

Abstract
Apigenin, an aromatic compound, exhibits antioxidant, anti-inflammatory and anti-viral effects. The present study aimed to investigate the effects of apigenin on cell proliferation and apoptosis of human melanoma cells A375P and A375SM. Therefore, melanoma cells were treated with apigenin to determine its anti-proliferative and survival effects, using wound healing and MTT assays. The results revealed that melanoma cell viability was decreased in a dose-dependent manner. Furthermore, chromatin condensation, indicating apoptosis, was significantly increased in a dose-dependent manner, as demonstrated by DAPI staining. In addition, increased apoptosis rate following treatment with apigenin was confirmed by Annexin V-propidium iodide staining. The changes in the expression levels of apoptosis-related proteins in A375P and A375SM melanoma cells were subsequently detected using western blot analysis. The results demonstrated that the protein expression levels of Bcl-2 were decreased, whereas those of Bax, cleaved poly ADP-ribose polymerase, cleaved caspase-9 and p53 were upregulated in a dose-dependent manner in apigenin-treated cells compared with those noted in untreated cells. In addition, in apigenin-treated A375P cells, phosphorylated (p)-p38 was upregulated and p-extracellular signal-regulated kinase (ERK), p-c-Jun N-terminal kinase (JNK) and p-protein kinase B (Akt) were downregulated. However, in A375SM cells, apigenin treatment increased p-ERK and p-JNK and decreased p-p38 and p-Akt protein expression levels. Subsequently, the inhibitory effect of apigenin on tumor growth was investigated in vivo. Tumor volume was significantly reduced in the 25 and 50 mg/kg apigenin-treated groups compared with the control group. Additionally, a TUNEL assay was performed to detect apoptotic cells. Immunohistochemical staining also revealed elevated p-ERK expression in the apigenin-treated group compared with the control group. Overall, the findings of the present study indicated that apigenin attenuated the growth of A375SM melanoma cells by inducing apoptosis via regulating the Akt and mitogen-activated protein kinase signaling pathways.
Keywords: melanoma, A375P, A375SM, apigenin, apoptosis, Akt pathway, MAPK pathway, xenograft
Introduction
The cancer-related mortality rate increases annually on a global scale, making cancer the leading cause of death in South Korea (1,2). The incidence of skin cancer is rising faster compared with other types of cancer, concurrent with increasing outdoor activity and subsequent exposure to ultraviolet radiation. Comprising only 4% of all types of cancer, but with a mortality rate approaching 80%, melanoma is considered to be highly malignant, and it is caused by the malignant alteration of melanin cells (3,4). Melanoma spreads rapidly through the lymphatic ducts and blood vessels to internal organs, such as the liver, lungs and bones, and it is highly resistant to chemotherapy and radiotherapy. Therefore, surgical excision following early diagnosis is considered as the only treatment for melanoma. In addition, melanoma is characterized as an intractable disease, as there is currently no evidence to support any treatment to prevent disease recurrence (5). Due to an increasing melanoma occurrence and high mortality rate, a number of studies on melanoma have been conducted in the western world; however, measures should be taken to encourage further studies in Korea, where the number of studies is limited (6). Previously, several international and Korean studies have been performed aiming to reduce the side effects and improve the anticancer effects of chemotherapy using carcinostatic substances made of natural compounds isolated from animals and plants (7–11). Widely distributed throughout the vegetable kingdom, flavonoids are known to exist in ~4,000 types of water-soluble pigments in various vegetables and fruits, such as green tea, grapes and onions. Functional studies have reported that flavonoids exhibit several bioactivities (12–14). The flavonoid 4′,5,7,-trihydroxyflavone (apigenin) has been demonstrated to exert an inhibitory effect on cancer cell growth and several other effects including anti-oxidant (15), anti-inflammatory (16) and anti-tumor (17) effects in vitro and in vivo.
Apigenin, an aromatic compound, is found in various fruits and plants, including parsley, onions, oranges, tea, chamomile, wheat and other seasonings, and has no evident toxicity (18). Several studies have been conducted on the antioxidant (19), anti-inflammatory (20) and anti-viral activities (21) of apigenin. Apigenin has been reported to affect cancer cell growth by promoting apoptosis and inhibiting the cell cycle of colorectal cancer cells (22), and inducing apoptosis via inhibiting angiogenesis in breast and prostate cancer cells (23,24). These findings suggest that apigenin could be considered as a potential carcinostatic substance. In addition, apigenin has been demonstrated to have cytotoxic and anti-proliferative activities by promoting G2/M cell cycle arrest in melanoma cells (25), as well as the ability to attenuate tumor invasion and metastatic potential via inhibiting lung colonization (26). Furthermore, apigenin can impede metastasis of melanoma cells by impairing interactions between tumor cells and endothelial cells (27).
Apoptosis is known to inhibit the development and progression of cancer, and is distinguished from necrosis as a defense mechanism that voluntarily eliminates cells with accumulated DNA damage caused by intracellular DNA damage and viral infection (28). The Bcl-2 family, which is commonly known to suppress the genesis and progression of cancer through apoptosis, is classified into pro- and anti-apoptotic proteins (29). The pro-apoptotic proteins include Bax, BH3-interacting domain death agonist (Bid) and Bad, which induce apoptosis via rupturing the outer mitochondrial membrane, whereas anti-apoptotic Bcl-2, Bcl-xL and A1 proteins inhibit apoptosis by preserving the outer mitochondrial membrane (30–32). The phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and mitogen-activated protein kinase (MAPK) pathways are among the signals associated with cell viability; they activate various intracellular signaling pathways to regulate cell proliferation and angiogenesis (33,34). As an enzyme that phosphorylates serine/threonine residues, Akt is known to be involved in cell cycle progression and survival-related proliferation, and inhibits apoptosis via downregulating the expression of the pro-apoptotic proteins Bcl-2 and caspase-9 via the PI3K/Akt pathway (35). The MAPK pathway includes three main kinases, namely extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 MAPK kinase, each with different activities, that suppress tumor growth and cell differentiation (36).
The present study aimed to explore the inhibitory effect of apigenin on human melanoma A375P and A375SM cell proliferation and its inductive activity on apoptosis, and to further elucidate whether the Akt and MAPK signaling pathways mediated the apigenin-induced cell apoptosis.
