

Abstract
Molecular, cellular, and clinical studies of human inborn errors of immunity have revolutionized our understanding of their pathogenesis, considerably broadened their spectrum of immunological and clinical phenotypes, and enabled successful targeted therapeutic interventions. These studies have also been of great scientific merit, challenging a number of immunological notions initially established in inbred mice, while revealing previously unrecognized mechanisms of host defense by leukocytes and other cells, and of both innate and adaptive tolerance to self.
Introduction
Primary Immune Deficiencies (PIDs) have been classically characterized by increased susceptibility to infections, due to genetic defects affecting development and/or function of the immune system. The history of PIDs is typically dated back to 1952, when Bruton described a boy who had suffered from 19 episodes of pneumococcal infections, lacked serum gamma globulins, and recovered upon intramuscular administration of gamma globulins (1). Two years earlier, Glanzmann and Riniker had described an infant with severe lymphopenia and atrophy of lymphoid tissues who succumbed to infections early in life (2). Subsequently, Hitzig reported other infants with early-onset life-threatening infections who lacked both gamma globulins and lymphocytes (3). This condition, which was initially named Swiss-type agammaglobulinemia (4) (to distinguish it from Bruton’s agammaglobulinemia), is now referred to as Severe Combined Immune Deficiency (SCID). Together, isolated agammaglobulinemia and SCID provided evidence of the critical role played by humoral and cellular immunity, respectively, in protection against infections, paving the way to the discovery of T and B cells few years later (5).
The first inborn errors of innate immunity defects were also reported in the 1950s. In 1950, Kostmann described the first patients with severe congenital neutropenia (6). While searching for other cases of hypogammaglobulinemia, Janeway et al. reported in 1954 a patient with recurrent infections and, paradoxically, elevated serum immunoglobulins (7). The patient was found in 1957 to suffer from a congenital defect of phagocyte function, designated as chronic granulomatous disease in 1957 (8). Inborn errors of complement were described from the 1960s onward. It was soon recognized that the nature of pathogens (viruses, bacteria, fungi, or parasites, “opportunistic” or not) causing infections in patients with PID is largely determined by the arm of immunity that is affected (T lymphocytes, B lymphocytes, phagocytes, complement). PIDs in any category have long been defined by susceptibility to a broad range of pathogens. Although clinically and immunologically homogeneous forms of PIDs (agammaglobulinemia, SCID, CGD) were evidently transmitted with different patterns (X-linked, autosomal recessive), too few immunological reagents were then available to distinguish different forms of PID in each category. Consequently, only a handful of distinct disorders were included in the first classification of PIDs in 1968, which, incidentally, did not include neutropenia as it was regarded by immunologists of the time as a hematological condition (9).
Over the years, it has been recognized that depending upon the specific nature of the disease, autoimmunity, autoinflammation, allergy, and malignancy can be common, and in some cases, predominant, clinical manifestations associated with monogenic defects of immunity (13). To capture this broad range of phenotypes associated with these disorders, the term “inborn errors of immunity” (IEI) has been proposed. Importantly, advances in molecular genetics and cellular immunology have allowed a much more precise definition of various forms of IEI (Fig. 1). Use of next generation sequencing (NGS) has permitted the identification of a growing number of IEI (Fig. 2A), whose number has now reached 431 in the 2020 classification from the International Union of Immunological Societies Committee on Inborn Errors of Immunity (10). At the molecular level, it has been shown that most PIDs can be caused by mutations in different genes (locus heterogeneity), which typically govern a certain pathway. More surprisingly, distinct pathogenic variants at the same locus have also been shown to cause different forms of IEI, usually but not necessarily because of different genotypes (allelic heterogeneity: mono-allelic vs. bi-allelic lesions, loss-of-function (or hypomorphic) vs. gain-of-function (or hypermorphic) variations, dominant-negative vs. haploinsufficient mode of dominance).
Figure 1.
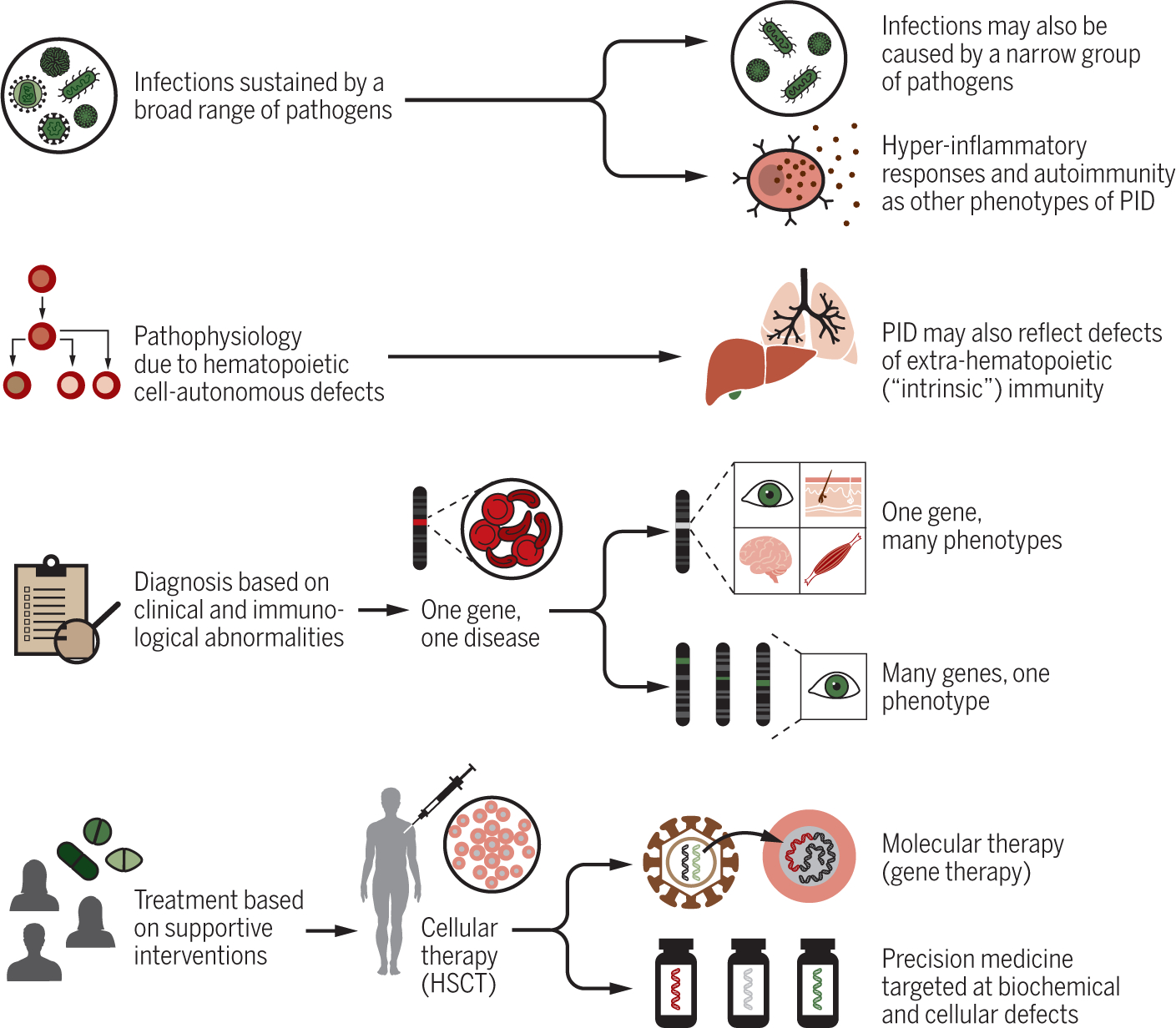
Evolution of clinical, pathophysiological diagnostic and therapeutic approach to inborn errors of immunity.
