

Abstract
Disruptor of Telomeric Silencing 1-Like (DOT1L), the sole histone H3 lysine 79 (H3K79) methyltransferase, is required for leukemogenic transformation in a subset of leukemias bearing chromosomal translocations of the Mixed Lineage Leukemia (MLL) gene, as well as other cancers. Thus, DOT1L is an attractive therapeutic target and discovery of small molecule inhibitors remain of high interest. Herein, we are presenting screening results for a unique focused library of 1,200 nucleoside analogs originally produced under the aegis of the NIH Pilot Scale Library Program. The complete nucleoside set was screened virtually against DOT1L, resulting in 210 putative hits. In vitro screening of the virtual hits resulted in validation of 11 compounds as DOT1L inhibitors clustered into two distinct chemical classes, adenosine-based inhibitors and a new chemotype that lacks adenosine. Based on the developed DOT1L ligand binding model, a structure-based design strategy was applied and a second-generation non-nucleoside DOT1L inhibitors was developed. Newly synthesized compound 25 was the most potent DOT1L inhibitor in the new series with an IC50 of 1.0 μM, showing 40-fold improvement in comparison with hit 9 and exhibiting reasonable on target effects in a DOT1L dependent murine cell line. These compounds represent novel chemical probes with unique non-nucleoside scaffold that bind and compete with the SAM binding site of DOT1L, thus providing foundation for further medicinal chemistry efforts and developing more potent compounds.
Keywords: Small-molecule inhibitors, Structure-based virtual screening, Histone methyltransferase, DOT1L, Synthesis, Molecular modeling
Graphical Abstract
1. INTRODUCTION
Within cells, DNA is condensed into chromatin by histone proteins.1 Covalent posttranslational modifications of histones such as methylation, acetylation, and ubiquitination of basic histone residues regulate the accessibility of gene loci activating or repressing their transcription.2 The combination of histone modifications across the genome creates an “epigenetic landscape” that maintains coordinated expression of transcriptional programs required for cell fate determination during development.3
Dysregulation of histone methylation leads to aberrant gene expression that is a driving force behind the development of various human cancers. Hence, the roles of methyltransferases in human disease remain an area of intense research especially relating to drug discovery pursuing the myriad of potential epigenetic targets.4 The histone H3 lysine 79 (H3K79) methyltransferase, Disruptor of Telomeric Silencing 1-Like (DOT1L),5 has been well characterized6 as an essential mediator of leukemias involving chromosomal translocations of the Mixed Lineage Leukemia (MLL) gene.6, 7-11 MLL translocations are among the most common translocations in hematologic malignancies leading to particularly aggressive forms of leukemia with poor prognosis.12 Several of the most common MLL-fusion proteins expressed because of MLL gene translocations, including ENL, AF9, and AF10, directly interact with DOT1L8, 13-18 and recruit DOT1L to MLL target genes including HOXA9 and MEIS1.19-21 Once recruited to MLL target genes, DOT1L methylation of H3K79 leads to increased gene expression, uncontrolled proliferation, and blocked differentiation resulting in leukemogenesis.6, 22
The role of DOT1L methylation in MLL-driven leukemia has led to further investigation of its role in other cancers.23 DOT1L promotes colon cancer stemness and increases tumorigenic potential by inducing expression of the core stem cell genes, NANOG, SOX2, and Pou51.24 H3K79, H3K4 methylation and H3 acetylation are linked to c-Myc transcription factor’s ability to recognize and bind target gene promoters.25 Furthermore, DOT1L interaction with c-Myc-p300 may be critical for promoting EMT/CSC leading to an aggressive phenotype in breast cancer.25-27 Overall, these studies suggest DOT1L may become a critical therapeutic target for combination therapy in several cancer types.28-30
Based on the reputed role of DOT1L in MLL and H3K79 methylation in cancer, there has been great interest in developing small molecule inhibitors and probes of DOT1L as well as other protein methyltransferases.31-35 DOT1L is an attractive target for inhibitor development because it has a natural small molecule binding partner, S-adenosylmethionine (SAM), the methyl donor in DOT1L catalysis. Hence, the design of SAM analogs is one obvious approach for drug targeting. There are, however, concerns about this approach considering that SAM is ubiquitous throughout methylation reactions in the one-carbon metabolic pathway and nucleoside-based inhibitors might be subject to common nucleoside metabolic and degradative pathways. Still, selectively targeting the SAM cofactor-binding site of DOT1L, as well as other histone methyltransferases, has been an attractive strategy for this family of enzymes, further inspired by the numerous examples of small molecule probes and drugs selectively targeting the ATPbinding site of specific oncogenic and other protein kinases.36
Structural information about the catalytic domain of DOT1L (1-416 amino acids) showed that the target has a unique structure with a protein fold similar to protein arginine methyltransferases, and absence of the SET domain conserved in other histone lysine methyltransferases.37 Several potent, SAM-competitive DOT1L inhibitors are reported to show selectivity versus other histone lysine and arginine methyltransferases.38-43 A common substructure of reported compounds is an adenosine moiety, which is potentially metabolically labile with attendant poor pharmacokinetics (PK). Utilizing a cyclopentane replacement of the ribose moiety showed some improvement in compound stability in plasma and liver microsome assays.44 Also, the first reported potent DOT1L inhibitor EPZ004777 has a substitution of carbon for N-7 resulting in a deazapurine nucleoside analog.38 Further attempts to improve the PK properties of this compound resulted in a more stable analog, EPZ-5676, with an adenosine scaffold and extensive modification of the substituents appended at the 5’-ribose position.42 Despite PK improvements, EPZ-5676 required administration via osmotic mini pump in order to maintain effective concentrations in vivo due to the metabolically labile adenosine scaffold and oxidative dealkylation at the basic amine attached at the 5’-position of the nucleoside.45 Alternative, nucleoside46-47 and non-nucleoside scaffolds, have been reported to potentially circumvent these issues.34, 48-58 Still, there remains significant interest in alternative, novel chemotypes for future development of DOT1L probes and inhibitors that demonstrate increased metabolic stability and high efficacy in vivo.
In this study, we applied a virtual screening strategy on a focused library of 1,200 nucleoside analogs designed as a nucleoside antibiotic-like library through the NIH Pilot Scale Library Program.59 A subset of this library contains basic 5’-amine linkages, suitable for mimicking the trivalent, positively charged sulfur atom in SAM. Other, non-nucleosides in this library have chemical features and structural motifs that may serve as nucleoside mimetics. This library provided the opportunity to screen adenosine-based as well as novel, small molecule ‘nucleoside-like’ scaffolds distinct from the current DOT1L clinical candidates and other reported scaffolds.33
The virtual screening strategy utilized the crystal structure of the DOT1L catalytic domain in complex with SAM (PDB: 1NW3)37 and a modelled complex between DOT1L and the first reported selective DOT1L inhibitor with cellular activity, EPZ004777.38 Biological evaluation of the obtained virtual screening hits led to the identification of new chemical scaffolds as DOT1L inhibitors. Among these, a class of second-generation non-nucleoside DOT1L inhibitors was optimized with improved enzymatic inhibition using structure-based design and molecular modeling studies.
2. RESULTS AND DISCUSSION
2.1. Discovery of novel chemical scaffolds as DOT1L inhibitors
2.1.1. Structure-based computational screening approach for discovery of novel chemical scaffolds as DOT1L inhibitors
To facilitate the structure-based virtual screening, two DOT1L structures were utilized: the crystal structure of the DOT1L catalytic domain in complex with SAM (PDB: 1NW3)37, and a structure with modelled binding mode of the first reported selective DOT1L inhibitor, a SAM analog with high potency (Ki 0.3 nM) and cellular activity, EPZ004777.38 At the time when we initiated the virtual screening, the structural data of EPZ004777 binding to DOT1L was not available and thus we modeled the complex structure. The strategy and applied methodology, as well as validation of the selected model complex structure are provided in the Supplemental Material (Supplementary Figure S1). The virtual screening strategy and biochemical evaluation of the hits is outlined in Figure 1. A focused library of 1,200 nucleoside and nucleoside-like analogs, prepared under the Pilot Scale Library Program of the NIH Roadmap initiative,59 was used and docked into the DOT1L - SAM crystal structure and the DOT1L - EPZ004777 model using Glide (version 5.9) in standard precision mode, applying extensive Monte Carlo conformational sampling. The top 50% of scored compounds were re-docked into both DOT1L structures using Glide in extra precision (XP) mode to enrich the molecular recognition, thus separating the active and inactive ligands and minimizing false positives.60 Out of 50% of the top scoring hits, 199 compounds were selected based on the DOT1L - SAM crystal structure and 95 compounds based on the DOT1L - EPZ004777 model. In total, 210 virtual hits were obtained and evaluated experimentally after combining the two sets obtained from docking into the two DOT1L structures with exclusion of duplicates.
Figure 1.
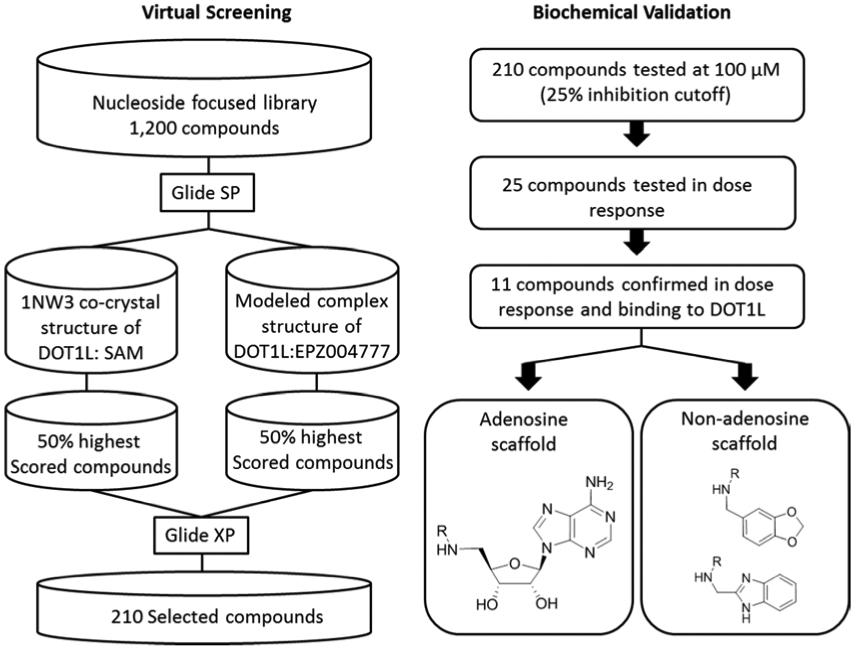
Schematic of the virtual screening strategy and biochemical validation of the identified DOT1L inhibitors.
2.1.2. Biological characterization and validation of the identified hits
The selected 210 in silico-screened compounds were tested in vitro for inhibitory activity against DOT1L by using a radio-isotope labeling histone methyltransferase (HMTase) assay with 3H-methyl-SAM as the methyl donor in the HMTase reaction of GST-DOT1L incubated with recombinant nucleosome as a substrate. From the in vitro DOT1L radioassay, 25 compounds inhibited DOT1L activity by > 25% at a concentration of 100 μM. The relatively low inhibition cutoff was selected in order to improve the likelihood of identifying inhibitors with a greater variety of novel chemical scaffolds for future development. A follow-up quantitative analysis measuring DOT1L methyltransferase activity resulted in the identification of two distinct classes of DOT1L inhibitors showing dose-dependent inhibition (Figure 2 and Supplementary Figure S2). Of these active compounds, seven were adenosine analogs similar to EPZ004777 with IC50 values ranging from 32 to 168 μM (Class I, compounds 1-7) and four compounds had a unique non-adenosine scaffold with IC50 values ranging from 83 to 710 μM (Class II, compounds 8-11). Interestingly, all eleven confirmed hits originated from docking into the DOT1L - EPZ004777 model, indicating the crucial role of the induced hydrophobic pocket in the modelled complex, which can accommodate bulkier non-polar groups and thus contribute to the binding. This observation further underlines the importance of using DOT1L structures with the opened/induced hydrophobic pocket region, a key element for the identification of new hits through our virtual screen. The putative binding modes of the Class I and II hits and their analysis showed that all have numerous DOT1L contacts recapitulating the SAM and EPZ004777 binding interactions and superimpose well with their binding site (Supplemental Material and Supplemental Figure S3). Importantly, the benzimidazole ring in 8 and the 1,3-benzodioxole group in compound 9 occupy overlapping space with the adenine group in EPZ004777 (Supplemental Figure S3D and S3E), suggesting possible replacements for an adenine in DOT1L inhibitors and potentially leading to improved metabolic stability.
Figure 2. Identified DOT1L inhibitors and potency of in vitro DOT1L HMTase activity inhibition.
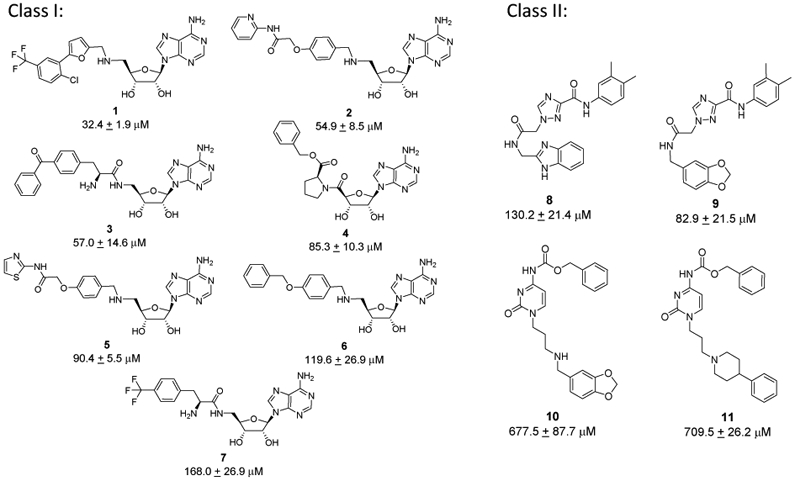
Chemical structures of two classes of identified DOT1L inhibitors: Class I - adenosine-based inhibitors and Class II compounds - non-adenosine chemical scaffolds.
Saturation transfer difference (STD) NMR and thermal stability shift assays were used to confirm the binding of the identified inhibitors to DOT1L (Figure 3 and Supplementary Figure S4). The proton signals in the 1H NMR spectrum of 2 were assigned (Figure 3A) and observed in the STD NMR spectrum (Figure 3B), which demonstrates that 2 binds to DOT1L. By comparing the relative signal intensity of individual protons between the STD NMR spectrum to the 1H NMR spectrum, we can determine which portions of a small molecule ligand bind to a protein target.61 Based on this comparison, we observed that the aromatic protons from the terminal pyridine and linker benzyl ring from the 5’-adenosine position both provide a high ratio of STD to the 1H NMR signal, indicating that they are critical mediators of the binding interactions between 2 and DOT1L. These results are consistent with the predicted binding pose of 2 (Supplementary Figure S3B). To further elucidate the binding site of 2, a competitive format of STD NMR was used. S-adenosylhomocysteine (SAH), which inhibits and binds to the SAM binding site of DOT1L, was added to the mixture of 2 and DOT1L protein in five-fold molar excess and additional peaks were observed in the 1H NMR spectrum from SAH protons (Figure 3C and Figure S5). In the presence of SAH, only a STD NMR signal for SAH was observed, while the STD NMR signal from 2 was diminished, indicating that 2 was competed off by SAH (Figure 3D and Figure S5). Overall, these results demonstrate that 2 binds to the SAM binding site as predicted through in silico methods. Binding of the identified DOT1L inhibitors was further assessed by a ligand-dependent thermal stability shift assay. Identified inhibitors bind to GST-DOT1L recombinant protein as indicated by stabilization of the protein, which increases the melting temperature of GST-DOT1L in the presence of compounds compared with DMSO (Figure 3E).
Figure 3. DOT1L inhibitors bind to the SAM binding site.

