

Abstract
The differential diagnosis of atypical parkinsonian syndromes is challenging. These severe and often rapidly progressive neurodegenerative disorders are clinically heterogeneous and show significant phenotypic overlap. Here, clinical, imaging, neuropathological and genetic features of multiple system atrophy, progressive supranuclear palsy, corticobasal degeneration and frontotemporal lobar degeneration (FTLD) are reviewed. The terms corticobasal degeneration and FTLD refer to pathologically confirmed cases of corticobasal syndrome and frontotemporal dementia (FTD). Frontotemporal lobar degeneration clinically presents as the behavioral variant FTD, semantic variant primary progressive aphasia (PPA), non-fluent agrammatic variant PPA, logopenic variant PPA and FTD associated with motor neuron disease. While progressive supranuclear palsy and corticobasal syndrome have been called Parkinson-plus syndromes in the past, they are now classified as FTD-related disorders, reflecting that they pathologically differ from α-synucleinopathies like multiple system atrophy and Parkinson disease. The contribution of genetic factors to atypical parkinsonian syndromes is increasingly recognized. Genes involved in the etiology of FTLD include MAPT, GRN and C9orf72. Novel neuroimaging techniques, including tau positron emission tomography imaging, are being investigated. Multimodal magnetic resonance imaging approaches and automated magnetic resonance imaging volume segmentation techniques are being evaluated for optimized differential diagnosis. Current treatment options are symptomatic, and disease modifying therapies are under active investigation.
Keywords: α-synucleinopathy, corticobasal syndrome, frontotemporal dementia, multiple system atrophy, primary progressive aphasia, progressive supranuclear palsy, tauopathy
Introduction
The accurate diagnosis of atypical parkinsonism is challenging even for movement disorder specialists, especially in early disease stages. Parkinsonism can be the main presentation or an accompanying sign of multiple system atrophy (MSA), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and frontotemporal lobar degeneration (FTLD). The terms CBD and FTLD are used to describe pathologically confirmed cases of corticobasal syndrome (CBS) and frontotemporal dementia (FTD). It may even be challenging to differentiate atypical parkinsonism from Parkinson disease (PD). The current clinical classification of FTLD consists of behavioral variant frontotemporal dementia (bvFTD), semantic variant primary progressive aphasia (svPPA), non-fluent agrammatic variant PPA (navPPA), logopenic variant PPA (lvPPA) and FTD associated with motor neuron disease (FTD-MND). CBD and PSP are classified as FTD-related disorders due to their underlying tau pathology. The wide variety of phenotypes encountered in atypical parkinsonism is increasingly recognized. Here the clinical, imaging, genetic and pathological aspects of MSA, PSP, CBD and FTLD are reviewed.
Epidemiology
Estimates of prevalence, age at onset and survival in rare atypical parkinsonian syndromes and FTD vary. In European studies, MSA had an incidence of 0.6/100 000, a prevalence of 1.9–4.9/100 000, a mean age of onset of 55 years and a median survival of 7–10 years [1–3]. In the USA, the incidence was 0.6–0.7/100 000, and the prevalence was 3.4–4.9/100 000 [4,5]. A study from Iceland found similar estimates (incidence 0.7/100 000, prevalence 3.4/100 000) [6]. The sex distribution is even. FTD is the second most common cause of neurodegenerative dementia with early onset (i.e. before age 65) after Alzheimer’s disease (AD) [7]. The prevalence of FTD, comprising bvFTD and PPA, was highest in patients aged 45–64 years (15–22/100 000 individuals); 30% of FTD patients were above age 65 years, and 10% were below age 45 years [7]. Its prevalence may be underestimated [7,8]. The incidence of FTD was estimated between 1.61–4.1/100 000 [7,9]. Both sexes were affected approximately equally [10]. The diagnosis of bvFTD was found to be four times more common than PPA [10]. The prevalence of FTD, CBS and PSP was 10.8/100 000, with a peak age-adjusted prevalence of 42.6/100 000 at ages 65–69 years [9]. PSP had an estimated prevalence of 5.8–6.5/100 000 [11], a mean age of onset of 65–68 years and also an even sex distribution. Median survival is 6–10 years in PSP, with wide variations observed in PSP subtypes [12].
Clinical features
Atypical parkinsonian syndromes and FTLD are clinically heterogeneous. Clinical subtypes and characteristic clinical features of MSA, PSP, CBD and FTLD are listed in Tables 1 and 2.
Table 1.
Clinical subtypes of MSA, PSP, CBD and FTLD
| Disorder | Clinical phenotypes | Comment |
|---|---|---|
| MSA | MSA-P | Parkinsonian variant |
| MSA-C | Cerebellar variant | |
| PSP | PSP-RS | Richardson’s syndrome (classical phenotype) |
| PSP-P | Parkinsonian variant | |
| PSP-OM | Predominance of ocular motor symptoms | |
| PSP-PI | Predominance of postural instability | |
| PSP-PPFGa | Primary progressive freezing of gait | |
| PSP-PA | Pure akinesia | |
| PSP-F | PSP with frontal lobe dysfunction, including: | |
| bvFTD | PSP presenting as bvFTD | |
| PSP-CBS | PSP presenting as CBS | |
| PSP-SL | PSP with speech or language impairment, including: | |
| PSP-navPPA | PSP presenting as non-fluent agrammatic variant PPA | |
| PSP-AOS | PSP presenting as primary progressive apraxia of speech | |
| PSP-C | Cerebellar variant | |
| PSP-PLS | Primary lateral sclerosis variant | |
| CBD | CBS | Classical phenotype (possible versus probable CBS) |
| FBS | Frontal behavioral-spatial syndrome | |
| PSPS | PSP syndrome with CBD pathology | |
| navPPA | Non-fluent agrammatic variant PPA with CBD pathology | |
| FTLD | bvFTD | Behavioral variant FTD |
| svPPA | Semantic variant PPA | |
| navPPA | Non-fluent agrammatic variant PPA | |
| lvPPA | Logopenic variant PPA | |
| FTD-MND | FTD associated with motor neuron disease | |
| CBS | CBS with FTLD pathology | |
| PSP | PSP with FTLD pathology |
PSP-PPFG is also called PSP-PAGF: pure akinesia with gait freezing.
Table 2.
Clinical features
| Disorder | Clinical features |
|---|---|
| MSA | Age at onset >30 years Parkinsonism (hypokinesia, rigidity > tremor) Cerebellar signs: gait ataxia, dysarthria, limb ataxia Ocular motor signs: impaired vestibulo-ocular reflex suppression, gaze-evoked nystagmus, downbeat nystagmus, square wave jerks Pyramidal signs: positive Babinski sign, hyperreflexia > spasticity Autonomic signs: orthostatic hypotension, urinary incontinence/urgency, erectile dysfunction, anhidrosis Dysphagia Dystonia (laryngeal stridor), antecollis (DD: dystonic, due to myopathy) Sleep apnea, rapid eye movement sleep behavior disorder Executive dysfunction, cognitive impairment Depression, anxiety Pain |
| PSP | Age at onset ≥40 years (carriers of rare MAPT mutations may have an earlier onset) Vertical supranuclear gaze palsy (downward > upward) Decreased velocity of saccades (vertical > horizontal), frequent square wave jerks Impaired postural reflexes with falls (typically backwards) Rigidity (axial > limbs), hypokinesia (symmetric, can be asymmetric in PSP-P) > tremor Dysarthria, dysphagia, pathological crying/laughter Freezing of gait, start hesitation ‘Astonished facies’ (Cowper’s sign), overactivity of the frontalis muscle, ‘apraxia of eyelid opening’, reducing blinking Behavioral changes, executive dysfunction, impaired inhibition, apathy, memory dysfunction, slowed thinking Speech apraxia (effortful slow speech with dysprosody and buccofacial apraxia), non-fluent aphasia (PSP-SL) Alien limb phenomenon, limb rigidity/hypokinesia/apraxia/myoclonus, cortical sensory deficit (PSP-CBS) |
| CBD | Age at onset ≥50 for probable sporadic CBD, no minimum age for possible CBD Parkinsonism (rigidity and bradykinesia typically starting in one limb > tremor, asymmetric) Dystonia (typically of one limb), contractures Stimulus-sensitive myoclonus Limb and ideomotor apraxia, oro-buccal apraxia Alien limb phenomenon (arm > leg) Cortical sensory deficits Depression, behavioral changes, compulsive behavior Executive dysfunction, speech apraxia, non-fluent aphasia, visuospatial deficits |
| bvFTD | Behavioral changes (disinhibition, apathy/inertia, loss of empathy, reduced social interest) Perseverative stereotyped movements, including language stereotypes Hyperorality (binge eating, consumption of inedible objects, altered food preferences) Executive dysfunction Parkinsonism (rigidity, bradykinesia, gait impairment, postural tremor) |
| svPPA | Impaired single-word comprehension, impaired object naming (anomia) Impaired object knowledge Dyslexia, dysgraphia |
| navFTD | Speech apraxia, agrammatism, effortful speech, impaired articulation and prosody |
| lvPPA | Impaired single-word retrieval, reduced speech rate, pauses, vague sentences, erroneous repetition due to short-term memory deficits |
| FTD-MND | FTD associated with signs of motor neuron disease |
Multiple system atrophy
Core symptoms of MSA are parkinsonism, cerebellar and pyramidal signs and autonomic dysfunction. In MSA-C, cerebellar symptoms prevail, while parkinsonism is predominant in MSA-P. When parkinsonian and cerebellar signs are seen equally, the term MSA-mixed is used. Consensus criteria for the diagnosis of MSA were published in 2008 [13]. The akinetic-rigid form of parkinsonism is typical for MSA, while tremor is rare. If tremor occurs, it is rather postural or action tremor, and it may incorporate myoclonus; resting tremor is not frequent. Parkinsonism is less responsive to levodopa and progresses faster compared to PD. Ocular motor dysfunction includes impaired suppression of the vestibulo-ocular reflex, downbeat and gaze-evoked nystagmus. Smooth pursuit eye movements are impaired, and saccades are frequently dysmetric. Pyramidal signs include extensor plantar responses and hyperreflexia. Spasticity occurs only in about 10% of patients. Orthostatic hypotension, with a drop in systolic (≥30 mmHg) and/or diastolic (≥15 mmHg) blood pressure within 3 min of standing, is the most frequent cardiovascular autonomic symptom. Syncopes may occur. Cognitive impairment, especially executive dysfunction, can be seen in later stages; dementia can develop [14]. Depression and anxiety occur in about 40% of MSA patients [15]. The courses of MSA-P and MSA-C are similar. Severe autonomic failure at diagnosis is associated with a worse prognosis [16].