Figure 2. History and current classification of monogenic inborn errors of immunity.
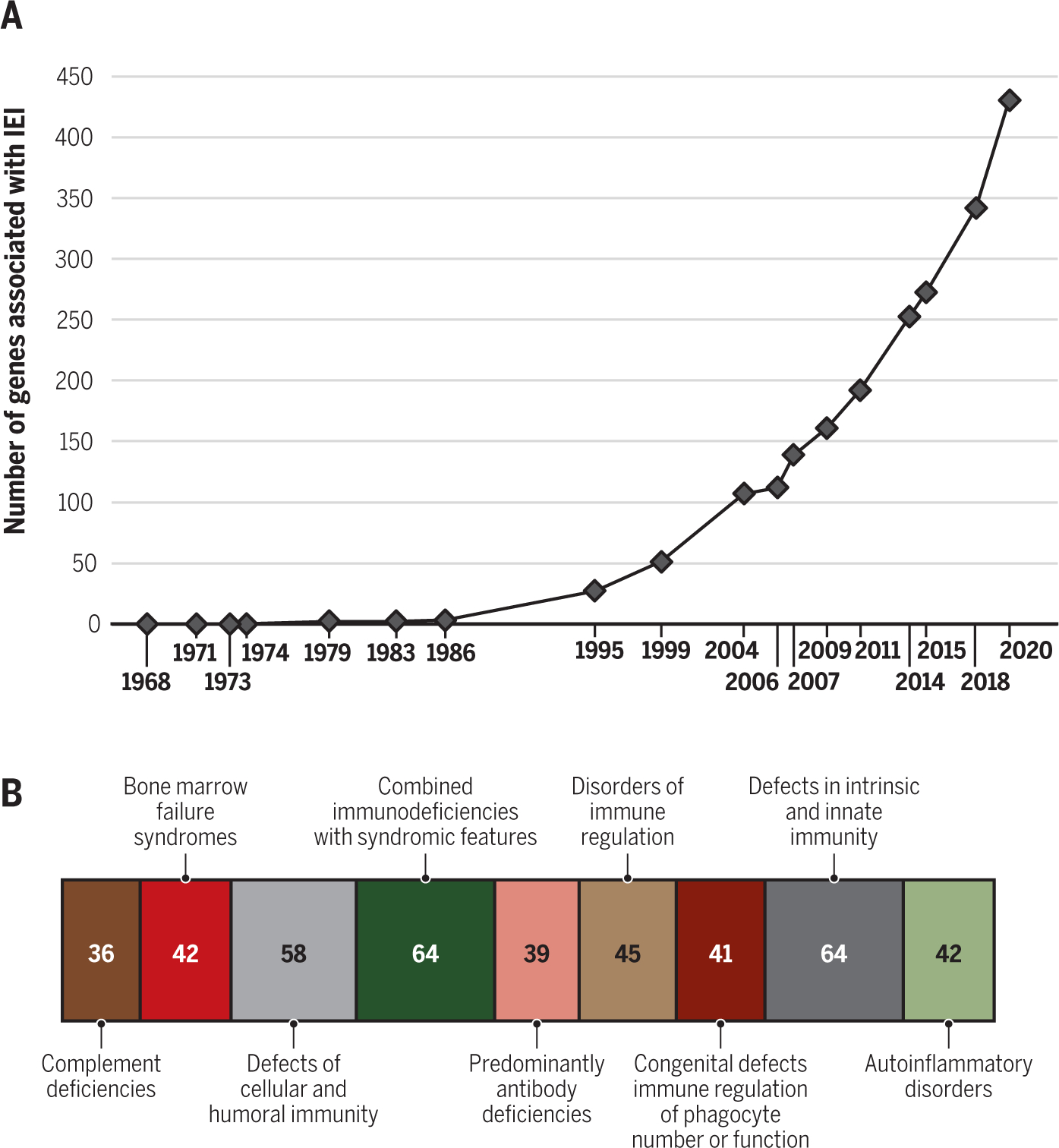
(A) Number of genes associated with inborn errors of immunity known at the time of various editions of the classification of these disorders; (B) Number of genes responsible for each category of inborn errors of immunity, as reported in the 2020 classification (10).
Following up on the surprising observation that patients with autosomal recessive (AR) defects in the terminal components of complement were selectively prone to Neisseria infections, it was gradually realized that many other IEI manifest as susceptibility to a narrow group of pathogens (11). Furthermore, some of these PIDs have been shown to reflect genetic defects that are extrinsic to the hematopoietic system, reflecting instead functional abnormalities of cell types other than leukocytes (12).
Importantly, it has also been recognized that depending upon the specific IEI, autoimmunity, autoinflammation, allergy, and malignancy can be common, and in some cases, predominant, clinical manifestations (13). Finally, the in-depth characterization of the molecular, cellular, and immunological mechanisms of disease has permitted a shift from supportive (mostly focusing on prevention and treatment of infections or inflammation) to definitive (hematopoietic stem cell transplantation, gene therapy) care and to targeted pharmacological interventions (enzyme replacement therapy, cytokine administration, therapeutic antibodies, small molecule inhibitors) (14–16). Here, we will review recent advances in the study of IEI, and how this has been of both clinical and immunological importance.
