PubMed] [Google Scholar]
- Tang Y, Xia H and Li D, 2018. Membrane Phospholipid Biosynthesis in Bacteria, in: Cao Y (Ed.), Advances in Membrane Proteins: Part I: Mass Processing and Transportation. Springer Singapore, Singapore, pp. 77–119. doi: 10.1007/978-981-13-0532-0_4. [DOI] [Google Scholar]
- Taylor WA, Mejia EM, Mitchell RW, Choy PC, Sparagna GC and Hatch GM, 2012. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS One 7, e48628. doi: 10.1371/journal.pone.0048628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto T, Shirai K, Daida H and Yamada N, 2012. Effects of bezafibrate on lipid and glucose metabolism in dyslipidemic patients with diabetes: the J-BENEFIT study. Cardiovasc Diabetol 11, 29. doi: 10.1186/1475-2840-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WR, DeCroes B, McClellan R, Rubens J, Vaz FM, Kristaponis K, Avramopoulos D and Vernon HJ, 2016. New targets for monitoring and therapy in Barth syndrome. Genet Med 18, 1001–10. doi: 10.1038/gim.2015.204. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Kini V, Tyurin VA, Vlasova II, Jiang J, Kapralov AA, Belikova NA, Yalowich JC, Kurnikov IV and Kagan VE, 2006. Mechanisms of cardiolipin oxidation by cytochrome c: relevance to pro- and antiapoptotic functions of etoposide. Mol Pharmacol 70, 706–17. doi: 10.1124/mol.106.022731. [DOI] [PubMed] [Google Scholar]
- Vaz FM, Houtkooper RH, Valianpour F, Barth PG and Wanders RJ, 2003. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J Biol Chem 278, 43089–94. doi: 10.1074/jbc.M305956200. [DOI] [PubMed] [Google Scholar]
- Vernon HJ, Sandlers Y, McClellan R and Kelley RI, 2014. Clinical laboratory studies in Barth Syndrome. Mol Genet Metab 112, 143–7. doi: 10.1016/j.ymgme.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang DZ, Li K, Wang J, Wanders RJ, Kulik W, Vaz FM, Laflamme MA, Murry CE, Chien KR, Kelley RI, Church GM, Parker KK and Pu WT, 2014. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 20, 616–23. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R and Schwede T, 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whited K, Baile MG, Currier P and Claypool SM, 2013. Seven functional classes of Barth syndrome mutation. Hum Mol Genet 22, 483–92. doi: 10.1093/hmg/dds447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woiewodski L, Ezon D, Cooper J and Feingold B, 2017. Barth Syndrome with Late-Onset Cardiomyopathy: A Missed Opportunity for Diagnosis. J Pediatr 183, 196–198. doi: 10.1016/j.jpeds.2016.12.070. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Duran M, Anikster Y, Barth PG, Sperl W, Zschocke J, Morava E and Wevers RA, 2013. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis 36, 923–8. doi: 10.1007/s10545-012-9580-0. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Kluijtmans LA, Engelke UF, Wevers RA and Morava E, 2012. The 3-methylglutaconic acidurias: what’s new? J Inherit Metab Dis 35, 13–22. doi: 10.1007/s10545-010-9210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Anjaneyulu M, Donelian A, Yu W, Greenberg ML, Ren M, Owusu-Ansah E and Schlame M, 2019. Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin. Proc Natl Acad Sci U S A 116, 11235–11240. doi: 10.1073/pnas.1900890116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Malhotra A, Claypool SM, Ren M and Schlame M, 2015. Tafazzins from Drosophila and mammalian cells assemble in large protein complexes with a short half-life. Mitochondrion 21, 27–32. doi: 10.1016/j.mito.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Malhotra A, Ren M and Schlame M, 2006. The enzymatic function of tafazzin. J Biol Chem 281, 39217–24. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- Xu Y, Sutachan JJ, Plesken H, Kelley RI and Schlame M, 2005. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest 85, 823–30. doi: 10.1038/labinvest.3700274. [DOI] [PubMed] [Google Scholar]