Materials and methods
Reagents
The human melanoma cell lines, A375P and A375SM, were obtained from the Korean Cell Line Bank (Korean Cell Line Research Foundation). Dulbecco's modified Eagle's medium (DMEM), minimum essential medium (MEM), fetal bovine serum (FBS) and penicillin-streptomycin were purchased from Welgene, Inc. Apigenin (Fig. 1), MTT cell lysis buffer, DAPI and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Merck KGaA). Fluoresceinisothiocyanate (FITC) Annexin V Apoptosis Detection kit was obtained from BD Pharmingen (BD Biosciences). Anti-β-actin (cat. no. 4967), anti-Bax (cat. no. 2772), anti-Bcl-2 (cat. no. 2876), anti-cleaved caspase-9 (cat. no. 9501), anti-caspase-9 (cat. no. 9502), anti-poly ADP-ribose polymerase (PARP; cat. no. 9542), anti-cleaved PARP (cat. no. 5625), anti-p53 (cat. no. 2527), anti-phosphorylated (p)-p38 (cat. no. 4631), anti-p38 (cat. no. 9212), anti-p-JNK (cat. no. 4668), anti-JNK (cat. no. 9252), anti-p-ERK (cat. no. 4376), anti-ERK (cat. no. 4695), anti-p-Akt (cat. no. 4060), anti-Akt (cat. no. 9272) and goat anti-rabbit IgG (cat. no. 7074) antibodies were obtained from Cell Signaling Technology, Inc.
Figure 1.
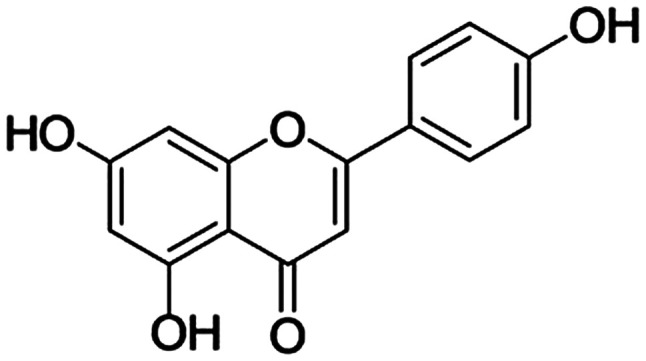
Molecular structure of apigenin (4′,5,7-trihydroxyflavone).
Cell line and culture
Melanoma cells were maintained in MEM and DMEM supplemented with 5% FBS and 1% penicillin/streptomycin under standard culture conditions at 37°C in a humidified atmosphere of 95% air and 5% CO2. The culture medium was replaced every 2–3 days. For apigenin treatment, melanoma cells were seeded into a 175 cm2 flask (Nalge Nunc International; Thermo Fisher Scientific, Inc.) when density reached ~80–90% and allowed to attach overnight.
Cell viability assay
The anticancer effects of apigenin were assessed using an MTT assay. A375P and A375SM cells were seeded onto a 96-well plate at a density of 2×104 cells/ml and a volume of 200 µl/well. Following incubation for 24 h, cells were treated with apigenin (0, 25, 50, 75 and 100 µM) for 24 h in triplicate. After apigenin treatment, the medium was discarded and a total of 40 µl MTT solution (5 mg/ml) was added to each well followed by incubation for an additional 2 h. Subsequently, the medium was aspirated and the formazan product generated by viable cells was solubilized with the addition of 100 µl DMSO. The absorbance of the solution was determined at 595 nm using a microplate reader (Bio-Rad Laboratories, Inc.). The percentage of viable cells in the apigenin treatment group was estimated in comparison with the untreated control cells.
Wound healing migration assay
A375P and A375SM cells were seeded in 60-mm culture dishes at a density of 1×105 cells/ml and allowed to grow for 24 h. A uniform wound was introduced by scraping the monolayers with a sterile blue-pipette tip. Subsequently, the culture medium was replaced with non-FBS fresh medium supplemented with increasing concentrations of apigenin (0, 50 and 100 µM) and cells were cultured for an additional 24 h. The rate of wound closure in apigenin-treated and untreated cells was monitored by images captured with a phase-contrast microscope (magnification, ×100) immediately after wound incision (0 h) and following 24 h. The percentage of the migrated cells was estimated compared with the cell density in the wound of the untreated control group.
DAPI staining
Apoptotic cell death was determined by observing morphological changes using DAPI fluorescent nuclear dye. DAPI stains cells undergoing apoptosis characterized by chromatin condensation and nuclear fragmentation. A375P and A375SM cells were treated with PBS or various concentrations of apigenin (0, 50 and 100 µM) for 24 h, harvested by trypsinization and fixed in 70% ethanol overnight at 4°C. The following day, cells were stained with DAPI at room temperature for 1 min, deposited onto slides and observed under a fluorescence microscope (magnification, ×200) to detect characteristics of apoptosis. The apoptotic rate was measured using the following equation: Number of positive cells/total number of cells in three random fields from each sample.
Annexin V-propidium iodide (PI) staining
Apoptosis rate was also determined using a FITC-Annexin V Apoptosis Detection kit (BD Pharmingen; BD Biosciences). For the Annexin V-PI staining, A375P and A375SM human melanoma cells were treated with 0, 50 and 100 µM apigenin for 24 h. Following treatment, cells were washed with PBS, suspended in trypsin-EDTA solution and centrifuged (200 × g) at 4°C for 5 min to obtain cell pellet. The harvested cells were diluted in 1X binding buffer at a density of 1×106 cells/ml. Subsequently, cells were treated with 5 µl/100 µl FITC-conjugated Annexin V and phycoerythrin-conjugated PI for 15 min and apoptosis was measured by flow cytometry (BD FACSVerse™ Flow cytometer, BD Life Sciences). BD FACSuite (v1.0.6; BD Life Sciences) was used for analysis. The early and late apoptosis rate was indicated by the percentage of Annexin V positive cells.
Western blot analysis
Cells were treated with various concentrations of apigenin (0, 50 and 100 µM) for 24 h, total proteins were extracted using PRO-PREP protein extraction solution (Intron Biotechnology, Inc., cat. no. 17081), and then the protein concentration was determined using a Bradford protein assay (Bio-Rad Laboratories, Inc.). Total proteins (30 µg per lane) in each cell lysate were resolved via SDS-PAGE on 6–14% gels, and subsequently electrotransferred onto nitrocellulose membranes. Following blocking with 5% non-fat dry milk in Tris-buffered saline with 0.5% Tween-20 (TBST) buffer for 1 h at room temperature, membranes were incubated with specific primary antibodies diluted in blocking solution at 4°C overnight. After washing with TBST, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Following washing, bands were visualized using an enhanced chemiluminescence detection reagent (Pierce; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. The following antibodies were used: β-actin (1:1,000), Bax (1:1,000), Bcl-2 (1:1,000), caspase-9 (1:1,000), cleaved-caspase-9 (1:1,000), PARP (1:1,000), cleaved-PARP (1:1,000), p38 (1:1,000), p-p38 (1:1,000), JNK (1:1,000), p-JNK (1:1,000), ERK (1:1,000), p-ERK (1:1,000), Akt (1:1,000) and p-Akt (1:1,000) as primary antibodies and rabbit IgG (1:1,000) as the secondary antibody. Protein intensity was semi-quantified by the ImageJ software (National Institutes of Health, v1.8.0).