The spectrum of IEI: Phenotypes and Genotypes
Combined immunodeficiencies: Uncovering Pathways to T Cell Development and Immune Regulation
Following the first descriptions of SCID in the 1950s, a growing number of patients with early-onset life-threatening infections and lymphopenia were described in the next decades. A first evidence of genetic heterogeneity came with the observation that SCID was inherited as an X-recessive (XR) (17) or autosomal recessive (AR) trait (3). After T and B lymphocytes (and later on, NK cells) were described, it was further found that SCID comprises different conditions, all of which are characterized by lack of autologous T cells, whereas B and/or NK cells may either be present or absent.
Eventually, cloning of SCID-causing genes revealed an even higher level of heterogeneity. The first SCID disorders that were defined at the molecular level were adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) deficiency. The metabolic basis of these disorders was established by Eloise Giblett in 1972 and in 1975, respectively (18, 19), while ADA and PNP mutations were reported in 1985 and 1987. Along with mutations in CYBB, causing X-linked granulomatous disease (20), mutations underlying ADA deficiency were actually the first identified in the entire field of IEI.
The molecular pathogenesis of XR SCID was revealed in 1993, with mutations in the IL2RG gene (21). This gene was initially thought to code only for the γ chain of the IL-2 receptor; however, it was soon found that the γ chain is also a subunit of receptors for IL-4, IL-7, IL-9, IL-15, and IL-21 (and for this reason is also referred to as the common γ chain, γc), and that it is constitutively associated with the intracellular tyrosine kinase JAK3 (22). These observations led to the identification of JAK3 deficiency as an AR form of SCID, which is clinically and immunologically indistinguishable from XR SCID (23). Important differences emerged when comparing the immunological phenotype of patients and mice with genetic defects of γc signaling. In particular, γc and Jak3 defects in mice are associated with a T− B− NK− SCID phenotype, whereas in humans they cause T− B+ NK− SCID. The discovery of patients with AR IL-7R deficiency as an etiology of T− B+ NK+ revealed that abolished IL-7 signaling plays a critical role in the SCID phenotype of IL2RG- and JAK3-deficient patients, while it has no impact on B cell development in humans, unlike in mice (24). The more recent discovery that patients with AR IL-7 deficiency display a milder T cell deficiency suggests that abrogated responses to TSLP, which also engages the IL-7R, contributes to the SCID phenotype of AR IL-7R deficiency (25, 26).
Many other gene defects accounting for defects of T-cell development were identified from the late 1990s onward. In some cases, these studies revealed previously unappreciated molecules and mechanisms that govern T-cell development, preceding the generation of animal mutants. This was the case for ARTEMIS and Cernunnos/XLF defects that compromise V(D)J recombination and DNA repair (27–29), for AK2 deficiency that compromises mitochondrial function and cell survival in reticular dysgenesis (30, 31), as well as for various defects of transcription factors that account for HLA class II deficiency (32–35). In other cases, mutations were found in human orthologs of genes previously studied in mice, such as RAG1 (36) and RAG2 (37).
Advances in molecular genetics have enabled the identification of a growing number of genetic etiologies of combined immune deficiency (CID). At variance with SCID, CID disorders are characterized by a less severe numerical and/or functional T-cell defect. Furthermore, while early-onset, life-threatening infections are the hallmark of SCID, various forms of immune dysregulation (such as autoimmune cytopenias, organ-specific autoimmunity, and lymphoproliferation) are commonly seen in CID patients (38). This led to the identification of previously undescribed disorders of immune regulation, as in the case of calcium release-activated calcium (CRAC) channel defects due to ORAI1 and STIM1 deficiency (39, 40).
The most recent classification of IEI includes 64 disorders underlying SCID or CID (Fig. 2B). Mutations in the same gene can underlie a broad range of clinical and immunological phenotypes, as neatly exemplified by RAG deficiency (41). Different levels of V(D)J recombination activity driven by distinct gene mutations allow for the development of variable numbers of T and B cells. Patients with loss-of-function RAG1 or RAG2 mutations display a T− B− NK+ SCID phenotype, whereas severely hypomorphic mutations often underlie Omenn syndrome, in which oligoclonal T cells infiltrate the skin and other target tissues. By contrast, less severely hypomorphic RAG gene variants are typically associated with autoimmunity and granulomatous lesions (42). The immune dysregulation of RAG deficiency (and of other forms of CID) reflects perturbations in the mechanisms of negative selection of self-reactive T and B lymphocytes, and poor function (or restricted TCR repertoire) of regulatory T cells (41).
Major progress has been made in the screening of inherited T cell disorders. Measurement of T-cell receptor excision circles (TRECs), a by-product of VDJ recombination at the T cell receptor α/δ locus (43), in dried blood spots collected at birth can identify newborns with severe T-cell lymphopenia (TCL). Immunological and genetic studies are needed to diagnose SCID as opposed to other causes of TCL, such as prematurity, some syndromic conditions, chylothorax and other causes of T-cell loss, and use of some immunosuppressive drugs during pregnancy (44). The implementation of universal newborn screening (NBS) for SCID in the USA has revealed an incidence of SCID of approximately 1:65,000, which is significantly higher than previously thought (44). By allowing early identification of SCID babies, NBS facilitates interventions aimed at preventing infections and rapid referral to hematopoietic stem cell transplantation (HSCT). This is especially important, because it has been shown that younger age and infection status at transplantation are key factors that determine improved outcome after HSCT for SCID (45, 46).
Monogenic errors of immune regulation
The understanding of immunological self-tolerance and autoimmunity greatly benefited from studies of monogenic inborn errors of T-cell tolerance, T-cell apoptosis, or regulatory T cells (Tregs), each presenting with distinctive immunological and clinical manifestations but unified by pathological self-reactive T-cell responses.
One of the longest-studied examples of such disorders, first reported by Leonard in 1929, as an AR condition known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome (APECED; also known as Type 1 autoimmune polyglandular syndrome, APS1) (reviewed in (47)). From a clinical standpoint, APECED is characterized by pleiotropic autoimmune manifestations and the presence of tissue- and cytokine-specific autoantibodies. The disease is caused by mutations in AIRE, a key transcription factor that regulates tissue-restricted self-antigen expression in medullary thymic epithelial cells, thereby governing negative selection of autoreactive T cells (48). In APECED patients, thymic escape of self-reactive TCRαβ+ CD4+ T cells causes autoimmunity. Furthermore, an altered positive selection of thymic Tregs, increased autoreactive B cells and production of autoantibodies further aggravate the autoimmunity of the disease (49–51). Adoptive transfer experiments in mouse models of APECED support a predominant pathogenetic role of self-reactive T cells, whereas autoantibody formation appears largely secondary to, rather than causative of, tissue damage (52).
