

Abstract
Coronavirus disease 2019 (COVID 19) was first identified in Wuhan, China near the end of 2019. To date, COVID‐19 had spread to almost 235 countries and territories due to its highly infectious nature. Moreover, there is no vaccine or Food and Drug Administration (FDA)‐approved drug. More time is needed to establish one of them. Consequently, the drug repurposing approach seems to be the most attractive and quick solution to accommodate this crisis. In this regard, we performed molecular docking‐based virtual screening of antiplatelet FDA‐approved drugs on the key two viral target proteins: main protease (Mpro) and spike glycoprotein (S) as potential inhibitor candidates for COVID‐19. In the present study, 15 antiplatelet FDA‐approved drugs were investigated against the concerned targets using the Molecular Docking Server. Our study revealed that only cilostazol has the most favorable binding interaction on Mpro (PDB ID: 6LU7) and cilostazol, iloprost, epoprostenol, prasugrel, and icosapent ethyl have a higher binding affinity on spike glycoprotein (S) (PDB ID: 6VYB) compared with recent anti‐CoVID‐19. Therefore, cilostazol is a promising FDA drug against COVID‐19 by inhibiting both Mpro and S protein. The insights gained in this study may be useful for quick approach against COVID‐19 in the future.
Keywords: antiplatelet, COVID‐19, molecular docking, Mpro , SARS‐CoV‐2, spike glycoprotein
1. INTRODUCTION
Coronaviruses (CoVs) are an etiologic factor of mild to severe respiratory tract infections in both animals and humans. Previous studies of CoVs revealed that more surely pathogenic viruses associated with high mortality rates, the severe acute respiratory syndrome coronavirus (SARS‐CoV) in 2003, and the Middle East respiratory syndrome coronavirus (MERS‐CoV) in 2012 (Paules, Marston, & Fauci, 2020). The novel coronavirus was reported on December 30, 2019, in Wuhan City, Hubei Province, P.R. China (Xu et al., 2020). At first, it was identified as 2019 novel coronavirus (2019‐nCoV) and renamed as severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) by the World Health Organization (WHO). As of March 11, 2020, WHO has stated that COVID‐19 has been categorized as a pandemic (Ramphul & Mejias, 2020). By April 23, 2020, 2,397,217 confirmed cases and 162,956 deaths in 235 countries and territories were recorded [World Health Organization (WHO), 2020].
SARS‐CoV‐2 is an enveloped positive single‐stranded RNA virus classified from betacoronavirus (β‐CoV) family, which contains other members including SARS‐CoV and MERS‐CoV (Chan et al., 2015; Salata, Calistri, Parolin, & Palù, 2019). The viral nucleocapsid consists of genomic genome RNA and nucleocapsid protein (N), which is embedded inside phospholipid bilayers and is protected by two separate forms of spike proteins: spike glycoprotein trimmer (S) in all CoVs, and the hemagglutinin‐esterase HE in some CoVs. The membrane protein (M) (transmembrane glycoprotein type III) and the envelope protein (E) are positioned among the S proteins in the viral envelope. CoVs were named based on the crown‐shaped appearance (Li et al., 2020). The estimated structure of SARS‐CoV‐2 is shown in Figure 1.
FIGURE 1.

The estimated structure of SARS‐CoV‐2
SARS‐CoV‐2 causes severe respiratory tract infection in humans utilizing angiotensin‐converting enzyme 2 (ACE2) receptors as a gate to infect epithelial cells of the lungs by attachment of spike glycoprotein (S) (Chen, Guo, Pan, & Zhao, 2020). The genomic sequence of SARS‐CoV‐2 was isolated and obtained by Lu et al. (2020) also the crystal structure of COVID‐19 main protease (Mpro) was confirmed by Jin et al. that considered as a potential drug target protein for inhibition of SARS‐CoV‐2 replication. The Mpro is a key protein in preventing virus maturation (Jin et al., 2020). Hence, targeting nonstructural (Mpro) and structural (S) proteins has a promising approach for effective treatment against SARS‐CoV‐2 (Sohag et al., 2020).
Scientists investigate alternative therapies for COVID‐19 using artificial intelligence for identification of possible candidates. Many researchers working in the field of drug repurposing use Drug bank and molecular docking software to hopefully find potential treatment. Drug repurposing (also commonly named as drug repositioning) is a drug development strategy used to identify novel uses for existing approved and investigational drugs outside of their original indication. In comparison to conventional pipelines for drug production, this approach has many advantages. Unlike conventional drug production, which could be ineffective in preclinical and early stage clinical trials based on safety issues, this risk is mitigated by the use of drugs that have demonstrated safety records in previous studies. Accordingly, drug repurposing is also significantly more efficient and cost‐effective than traditional drug development since preclinical and early stage clinical trials do not need to be repeated (Pushpakom et al., 2018).
In past respiratory virus pandemics, such as H1N1 influenza, therapeutical anticoagulants have been used (Obi et al., 2019). A recent study suggests that the use of heparin as a prophylactic agent in 99 patients has been associated with an improvement in mortality in a cohort study of 449 COVID‐19 patients from Wuhan, China. However, the rate of prophylactic anticoagulants was low, further prospective studies are needed to confirm this hypothesis (Tremblay et al., 2020). Also, Xijing Hospital started the clinical trial proposing the early usage of aspirin is expected to reduce the incidence of severe and critical COVID‐19 patients, minimize their hospital staying, and avoid the occurrence of cardiovascular complications based on aspirin role as antivirus replication, antiplatelet aggregation, antiinfection, and antilung injury (NCT04365309, 2020); which raises the question of whether the antiplatelets may play a role in the treatment of COVID‐19.
To answer this question, we performed molecular docking‐based virtual screening of antiplatelet Food and Drug Administration (FDA)‐approved drugs on the following two viral target proteins: main protease (Mpro) and spike glycoprotein (S) as potential inhibitor candidates for COVID‐19.
2. MATERIAL AND METHODS
2.1. Molecular docking platform
The computational investigations were performed using the Molecular Docking Server (Bikadi & Hazai, 2009) (https://www.dockingserver.com) based on AutoDock 4 for docking calculation. In cases where protein and ligand partial charges were calculated with the PM6 method using MOPAC2009 software (Huey, Morris, Olson, & Goodsell, 2007; Stewart, 2009).
2.2. Ligand determination and preparation
According to Drugbank database, 47 antiplatelet drugs (DBCAT000149), agents which antagonize any mechanism leading to blood platelet aggregation, were selected. Only small FDA‐approved molecules were summarized into 15 candidates (Table 1) comparing with the recently used and predictable COVID‐19 inhibitors outlined in Table 2. PubChem database was used to extract out the three‐dimensional (3D) chemical structures of the selected molecules. The 3D and geometry optimizations with energy minimization of ligands were executed using algorithms monitored in Docking Server. Ligand preparation module used the included Merck Molecular Force Field 94 (MMFF94) as Geometry optimization method and Gasteiger as Charge calculation method at pH 7.
TABLE 1.
List of antiplatelet FDA‐approved drugs docked against COVID‐19
| Name | Accession number | Molecular weight (g/mol) | Structure |
|---|---|---|---|
| Anagrelide | DB00261 | 256.079 |