Animals and in vivo xenograft tumor model
A total of 15 male BALB/c nude (nu/nu) mice (age, 5 weeks, body weight, 17–19 g) were purchased from Orient Bio Inc. Animal experiments were performed in accordance with the Guidelines for the Kongju National University Institutional Animal Care and Use Committee (Chungcheongnam, Korea) and approved by the Ethics Committee of Kongju National University (approval no. KNU_2018-5). Equipment was provided by the Laboratory Animal Resource Center (Gwangju, Korea). Mice were maintained under a 12 h light/dark cycle and housed under a controlled temperature of 23±3°C and humidity of 40±10%. Mice were allowed ad libitum access to laboratory pellet food and water. A375SM cells at 80–90% density were maintained in DMEM and MEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in a humidified atmosphere of 5% CO2. A375SM cells were harvested from cultures using 0.25% trypsin. Trypsinization was stopped using a solution containing 10% FBS, cells were then rinsed twice and resuspended in DMEM and MEM. Subsequently, a total of 2×107 cells in 0.2 ml culture medium were injected subcutaneously into the right flank of donor nude mice. On day 7 following injection, A375SM cells growing under the skin of nude mice developed tumors. When the tumors became palpable, mice were assigned randomly into three groups (n=5), namely the vehicle-treated control group and the apigenin-treated groups (25 or 50 mg/kg body weight). Apigenin was orally administrated five times/week for 3 weeks at a dose of 25 or 50 mg/kg body weight, while control mice were treated with the vehicle only. Oral administration was performed using an oral zonde needle. Animal health and behavior were monitored daily. Body weight and tumor volume were monitored twice weekly. The tumor volumes were calculated using the following equation: Tumor volume (mm3)=0.5 × length × width2. Then, three weeks after the start of apigenin injection, the final tumor size was measured. All mice were sacrificed using CO2 gas (30% per min, 3 min) and tumors were excised to measure tumor weight. A section of the tumor tissue was embedded in paraffin and fixed with 10% formalin at room temperature for 12 h was subsequently used for TUNEL and immunohistochemistry (IHC) assays. The criteria used to determine when an animal should be euthanized were set as follows: i) Mice showed a weight loss of ≥20% of its normal weight; ii) tumor grew to ≥10% of its normal weight; iii) mice developed ulcers or infections in the tumor area; or iv) erosion of surrounding tissues.
TUNEL assay
TUNEL staining was performed in paraffin-embedded 5-µm-thick tumor sections using the DeadEnd™ Colorimetric TUNEL System (Promega Corporation), according to the manufacturer's protocol. Briefly, sections were deparaffinized in xylene, dehydrated via a series of graded alcohol rinses (100, 95, 85, 70 and 50% ethanol (v/v) in double-distilled H2O) and rehydrated in PBS (pH 7.5). Subsequently, the tissue samples were permeabilized with a proteinase K solution following refixing in 4% paraformaldehyde solution at room temperature for 15 min. Slides were treated with the rTdT reaction mix and incubated at 37°C for 1 h. Reactions were terminated by immersing the slides in 2X SSC solution for 15 min at room temperature. Following blocking of endogenous peroxidase activity with 0.3% hydrogen peroxide, slides were washed with PBS, and then incubated with streptavidin HRP solution for 30 min at room temperature. After washing, slides were incubated with a 3,3-diaminobenzidine (DAB; substrate) solution until a light brown background appeared (10 min) and rinsed several times in deionized water. Following mounting, slides were observed under a light microscope. The number of positive cells in three random fields from each sample was counted indicating the number of apoptotic cells.
Immunohistochemistry
The paraffin-embedded sections were deparaffinized and dehydrated by sequential immersion in xylene and graded alcohol solutions, respectively. Sections were blocked using 1X Animal-Free Blocking Solution (Cell Signaling Technology, Inc., cat. no. 15019) at room temperature for 1 h. The sections were incubated with an antibody against p-ERK (1:100) at 4°C overnight, and subsequently incubated with a HRP-conjugated goat anti-rabbit antibody for 1 h at room temperature. The tumor sections were visualized using a DAB solution, treated with mounting reagent and observed under a routines light microscope (magnification, ×200). Finally, p-ERK positive cells were counted in three random fields from each sample.
Histological examination
The excised liver and kidney specimens were immediately fixed in 10% neutral buffered formalin at room temperature for 72 h, embedded in paraffin and cut into 5-µm-thick sections. Following hematoxylin and eosin (H&E) staining at room temperature (hematoxylin for 5 min, eosin for 1 min), the sections were examined under a light microscope (magnification, ×200).
Statistical analysis
Results are presented as the mean ± standard error of the mean for tumor volume, tumor weight, and body weight in vivo, while others are presented as the mean ± standard deviation. Differences in the mean values between control and apigenin-treated groups were assessed by one-way ANOVA followed by a Dunnett's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Apigenin inhibits the survival of melanoma cells
To investigate the effects of apigenin on A375P and A375SM cell viability, cells were treated with different concentrations (0, 25, 50, 75 and 100 µM) of apigenin and viability was assessed using an MTT assay. When A375P and A375SM cells were treated with 0, 25, 50, 75 and 100 µM of apigenin for 24 h, the cell survival rates were 100, 90, 71, 55 and 35%, and 100, 89, 78, 69 and 55% for A375P and A375SM cells, respectively (Fig. 2). The decrease in cell viability was dose-dependent, as it decreased with increasing concentrations of apigenin. Significant effects were observed at concentrations ≥50 µM.
Figure 2.

Effect of apigenin on melanoma cell viability. A375P and A375SM cells (density, 2×104 cells/ml) were treated with apigenin (0, 25, 50, 75, 100 µM) in DMEM and MEM supplemented with 5% FBS for 24 h. Cell viability was measured by a MTT assay. Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group.
Apigenin affects the migratory ability of melanoma cells
Subsequently, to explore the effect on apigenin on A375P and A375SM cell migration, melanoma cells were treated with various concentrations of apigenin (0, 50 and 100 µM) that significantly affected the viability of A375P and A375SM cells as demonstated by the MTT assay. The results revealed that cell migration was attenuated in a dose-dependent manner compared with that noted in the control group (Fig. 3A and C). The migration rates in A375P and A375SM cells treated with 0, 50 and 100 µM apigenin were 100, 53 and 25%, and 100, 34 and 4%, respectively (Fig. 3B and D).