Patients with APECED typically suffer from hypoparathyroidism, adrenal insufficiency (Addison’s disease), and chronic mucocutaneous candidiasis (CMC). Other autoimmune endocrine problems (hypothyroidism, ovarian and testicular failure hypopituitarism), as well skin lesions (urticarial eruption, alopecia, vitiligo), and pulmonary, gastrointestinal, liver, renal, and ocular manifestations are frequently seen (47). Neutralizing anti-IL-17A/F antibodies are often present in APECED patients and play a pathogenic role in CMC (53, 54). This is an example of increased susceptibility to infection due to an autoimmune mechanism. In addition to anti-IL-17A/F autoantibodies, APECED patients have anti-IFN-α and anti-IFN-ω autoantibodies, which are not known to be pathogenic, yet represent useful diagnostic biomarkers. As the defect is in the thymic epithelial cells and not in hematopoietic cells, there are currently no curative approaches for APECED, and patients with this disease have an increased mortality rate and a poor quality of life.
Autoimmune lymphoproliferative disease (ALPS) is another form of monogenic autoimmunity, which is caused by germline or somatic mutations in TNFRSF6, TNFSF6, and CASP10, coding for FAS/CD95, FAS ligand (FasL), and caspase 10, respectively. These defects impair FAS/CD95-mediated cell death of lymphocytes, leading to the main clinical manifestations of lymphadenopathy, splenomegaly and autoimmune cytopenias (reviewed in (55, 56)). An unusual population of TCRαβ+ CD4− CD8− double negative T (DNT) cells infiltrates and accumulates within lymphoid organs. These DNT cells resemble TEMRA cells, as they express the naïve marker CD45RA but also activation and exhaustion markers. In some patients DNT cells are the only cell type carrying the mutation, supporting their important pathogenic role. B cells are also altered in ALPS, with decreased marginal zone and memory B cells, abundant plasma cells, and increased levels of IgG and IgA. Other diagnostic biomarkers include elevated serum levels of IL-10, soluble FasL, and vitamin B12. Although lymphoproliferation tends to improve in adulthood, ALPS patients usually require immunosuppression and have an increased risk of lymphoma. Related disorders of defective apoptosis are caused by Caspase-8 (57) or FADD deficiencies (56), or activating mutations in NRAS/KRAS (58, 59), which are associated with other immunological and clinical phenotypes.
A third form of monogenic autoimmune disorders is inborn errors of Tregs, collectively called Tregopathies (reviewed in (60)). Immune dysregulation - Polyendocrinopathy - Enteropathy X-linked (IPEX) syndrome (reviewed in (61)), affecting CD4+CD25hiCD127lo thymic derived Tregs, represents the prototype of this group of disorders. IPEX was first clinically described in 1982 by Powell (62), and its successful treatment with HSCT (proving the hematopoietic intrinsic nature of the disease) was reported in 2001 (63). Around the same time, three groups described mutations in human FOXP3 (64–66). In 2003, several publications connected Foxp3 mutations, defective Tregs, and autoimmunity in mice (67–69). Defective Treg function in patients with IPEX (with impaired capacity to suppress proliferation and cytokine production of T effector cells) was reported in 2006 (70), establishing Treg dysfunction as the primary pathogenic cause of IPEX. FOXP3 mutated effector T cells have an increased proliferative capacity, and their cytokine expression profile is skewed toward Th2 and Th17 cells, which may contribute to the autoimmune pathology and probably to atopic disease as well (61, 71).
In most IPEX patients, the disease manifests within the first months of life (72), with severe enteropathy, type 1 diabetes and skin disease resembling atopic dermatitis. Cytopenias and autoimmune hepatitis are also very common. In some cases, however, FOXP3 mutations are detected in adolescents with milder or atypical autoimmune manifestations, including thyroiditis, glomerulonephritis, alopecia, psoriasis or arthritis. An increased CD4:CD8 ratio, eosinophilia, and elevated serum IgE are typical biomarkers of the disease. Tissue-specific auto-Abs are frequently detected; in particular, anti-harmonin and anti-villin Abs are associated with enteropathy (73). Enumeration of circulating Treg cells and evaluation of FOXP3 expression have limited diagnostic value, since some patients express fairly normal amounts of mutated FOXP3 protein. DNA demethylation at the Treg-specific-region (TSDR) of the FOXP3 gene promoter is a requirement for stable FOXP3 expression (74). Increased TSDR demethylation (suggesting a possible compensatory mechanism) has been reported in IPEX patients, and may represent an important diagnostic aid (75). IPEX is a life-threatening disease, with limited response to immunosuppression and poor long-term disease-free survival. HSCT can be curative, although the clinical status of the patients at the time of transplant can negatively affect the outcome (72).
IPEX-like diseases are caused by mutations in genes encoding molecules involved in survival, development, signaling or function of Tregs, but also of T effector (Teff) cells (60, 76). This group of disorders comprises defects in several genes: i) CD25, encoding the α chain of the IL-2 receptor (IL2-Rα) complex, which is essential for Treg peripheral survival, Teff expansion, and for sustaining Ag-specific T-cell immune responses. CD25 deficiency manifests with enteropathy, other autoimmune features, and increased susceptibility to infections, in particular to CMV. In a few cases, all carrying the same mutation, autoimmunity has been reported as the only clinical feature (77). Biallelic mutations in CD122, encoding for IL-2Rβ, cause similar clinical manifestations (78, 79), and mutations in STAT5B, which lead to growth hormone insensitivity and short stature, also impair IL-2 signaling and Treg fitness (80, 81); ii) CTLA4, encoding a negative regulator of T cell responses. CTLA-4 is constitutively expressed by Treg, and directly regulated by FOXP3, in contrast to its inducible expression in Teff cells. By competing with CD28 for binding to the costimulatory molecules CD80 and CD86, expressed on the surface of APCs, CTLA-4 blocks T cell activation. CTLA4 haploinsufficiency is characterized by lymphoproliferation (including lymphoid infiltrates in the lungs and in the CNS), increased susceptibility to infections, and autoimmunity (enteropathy, cytopenias, skin lesions) (82, 83). The disease has incomplete penetrance; iii) LRBA, encoding an anchor-protein involved in membrane recycling of CTLA-4 (84). LRBA deficiency is an AR disease with similar manifestations to CTLA4 haploinsufficiency, but with earlier clinical onset and complete penetrance (85). Recently, mutations in DEF6, also involved in CTLA-4 membrane trafficking, have been described (86); iv) STAT3, encoding a signaling mediator of many cytokine receptors. Gain-of-function (GOF) STAT3 mutations are associated with reduced STAT5B signaling, and impaired FOXP3 and IL17A expression. The clinical manifestations are heterogeneous, including autoimmune polyendocrinopathy, cytopenias, enteropathy and lung disease, as well as recurrent infections and short stature (87). Related to altered STAT3 signaling are also IL10, IL10RA, and IL10RB mutations, which impact production of, and response to, the immunosuppressive cytokine IL-10. IL-10 antagonizes IFN-γ and is also important for gut immune homeostasis and for the induction of Type 1 regulatory T cells (88). Loss of IL-10 or IL-10 responsiveness causes a disease that manifests primarily with very early onset inflammatory bowel disease (89, 90). Together, this third group of Tregopathies illustrates the many pathways regulating this cell subset that is important for maintaining immune homeostasis and preventing autoimmunity.