Progressive supranuclear palsy
There are several subtypes of PSP. The ‘classic’ and most frequent form is designated Richardson’s syndrome (PSP-RS) [17]. In an autopsy-confirmed case series, PSP-RS accounted for 24% of 100 PSP cases [18]. Other common subtypes are the parkinsonian variant (PSP-P) and variants with initial predominance of ocular motor dysfunction (PSP-OM) or postural instability (PSP-PI). Further variants are PSP-pure akinesia (PSP-PA) and PSP-primary progressive freezing of gait (PPGF), which closely resemble each other, as well as ‘cortical PSP variants’ including PSP-CBS, presentations with frontal lobe dysfunction (PSP-F) presenting with cognitive or behavioral symptoms (mostly bvFTD) and variants with speech or language disorders (PSP-SL, including navPPA and primary apraxia of speech). Infrequent variants are PSP-C, with predominant cerebellar ataxia, and PSP-primary lateral sclerosis (PLS). National Institute of Neurological Disorders and Stroke (NINDS) diagnostic guidelines existed only for PSP-RS until very recently [19]. In 2017, diagnostic guidelines have been published with the aim of including all PSP subtypes early with high sensitivity and specificity [20]. Core features of PSP-RS are vertical gaze palsy (especially downward gaze, preceded by slowing of vertical saccades), postural instability with falls and axial rigidity. The vertical gaze palsy can be overcome by the vestibulo-ocular reflex. Horizontal gaze and saccades can be affected to a lesser degree. Hypokinesia is symmetric in PSP-RS, and the response to levodopa is limited or absent. Dyskinesias during levodopa therapy do not develop. In the final disease stage akinetic mutism, complete ophthalmoplegia and immobility develop. In PSP-P, bradykinesia and rigidity can be asymmetric, and there is a better response to levodopa. In PSP-PPGF, severe start hesitation and freezing of gait can remain the predominant sign for several years. PSP-P and PPGF are also referred to as brainstem variants, associated with a slower disease progression than PSP-RS. The cortical PSP variants present with non-fluent aphasia, speech apraxia or behavioral symptoms.
Corticobasal degeneration
The clinical diagnostic accuracy of CBD is the lowest amongst all diseases discussed here (about 50%), which is due to variable clinical presentations and substantial overlap of clinical features with other neurodegenerative conditions including AD. Parkinsonism is the most frequently observed motor symptom, typically asymmetric and starting in one arm. Rigidity and bradykinesia are common. Tremor is rare and, if present, it is rather action/postural than resting tremor and more irregular and jerky than in PD. Impaired postural reflexes may be present. Dystonia is also frequent, affecting most often one limb, leading to contractures, pain and immobility of this limb. While levodopa-responsive dystonic posturing mostly of a foot is frequently seen in PD, dystonia in atypical parkinsonism (CBD and less frequently MSA) does not respond well to levodopa and is not bound to off-periods. One classic sign of CBS is the alien limb phenomenon, which usually occurs in one arm. Several variants have been described [21]. The affected limb does not obey the patient’s commands and assumes involuntary positions, including elevation. Alien foot syndrome may be underdiagnosed. Neuropsychiatric features include depression and compulsive behaviors. Proposed clinical criteria for CBS have been published [22]. Accordingly, in ‘probable CBS’ asymmetric presentation of rigidity, hypokinesia and dystonia is observed, with each of these signs typically being limited to one limb; asymmetric limb myoclonus may occur. When these signs occur symmetrically, ‘possible CBS’ can be diagnosed. Additional syndromes have been described: frontal behavioral-spatial syndrome (FBS), navPPA, PSP syndrome and posterior cortical atrophy syndrome. Thus, behavioral changes, executive dysfunction, visuospatial deficits, agrammatism, speech apraxia, supranuclear vertical gaze palsy and visuospatial deficits can all be observed in CBD; urinary incontinence may occur in PSP syndrome [22,23].
Frontotemporal lobar degeneration
In FTLD, clinical features correspond to affected brain areas, with the non-dominant hemisphere being affected more severely in bvFTD and the dominant hemisphere in the PPAs. The most common clinical presentation of FTLD is bvFTD. Core symptoms of bvFTD are behavioral changes (e.g. disinhibition), apathy/inertia and changes in emotional modulation with loss of empathy. Further important symptoms are perseverative, sometimes compulsive, stereotyped movements, hyperorality and executive dysfunction. Behavioral disinhibition manifests as socially inappropriate behavior, loss of manners and impulsivity. Because of apathy, patients may not speak spontaneously. Hyperorality manifests as binge eating with weight gain, consumption of inedible objects and altered food preferences. Patients may start smoking, drinking alcohol or using illicit drugs. Memory functions are relatively spared in early disease stages. Hallucinations are particularly common in FTD due to GRN mutations (about 25%). International consensus criteria for bvFTD were published in 2011 [24]; the diagnosis of possible bvFTD is made on clinical grounds, while probable bvFTD includes neuroimaging criteria and a significant decline in functioning.
Semantic variant primary progressive aphasia, previously called semantic dementia, is characterized by the loss of word meaning. Both object naming (anomia) and single-word comprehension are impaired. Dyslexia, dysgraphia and impaired object knowledge are common [25]. Vague words replace nouns. Verbal fluency and grammaticality are intact. Symptoms of bvFTD can be observed. NavPPA presents with agrammatism and speech apraxia [25]. Impaired motor speech planning causes effortful speech, impaired articulation and altered prosody. Comprehension is generally only disturbed in syntactically complex sentences, and single-word comprehension is intact. LvPPA is characterized by impaired single-word retrieval with reduced speech rate and pauses and by erroneous repetition due to short-term memory deficits. Single-word comprehension and object knowledge are intact; there is no agrammatism and no speech apraxia. Sentences lack descriptive detail. When PPA spreads to posterior temporal regions, visual agnosia and prosopagnosia can occur.
There is significant overlap between FTD and MND (FTD-MND/FTD and amyotrophic lateral sclerosis, FTD-ALS). Motor neuron signs are mostly seen in bvFTD, with subtle or subclinical motor neuron signs found in about 50% of bvFTD patients. FTD symptoms occur in about 30% of patients with ALS, including subtle symptoms of speech or executive dysfunction as well as the full bvFTD phenotype. FTD-ALS due to C9orf72 mutations shows parkinsonism in about one-third of patients, and it may even mimic PSP. Hallucinations, delusions and psychosis occur more frequently and may be mistaken for bipolar disorder and schizophrenia [26].
The term ‘FTD and parkinsonism linked to chromosome 17’ (FTDP-17) stems from a consensus conference in 1996 and relates to families with FTD inherited in an autosomal dominant mode later shown to be due to mutations in MAPT or GRN, both located on chromosome 17. These patients usually present with bvFTD and parkinsonism with an early age of onset (MAPT mutations, early 40s; GRN, early 50s). The pathology is different in FTDP-17 due to MAPT mutations (tauopathy) versus FTDP-17 due to GRN mutations (TDP-43 proteinopathy).
Differential diagnoses
Atypical parkinsonian syndromes first have to be differentiated from PD. In PD, onset of parkinsonism is mostly unilateral, and over time asymmetric bilateral symptoms develop. If tremor is the dominant symptom, especially as regular ‘pill rolling’ resting tremor, atypical parkinsonism is unlikely. The levodopa response is good in PD, while it is limited or even absent in atypical parkinsonism. Hyperkinesia and dyskinesias are seen in later stages of PD, but not in atypical parkinsonism. The disease course is far more benign in PD. Cerebellar symptoms and extrapyramidal motor signs are not present in typical PD, while autonomic symptoms do occur, primarily cardiovascular (orthostasis). Olfactory dysfunction is common in PD, rare in CBD and PSP, and mildly impaired in MSA. When the distinction between atypical parkinsonism and PD cannot be reached on clinical grounds, neuroimaging techniques, especially investigations of the integrity of pre- and postsynaptic nigrostriatal dopaminergic neurons, can be of assistance. Important differential diagnoses of parkinsonism include drug-induced parkinsonism (e.g. neuroleptics, valproate). Patients with normal pressure hydrocephalus present with a typical gait impairment, cognitive dysfunction and urinary incontinence. Patients with AD may also show some degree of parkinsonism, both in the sporadic form and due to mutations (mostly in PSEN1).
There are many rare genetic disorders in which parkinsonism may be present. Perry syndrome is caused by mutations in the dynactin (DCTN1) gene and is inherited in an autosomal dominant mode [27,28]. Patients are affected by parkinsonism, central hypoventilation, weight loss and psychiatric symptoms (e.g. hallucination, depression). DCTN1 mutations may rarely present as PSP look-alikes [29]. Kufor–Rakeb syndrome is caused by ATP13A2 mutations and is inherited in an autosomal recessive mode. It presents with parkinsonism, pyramidal signs, dementia and hallucinations. Importantly, it has an earlier onset than the atypical parkinsonian syndromes reviewed here. Parkinsonism may be present in other forms of neurodegeneration with brain iron accumulation and, while there are adult-onset forms, these disorders also have an earlier onset age. Parkinsonism has been described in the fragile X-associated tremor/ataxia syndrome caused by pathologically expanded repeat expansion in the FMR1 gene and in Huntington’s disease. Parkinsonian variants and variants mimicking MSA in spinocerebellar ataxias (SCA) have been described for SCA1, SCA2, SCA3, SCA6, SCA7 and SCA17. Late-onset Friedreich ataxia can mimic MSA-C. Patients with the rare white matter disorder adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) due to CSF1R mutations may show parkinsonism and psychiatric symptoms, including behavioral changes and dementia of a frontotemporal type [30–32]. Mitochondrial disorders presenting with parkinsonism include polymerase gamma (POLG)-related parkinsonism.
Several metabolic autosomal recessive disorders can present with parkinsonism, e.g. Gaucher’s disease (due to mutations in the GBA gene), Niemann–Pick C (mutations in NPC1 and NPC2) and cerebrotendinous xanthomatosis (CYP27A1 mutations). In younger patients (<45 years), Wilson’s disease must be excluded, and neuroacanthocytosis is another differential diagnosis. Supranuclear gaze palsy has been reported in Niemann–Pick C, Kufor–Rakeb disease and Perry syndrome [33]. Paraneoplastic disorders should be ruled out in atypical parkinsonism (paraneoplastic disease autoantibodies, including antibodies for stiff person syndrome in PSP). In some instances prion disease (14-3-3 protein in the cerebrospinal fluid, CSF) will have to be excluded (e.g. PSP with rapid course). Midbrain tumors and manganese intoxication [magnetic resonance imaging (MRI) T1 hypointensities in the globus pallidus] can present with a PSP phenotype.
Importantly, the prevailing symptoms in FTLD and its related disorders can be cognitive impairment and psychiatric symptoms, including confusion and apathy. Differential diagnoses thus include other types of dementia (AD, vascular dementia), metabolic and endocrinological causes (e.g. thiamin deficiency, hypothyroidism) as well as infections of the central nervous system (e.g. neurosyphilis, HIV infection, Lyme disease, rarely Whipple’s disease). Autoimmune and paraneoplastic disorders have to be ruled out. Psychiatric diseases which may be misdiagnosed in FTLD patients have a wide spectrum and include minor and major depressive disorder, bipolar affective disorder, obsessive–compulsive disorder and schizophrenia. FTLD due to MAPT, GRN and C9orf72 mutations may present as PSP look-alike, and FTLD due to GRN and C9orf72 mutations as CBD look-alike [33]. However, most patients with genetic forms of FTLD will have a positive family history of dementia or parkinsonism.