|
| Aspirin | DB00945 | 180.16 |

|
| Cangrelor | DB06441 | 776.4 |

|
| Cilostazol | DB01166 | 369.5 |

|
| Clopidogrel | DB00758 | 321.8 |

|
| Dipyridamole | DB00975 | 504.6 |

|
| Epoprostenol | DB01240 | 352.5 |

|
| Icosapent ethyl | DB08887 | 330.5 |

|
| Iloprost | DB01088 | 360.5 |
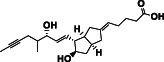
|
| Pentoxifylline | DB00806 | 278.31 |

|
| Prasugrel | DB06209 | 373.4 |
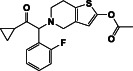
|
| Ticagrelor | DB08816 | 522.6 |

|
| Ticlopidine | DB00208 | 263.8 |

|
| Tirofiban | DB00775 | 440.6 |

|
| Vorapaxar | DB09030 | 492.6 |

|
TABLE 2.
List of recent inhibitors against COVID‐19
| Name | Accession number | Molecular weight (g/mol) | Structure | Mechanism of action |
|---|---|---|---|---|
| Darunavir | DB01264 | 547.7 |

|
An inhibitor of HIV protease |
| Hydroxychloroquine | DB01611 | 335.9 |

|
Inhibits antigen processing, and reduces the inflammatory response |
| Nelfinavir | DB00220 | 567.8 |