Autoinflammatory disorders: IL-1and Type I Interferon-associated diseases
Autoinflammatory diseases are defined by fever, skin rashes or other manifestations of inflammation, in the absence of autoimmunity and infection, or disproportionate to a precipitating infection (reviewed in (91)). Two major groups of monogenic autoinflammatory disorders are the IL-1 inflammasomopathies and the type I interferonopathies. In inflammasomopathies, overactivation of IL-1β and related cytokines like IL-18, results from dysregulated inflammasome activation. Normally, various stimuli which cells interpret as danger or damage, including microbial products or microcrystals, trigger assembly of inflammasomes in the cytosol. The inflammasome is a multimeric signaling complex that brings together through homotypic interactions NLRP1, NLRP3, or NLRC4 sensor proteins with pyrin and ASC. Entry of procaspase-1 into this complex promotes self-cleavage for activation. The activated caspase-1 in turn cleaves to activate the proinflammatory cytokines IL-1β and IL-18, which unleashes a cascade of diverse biological effects including leukocyte extravasation into tissues, modulation of immune cell functions, and induction of fever, acute phase reactants, and other metabolic changes. Additionally, activated caspase-1 cleaves and activates gasdermin D, which forms pores in the membrane to cause pyroptotic cell death.
Monogenic autoinflammatory disorders caused by dysregulated inflammasome activation and/or hyperactivation of the IL-1 pathway are exemplified by Familial Mediterranean Fever due to mutations in MEFV encoding pyrin. However, inflammasomopathies have since broadened to include those stemming from GOF mutations in other inflammasome components (NLRP3, NLRC4, and NLRP1), or in molecules upstream that regulate inflammasome activation (LPIN2, PSTPIP1, MVK, WDR1). Additionally, inflammasomopathies include LOF mutations that decrease IL-1 antagonism (IL1RN). In contrast, genetic loss of human IL-18 antagonism (mutations in IL18BP) does not underlie autoinflammation, but excessive inflammation and liver destruction in response to hepatitis virus A infection (92). As would be expected from their shared pathogenic mechanism, most inflammasomopathies are amenable to primary or adjunctive treatments targeting production of or responsiveness to IL-1 (91).
By contrast, interferonopathies, which constitute a second group of monogenic autoinflammatory disorders, are caused by overactivation of type I IFNs and IFN-stimulated genes (ISG) or IFN-regulated genes (IRG) (93). Normally, the accumulation of virus products – such as nucleic acid structures not normally found in uninfected cells or found in inappropriate cellular compartments – are detected by various intracellular sensors. These sensors encompass the RIG-I-like receptors (RLRs), certain TLRs, and cyclic GMP-AMP synthase (cGAS). Their binding to virus products triggers the assembly of signaling platforms that turn on transcription of antiviral type I IFNs and proinflammatory cytokines. Interferonopathies can result from GOF mutations in the virus sensors or associated adaptors (IFIH1, DDX58, TMEM173), or LOF mutations in nucleases or in RNA-editing enzymes (TREX1, SAMHD1, ADAR1, RNASEH2A, RNASEH2B, RNASEH2C) that lead to abnormal accumulation of endogenous nucleic acids. Alternatively, LOF mutations that negatively regulate the IFN responsive pathway (USP18, ISG15), or GOF mutations in the same pathway (STAT1, STAT2, JAK2), can also result in type I interferonopathy (94). The latter type of condition was recently shown to be controlled by a JAK inhibitor (95). Finally, LOF mutations that impair proteasomal function (PSMB8, PBSM4, PSMA3) and lead to accumulation of ubiquitinated proteins also result in inappropriate IFN induction through an unclear mechanism.
Interferonopathies tend to show more central nervous system and skin involvement, often leading to a picture resembling congenital infection or a diagnosis of Aicardi-Goutière syndrome during infancy. However, when vasculitis and other manifestations of systemic inflammation are prominent, treatment with JAK inhibitors aimed at interrupting IFN receptor-mediated signaling has been shown to ameliorate disease (96).
Finally, recently it has been recognized that autoinflammatory disorders can result from dysregulation of pathways that impact protein ubiquitination, a post-translational modification process (97). Ubiquitin is a small protein that is attached to substrate proteins usually through a lysine linkage, sometimes by itself and sometimes in chains, through a series of enzymatic reactions involving activation, conjugation, and ligation; this process is countered by deubiquitinases that remove ubiquitin from proteins. Normally, the ubiquitin tags mark substrate proteins for degradation via proteasomes. However, ubiquitination is also important for non-degradative functions involved in cellular homeostasis including intracellular signaling.
Autoinflammatory disorders of the third group of dysregulated ubiquitination can result from LOF mutations in components of the linear ubiquitin assembly complex (LUBAC) that is required for ubiquitination (RNF31 encoding the catalytic HOIP, and RBCK1 encoding HOIL-1 accessory protein). Alternatively, disease can arise from LOF mutations in genes whose products function as deubiquitinases (TNFAIP3, OTULIN). Collectively, these mutations result in the abnormal accumulation of signaling intermediates downstream of IL-1 or TNF, which effectively prolong their functional activities. Through effects on ubiquitinated targets including IKKγ and RIPK1, these mutations can contribute to hyperactivation of NF-κB, mitogen-activated protein kinases (MAPK), and other signaling pathways to reinforce autoinflammatory effects. Furthermore, mutations in the ubiquitinated target RIPK1 can also phenocopy disease through complex effects that cause increased programmed cell death to secondarily promote inflammation (98). Interestingly, in the case of LUBAC deficiencies, NF-κB is hyperactivated in monocytes, but simultaneously impaired in fibroblasts and B cells; this divergence can help explain the mixed picture of immunodeficiency with autoinflammation in such patients. Overall, this new group of autoinflammatory disorders reveals new molecular targets in the ubiquitin-proteasome system for potential future interventional therapies.