Investigations
A thorough history is required, and family members or caregivers often have to be included. A full neurological examination is performed and includes careful assessment of ocular motor function, cortical sensory testing (graphesthesia, stereognosis), testing for apraxia (ideomotor, speech) and neuropsychiatric/cognitive assessment including frontal lobe functions and language skills (e.g. verbal fluency tasks). Frontal release signs include the grasping reflex, palmomental reflex and the applause test (pathological, if there is an inability to stop after three claps). The Luria sequence (fist-edge-palm series) tests executive function. A recommended scale for overall cognitive testing is the Montreal Cognitive Assessment. In FTD, standard questionnaires for evaluating behavioral changes include the Frontal Behavioral Inventory, the Frontotemporal Dementia Rating Scale and the Frontotemporal Lobar Degeneration-Modified Clinical Dementia Rating Scale.
Laboratory investigations are mainly performed to exclude other diseases. CSF biomarkers in AD are increased tau, increased phospho-tau and reduced β-amyloid (Aβ42). In FTD, Aβ42 is moderately reduced, phospho-tau is normal and total tau is moderately increased; overlap with the AD profile, however, is not uncommon. Further, Aβ40 may show lower levels in FTD patients than in controls. Patients with FTD due to GRN mutations may have reduced progranulin levels (plasma and CSF). In PSP patients, the level of homovanillic acid, a catabolic product of dopamine, and of tau may be reduced in the CSF. The C- and N-terminal fragments of tau may be promising biomarkers in PSP. Further tests of assistance include the following. Heart rate variability is frequently pathological in MSA. Using transcranial sonography, concurrent findings of normal substantia nigra hyperechogenicity and increased lentiform nucleus hyperechogenicity support a diagnosis of atypical parkinsonism. MSA patients may show phosphorylated α-synuclein deposits in somatosensory fibers of the subepidermal plexus. Certain electroencephalography techniques may be used to investigate myoclonus in CBD. 123I-MIBG scintigraphy investigates postganglionic sympathetic denervation, which is seen in PD with autonomic failure [34]. Patients with MSA, however, may also show a decrease of cardiac MIBG uptake [35]; in PSP and CBD cardiac MIBG uptake is normal or slightly reduced.
Neuroimaging
The structural imaging modality of choice is MRI, and this modality is reviewed here in detail. Computed tomography can occasionally be of additional assistance, e.g. to demonstrate the presence of brain calcifications in the basal ganglia or the cerebellum (Fahr’s disease) or in the pericallosal region (ALSP) [36].
Magnetic resonance imaging
Using MRI, characteristic differential structural brain changes of the neurodegenerative disorders reviewed here can be seen (Fig. 1, Table 3), and unrelated structural pathologies can be excluded, e.g. tumors, malformations, vascular lesions, normal pressure or obstructive hydrocephalus. All patients presented in this paper were participants of Mayo Clinic International Review Board approved protocols.
Figure 1.
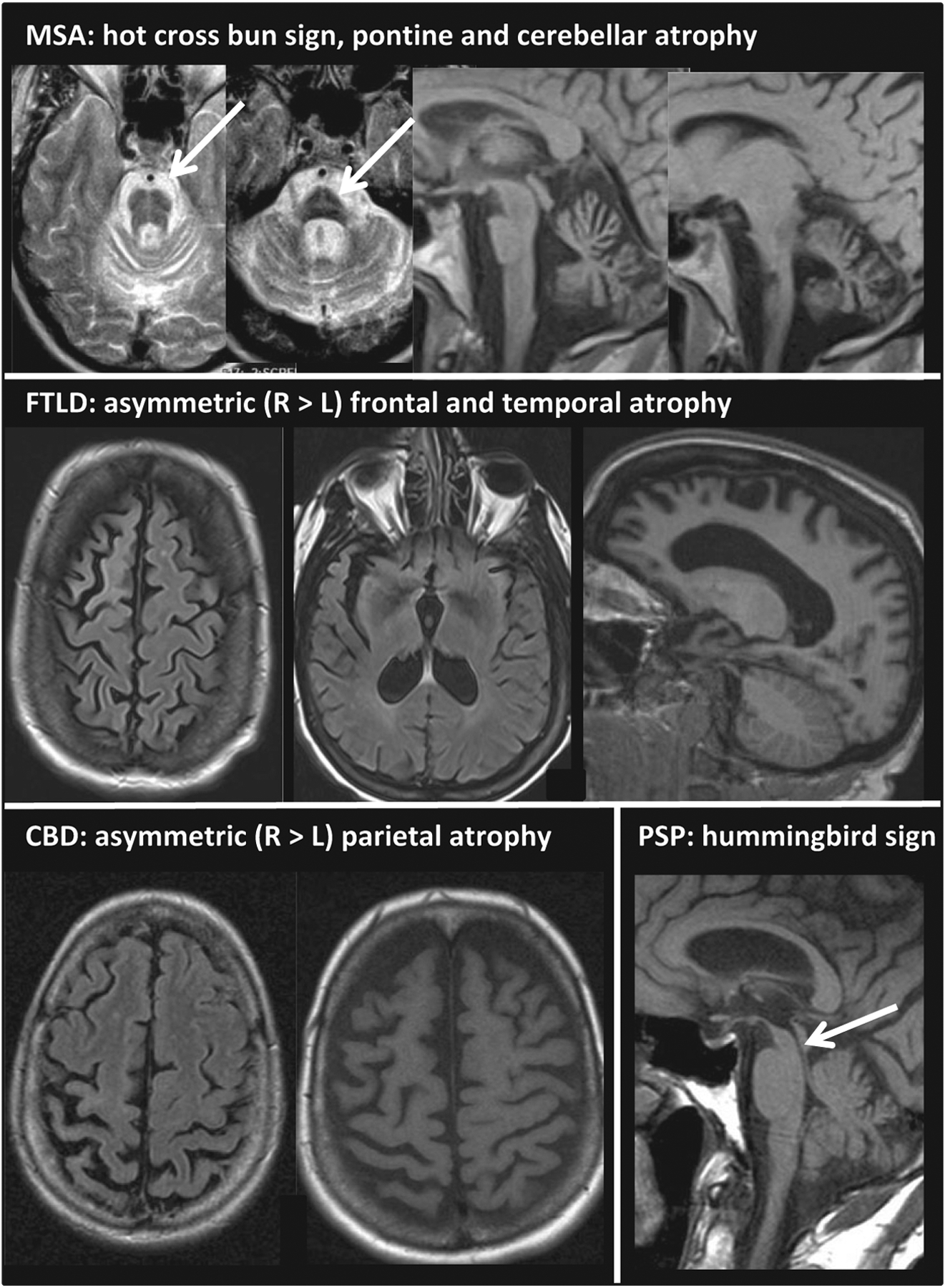
Cranial MRIs of patients with MSA-C, FTLD, CBD and PSP-RS. Upper row: 52-year-old male patient with MSA-C: MRI shows the pontine ‘hot-cross bun’ sign (arrow) and severe cerebellar and pontine atrophy. Clinical features were pancerebellar dysfunction including limb ataxia for 7 years, mild parkinsonism, orthostasis, urinary dysfunction, vasomotor, sudomotor and trophic changes of distal extremities, corticospinal tract signs and rapid eye movement sleep behavior disorder. Middle row: 46-year-old female patient with FTLD [pallido-ponto-nigral degeneration (PPND) family] due to an MAPT gene mutation (N279K): MRI depicts asymmetric frontotemporal atrophy (right > left). Clinical features included mild behavioral problems, depression, memory loss (leading to dementia) and parkinsonism with severe rigidity and bradykinesia (L > R), tremor, hypophonia and impaired postural reflexes with falls. Further, eyelid apraxia, downward gaze palsy and limb dystonia with contractures developed. Disease course was rapidly progressive with early age at disease onset (38 years). Age at death was 46 years. Autopsy showed FTLD with extensive tauopathy. Lower row, left: cranial MRI of a 60-year-old patient with CBD showing parietal > frontal asymmetric atrophy (right > left). The patient had shown progressive limb apraxia, dystonia, cortical sensory impairment and myoclonus of her left arm (alien limb phenomenon) since age 56 years. Further, left-sided bradykinesia and rigidity were seen. Treatment with levodopa and intramuscular botulinum toxin injections provided some benefit. Age at death was 62 years. Autopsy showed typical CBD pathology. Lower row, right: 70-year-old male patient with PSP-RS: MRI depicts the ‘hummingbird sign’ (arrow) due to severe midbrain atrophy. Clinical features were supranuclear vertical gaze palsy, slow horizontal saccades, Cowper’s sign, severely impaired postural reflexes with falls causing severe injuries, dysphagia, mild parkinsonism (bradykinesia and rigidity, axial > limbs) and cognitive impairment. Disease course was rapidly progressive. Age at death was 71. Pathology was consistent with PSP and additionally showed pallido-nigral pigment spheroid degeneration.
Table 3.
Neuroimaging
| Characteristic MRI findings | |
|---|---|
| MSA | ‘Hot-cross bun’ sign (pontine T2 hyperintensity) Putaminal rim sign (T2 hyperintense rim of dorsolateral putaminal border) Atrophy of pons, cerebellum, middle cerebellar peduncle |
| PSP | ‘Hummingbird sign’ (more severe midbrain than pontine atrophy) ‘Morning glory sign’ (atrophy of midbrain tegmentum) Atrophy of midbrain, tegmentum, superior cerebellar peduncles |
| CBS | Asymmetric atrophy in parietal and (pre)frontal cortices |
| bvFTD | Frontal and anterior temporal atrophy (more pronounced in non-dominant hemisphere) |
| svPPA | Anterior temporal atrophy (dominant hemisphere) |
| navPPA | Atrophy of the inferior frontal cortex (dominant hemisphere) |
| lvPPA | Posterior temporal and parietal atrophy (dominant hemisphere) |
In MSA, MRI can be normal in the early stages. The pontine ‘hot-cross bun’ sign is highly suggestive of MSA [37]. The putamen shows atrophy, T2 hypointensity and a T2 hyperintense rim of its dorsolateral border (‘putaminal rim sign’). Atrophy of pons, cerebellum and the middle cerebellar peduncle (MCP) may be severe. Using volumetric studies such as voxel based morphometry, atrophy is further seen in the substantia nigra, inferior olive, prefrontal cortex, motor cortex, supplementary motor area (SMA) and insular cortex. Volumetric loss in the olfactory bulb and tract may distinguish between PD and MSA [38].
Characteristic changes in PSP include atrophy of the midbrain (reduced midbrain diameter), tegmentum and the superior cerebellar peduncle (SCP). Some patients show frontal and prefrontal cortical atrophy. Due to more severe midbrain atrophy relative to the pons, the ‘hummingbird sign’ is found on midsagittal slices. Atrophy of the midbrain tegmentum underlies the ‘morning glory sign’. Volumetric studies found volume loss in further areas including the thalamus, striatum, insula, SMA and the (para-)hippocampal gyri.