- Ye C, Shen Z and Greenberg ML, 2016. Cardiolipin remodeling: a regulatory hub for modulating cardiolipin metabolism and function. J Bioenerg Biomembr 48, 113–23. doi: 10.1007/s10863-014-9591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda E, Hachisu K, Taketomi Y, Yoshida K, Nakamura M, Ikeda K, Taguchi R, Nakatani Y, Kuwata H, Murakami M, Kudo I and Hara S, 2010. Mitochondrial dysfunction and reduced prostaglandin synthesis in skeletal muscle of Group VIB Ca2+-independent phospholipase A2gamma-deficient mice. J Lipid Res 51, 3003–15. doi: 10.1194/jlr.M008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapala B, Platek T and Wybranska I, 2015. A novel TAZ gene mutation and mosaicism in a Polish family with Barth syndrome. Ann Hum Genet 79, 218–24. doi: 10.1111/ahg.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E and Dowhan W, 2002. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277, 43553–6. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
References Cited: Clinical Case Reports
- Adwani SS, Whitehead BF, Rees PG, Morris A, Turnball DM, Elliott MJ and de Leval MR, 1997. Heart transplantation for Barth syndrome. Pediatr Cardiol 18, 143–5. doi: 10.1007/s002469900135. [DOI] [PubMed] [Google Scholar]
- Aljishi E and Ali F, 2010. Barth syndrome: an X-linked cardiomyopathy with a novel mutation. Indian J Pediatr 77, 1432–3. doi: 10.1007/s12098-010-0222-y. [DOI] [PubMed] [Google Scholar]
- Alter P and Maisch B, 2007. Non-compaction cardiomyopathy in an adult with hereditary spherocytosis. Eur J Heart Fail 9, 98–9. doi: 10.1016/j.ejheart.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Ances BM, Sullivan J, Weigele JB, Hwang V, Messe SR, Kasner SE and Liebeskind DS, 2006. Stroke associated with Barth syndrome. J Child Neurol 21, 805–7. doi: 10.1177/08830738060210090901. [DOI] [PubMed] [Google Scholar]
- Bachou T, Giannakopoulos A, Trapali C, Vazeou A and Kattamis A, 2009. A novel mutation in the G4.5 (TAZ) gene in a Greek patient with Barth syndrome. Blood Cells Mol Dis 42, 262–4. doi: 10.1016/j.bcmd.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Baksiene M, Benusiene E, Morkuniene A, Ambrozaityte L, Utkus A and Kucinskas V, 2016. A novel intronic splice site tafazzin gene mutation detected prenatally in a family with Barth syndrome. Balkan J Med Genet 19, 95–100. doi: 10.1515/bjmg-2016-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PG, Van den Bogert C, Bolhuis PA, Scholte HR, van Gennip AH, Schutgens RB and Ketel AG, 1996. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratorychain abnormalities in cultured fibroblasts. J Inherit Metab Dis 19, 157–60. [DOI] [PubMed] [Google Scholar]
- Borna NN, Kishita Y, Ishikawa K, Nakada K, Hayashi JI, Tokuzawa Y, Kohda M, Nyuzuki H, Yamashita-Sugahara Y, Nasu T, Takeda A, Murayama K, Ohtake A and Okazaki Y, 2017. A novel mutation in TAZ causes mitochondrial respiratory chain disorder without cardiomyopathy. J Hum Genet 62, 539–547. doi: 10.1038/jhg.2016.165. [DOI] [PubMed] [Google Scholar]
- Bowron A, Honeychurch J, Williams M, Tsai-Goodman B, Clayton N, Jones L, Shortland GJ, Qureshi SA, Heales SJ and Steward CG, 2015. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J Inherit Metab Dis 38, 279–86. doi: 10.1007/s10545-014-9747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady AN, Shehata BM and Fernhoff PM, 2006. X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn 26, 462–5. doi: 10.1002/pd.1438. [DOI] [PubMed] [Google Scholar]
- Brion M, de Castro Lopez MJ, Santori M, Perez Munuzuri A, Lopez Abel B, Bana Souto AM, Martinez Soto MI and Couce ML, 2016. Prospective and Retrospective Diagnosis of Barth Syndrome Aided by Next-Generation Sequencing. Am J Clin Pathol 145, 507–13. doi: 10.1093/ajcp/aqw025. [DOI] [PubMed] [Google Scholar]
- Cardonick EH, Kuhlman K, Ganz E and Pagotto LT, 1997. Prenatal clinical expression of 3-methylglutaconic aciduria: Barth syndrome. Prenat Diagn 17, 983–8. [DOI] [PubMed] [Google Scholar]