Inborn errors of immunity underlying allergy: the hyper IgE syndrome
Severe allergy can also be a manifestation of inborn errors of immunity. While it is now well established that many IEI can manifest with elevated serum IgE as a sign of immune dysregulation, in this review we will focus primarily on “the” hyper IgE syndrome (HIES), whose clinical and immunological features were described between 1966 and 1999 (99, 100). These patients suffer from severe eczema and show elevated serum IgE levels, which can show reactivity toward a variety of allergens. The patients also suffer from severe infections, especially chronic mucocutaneous candidiasis (CMC) and staphylococcal diseases of the skin and lungs, often with pneumatoceles that are prone to superinfection. Another characteristic feature of HIES is the poor clinical and biological inflammation, with cold abscesses of the skin and lungs. A fourth defining phenotype includes a range of extra-hematopoietic manifestations, including skeletal disorders in particular, such as bone fragility, scoliosis, and decidual teeth retention. Two of these four cardinal features can be seen in similar but unrelated disorders: eczema, infection susceptibility, and elevated IgE are seen for example in patients with DOCK8 deficiency, while poor inflammation in the course of skin or systemic, but rarely pulmonary, staphylococcal infection is seen in patients with MyD88 or IRAK-4 deficiency. It is the combination of these four phenotypes that makes the HIES a distinctive clinical entity. This by no means implies that all patients are closely similar. There is actually great inter-individual variability among HIES patients, even within a given family.
The first step toward the molecular and cellular dissection of the HIES came in 2007 with the discovery of heterozygous HIES-causing mutations in STAT3 (101). The mutations are loss-of-function and dominant-negative. Even low amounts of STAT3 can exert negative dominance. An autosomal recessive (AR) form of HIES was found in 2018 with bi-allelic mutations in ZNF341, a transcription factor that governs the baseline and inducible transcription of STAT3, thereby also regulating its expression and activity (102, 103). Severely hypomorphic mutations of the IL6ST gene, were reported to underlie a severe AR form of HIES (104). The IL6ST gene encodes for the gp130 co-receptor of IL-6 family cytokines, including IL-6, IL-11, IL-27, OSM and LIF; all of these receptors signal via STAT3. A complete form of gp130 deficiency underlies an even more severe condition, evoking Stüve-Wiedemman syndrome (105). A second genetic etiology of autosomal dominant (AD) HIES is due to heterozygous, dominant-negative mutations in IL6ST, which impair cellular responses to IL-6 and IL-11 to a greater extent than other IL-6 family cytokines (106).
The immunological and clinical phenotypes of the patients with these three genetic etiologies of HIES largely but not completely overlap, possibly reflecting differences in the biochemical pathways affected. Cells from HIES patients mutated in STAT3, ZNF341, or IL6ST respond poorly to IL-6 family cytokines. Cells mutated in STAT3 or ZNF341 also respond poorly to IL-21 and IL-23. The discovery of other inborn errors of immunity has shed light onto the molecular and cellular basis of each of the cardinal HIES phenotypes. Their staphylococcal disease results mostly from impaired responses to IL-6, as inferred from the discovery of patients with AR IL-6R deficiency (107). Interestingly, their eczema and elevated IgE levels, as well as their poor inflammation, also seems to result mostly from insufficient responses to IL-6. In contrast, IL-6R-deficient patients do not display skeletal phenotypes. The skeletal manifestations seen in patients with HIES result at least in part from the poor cellular responses to IL-11, as inferred from the discovery of patients with IL-11R deficiency and Crouzon-like skeletal anomalies (108). Some other shared features of HIES remain unexplained in molecular terms, e.g. vascular anomalies, lymphomas.
Although the four genetic etiologies of HIES seem to be broadly responsible for phenocopies, a detailed and systematic clinical survey would be useful. Anecdotally, it seems that ZNF341-deficient patients have a better inflammatory response and fewer somatic manifestations than STAT3-mutated patients, but the underlying mechanisms for this remain unknown. Moreover, STAT3- and ZNF341-mutated patients frequently have chronic mucocutaneous candidiasis (CMC), unlike IL6R- and IL6ST-mutated patients. The former patients display CMC because of impaired development of Th17 and perhaps other IL-17-producing lymphocytes, as inferred from inborn errors of IL-17. While isolated defects of IL-6R, IL21R, and IL-23R do not or rarely underlie CMC (109, 110), their combined deficiency via mutations in STAT3 or ZNF341, but not IL6ST, commonly underlies CMC. Overall, core molecular deficiencies of these three groups of HIES patients involve an impaired ZNF341- and STAT3-dependent IL-6ST-mediated response to IL-6 (allergy and staphylococcal disease) and IL-11 (skeletal anomalies), while at least some specific features, such as CMC, are caused by ZNF341- and STAT3-dependent impairment of IL-17.
Inborn errors of immunity with increased susceptibility to Epstein-Barr virus (EBV) infection and malignancies
Some IEIs result from genomic instability, apoptosis defects, and other abnormalities that predispose to cancer, without necessarily interfering with immune responses (111). However, other PIDs have an increased risk of cancers due to inadequate control by the immune system of oncogenic viruses such as human papillomaviruses, human herpes virus 8, and Epstein-Barr virus (EBV). Of these, EBV is illustrative because it infects nearly all adults (reviewed in (112)). PID patients can develop EBV-associated cancers such as Hodgkin’s disease, non-Hodgkin lymphomas, and rare smooth muscle cancers, which reflect chronically increased virus loads due to impaired immune surveillance. EBV can be also associated in the general population with other cancers such as Burkitt’s lymphoma, nasopharyngeal carcinomas, and gastric cancers.
Primary infection with EBV is usually asymptomatic during childhood but can sometimes cause an acute mononucleosis-like illness in adolescents and adults. Subsequently, EBV establishes a lifelong infection where the virus persists in latent form within B cells. It is periodically reactivated to be shed as infectious virus, and its increased replication can also drive lymphoproliferation. Control of both the primary infection and subsequent EBV reactivation requires robust T and NK cells immunity. This requirement has been demonstrated in conditions characterized by severe immunosuppression such as following HSCT, or in CID that have defective lymphocyte functions and broad susceptibility to different viruses and other microbes. Illustrative PIDs in this category include deficiencies in RAS guanyl releasing protein 1 (RASGRP1), CTP synthase 1 (CTPS1), WASP actin nucleation promoting factor (WAS), serine/threonine kinase 4 (STK4), dedicator of cytokinesis 8 (DOCK8), coronin 1A (CORO1A), IL-21R, GATA2, as well as activating mutations in phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit delta (PIK3CD) or phosphoinositide-3-kinase-regulatory subunit 1 (PIK3R1) (113).