Corticobasal degeneration is characterized by asymmetric atrophy in parietal and (pre)frontal cortical areas including the precentral and postcentral gyri, extending into the posterior temporal areas; in autopsy proven CBD atrophy was also seen in the striatum, SMA and brainstem [39]. Patterns of atrophy are variable and correlate with the clinical presentation. Perisylvian atrophy is seen in CBD presenting with navPPA. A further study of pathology proven CBD found additional areas of atrophy in the insular cortex and globus pallidus; in this study only a small region in the dorsal ponto-medullary junction was affected in the brainstem [40].
The ‘MR parkinsonism index’, which calculates both the midbrain:pons and the SCP:MCP ratios, was found to be 100% specific and sensitive for differentiating PSP versus MSA-P [41]. Automated MRI-based segmentation of regions or volumes of interest are being developed for more accurate diagnosis of atypical parkinsonism [42,43]. The tractography technique diffusion tensor imaging proved useful in differentiating MSA-P and PSP from PD [44]. Diffusion-weighted brain imaging was successfully used for discriminating CBD, PSP and PD [45]. A multimodal approach with T2*, diffusion-weighted volume measurements and region of interest analyses was of assistance in discriminating parkinsonian syndromes [46].
In bvFTD, atrophy of the frontal and (anterior) temporal cortices is more pronounced in the non-dominant hemisphere. SvPPA presents with anterior temporal cortical atrophy of the dominant hemisphere. NavPPA is characterized by atrophy of the inferior frontal cortex of the dominant hemisphere. In lvPPA predominant left posterior temporal and parietal atrophy is seen, areas vulnerable also to AD pathology. When FTD progresses, atrophy can become bilateral and extend into further brain areas, in particular into posterior temporal areas.
Radioligand imaging
Single-photon emission computed tomography (SPECT) and positron emission tomography (PET) are frequently applied to investigate the integrity of the pre- and postsynaptic dopaminergic system in order to differentiate atypical parkinsonism (MSA, PSP) from PD.
The dopamine transporter (DAT) as a measure of presynaptic nigrostriatal integrity can be investigated using different radiotracers (e.g. SPECT tracers: 123I-FP-CIT, 123I-beta-CIT; PET tracers: 18F-FP-CIT, 18F-CFT). DAT density is reduced in MSA, PSP and PD. In PD, asymmetric affection of the striatum with a rostral–caudal gradient is characteristic; the posterior putamen is the most affected area. In PSP, DAT loss may be more symmetric and the putamen-to-caudate ratio may be higher compared to PD. This pattern of striatal DAT loss, however, may not be reliable on an individual basis. 18F-fluorodopa PET also measures presynaptic dopaminergic integrity.
To investigate postsynaptic striatal dopaminergic integrity, SPECT and PET ligands binding to dopamine receptors (DRD1, DRD2) are used. Tracers include 123I-IBZM (SPECT) as well as 11C-raclopride, 18F-fallypride and 18F-DMFP (PET). While reduced radioligand binding is well established in MSA and PSP, thus differentiating these disorders from PD, further studies with autopsy verified CBD are warranted. It is important to hold some medications which bind to dopamine receptors before performing these techniques. 11C-raclopride PET imaging combined with local tracer influx ratios in several brain areas reliably discriminated between MSA-P and PD [47]. SPECT studies also investigate cerebral perfusion in parkinsonian syndromes and dementia (e.g. 99Tc-HMPAO SPECT). PET studies further investigate non-dopaminergic neurotransmitter systems, especially the cholinergic and serotoninergic systems.
18F-Fluorodeoxyglucose (FDG) PET is used to identify areas of glucose hypometabolism in cortical areas. In patients with MSA, impaired glucose metabolism is most evident in the cerebellum, basal ganglia and pons. In PSP patients, basal ganglia, midbrain and to a lesser extent the thalamus are affected; supratentorial areas include the frontal cortex (precentral gyrus) and the anterior cingulate cortex. Asymmetrically reduced metabolism in frontoparietal regions, basal ganglia and thalamus is characteristic of CBD. In FTLD, hypometabolism in PET (as well as hypoperfusion in SPECT) is seen in areas which are also affected by atrophy; e.g., in bvFTD, atrophy of the frontal and the anterior temporal cortex of the non-dominant hemisphere is characteristic. By combining 18F-FDG PET resting state data and spatial covariance analysis, Eidelberg and colleagues identified differential CBD-, MSA- and PSP-related covariance patterns in addition to previously described PD covariance patterns [48,49].
The PET ligand 11C-(R)-PK11195 for translocator protein (TSPO) is upregulated in activated microglia and serves as a marker of neuroinflammation. Increased tracer binding occurs in MSA, PSP, CBD and PD [50]. A significant number of second generation tracers for TSPO, including 11C-DPA713 and 18F-FEPPA, have been developed. Using 11C-Pittsburgh compound B (PIB) PET, Aβ deposition can be depicted in cortical and subcortical regions including the cingulum and striatum. A further Aβ tracer is 18F-florbetaben. An important application of Aβ imaging is to discriminate between AD, Lewy body dementia and FTLD. Aβ imaging in patients with lvPPA found AD pathology in a subset of these patients. Rarely, patients with CBS have underlying AD pathology. Amyloid abnormalities are virtually absent in MSA. While tau PET imaging is an area under very active investigation, it is so far used only for research purposes. Ligands include 18F-T807, also known as 18F-AV-1451, 18F-FDDNP, 18F-THK523, 18F-THK5105 and 11C-PBB3. Challenges include the intracellular localization of tau aggregates and the existence of different tau subtypes and conformations.
Neuropathology and pathomechanisms
Pathologically, MSA is classified as α-synucleinopathy and has two variants, olivopontocerebellar atrophy (OPCA) and striatonigral degeneration (SND), corresponding to the clinical variants MSA-C and MSA-P. Macroscopically, the cerebellum, MCP and the pontine base show atrophy in OPCA, and atrophy and dark discoloration of the putamen are seen in SND. Histologically, MSA is characterized by oligodendroglial cytoplasmic inclusions (GCIs), which contain misfolded hyperphosphorylated α-synuclein (Fig. 2, Table 4). GCIs further contain ubiquitin, tau and LRRK2. Selected populations of neurons also have α-synuclein-positive inclusions in neuronal cytoplasm, nuclei and cell processes. There is neuronal loss, astrogliosis, myelin degeneration with myelin pallor, and axonal degeneration. Iron pigment is frequent in the striatum.
Figure 2.
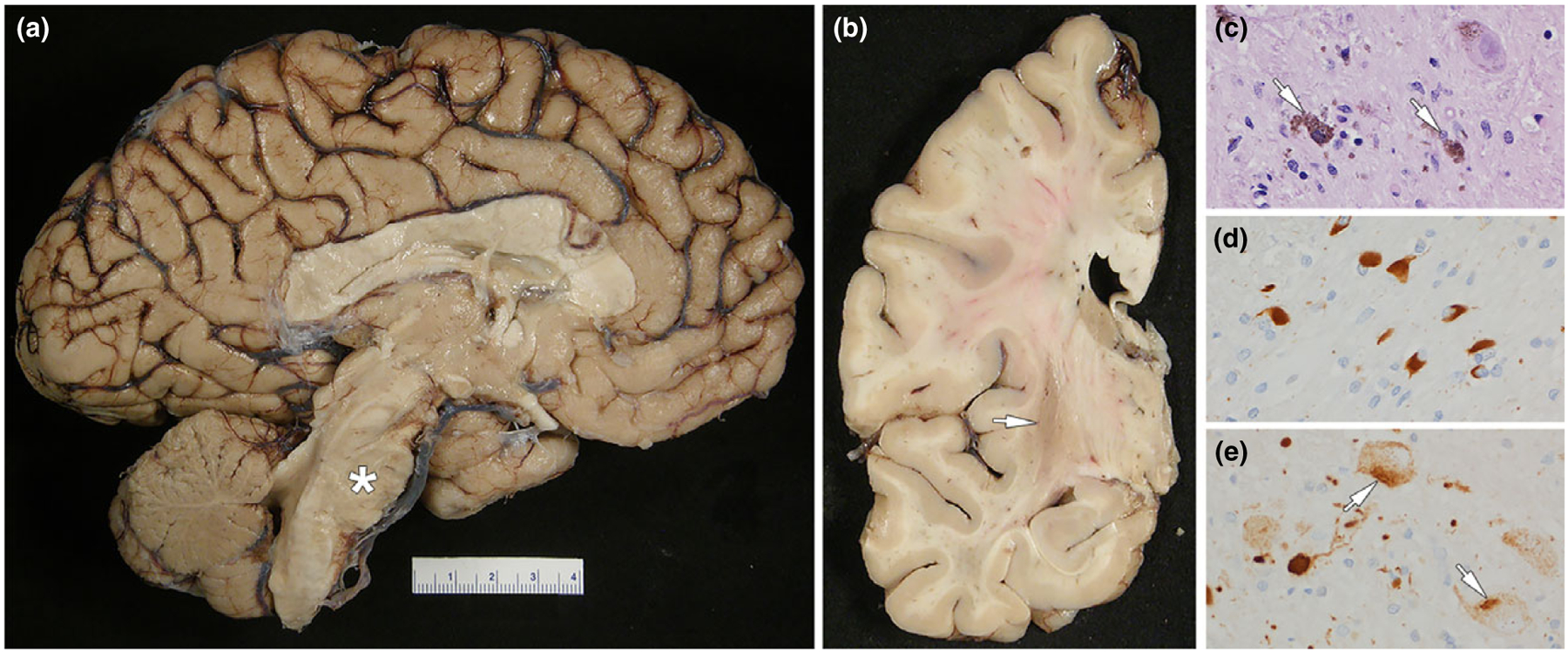
MSA: (a) medial view of the left cerebral hemisphere showing severe atrophy of the pontine base (asterisk); note also the cerebellar vermis atrophy and discoloration; (b) coronal section of the left cerebral hemisphere showing atrophy and dark brown discoloration of the posterior putamen (arrow); (c) neuronal loss in substantia nigra with neuromelanin containing macrophages (arrows) (d) fiber tracts in the pontine base have numerous oligodendroglial inclusions (GCIs) with α-synuclein immunohistochemistry; (e) neurons in the pontine base have neuronal cytoplasmic inclusions (arrows) with α-synuclein immunohistochemistry (note also the dystrophic neurites). The patient had MSA with a mixed subtype since age 55 years. She had limb ataxia, rigidity, urinary incontinence and anxiety. Further, rapid eye movement sleep behavior disorder and cognitive decline developed. Age at death was 64 years.
Table 4.