NMR spectra of 200 μM 2 in the presence of 5 μM GST-DOT1L. (A) 1H NMR of 2. Compound protons numbered according to corresponding 1H NMR signal. (B) STD NMR of 2 demonstrates binding to DOT1L. (C) 1H NMR with addition of 2 mM SAH. Additional peaks in spectra that do not correspond to 2 are from SAH. (D) STD NMR with addition of 2 mM SAH. Loss of STD NMR signal from 2 indicates that SAH competes with 2 for binding to the SAM binding site. (E) Thermal stability shift assay of 1μM GST-DOT1L in the presence of DMSO or 100 μM adenosine based DOT1L inhibitors (filled bars) or 200 μM non-adenosine DOT1L inhibitors (empty bars) showing increased melting temperature in the presence of compounds indicating binding to DOT1L. (F) 2 inhibits cellular H3K79 dimethylation in MLL-AF9 murine model cells over the time course of 3, 6 and 9 days.
To determine if the identified in vitro inhibitors of DOT1L are active in cells, we utilized a murine model cell line transformed with the MLL-translocation, MLL-AF9. This cell line was selected because it is well documented that MLL-AF9 transformed murine bone marrow cell lines require DOT1L for sustained proliferation.10, 20, 38 Therefore, we treated MLL-AF9 cells with 2 for 3 days and H3K79 methylation was analyzed by Western blot (Figure 3F). As predicted, methylation of H3K79 was reduced in the presence of 2 in a time-dependent manner. Inhibition of H3K79 dimethylation was most profound after nine days of inhibitor treatment consistent with other reports.38 Loss of H3K79 methylation requires prolonged treatment with inhibitors of DOT1L, as there is no currently known H3K79 demethylase, and the methylation turnover rate is similar to that of histones.42 Importantly, H3K4, H3K27, and H3K36 trimethylation states were not changed in the presence of 2 indicating that inhibition of HMTase activity is selective for DOT1L (Supplementary Figure S5). Together, these results indicate that the compounds identified as DOT1L inhibitors through virtual screening inhibit in vitro and cellular DOT1L HMTase activity and bind DOT1L in a SAM competitive manner, consistent with the applied virtual screening strategy.
2.2. Optimization of ‘non-nucleoside’ validated hit 9
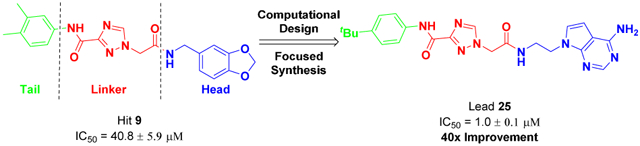
It is known that the adenosine moiety has potential issues, including metabolic lability and poor pharmacokinetics, and thus we focused on the unique Class II hit scaffold for further optimization. Related analogs, compounds 8 and 9, were the most potent non-nucleoside inhibitors, with 9 showing 1.5-fold better activity (IC50 = 82.9 μM) than 8 (Figure 2) in our initial assays. These two compounds only differ in an end group, benzodioxole (9) versus benzimidazole (8), suggesting that other, more optimal replacements may lead to further improvements in potency.
2.2.1. Design of Compound 9 analogs as DOT1L inhibitors
To facilitate the medicinal chemistry efforts, 9 was divided into head, linker and tail groups (Figure 4) mirroring the robust assembly chemistry.
Figure 4.

Dissection of validated hit 9 into three regions based on synthetic approaches and projected DOT1L binding.
Interactions contributed by each of these regions (head, linker, tail) were assessed in comparison with those formed by the highly potent co-crystallized EPZ004777 inhibitor, with the ultimate goal to identify regions in 9 needing improvement relative to an optimal and highly potent, SAM-based inhibitor. Utilizing the crystal structure of DOT1L with EPZ004777 bound in the SAM binding site (PDB: 4ER5)43, the predicted binding model of 9 was enhanced using the Induced Fit docking method (Schrodinger software). This method allows for protein flexibility during docking, and therefore is capable of reproducing small structural changes of active site residues induced by the binding of a ligand. Consistent with the virtual screening results, 9 occupies a region overlapping with the co-crystallized EPZ004777 inhibitor (Figure 5).
Figure 5. DOT1L modeling with 9 and complex structure with EPZ004777.

Protein carbons are colored gray, ligand carbons are colored green, all other atoms by atom type (O red, N blue, S yellow, H white). Interactions are indicated with dashed lines. (A) Co-crystal structure of EPZ004777 in complex with DOT1L (PDB code 4ER5). EPZ004777 head-group region forms hydrogen bonds with D222, F223(backbone) and K187 (backbone). In the linker region, the diol forms bidentate hydrogen bonding interactions with E186, the protonated tertiary nitrogen forms inductive interactions with Y128, and both nitrogens of the urea form hydrogen bonds to D161. Lastly, the tert-butylbenzene of the tail group region forms non-polar (not shown) and aromatic hydrogen bonding interactions. (B) Docked pose of 9. The tail group region maintains non-polar interactions similar to EPZ004777. In the linker region, the triazole forms strong pi-pi interactions with Y128, the oxygen of the carbonyl forms hydrogen bonding interactions with N241. 9 head group region forms hydrogen bonding interaction with G163 (backbone) and, in contrast to EPZ004777, forms hydrogen bonding interactions E129 and orients to form aromatic hydrogen bonding interactions with N241.
Analyzing the head group region, EPZ004777 occupies the adenine-binding region much more efficiently than hit 9, engaging in hydrogen bonding interactions and favorable π-stacking between its pyrrolopyrimidine ring and the electron rich phenyl of Phe223. Compound 9 contains a benzodioxole head group, lacking several of these favorable contacts. Both 9 and EPZ004777 form several comparable interactions in the linker region (Figure 5). The hydrophobic tail of 9 is accommodated in the same non-polar pocket induced upon binding of EPZ004777 as seen in the reported PDB crystal structure (Figure 5). In contrast, EPZ004777 has a bulkier 4-tert-butylphenyl tail group that better fills the induced hydrophobic pocket compared with tail region of 9. Hence, this region of 9 may accommodate tail groups longer/bulkier than the 3,4-dimethylphenyl. Solvent exclusion of larger hydrophobic tail groups should improve binding potency. Compound 9 lacks significant and optimal interactions in both the head and tail group binding regions. Hence, our optimization strategy focused on these two areas.
In a second-generation series based on the compound 9 scaffold, the reasonably efficient core linker was kept constant while the head and tail regions were substituted. A combinatorial library enumeration strategy was applied for the identification of the most promising head and hydrophobic tail groups from commercially available reagent libraries (Supplementary material and Supplementary Figure S6). Selected tail groups included that of the parent ligand (3,4-dimethylphenyl), a 4-tert-butylphenyl as in EPZ004777, a 6-methyl-benzimidazole and a hydrophobic, non-aromatic cyclohexyl tail. For head group replacements, a set of commercially available heterocyclic amines were enumerated onto the scaffold with distinct tail groups, followed by Glide XP docking of the full analogs into the DOT1L crystal structure (PDB: 4ER5). Based on predicted favorable interactions, reagent availability, synthetic considerations and structural diversity, our second-generation of inhibitors targeted small group of 14 analogs (including hit 9 as control), a strategy that combines four structurally diverse head and four tail groups.
2.2.2. Chemical synthesis of second-generation non-nucleoside DOT1L inhibitors
The synthesis of the second-generation potential DOT1L inhibitors (18-29, 31) evolved from the approach originally reported by Reynolds, et al. to prepare 9.59 The proclivity of 12 to undergo thermal decarboxylation at ambient temperature during attempted amidations (Scheme 1, Method A)62-63 prompted employment of trimethylaluminum-promoted aminolysis of methyl 1,2,4-triazole-3-carboxylate 15 (Scheme 1, Method B).64 The triazole-3-carboxamides 13a-13d were subsequently N-alkylated using ethyl bromoacetate,65-67 then saponified to afford 14a-14c. Acid 14d could not be prepared in satisfactory yield using this strategy due to poly-N-alkylation and difficulties in isolation and purification of the ionic polyaza carboxylic acid after saponification, vide infra.
Scheme 1.
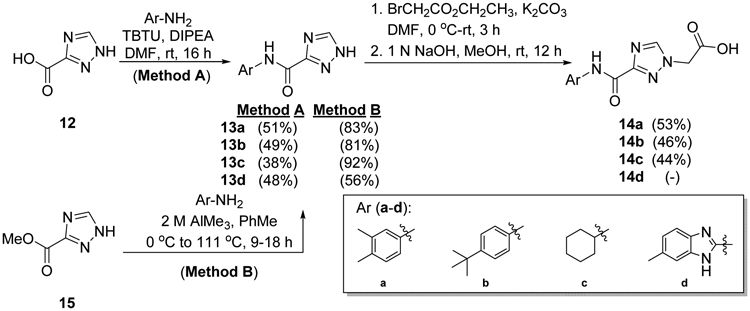
Preparation of 3-carbamoyl-1,2,4-triazolyl acetic acids 14a-14c.
Two N-aminoethyl head groups (D and E) were prepared via N-alkylation of commercial 7H-pyrrolo[2,3-d]pyrimidin-4-amine (16) and 7-deazahypoxanthine (17), the latter of which was not identified from our virtual screening efforts but was included due to its availability and varied electronics and functionality relative to head groups A-D. Treatment of both pyrrolopyrimidines with excess tert-butyl-2-(chloroethyl)carbamate in DMF followed by amine deprotection and generation of the free-based amine products using Amberlyst A-21 in EtOH afforded D and E as dry solids (Scheme 2).
Scheme 2.
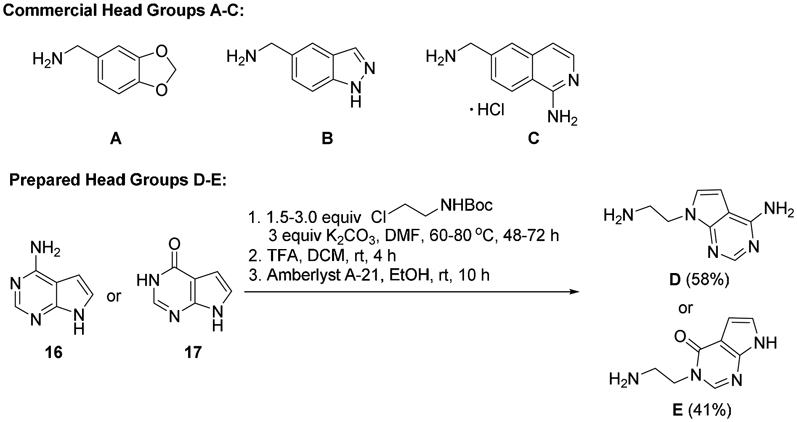
Commercial head groups A-C and route used to prepare N-aminoethyl pyrrolopyrimidine head groups D and E.
Commercial head groups A-C and the prepared head group D were each treated with 14a-14c using isobutyl chloroformate (IBCF) and N-methylmorpholine (NMM) in DMF at −10 °C.68 This provided markedly higher yields, cleaner reactions, and simplified isolations and purifications of targeted inhibitors 9, 18-20 and 22-29 (Table 1 Method B)69 relative to attempts employing TBTU (Table 1 Method A).59 Head group E was coupled only with 14a to synthesize 21 for a DOT1L inhibition comparison with original hit 9 and with its head group analogs B-D in products 18-20.
Table 1.
Amidations of 14a-14c with head group amines to form targeted inhibitors 9 and 18-29 and DOT1L methyltransferase Glo Assay Results.
 | |||||||
|---|---|---|---|---|---|---|---|
| product | Tail Group (TG) |
Head Group (HG) |
Method A Yield (%)a |
Method B Yield (%)a |
IC50 ± SD [μM]b |
||
| 9 | 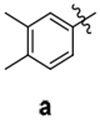 |
A | 25 | 94 | 40.8 ± 5.9 | ||
| 18 | B | 36 | 80 | 36.9 ± 1.7 | |||
| 19 | C | 23 | 58 | 69.2 ± 8.0 | |||
| 20 | D | 28 | 79 | 10.9 ± 2.6 | |||
| 21 | E | 20 | 66 | 137.8 ± 17.0 | |||
| 22 | 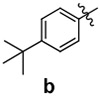 |
A | 22 | 93 | 23.5 ± 2.7 | ||
| 23 | B | 18 | 97 | 22.4 ± 2.3 | |||
| 24 | C | 46 | 83 | 19.5 ± 0.6 | |||
| 25 | D | 25 | 96 | 1.0 ± 0.1 | |||
| 26 |  |
A | 17 | 85 | > 100 | ||
| 27 | B | 16 | 63 | > 100 | |||
| 28 | C | 25 | 51 | > 100 | |||
| 29 | D | 18 | 84 | > 100 | |||
| 31 | 55.6 ± 3.3 | ||||||
| IC50 ± SD [nM]b | |||||||
| EPZ004777 | 0.86 ± 0.1 | ||||||
Isolated yields of purified products. Corresponding NMR spectra provided in Supplementary Material.
DOT1L methyltransferase-Glo assay.
A route avoiding formation of the carboxylic acid was explored to prepare benzodioxolyl head group derivative 31 (Scheme 3). Head group A was N-acylated in 76% yield using bromoacetylbromide and Et3N.70 Addition of bromoacetamide product 30 to 6-methylbenzimidazol-2-yl-1,2,4-triazole-3-carboxamide 13d in K2CO3 and DMF afforded a complex mixture of N-alkylated products from which polar product 31 was isolated and purified in 50% yield. Longer reaction times or higher temperatures lowered the yield of 31 due to increased formation of N-alkylated regioisomers and polyalkylation products.
Scheme 3.
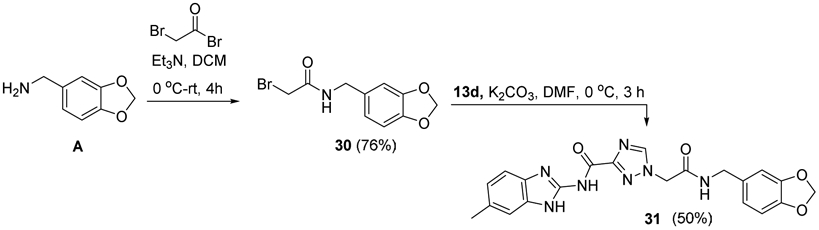
Synthesis of benzodioxolyl-6-methylbenzimidazolyl derivative 31.
2.2.3. Biochemical characterization and SAR of novel synthesized non-nucleoside compounds
A universal, bioluminescent, and homogenous methyltransferase (MTase Glo) assay, with recombinant DOT1L and core histones as substrate, was used to determine the DOT1L methyltransferase inhibition activity of novel non-nucleoside compounds. The structures and inhibitory activities of these compounds are shown in Table 1, together with those of EPZ004777, a potent and selective DOT1L inhibitor used as a positive control.
Analyzing the activity of the second-generation series and hit 9, showed that the compounds with a 4-t-butylphenyl as a tail group (22 - 25) have the most potent activity against DOT1L with IC50 values ranging from 1.0 to 23.5 μM (Supplementary Figure S7). Compounds bearing 3,4-dimethylphenyl tail group, 9, 18 – 21, showed IC50 values from 10.9 to 137.8 μM and compound 31 with methyl benzimidazole tail group had an IC50 of 55.6 μM. Interestingly, compounds with the cyclohexyl tail group, regardless of the head group (26 - 29), were not active up to 100 μM. These results showed that the 4-tert-butylphenyl tail group, which was predicted to occupy the adjacent induced fit hydrophobic binding pocket as in EPZ004777, provides the best activity. Compound 25 with a 4-tert-butylphenyl tail group and an N-aminoethyl-pyrrolopyrimidin-4-amine head group was the most potent DOT1L inhibitor in the new set with an IC50 of 1.0 μM showing 40-fold improvement in comparison with hit 9. Compounds 22 – 24 with head groups benzodioxolylmethanamine, indazolylmethanamine and aminomethylisoquinolin-1-amine, respectively, had considerably (20-fold) reduced activity in comparison with 25. The same pattern was observed in the subgroup having the 3,4-dimethylphenyl tail group, where 20 with an N-aminoethyl-pyrrolopyrimidin-4-amine head group was also the most potent DOT1L inhibitor of that subset. The direct binding of our most potent compound 25 was confirmed by thermal shift assay, showing dose-dependent stabilization of DOT1L and an increase of its melting temperature (Supplementary Figure S8).