Figure 3.

Effect of apigenin on melanoma cell migration ability. (A) A375P and (C) A375SM melanoma cells were treated with apigenin (0, 50, 100 µM) for 24 h and cell density in the wound area was measured using a wound healing assay (magnification, ×100). (B and D) The percentage of cell migration was estimated compared with the cell density in the wound area of the untreated control group. Cell density in the wound area for any field of view was measured at different locations. Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group.
Effect of apigenin on morphological changes in melanoma cells
To determine whether the decreased cell viability observed in apigenin-treated A375P and A375SM cells was mediated by apoptosis, the morphological changes and chromatin condensation in the nucleus of apigenin-treated cells were observed following DAPI staining. Therefore, A375P and A375SM cells were treated with 0, 50 and 100 µM of apigenin for 24 h, co-treated with DAPI staining and observed under a fluorescence microscope (Fig. 4A). Consistent with the MTT assays, the results revealed that apigenin exerted inhibitory effects on cancer cell growth by decreasing total cell count. The inhibitory effect of apigenin on cancer cell growth was mediated by apoptosis as confirmed by the presense of morphological features of apoptosis, such as apoptotic bodies and chromatin condensation. To quantitatively analyze apoptosis, DAPI-positive cells were counted (Fig. 4B) and the morphology of the apoptotic bodies was observed under a fluorescence microscope. The apoptosis rates of A375P and A375SM cells treated with 0, 50 and 100 µM apigenin were 3.8, 24.8 and 44.8%, and 6.6, 20.0 and 36.4%, respectively. Thus, this indicated that apigenin increased apoptosis in a dose-dependent manner.
Figure 4.

Effect of apigenin on chromatin condensation in melanoma cells (A375P, A375SM). (A) A375P and A375SM cells were treated with apigenin (0, 50, 100 µM) in DMEM and MEM supplemented with 5% FBS for 24 h and cells were stained with DAPI. The arrows indicate chromatin condensation in melanoma cells. Scale bar, 10 µm. (B) Apoptotic cells were counted under a light microscope and apoptosis rate was expressed as the percentage of apoptotic cells in five random fields. Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group.
Effect of apigenin on apoptosis in melanoma cells
To investigate whether the formation of apoptotic bodies observed with DAPI staining was mediated by apoptosis, Annexin V-PI staining was used. Therefore, A375P and A375SM cells were treated with 0, 50 and 100 µM apigenin for 24 h and apoptosis was confirmed by flow cytometry (Fig. 5A and B). The results showed that the apoptosis rates (percentage of Annexin V positive cells) of apigenin-treated melanoma cells increased in a dose-dependent manner. As shown in Fig. 5C and D, apoptosis rates of A375P and A375SM cells treated with 0, 50 and 100 µM apigenin were 8.7, 40.3 and 59.6%, and 11.6, 38.5 and 47.5%, respectively.
Figure 5.

Effect of apigenin-induced apoptosis rates on A375P and A375SM cells. (A) A375P and (B) A375SM cell were treated with or without apigenin (0, 50, 100 µM) for 24 h, and apoptotic and necrotic cells were stained with Annexin V and PI. Apoptosis and necrosis rates were analyzed by flow cytometry. Quantification of apoptosis rates (Annexin V-positive cells) in (C) A375P and (D) A357SM cells. Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group. LL, live cell; UL, necrotic cell; LR, early apoptotic cell; UR, late apoptotic cell; PI, propidium iodide.
Effect of apigenin on apoptosis-related protein expression in melanoma cells
DAPI and Annexin V-PI staining showed that apoptosis was significantly increased in A375P and A375SM cell lines treated with 50 and 100 µM of apigenin. Therefore, the expression levels of proteins that regulate apoptosis were measured using western blot analysis. To detect the expression levels of Bax, Bcl-2, PARP, p53 and caspase-9, which are known to regulate apoptosis, A375P and A375SM cells were treated with apigenin (0, 50 and 100 µM), and subsequently western blotting was performed. As the concentration of apigenin increased, the expression levels of PARP and caspase-9 decreased, and the expression levels of pro-apoptotic proteins Bax, p53, cleaved PARP and cleaved caspase-9 significantly increased, whereas the expression of Bcl-2, known to inhibit apoptosis, was downregulated (Fig. 6A and B).
Figure 6.

Effect of apigenin on the apoptotic pathway in melanoma cells. (A) A375P and (B) A375SM melanoma cells were treated with apigenin (0, 50, 100 µM) for 24 h and cells were harvested to measure protein levels of Bax, Bcl-2, PARP, p53 and caspase-9 by western blotting. Blots were also probed with β-actin antibody to confirm equal sample loading. Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group. PARP, poly ADP-ribose polymerase.
Effect of apigenin on the MAPK and Akt signaling pathways in melanoma cells
The phosphorylation status of specific proteins in cancer cells is widely recognized as an important process in determining cell fate, including apoptosis. Various kinases of the MAPK pathway, an intracellular signaling system, are involved in intercellular and intracellular reactions in response to changes in the intracellular environment (34). ERK, JNK and p38, which belong to the MAPK pathway, are known to regulate several biological functions including cell proliferation, differentiation and apoptosis. Furthermore, Akt plays a central role in cell growth and proliferation, and is active in the majority of tumor tissues (35,36). However, Akt expression significantly differs in response to different active cellular materials, biological functions and environment. Therefore, western blot analysis was performed to reveal the effects of apigenin (0, 50 and 100 µM) on the protein expression of members of the Akt and MAPK pathways in A375P and A375SM cells (Fig. 7A and B). The expression levels of the MAPK pathway-related proteins, p-JNK and p-p38, significantly decreased and increased, respectively, in A375P cells. Furthermore, expression of p-ERK and p-Akt decreased following treatment with 100 µM apigenin. However, the increase in p-p38 expression was not associated with the concentration of apigenin and p-ERK was not downregulated in a dose-dependent manner. In A375SM cells, the expression levels of p-JNK and p-ERK in the MAPK pathway increased, whereas those of p-p38 and p-Akt decreased, depending on the concentration of apigenin. However, the increase in p-ERK expression was not associated with the concentration of apigenin. The aforementioned results indicated that proteins in the signal transduction pathways were differentially expressed in the two melanoma cell lines.
Figure 7.

Effect of apigenin on the activation of the mitogen-activated protein kinase and Akt signaling pathways in melanoma cells. (A) A375P and (B) A375SM cells were treated with apigenin (0, 50, 100 µM) for 24 h. Cell lysates were prepared as previously described and analyzed via western blotting. Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group. Akt, protein kinase B; ERK, extracellular signal-regulated protein kinase; JNK, c-Jun N-terminal kinase; p-, phosphorylated.