Characteristic pathological features
| Main pathology | Histopathological findings | Areas of atrophy and neuronal loss; further characteristics | |
|---|---|---|---|
| MSA | α-synucleinopathy | α-synuclein(+) oligodendrocytic glial cytoplasmic inclusions (GCIs); α-synuclein(+) neuronal cytoplasmic and intranuclear inclusions; myelin loss | Atrophy in putamen, pons, middle cerebellar peduncle, cerebellum; neuronal loss in putamen, substantia nigra, inferior olivary nucleus, Onuf’s nucleus, intermediolateral cell column; demyelination – striato-pallidal fibers, transverse pontine fibers, olivocerebellar fibers, cerebellar white matter |
| PSP | 4R tauopathy | Tau(+) neurofibrillary globose tangles (4R tau); tufted astrocytes with tau(+) inclusions; tau(+) coiled bodies in oligodendrocytes; thread-like structures (diencephalon, brainstem) | Atrophy of midbrain, superior cerebellar peduncle, prefrontal cortex; neuronal loss in subthalamic nucleus, substantia nigra, cerebellar dentate nucleus; loss of pigment in substantia nigra |
| CBD | 4R tauopathy | Astrocytic plaques (4R tau in terminal processes); pleomorphic tau(+) cortical neuronal inclusions, achromatic ballooned neurons; pretangles (tau(+) cytoplasmic inclusions in neurons); coiled bodies (less than in PSP); sparse tau(+) oligodendroglial lesions; dense, widespread thread-like structures in gray and white matter (neuropil threads) | Atrophy of superior frontal, superior parietal and motor cortices, SMA, perisylvian region, corpus callosum; neuronal loss in globus pallidum, substantia nigra, subthalamic nucleus; loss of pigment in substantia nigra |
| FTLD-tau | 3R, 4R or 3R + 4R tauopathies | Tau(+) cytoplasmic inclusions in neurons and glia Pick’s disease: 3R tau(+) neuronal cytoplasmatic inclusions (Pick bodies); ballooned neurons (Pick cells); sparse glial inclusions Argyrophilic grain disease: 4R tau(+) grains, oligodendroglial coiled bodies, ballooned neurons NFT dementia: 3R + 4R tau(+) neuronal inclusions (neurofibrillary tangles) |
Bilateral atrophy with neuronal loss and astrogliosis of frontal and temporal cortices, including medial temporal regions, spreading to more posterior areas (parietal cortex) in disease course |
| FTLD-TDP | TDP-43 proteinopathy | TDP-43(+), ubiquitin(+), p62-sequestosome(+) neuronal cytoplasmic and intranuclear inclusions; oligodendroglial and astrocytic inclusions; superficial cortical laminar spongiosis subtypes A, B, C and D | Cortical atrophy (frontal > temporal and parietal lobes), hippocampus, amygdala, basal ganglia (caudate > putamen > globus pallidus) |
| FTLD-TDP with C9orf72 mutation | TDP-43 proteinopathy | TDP-43(+) inclusions; in addition to TDP-43 pathology: dipeptide repeat protein(+), ubiquitin(+), p62-sequestosome(+) neuronal and glial inclusions (cerebellar granule cells); nuclear RNA foci | Cortical atrophy (frontal > temporal and parietal lobes), motor cortex (some cases), hippocampus, amygdala, basal ganglia (caudate > putamen > globus pallidus); neuronal loss in brainstem, spinal cord motor neurons |
| FTLD-FET | Ubiquitinopathy | FUS(+), ubiquitin(+), p62-sequestosome(+) neuronal cytoplasmic and neuronal intranuclear inclusions | Cortical atrophy (frontal > temporal and parietal lobes), striatal atrophy (severe) |
The etiology of MSA is poorly understood and the origin of GCIs (oligodendroglial origin versus cell-to-cell transfer) is unknown. Oligodendroglial dysfunction is of critical importance in the etiology of MSA [51]. Prion-like spreading of α-synuclein from neurons to glia cells has been proposed to cause glial and myelin dysfunction, neuroinflammation with increased amount of microglia and astrocytes, and secondary neurodegeneration [52,53]. Inoculation of brain homogenates of MSA patients was found to induce motor impairment and α-synuclein deposits in transgenic M83+/− mice hemizygous for the PD-causing α-synuclein A53T mutation [54]. Oligodendrogenesis may be impaired by α-synuclein deposits, with increased numbers of oligodendroglial progenitor cells observed in brains of MSA patients [55]. Mitochondrial dysfunction, impaired autophagy and oxidative stress are further mechanisms proposed. A possible link between α-synuclein deposition and insulin resistance has recently been described [56].
COQ2 mutations have been reported to cause familial MSA, and variants in COQ2 conferred risk in sporadic MSA patients [57]. Genetic variants in MAPT, GBA, LRRK2 and SHC2 genes are inconsistently associated with MSA. SNCA variants were highly significantly associated with MSA risk (Table 5) [58]. The largest genome-wide association study (GWAS) of MSA, however, found no significant association at any genetic locus [59].
Table 5.
Genetic causes in MSA, PSP, CBD and FTD
| Gene | Gene product | Clinical phenotypes |
|---|---|---|
| FTLD | ||
| MAPT | Microtubule-associated protein tau | bvFTD, navPPA, svPPA, CBS, PSP, FTDP-17, PiD |
| GRN | Progranulin | bvFTD, navPPA, svPPA, CBS, FTDP-17 |
| C9orf72 | Chromosome 9 open reading frame 72 protein | bvFTD, MND, FTD-MND, svPPA, navPPA, lvPPA, akinetic-rigid syndrome |
| CHMP2B | Chromatin-modifying protein 2Ba | bvFTD, FTD-MND |
| VCP | Valosin-containing protein | IBMPFD, FTD-MND |
| hnRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | MSP (FTD-MND) |
| hnRNPA2B1 | Heterogeneous nuclear ribonucleoprotein A2B1 | MSP (FTD-MND) |
| TARDBP | TAR DNA binding protein | MND, bvFTD, svPPA |
| FUS/TLS | Fused in sarcoma/translocated in liposarcoma protein | MND, bvFTD, FTD-MND |
| SQSTM1 | Sequestosome 1 | FTD-MND, PPA, bvFTD |
| TBK1 | TANK binding kinase 1 | bvFTD, PPA, FTD-MND |
| CHCHD10 | Coiled-coil-helix coiled-coil-helix domain-containing protein 10 | bvFTD, FTD-MND |
| TMEM106Bb,c | Transmembrane protein 106B | |
| RAB38b | Ras-related protein Rab-38 | |
| HLA-locusb | Major histocompatibility complex, class II, DR alpha / DR beta 5 | |
| MSA | ||
| COQ2d | Coenzyme Q2 | |
| SNCAb | α-synuclein | |
| PSP | ||
| MAPT | Microtubule-associated protein tau | |
| STX-6b | Syntaxin 6 | |
| EIF2AK3b | Eukaryotic translation initiation factor 2α kinase 3 | |
| CBD | ||
| SOS1b | Son of sevenless homologue 1 | |
| lnc-KIF13B-1b | Long noncoding RNA, 8p12 locus | |
| MSA, PSP, CBD | ||
| MAPT H1b | Microtubule-associated protein tau (H1 haplotype) | |
| PSP, CBD | ||
| MOBPb | Myelin-associated oligodendrocyte basic protein |
IBMPFD, inclusion body myopathy with Paget disease of the bone and frontotemporal dementia; MSP, multisystem proteinopathy; PiD, Pick’s disease.
Synonymous with charged multivesicular body protein 2B;
these genes were associated with the disorders in GWAS and are considered risk factors for sporadic disease, while mutations in the other genes listed cause familial disease;
TMEM106B modifies disease (FTD with TDP-43 pathology) in GRN mutation carriers;
COQ2 variants both confer risk in sporadic MSA patients (GWAS locus) and cause familial MSA.
Corticobasal degeneration and PSP are considered 4R tauopathies with extensive overlap in clinical presentation and in pathology. While they were suggested to form a 4R tauopathy disease spectrum, neuropathological differences have been described in detail [60,61]. Tau protein has roles in microtubule stabilization and assembly as well as in axonal transport. It has alternative splice forms and post-translational modifications like phosphorylation. Tau proteins may contain three or four repeats of about 32 amino acids in the microtubule binding domain.
Histologically, PSP is characterized by tau-positive inclusions in neurons, astrocytes and oligodendrocytes. Neurofibrillary globose tangles are composed of misfolded and abnormally phosphorylated 4R tau in a conformation of single straight filaments. Astrocytes with tau-positive inclusions are called tufted astrocytes; they are frequent in the motor cortex and striatum. Tau-positive coiled bodies form in oligodendrocytes (Fig. 3, upper row; Table 4). Neuronal loss and gliosis occur especially in the substantia nigra, subthalamic nucleus and cerebellar areas (dentate nucleus). The clinical PSP subtypes have differential affection of cortical areas and brainstem sites, e.g. PPGF and PSP-P have more severe involvement of brainstem sites.
Figure 3.