2.2.4. Molecular modeling studies of non-adenosine series of DOT1L inhibitors
Molecular modeling studies were performed to further explore and understand the SAR findings of the new series of non-adenosine DOT1L inhibitors using docking and molecular dynamics (MD) simulations. Based on reported DOT1L inhibitors compiled from the literature and reported co-crystal structures in the Protein Data Bank, we identified five unique DOT1L binding modes, including structures characterized by Novartis and Epizyme in fragment-based screening and mechanism-guided inhibitor discovery, respectively. Co-crystal structures were used for building models: NOVA2, NOVA5, NOVA7, EPZ004777 and EPZ5676 (Supplementary Table 1). The NOVA2, NOVA5 and NOVA7 models were generated from Novartis’ reported structurally novel fragment screening hits: compound 2, compound 5 and compound 7, respectively.54, 55, 58 NOVA2 models an induced pocket adjacent to the SAM binding site.54 NOVA5 models interactions with Ser140 in a hydrophobic channel.55 NOVA7 models the fragment-linkage of these two binding sites.58 Both the EPZ47743 and EPZ567 models42 were generated from co-crystal structures of Epizyme’s DOT1L inhibitors of the same series, EPZ004777 and EPZ5676. To evaluate each docking model, non-adenosine series inhibitors were computationally prepared and docked in all binding models. Compounds were subsequently grouped by biological activity (pIC50 > 4.5, 4.0 < pIC50 < 4.5, and pIC50 < 4.0) and ranked based on docking score (Supplementary Table 2 and 3). Docking results demonstrated that the EPZ models most accurately reproduced biological activity rankings for the non-adenosine series based on hit 9, and provided consistent binding poses. Semi-quantitative structure-activity analysis of the docking results by characterizing compounds by head and tailgroup modifications, which significantly influenced DOT1L binding based on their IC50 values, showed that the EPZ477 model provided the best correlation between the docking score of compounds and their pIC50 values (Supplementary Figure S9). Furthermore, active compounds clustered together and the most active compound, 25, was the top docking compound in this model. Ultimately, the EPZ477 model was selected as most predictive and used for analysis of the interactions of this compound series.
Figure 6 illustrates the predicted binding modes and key interactions for compounds with a range of activities using the best model (Supplementary Table 2, 3). The most active compound bound favorably in the hydrophobic pocket via 4-t-butylaniline tail group and also positioned the head group for favorable hydrogen bonding interactions with Asp222 and the backbone of Phe223 in the adenosine binding site. In addition, the model predicts aromatic π interactions with Phe223 and Tyr128 and hydrogen bonds with Gly163 and Asn241. In contrast, for inactive compounds, the cyclohexylamine tail group appears too short to bind in the hydrophobic pocket while simultaneously interacting via the head group in the adenylyl binding site of SAM.
Figure 6. EPZ477 (best) docking model.

Docked pose of 25 in best docking model (EPZ004777). Protein carbons are colored gray, ligand carbons are colored green, all other atoms by atom type (O red, N blue, S yellow, H white). Dashed lines indicate favorable interactions. In contrast to 9, optimized 25 orients more similarly to the co-crystal ligand. The head group region reproduces pi-pi interactions with F223, inductive interactions with K187 and hydrogen bonding interactions with D222 and F223 (backbone). In the linker region, 25 forms hydrogen bonding interactions with G163 (backbone) and N241, as well as, pi-pi interactions with Y128. The tail group reproduces non-polar and aromatic-hydrogen interactions (D161) also seen in co-crystal ligand.
The analysis of docking results and predicted binding interactions for the other five models are provided in the supplemental material (Supplementary Figures S10-S14). Overall these results suggested that the closed binding site was too small to allow for the active compounds to adopt a favorable binding conformation due to significant (predicted) steric clashes between compounds and protein (NOVA7 model) or leaving the hydrophobic tail or head group solvent exposed in the NOVA5 and NOVA2 models, respectively (Supplementary Figures S10 - S12). Similar to the EPZ477 model, consistent poses were obtained in the EPZ567 model. However, the binding site was not as well resolved as in the EPZ477 model (Supplementary Figure S13), especially the SAM flexible binding loop, and EPZ477 outperformed the EPZ567 model in predicting the rank order of biological activity.
To further validate the binding pose of the most active compound 25 in the EPZ004777 model, we performed a 200 ns all atom explicit water molecular dynamic (MD) simulation (Supplementary Figure S15). The MD simulation results further support this binding hypothesis, including the hydrophobic interactions with the tert-butyl-phenyl tail group at the same time as hydrogen bonds and pi-stacking interactions with the head group and linger region. Based on this model, the addition of large hydrophobic tail groups would result in increased activity, because larger compounds preferentially adopted binding modes that aligned head groups in the SAM binding site, leveraging interactions that reproduced hydrogen bonding interactions with Phe223, Asp222, and Lys187 formed by adenine of SAM. Decreasing the size of the tail group (i.e. shortening the length of the compound) would result in less favorable binding, due to increased space in the pocket. In the same vein, head groups that more closely mimicked an adenylyl moiety generated more stabilizing interactions in the adenine binding site and had better docking scores suggesting increased binding. Based on these molecular modeling results, we proposed that the non-adenosine DOT1L inhibitors, designed based on hit 9, simultaneously bind in the co-factor binding site of DOT1L, as well as in an induced hydrophobic pocket adjacent to the SAM binding site.43
2.2.5. Cellular activity of improved and potent non-nucleoside compound 25
To test the cellular activity of the most potent non-nucleoside compound 25 and to demonstrate target specific phenotype, murine established leukemia cell lines harboring either the MLL-AF9 fusion or E2A-HLF fusion proteins were used.10, 20, 38 E2A-HLF cells are known to be independent of DOT1L’s presence or function for leukemogenesis and were used as a negative control. Figure 7A shows time- and dose-dependent activity of 25 on MLL-AF9 transformed cells at four different concentrations (1, 3, 10, and 30 μM). Consistent with the literature and requirement of prolonged treatment with epigenetic inhibitors, 25 works slowly in blocking the proliferation of the MLL-AF9 translocated cells with IC50 values of 8.8 μM, 4.2 μM and 1 μM on days 6, 9 and 11, respectively (Figure S11) and complete cell growth inhibition on day 11 at 30 μM (Figure 7A). Importantly, compound 25 selectively killed MLL-AF9 without showing any effect on the growth of E2A-HLF cells (Figure 7B). These findings are consistent with the effect of the EPZ004777 on the growth of these murine established leukemia cell lines which selectively kills only cells harboring MLL-AF9 fusion (Figure 7A and 7B), thus demonstrating that compound 25 gives a target specific phenotype. Consequently, the methylation of H3K79 was reduced in the presence of 25 in a dose-dependent manner on day 9 with complete inhibition at 30 μM and significant decrease at 10 and 3 μM (Figure 7C). Consistent with the inhibition of the cell proliferation results, the dose-dependent inhibition of H3K79 methylation was observed on day 6, with no significant changes in H3K4me3 and H4K20me2 (Figure S12). It is known that upon DOT1L enzymatic inhibition, cells harboring the MLL-AF9 fusion protein are driven to cell differentiation and apoptosis.38 Indeed 25 triggers dose-dependent cell differentiation as shown morphologically by Wright-Giemsa stain (Figure 7D), similar to EPZ00477.
Figure 7. Cellular activity of compound 25.

(A) 25 showing time- and dose-dependent killing of murine cell lines transformed with MLL-AF9 and (B) doesn’t show any effect on the growth of E2A-HLF cells, as EPZ004777, reported DOT1L inhibitor. (C) 25 inhibits cellular H3K79 dimethylation in MLL-AF9 murine model cells. (D) Wright-Giemsa stain of cells after treatment with 25 at various doses and EPZ00477 at 3 μM inducing cell differentiation (Olympus IX83 Inverted Microscope; Original magnification x400).
2.2.6. Profiling of selected inhibitors against a panel of methyltransferases
A select set of compounds based on diversity and bioactivity were screened in an independent contract lab (Reaction Biology Corporation, Malvern, PA) for inhibition of several methyltransferases including PKMTs and PRMTs (Table 2). In addition to MTs listed in Table 2, methyltransferase SUV39H2 was not inhibited by 20, 22, 23, 24 or 25 up to 250 μM. Methyltransferases PRMT1, PRMT4, PRMT5-MEP50 complex, and PRMT6 were not inhibited by 19, 24, 25 or 28 up to 250 μM. Compound 22 (omitted from table) showed reasonable activity versus DOT1L (Table 1), but it was inactive versus all MTs tested in the profiling assays. In general, our best DOT1L lead 25 showed very modest activity against DNMT1, PRMT3, PRMT5 (alone), and PRMT8 with selectivity (SI) for DOT1L (IC50 MT/IC50 DOT1L) ca. 250, 130, 175, and >250 respectively. The next best active versus DOT1L, 20, differs from 25 in the tail region (3,4-dimethylphenyl versus 4-tert-butylphenyl respectively) and shows very weak activity against only DNMT1. Thus, the tail region can potentially be altered to improve selectivity for DOT1L or to dial in activity for other MTs of interest. Likewise, the head group can alter both MT activity and selectivity. For example, compounds 19 and 20 differ in the head group and show different activity profiles, with 19 having modest activity versus PRMT7 and PRMT8, while 20 is very weakly active against DNMT1 only. Compounds 23, 24, and 25 all have the 4-tert-butylphenyl tail group with an indazole, isoquinoline, and deazaadenine head group, respectively. Compound 23 shows modest activity versus DOT1L and weak activity against EZH2 and G9a. Compounds 24 and 25, on the other hand, are more promiscuous although still somewhat selective for DOT1L. Compound 25 demonstrates sufficient aqueous solubility (687±26 μM) to indicate its selectivity profile is not solubility-related; however, the lower computed logS value of 24 suggests its reduced aqueous solubility could potentially affect its methyl transferase selectivity to some extent.71 Considering the weak activities against most profiled MTs, the selectivity results should not be overanalyzed, but the structure and activity differences may suggest that judicious alterations in both the head and tail groups could guide development of more active and selective compounds versus a variety of methyltransferases as probes for chemical biology applications. Changes in the linker region might also be useful for developing new probes, although we have yet to pursue that avenue. Our current hypothesis based on the DOT1L models and data is that this class of compound targets the SAM binding site, potentially in several MTs, and we are actively developing binding models for several clinically relevant MTs.
Table 2.
Profiling data for select inhibitors against a panel of Methyltransferases
| Methyltransferase | Compound Activity – IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| 19 | 20 | 23 | 24 | 25 | 28 | SAH | |
| DNMT1 (Poly dl-dC)a | ND | >250b | NA | NA | 248 | ND | 0.071 |
| EZH2 Complex (Core Histone)a | ND | NA | >250b | NA | NA | ND | 22.6 |
| G9a [Histone H3 (1-21)]a | ND | NA | >250b | 61.4 | NA | ND | 1.82 |
| SET 7/9 (Core Histone)a | ND | NA | NA | 79.8 | NA | ND | ND |
| PRMT3 (Histone H4)a | NA | NA | NA | 25.3 (41.1)* | 115 (149)* | NA | 0.76 (1.44)* |
| PRMT5 Alone (Histone H2A)a | NA | NA | NA | 227 (113)* | 187 (165)* | NA | ND |
| PRMT7 (GST-GAR)a | 69.4 | ND | ND | 61.5 | NA | NA | 0.11 |
| PRMT8 (Histone H4)a | 216 | ND | ND | 53.6 | >250b | 94.9 | 0.11 |
Screened by Reaction Biology Corporation (Malvern, PA) – for assays and assay conditions see http://www.reactionbiology.com/webapps/site/HMTProfiling.aspx. IC50 results were determined from 10-point dose response curves (test concentrations: 0.5 μM to 250 μM). IC50 values are single dose response determinations except
indicating a duplicate result. SAH was used as a reference compound throughout and data are presented for comparison in a common assay format.
Indicates assay substrate.
Indicates partial inhibition, but IC50 was not achieved up to 250 μM. NA = not active and ND = not determined.
3. CONCLUSION
We demonstrated the utility of applying structure-based virtual screening, followed by synthesis, biological activity, and molecular modeling studies for identifying and developing a non-adenosine series of DOT1L inhibitors. First, a nucleoside-focused library was computationally screened using two different models, which resulted in identification of 210 compounds for in vitro screening. Upon their biochemical and biophysical testing, 11 compounds in two distinct chemical classes were validated. All confirmed hits originated from the virtual screening selection obtained based on the DOT1L - EPZ004777 model, demonstrating the importance of using multiple protein structures in virtual screening to represent distinct and possible conformations of flexible regions in a targeted ligand binding site. Although the DOT1L inhibitors reported in this study are only modestly potent, they do include novel, non-nucleoside small molecules that may be readily modified using robust chemistry for further optimization. We expanded SAR around hit 9 through a small set of 14 second generation analogs designed to replace groups lacking favorable interactions with DOT1L as defined by the EPZ004777 PDB structure. Optimization led to 25 (IC50 = 1.0 μM) with a 40-fold greater DOT1L inhibition compared to 9. Analysis of these compounds showed that: (1) incorporating a bulky hydrophobic group (e.g. 4-t-butylphenyl) leads to a subgroup of inhibitors with improved potency, and (2) N-aminoethyl-pyrrolopyrimidin-4-amine may be utilized in future designs as a bioisostere of the adenosine moiety for the improvement of activity. Next, we developed a computational model to predict scaffold-DOT1L binding and which can be used to guide future optimization of this class of inhibitors. Finally, compound 25 demonstrated dose-dependent cellular activity against the proliferation of murine MLL-AF9 transformed cells, translating to a dose-dependent decrease of H3K79 dimethylation and induction of cell differentiation. We are currently developing binding models for other MTs based on profiling results and the hypothesis that this scaffold may associate and compete with the SAM binding site of DOT1L and possibly other MTs of interest.
4. METHODS
4.1. Biological Methods
4.1.1. Expression and purification of recombinant DOT1L.
The catalytic domain of DOT1L (1-420) was cloned into a pMCSG vector with an N-terminal His6–GST tag. GST-DOT1L (1-420) was expressed and purified in Escherichia coli strain BL21 (DE3) (Invitrogen). The proteins were induced with 200 μM IPTG at 16°C. The cells were harvested after 20 hr induction and resuspended in cold lysis buffer (50mM Tris HCl, pH 8.0, 150 mM NaCl, 0.01% B-mercaptoethanol, 20 mM imidazole, 10% glycerol, 1X protease inhibitor cocktail (Bimake)) and purified by affinity chromatography using Ni-agarose (Qiagen). The purified protein was dialyzed into storage buffer (50 mM Tris HCL, pH 8.0, 150 mM NaCl, 0.01% B-mercaptoethanol) and stored in 10% glycerol at −80 °C.
4.1.2. In vitro radio-isotope labeling DOT1L histone methyltransferase assay.
Enzymatic activity of DOT1L was assessed by incubating 125 nM of recombinantly expressed and purified GST-DOT1L in the presence of 125 nM (0.28 μCi) 3H-S-adenosyl methionine (NET155250UC; Perkin Elmer), and 0.7 μg of recombinant nucleosomes in HMTase buffer (20 mM Tris pH 7.9, 4 mM EDTA, 1 mM DTT, 0.01% Triton X-100) at a final volume of 26.5 μL. For determination of inhibitor potency, compounds were added to the reaction mixture prior to initiation of the HMTase reaction with 3H-SAM. The HMTase reaction was allowed to proceed for 1 hr at room temperature and was stopped by transferring 5 μL of reaction mixture to P81 filter paper. After drying, filter papers were washed three times with 50 mM NaHCO3 pH 9.0. Filter papers were then dried and radioactivity measured in vials with 10 mL liquid scintillation fluid on a Tri-Carb 2800 TR liquid scintillation counter (Perkin Elmer).