Effect of apigenin on tumor growth in an in vivo animal model
Based on the in vitro experiments, a A375SM cell line-derived xenograft mouse model was established. A375SM melanoma cells were transplanted into nude mice and the effect of apigenin on tumor growth was investigated. Apigenin was diluted in PBS and orally administered at concentrations of 25 and 50 mg/kg five times per week for 3 weeks. Tumor size was measured twice per week. Tumor size was decreased in the apigenin-treated group after two weeks of treatment compared with that observed in the control group (Fig. 8A). The tumor inhibition rate was 35.0% on the 21st day post-administration in the low-concentration group (25 mg/kg) and 15.4% in the high-concentration group (50 mg/kg), as shown in Table I. Tumor weights were 0.64±0.1, 0.46±0.1 and 0.57±0.07 g in the control group, low-concentration group and high-concentration group, respectively. Therefore, tumor weight was decreased in both groups following treatment with apigenin compared with the control group (Fig. 8B). However, there were no statistically significant changes in body weight following apigenin administration (Fig. 8C).
Figure 8.

Effects of apigenin on melanoma tumor growth and apoptosis. Nude mice bearing A375SM cells as xenograft models were treated with apigenin for 21 days and (A) tumor volume, (B) tumor weight and (C) body weight were measured. Data are presented as the mean ± standard error of the mean. (D) Apoptosis was determined in tumor tissues using a TUNEL assay. Slides were observed under a microscope (magnification, ×200; scale bar, 10 µm). (E) TUNEL assay results are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group.
Table I.
Tumor inhibtion rate in mice implanted with A375SM melanoma cells treated with apigenin.
| Apigenin (mg/kg) | Pre-experiment size, mm3 | Post-experiment size, mm3 | Inhibition rateb, % |
|---|---|---|---|
| 0a | 83.3 | 1,724.6 | |
| 25 | 93.6 | 1,120.8 | 35.0 |
| 50 | 95.1 | 1,458.3 | 15.4 |
Control group
data are expressed as percentage relative to control.
Effect of apigenin on melanoma tumor cell apoptosis in vivo
A TUNEL assay was performed to determine whether the inhibitory effect of apigenin on the growth of A375SM melanoma cells isolated from a melanoma xenograft model was mediated by apoptosis. The results revealed that the number of TUNEL-positive cells was increased in the apigenin-treated group compared with the control group (Fig. 8D and E).
Effect of apigenin on p-ERK expression in melanoma tumor tissue
The in vitro experiments demonstrated that p-ERK was upregulated in apigenin-treated melanoma cells. Therefore, melanoma tumor samples were isolated from the melanoma mouse xenograft model 3 weeks following apigenin administration, and IHC revealed an increase in p-ERK expression compared with the control group (Fig. 9).
Figure 9.

Effect of apigenin on p-ERK expression in A375SM-induced melanoma tumors. Nude mice were administered with apigenin for 3 weeks and the expression of p-ERK was assessed by immunohistochemistry. Paraffin-embedded tumor tissues were cut into 5-µm-thick sections and tissues were observed under a microscope and photographed (magnification, ×200; scale bar, 10 µm). Each bar represents the p-ERK-positive cells (relative to control). Data are presented as the mean ± standard deviation from three independent experiments. Significance was determined using ANOVA followed by a Dunnett's post hoc test. *P<0.05 vs. untreated control group. ERK, extracellular signal-regulated protein kinase; p-, phosphorylated.
Apigenin induces histopathological changes in melanoma tumor tissues
Subsequently, to assess apigenin-induced organ toxicity, liver and kidney tissues derived from tumor-xenografted mice were histologically examined by H&E staining under a light microscope (Fig. 10). However, no histopathological abnormalities were observed, indicating that apigenin had no detectable toxic effects.
Figure 10.

Histological observation of apigenin-treated nude mice. The microscopic analysis of murine liver and kidney specimens showed no evidence of adverse systemic toxicity following apigenin treatment. Slides were observed under a microscope (magnification, ×200; scale bar, 10 µm).
Discussion
The present study suggested a mechanism for the inhibitory effect of apigenin on A375P and A375SM melanoma cell growth and proliferation. The two melanoma cell lines differ in their invasive and metastatic properties, with A375P cells exhibiting decreased properties compared with A375SM cells (37). Cancer cells induce tumorigenesis via invasion to healthy tissues from blood and lymphatic ducts after their proliferation into the adjacent tissues (38). To investigate the inhibitory effects of apigenin in vitro, A375P and A375SM cells were treated with different concentrations of apigenin, and MTT and wound healing assays were performed. The results of the MTT assay showed reduced cell viability of A375P and A375SM melanoma cells treated with apigenin in a dose-dependent manner. These results were consistent with previous studies, indicating that apigenin decreased tumor growth in KB oral cavity cancer cells (39), HCT-16 colorectal cancer cells (22) and HL-60 human myeloma cancer cells (40) in a dose-dependent manner. The wound healing assay in the present study demonstrated that apigenin attenuated the migratory ability of melanoma cells. Previous studies revealed that apigenin inhibits cell migration of CD44+ prostate cancer cells (41), and DLD1 and SW480 colorectal cancer cells (42). These previous findings were consistent with the results from the present study, demonstrating the inhibitory effects of apigenin on A375P and A375SM melanoma cell migration when applied at different concentrations.
Apoptosis may occur in response to normal cell injury or pathological factors (28) and is accompanied by various features, such as cytoplasmic or chromatin condensation, cell membrane alteration and DNA fragmentation. These features play important roles in biological generation and maintenance of homeostasis. Therefore, DAPI staining was performed to clarify whether the inhibitory effect of apigenin on melanoma cell growth and proliferation was mediated by apoptosis. Cancer cell growth and count were decreased following apigenin treatment. In addition, characteristics of apoptosis were observed, including chromatin condensation and the presence of apoptotic bodies. A previous study also demonstrated that apigenin induces apoptosis in KB oral cavity cancer cells in a dose-dependent manner (43). Apoptosis was triggered by DNA fragmentation and subsequent apoptosis in the nucleosome of A375P and A375SM melanoma cells. DAPI staining showed a significant increase in apoptotic cell count. Annexin V is a representative staining agent for apoptosis. Annexin V positive cells are early and late apoptotic cells, and Annexin V negative PI positive cells are necrotic cells. Apoptosis rate was measured by Annexin V-PI staining and supported the results of DAPI staining. Therefore, these findings suggested that apigenin could induce apoptosis in A375P and A375SM melanoma cells.