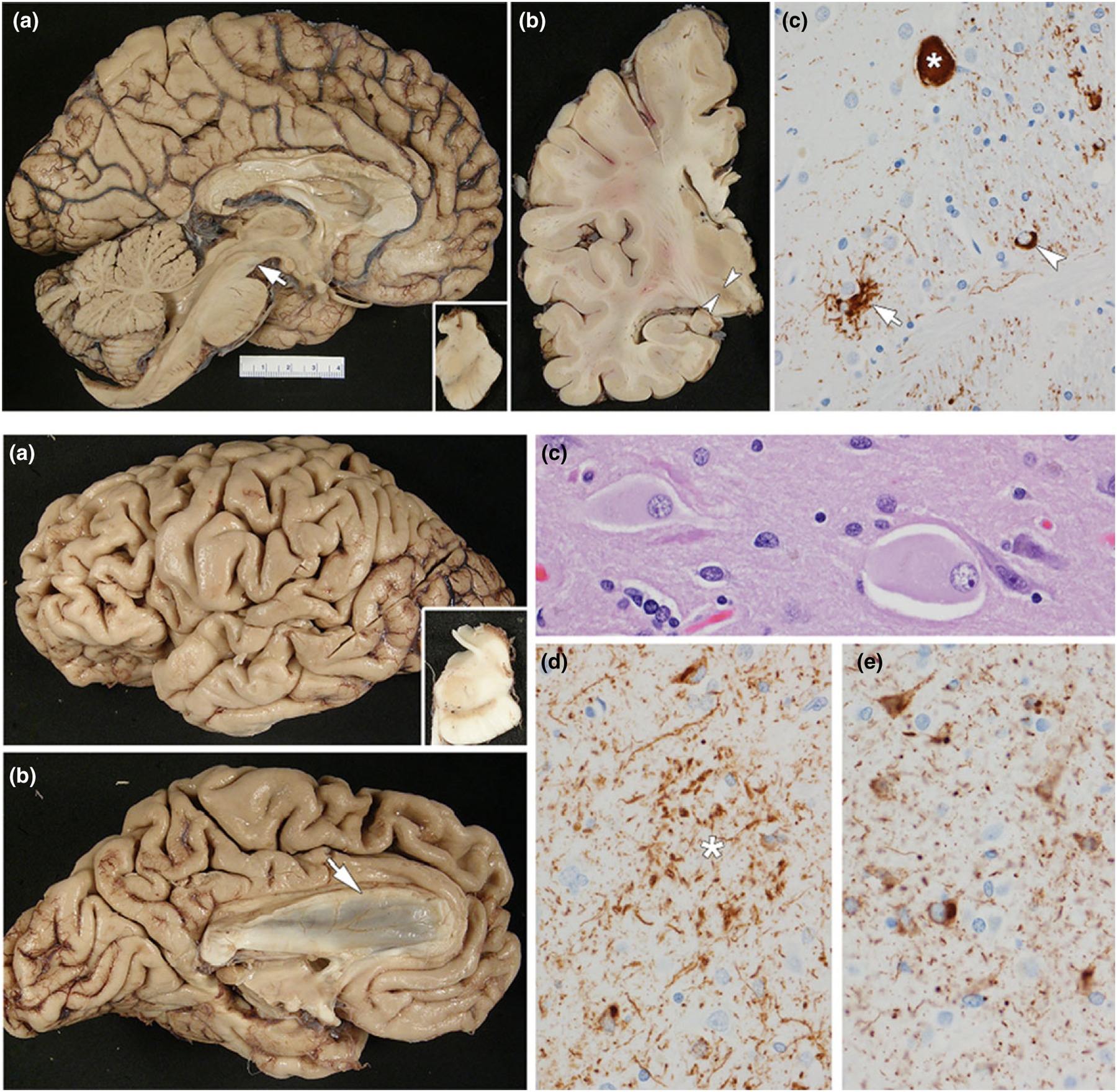
Tauopathies. Upper row: PSP: (a) medial view of the left cerebral hemisphere showing severe midbrain atrophy (arrow); note also the dilation of the aqueduct of Sylvius; inset shows hemi-midbrain with loss of pigment in the substantia nigra; (b) coronal section of the left cerebral hemisphere demonstrating subthalamic nucleus atrophy (arrowheads); (c) phospho-tau immunohistochemistry shows a globose neurofibrillary tangle (asterisk), tufted astrocyte (arrow) and oligodendroglial ‘coiled body’ (arrowhead). The patient presented with supranuclear gaze palsy, dysarthria, truncal rigidity and impaired postural reflexes with falls since age 64. He had a slight levodopa response. The disease course was 5 years. Lower row: CBD: (a) left cerebral hemisphere showing premotor cortical atrophy (dorsal and ventral); inset shows hemi-midbrain with loss of pigment in the substantia nigra; (b) medial view of the left cerebrum showing superior frontal gyrus atrophy and atrophy of the corpus callosum (arrow); (c) ballooned neurons in the superior frontal gyrus (hematoxylin and eosin stain); (d) astrocytic plaque (asterisk) with phospho-tau immunohistochemistry (note also many neuropil threads); (e) pleomorphic cortical neuronal inclusions with phospho-tau immunohistochemistry. At age 67 years, the patient developed progressive speech difficulties, attention deficits and a gait disorder. Executive dysfunction, apraxia, emotional changes (indifference) and mild hypokinesia (left > right) consecutively developed. He died at age 73 years. While he was clinically diagnosed with FTD, autopsy showed CBD.
The etiology of PSP is unknown. Advanced age is the only established risk factor. PSP-tau pathology was found to be transmissable in transgenic mouse brains supporting a theory of a cell-to-cell propagation mechanism of different fibrillary ‘tau strains’ [62]. Patients with PSP very rarely have a positive family history [63]. PSP is highly associated with the common H1 haplotype of the MAPT gene [64]. GWAS loci showing association with PSP are MAPT, EIF2AK3, MOBP and STX-6 (Table 5) [65]. Association with MOBP and STX-6 loci supports the notion that altered myelin, oligodendrocyte dysfunction (MOBP) and impaired fusion of vesicles with membranes (STX-6) may be involved in PSP pathogenesis.
At autopsy, CBD typically has focal and asymmetric cortical atrophy, which is often associated with ventriculomegaly and thinning of the corpus callosum (Table 4). Histopathology of CBD shows ballooned neurons, tau-positive neuronal cytoplasmic inclusions, pre-tangles and tau-positive astrocytic plaques. Tau-positive oligodendroglial lesions are sparse. The dense thread-like structures in the white and gray matter of CBD are far more numerous and widespread than in PSP (Fig. 3, lower row; Table 4) [61]. The brain areas most affected by tau pathology are the parietal and frontal cortices, the basal ganglia, thalamus and the subthalamic nucleus, with less involvement of the brainstem.
MAPT, GRN and C9orf72 mutations may in some instances cause a phenotype of CBS. Like PSP, CBD is associated with homozygosity for the MAPT H1 haplotype. The largest GWAS performed in CBD found associations at MAPT, SOS1 and at a chromosome 8p12 locus (including the genes lnc-KIF13B-1, DUSP4 and KIF13B) [66]. Testing for association of CBD with top PSP GWAS hits identified further associations at MOBP and MAPT H1c, indicating an overlap of CBD and PSP genetic risk factors (Table 5) [66]. A recent GWAS meta-analysis found genetic overlap at the MAPT locus, MOBP, CXCR4, EGFR and GLDC between CBD and PSP. Functions of these genes include brain development, neuronal migration and forebrain cell migration. Genetic overlap of CBD and FTD was seen only at the MAPT locus [67].
Frontotemporal lobar degeneration is pathologically classified based on the predominant protein aggregates of pathological inclusions (tau, TDP-43 or FET). The FET family of proteins includes fused in sarcoma (FUS), Ewing’s sarcoma protein and TATA-binding protein-associated factor 15. FTLD-TDP (Fig. 4) accounts for about 50% and FTLD-tau for about 45% of FTLD cases, while FTLD-FET is uncommon [68]. FTLD-tau includes Pick’s disease, argyrophilic grain disease and some cases of FTDP-17. Histopathologically, FTLD-tau is characterized by intracellular insoluble aggregates of hyperphosphorylated tau protein; it may underlie bvFTD, navPPA and rarely svPPA. FTLD-TDP and FTLD-FET have neuronal inclusions positive for ubiquitin and p62-sequestosome that are composed of either TDP-43 or FUS protein. In FTLD associated with C9orf72 repeat expansions, inclusions contain TDP-43, but there are also neuronal inclusions composed of aberrantly transcribed hexanucleotide (GGGGCC) repeats, producing five distinct dipeptide repeat polymers (Table 4).
Figure 4.
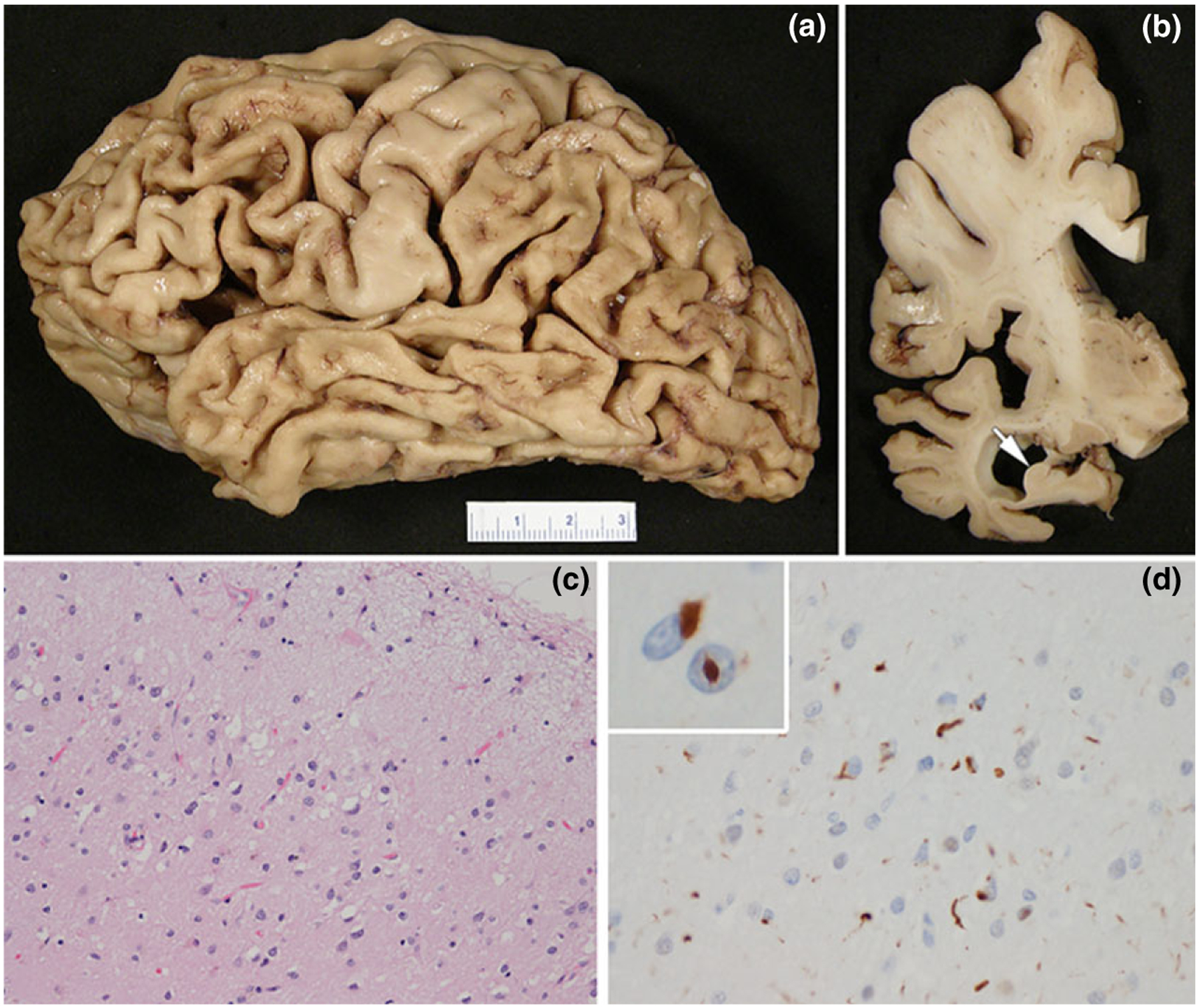
FTLD-TDP: (a) left cerebral hemisphere showing severe cortical atrophy affecting frontal and temporal lobes, as well as the inferior parietal lobule; (b) coronal section of the left cerebral hemisphere demonstrating ventricular enlargement and hippocampal atrophy (arrow); (c) the temporal cortex has neuronal loss and superficial laminar spongiosis (hematoxylin and eosin stain); (d) TDP-43 immunohistochemistry shows neuronal cytoplasmic inclusions and dystrophic neurites; inset shows neurons with neuronal cytoplasmic and neuronal intranuclear inclusions. The male patient showed memory impairment, language difficulties and behavioral changes (compulsiveness, irritability) with a slowly progressive course over more than 10 years. He died at age 76years.