4.1.3. Preparation of recombinant nucleosomes.
Histone octamer was purified based on our optimized method.72 Briefly, for rapid purification of histone octamer, a polyhistidine-tag was added at the N-terminus of histone H2A. Inclusion bodies from all four histone proteins were mixed together for denaturation followed by histone octamer refolding in 2 M salt. Bacterial impurities were removed on the nickel-affinity resin. After cleaving off the polyhistidine-tag, histone octamer was separated from histone dimer and the tag on the Superdex S200 FPLC column. Plasmid DNA containing 12 copies of strong positioning Widom 601 DNA sequence73 was purified using Qiagen Plasmid Giga Kit. After excision from the plasmid, the Widom 601 fragment was purified by PEG fractionation. Purified histone octamer and DNA were mixed with 1:1 ratio and reconstituted by high to low salt dialysis as described previously.74
4.1.4. MTase-Glo DOT1L Methyltransferase Assay
The MTase-Glo methyltransferase assay (Promega) was optimized for testing DOT1L activity using white, flat bottom, non-treated, 384 well platers (Corning 3572) in an assay volume of 10 μL for component optimization and 11 μL for testing compounds. Luminescence was read using a Biotek Hi hybrid plate reader after a 1 hr incubation of SAH standard, a 30 min incubation with 10x MTase-Glo reagent and 30 min incubation with MTase-Glo Detection reagent. For the SAH standard curve, maximum luminescence signal based on concentration was calculated in reference to a buffer only blank using GraphPad Prism 7.0.
To determine the optimal GST-DOT1L concentration, the protein was prepared and titrated in reaction buffer (80 mM Tris HCl, pH 8.0, 200 mM NaCl, 4 mM EDTA, 12 mM MgCl2, 0.4 mg/mL BSA, 4 mM dithiothreitol (DTT) in a concentration range from 2.5 μM – 500 pM. Substrate was prepared with a final concentration of 0.45 μg/rxn calf thymus core histones (Sigma-Aldrich) in reaction buffer, as well as 200 nM co-factor SAM and DEPC treated water (Invitrogen). Five (5.0) μL of each solution at constant substrate and variable enzyme were incubated together for 1hr. Each reaction condition was tested in triplicate. After incubation with MTase-Glo reagent and detection solutions as noted above, luminescence was read using a Biotek Hi hybrid plate reader and plotted using a nonlinear regression in GraphPad Prism 7.0.
The compounds were tested in triplicate with 200 nM GST-DOT1L with fixed substrate conditions with a final DMSO concentration of 9% and overall final reaction volume of 11 μL. GST-DOT1L, prepared in the reaction buffer, was added to the wells. GST-DOT1L (5 μL) and compounds (1 μL) were briefly pre-incubated, followed by adding of 5 μL substrate and left to incubate for 1 hour. As a negative control, protein and substrate was added to 1 μL of DMSO (0% inhibition) and positive control with no enzyme or SAM (100% inhibition). After incubation with MTase-Glo reagent and detection solutions as noted above, luminescence was read using a Biotek Hi hybrid plate reader. IC50 values were determined by nonlinear regression fitting of the competitive curves using GraphPad Prism 7.0.
4.1.5. Saturation transfer difference NMR
All STD NMR experiments were performed on a 600 MHz Bruker Avance III equipped with a CryoProbe at 25 °C as previously described.61 Samples were prepared in HMTase buffer with 7% D2O, and 1% DMSO and contained 200 μM ligand and 5 μM GST-DOT1L with or without 1 mM SAH (Sigma-Aldrich). The on-resonance irradiation of the protein was performed using a 2 s pulse train of 50 ms Gaussian pulses centered at 0 ppm and off-resonance irradiation was applied at 30 ppm as the reference. Experiments used 16 scans with 16 dummy scans and were processed using TOPSPIN 2.1 (Bruker).
4.1.6. Thermal stability shift assay
Thermal stability shift experiments for validation of the virtual screening identified hits were performed as previously described75 using a Thermo-Fluor 384-well plate reader (Johnson & Johnson). The melting temperature (Tm) of GST-DOT1L was determined by measuring the fluorescence of 1-Anilinonaphthalene-8-Sulfonic Acid (1,8-ANS) (Cayman Chemical) upon thermal denaturing of DOT1L heated in a continuous gradient with 1 min incubations at 1°C increments. Samples contained 4 μM DOT1L, 100 μM ligand, and 100 μM 1,8-ANS in 25 mM Tris pH 7.5, 100 mM NaCl, 3 mM DTT in a total volume of 11 μL overlaid with 2 μL of silicone oil in ABgene 384-well PCR microtiter plates (Thermo Scientific).
The thermal stability shift assay for the synthesized analogs of the validated non-adenosine hit 9 was performed in a 10 μL reaction mixture using 10 μM of recombinant GST-DOT1L (aa 1-420) protein in MTase assay reaction buffer, various concentrations of compounds at 5% DMSO, and 1x (Protein Thermal Shift; cat no: 4461146; Applied Biosystems) dye. Raw data was analyzed using Protein Thermal Shift software (Applied Biosystems) to determine Tm of the protein alone and in the presence of tested compounds.
4.1.7. Cell proliferation assay of murine transformed MLL-AF9 or E2A-HLF established cell lines
MLL-AF915 and E2A-HLF9 transduced murine cell lines were treated with 25, in duplicate, at a density of 4 x 103 cells/mL in 2.5 mL of media (IMDM +15% Stem Cell FBS + IL-3) in 12 well non-treated TC plates. Cell numbers were assessed using a hemocytometer. Viability was checked every 3-4 days up to 15 days. After the viability check, the cells were normalized to a density of 4 x 103 cells/mL in 2.5 mL of media and retreated with compound or DMSO at 0.3% DMSO concentration. Total cell numbers are adjusted for the dilution factor for each split of viable cells. IC50 and percent live cells were plotted using GraphPad Prism 7.
4.1.8. Western blot analysis of H3 lysine methylation
Murine bone marrow cells transformed with MLL-AF976 were plated at 2 x 105 cells/mL in 12 well plates and treated with compounds. Cells were harvested and processed using a previously reported method.38 The concentration of extracted histones was determined using the Bradford assay (BioRad). Equal quantities of histones were separated on a 4-20% Tris-glycine gel (Life Technologies) and transferred to PVDF membrane (Thermo Scientific). Membranes were probed with primary rabbit polyclonal antibodies against histone H3 (ab1791), H3K79Me1 (ab2886), H3K79Me2 (ab3594, Cell Signaling), H3K79Me3 (ab2621), H3K4Me3 (ab8580), H3K9Me3 (ab8898), H2K27Me3 (Millipore ABE44), H3K36Me3 (ab9050). Membranes were subsequently probed with HRP-conjugated goat anti rabbit secondary antibody (GenScript) and signal detected with Lumi-Light western blotting substrate (Roche) and exposure to autoradiography film (Denville), or incubated with Amersham ECL Prime western blotting detecting reagent and imaged on the ThermoFisher iBright FL1000.
4.1.9. Differentiation
Cells were harvested from the cell proliferation assay on day 6 and cytospun onto microscope slides. Cells were then stained with Wright-Giemsa stain (Millipore) and mounted on a slide with a coverslip using Permount (Fisher Chemical) mounting medium. Images were captured using bright field condition with an Olympus IX83 inverted microscope.
4.2. Synthetic Methods
4.2.1. General Information
All reactions were performed under a dry nitrogen atmosphere unless otherwise noted, and reaction temperatures were measured externally. All reagents were used directly as obtained from commercial sources. Anhydrous DMF was purchased from TCI and used as received. Toluene was distilled from sodium benzophenone ketyl radical. Reactions were monitored by thin-layer chromatography (TLC) on pre-coated silica gel (60F254) glass plates (0.25 mm) purchased from Silicycle and visualized using UV light (254 nm), an I2 chamber, and/or ninhydrin stain. Flash chromatography was conducted using a Teledyne ISCO CombiFlash Rf 200i with the listed eluents, unless noted otherwise. 1H and 13C NMR spectra were recorded on a Bruker Avance-500 spectrometer at 500 MHz and 125.4 MHz, respectively. Chemical shifts in CDCl3 and d6-DMSO are expressed in ppm relative to the deuterated solvent signal(s).77 Mass spectra were recorded on an AutoSpec-Ultima_NT mass spectrometer using electron ionization (EI) at 70 eV and an EBE sector mass analyzer. Melting points were determined with a Mel-Temp 1001D capillary melting point apparatus and are uncorrected.
4.2.2. General Method of Amidation with Tail Group Amine.59, 64
Method A: The tail group amines Ar-NH2 (Ar = a-d) (19.5 mmol, 1.1 equiv) were added to a stirring solution of 1,2,4-triazole-3-carboxylic acid 12 (2.00 g, 17.7 mmol, 1.0 equiv) in dry DMF (30 mL) at room temperature. After 5 min, TBTU (6.0 g, 18.6 mmol, 1.0 equiv) and DIPEA (11.0 mL, 61.9 mmol, 3.5 equiv) were added in sequence. The resulting solution was stirred at room temperature for 16 h. After completion of the reaction, as indicated by TLC, DMF was removed by rotary evaporation at 30 torr and 70 °C. The crude material was purified by methods described in each individual entry.
Method B: Methyl-1H-1,2,4-triazole-3-carboxylate 15 (0.500 g, 3.93 mmol, 1.0 equiv) and tail group amines Ar-NH2 (Ar = a-c) (5.90 mmol, 1.5 equiv) were suspended in freshly distilled toluene (60 mL). The suspension was cooled to 0 °C before a 2 M solution of trimethylaluminum in toluene (4.0 mL, 7.9 mmol, 2.0 equiv) was added dropwise by syringe. The resulting suspension was warmed to room temperature at which point it became homogenous. The solution was subsequently refluxed for 9 h. After the reaction was complete, as indicated by TLC, the solution was cooled to 0 °C, quenched with 1 N HCl, and extracted with ethyl acetate (250 mL x 5). The combined organic layers were dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude material was purified by methods described in each individual entry.
4.2.2.1. N-(3,4-dimethylphenyl)-1H-1,2,4-triazole-3-carboxamide 13a.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration of the solid with dichloromethane afforded pure 13a as a white solid in 51% yield (1.95 g, 9.02 mmol).
Method B: The desired compound was eluted using a 20-25% gradient elution with ammonia-saturated methanol/dichloromethane to afford pure 13a as a white solid in 83% yield (0.706 g, 3.26 mmol).
Characterization: m.p. >260 °C; 1H NMR (d6-DMSO, 500 MHz): δ 14.44 (1H, br, s), 10.27 (1H, s), 8.56 (1H, s), 7.61 (1H, s), 7.53 (1H, d, J = 7.9 Hz), 7.08 (1H, d, J = 7.9 Hz), 2.20 (3H, s), 2.17 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 156.78, 145.19, 136.21, 135.89, 131.89, 129.47, 121.66, 118.02, 19.62, 18.80; HRMS: m/z calcd for C11H12N4O, 216.1011; found 216.1015.
4.2.2.2. N-(4-(tert-butyl)phenyl)-1H-1,2,4-triazole-3-carboxamide 13b.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration of the solid with dichloromethane afforded the pure 13b as a white solid in 49% yield (2.14 g, 8.66 mmol).
Method B: The desired compound was eluted using a 18-22% gradient elution with ammonia-saturated methanol/dichloromethane to afford pure 13b as a white solid in 81% yield (0.782 g, 3.20 mmol).
Characterization: m.p. >260 °C; 1H NMR (d6-DMSO, 500 MHz): δ 14.78 (1H, br, s), 10.41 (1H, s), 8.57 (1H, s), 7.74 (2H, d, J = 8.5 Hz), 7.35 (2H, d, J = 8.5 Hz), 1.27 (9H, s); 13C NMR (d6-DMSO, 125 MHz): δ 156.68, 146.42, 135.58, 125.21, 120.33, 34.04, 31.14; HRMS: m/z calcd for C13H16N4O, 244.1324; found 244.1325.
4.2.2.3. N-cyclohexyl-1H-1,2,4-triazole-3-carboxamide 13c.
Method A: The crude material was dissolved in dichloromethane (100 mL), and the organic layer was extracted with satd. aqueous NH4Cl solution (50 mL x 3). When the combined aqueous phase was neutralized with dropwise addition of aq. 10% NH4OH solution to pH 7, pure 13c precipitated as a white solid in 38% yield (0.981 g, 5.05 mmol (on a 1.50 g scale)).
Method B: The desired compound was eluted using a 30-35% gradient elution with ammonia-saturated methanol/dichloromethane to afford pure 13c as a white solid in 92% yield (0.702 g, 3.61 mmol).
Characterization: m.p. 250-251 °C; 1H NMR (d6-DMSO, 500 MHz): δ 14.61 (1H, br, s), 8.40 (1H, s), 8.31 (1H, d, J = 7.8 Hz), 3.74 (1H, m), 1.76-1.57 (5H, m), 1.39-1.06 (5H, m); 13C NMR (d6-DMSO, 125MHz): δ 157.00, 47.82, 32.00, 25.04, 24.81; HRMS: m/z calcd for C9H14N4O, 194.1168; found 194.1174.
4.2.2.4. N-(6-methyl-1H-benzo[d]imidazol-2-yl)-1H-1,2,4-triazole-3-carboxamide 13d.
Method A: The crude material was suspended in dichloromethane (100 mL), sonicated at 25 °C for 2-3 min then filtered. The precipitate was triturated with dichloromethane (20 mL x 4) to afford pure 13d as a light brown solid in 48% yield (0.329 g, 1.36 mmol (on a 0.320 g scale)).
Method B: Methyl-1H-1,2,4-triazole-3-carboxylate 15 (0.480 g, 3.78 mmol, 1.0 equiv) and 6-methyl-1H-benzo[d]imidazol-2-amine (0.58 g, 3.97 mmol, 1.0 equiv) were suspended in freshly distilled toluene (60 mL). The suspension was cooled to 0 °C before a 2 M solution of trimethylaluminum in toluene (3.8 mL, 7.6 mmol, 2.0 equiv) was added dropwise by syringe. The resulting suspension was warmed to room temperature at which point the suspension became homogenous. The solution was subsequently refluxed for 18 h. After the reaction was complete, as indicated by TLC, the solution was cooled to 0 °C and quenched with 1 N HCl. The aqueous phase was washed with dichloromethane (200 mL x 2) before the pH was adjusted to 7 by dropwise addition of 1 N NaOH. The neutral aqueous phase was extracted with 10% isopropanol in dichloromethane (v/v) (250 mL x 15), and the combined organic layers were evaporated to a brown solid. Purification was carried out using a 30-35% gradient elution with ammonia-saturated methanol/dichloromethane to afford amide 13d as a light brown solid in 56% yield (0.511 g, 2.11 mmol).
Characterization: m.p. >260 °C 1H NMR (d6-DMSO, 500 MHz): δ 12.93 (3H, br, s), 8.47 (1H, s), 7.34 (1H, d, J = 8.05 Hz), 7.26 (1H, s), 6.98 (1H, d, J = 8.05 Hz), 2.38 (3H, s); 13C NMR (d6-DMSO, 125MHz,): δ 161.05, 154.69, 148.63, 147.65, 133.46, 131.47, 131.09, 123.11, 112.99, 21.22; HRMS: m/z calcd for C11H10N6O, 242.0916; found 242.0907.
4.2.3.1. 2-(3-((3,4-dimethylphenyl)carbamoyl)-1H-1,2,4-triazol-1-yl)acetic acid 14a.59
Solid K2CO3 (0.575 g, 4.16 mmol, 1.5 equiv) was added to a stirring solution of 13a (0.600 g, 2.77 mmol, 1.0 equiv) in dry DMF (40 mL) at 0 °C. After 1 h, a solution of ethyl bromoacetate (0.556 g, 3.33 mmol, 1.2 equiv) in dry DMF (5 mL) was added dropwise. The resulting heterogeneous solution was brought to room temperature and stirred for another 3 h. After the reaction was complete, as indicated by TLC, the solvent was removed by rotary evaporation at 30 torr and 70 °C. Purification of the crude material was carried out using 50% ethyl acetate/hexane as the eluent to provide the corresponding ethyl ester in 60% yield (0.502 g, 1.66 mmol). m.p. 115-116 °C; 1H NMR (CDCl3, 500 MHz): δ 8.32 (1H, s), 8.26 (1H, s), 7.46 (1H, s), 7.45 (1H, d, J = 8.2 Hz), 7.12 (1H, d, J = 8.2 Hz), 5.06 (2H, s), 4.27 (2H, q, J = 7.12 Hz), 2.28 (3H, s), 2.24 (3H, s), 1.3 (3H, t, J = 7.12 Hz ); 13C NMR (CDCl3, 125 MHz,): δ 166.10, 157.61, 156.32, 145.24, 137.48, 135.14, 133.23, 130.23, 121.32, 117.58, 62.79, 51.28, 20.04, 19.37, 14.20; HRMS: m/z calcd for C15H18N4O3, 302.1379; found 302.1371.