By contrast, several other PIDs display a narrower defect with a heightened susceptibility to EBV relative to other viruses. Studies into these latter disorders have revealed mechanisms that are particularly important for immunity against EBV (reviewed in (113)). In this category of PIDs are the X-linked lymphoproliferative diseases, caused by hemizygous loss-of-function mutations in SH2D1A encoding the signaling adaptor SLAM-associated protein (SAP), or in XIAP encoding the X-linked inhibitor of apoptosis protein. These, or several other mutations that impair cytotoxic function due to deficiency in perforin or molecules involved in lytic granule exocytosis, result in prolonged immune activation that however is ineffective in suppressing EBV replication, and/or can impair cell-cell interaction between EBV-infected B cells and cognate cytotoxic T lymphocytes. When initially infected with EBV, such patients typically present with hemophagocytic lymphohistiocytosis, characterized by life-threatening fulminant lymphoproliferation, macrophage activation and end-organ damage.
Finally, among PIDs that display a relatively selective susceptibility to EBV are those that impair proximal signaling molecules for T cell activation. Such PIDs include deficiencies in IL2 inducible T cell kinase (ITK), magnesium transporter 1 (MAGT1), CD70, CD27, and CD137 (4–1BB) (113). MAGT1 promotes T cell activation through magnesium second messenger signaling for calcium-dependent responses through its effects on ITK, and ITK activity is directly regulated by Mg2+ (114). Additionally, MAGT1 promotes NK cell cytotoxicity against EBV-infected cells through its ability to upregulate NKG2D on NK cells and CD8 T cells. Recent studies have identified that MAGT1 participates in an N-glycosylation complex, which is necessary for maturation of fully functional glycosylated NKG2D and CD70 (115, 116). CD70 is normally highly expressed on EBV-infected B cells, and this ligand engages the CD27 costimulatory molecule on T cells. Deficiency in either CD70 or CD27 impairs priming and expansion of EBV-specific CD8 T cells, as well as expression of NKG2D and other molecules important for such cells to interact with and respond to EBV-infected cells. The recent discovery of CD137-deficient patients with poor EBV control indicates that other costimulatory pathways can also contribute to anti-EBV immunity (117). Thus, the EBV susceptibility in this category of PIDs may reflect defects in multiple overlapping pathways.
Phenotypic and genetic heterogeneity of inborn errors of immunity: Converging pathways and allelic heterogeneity
Inborn errors of innate-adaptive immunity: MSMD and CMC
Inborn errors that selectively prevent the development of neutrophils are probably the only pure disorders of innate immunity, echoing the selective disorders of development of T and/or B cells that are pure disorders of adaptive immunity. Only a few inborn errors (such as reticular dysgenesis and telomeropathies) disrupt the development of both innate and adaptive cell types. Most other IEI affect leukocyte function, and typically impact both innate and adaptive leukocytes. Two good examples are the inborn errors of IFN-γ and IL-17A/F immunity (118). These are the prototypic type 1 and type 17 signature cytokines, and can be secreted by both innate and adaptive leukocytes. In particular, IFN-γ can be secreted by αβ+ and γδ+ T cells, and among αβ+ T cells by NKT, mucosa-associated invariant T (MAIT) cells, and conventional CD4+ and CD8+ T cells. It is produced by memory CD4+ T cells in particular by the Th1 and Th1* subsets. However, IFN-γ is also produced by B cells, NK cells and some innate lymphoid cells (ILCs) (119). It serves as the macrophage-activating factor, more so than an anti-viral interferon. Indeed, patients with any of the 31 known inborn errors of IFN-γ affecting 16 distinct genes, are prone to mycobacterial disease and more rarely to related infections by intra-macrophagic microbes (120).
These inborn errors were discovered by forward genetics from the study of Mendelian susceptibility to mycobacterial disease (MSMD), a “Mendelian infection” characterized by severe disease caused by poorly virulent mycobacteria, e.g., BCG vaccines and environmental mycobacteria, in otherwise healthy individuals (118). There is genetic heterogeneity but physiological homogeneity, as all gene products are involved in IFN-γ immunity (Fig. 3). Interestingly, three IFN-γ-inducing monokines are each essential for optimal production of IFN-γ, as AR IL-12R, IL-23R, and ISG15 deficiencies are all genetic etiologies of MSMD, albeit with incomplete penetrance, which is higher in patients unresponsive to both IL-12 and IL-23 (110, 118, 121). It is interesting that etiologies with low penetrance for MSMD led to the discovery of genetic etiologies of bona fide tuberculosis (122, 123). While the defects that impair responses to IFN-γ are most likely disorders of mononuclear phagocytes, in which mycobacterial reside, the cellular basis of inborn errors of IFN-γ production has proven much more difficult to decipher. A lack of NK, NKT, or B cells alone is not sufficient to be prone to mycobacterial disease, unlike a lack of all T cells. Patients with RORC deficiency have absent (NKT, MAIT) or defective (γδ+ and Th1*) IFN-γ-producing cells (124). The discovery of additional genetic etiologies of MSMD is required to better delineate the cellular basis of anti-mycobacterial IFN-γ immunity in humans.
Figure 3. Genetic etiologies of Mendelian susceptibility to mycobacterial disease (MSMD).