Pick bodies are argyrophilic, globose neuronal inclusions composed of hyperphosphorylated 3R tau. They are frequent in frontal and temporal cortices, (para-) limbic areas (amygdala and hippocampus, including the dentate gyrus) and ventral temporal cortical areas. They may also be seen in the striatum, substantia nigra and locus ceruleus. Swollen (ballooned) neurons are called Pick cells. The term Pick disease denotes one of the FTLD-tau subtypes and is meanwhile no longer used to refer to a clinical syndrome. In ‘argyrophilic grain disease’ 4R tau-positive astrocytes, coiled bodies and grains are found in medial temporal and limbic regions. FTLD associated with MAPT mutations shows both 3R and 4R tau pathologies and can mimic all forms of tauopathies including Pick’s disease, PSP, CBD and tangle predominant dementia.
Four types of TDP-43 pathology (types A–D) with differential patterns of neuronal cytoplasmic and intranuclear inclusions (compact/granular) and neurite length have been described. TDP-43 types correlate with clinical and genetic subtypes. Type A, the most common type, is most frequently associated with bvFTD and navPPA. It has the most neuronal cytoplasmic and intranuclear inclusions, cortical neurites are short, and neurites in the hippocampus are fine. It is the most common finding in FTLD due to GRN mutations. Type B is also common and associated with bvFTD and FTD-MND. It has predominantly neuronal cytoplasmic inclusions with sparse neurites. It is the most common subtype associated with C9orf72 mutations. Type C has long thick cortical neurites and compact, Pick-body-like neuronal inclusions in the dentate gyrus. It is most often associated with svPPA, and it is usually sporadic. Type D has many neuronal intranuclear inclusions and is characteristic of FTLD due to mutations in VCP. FTLD-TDP is further associated with mutations in TBK1, SQSTM1 and UBQLN2. FTLD-FET presents at a young age with bvFTD and is associated with severe cortical and striatal atrophy. Neurons contain TDP-43-negative, FUS-, ubiquitin-and p62-sequestosome-positive inclusions. FUS inclusions are also seen in some cases with MND, basophilic inclusion body disease, atypical ubiquitin-positive FTLD and neuronal intermediate filament inclusion disease.
The heritability for FTLD is high, with an estimated 40% of patients reporting a family history of dementia or psychiatric symptoms and an autosomal dominant inheritance pattern in 10%–25%. Mutations in several genes have been linked to FTLD (Table 5), with MAPT, GRN and C9orf72 mutations being the most frequent causes for familial FTLD [69–73]. FTD-MND is assumed to be the subtype with the highest heritability [74]. Mutations in known FTLD genes explain the disease in almost all FTLD families with a clear mode of autosomal dominant inheritance. In sporadic FTD and in FTD with a less clear family history, these genes are found to be causative only in a minority of patients. A GWAS of autopsy-confirmed FTLD-TDP discovered risk and disease modifying variants [75]. A variant in TMEM106B was found to be protective in GRN mutation carriers [76]. FTD associated with MAPT mutations can present with a wide variety of phenotypes, and motor, speech, cognitive or behavioral symptoms may prevail; age of onset is relatively early. Patients with C9orf72 mutations mainly present with bvFTD and FTD-MND. All pathogenic GRN mutations result in a haploinsufficiency resulting in a 50% loss of progranulin levels; main clinical presentations are bvFTD and navPPA.
Management
Treatment options for motor and neuropsychiatric symptoms in atypical parkinsonian syndromes and FTD are currently limited to symptomatic therapies. Parkinsonism in atypical parkinsonian syndromes is generally far less levodopa responsive than in PD. Significant improvement may still occur for rigidity, hypokinesia and tremor. Treatment efficacy is not limited to PSP-P and MSA-P. Thus, a trial of levodopa is generally indicated, with slowly increasing levodopa doses up to 900–1000 mg daily (temporarily accompanied by carbidopa) for about 2–3 months. If well tolerated, the dosage can be further increased. Focal dystonia (e.g. limb dystonia in CBS) and drooling can be treated with botulinum toxin injections. Treatment of autonomic symptoms such as urinary dysfunction in MSA has to take into account that the use of anticholinergic drugs including amitriptyline is limited by adverse effects, especially by a possible negative impact on cognition. Droxidopa and fludrocortisone acetate may improve orthostatic hypotension in some patients with MSA.
Psychotic symptoms including hallucinations, delusions and aggressive behavior are treated with neuroleptic medication while carefully assessing development or progression of extrapyramidal motor signs. Atypical neuroleptics are much preferred and are given at the lowest dose possible. Depressive symptoms have an estimated prevalence of 33% in FTD and should be treated following guidelines [77]. Serotonin reuptake inhibitors (fluoxetine, fluvoxamine, sertraline, paroxetine and citalopram) as well as trazodone have been shown to be beneficial in the treatment of behavioral symptoms including disinhibition, stereotypic movements and hyperorality in FTD in clinical studies [78]. Oxytocin showed mild improvement of emotional changes including loss of empathy and social cognition and was well tolerated, but its routine use is not yet recommended [79,80]. Psychostimulants, e.g. methylphenidate and dextroamphetamine, may improve behavioral symptoms such as risk-tasking [81]. Dopaminergic medication and levodopa may improve executive dysfunction and apathy. Rapid eye movement sleep behavior disorder seen in MSA is treated with clonazepam and melatonin.
For cognitive impairment the use of acetyl-cholinesterase inhibitors in FTD is generally not recommended since symptoms may worsen and agitation may occur. Since a few patients with CBS and PPA may have AD pathology, a trial of cholinesterase inhibitors over 2–3 months can be indicated if cognition declines. Memantine was well tolerated but not effective in treating cognitive and behavioral symptoms in FTD [82]. Serotonin reuptake inhibitors, trazodone and amphetamines did not improve cognition [78]. Physical exercise, if tolerated, may delay the onset of cognitive impairment in FTD. Further non-pharmaco-logical approaches are speech therapy and caregiver education.
Randomized placebo-controlled trials of the GSK-3 inhibitor tideglusib [83] or davunetide [84], which are assumed to reduce tau phosphorylation, stabilize microtubule function and/or inhibit neuronal loss, showed no effect in patients with PSP. A phase 2 study on davunetide in tauopathies (ClinicalTrials.gov identifier NCT01056965) is not recruiting yet. Other therapeutic approaches target an increase of the reduced progranulin levels in FTLD due to GRN mutations or the reduction of either RNA foci or the C9orf72 expanded repeats dipeptide production, e.g. using antisense oligonucleotide therapies. Active immunization against pathological tau was assessed in a phase 1 clinical trial using AADvac1 in AD [85]. A phase 2 clinical trial using humanized monoclonal anti-tau antibodies (ABBV-8E12; AbbVie) is currently recruiting patients with early PSP (NCT02985879) and AD (NCT02880956) after positive results of a phase 1 study [86]. A phase 2 study on low dose lithium in FTD is currently recruiting.
High dose ubiquinol supplementation may be efficient in familial MSA with COQ2 mutations; further clinical trials are warranted [87]. High doses of coenzyme Q10 did not modify disease progression in patients with PSP [88]. In MSA patients, rifampicin and rasagiline were not effective as neuroprotective agents [89–91]. A trial with fluoxetine in MSA patients has been completed (NCT01146548). The administration of lithium was not well tolerated in MSA, and the trial was stopped. An ongoing trial in MSA is investigating the CSF delivery of autologous mesenchymal stem cells (phase 1 study, Mayo Clinic). Preclinical studies in MSA focused on the inhibition of α-synuclein aggregation, degradation and clearance as well as on inhibition of α-synuclein cell-to-cell propagation. These approaches included active and passive immunization, reducing truncated α-synuclein (VX-765), overexpression of proteases that cleave α-synuclein and degrading extracellular α-synuclein by modifying neurosin [92]. Active immunization has been tested in patients with early MSA in a phase 1 study, which has recently been completed. (AFFITOPE PD01A and PD03A, Affiris AG, Vienna, Austria). A phase 2 study on the effect of the myeloperoxidase inhibitor AZD3241 on microglial activation as measured by PET and a phase 3 study on targeting α-synuclein aggregation by epigallocatechin-gallate supplementation (PROMESA) [93] have recently been completed (NCT02388295, NCT02008721).
Future prospects
The wide clinical and pathological overlap of atypical parkinsonian syndromes is increasingly recognized. FTLD has also been recognized to be highly heterogeneous, both clinically and pathologically. Novel neuroimaging techniques are being developed at a fast pace, including radioligand imaging. Genetics will continue to provide information about the pathophysiology and disease pathways of these disorders. With the use of next generation sequencing techniques and novel bioinformatics approaches, additional diseasecausing genes and risk genes are likely to be identified in the near future. Emergent neuroimaging, genetic and biochemical biomarkers may allow for early and accurate diagnoses in patients with rare parkinsonian syndromes, which is a prerequisite for clinical trials. With positive results of preclinical studies on a wide variety of disease modifying drugs, there is hope that neuroprotective agents will also prove efficacious in clinical trials.
Acknowledgements
AD is supported by the Max Kade Foundation and a Gift from Carl Edward Bolch, Jr, and Susan Bass Bolch. ZKW is partially supported by the NIH/NINDS P50 NS072187, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at the Mayo Clinic in Florida), the Sol Goldman Charitable Trust and a Gift from Donald G. and Jodi P. Heeringa. OAR is supported by the Morris K. Udall Parkinson’s Disease Research Center of Excellence (P50-NS072187), R01-NS078086 and U54-NS10069 and the Mayo Clinic Neuroscience Focused Research Team.
Footnotes
Disclosure of conflicts of interest
The authors declare no financial or other conflicts of interest.