|
A potent HIV‐1 protease inhibitor |
| Umifenovir | DB13609 | 477.4 |

|
Direct virucidal effects and a host‐targeting agent (HTA) |
2.3. Protein determination and preparation
Two SARS‐CoV‐2 proteins were chosen as drug inhibition targets: main protease (Mpro) (PDB ID: 6LU7) (Jin et al., 2020) and spike glycoprotein (S) (PDB ID: 6VYB) (Walls et al., 2020) and obtained from RCSB Protein Data Bank (http://www.rscb.org). Protein structures were prepared using protein preparation wizard in Molecular Docking Server panel. Bond orders were assigned and hydrogen atoms were added as well. Water molecules and other nonspecific molecules were removed. Affinity (grid) maps of 20 × 20 × 20 Å grid points and 0.375 Å spacing were generated using the Autogrid program (Morris et al., 1998). AutoDock parameter set‐ and distance‐dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively.
2.4. Computational methods
Docking simulations were performed using the Lamarckian genetic algorithm (LGA) and the Solis and Wets local search method (Solis & Wets, 1981). Initial position, orientation, and torsions of the ligand molecules were set randomly. Each docking experiment was derived from 100 different runs that were set to terminate after a maximum of 2,500,000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied. After each docking calculation, the root mean square deviation (RMSD) between the lowest energy docked ligand pose and the complex crystal structure ligand pose was evaluated. For pose selection, the pose with the lowest RMSD was determined from all poses performed by the docking program. Molecular Docking Server output results represented the estimated free energy of binding (kcal/mol) as ΔG values. They were further converted to the estimated inhibition constants (K i). The K i values for analyzed docking poses were calculated from the ΔG parameters as follows (Huey et al., 2007):
where, R (gas constant) is 1.98 cal (mol K)−1, and T (room temperature) is 298.15 K.
After docking, the complexes were analyzed using the Protein–Ligand Interaction Profiler (PLIP) web server (Technical University of Dresden) (Salentin, Schreiber, Haupt, Adasme, & Schroeder, 2015).
3. RESULTS AND DISCUSSION
In the current study, the parameters of estimated free energy of binding, inhibition constant (K i), total estimated energy of vdW + Hbond + desolv (EVHD), electrostatic energy, total intermolecular energy, frequency of binding, and interacting surface area were evaluated to estimate the favorable binding of antiplatelet FDA‐approved drugs against COVID‐19 (Mpro) and spike glycoprotein (S).
3.1. Molecular docking of antiplatelet FDA‐approved drugs against COVID‐19 (Mpro)
The results of molecular docking showed that cilostazol has the most favorable binding interaction on Mpro (PDB ID: 6LU7) with estimated free energy of binding −8.48 kcal/mol, and inhibition constant (K i) 612.08 nM while nelfinavir with estimated free energy of binding −7.69 kcal/mol, and inhibition constant (K i) 2.31 μM. However, hydroxychloroquine, umifenovir, and darunavir were recently reported to have a potent inhibition effect against SARS‐CoV‐2 (Devaux, Rolain, Colson, & Raoult, 2020; Harrison, 2020; Wang, Chen, Lu, Chen, & Zhang, 2020). Those revealed lower binding affinity to Mpro with estimated free energy of binding −7.06, −6.51, and −6.47 kcal/mol, respectively, than ticagrelor, ticlopidine, and prasugrel, estimated free energy of binding −7.51, −7.34, and −7.29 kcal/mol, respectively (Table 3).
TABLE 3.
Results of the docking of antiplatelet FDA‐approved drugs versus common inhibitors on the crystal structure of COVID‐19 (Mpro) (PDB ID: 6LU7)
| No. | Drug name | Est. free energy of binding kcal/mol | Est. inhibition constant, K i | vdW + Hbond + desolv energy kcal/mol | Electrostatic energy kcal/mol | Total intermolec. energy kcal/mol | Frequency | Interact. Surface |
|---|---|---|---|---|---|---|---|---|
| 1 | Cilostazol | −8.48 | 612.08 nM | −9.77 | +0.04 | −9.73 | 4% | 736.864 |
| 2 | Nelfinavir | −7.69 | 2.31 μM | −9.87 | −0.44 | −10.30 | 1% | 870.696 |
| 3 | Ticagrelor | −7.51 | 3.13 μM | −8.82 | −0.09 | −8.91 | 5% | 769.423 |
| 4 | Ticlopidine | −7.34 | 4.18 μM | −7.24 | −0.39 | −7.63 | 49% | 649.363 |
| 5 | Prasugrel | −7.29 | 4.57 μM | −7.67 | −0.39 | −8.06 | 1% | 707.148 |
| 6 | Hydroxychloroquine | −7.06 | 6.63 μM | −7.85 | −1.59 | −9.44 | 6% | 654.874 |
| 7 | Umifenovir | −6.51 | 16.85 μM | −8.37 | +0.01 | −8.36 | 9% | 782.885 |
| 8 | Clopidogrel | −6.50 | 17.27 μM | −6.03 | −0.42 | −6.45 | 25% | 550.592 |
| 9 | Vorapaxar | −6.48 | 17.74 μM | −8.25 | +0.01 | −8.23 | 10% | 757.885 |
| 10 | Darunavir | −6.47 | 18.02 μM | −7.31 | −0.06 | −7.37 | 1% | 678.669 |
| 11 | Epoprostenol | −6.36 | 21.66 μM | −9.14 | −0.11 | −9.25 | 19% | 771.183 |
| 12 | Iloprost | −6.20 | 28.63 μM | −9.45 | +0.05 | −9.40 | 4% | 793.819 |
| 13 | Tirofiban | −6.02 | 38.42 μM | −6.80 | −1.30 | −8.10 | 3% | 658.473 |
| 14 | Anagrelide | −5.93 | 44.85 μM | −5.92 | −0.02 | −5.93 | 11% | 575.394 |
| 15 | Pentoxifylline | −5.31 | 127.89 μM | −6.74 | +0.01 | −6.74 | 12% | 691.698 |
| 16 | Icosapent ethyl | −5.20 | 154.88 μM | −8.34 | −0.06 | −8.41 | 14% | 805.185 |
| 17 | Aspirin | −3.94 | 1.29 mM | −4.99 | +0.14 | −4.85 | 95% | 447.578 |
| 18 | Dipyridamole | −3.55 | 2.48 mM | −7.31 | −0.14 | −7.45 | 17% | 631.259 |
| 19 | Cangrelor | −2.90 | 7.49 mM | −9.72 | +0.50 | −9.22 | 1% | 983.638 |
Figure 2 demonstrates the PLIP analysis for the docked structures of Mpro to cilostazol (Figure 2a) and nelfinavir (Figure 2b). The Mpro is shown in colored ribbon surface model. The ligands are shown in orange sticks, where it seems to fit into the Mpro binding site pocket. Enlarged views of the binding sites show how the interactions are established when docking. Binding site residues of Mpro are represented in blue sticks and labeled with its three‐letter code. In Figure 2, the hydrophobic interactions are described in dashed‐gray lines, while H‐bonds are illustrated in solid blue lines. Interestingly, most residues are predominantly non‐hydrophobic, whereas all docking complexes are dominated by hydrophobic interactions.
FIGURE 2.