Aqueous 1 N NaOH (2.5 mL, 2.5 mmol, 1.5 equiv) was added to a solution of the ethyl ester (0.502 g, 1.66 mmol, 1.0 equiv) in methanol (10 mL) at 0 °C and stirred at room temperature for 16 h. The methanol was removed by rotary evaporation to obtain the sodium carboxylate as a hydrated amorphous solid. This was dissolved in water (5 mL), cooled to 0 °C, and acidified to pH 1 using 1 N HCl. The resultant acid 14a precipitated from the aqueous solution and was recovered by vacuum filtration as a pure white solid in 88% yield (0.400 g, 1.46 mmol). The two-step overall yield was 53%. m.p. 250-251 °C; 1H NMR (d6-DMSO, 500 MHz): δ 13.45 (1H, br, s), 10.17 (1H, s), 8.71 (1H, s), 7.6 (1H, s), 7.51 (1H, d, J = 7.25 Hz), 7.09 (1H, d, J = 7.25 Hz), 5.2 (2H, s), 2.21 (3H, s), 2.19 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 168.51, 156.98, 156.82, 146.46, 136.15, 135.96, 131.77, 129.43, 121.61, 117.97, 50.65, 19.62, 18.79; HRMS: m/z calcd for C13H14N4O3, 274.1066; found 274.1066.
4.2.3.2. 2-(3-((4-(tert-butyl)phenyl)carbamoyl)-1H-1,2,4-triazol-1-yl)acetic acid 14b.59
Solid K2CO3 (0.509 g, 3.68 mmol, 1.5 equiv) was added to a stirring solution of 13b (0.600 g, 2.46 mmol, 1.0 equiv) in dry DMF (40 mL) at 0 °C. After 1 h, a solution of ethyl bromoacetate (0.492 g, 2.95 mmol, 1.2 equiv) in dry DMF (5 mL) was added dropwise. The resulting suspension was brought to room temperature and stirred for another 3 h. After the reaction was complete, as indicated by TLC, the solvent was removed by rotary evaporation at 30 torr and 70 °C. Purification of the crude material was carried out using 50% ethyl acetate/hexane as the eluent to provide the corresponding ethyl ester in 52% yield (0.423 g, 1.28 mmol); m.p. 150-151 °C; 1H NMR (CDCl3, 500 MHz): δ 8.87 (1H, s), 8.28 (1H, s), 7.64 (2H, dt, J = 9.4 Hz and 2.7 Hz), 7.12 (2H, dt, J = 9.4 Hz and 2.7 Hz), 5.06 (2H, s), 4.28 (2H, q, J = 7.15 Hz), 1.32 (9H, s), 1.3 (3H, t, J = 7.15 Hz ); 13C NMR (CDCl3, 125 MHz): δ 166.09, 157.55, 156.40, 147.86, 145.26, 134.78, 126.08, 119.87, 62.80, 51.29, 34.58, 31.49, 14.20; HRMS: m/z calcd for C17H22N4O3, 330.1692; found 330.1688.
Aqueous 1 N NaOH (1.9 mL, 1.9 mmol, 1.5 equiv) was added to a stirring solution of the ethyl ester (0.423 g, 1.28 mmol, 1.0 equiv) in methanol (10 mL) at 0 °C and stirred at room temperature for 16 h. The methanol was removed by rotary evaporation to obtain the sodium carboxylate as a hydrated amorphous solid. This was dissolved in water (5 mL), cooled to 0 °C, and acidified to pH 1 using 1 N HCl. The resultant acid 14b precipitated from the aqueous solution and was recovered by vacuum filtration as a pure white solid in 88% yield (0.338 g, 1.12 mmol). The two-step overall yield was 46%. m.p. > 260 °C; 1H NMR (d6-DMSO, 500 MHz): δ 13.47 (1H, br, s), 10.30 (1H, s), 8.72 (1H, s), 7.71 (2H, d, J = 8.7 Hz), 7.36 (2H, d, J = 8.7 Hz), 5.2 (2H, s), 1.28 (9H, s); 13C NMR (d6-DMSO, 125 MHz): δ 168.51, 157.05, 156.78, 146.48, 146.32,135.65, 125.18, 120.30, 50.64, 34.04, 31.15; HRMS: m/z calcd for C15H18N4O3, 302.1379; found 302.1376.
4.2.3.3. 2-(3-(cyclohexylcarbamoyl)-1H-1,2,4-triazol-1-yl)acetic acid 14c.59
Solid K2CO3 (0.640 g, 4.63 mmol, 1.5 equiv) was added to a stirring solution of 13c (0.600 g, 3.09 mmol, 1.0 equiv) in dry DMF (40 mL) at 0 °C. After 1 h, a solution of ethyl bromoacetate (0.619 g, 3.71 mmol, 1.2 equiv) in dry DMF (5 mL) was added dropwise. The resulting suspension was brought to room temperature and stirred another 3 h. After the reaction was complete, as indicated by TLC, the solvent was removed by rotary evaporation at 30 torr and 70 °C. Purification of the crude material was carried out using 70% ethyl acetate/hexane as the eluent to provide the corresponding ethyl ester in 57% yield (0.493 g, 1.76 mmol); m.p. 121-122 °C; 1H NMR (CDCl3, 500 MHz): δ 8.20 (1H, s), 7.01 (1H, d, J = 8.0 Hz),5.02 (2H, s), 4.24 (2H, q, J = 7.15 Hz), 3.98 (1H, m), 2.01-1.61 (5H, m), 1.45-1.16 (8H, m); 13C NMR (CDCl3, 125 MHz): δ 166.18, 157.84, 157.69, 145.08, 62.68, 51.13, 48.16, 33.15, 25.63, 24.91, 14.17; HRMS: m/z calcd for C13H20N4O3, 280.1535; found 280.1538.
Aqueous 1 N NaOH (2.6 mL, 2.6 mmol, 1.5 equiv) was added to a stirring solution of the ethyl ester (0.493 g, 1.76 mmol, 1.0 equiv) in methanol (10 mL) at 0 °C and stirred at room temperature for 16 h. The methanol was removed by rotary evaporation to obtain the sodium carboxylate as a hydrated amorphous solid. This was dissolved in water (5 mL), cooled to 0 °C, and acidified to pH 1 using 1 N HCl. The aqueous solution was extracted with dichloromethane (100 mL x 5), then the combined organic layers were dried over anhydrous Na2SO4 and concentrated by rotary evaporation to afford pure 14c as a white solid in 78% yield (0.346 g, 1.37 mmol). Overall two-step yield was 44%. m.p. 190-191 °C; 1H NMR (d6-DMSO, 500 MHz): δ 13.41 (1H, br, s) 8.60 (1H, s), 8.13 (1H, d, J = 8.4 Hz), 5.14 (2H, s), 3.73 (1H, m), 1.76-1.57 (5H, m), 1.40-1.11 (5H, m); 13C NMR (d6-DMSO, 125 MHz): δ 168.57, 157.67, 156.91, 146.16, 50.49, 47.78, 32.05, 25.06, 24.84; HRMS: m/z calcd for C11H16N4O3, 252.1222; found 252.1213.
4.2.4.1. 7-(2-aminoethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine D.
Solid K2CO3 (0.930 g, 6.71 mmol, 3.0 equiv) was added to a stirring solution of 7H-pyrrolo[2,3-d]pyrimidin-4-amine 16 (0.300 g, 2.24 mmol, 1.0 equiv) in dry DMF (15 mL) at room temperature. After 1 h, a solution of tert-butyl (2-chloroethyl)carbamate (0.602 g, 3.35 mmol, 1.5 equiv) in dry DMF (5 mL) was added dropwise. The resulting suspension was stirred for 48 h at 80 °C. After the reaction was complete, as indicated by TLC, the crude material was filtered through a plug of Celite to remove inorganic salts and then washed with dichloromethane (50 mL x 4). The filtrate was concentrated by rotary evaporation and purification was carried out using 9% methanol/dichloromethane as the eluent to provide a light brown solid of the corresponding tert-butyl carbamate. This material was dissolved in 15 mL of a 1:1 (v/v) mixture of trifluoroacetic acid and dichloromethane. The resulting solution was stirred at room temperature for 4 h before it was concentrated by rotary evaporation at 35 °C. The resulting trifluoroacetic acid salt of the deprotected amine was dissolved in absolute ethanol (20 mL) and stirred with Amberlyst® A-21 free base resin (4 g) at room temperature for 10 h. The heterogeneous reaction mixture was filtered and washed with methanol (50 mL x 4) until the chromophore was no longer evident in the dripping wash by TLC. The filtrate was concentrated by rotary evaporation and purified using 15% ammonia-saturated methanol/dichloromethane as the eluent to provide product D as a light brown solid in 58% yield (0.231 g, 1.30 mmol) from 16. m.p. 145-146 °C; 1H NMR (d6-DMSO, 500 MHz): δ 8.03 (1H, s), 7.14 (1H, d, J = 3.4 Hz), 6.90 (2H, br, s), 6.50 (1H, d, J = 3.4 Hz), 4.07 (2H, t, J = 6.6 Hz), 2.86 (2H, t, J = 6.6 Hz), 1.57 (2H, br, s); 13C NMR (d6-DMSO, 125 MHz): δ 157.37, 151.40, 149.55, 124.38, 102.35, 98.07, 47.16, 41.99; HRMS: m/z calcd for C8H11N5, 177.1014; found 177.1010.
4.2.4.2. 3-(2-aminoethyl)-3,7-dihydro-4H-pyrrolo[2,3-d]pyrimidin-4-one E.
Solid K2CO3 (0.613 g, 4.44 mmol, 3.0 equiv) was added to a stirring solution of 7-deazahypoxanthine 17 (0.200 g, 1.48 mmol, 1.0 equiv) in dry DMF (10 mL) at room temperature. After 1 h, a solution of tert-butyl (2-chloroethyl)carbamate (0.399 g, 2.22 mmol, 1.5 equiv) in dry DMF (5 mL) was added dropwise. The resulting suspension was stirred at 60 °C for 72 h, at which time an additional 1.0 equiv and later 0.5 equiv of tert-butyl (2-chloroethyl)carbamate in dry DMF (0.267 g (1.48 mmol) in 4 mL and 0.134 g (0.74 mmol) in 2 mL) were added dropwise to the suspension after 24 h and 48 h additional reaction time, respectively. The crude material was filtered through a plug of Celite to remove inorganic salts and then washed with dichloromethane (50 mL x 4). The filtrate was concentrated using rotary evaporation. Purification was carried out using 9% methanol/dichloromethane as the eluent to provide the corresponding tert-butyl carbamate as an off-white solid. This was subsequently dissolved in 10 mL of a 1:1 (v/v) mixture of trifluoroacetic acid and dichloromethane. The solution was stirred at room temperature for 4 h before it was concentrated to dryness by rotary evaporation at 35 °C. The resulting trifluoroacetic acid salt of the deprotected amine was dissolved in absolute ethanol (15 mL) and stirred with Amberlyst® A-21 free base resin (4 g) at room temperature for 10 h. The heterogeneous reaction mixture was filtered and washed with methanol (50 mL x 4) until the chromophore was no longer evident in the dripping wash by TLC. The filtrate was concentrated by rotary evaporation and purified using 15% ammonia-saturated methanol/dichloromethane as the eluent to provide the corresponding product E as a light brown solid in 41% yield (0.109 g, 0.61 mmol) from 17. m.p. 150-151 °C; 1H NMR (d6-DMSO, 500 MHz): δ 11.82 (1H, br, s), 8.08 (1H, s), 7.03 (1H, d, J = 3.3 Hz), 6.44 (1H, d, J = 3.3 Hz), 3.91 (2H, t, J = 6.12 Hz), 2.86 (2H, t, J = 6.12 Hz), 1.69 (2H, br, s); 13C NMR (d6-DMSO, 125 MHz): δ 157.70, 147.55, 146.49, 120.63, 106.95, 101.96, 48.23, 40.72; HRMS: m/z calcd for C8H10N4O, 178.0855; found 178.0854.
4.2.5. General Procedure for the Amidation of Carboxylic Acid Scaffolds 14a-c:59, 68
Method A: Corresponding head group amines A-E (1.5 equiv) were added to a stirring solution of 14a-c (50.0 mg, 1.0 equiv) in dry DMF (3 mL) at room temperature. After 5 min, TBTU (1.0 equiv) and DIPEA (3.5 equiv)* were added in sequence. The resulting solution was stirred at room temperature for 24 h before it was concentrated to dryness by rotary evaporation at 30 torr and 70 °C. The crude material was purified by methods described in each individual entry.
Method B: Using a gas tight syringe, N-methylmorpholine (1.1 equiv) was added dropwise to a stirring solution of 14a-c (40.0 mg, 1.0 equiv) in dry DMF (6 mL) at −10 °C. The resulting solution was stirred for 10 min before isobutylchloroformate (1.1 equiv) was added dropwise. After 10 min, a solution of corresponding head group amines A-E (1.5 equiv) and N-methylmorpholine (1.1 equiv)* in dry DMF (3 mL) were added dropwise. The reaction mixture was stirred for another 30 min before it was warmed to room temperature and stirred for an additional 12 h. After the reaction was complete, as indicated by TLC, the DMF was removed by rotary evaporation at 30 torr and 70 °C. The crude material was purified by methods described in each individual entry.
*Since head group amine C was received as a hydrochloride salt, one extra equivalent of DIPEA (in Method A) or N-methylmorpholine (in Method B) was added to free base the amine in situ.
4.2.5.1. 1-(2-((benzo[d][1,3]dioxol-5-ylmethyl)amino)-2-oxoethyl)-N-(3,4-dimethylphenyl)-1H-1,2,4-triazole-3-carboxamide 9.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration of the solid with dichloromethane (5 mL x 3) afforded 9 as a white solid in 25% yield (19.0 mg, 0.047 mmol).
Method B: Purification was carried out using 5% methanol/dichloromethane as the eluent to provide 9 as a white solid in 94% yield (56.0 mg, 0.137 mmol).
Characterization: m.p. 226-227 °C; 1H NMR (d6-DMSO, 500 MHz): δ 10.15 (1H, s), 8.75 (1H, t, J = 5.75 Hz), 8.69 (1H, s), 7.60 (1H, d, J = 1.85 Hz), 7.51 (1H, dd, J = 8.2 Hz, 1.85 Hz),_7.08 (1H, d, J = 8.2 Hz), 6.87 (1H, d, J = 7.92 Hz), 6.86 (1H, d, J = 1.47 Hz), 6.77 (1H, dd, J = 7.92 Hz, 1.47 Hz), 5.99 (2H, s), 5.07 (2H, s), 4.23 (2H, d, J = 5.75 Hz), 2.21 (3H, s), 2.19 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.27, 157.03, 156.82, 147.27, 146.65, 146.21, 136.14, 135.98, 132.51, 131.74, 129.42, 121.59, 120.68, 117.95, 108.06, 108.03, 100.84, 51.73, 42.20, 19.62, 18.79; HRMS: m/z calcd for C21H21N5O4 407.1594; found 407.1610.
4.2.5.2. 1-(2-(((1H-indazol-5-yl)methyl)amino)-2-oxoethyl)-N-(3,4-dimethylphenyl)-1H-1,2,4-triazole-3-carboxamide 18.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration of the solid with dichloromethane (5 mL x 3) afforded 18 as a white solid in 36% yield (26.5 mg, 0.065 mmol).
Method B: Purification was carried out using 8% methanol/dichloromethane as the eluent to provide 18 along with a minor impurity, which was removed by triturating the solid with diethyl ether (5 mL x 3). Pure 18 was obtained as a white solid in 80% yield (47.0 mg, 0.116 mmol).
Characterization: m.p. >260 °C; 1H NMR (d6-DMSO, 500 MHz): δ 13.04 (1H, s), 10.17 (1H, s), 8.84 (1H, t, J = 5.7 Hz), 8.71 (1H, s), 8.05 (1H, s), 7.67 (1H, s), 7.60 (1H, s), 7.51 (2H, d, J = 8.45 Hz), 7.29 (1H, d, J = 8.45 Hz), 7.08 (1H, d, J = 8.2 Hz), 5.1 (2H, s), 4.43 (2H, d, J = 5.7 Hz), 2.21 (3H, s), 2.18 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.27, 157.07, 156.86, 146.69, 139.20, 136.17, 136.00, 133.30, 131.77, 130.54, 129.44, 126.32, 122.81, 121.62, 118.91, 117.98, 110.10, 51.82, 42.64, 19.64, 18.81; HRMS: m/z calcd for C21H21N7O2, 403.1757; found 407.1772.