- Christodoulou J, McInnes RR, Jay V, Wilson G, Becker LE, Lehotay DC, Platt BA, Bridge PJ, Robinson BH and Clarke JT, 1994. Barth syndrome: clinical observations and genetic linkage studies. Am J Med Genet 50, 255–64. doi: 10.1002/ajmg.1320500309. [DOI] [PubMed] [Google Scholar]
- Cosson L, Toutain A, Simard G, Kulik W, Matyas G, Guichet A, Blasco H, Maakaroun-Vermesse Z, Vaillant MC, Le Caignec C, Chantepie A and Labarthe F, 2012. Barth syndrome in a female patient. Mol Genet Metab 106, 115–20. doi: 10.1016/j.ymgme.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Dedieu N, Giardini A, Steward CG, Fenton M, Karimova A, Hsia TY and Burch M, 2013. Successful mechanical circulatory support for 251 days in a child with intermittent severe neutropenia due to Barth syndrome. Pediatr Transplant 17, E46–9. doi: 10.1111/petr.12027. [DOI] [PubMed] [Google Scholar]
- Fan Y, Steller J, Gonzalez IL, Kulik W, Fox M, Chang R, Westerfield BA, Batra AS, Wang RY, Gallant NM, Pena LS, Wang H, Huang T, Bhuta S, Penny DJ, McCabe ER and Kimonis VE, 2013. A Novel Exonic Splicing Mutation in the TAZ (G4.5) Gene in a Case with Atypical Barth Syndrome. JIMD Rep 11, 99–106. doi: 10.1007/8904_2013_228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri L, Dionisi-Vici C, Taurisano R, Vaz FM, Guerrini R and Morrone A, 2016. When silence is noise: infantile-onset Barth syndrome caused by a synonymous substitution affecting TAZ gene transcription. Clin Genet 90, 461–465. doi: 10.1111/cge.12756. [DOI] [PubMed] [Google Scholar]
- Ferri L, Donati MA, Funghini S, Cavicchi C, Pensato V, Gellera C, Natacci F, Spaccini L, Gasperini S, Vaz FM, Cooper DN, Guerrini R and Morrone A, 2015. Intra-individual plasticity of the TAZ gene leading to different heritable mutations in siblings with Barth syndrome. Eur J Hum Genet 23, 1708–12. doi: 10.1038/ejhg.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri L, Donati MA, Funghini S, Malvagia S, Catarzi S, Lugli L, Ragni L, Bertini E, Vaz FM, Cooper DN, Guerrini R and Morrone A, 2013. New clinical and molecular insights on Barth syndrome. Orphanet J Rare Dis 8, 27. doi: 10.1186/1750-1172-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsi V, Miglietti N, Lombardi A, Boccacci S, Utyatnikova T, Donati C, Squassabia L, Gazzola L, Bosio I, Borghi A, Grassi V, Notarangelo LD and Plebani A, 2014. Cardiomyopathy in a male patient with neutropenia and growth delay. Ital J Pediatr 40, 45. doi: 10.1186/1824-7288-40-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke SP, Gardner AB, Lombardi JP, Manning PB, Nelson DP, Towbin JA, Jefferies JL and Lorts A, 2012. Left ventricular noncompaction cardiomyopathy in Barth syndrome: an example of an undulating cardiac phenotype necessitating mechanical circulatory support as a bridge to transplantation. Pediatr Cardiol 33, 1430–4. doi: 10.1007/s00246-012-0258-z. [DOI] [PubMed] [Google Scholar]
- Huang SC, Wu ET, Chiu SN, Hwu WL, Wu MH and Wang SS, 2008. Mitral annuloplasty in an infant with Barth syndrome and severe mitral insufficiency: first case report and determination of annular diameter. J Thorac Cardiovasc Surg 136, 1095–7. doi: 10.1016/j.jtcvs.2008.01.031. [DOI] [PubMed] [Google Scholar]
- Huhta JC, Pomerance HH and Barness EG, 2005. Clinicopathologic conference: Barth Syndrome. Fetal Pediatr Pathol 24, 239–54. doi: 10.1080/15227950500405429. [DOI] [PubMed] [Google Scholar]
- Imai-Okazaki A, Kishita Y, Kohda M, Yatsuka Y, Hirata T, Mizuno Y, Harashima H, Hirono K, Ichida F, Noguchi A, Yoshida M, Tokorodani C, Nishiuchi R, Takeda A, Nakaya A, Sakata Y, Murayama K, Ohtake A and Okazaki Y, 2018. Barth Syndrome: Different Approaches toDiagnosis. J Pediatr 193, 256–260. doi: 10.1016/j.jpeds.2017.09.075. [DOI] [PubMed] [Google Scholar]