Predicted binding models obtained from the docking simulation analysis of cilostazol and nelfinavir against COVID‐19 main protease (Mpro). Structure of Mpro is shown as ribbon surface model. The cilostazol and nelfinavir are represented as orange stick model. (a) SARS‐CoV‐2 Mpro–cilostazol complex. (b) SARS‐CoV‐2 Mpro–nelfinavir complex. The active site residues in the expanded panels are represented in blue sticks. H‐bonds and hydrophobic interactions are shown by blue lines, dashed‐gray lines, respectively
3.2. Molecular docking of antiplatelet FDA‐approved drugs against COVID‐19 spike glycoprotein (S)
SARS‐CoV‐2 can enter to the host cell by binding on human angiotensin converting enzyme 2 (hACE2) receptor by spike glycoprotein (S). Subsequently, targeting the protein (S) plays a key role in treatment of COVID‐19 (Ou et al., 2020). Furthermore, molecular docking results of antiplatelet FDA‐approved drugs against COVID‐19 spike glycoprotein (S) showed that cilostazol, iloprost, epoprostenol, prasugrel, and icosapent ethyl have the most promising binding interaction with spike glycoprotein (S) (PDB ID: 6VYB), estimated free energy of binding −9.97, −9.68, −9.07, −9.00, and −8.29 kcal/mol, respectively, and inhibition constant (K i) 48.86, 80.24, 222.84, 251.80, and 842.93 nM, respectively. While nelfinavir showed lowest binding affinity to (PDB ID: 6VYB) with estimated free energy of binding −7.78 kcal/mol, and inhibition constant (K i) 1.98 μM (Table 4).
TABLE 4.
Results of the docking of antiplatelet FDA‐approved drugs versus common inhibitors on the crystal structure of COVID‐19 spike glycoprotein (S) (PDB ID: 6VYB)
| No. | Drug name | Est. free energy of binding kcal/mol | Est. inhibition constant, K i | vdW + Hbond + desolv energy kcal/mol | Electrostatic energy kcal/mol | Total intermolec. energy kcal/mol | Frequency | Interact. Surface |
|---|---|---|---|---|---|---|---|---|
| 1 | Cilostazol | −9.97 | 48.86 nM | −11.54 | −0.06 | −11.60 | 14% | 812.91 |
| 2 | Iloprost | −9.68 | 80.24 nM | −12.05 | −0.89 | −12.94 | 13% | 923.442 |
| 3 | Epoprostenol | −9.07 | 222.84 nM | −10.62 | −0.63 | −11.25 | 4% | 853.329 |
| 4 | Prasugrel | −9.00 | 251.80 nM | −10.69 | +0.60 | −10.09 | 27% | 815.205 |
| 5 | Icosapent ethyl | −8.29 | 842.93 nM | −12.08 | +0.00 | −12.08 | 10% | 917.712 |
| 6 | Nelfinavir | −7.78 | 1.98 μM | −12.95 | +0.75 | −12.20 | 8% | 1,180.62 |
| 7 | Clopidogrel | −7.77 | 2.01 μM | −8.74 | +0.66 | −8.09 | 46% | 718.458 |
| 8 | Ticagrelor | −7.77 | 2.01 μM | −8.74 | +0.66 | −8.09 | 46% | 718.458 |
| 9 | Ticlopidine | −7.74 | 2.13 μM | −8.76 | +0.73 | −8.04 | 89% | 669.71 |
| 10 | Anagrelide | −7.31 | 4.36 μM | −7.30 | −0.02 | −7.31 | 57% | 630.015 |