It has been established that apoptosis inhibits tumor development and progression. Pro-apoptotic proteins, such as Bax, Bid and Bad, promote apoptosis by rupturing the outer mitochondrial membrane, whereas anti-apoptotic proteins, including Bcl-2, Bcl-xL and A1, inhibit apoptosis by preserving the ruprure of the outer mitochondrial membrane (30–32). Akt is known to be involved in cell cycle progression and survival-related proliferation. In addition, Akt inhibits apoptosis via downregulating the expression of the pro-apoptotic proteins Bcl-2 and caspase-9 via the PI3K/Akt pathway (35). Three types of MAPKs have been identified, each with different activities, namely ERK, stress-activated JNK and p38 kinase MAPK, that attenuate the growth and differentiation of cancer cells (36).
In the present study, western blot analysis was performed to detect the expression levels of the apoptosis-related proteins. The reuslts demonstrated that the expression of Bax, p53, cleaved PARP and cleaved caspase-9 increased in a dose-dependent manner following treatment of A375P and A375SM cells with apigenin. However, Bcl-2 was downregulated. It has been reported that apigenin upregulates the expression of various proteins in different ypes of cancer cells in a dose-dependent manner, including p53 and Bax in SK-BR-3 breast cancer cells (44), and cleaved PARP and cleaved caspase-9 in PC-3 cancer cells (24). Apigenin has also been demonstrated to increase the expression of Bax, cleaved PARP and cleaved caspase-9, and decrease that of Bcl-2 in DU-145 prostate cancer cells in a dose-dependent manner (45). These findings suggested that apigenin induced apoptosis via regulating the expression of the apoptosis-related proteins, Bax, Bcl-2, PARP, p53 and caspase-9 in A375P and A375SM melanoma cells.
Subsequently, western blot analysis was also performed to assess the effects of apigenin on the MAPK and Akt pathway in A375P and A375SM cells. It was demonstrated that treatment with 100 µM apigenin led to a significant downregulation of p-ERK, p-JNK and p-Akt expression in A375P cells, and a downregulation of p-p38 and p-Akt expression in A375SM cells. Results in A375SM were consistent with those observed in previous studies demonstrating that apigenin increased the expression of p-ERK and p-JNK and decreased that of p-p38 in PC-3 prostate (46) and MDA-MB-231 breast cancer cells (47). Although p-ERK has been known to affect cell proliferation and migration in previous studies, the cellular signaling pathways are complex and the mechanisms of synergy or antagonism between intracellular signaling pathways are still unclear (48–50). Therefore, the change in p-ERK in A375P following apigenin treatment appeared to be due to complex interactions of intracellular signaling pathways, and further studies are needed to clarify the role of signaling pathways in cellular processes. Apigenin presumably inhibited the proliferation of A375P and A375SM cells in vitro via promoting apoptosis mediated by the MAPK and Akt signaling pathways. However, the expression levels of MAPKs were different between the two melanoma cell lines. It has been reported that A375P and A375SM melanoma cell lines exert different metastatic and invasive properties (37). Therefore, consistent with previous studies, apigenin induced apoptosis in vitro in A375SM melanoma cells via the Akt and MAPK signal transduction proteins. However, the protein expression of A375P under the same conditions was consistent with that observed in previous studies showing that apigenin decreased expression of p-ERK and p-AKT in A375, C8161 human melanoma cells (51), U266, RPMI8226 human multiple myeloma cells (52) and decreased expression of p-Akt in MDA-MB-231 human breast cancer cell (53). The aforementioned findings suggested that the different effects of apigenin on A375P and A375SM cells should be further investigated.
Based on the in vitro results, A375SM cells were transplanted into nude mice, which were in turn treated with apigenin to determine its effect on tumor growth. Tumor size and weight decreased in a dose-dependent manner in the apigenin-treated groups compared with the control group. Previous studies also demonstrated that intraperitoneally administrated apigenin at a concentration of 50 and 5.25 mg/kg, attenuated tumor growth of MIA PaCa-2 pancreatic (54) and MDA-MB-231 breast cancer cells (23), respectively, when transplanted into mice compared with non-treated control mice. These results were in agreement with those of the present study revealing the inhibitory effect of apigenin on A375SM tumor growth.
DNA fragmentation is a hallmark of apoptosis (55). In the present study a TUNEL assay was performed to determine whether the inhibitory effect of apigenin on A375SM tumor growth was mediated by apoptosis. TUNEL-positive cells were elevated in the apigenin-treated group compared with the control group. Furthermore, the apoptosis rate was increased in a concentration-dependent manner in the low-concentration and high-concentration groups, respectively. It has been also reported that apigenin increases apoptosis in KB oral cavity cancer cells in a dose-dependent manner (39). Therefore, these results indicated that apigenin inhibited the growth of A375SM cells via inducing apoptosis.
Currently, three MAPK pathways have been identified, namely ERK1/2, p38 MAPK and JNK/stress-activated protein kinase. Among them, p38 and JNK pathways are activated in response to stress-like stimuli to induce apoptosis. The ERK1/2 pathway functions as an anti- or pro-apoptotic factor depending on cellular characteristics (56–58). In the present study IHC was performed to explore the effect of apigenin on p-ERK expression. Consistent with the in vitro western blotting results, p-ERK expression was increased in A375SM tumor tissues. It was also previously revleaed that apigenin upregulated p-ERK expression in a dose-dependent manner in PC-3 prostate cancer cells (46). These results indicated that apigenin inhibited A375SM tumor growth and proliferation via regulating p-ERK expression.
In vivo results showed that apigenin did not induce concentration-dependent inhibition of tumor growth. However, tumor weight was attenuated in both concentration groups compared with the control group. Subsequent experiments demostrated that the apigenin-treated group exhibited a significant increase in apoptosis compared with the control group. In view of the differences between tumor volume and weight, and the results obtained by TUNEL assays, the aforementioned findings suggested that tumor volume and weight were not proportional to the induction of apoptosis. Therefore, further studies on the associasion between tumor weight and apoptosis-related tumor suppression are necessary.
In conclusion, the results of the in vitro and in vivo experiments suggested that apigenin induced apoptosis via regulating the Akt and MAPK signaling pathways, thus inhibiting the growth and proliferation of A375SM melanoma cells. Therefore, apigenin may be considered as an alternative therapeutic target to modulate the Akt and MAPK pathways in A375SM melanoma cells.
Acknowledgements
Not applicable.
Funding
This research was supported by a research grant of the Kongju National University in 2017 and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grant no. NRF 2017R1A2B4005516).