- Karkucinska-Wieckowska A, Trubicka J, Werner B, Kokoszynska K, Pajdowska M, Pronicki M, Czarnowska E, Lebiedzinska M, Sykut-Cegielska J, Ziolkowska L, Jaron W, Dobrzanska A, Ciara E, Wieckowski MR and Pronicka E, 2013. Left ventricular noncompaction (LVNC) and low mitochondrial membrane potential are specific for Barth syndrome. J Inherit Metab Dis 36, 929–37. doi: 10.1007/s10545-013-9584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsushima Y, Fujiwara I, Sakamoto O, Ohura T, Miyabayashi S, Ohnuma A, Yamaguchi S and Iinuma K, 2002. Normal pituitary function in a Japanese patient with Barth syndrome. Eur J Pediatr 161, 67–8. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Cheatham JP, Clark BJ, Nigro MA, Powell BR, Sherwood GW, Sladky JT and Swisher WP, 1991. X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr 119, 738–47. [DOI] [PubMed] [Google Scholar]
- Kim GB, Kwon BS, Bae EJ, Noh CI, Seong MW and Park SS, 2013. A novel mutation of the TAZ gene in Barth syndrome: acute exacerbation after contrast-dye injection. J Korean Med Sci 28, 784–7. doi: 10.3346/jkms.2013.28.5.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwin SM, Vinette KM, Schwartz SB, Funanage VL and Gonzalez IL, 2007. Multiple transmissions of Barth syndrome through an oocyte donor with a de novo TAZ mutation. Fertil Steril 87, 976.e5–7. doi: 10.1016/j.fertnstert.2006.07.1543. [DOI] [PubMed] [Google Scholar]
- Lindenbaum RH, Andrews PS and Khan AS, 1973. Two cases of endocardial fibroelastosis--possible x-linked determination. Br Heart J 35, 38–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man E, Lafferty KA, Funke BH, Lun KS, Chan SY, Chau AK and Chung BH, 2013. NGS identifies TAZ mutation in a family with X-linked dilated cardiomyopathy. BMJ Case Rep 2013. doi: 10.1136/bcr-2012-007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marziliano N, Mannarino S, Nespoli L, Diegoli M, Pasotti M, Malattia C, Grasso M, Pilotto A, Porcu E, Raisaro A, Raineri C, Dore R, Maggio PP, Brega A and Arbustini E, 2007. Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am J Med Genet A 143a, 907–15. doi: 10.1002/ajmg.a.31653. [DOI] [PubMed] [Google Scholar]
- Mazurova S, Tesarova M, Magner M, Houstkova H, Hansikova H, Augustinova J, Tomek V,Vondrackova A, Zeman J and Honzik T, 2013. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med Rep 114, 139–53. doi: 10.14712/23362936.2014.16. [DOI] [PubMed] [Google Scholar]
- McCanta AC, Chang AC and Weiner K, 2008. Cardiomyopathy in a child with neutropenia and motor delay. Curr Opin Pediatr 20, 605–7. doi: 10.1097/MOP.0b013e32830a990a. [DOI] [PubMed] [Google Scholar]
- Momoi N, Chang B, Takeda I, Aoyagi Y, Endo K and Ichida F, 2012. Differing clinical courses and outcomes in two siblings with Barth syndrome and left ventricular noncompaction. Eur J Pediatr 171, 515–20. doi: 10.1007/s00431-011-1597-0. [DOI] [PubMed] [Google Scholar]
- Rigaud C, Lebre AS, Touraine R, Beaupain B, Ottolenghi C, Chabli A, Ansquer H, Ozsahin H, Di Filippo S, De Lonlay P, Borm B, Rivier F, Vaillant MC, Mathieu-Dramard M, Goldenberg A, Viot G, Charron P, Rio M, Bonnet D and Donadieu J, 2013. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis 8, 70. doi: 10.1186/1750-1172-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronghe MD, Foot AB, Martin R, Ashworth M and Steward CG, 2001. Non-Epstein-Barr virusassociated T-cell lymphoma following cardiac transplantation for Barth syndrome. Acta Paediatr 90, 584–6. [PubMed] [Google Scholar]
- Ronvelia D, Greenwood J, Platt J, Hakim S and Zaragoza MV, 2012. Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol Genet Metab 107, 428–32. doi: 10.1016/j.ymgme.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugolotto S, Prioli MD, Toniolo D, Pellegrino P, Catuogno S and Burlina AB, 2003. Long-term treatment of Barth syndrome with pantothenic acid: a retrospective study. Mol Genet Metab 80, 408–11. [DOI] [PubMed] [Google Scholar]