| 11 | Vorapaxar | −7.10 | 6.23 μM | −8.83 | +0.02 | −8.81 | 43% | 1,076.541 |
| 12 | Umifenovir | −6.98 | 7.64 μM | −9.20 | −0.03 | −9.23 | 21% | 903.724 |
| 13 | Hydroxychloroquine | −6.61 | 14.22 μM | −9.59 | +1.13 | −8.46 | 10% | 798.232 |
| 14 | Tirofiban | −6.16 | 30.44 μM | −10.20 | +0.79 | −9.41 | 13% | 1,109.697 |
| 15 | Darunavir | −6.01 | 39.03 μM | −10.21 | +0.06 | −10.14 | 2% | 1,017.412 |
| 16 | Pentoxifylline | −5.96 | 42.88 μM | −7.35 | −0.02 | −7.38 | 59% | 735.023 |
| 17 | Aspirin | −5.02 | 209.08 μM | −4.84 | −1.07 | −5.91 | 29% | 574.815 |
| 18 | Dipyridamole | +32.75 | – | +29.36 | −0.11 | +29.25 | 18% | 1,036.243 |
| 19 | Cangrelor | +22.08 | – | +16.00 | −2.53 | +13.46 | 5% | 1,075.65 |
On the other hand, other investigated antiplatelet FDA‐approved drugs like: clopidogrel, ticagrelor, ticlopidine, anagrelide, and vorapaxar showed better binding affinity with an estimated free energy of binding −7.77, −7.77, −7.74, −7.31, and −7.10 kcal/mol, respectively, than umifenovir, hydroxychloroquine, and darunavir with an estimated free energy of binding −6.98, −6.61, and −6.01 kcal/mol, respectively (Table 4).
Figures 3 and 4 illustrate the PLIP analysis for the docked structures of spike glycoprotein (S) to cilostazol, iloprost, epoprostenol, prasugrel, icosapent ethyl and nelfinavir, respectively. The spike glycoprotein (S) is shown in colored ribbon surface model. The ligands are shown in orange sticks, where it seems to fit into the spike glycoprotein (S) binding site pocket. Enlarged views of the binding sites show how the interactions are established when docking. Binding site residues of spike glycoprotein are demonstrated in blue sticks and labeled with its three‐letter code. The hydrophobic interactions are described in dashed‐gray lines, while H‐bonds are illustrated in solid blue lines. As shown in Mpro ligand interactions, also hydrophobic interactions are dominant in all spike protein docked complexes. For prasugrel, only one hydrogen bond is constructed with Q 1010C, while seven hydrogen bonds are reported in nelfinavir.
FIGURE 3.
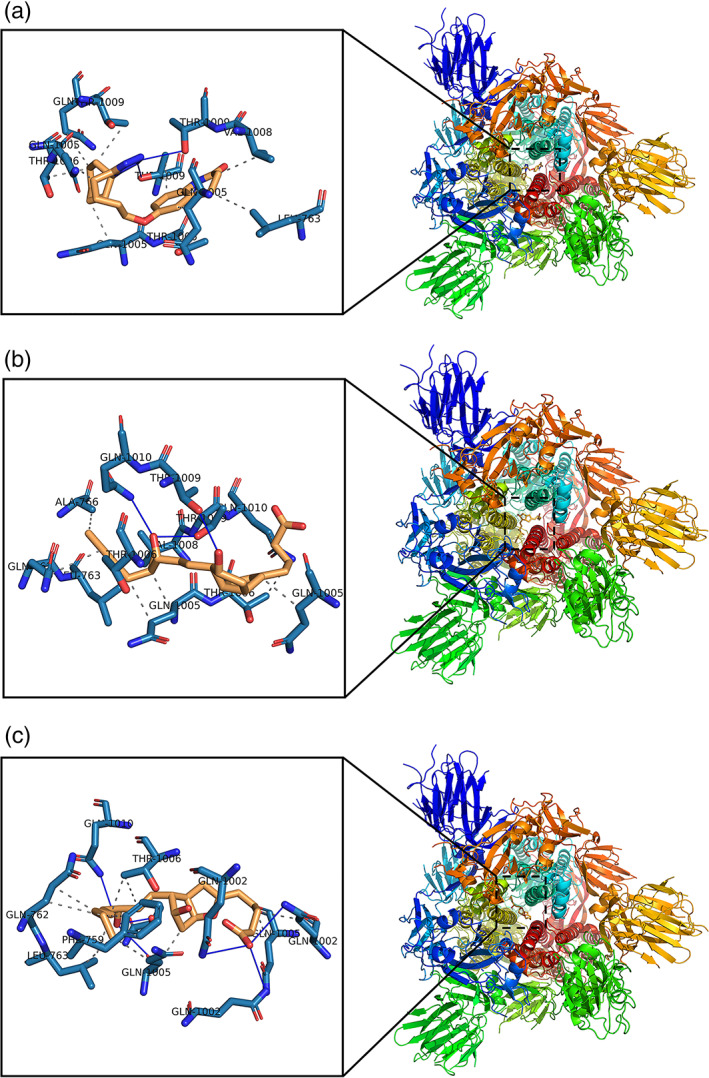
Predicted binding models obtained from the docking simulation analysis of cilostazol, iloprost, and epoprostenol against COVID‐19 Spike Glycoprotein (S). Structure of (S) protein is shown as ribbon surface model. The cilostazol, iloprost, and epoprostenol are represented as orange stick model. (a) SARS‐CoV‐2 (S)–cilostazol complex. (b) SARS‐CoV‐2 (S)–iloprost complex. (c) SARS‐CoV‐2 (S)–epoprostenol complex. The active site residues in the expanded panels are represented in blue sticks. H‐bonds and hydrophobic interactions are shown by blue lines, dashed‐gray lines, respectively
FIGURE 4.