4.2.5.3. 1-(2-(((1-aminoisoquinolin-6-yl)methyl)amino)-2-oxoethyl)-N-(3,4-dimethylphenyl)-1H-1,2,4-triazole-3-carboxamide 19.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration with dichloromethane (5 mL x 3) afforded 19 as an off-white solid in 23% yield (18.0 mg, 0.042 mmol).
Method B: Purification was carried out using 10% ammonia-saturated methanol/dichloromethane as the eluent to provide 19 as a light yellow solid along with a minor impurity. The material was suspended in biphasic 1:1 dichloromethane:water (v/v) and sonicated for 2-3 min at room temperature. The resulting suspension was filtered and triturated with cold dichloromethane (5 mL x 3) to obtain pure 19 as an off-white solid in 58% yield (36.0 mg, 0.084 mmol).
Characterization: m.p. 239-240 °C; 1H NMR (d6-DMSO, 500 MHz): δ 10.16 (1H, s), 8.92 (1H, t, J = 5.87 Hz), 8.72 (1H, s), 8.15 (1H, d, J = 8.65 Hz), 7.76 (1H, d, J = 5.75 Hz), 7.60 (1H, d, J = 1.07 Hz), 7.56 (1H, s), 7.52 (1H, d, J = 7.9 Hz), 7.36 (1H, dd, J = 8.65 Hz, 1.07 Hz), 7.09 (1H, d, J = 7.9 Hz), 6.87 (1H, d, J = 5.75 Hz), 6.73 (2H, s), 5.14 (2H, s), 4.48 (2H, d, J = 5.87 Hz), 2.21 (3H, s), 2.19 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.58, 157.16, 157.05, 156.90, 146.69, 142.09, 140.34, 136.93, 136.15, 135.99, 131.75, 129.43, 124.92, 124.25, 123.95, 121.60, 117.97, 116.23, 109.66, 51.84, 42.34, 19.63, 18.80; HRMS: m/z calcd for C23H23N7O2 429.1913; found 429.1920.
4.2.5.4. 1-(2-((2-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)amino)-2-oxoethyl)-N-(3,4-dimethylphenyl)-1H-1,2,4-triazole-3-carboxamide 20.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration of the solid with dichloromethane (5 mL x 3) afforded 20 as an off-white solid in 28% yield (22.2 mg, 0.051 mmol).
Method B: Purification was carried out using 10% ammonia-saturated methanol/dichloromethane as the eluent to provide 20 as a light brown solid along with a minor impurity. The material was suspended in biphasic dichloromethane:water (v/v) and sonicated for 2-3 min at room temperature. The resulting suspension was filtered and triturated with cold dichloromethane (5 mL x 3) to obtain pure 20 as an off-white solid in 79% yield (50.0 mg, 0.115 mmol).
Characterization: m.p. 160-161 °C; 1H NMR (d6-DMSO, 500 MHz): δ 10.14 (1H, s), 8.65 (1H, s), 8.46 (1H, t, J = 5.7 Hz), 8.07 (1H, s), 7.60 (1H, s), 7.51 (1H, d, J = 7.82 Hz), 7.13 (1H, d, J = 3.25 Hz), 7.08 (1H, d, J = 7.82 Hz), 6.94 (2H, br, s), 6.53 (1H, d, J = 3.25 Hz), 4.96 (2H, s), 4.20 (2H, t, J = 5.7 Hz), 3.47 (2H, t, J = 5.7 Hz), 2.21 (3H, s), 2.18 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.61, 157.42, 157.02, 156.79, 151.56, 149.65, 146.59, 136.14, 135.98, 131.73, 129.42, 124.31, 121.59, 117.96, 102.48, 98.52, 51.72, 43.13, 19.62, 18.79. HRMS: m/z calcd for C21H23N9O2 433.1975; found 433.1985.
4.2.5.5. N-(3,4-dimethylphenyl)-1-(2-oxo-2-((2-(4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-3-yl)ethyl)amino)ethyl)-1H-1,2,4-triazole-3-carboxamide 21.
Method A: The crude material was suspended in biphasic 1:1 dichloromethane:water (v/v), and a precipitate was recovered by vacuum filtration. Trituration of the solid with dichloromethane (5 mL x 3) afforded 21 as an off-white solid in 20% yield (16.0 mg, 0.037 mmol).
Method B: Purification was carried out using 16% ammonia-saturated methanol/dichloromethane as the eluent to provide 21 as a light brown solid along with a minor impurity. The material was suspended in a biphasic solvent of 1:1 dichloromethane:water (v/v) and sonicated for 2-3 min at room temperature. The resulting suspension was filtered and triturated with cold dichloromethane (5 mL x 3) to obtain pure 21 as an off-white solid in 66% yield (42.0 mg, 0.097 mmol).
Characterization: m.p. 165-166 °C; 1H NMR (d6-DMSO, 500 MHz): δ 11.87 (s, 1H), 10.15 (1H, s), 8.64 (1H, s), 8.46 (1H, t, J = 5.6 Hz ), 8.03 (1H, s), 7.60 (1H, s), 7.51 (1H, d, J = 8.1 Hz), 7.08 (1H, d, J = 8.1 Hz), 7.06 (1H, t, J = 2.62 Hz), 6.47 (1H, t, J = 2.62 Hz), 4.98 (2H, s), 4.04 (2H, t, J = 5.6 Hz), 3.44 (2H, q, J = 5.6 Hz), 2.20 (3H, s), 2.18 (3H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.75, 157.75, 157.03, 156.81, 147.51, 146.61 146.07, 136.15, 135.99, 131.73, 129.43, 121.57, 120.85, 117.93, 106.93, 102.08, 51.71, 44.95, 19.62, 18.79. HRMS: m/z calcd for C21H22N8O3, 434.1815; found 434.1812.
4.2.5.6. 1-(2-((benzo[d][1,3]dioxol-5-ylmethyl)amino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)-1H-1,2,4-triazole-3-carboxamide 22.
Method A: Purification was carried out using 5% methanol/dichloromethane as the eluent, and 22 was isolated as an off-white solid in 22% yield (16.0 mg, 0.037 mmol).
Method B: Purification was carried out using 5% methanol/dichloromethane as the eluent, and 22 was isolated as a white solid in 93% yield (53.5 mg, 0.123 mmol).
Characterization: m.p. 203-204 °C; 1H NMR (d6-DMSO, 500 MHz): δ 10.29 (1H, s), 8.77 (1H, t, J = 5.75 Hz), 8.70 (1H, s), 7.71 (2H, d, J = 8.7 Hz), 7.35 (2H, d, J = 8.7 Hz), 6.86 (1H, d, J = 7.95 Hz), 6.86 (1H, s), 6.77 (1H, d, J = 7.95 Hz), 5.99 (2H, s), 5.07 (2H, s), 4.23 (2H, d, J = 5.75 Hz), 1.28 (9H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.28, 157.12, 156.80, 147.28, 146.70, 146.31, 146.22, 135.69, 132.53, 125.20, 120.70, 120.31, 108.07, 100.86, 51.74, 42.22, 34.05, 31.16; HRMS: m/z calcd for C23H25N5O4, 435.1907; found 435.1906.
4.2.5.7. 1-(2-(((1H-indazol-5-yl)methyl)amino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)-1H-1,2,4-triazole-3-carboxamide 23.
Method A: Purification was carried out using 7% ammonia-saturated methanol/dichloromethane as the eluent, and 23 was isolated as an off-white solid in 18% yield (13.0 mg, 0.030 mmol).
Method B: Purification was carried out using 10% methanol/dichloromethane as the eluent, and 23 was isolated as a white solid in 97% yield (55.5 mg, 0.129 mmol).
Characterization: m.p. 255-256 °C; 1H NMR (d6-DMSO, 500 MHz): δ 13.03 (1H, s), 10.29 (1H, s), 8.84 (1H, t, J = 5.7 Hz), 8.71 (1H, s), 8.04 (1H, s), 7.72 (2H, d, J = 8.67 Hz), 7.67 (1H, s), 7.51 (1H, d, J = 8.57 Hz), 7.36 (2H, d, J = 8.67 Hz), 7.29 (1H, d, J = 8.57 Hz), 5.1 (2H, s), 4.42 (2H, d, J = 5.7 Hz), 1.27 (9H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.25, 157.13, 156.81, 146.69, 146.30, 139.19, 135.68, 133.28, 130.52, 126.30, 125.19, 122.80, 120.30, 118.90, 110.09, 51.81, 42.62, 34.04, 31.15; HRMS: m/z calcd for C23H25N7O2, 431.2070; found 431.2072.
4.2.5.8. 1-(2-(((1-aminoisoquinolin-6-yl)methyl)amino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)-1H-1,2,4-triazole-3-carboxamide 24.
Method A: Purification was carried out using 8% ammonia-saturated methanol/dichloromethane as the eluent, and 24 was isolated as an off-white solid in 46% yield (35.0 mg, 0.076 mmol).
Method B: Purification was carried out using 8% ammonia-saturated methanol/dichloromethane as the eluent to obtain slightly impure compound, which was triturated with diethyl ether (4 mL x 3) to afford 24 as an off-white solid in 83% yield (50.0 mg, 0.109 mmol).
Characterization: m.p. >260 °C; 1H NMR (d6-DMSO, 500 MHz): δ 10.31 (1H, s), 8.93 (1H, t, J = 5.4 Hz), 8.73 (1H, s), 8.15 (1H, d, J = 8.5 Hz), 7.76 (1H, d, J = 5.6 Hz), 7.72 (2H, d, J = 8.6 Hz), 7.56 (1H, s), 7.38-7.34 (3H, m), 6.87 (1H, d, J = 5.6 Hz), 6.73 (2H, s), 5.14 (2H, s), 4.48 (2H, d, J = 5.4 Hz), 1.28 (9H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.59, 157.13, 157.07, 156.87, 146.72, 146.31, 141.75, 140.48, 136.94, 135.68, 125.19, 124.99, 124.28, 123.98, 120.30, 116.22, 109.69, 51.84, 42.33, 34.05, 31.16; HRMS: m/z calcd for C25H27N7O2 457.2226; found 457.2222.
4.2.5.9. 1-(2-((2-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)amino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)-1H-1,2,4-triazole-3-carboxamide 25.
Method A: Purification was carried out using 10% ammonia-saturated methanol/dichloromethane as the eluent, and 25 was isolated as an off-white solid in 25% yield (19.0 mg, 0.041 mmol).
Method B: Purification was carried out using 10% ammonia-saturated methanol/dichloromethane as the eluent to obtain slightly impure compound, which was dissolved in dichloromethane (20 mL) and washed with ice-cold water (5 mL x 2). The organic layer was dried over anhydrous Na2SO4 and concentrated by rotary evaporation to afford pure 25 as an off-white solid in 96% yield (58.5 mg, 0.127 mmol).
Characterization: m.p. 139-140 °C; 1H NMR (d6-DMSO, 500 MHz): δ 10.29 (1H, s), 8.66 (1H, s), 8.46 (1H, t, J = 5.7 Hz), 8.07 (1H, s), 7.71 (2H, d, J = 8.75 Hz), 7.35 (2H, d, J = 8.75 Hz), 7.14 (1H, d, J = 3.45 Hz), 6.95 (2H, br, s), 6.54 (1H, d, J = 3.45 Hz ) 4.97 (2H, s), 4.20 (2H, t, J = 6.02 Hz), 3.47 (2H, q, J = 6.02 Hz), 1.27 (9H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.62, 157.44, 157.11, 156.77, 151.58, 149.66, 146.63, 146.30, 135.69, 125.19, 124.31, 120.31, 102.49, 98.53, 51.73, 43.14, 34.04, 31.16; HRMS: m/z calcd for C23H27N9O2 461.2288; found 461.2289.
4.2.5.10. 1-(2-((benzo[d][1,3]dioxol-5-ylmethyl)amino)-2-oxoethyl)-N-cyclohexyl-1H-1,2,4-triazole-3-carboxamide 26.
Method A: Purification was carried out using 7% methanol/dichloromethane as the eluent, and 26 was isolated as a white solid in 17% yield (13.0 mg, 0.034 mmol).
Method B: Purification was carried out using 5% methanol/dichloromethane as the eluent, and 26 was isolated as a white solid in 85% yield (52.0 mg, 0.135 mmol).
Characterization: m.p. 188-189 °C; 1H NMR (d6-DMSO, 500 MHz): δ 8.72 (1H, t, J = 5.77 Hz), 8.59 (1H, s), 8.13 (1H, d, J = 8.45 Hz), 6.87 (1H, s), 6.85 (1H, dd, J = 7.9 Hz, 1.45 Hz), 6.75 (1H, dd, J = 7.9 Hz, 1.45 Hz), 5.99 (2H, s), 5.01 (2H, s), 4.21 (2H, d, J = 5.77 Hz), 3.74-3.60 (1H, m), 1.76-1.57 (5H, m), 1.40-1.09 (5H, m); 13C NMR (d6-DMSO, 125 MHz): δ 165.32, 157.72, 156.92, 147.26, 146.35, 146.19, 132.52, 120.67, 108.05, 108.02, 100.84, 51.58, 47.75, 42.18, 32.05, 25.06, 24.83; HRMS: m/z calcd for C19H23N5O4 385.1750; found 385.1751.
4.2.5.11. 1-(2-(((1H-indazol-5-yl)methyl)amino)-2-oxoethyl)-N-cyclohexyl-1H-1,2,4-triazole-3-carboxamide 27.
Method A: Purification was carried out using 8% methanol/dichloromethane as the eluent, and 27 was isolated as a white solid in 16% yield (12.0 mg, 0.031 mmol).
Method B: Purification was carried out using 10% methanol/dichloromethane as the eluent to obtain slightly impure compound, which was triturated with diethyl ether (4 mL x 3) to afford pure 27 as a white solid in 63% yield (38.0 mg, 0.996 mmol).
Characterization: m.p. > 260 °C; 1H NMR (d6-DMSO, 500 MHz): δ 13.02 (1H, s), 8.81 (1H, t, J = 5.75 Hz), 8.60 (1H, s), 8.12 (1H, d, J = 8.5 Hz), 8.04 (1H, s), 7.65 (1H, s), 7.5 (1H, d, J = 8.52 Hz), 7. 28 (1H, d, J = 8.52 Hz), 5.02 (2H, s), 4.40 (2H, d, J = 5.75 Hz), 3.74-3.71 (1H, m), 1.76-1.57 (5H, m), 1.40-1.06 (5H, m); 13C NMR (d6-DMSO, 125 MHz): δ 165.30, 157.74, 156.94, 146.35, 139.18, 133.28, 130.52, 126.29, 122.79, 118.87, 110.07, 51.66, 47.75, 42.59, 32.06, 25.06, 24.84. HRMS: m/z calcd for C19H23N7O2 381.1913; found 381.1917.
4.2.5.12. 1-(2-(((1-aminoisoquinolin-6-yl)methyl)amino)-2-oxoethyl)-N-cyclohexyl-1H-1,2,4-triazole-3-carboxamide 28.
Method A: Purification was carried out using 11% ammonia-saturated methanol/dichloromethane as the eluent, and 28 was isolated as an-off white solid in 25% yield (20.2 mg, 0.050 mmol).
Method B: The crude material was dissolved in dichloromethane (50 mL) and washed with ice-cold water (10 mL x 2). The organic layer was dried over anhydrous Na2SO4 and concentrated by rotary evaporation to obtain the slightly impure compound This was purified using 12% ammonia-saturated methanol/dichloromethane as the eluent to afford pure 28 as a light-yellow solid in 51% yield (33.0 mg, 0.081 mmol).