Availability of data and materials
The datasets used and/or analysed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
JSW, GSC, SDC, JHC, JSN, YSP, CSC, SKK and JYJ conceived and designed the experiments. JSW, ESY, SHK, JHL, SHH, HJK, SHJ and BSK performed the experiments. JSW and BKP analyzed the data. JSW and GSC wrote the paper. JYJ supervised the entire project. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The animal experiments were conducted in accordance with the regulations of the Kongju National University Institutional Animal Care and Use Committee with the approval of the Ethics Committee of Kongju National University (approval no. KNU_2018-5).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Doll SR. The lessons of life: Keynote address to the nutrition and cancer conference. J Cancer Res. 1992;52(Suppl 7):S2024–S2029. [PubMed] [Google Scholar]
- 2.Jung KW, Won YJ, Kong HJ, Oh CM, Lee DH, Lee JS. Cancer statistics in korea: Incidence, mortality, survival, and prevalence in 2011. J Cancer Res. 2014;46:109–123. doi: 10.4143/crt.2014.46.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 4.Megahed M, Schön M, Selimovic D, Schön MP. Reliability of diagnosis of melanoma in situ. Lancet. 2002;359:1921–1922. doi: 10.1016/S0140-6736(02)08741-X. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, He X, Peng H, Yang XO. Research on the antitumor effect of ginsenoside Rg3 in B16 melanoma cells. Melanoma Res. 2008;18:322–329. doi: 10.1097/CMR.0b013e32830b3536. [DOI] [PubMed] [Google Scholar]
- 6.Meier F, Satyamoorthy K, Nesbit M, Hsu MY, Schittek B, Garbe C, Herlyn M. Molecular events in melanoma development and progression. Front Bio Sci. 1998;3:D1005–D1010. doi: 10.2741/A341. [DOI] [PubMed] [Google Scholar]
- 7.Changmin K, Bonglee K. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: A review. Nutrients. 2018;10:1021. doi: 10.3390/nu10081021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simone F. Modulation of apoptosis by natural products for cancer therapy. Planta Med. 2010;76:1075–1079. doi: 10.1055/s-0030-1249961. [DOI] [PubMed] [Google Scholar]
- 9.Aung TN, Qu Z, Kortschak RD, Adelson DL. Understanding the effectiveness of natural compound mixtures in cancer through their molecular mode of action. Int J Mol Sci. 2017;18:656. doi: 10.3390/ijms18030656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin YJ, Liang WM, Chen CJ, Tsang H, Chiou JS, Liu X, Cheng CF, Lin TH, Liao CC, Huang SM, et al. Network analysis and mechanisms of action of Chinese herb-related natural compounds in lung cancer cells. Phytomedicine. 2019;58:152893. doi: 10.1016/j.phymed.2019.152893. [DOI] [PubMed] [Google Scholar]
- 11.Anna L, Krzysztof G. Anticancer activity of natural compounds from plant and marine environment. Int J Mol Sci. 2018;19:3533. doi: 10.3390/ijms19113533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Vizcaino F, Fraga CG. Research trends in flavonoids and health. Arch Biochem Biophys. 2018;646:107–112. doi: 10.1016/j.abb.2018.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Fraga CG, Croft KD, Kennedy DO, Tomás-Barberán FA. The effects of polyphenols and other bioactives on human health. Food Funct. 2019;10:514–528. doi: 10.1039/C8FO01997E. [DOI] [PubMed] [Google Scholar]
- 14.Wang M, Firrman J, Liu L, Yam K. A review on flavonoid apigenin: Dietary intake, ADME, antimicrobial effects, and interactions with human gut microbiota. Biomed Res Int. 2019;2019:7010467. doi: 10.1155/2019/7010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon RA, Steele CL. Flavonoids and isoflavonoids-a gold mine for metabolic engineering. Trends Plant Sci. 1999;4:394–400. doi: 10.1016/S1360-1385(99)01471-5. [DOI] [PubMed] [Google Scholar]
- 16.Yin Y, Gong FY, Wu XX, Sun Y, Li YH, Chen T, Xu Q. Antiinflammatory and immunosuppressive effect of flavones isolated from artemisia vestita. J Ethnopharmacol. 2008;120:1–6. doi: 10.1016/j.jep.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 17.Chuang CM, Monie A, Wu A, Hung CF. Combination of apigenin treatment with therapeutic HPV DNA vaccination generates enhanced therapeutic antitumor effects. J Biomed Sci. 2009;16:49. doi: 10.1186/1423-0127-16-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel D, Shukla S, Gupta S. Apigenin and cancer chemoprevention: Progress, potential and promise. Int J Oncol. 2007;30:233–245. [PubMed] [Google Scholar]
- 19.Romanová D, Vachálková A, Cipák L, Ovesná Z, Rauko P. Study of antioxidant effect of apigenin, luteolin and quercetin by DNA protective method. Neoplasma. 2001;48:104–107. [PubMed] [Google Scholar]
- 20.Fuchs J, Milbradt R. Skin anti-inflammatory activity of apigenin-7-glucoside in rats. Arzneimittelforschung. 1993;43:370–372. [PubMed] [Google Scholar]
- 21.Zhang W, Qiao H, Lv Y, Wang J, Chen X, Hou Y, Tan R, Li E. Apigenin inhibits enterovirus-71 infection by disrupting viral RNA association with trans-acting factors. PLoS One. 2014;9:e110429. doi: 10.1371/journal.pone.0110429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon TS, Cui LG, Yang H. Involvement of early growth response gene 1 (EGR-1) in growth suppression of the human colonic tumor cells by apigenin and its derivative isovitexin. J Life Sci. 2007;17:100–115. doi: 10.5352/JLS.2007.17.1.110. [DOI] [Google Scholar]
- 23.Tseng YH, Chien MH, Lin WL, Wen YC, Chow JM, Chen CK, Kuo TC, Lee WJ. Inhibition of MDA-MB-231 breast cancer cell proliferation and tumor growth by apigenin through induction of G2/M arrest and histone H3 acetylation-mediated p21WAF1/CIP1 expression. Environ Toxicol. 2017;32:434–444. doi: 10.1002/tox.22247. [DOI] [PubMed] [Google Scholar]
- 24.Kaur P, Shukla S, Gupta S. Plant flavonoid apigenin inactivates Akt to trigger apoptosis in human prostate cancer: An in vitro and in vivo study. Carcinogenesis. 2008;29:2210–2217. doi: 10.1093/carcin/bgn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghițu A, Schwiebs A, Radeke HH, Avram S, Zupko I, Bor A, Pavel IZ, Dehelean CA, Oprean C, Bojin F, et al. A comprehensive assessment of apigenin as an antiproliferative, proapoptotic, antiangiogenic and immunomodulatory phytocompound. Nutrients. 2019;11:858. doi: 10.3390/nu11040858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caltagirone S, Rossi C, Poggi A, Ranelletti FO, Natali PG, Brunetti M, Aiello FB, Piantelli M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int J Cancer. 2000;87:595–600. doi: 10.1002/1097-0215(20000815)87:4<595::AID-IJC21>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 27.Piantelli M, Rossi C, Iezzi M, La Sorda R, Iacobelli S, Alberti S, Natali PG. Flavonoids inhibit melanoma lung metastasis by impairing tumor cells endothelium interactions. J Cell Physiol. 2006;207:23–29. doi: 10.1002/jcp.20510. [DOI] [PubMed] [Google Scholar]