Predicted binding models obtained from the docking simulation analysis of prasugrel, icosapent ethyl, and nelfinavir against COVID‐19 spike glycoprotein (S). Structure of (S) protein is shown as ribbon surface model. The prasugrel, icosapent ethyl, and nelfinavir are represented as orange stick model. (a) SARS‐CoV‐2 (S)–prasugrel complex. (b) SARS‐CoV‐2 (S)–icosapent ethyl complex. (c) SARS‐CoV‐2 (S)–nelfinavir complex. The active site residues in the expanded panels are represented in blue sticks. H‐bonds and hydrophobic interactions are shown by blue lines, dashed‐gray lines, respectively
Tables 5 and 6 summarize the interactions established between antiplatelet FDA‐approved drugs and target proteins Mpro and protein S, respectively. Two kinds of interactions are dominant, the H‐bonding and the hydrophobic interactions. Additionally, halogen bond (residues in bold in the tables) is reported between the Mpro residues E 166, Q 189, and F 140 with prasugrel, clopidogrel, and anagrelide, respectively, or between the spike protein residue Q 1002C with anagrelide. Hydrophobic interactions are more dominant compared with the H‐bonding, as can be seen from almost all selected antiplatelet FDA‐approved drugs with Mpro and S protein.
TABLE 5.
The interactions constructed between antiplatelet FDA‐approved drugs and SARS‐CoV‐2 Mpro
| Compound | Est. free energy of binding kcal/mol | H‐bonding | Hydrophobic interaction and others | ||
|---|---|---|---|---|---|
| Number | Residues of SARS‐CoV‐2 Mpro involved | Number | Residues of SARS‐CoV‐2 Mpro involved | ||
| Cilostazol | −8.48 | 2 | Q 192(2) | 3 | E 166 and Q 189(2) |
| Nelfinavir | −7.69 | 3 | E 166 (3) | 6 | M 165, E 166, L 167, P 168(2), and A 191 |
| Ticagrelor | −7.51 | 1 | A 191 | 3 | E 166(2), Q 189 |
| Ticlopidine | −7.34 | 1 | E 166 | 2 | M 165 and E 166 |
| Prasugrel | −7.29 | 3 | T 190, Q 192 (2) | 4 | F 140, M 165, L 167, and Q 192, E 166 |
| Hydroxychloroquine | −7.06 | 5 | E 166 (2), R 188, T 190, and Q 192 | 0 | – |
| Umifenovir | −6.51 | 0 | – | 3 | E 166 and Q 189(2) |
| Clopidogrel | −6.50 | 0 | – | 1 | P 168, Q 189 |
| Vorapaxar | −6.48 | 0 | – | 5 | M 165, P168(2) Q 189, and A 191 |
| Darunavir | −6.47 | 0 | – | L 50, L 167, A 191, and Q 192 | |
| Epoprostenol | −6.36 | 5 | Q 189(2), T 190, A191, and Q 192 | 5 | M 165, P 168, Q 189(2), and A 191 |
| Iloprost | −6.20 | 3 | Q 189, T 190, and Q 192 | 7 | L 50, F 140, M 165, E 166, P 168, and Q 189(2) |
| Tirofiban | −6.02 | 4 | E 166(3) and Q 189 | 5 | M 165, E 166, P 168, and Q 189(2) |
| Anagrelide | −5.93 | 2 | E 166 (2) | 1 | E 166 (1), F 140 |
| Pentoxifylline | −5.31 | 1 | Q 192 | 1 | F 140 |
| Icosapent ethyl | −5.20 | 0 | – | 5 | F 140, M 165, E 166, P 168, and Q 189 |
| Aspirin | −3.94 | 2 | E 166, T 190 | 5 | M 165, L 167, P 168, Q 189, and Q192 |
| Dipyridamole | −3.55 | 5 | E 166(3), G 170, and T 190 | 1 | Q189 |
| Cangrelor | −2.90 | 3 | S 46, L 50, and Q 189 | 1 | E 166 |
Notes: Bold residues are interacting through halogen bond.
TABLE 6.
The interactions constructed between antiplatelet FDA‐approved drugs and SARS‐CoV‐2 Spike protein
| Compound | Est. free energy of binding kcal/Mol | H‐bonding | Hydrophobic interaction and others | ||
|---|---|---|---|---|---|