Characterization: m.p. 250-251 °C; 1H NMR (d6-DMSO, 500 MHz): δ 8.89 (1H, t, J = 5.55 Hz), 8.61 (1H, s), 8.15 ( 1H, d, J = 8.55 Hz), 8.12 (1H, d, J = 8.52 Hz), 7.77 (1H, d, J = 5.77 Hz), 7.55 (1H, s), 7.36 (1H, d, J = 8.52 Hz), 6. 87 (1H, d, J = 5.77 Hz), 6.76 (2H, br, s), 5.08 (2H, s), 4.46 (2H, d, J = 5.55 Hz), 3.76-3.73 (1H, m), 1.77-1.57 (5H, m), 1.40-1.09 (5H, m); 13C NMR (d6-DMSO, 125 MHz): δ 165.59, 157.72, 157.07, 156.97, 146.33, 141.83, 140.40, 136.90, 124.93, 124.22, 123.93, 116.20, 109.66, 51.68, 47.74, 42.29, 32.03, 25.04, 24.8; HRMS: m/z calcd for C21H25N7O2 407.2070; found 407.2056.
4.2.5.13. 1-(2-((2-(4-amino-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)amino)-2-oxoethyl)-N-cyclohexyl-1H-1,2,4-triazole-3-carboxamide 29.
Method A: Purification was carried out using 10% ammonia-saturated methanol/dichloromethane as the eluent, and 29 was isolated as a light yellow solid in 18% yield (14.7 mg, 0.036 mmol).
Method B: Purification was carried out using 10% ammonia-saturated methanol/dichloromethane as the eluent to obtain slightly impure compound, which was dissolved in dichloromethane (20 mL) and washed with ice-cold water (5 mL x 2). The organic layer was dried over anhydrous Na2SO4 and concentrated by rotary evaporation to afford pure 29 as an off-white solid in 84% yield (55.0 mg, 0.134 mmol).
Characterization: m.p. 144-145 °C; 1H NMR (d6-DMSO, 500 MHz):δ 8.55 (1H, s), 8.43 (1H, t, J = 5.65 Hz), 8.11 (1H, d, J = 8.45 Hz), 8.06 (1H, s), 7.12 (1H, d, J = 3.47 Hz), 6.94 (2H, br, s), 6.52 (1H, d, J = 3.47 Hz), 4.90 (2H, s), 4.17 (2H, t, J = 6.02 Hz), 3.77-3.69 (1H, m), 3.46 (2H, q, J = 6.02 Hz), 1.76-1.57 (5H, m), 1.40-1.08 (5H, m). 13C NMR (d6-DMSO, 125MHz): δ 165.67, 157.72, 157.43, 156.90, 151.56, 149.64, 146.30, 124.30, 102.48, 98.51, 51.58, 47.76, 43.13, 32.06, 25.06, 24.84; HRMS: m/z calc. for C19H25N9O2, 411.2131 found: 411.2121.
4.2.6. N-(benzo[d][1,3]dioxol-5-ylmethyl)-2-bromoacetamide 3070
Triethylamine (0.6 mL, 3.97 mmol, 1.2 equiv) was added to a stirring solution of head group amine A (0.500 g, 3.30 mmol, 1.0 equiv) in dry dichloromethane (20 mL) at 0 °C. After 10 min, a solution of bromoacetyl bromide (0.35 mL, 3.97 mmol, 1.2 equiv) in dichloromethane (5 mL) was added dropwise. The resulting solution was stirred at 0 °C for 2 h before it was warmed to room temperature and stirred for an additional 2 h. After the reaction was complete, as indicated by TLC, the solution was concentrated by rotary evaporation to obtain a light brown solid, which was purified using 20% ethyl acetate/hexane as the eluent to provide 30 as a white crystalline solid in 76% yield (0.683 g, 2.51 mmol). m.p. 114-115 °C; 1H NMR (d6-DMSO, 500 MHz): δ 8.70 (1H, t, J = 5.5 Hz), 6.85 (1H, d, J = 7.95 Hz), 6.82 (1H, d, J = 1.62 Hz), 6.73 (1H, dd, J = 7.95 Hz and 1.62 Hz), 5.98 (2H, s), 4.19 (2H, d, J = 5.5 Hz), 3.89 (2H, s); 13C NMR (d6-DMSO, 125 MHz): δ 165.92, 147.24, 146.14, 132.64, 120.50, 108.01, 107.88, 100.82, 42.29, 29.45; HRMS: m/z calcd for C10H10NO3Br, 270.9844; found 270.9839.
4.2.7. 1-(2-((benzo[d][1,3]dioxol-5-ylmethyl)amino)-2-oxoethyl)-N-(6-methyl-1H-benzo[d]imidazol-2-yl)-1H-1,2,4-triazole-3-carboxamide 31.
Solid K2CO3 (17.1 mg, 0.124 mmol, 1.0 equiv). was added to a stirring solution of 13d (30.0 mg, 0.124 mmol, 1.0 equiv) in dry DMF (32 mL) at 0 °C. After 30 min, a solution of 30 (33.7 mg, 0.124 mmol, 1.0 equiv) in dry DMF (8 mL) was added dropwise. The resulting suspension was stirred for 3 h at 0 °C. After the reaction was complete, as indicated by TLC, the crude material was filtered through a plug of Celite to remove inorganic salts and then washed with dichloromethane (50 mL x 4). The filtrate was concentrated by rotary evaporation. Purification of the resulting light-yellow material was carried out using 15-18% gradient elution with ammonia-saturated methanol/dichloromethane to provide 31 in 50% yield (26.8 mg, 0.062 mmol) as a white solid. m.p. 238-239 °C; 1H NMR (d6-DMSO, 500 MHz): δ 11.95 (2H, br, s), 8.75 (1H, t, J = 5.7 Hz), 8.68 (1H, s), 7.34 (1H, d, J = 8.05 Hz), 7.26 (1H, s), 6.96 (1H, d, J = 8.05 Hz), 6.87 (1H, d, J = 7.9 Hz), 6.86 (1H, s), 6.77 (1H, d, J = 7.9 Hz), 5.99 (2H, s), 5.08 (2H, s), 4.24 (2H, d, J = 5.7 Hz), 2.38 (3H, s); 13C NMR (d6-DMSO, 125 MHz): 165.30, 147.26, 146.74, 146.20, 132.53, 130.71, 122.85, 120.67, 108.05, 108.03, 100.84, 51.78, 42.20, 21.24; HRMS: m/z calcd for C21H19N7O4, 433.1499; found 433.1502.
4.3. In silico Methods
4.3.1. In silico methods applied for virtual screening.
The 1200 chemical structures of the focused library used in this study were downloaded in sdf format from the Substance database of PubChem,78 maintained by the National Library of Medicine. These compounds were converted to 3D structures using Schrodinger LigPrep 2.5. All parameters were set to the default values except that the “Ionization” was set to “Epik” The DOT1L:SAM complex (PDB ID: 1NW3) was downloaded from the PDB. Hydrogen atoms were added, bond orders assigned, hydrogen bonds optimized, followed by constraint energy minimization of the complex using the OPLS-AA 2005 force field within the Protein Preparation Wizard of Schrodinger Maestro.
To obtain a reasonable binding pose of EPZ004777 to the target of DOT1L, a molecule which is derived from EPZ004777, but does not have the 4-tert-butylphenyl group was docked first into the active site of 1NW3, using Schrodinger Glide. The 4-tert-butylphenyl group was then manually built into the docked molecule. The resulting modeled DOT1L:EPZ004777 complex structure was soaked in a box of TIP3P water molecules with a margin of 10 A along each dimension. An appropriate number of counter ions were added to neutralize the whole system. The obtained structure was minimized in three rounds, each of which consisted of 1000 steps, with harmonic constraints on all non-hydrogen atoms by employing the AMBER (version 11.0) program. The force constant was set to 100, 10 and 0 kcal/(mol*Å2) respectively. The molecular dynamics simulation was started by heating the entire system from 0 to 300 K in 100 ps and equilibrating at 300 K for another 100 ps. A subsequent 2 ns production run was performed under a constant temperature of 300 K and a constant pressure of 1 atm. No other constraint was applied to either the protein or the ligand during the entire MD simulation. The final snapshot of DOT1L:EPZ004777 complex produced from the molecular dynamics was minimized again and then used in the virtual screening.
The compounds of the library were docked and scored using Glide 5.7 in standard precision (SP) and extra precision (XP) modes. The receptor grids were prepared with a 20 Å side length with the centroids in the centers of SAM (1NW3) and EPZ004777 (simulated DOT1L:EPZ004777 complex) respectively. All Glide options were kept at default settings except that for Glide SP, at most one pose per ligand was written out; for Glide XP, at most five poses per ligand were written out for analysis.
4.3.2. In silico methods for the molecular modeling and optimization of DOT1L binding model
Protein co-crystal structures were attained from the Protein Data Bank. PDB structures 3QOW, 5MW4, 5DRT, 5DTR, 4HRA, and 4ER5, were used for modeling SAM, Novartis’ Compound 7, Compound 2, and Compound 5, EPZ-5676 and EPZ004777 bound to DOT1L, respectively.
Protein Preparation & Alignment:
Protein models were prepared using “Protein Preparation Wizard” in Schrödinger Maestro v11.4. PDB files of DOT1L co-crystal structures were imported and processed using default settings. Schrödinger Prime was used to fill in missing side chains and cap termini. Bond orders were assigned using the CCD database. Default pre-processing included adding hydrogens, and where applicable, assigning bond orders for ligands, capping termini and deleting waters beyond 5 angstroms from heteroatoms. The workspace was analyzed, and all molecules were deleted except for the protein chain, the co-crystal ligand and all waters in the binding site. The most stable state was selected for the co-crystal ligand. H-bond assignments were refined by sampled water orientations in Epik using PROPKA pH. Waters with less than three hydrogen bonds to non-waters were removed. The model underwent retrained minimization, converging heavy atoms to RMSD 0.30 angstroms. Using Maestro “Protein Structure Alignment,” we all prepared protein structures were aligned to the SAM co-crystal structure (PDB: 3QOW) by referencing backbone residues and excluding ligand and flexible loops.
Receptor Grid Generation:
Docking grids were generated using GLIDE “Receptor Grid Generation” in default settings. Co-crystals were excluded from docking grids. A scaling factor of 1.0 and partial charge cutoff of 0.25 for Van der Walls radius scaling were used. No sites or constraints and no excluded volumes were specified (default settings). Rotatable hydroxyl and thiol groups for residues in the SAM flexible loop (126-140) were allowed for those applicable receptor grids.
Ligand Preparation:
Canonical Smiles were converted to SDF file format using BIOVIA Pipeline Pilot (8.0). The structure file was input to Schrodinger LigPrep using the default settings. Ionization states at pH 7.0 +/− 2.0 were generated using Epik; the original state was also kept. Ligands were desalted and tautomeric forms were generated. Specified chiral centers of ligands were maintained and at most 32 representations were generated per ligand and original ligand representations were retained. Using these settings, 22 distinct representations were generated from the 14 compounds.
Ligand Docking:
Ligands, prepared as described above, were docked using Schrodinger Glide XP (precision docking) and flexible ligand sampling under default settings with the previously generated receptor grid files. Van der Waals radii had a scaling factor of 0.8 and a partial charge cutoff of 0.15. Standard Glide settings include sampling of nitrogen inversions, ring conformations, and partial sampling of torsions for amides (penalize nonplanar conformation). Epik state penalties were added to docking score intramolecular hydrogen bonds were rewarded. At most 10 pose per ligand were written out; post-docking minimization included 25 poses per ligand. Generated docking scores were represented by their absolute value.
Molecular Dynamic Simulations:
All atom explicit water simulations were setup using the Desmond “System Builder”. The OPLS3 force field and TIP4P solvent model was used with orthorhombic boundary conditions of 10 Å in each direction. The system was neutralized by adding Na+ ions, in a 0.15 M NaCl buffer solution. The specific parameters are: SAM model: box volume=456770 Å3, 5 Na+ ions; EPZ004777 Model: box volume= 510017 Å3, 6 Na+ ions. The all atom explicit water molecular dynamics simulations were run using as NPT ensemble with default temperature (300K) and pressure (1.01325 bar) using the Desmond Molecular Dynamics workflow with multiple rounds of energy minimization and model relaxation before the production run. Production simulation time was 200 ns and trajectory were recorded in intervals of 200 ps (with otherwise default settings) generating 1000 frames. For analysis, reports were generated using Desmond Simulation Interactions Diagram with standard setting (Supplementary Figure S15).
Supplementary Material
Highlights.
Virtual screening of unique 1,200 nucleoside analogs library against DOT1L.
11 compounds were confirmed and clustered into two distinct chemical classes.
A series of non-nucleoside DOT1L inhibitors was developed by structure-based design.
25 with N-aminoethyl-pyrrolopyrimidin-4-amine scaffold has 40-fold improvement.
Acknowledgments
We are thankful and acknowledge the financial support from the Leukemia Research Foundation and the Cancer Research Committee Fund of the University of Michigan Comprehensive Cancer Center (UMCC) award to Z. Nikolovska-Coleska. The preparation of the reported compounds in this investigation was partially supported by NIH grant No. 1P41 GM086163-01 (entitled: Pilot-Scale Libraries Based on Nucleoside Templates for the ML Initiative, R. C. Reynolds, P.I.). Dr. Robert C. Reynolds would like to thank Drs. Boris Pasche and Ravi Bhatia for support of this research project via start-up funds provided through the Division of Hematology and Oncology at the University of Alabama at Birmingham. Additionally, Judith V. Hobrath of the Drug Discovery Unit in the College of Life Sciences at the University of Dundee gave critical input into manuscript preparation and the DOT1L binding models presented in this work during her time at Southern Research and the University of Alabama at Birmingham. Afoma C. Umeano is supported by the National Cancer Institute, Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship under NIH award number 1F31CA228331-01A1. We would like to thank graduate student Michael Ide at the University of Alabama who was incredibly helpful with the solubility studies. Dr. Stephan Schürer acknowledges support from NIH grants U54HL127624 (Data Coordination and Integration Center for LINCS-BD2K) and U24TR002278 (Illuminating the Druggable Genome Resource Dissemination and Outreach Center, IDG-RDOC). Dr. Schürer thanks OpenEye Scientific Software (https://www.eyesopen.com/) and ChemAxon (https://chemaxon.com/) for making their software tools accessible to the research group via their academic software licenses.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary Data
Supplementary data related to this article are provided.