- 28.Han SI, Kim YS, Kim TH. Role of apoptotic and necrotic cell death under physiologic conditions. BMB Rep. 2008;41:1–10. doi: 10.5483/BMBRep.2008.41.1.001. [DOI] [PubMed] [Google Scholar]
- 29.Evans VG. Multiple pathways to apoptosis. Cell Biol Int Rep. 1993;17:461–476. doi: 10.1006/cbir.1993.1087. [DOI] [PubMed] [Google Scholar]
- 30.Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 31.Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–219. doi: 10.1016/S0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 32.Song Q, Kuang Y, Dixit VM, Vincenz C. Boo, a novel negative regulator of cell death, interacts with Apaf 1. EMBO J. 1999;18:167–178. doi: 10.1093/emboj/18.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 34.Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des. 2010;16:34–44. doi: 10.2174/138161210789941865. [DOI] [PubMed] [Google Scholar]
- 35.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis. 2004;9:667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 36.Dhillon AS, Hangan S, Rath O, Kolch W. MAP kinase signaling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 37.Ham SA, Yoo T, Hwang JS, Kang ES, Lee WJ, Paek KS, Park C, Kim JH, Do JT, Lim DS, Seo HG. Ligand-activated PPARδ modulates the migration and invasion of melanoma cells by regulating Snail expression. Am. J Cancer Res. 2014;4:674–682. [PMC free article] [PubMed] [Google Scholar]
- 38.Rao KM. MAP kinase activation in macrophages. J Leukoc Biol. 2001;69:3–10. [PubMed] [Google Scholar]
- 39.Lee JS, Seo HS, Kim SJ, Kim HJ, Kim J, Lee SH, Park YS, Park BK, Kim BS, Kim SK, Jung JY. Studies on the anticancer effect of apigenin in KB cell xenograft nude mouse model. J Life Sci. 2010;20:1519–1524. doi: 10.5352/JLS.2010.20.10.1519. [DOI] [Google Scholar]
- 40.Wang IK, Lin-Shiau SY, Lin JK. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. Eur J Cancer. 1999;35:1517–1525. doi: 10.1016/S0959-8049(99)00168-9. [DOI] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Erdogan S, Doganlar O, Doganlar ZB, Serttas R, Turkekul K, Dibirdik I, Bilir A. The flavonoid apigenin reduces prostate cancer CD44(+) stem cell survival and migration through PI3K/Akt/NF-κB signaling. Life Sci. 2016;162:77–86. doi: 10.1016/j.lfs.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Toker A, Yoeli-Lerner M. Akt signaling and cancer: Surviving but not moving on. Cancer Res. 2006;66:3963–3966. doi: 10.1158/0008-5472.CAN-06-0743. [DOI] [PubMed] [Google Scholar]
- 44.Choi EJ, Kim GH. Apigenin causes G(2)/M arrest associated with the modulation of p21(Cip1) and Cdc2 and activates p53-dependent apoptosis pathway in human breast cancer SK-BR-3 cells. J Nutr Biochem. 2009;20:285–290. doi: 10.1016/j.jnutbio.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Shukla S, Gupta S. Molecular mechanisms for apigenin-induced cell-cycle arrest and apoptosis of hormone refractory human prostate carcinoma DU145 cells. Mol Carcinog. 2004;39:114–126. doi: 10.1002/mc.10168. [DOI] [PubMed] [Google Scholar]
- 46.Shukla S, Gupta S. Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma dephosphorylation in human prostate cancer cells. Cell Cycle. 2007;6:1102–1114. doi: 10.4161/cc.6.9.4146. [DOI] [PubMed] [Google Scholar]
- 47.Lee WJ, Chen WK, Wang CJ, Lin WL, Tseng TH. Apigenin inhibits HGF-promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and beta 4 integrin function in MDA-MB-231 breast cancer cells. Toxicol Appl Pharmacol. 2008;226:178–191. doi: 10.1016/j.taap.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 48.Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19:1997–2007. doi: 10.3892/etm.2020.8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S, Liu G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett. 2017;13:1041–1047. doi: 10.3892/ol.2017.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel AL, Shvartsman SY. Outstanding questions in developmental ERK signaling. Development. 2018;145:dev143818. doi: 10.1242/dev.143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao G, Han X, Cheng W, Ni J, Zhang Y, Lin J, Song Z. Apigenin inhibits proliferation and invasion, and induces apoptosis and cell cycle arrest in human melanoma cells. Oncol Rep. 2017;37:2277–2285. doi: 10.3892/or.2017.5450. [DOI] [PubMed] [Google Scholar]
- 52.Zhao M, Ma J, Zhu HY, Zhang XH, Du ZY, Xu YJ, Yu XD. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol Cancer. 2011;10:104. doi: 10.1186/1476-4598-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin XY, Ren CS. Effect and mechanism of apigenin on VEGF expression in human breast cancer cells. Zhonghua Zhong Liu Za Zhi. 2007;29:495–499. (In Chinese) [PubMed] [Google Scholar]
- 54.Lee SH, Ryu JK, Lee KY, Woo SM, Park JK, Yoo JW, Kim YT, Yoon YB. Enhanced anti-tumor effect of combination therapy with gemcitabine and apigenin in pancreatic cancer. Cancer Lett. 2008;259:39–49. doi: 10.1016/j.canlet.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 55.Pecorino L. Oxford University Press; 2012. Molecular biology of cancer: Mechanisms, targets, and therapeutics; pp. 157–182. [Google Scholar]
- 56.Yang Y, Zhu X, Chen Y, Wang X, Chen R. p38 and JNK MAPK, but not ERK1/2 MAPK, play important role in colchicine-induced cortical neurons apoptosis. Eur J Pharmacol. 2007;576:26–33. doi: 10.1016/j.ejphar.2007.07.067. [DOI] [PubMed] [Google Scholar]
- 57.Ballif BA, Blenis J. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 2001;12:397–408. [PubMed] [Google Scholar]
- 58.Moos PJ, Fitzpatrick FA. Taxanes propagate apoptosis via two cell populations with distinctive cytological and molecular traits. Cell Growth Differ. 1998;9:687–697. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the present study are available from the corresponding author on reasonable request.