| Number | Residues of SARS‐CoV‐2 spike involved | Number | Residues of SARS‐CoV‐2 spike involved | ||
| Cilostazol | −9.97 | 3 | T 1006C, T 1009A, and T 1009C | 9 | L 763A, Q 1005A, Q 1005B, Q 1005C, T 1006B, V 1006A, T 1009B, T 1009C, and Q 1010B |
| Iloprost | −9.68 | 3 | T 1009B, T 1009C, and Q 1010B | 10 | Q 762C, L 763C, A 766C, Q 1005A, Q 1005C(2), T 1006B, T 1006C, V 1008C, and Q 1010C |
| Epoprostenol | −9.07 | 6 | Q 1002A, Q 1002B, Q 1002C, Q 1005B, T 1009B, and Q 1010A | 11 | F 759B, Q 762B(2), L 763B(2), Q 1002C, Q 1005B(2), Q 1004C, and T 1006A(2) |
| Prasugrel | −9.00 | 1 | Q 1010C | 10 | Q 1005A, Q 1005B, Q 1005C, T 1006A, T 1006B, T 1006C, V 1008A, T 1009B, T 1009C, and L 1012A |
| Icosapent ethyl | −8.29 | 3 | T 1009B(2) and Q 1010A | 13 | Q 762B, L 763A, L 763B, A 766B, Q 1002B, Q 1002C, Q 1005A, Q 1005C, T 1006A, T 1006C, V 1008A, T1009C, and L 1012A |
| Nelfinavir | −7.78 | 7 | Q 762B, T 1006A, T 1009A, T 1009B(3), and T 1009C | 17 | F 759B, Q 762B, L 763A, L 763B, Q 1002A, Q 1002B, Q 1005A, Q 1005B, Q 1005C, T 1006A, T 1006B, T 1006C, V 1008A, V 1008B, T 1009 B, T 1009C, and Q 1010C |
| Clopidogrel | −7.77 | 2 | Q 1005A and T 1006C | 3 | Q 1005B, T 1009A, and T 1009B |
| Ticagrelor | −7.77 | 2 | Q 1005A and T 1006C | 3 | Q 1005B, T 1009A, and T 1009B |
| Ticlopidine | −7.74 | 2 | Q 1005A and T 1009A | 6 | L 763A, Q 1002A, Q 1005A, T 1006C, T 1009C, and Q 1010C |
| Anagrelide | −7.31 | 3 | T 1006A, T 1009A and Q 1010A | 1 | Q 1005A, Q 1002C |
| Vorapaxar | −7.10 | 1 | T 1009B | 14 | Q 762C, L 763A, L 763C, A 766A, A 766C, Q 1002B, Q 1005A (2), Q 1005C, T 1006B, T 1006C, V 1008A, V 1008C, and Q 1010B |
| Umifenovir | −6.98 | 4 | Q 1002B, T 1006B, T 1009A, and T 1009C | 7 | L 763A, A 766A, Q 1002C, T 1006A, V 1008A, Q 1010C, and L 1012A |
| Hydroxychloroquine | −6.61 | 5 | Q 1002A, Q 1005A, Q 1005B, and T 1009C(2) | 6 | Q 1005A, T 1006A, T 1006B, T 1006C, T 1009A, and T 1009C |
| Tirofiban | −6.16 | 6 | Q 1005B(2), T 1009A, Q 1010B(2), and R 1014B | 11 | Q 762C (2), L 763C, Q 1005B, Q 1005C, T 1006A, T 1006B, V 1008B, V 1008C, T 1009B, and Q 1010B |
| Darunavir | −6.01 | 5 | Q 1005C, T 1009A, T 1009C(2), and Q 1010C | 12 | L 763B, Q 1002A, Q 1002C, Q 1005A, Q 1005B (2), Q 1005C, T 1006A, T 1009C, Q 1010C, L 1012A, and I 1013C |
| Pentoxifylline | −5.96 | 4 | Q 1002B, T 1006C, T 1009A, and T 1009C | 4 | Q 1005A, T 1006C, V 1008A, and T 1009C |
| Aspirin | −5.02 | 2 | T 1009A and T 1009C | 3 | Q 1002C, Q 1005C, and T 1009C |
| Cangrelor | +22.08 | 10 | Q 762B, L 1001A, Q 1002C, Q 1005A, Q 1005B, T 1006A, T 1006B, T 1009A, T 1009C, and Q 1010A | 1 | Q 1005A |
| Dipyridamole | +32.75 | 7 | C 760C, Q 762C, Q 1005C, T 1006A, T 1009A, and T 1009C(2) | 5 | Q 1002B, Q 1005B, V 1008C, L 1012C, and I 1013B |
Notes: Bold residues are interacting through halogen bond.
Figure 5 summarizes the estimated free binding energy of antiplatelet FDA‐approved drugs against COVID‐19 (Mpro) and spike glycoprotein (S) and shows that cilostazol has the lowest free binding energy −8.48 and −9.97 kcal/mol among Mpro and spike glycoprotein, respectively, which suggests that cilostazol is a promising drug for inhibition of both Mpro and S protein in the treatment of COVID‐19.
FIGURE 5.