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Kornberg RD, Chromatin structure: a repeating unit of histones and DNA. Science 1974, 184 (4139), 868–71. [DOI] [PubMed] [Google Scholar]
- 2.Berger SL, Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev 2002, 12 (2), 142–148. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg AD; Allis CD; Bernstein E, Epigenetics: A landscape takes shape. Cell 2007, 128 (4), 635–638. [DOI] [PubMed] [Google Scholar]
- 4.Albert M; Helin K, Histone methyltransferases in cancer. Semin. Cell Dev. Biol 2010, 21 (2), 209–20. [DOI] [PubMed] [Google Scholar]
- 5.Feng Q; Wang H; Ng HH; Erdjument-Bromage H; Tempst P; Struhl K; Zhang Y, Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 2002, 12 (12), 1052–8. [DOI] [PubMed] [Google Scholar]
- 6.Wood K; Tellier M, Murphy S, DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muntean AG; Hess JL, The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol 2012, 7, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada Y; Feng Q; Lin YH; Jiang Q; Li YQ; Coffield VM; Su LS; Xu GL; Zhang Y, hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121 (2), 167–178. [DOI] [PubMed] [Google Scholar]
- 9.Jo SY; Granowicz EM; Maillard I; Thomas D; Hess JL, Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 2011, 117 (18), 4759–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernt KM; Zhu N; Sinha AU; Vempati S; Faber J; Krivtsov AV; Feng Z; Punt N; Daigle A; Bullinger L; Pollock RM; Richon VM; Kung AL; Armstrong SA, MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20 (1), 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller D; Garcia-Cuellar MP; Bach C; Buhl S; Maethner E; Slany RK, Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009, 7 (11), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilden JM; Dinndorf PA; Meerbaum SO; Sather H; Villaluna D; Heerema NA; McGlennen R; Smith FO; Woods WG; Salzer WL; Johnstone HS; Dreyer Z; Reaman GH, Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 2006, 108 (2), 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueller D; Bach C; Zeisig D; Garcia-Cuellar MP; Monroe S; Sreekumar A; Zhou R; Nesvizhskii A; Chinnaiyan A; Hess JL; Slany RK, A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110 (13), 4445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen AT; Taranova O; He J; Zhang Y, DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 2011, 117 (25), 6912–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen C; Jo SY; Liao C; Hess JL; Nikolovska-Coleska Z, Targeting recruitment of Disruptor of Telomeric silencing 1-Like (DOT1L): Characterizing the interactions between DOT1L and Mixed Lineage Leukemia (MLL) fusion proteins. J. Biol. Chem 2013, 288 (42), 30585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H; Zhou B; Qin S; Xu J; Harding R; Tempel W; Nayak V; Li Y; Loppnau P; Dou Y; Min J, Structural and functional analysis of the DOT1L-AF10 complex reveals mechanistic insights into MLL-AF10-associated leukemogenesis. Genes Dev. 2018, 32 (5-6), 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerry J; Godfrey L; Repapi E; Tapia M; Blackledge NP; Ma H; Ballabio E; O’Byrne S; Ponthan F; Heidenreich O; Roy A; Roberts I; Konopleva M; Klose RJ; Geng H; Milne TA, MLL-AF4 Spreading Identifies Binding Sites that Are Distinct from Super-Enhancers and that Govern Sensitivity to DOT1L Inhibition in Leukemia. Cell Rep. 2017, 482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okuda H; Stanojevic B; Kanai A; Kawamura T; Takahashi S; Matsui H; Takaori-Kondo A; Yokoyama A, Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Invest 2017, 127 (5), 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krivtsov AV; Feng Z; Lemieux ME; Faber J; Vempati S; Sinha AU; Xia X; Jesneck J; Bracken AP; Silverman LB; Kutok JL; Kung AL; Armstrong SA, H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14 (5), 355–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monroe SC; Jo SY; Sanders DS; Basrur V; Elenitoba-Johnson KS; Slany RK; Hess JL, MLL-AF9 and MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp. Hematol 2011, 39 (1), 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milne TA; Martin ME; Brock HW; Slany RK; Hess JL, Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005, 65 (24), 11367–74. [DOI] [PubMed] [Google Scholar]
- 22.Saygin C, Carraway HE. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol 2017, 10 (1), 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guppy BJ; Jeusset LMP; McManus KJ, The Relationship Between DOT1L, Histone H3 Methylation, and Genome Stability in Cancer. Curr. Mol. Biol. Rep 2017, 3 (1), 18–27. [Google Scholar]
- 24.Kryczek I; Lin Y; Nagarsheth N; Peng D; Zhao L; Zhao E; Vatan L; Szeliga W; Dou Y; Owens S; Zgodzinski W; Majewski M; Wallner G; Fang J; Huang E; Zou W, IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40 (5), 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho MH; Park JH; Choi HJ; Park MK; Won HY; Park YJ; Lee CH; Oh SH; Song YS; Kim HS; Oh YH; Lee JY; Kong G, DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun 2015, 6, 7821–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L; Deng L; Chen F; Yao Y; Wu B; Wei L; Mo Q; Song Y, Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer. Oncotarget 2014, 5 (21), 10665–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oktyabri D; Ishimura A; Tange S; Terashima M; Suzuki T, DOT1L histone methyltransferase regulates the expression of BCAT1 and is involved in sphere formation and cell migration of breast cancer cell lines. Biochimie 2016, 123, 20–31. [DOI] [PubMed] [Google Scholar]
- 28.Bewersdorf JP; Shallis R; Stahl M; Zeidan AM, Epigenetic therapy combinations in acute myeloid leukemia: what are the options? Ther. Adv. Hematol 2019, 10, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J-Y; Kong G DOT1L: a new therapeutic target for aggressive breast cancer. Oncotarget 2015, 6 (31), 30451–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dafflon C; Tiedt R; Schwaller J, Targeting multiple nodes of MLL complexes to improve leukemia therapy. Oncotarget 2017, 8 (53), 90614–90615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anglin JL; Song Y, A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J. Med. Chem 2013, 56 (22), 8972–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLean CM; Karemaker ID; van Leeuwen F, The emerging roles of DOT1L in leukemia and normal development. Leukemia 2014, 28 (11), 2131–8. [DOI] [PubMed] [Google Scholar]
- 33.Kaniskan HϜ; Martini ML; Jin J, Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev 2018, 118 (3), 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheer S; Ackloo S; Medina TS; Schapira M; Li F; Ward JA; Lewis AM; Northrop JP; Richardson PL; Kaniskan HϜ; Shen Y; Liu J; Smil D; McLeod D; Zepeda-Velazquez CA; Luo M; Jin J; Barsyte-Lovejoy D; Huber KVM; De Carvalho DD; Vedadi M; Zaph C; Brown PJ; Arrowsmith CH, A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun 2019, 10 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh PK Histone methyl transferases: A class of epigenetic opportunities to counter uncontrolled cell proliferation. Eur. J. Med. Chem 2019, 166, 351–368. [DOI] [PubMed] [Google Scholar]
- 36.Zhang JM; Yang PL; Gray NS, Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9 (1), 28–39. [DOI] [PubMed] [Google Scholar]
- 37.Min J; Feng Q; Li Z; Zhang Y; Xu RM, Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 2003, 112 (5), 711–23. [DOI] [PubMed] [Google Scholar]
- 38.Daigle SR; Olhava EJ; Therkelsen CA; Majer CR; Sneeringer CJ; Song J; Johnston LD; Scott MP; Smith JJ; Xiao Y; Jin L; Kuntz KW; Chesworth R; Moyer MP; Bernt KM; Tseng JC; Kung AL; Armstrong SA; Copeland RA; Richon VM; Pollock RM, Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20 (1), 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Y; Chen P; Diao J; Cheng G; Deng L; Anglin JL; Prasad BV; Song Y, Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J. Am. Chem. Soc 2011, 133 (42), 16746–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anglin JL; Deng L; Yao Y; Cai G; Liu Z; Jiang H; Cheng G; Chen P; Dong S; Song Y, Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J. Med. Chem 2012, 55 (18), 8066–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu WY; Smil D; Li FL; Tempel W; Fedorov O; Nguyen KT; Bolshan Y; Al-Awar R; Knapp S; Arrowsmith CH; Vedadi M; Brown PJ; Schapira M, Bromo-deaza-SAH: A potent and selective DOT1L inhibitor. Bioorg. Med. Chem 2013, 21 (7), 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daigle SR; Olhava EJ; Therkelsen CA; Basavapathruni A; Jin L; Boriack-Sjodin PA; Allain CJ; Klaus CR; Raimondi A; Scott MP; Waters NJ; Chesworth R; Moyer MP; Copeland RA; Richon VM; Pollock RM, Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122 (6), 1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu W; Chory EJ; Wernimont AK; Tempel W; Scopton A; Federation A; Marineau JJ; Qi J; Barsyte-Lovejoy D; Yi J; Marcellus R; Iacob RE; Engen JR; Griffin C; Aman A; Wienholds E; Li F; Pineda J; Estiu G; Shatseva T; Hajian T; Al-awar R; Dick JE; Vedadi M; Brown PJ; Arrowsmith CH; Bradner JE; Schapira M, Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun 2012, 3, 1288. [DOI] [PubMed] [Google Scholar]
- 44.Deng L; Zhang L; Yao Y; Wang C; Redell MS; Dong S; Song Y, Synthesis, Activity and Metabolic Stability of Non-Ribose Containing Inhibitors of Histone Methyltransferase DOT1L. MedChemComm 2013, 4 (5), 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waters NJ Preclinical Pharmacokinetics and Pharmacodynamics of Pinometostat (EPZ-5676), a First-in-Class, Small Molecule S-Adenosyl Methionine Competitive Inhibitor of DOT1L. Eur. J. Drug Metab. Pharmacokinet 2017, 42 (6), 891–901. [DOI] [PubMed] [Google Scholar]
- 46.Liu T; Xie W; Li C; Ren H; Mao Y; Chen G; Cheng M; Zhao D; Shen J; Li J; Zhou Y; Xiong B; Chen YL Preparation of 5'-deoxy-5'-amino-5'-C-methyl adenosine derivatives and their activity against DOT1L. Bioorg. Med. Chem. Lett 2017, 27 (22), 4960–4963. [DOI] [PubMed] [Google Scholar]
- 47.Spurr SS; Bayle ED; Yu W; Li F; Tempel W; Vedadi M; Schapira M; Fish PV New small molecule inhibitors of histone methyl transferase DOT1L with a nitrile as a nontraditional replacement for heavy halogen atoms. Bioorg. Med. Chem. Lett 2016, 26 (18), 4518–4522. [DOI] [PubMed] [Google Scholar]
- 48.Chen J; Park HJ, Computer-Aided Discovery of Massonianoside B as a Novel Selective DOT1L Inhibitor. ACS Chem. Biol 2019, 14 (5), 873–881. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L; Chen YT; Liu N; Li LJ; Xiao SH; Li XL; Chen KX; Luo C; Chen SJ; Chen H, Design, synthesis and anti leukemia cells proliferation activities of pyrimidylaminoquinoline derivatives as DOT1L inhibitors. Bioorg. Chem 2018, 80, 649–654. [DOI] [PubMed] [Google Scholar]
- 50.Song Y; Li L; Chen Y; Liu J; Xiao S; Lian F; Zhang N; Ding H; Zhang Y; Chen K; Jiang H; Zhang C; Liu YC; Chen S; Luo C, Discovery of potent DOT1L inhibitors by AlphaLISA based High Throughput Screening assay. Bioorg. Med. Chem 2018, 26 (8), 1751–1758. [DOI] [PubMed] [Google Scholar]
- 51.Sabatino M; Rotili D; Patsilinakos A; Forgione M; Tomaselli D; Alby F; Arimondo PB; Mai A; Ragno R, Disruptor of telomeric silencing 1-like (DOT1L): disclosing a new class of non-nucleoside inhibitors by means of ligand-based and structure-based approaches. J. Comput. Aid. Mol. Des 2018, 32 (3), 435–458. [DOI] [PubMed] [Google Scholar]
- 52.Du L; Grigsby SM; Yao A; Chang Y; Johnson G; Sun H; Nikolovska-Coleska Z, Peptidomimetics for Targeting Protein-Protein Interactions between DOT1L and MLL Oncofusion Proteins AF9 and ENL. ACS Med. Chem. Lett 2018, 9 (9), 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y; Li L; Zhang B; Xing J; Chen S; Wan W; Song Y; Jiang H; Jiang H; Luo C; Zheng M, Discovery of Novel Disruptor of Silencing Telomeric 1-Like (DOT1L) Inhibitors using a Target-Specific Scoring Function for the (S)-Adenosyl-L-methionine (SAM)-Dependent Methyltransferase Family. J. Med. Chem 2017, 60 (5), 2026–2036. [DOI] [PubMed] [Google Scholar]
- 54.Mobitz H; Machauer R; Holzer P; Vaupel A; Stauffer F; Ragot C; Carayatti G; Scheufler C; Fernandez C; Hommel U; Tiedt R; Beyer KS; Chen C; Zhu H; Gaul C, Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach. ACS Med. Chem. Lett 2017, 8 (3), 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheufler C; Mobitz H; Gaul C; Ragot C; Be C; Fernandez C; Beyer KS; Tiedt R; Stauffer F, Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett 2016, 7 (8), 730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo MH; Wang H; Zou Y; Zhang SP; Xiao JH; Jiang GD; Zhang YH; Lai YS, Identification of phenoxyacetamide derivatives as novel DOT1L inhibitors via docking screening and molecular dynamics simulation. J. Mol. Graph. Model 2016, 68, 128–139. [DOI] [PubMed] [Google Scholar]
- 57.Chen SJ; Li LJ; Chen YT; Hu JC; Liu JQ; Liu YC; Liu RF; Zhang YY; Meng FW; Zhu KK; Lu JY; Zheng MY; Chen KX; Zhang J; Jiang HL; Yao ZY; Luo C, Identification of Novel Disruptor of Telomeric Silencing 1-like (DOT1L) Inhibitors through Structure-Based Virtual Screening and Biological Assays. J. Chem. Inf Model 2016, 56 (3), 527–534. [DOI] [PubMed] [Google Scholar]
- 58.Chen C; Zhu H; Stauffer F; Caravatti G; Vollmer S; Machauer R; Holzer P; Mobitz H; Scheufler C; Klumpp M; Tiedt R; Beyer KS; Calkins K; Guthy D; Kiffe M; Zhang J; Gaul C, Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett 2016, 7 (8), 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moukha-chafiq O; Reynolds RC, Parallel Solution-Phase Synthesis of an Adenosine Antibiotic Analog Library. ACS Comb. Sci 2013, 15 (3), 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49 (21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- 61.Mayer M; Meyer B, Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc 2001, 123 (25), 6108–6117. [DOI] [PubMed] [Google Scholar]
- 62.Ellis D, Alternative Approach to 1,2,4-Triazole-3-carboxamides. Synth. Commun 2009, 39 (14), 2585–2595. [Google Scholar]
- 63.Brown AD; Ellis D; Smith CR, Substituted triazole derivatives as oxytocin antagonists. WO 2005028452, 2005. [Google Scholar]
- 64.Koubachi J; Berteina-Raboin S; Mouaddib A; Guillaumet G, Intramolecular arylation reactions: first efficient synthesis of novel fused pyridoimidazoquinolinones or pyridoimidazoazepinones libraries. Tetrahedron 2010, 66 (10), 1937–1946. [Google Scholar]
- 65.Bulger PG; Cottrell IF; Cowden CJ; Davies AJ; Dolling U-H, An investigation into the alkylation of 1,2,4-triazole. Tet. Lett 2000, 41 (8), 1297–1301. [Google Scholar]
- 66.Holm SC; Straub BF, Synthesis of N-Substituted 1,2,4-Triazoles. A Review. Org. Prep. Proced. Int 2011, 43 (4), 319–347. [Google Scholar]
- 67.N-1 Alkylation was confirmed for the ethyl ester of 14a using HMBC and HSQC NMR analyses. See Supplementary Material for spectra.
- 68.Perich JW; Johns RB, The synthesis of multiple O-phosphoseryl-containing peptides via phenyl phosphate protection. J. Org. Chem 1988, 53 (17), 4103–4105. [Google Scholar]
- 69.The yields associated with aminoisoquinoline head group C were uniformly lower than those involving the other head groups due to competitive isobutyl carbamate formation at the exocyclic amine. Interestingly, corresponding byproducts were not observed from coupling reactions involving D.
- 70.Zbinden KG; Anselm L; Banner DW; Benz J; Blasco F; Décoret G; Himber J; Kuhn B; Panday N; Ricklin F; Risch P; Schlatter D; Stahl M; Thomi S; Unger R; Haap W, Design of novel aminopyrrolidine factor Xa inhibitors from a screening hit. Eur. J. Med. Chem 2009, 44 (7), 2787–2795. [DOI] [PubMed] [Google Scholar]
- 71.For a list of calculated physicochemical properties see Supplementary Table S4. Compound 25 was experimentally determined to have a kinetic solubility of 686.7±25.7 μM in 4% DMSO/96% 80 mM tris HCl, pH 8.0 buffer at 37 oC—see Supplementary Figures S20 and S21 and corresponding procedures for details.
- 72.Lee YT; Gibbons G; Lee SY; Nikolovska-Coleska Z; Dou Y, One-pot refolding of core histones from bacterial inclusion bodies allows rapid reconstitution of histone octamer. Protein Expr. Purif 2015, 110, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lowary PT; Widom J, New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 1998, 276 (1), 19–42. [DOI] [PubMed] [Google Scholar]
- 74.Dyer PN; Edayathumangalam RS; White CL; Bao YH; Chakravarthy S; Muthurajan UM; Luger K, Reconstitution of nucleosome core particles from recombinant histones and DNA. Method Enzymol. 2004, 375, 23–44. [DOI] [PubMed] [Google Scholar]
- 75.Mezzasalma TM; Kranz JK; Chan W; Struble GT; Schalk-Hihi C; Deckman IC; Springer BA; Todd MJ, Enhancing recombinant protein quality and yield by protein stability profiling. J. Biomol. Screen 2007, 12 (3), 418–428. [DOI] [PubMed] [Google Scholar]
- 76.Muntean AG; Giannola D; Udager AM; Hess JL, The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood 2008, 112 (12), 4690–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29 (9), 2176–2179. [Google Scholar]
- 78.Li QL; Chen TJ; Wang YL; Bryant SH, PubChem as a public resource for drug discovery. Drug Discov. Today 2010, 15 (23-24), 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.