- Sabater-Molina M, Guillen-Navarro E, Garcia-Molina E, Ballesta-Martinez MJ, Escudero F and Ruiz-Espejo F, 2013. Barth syndrome in adulthood: a clinical case. Rev Esp Cardiol (Engl Ed) 66, 68–70. doi: 10.1016/j.recesp.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Sakamoto O, Kitoh T, Ohura T, Ohya N and Iinuma K, 2002. Novel missense mutation (R94S) in the TAZ ( G4.5) gene in a Japanese patient with Barth syndrome. J Hum Genet 47, 229–31. doi: 10.1007/s100380200030. [DOI] [PubMed] [Google Scholar]
- Schmidt MR, Birkebaek N, Gonzalez I and Sunde L, 2004. Barth syndrome without 3-methylglutaconic aciduria. Acta Paediatr 93, 419–21. [DOI] [PubMed] [Google Scholar]
- Singh HR, Yang Z, Siddiqui S, Pena LS, Westerfield BH, Fan Y, Towbin JA and Vatta M, 2009. A novel Alu-mediated Xq28 microdeletion ablates TAZ and partially deletes DNL1L in a patient with Barth syndrome. Am J Med Genet A 149a, 1082–5. doi: 10.1002/ajmg.a.32822. [DOI] [PubMed] [Google Scholar]
- Steward CG, Newbury-Ecob RA, Hastings R, Smithson SF, Tsai-Goodman B, Quarrell OW, Kulik W, Wanders R, Pennock M, Williams M, Cresswell JL, Gonzalez IL and Brennan P, 2010. Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenat Diagn 30, 970–6. doi: 10.1002/pd.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney RT, Davis GJ and Noonan JA, 2008. Cardiomyopathy of unknown etiology: Barth syndrome unrecognized. Congenit Heart Dis 3, 443–8. PMID: 19037987. doi: 10.1111/j.1747-0803.2008.00226.x. [DOI] [PubMed] [Google Scholar]
- Takeda A, Sudo A, Yamada M, Yamazawa H, Izumi G, Nishino I and Ariga T, 2011. Barth syndrome diagnosed in the subclinical stage of heart failure based on the presence of lipid storage myopathy and isolated noncompaction of the ventric ventricular myocardium. Eur J Pediatr 170, 1481–4.doi: 10.1007/s00431-011-1576-5. [DOI] [PubMed] [Google Scholar]
- Thiels C, Fleger M, Huemer M, Rodenburg RJ, Vaz FM, Houtkooper RH, Haack TB, Prokisch H, Feichtinger RG, Lucke T, Mayr JA and Wortmann SB, 2016. Atypical Clinical Presentations of TAZ Mutations: An Underdiagnosed Cause of Growth Retardation? JIMD Rep 29, 89–93. doi: 10.1007/8904_2015_525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valianpour F, Wanders RJ, Overmars H, Vreken P, Van Gennip AH, Baas F, Plecko B, Santer R, Becker K and Barth PG, 2002. Cardiolipin deficiency in X-linked cardioskeletal myopathy and neutropenia (Barth syndrome, MIM 302060): a study in cultured skin fibroblasts. J Pediatr 141, 729–33. doi: 10.1067/mpd.2002.129174. [DOI] [PubMed] [Google Scholar]
- Vesel S, Stopar-Obreza M, Trebusak-Podkrajsek K, Jazbec J, Podnar T and Battelino T, 2003. A novel mutation in the G4.5 (TAZ) gene in a kindred with Barth syndrome. Eur J Hum Genet 11, 97–101. doi: 10.1038/sj.ejhg.5200926. [DOI] [PubMed] [Google Scholar]
- Wang J, Guo Y, Huang M, Zhang Z, Zhu J, Liu T, Shi L, Li F, Huang H and Fu L, 2017. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J Rare Dis 12, 26. doi: 10.1186/s13023-016-0562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woiewodski L, Ezon D, Cooper J and Feingold B, 2017. Barth Syndrome with Late-Onset Cardiomyopathy: A Missed Opportunity for Diagnosis. J Pediatr 183, 196–198. doi: 10.1016/j.jpeds.2016.12.070. [DOI] [PubMed] [Google Scholar]
- Yen TY, Hwu WL, Chien YH, Wu MH, Lin MT, Tsao LY, Hsieh WS and Lee NC, 2008. Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur J Pediatr 167, 941–4. doi: 10.1007/s00431-007-0592-y. [DOI] [PubMed] [Google Scholar]
- Yoo TY, Kim MR, Son JS, Lee R, Bae SH, Chung S, Kim KS, Seong MW and Park SS, 2016. Identification of a Novel De Novo Mutation of the TAZ Gene in a Korean Patient with Barth Syndrome. J Cardiovasc Ultrasound 24, 153–7. doi: 10.4250/jcu.2016.24.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapala B, Platek T and Wybranska I, 2015. A novel TAZ gene mutation and mosaicism in a Polish family with Barth syndrome. Ann Hum Genet 79, 218–24. doi: 10.1111/ahg.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]