References
- 1.Vanacore N, Bonifati V, Fabbrini G, et al. Epidemiology of multiple system atrophy. ESGAP Consortium. European Study Group on Atypical Parkinsonisms. Neurol Sci 2001; 22: 97–99. [DOI] [PubMed] [Google Scholar]
- 2.Vanacore N Epidemiological evidence on multiple system atrophy. J Neural Transm (Vienna) 2005; 112: 1605–1612. [DOI] [PubMed] [Google Scholar]
- 3.Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013; 12: 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med 2015; 372: 249–263. [DOI] [PubMed] [Google Scholar]
- 5.Stefanova N, Bücke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009; 8: 1172–1178. [DOI] [PubMed] [Google Scholar]
- 6.Bjornsdottir A, Gudmundsson G, Blondal H, Olafsson E. Incidence and prevalence of multiple system atrophy: a nationwide study in Iceland. J Neurol Neurosurg Psychiatry 2013; 84: 136–140. [DOI] [PubMed] [Google Scholar]
- 7.Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci 2011; 45: 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry 2013; 25: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coyle-Gilchrist IT, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016; 86: 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogan DB, Jetté N, Fiest KM, et al. The prevalence and incidence of frontotemporal dementia: a systematic review. Can J Neurol Sci 2016; 43(Suppl. 1): S96–S109. [DOI] [PubMed] [Google Scholar]
- 11.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 1999; 354: 1771–1775. [DOI] [PubMed] [Google Scholar]
- 12.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8: 270–279. [DOI] [PubMed] [Google Scholar]
- 13.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71: 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown RG, Lacomblez L, Landwehrmeyer BG, et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain 2010; 133: 2382–2393. [DOI] [PubMed] [Google Scholar]
- 15.Schrag A, Sheikh S, Quinn NP, et al. A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Mov Disord 2010; 25: 1077–1081. [DOI] [PubMed] [Google Scholar]
- 16.Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015; 14: 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richardson JC, Steele J, Olszewski J. Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of ‘heterogenous system degeneration’. Trans Am Neurol Assoc 1963; 88: 25–29. [PubMed] [Google Scholar]
- 18.Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014; 29: 1758–1766. [DOI] [PubMed] [Google Scholar]
- 19.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47: 1–9. [DOI] [PubMed] [Google Scholar]
- 20.Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017; 32: 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hassan A, Josephs KA. Alien hand syndrome. Curr Neurol Neurosci Rep 2016; 16: 73. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80: 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray R, Neumann M, Forman MS, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007; 68: 1274–1283. [DOI] [PubMed] [Google Scholar]
- 24.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134: 2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76: 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 2017; 18: 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konno T, Ross OA, Teive HAG, Sławek J, Dickson DW, Wszolek ZK. DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat Disord 2017; 41: 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vilariño-Güell C, Wider C, Soto-Ortolaza AI, et al. Characterization of DCTN1 genetic variability in neurodegeneration. Neurology 2009; 72: 2024–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caroppo P, Le Ber I, Clot F, et al. DCTN1 mutation analysis in families with progressive supranuclear palsy-like phenotypes. JAMA Neurol 2014; 71: 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konno T, Yoshida K, Mizuno T, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol 2017; 24: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundal C, Wszolek Z. Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. 2012 [Updated 2014] In: Pagon RA, Adam MP, Ardinger HH, et al. , editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, 1993–2017; Available from: https://www.ncbi.nlm.nih.gov/books/NBK100239/ [Google Scholar]
- 32.Sundal C, Fujioka S, Van Gerpen JA, et al. Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Parkinsonism Relat Disord 2013; 19: 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamelou M, Quinn NP, Bhatia KP. ‘Atypical’ atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy – a diagnostic guide. Mov Disord 2013; 28: 1184–1199. [DOI] [PubMed] [Google Scholar]
- 34.Orimo S, Suzuki M, Inaba A, Mizusawa H. 123I-MIBG myocardial scintigraphy for differentiating Parkinson’s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat Disord 2012; 18: 494–500. [DOI] [PubMed] [Google Scholar]
- 35.Nagayama H, Ueda M, Yamazaki M, et al. Abnormal cardiac [(123)I]-meta-iodobenzylguanidine uptake in multiple system atrophy. Mov Disord 2010; 25: 1744–1747. [DOI] [PubMed] [Google Scholar]
- 36.Konno T, Broderick DF, Mezaki N, et al. Diagnostic value of brain calcifications in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. AJNR Am J Neuroradiol 2017; 38: 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrag A, Kingsley D, Phatouros C, et al. Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry 1998; 65: 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Tan HY, Wu ZH, et al. Imaging of olfactory bulb and gray matter volumes in brain areas associated with olfactory function in patients with Parkinson’s disease and multiple system atrophy. Eur J Radiol 2014; 83: 564–570. [DOI] [PubMed] [Google Scholar]
- 39.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011; 70: 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 2008; 29: 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology 2008; 246: 214–221. [DOI] [PubMed] [Google Scholar]
- 42.Huppertz HJ, Möller L, Südmeyer M, et al. Differentiation of neurodegenerative parkinsonian syndromes by volumetric magnetic resonance imaging analysis and support vector machine classification. Mov Disord 2016; 31: 1506–1517. [DOI] [PubMed] [Google Scholar]
- 43.Scherfler C, Göbel G, Müller C, et al. Diagnostic potential of automated subcortical volume segmentation in atypical parkinsonism. Neurology 2016; 86: 1242–1249. [DOI] [PubMed] [Google Scholar]
- 44.Worker A, Blain C, Jarosz J, et al. Diffusion tensor imaging of Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy: a tract-based spatial statistics study. PLoS One 2014; 9: e112638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizzo G, Martinelli P, Manners D, et al. Diffusion-weighted brain imaging study of patients with clinical diagnosis of corticobasal degeneration, progressive supranuclear palsy and Parkinson’s disease. Brain 2008; 131: 2690–2700. [DOI] [PubMed] [Google Scholar]
- 46.Barbagallo G, Sierra-Peña M, Nemmi F, et al. Multimodal MRI assessment of nigro-striatal pathway in multiple system atrophy and Parkinson disease. Mov Disord 2016; 31: 325–334. [DOI] [PubMed] [Google Scholar]
- 47.Van Laere K, Clerinx K, D’Hondt E, et al. Combined striatal binding and cerebral influx analysis of dynamic 11C-raclopride PET improves early differentiation between multiple-system atrophy and Parkinson disease. J Nucl Med 2010; 51: 588–595. [DOI] [PubMed] [Google Scholar]
- 48.Eckert T, Tang C, Ma Y, et al. Abnormal metabolic networks in atypical parkinsonism. Mov Disord 2008; 23: 727–733. [DOI] [PubMed] [Google Scholar]
- 49.Niethammer M, Tang CC, Feigin A, et al. A disease-specific metabolic brain network associated with corticobasal degeneration. Brain 2014; 137: 3036–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerhard A TSPO imaging in parkinsonian disorders. Clin Transl Imaging 2016; 4: 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ettle B, Schlachetzki JCM, Winkler J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol 2016; 53: 3046–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jellinger KA. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov Disord 2014; 29: 1720–1741. [DOI] [PubMed] [Google Scholar]
- 53.Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 2015; 112: E5308–E5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watts JC, Giles K, Oehler A, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA 2013; 110: 19555–19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.May VE, Ettle B, Poehler AM, et al. Alpha-synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol Aging 2014; 35: 2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bassil F, Canron MH, Vital A, et al. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain 2017; 140: 1420–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.The Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 2013; 369: 233–244. [DOI] [PubMed] [Google Scholar]
- 58.Scholz SW, Houlden H, Schulte C, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 2009; 65: 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sailer A, Scholz SW, Nalls MA, et al. A genome-wide association study in multiple system atrophy. Neurology 2016; 87: 1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 1999; 246(Suppl. 2): II6–II15. [DOI] [PubMed] [Google Scholar]
- 61.Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration (FTLD-tau). J Mol Neurosci 2011; 45: 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11: 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujioka S, Sanchez-Contreras MY, Strongosky AJ, et al. Three sib-pairs of autopsy-confirmed progressive supranuclear palsy. Parkinsonism Relat Disord 2015; 21: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rademakers R, Melquist S, Cruts M, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet 2005; 14: 3281–3292. [DOI] [PubMed] [Google Scholar]
- 65.Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 2011; 43: 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kouri N, Ross OA, Dombroski B, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun 2015; 6: 7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yokoyama JS, Karch CM, Fan CC, et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol 2017; 133: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012; 8: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916–919. [DOI] [PubMed] [Google Scholar]
- 70.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006; 442: 920–924. [DOI] [PubMed] [Google Scholar]
- 71.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702–705. [DOI] [PubMed] [Google Scholar]
- 73.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005; 65: 1817–1819. [DOI] [PubMed] [Google Scholar]
- 75.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010; 42: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrari R, Hernandez DG, Nalls MA, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol 2014; 13: 686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chakrabarty T, Sepehry AA, Jacova C, Hsiung GY. The prevalence of depressive symptoms in frontotemporal dementia: a meta-analysis. Dement Geriatr Cogn Disord 2015; 39: 257–271. [DOI] [PubMed] [Google Scholar]
- 78.Nardell M, Tampi RR. Pharmacological treatments for frontotemporal dementias: a systematic review of randomized controlled trials. Am J Alzheimers Dis Other Demen 2014; 29: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jesso S, Morlog D, Ross S, et al. The effects of oxytocin on social cognition and behaviour in frontotemporal dementia. Brain 2011; 134: 2493–2501. [DOI] [PubMed] [Google Scholar]
- 80.Tampi RR, Maksimowski M, Ahmed M, Tampi DJ. Oxytocin for frontotemporal dementia: a systematic review. Ther Adv Psychopharmacol 2017; 7: 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rahman S, Robbins TW, Hodges JR, et al. Methylphenidate (‘ritalin’) can ameliorate abnormal risk-taking behavior in the frontal variant of frontotemporal dementia. Neuropsychopharmacology 2006; 31: 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boxer AL, Knopman DS, Kaufer DI, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2013; 12: 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tolosa E, Litvan I, Höglinger GU, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord 2014; 29: 470–478. [DOI] [PubMed] [Google Scholar]
- 84.Boxer AL, Lang AE, Grossman M, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol 2014; 13: 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol 2017; 16: 123–134. [DOI] [PubMed] [Google Scholar]
- 86.Fogelman I, Boxer AL, Hu H, et al. Safety, tolerability and pharmacokinetics of ABBV-8E12, a humanized anti-tau monoclonal antibody, in a phase 1, single ascending dose, placebo-controlled study in subjects with progressive supranuclear palsy 9th Clinical Trials on Alzheimer’s Disease, San Diego, CA, 2016: 285 http://www.ctad-alzheimer.com (accessed 11/09/2016). [Google Scholar]
- 87.Mitsui J, Koguchi K, Momose T, et al. Three-year follow-up of high-dose ubiquinol supplementation in a case of familial multiple system atrophy with compound heterozygous COQ2 mutations. Cerebellum 2017; 16: 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Apetauerova D, Scala SA, Hamill RW, et al. CoQ10 in progressive supranuclear palsy: a randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm 2016; 3: e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Low PA, Robertson D, Gilman S, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014; 13: 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Poewe W, Mahlknecht P, Krismer F. Therapeutic advances in multiple system atrophy and progressive supranuclear palsy. Mov Disord 2015; 30: 1528–1538. [DOI] [PubMed] [Google Scholar]
- 91.Poewe W, Seppi K, Fitzer-Attas CJ, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol 2015; 14: 145–152. [DOI] [PubMed] [Google Scholar]
- 92.Spencer B, Valera E, Rockenstein E, et al. A brain-targeted, modified neurosin (kallikrein-6) reduces α-synuclein accumulation in a mouse model of multiple system atrophy. Mol Neurodegener 2015; 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Levin J, Maaß S, Schuberth M, et al. The PROMESA-protocol: progression rate of multiple system atrophy under EGCG supplementation as anti-aggregation-approach. J Neural Transm (Vienna) 2016; 123: 439–445. [DOI] [PubMed] [Google Scholar]