Estimated free energy of binding (−kcal/mol) for antiplatelet FDA‐approved drugs with COVID‐19 main protease (PDB ID: 6LU7) and spike protein (PDB ID: 6VYB)
4. CONCLUSION
In the past few months, COVID‐19's rapidly spread outbreak has raised challenges to the global health market. To date, there is no effective vaccine or approved medication to treat this disease. Given the time needed to establish one of these alternatives, the drug repurposing approach appears to be the most attractive and quick. To help counter COVID‐19, the virtual molecular screening was carried out to classify antiplatelet FDA‐approved drugs that are capable of linking COVID‐19 with the Mpro and S protein. Among all antiplatelet FDA‐approved drugs, cilostazol showed a promising FDA drug against COVID‐19 by inhibiting both Mpro and S protein. In order to turn these potential inhibitors into therapeutic medicines, more in vitro and in vivo tests are required. The insights gained in this study may be useful for studying and designing new therapeutic anti‐COVID‐19 agents in the future.
ETHICS STATEMENT
The authors declare no conflicts of interest regarding financial and/or personal relationships with other people or organizations that could inappropriately influence (bias) this work. No work was done on animals and/or humans.
Abosheasha MA, El‐Gowily AH. Superiority of cilostazol among antiplatelet FDA‐approved drugs against COVID 19 Mpro and spike protein: Drug repurposing approach. Drug Dev Res. 2021;82:217–229. 10.1002/ddr.21743
Mohammed A. Abosheasha and Afnan H. El‐Gowily contributed equally to this study.
Contributor Information
Mohammed A. Abosheasha, Email: mohammed-abosheasha@ed.tmu.ac.jp, Email: abosheasha@gmail.com.
Afnan H. El‐Gowily, Email: afnan.hamdy@science.tanta.edu.eg, Email: a.elgowily.mc@juntendo.ac.jp.
DATA AVAILABILITY STATEMENT
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Bikadi, Z. , & Hazai, E. (2009). Application of the PM6 semi‐empirical method to modeling proteins enhances docking accuracy of AutoDock. Journal of Cheminformatics, 1(1), 15. 10.1186/1758-2946-1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J. F. W. , Lau, S. K. P. , To, K. K. W. , Cheng, V. C. C. , Woo, P. C. Y. , & Yue, K. Y. (2015). Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS‐like disease. Clinical Microbiology Reviews, 28(2), 465–522. 10.1128/CMR.00102-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Guo, Y. , Pan, Y. , & Zhao, Z. J. (2020). Structure analysis of the receptor binding of 2019‐nCoV. Biochemical and Biophysical Research Communications, 525(1), 135–140. 10.1016/j.bbrc.2020.02.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux, C. A. , Rolain, J.‐M. , Colson, P. , & Raoult, D. (2020). New insights on the antiviral effects of chloroquine against coronavirus: What to expect for COVID‐19? International Journal of Antimicrobial Agents, 55, 105938. 10.1016/j.ijantimicag.2020.105938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, C. (2020). Coronavirus puts drug repurposing on the fast track. Nature Biotechnology, 38, 379–381. 10.1038/d41587-020-00003-1 [DOI] [PubMed] [Google Scholar]
- Huey, R. , Morris, G. M. , Olson, A. J. , & Goodsell, D. S. (2007). A semiempirical free energy force field with charge‐based desolvation. Journal of Computational Chemistry, 28(6), 1145–1152. 10.1002/jcc.20634 [DOI] [PubMed] [Google Scholar]
- Jin, Z. , Du, X. , Xu, Y. , Deng, Y. , Liu, M. , Zhao, Y. , … Yang, H. (2020). Structure of Mpro from COVID‐19 virus and discovery of its inhibitors. Nature, 582, 1–9. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Li, G. , Fan, Y. , Lai, Y. , Han, T. , Li, Z. , Zhou, P. , … Wu, J. (2020). Coronavirus infections and immune responses. Journal of Medical Virology, 92(4), 424–432. 10.1002/jmv.25685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R. , Zhao, X. , Li, J. , Niu, P. , Yang, B. , Wu, H. , … Tan, W. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. The Lancet, 395(10224), 565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G. M. , Goodsell, D. S. , Halliday, R. S. , Huey, R. , Hart, W. E. , Belew, R. K. , & Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry, 19(14), 1639–1662. 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B [DOI] [Google Scholar]
- NCT04365309 . (2020). Protective effect of aspirin on COVID‐19 patients. Retrieved from https://clinicaltrials.gov/ct2/show/study/NCT04365309
- Obi, A. T. , Tignanelli, C. J. , Jacobs, B. N. , Arya, S. , Park, P. K. , Wakefield, T. W. , … Napolitano, L. M. (2019). Empirical systemic anticoagulation is associated with decreased venous thromboembolism in critically ill influenza a H1N1 acute respiratory distress syndrome patients. Journal of Vascular Surgery: Venous and Lymphatic Disorders, 7(3), 317–324. 10.1016/j.jvsv.2018.08.010 [DOI] [PubMed] [Google Scholar]
- Ou, X. , Liu, Y. , Lei, X. , Li, P. , Mi, D. , Ren, L. , … Qian, Z. (2020). Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nature Communications, 11(1), 1620. 10.1038/s41467-020-15562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules, C. I. , Marston, H. D. , & Fauci, A. S. (2020). Coronavirus infections‐more than just the common cold. JAMA ‐ Journal of the American Medical Association, 323, 707–708. 10.1001/jama.2020.0757 [DOI] [PubMed] [Google Scholar]
- Pushpakom, S. , Iorio, F. , Eyers, P. A. , Escott, K. J. , Hopper, S. , Wells, A. , … Pirmohamed, M. (2018). Drug repurposing: Progress, challenges and recommendations. Nature Reviews Drug Discovery, 18, 41–58. 10.1038/nrd.2018.168 [DOI] [PubMed] [Google Scholar]
- Ramphul, K. , & Mejias, S. G. (2020). Coronavirus disease: A review of a new threat to public health. Cureus, 12(3), e7276. 10.7759/cureus.7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salata, C. , Calistri, A. , Parolin, C. , & Palù, G. (2019). Coronaviruses: A paradigm of new emerging zoonotic diseases. Pathogens and Disease, 77(9), ftaa006. 10.1093/FEMSPD/FTAA006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin, S. , Schreiber, S. , Haupt, V. J. , Adasme, M. F. , & Schroeder, M. (2015). PLIP: Fully automated protein‐ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–W447. 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohag, A. , Hannan, M. , Rahman, S. , Hossain, M. , Hasan, M. , Khan, M. , … Uddin, M. (2020). Revisiting potential druggable targets against SARS‐CoV‐2 and repurposing therapeutics under preclinical study and clinical trials: A comprehensive review. Drug Development Research, 21709. 10.1002/ddr.21709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis, F. J. , & Wets, R. J. B. (1981). Minimization by random search techniques. Mathematics of Operations Research, 6(1), 19–30. 10.1287/moor.6.1.19 [DOI] [Google Scholar]
- Stewart, J. J. P. (2009). Application of the PM6 method to modeling proteins. Journal of Molecular Modeling, 15(7), 765–805. 10.1007/s00894-008-0420-y [DOI] [PubMed] [Google Scholar]
- Tremblay, D. , van Gerwen, M. , Alsen, M. , Thibaud, S. , Kessler, A. J. , Venugopal, S. , … Naymagon, L. (2020). Impact of anticoagulation prior to COVID‐19 infection: A propensity score‐matched cohort study. Blood, 136(1), 144–147. 10.1182/blood.2020006941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y.‐J. , Tortorici, M. A. , Wall, A. , Mcguire, A. T. , & Correspondence, D. V. (2020). Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell, 181, 281–292. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Chen, X. , Lu, Y. , Chen, F. , & Zhang, W. (2020). Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Bioscience Trends, 14(1), 64–68. 10.5582/BST.2020.01030 [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) . (2020). Coronavirus disease (COVID‐2019) situation reports: Situation Report‐92. Retrieved from https://www.who.int/emergencies/diseases/novel‐coronavirus‐2019/situation‐reports
- Xu, X. , Chen, P. , Wang, J. , Feng, J. , Zhou, H. , Li, X. , … Hao, P. (2020). Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences, 63, 457–460. 10.1007/s11427-020-1637-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.