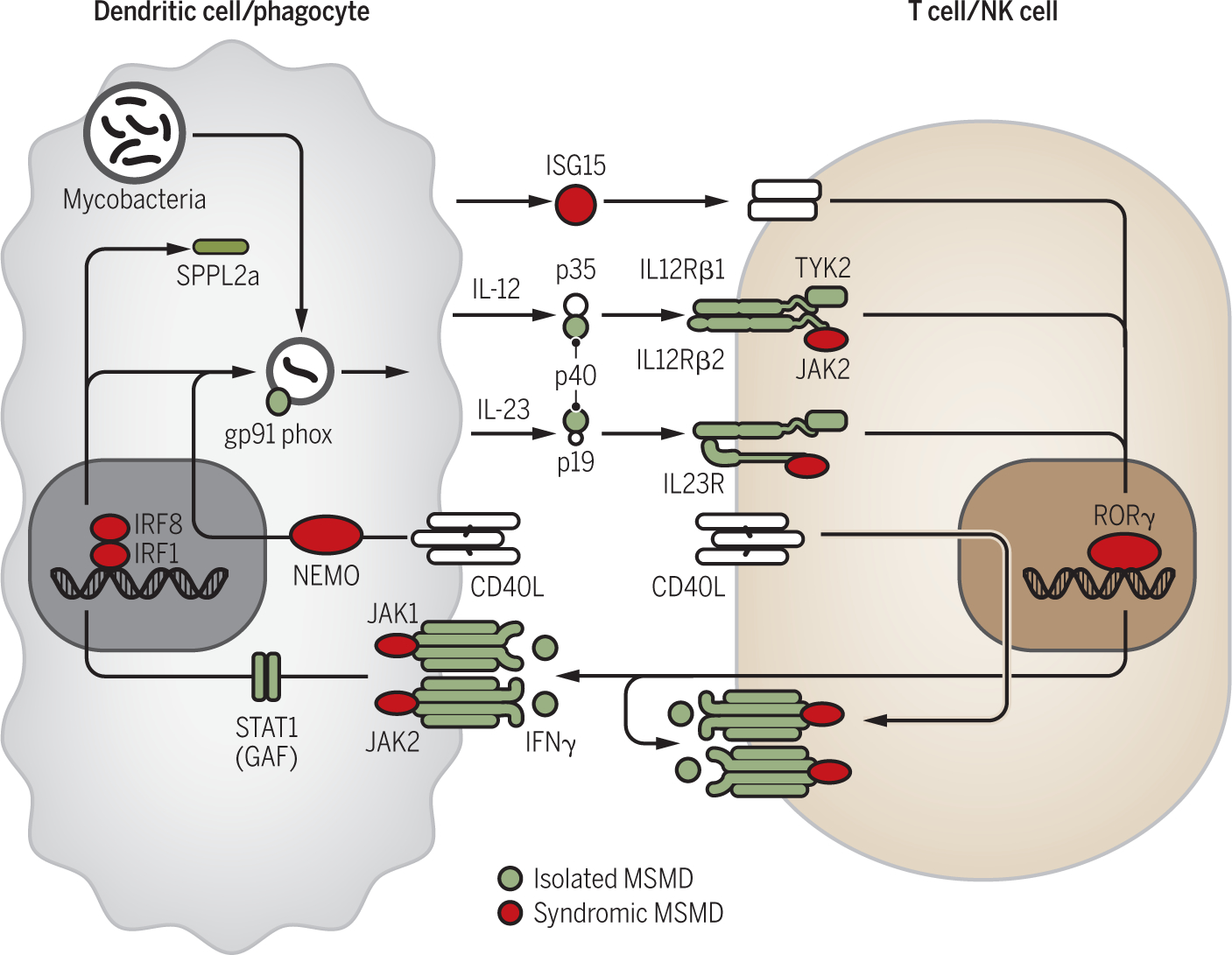
Interactions between cells involved in the production of (T cells/NK cells) and in the response to IFN-γ (dendritic cells, phagocytes). Proteins whose mutations in the corresponding gene cause isolated MSMD are depicted in green, while those responsible for syndromic MSMD are depicted in red.
While IFN-γ is the classic Th1 signature cytokine, IL-17A/F is the prototypic Th17 signature cytokine. The role of human IL-17 cytokines in host defense was first suspected when it was shown that patients with autoimmune polyendocrinopathy type 1 (APS-1), whose only infectious disease is chronic mucocutaneous candidiasis (CMC), have neutralizing autoantibodies against IL-17A and IL-17F (53, 54), Patients with inborn errors of IL-17 immunity were discovered from the forward genetic study of isolated and familial forms of CMC (125). The study of patients harboring mutations in IL17RA and IL17A/F genes showed that IL-17 is required for mucocutaneous immunity to Candida albicans, while being redundant for host defense against other microbes, with the exception of Staphylococcus aureus, which can also cause peripheral lesions in the patients. Interestingly, gain of function (GOF) mutations in STAT1 underlie CMC by impairing the production of IL-17A/F- by CD4+ cells, and perhaps by other lymphocytes, probably because of enhanced responses to interferons (IFNs)-producing T cells (126).
IL-17A and IL-17F multimerize to signal via IL-17RA and IL-17RC. Interestingly, patients with IL-17RC deficiency are prone to CMC but not staphylococcal disease, perhaps because responses to IL-25 (IL-17E) are maintained (127). Mutations downstream of the IL-17RA/RC receptor include ACT1 deficiency and JNK1 haploinsufficiency (128). The latter defect also impairs the homeostasis of connective tissues, probably in part by disrupting cellular responses to TGF-β. The nature of the cells that are responsible for the CMC phenotype in patients with inborn errors of the IL-17-responsive pathway remains unclear, as IL-17A/F can activate a variety of conjunctive and epithelial cells in the skin and mucosae, promoting their secretion of chemo-attractants for leukocytes and also inducing the production of effector genes against fungi. As a matter of fact, the nature of the IL-17-producing lymphocytes that are required for mucocutaneous immunity to Candida is also elusive, as IL-17A/F can be secreted by αβ+ and γδ+ T cells, including at least the Th17 and Th1* subsets, as well as innate lymphocytes. The discovery of additional genetic etiologies of isolated or syndromic CMC will shed light onto the cellular basis of human CMC, including the roles of IL-17-producing leukocyte subsets and that of IL-17-responding cells other than leukocytes.
Inborn errors of non-hematopoietic cell-intrinsic immunity: from EV to HSE
The study of inborn errors of immunity has also revealed the importance of non-hematopoietic cell-intrinsic immunity for host defense (12). The importance of cells other than leukocytes was already established from the study of thymic disorders, which were shown from DiGeorge onward to be stromal phenocopies of T cell-intrinsic severe combined immunodeficiency (129). The discovery of the importance of non-hematopoietic cell-intrinsic immunity to viruses came from the study of patients with monogenic infections, whether Mendelian or not. Epidermodysplasia verruciformis (EV) was with hindsight the first described inborn error of immunity, in 1946 (130). It is precisely the lack of leukocytic phenotype that prevented its inclusion in the international list of inborn errors of immunity until 2004. These patients are prone to flat warts and pityriasis-like skin lesions that evolve into non-melanoma skin cancer. The lesions are caused by defective beta-human papillomaviruses (HPV), which lack the E5 and E8 virulence genes present in other, more pathogenic HPVs. HPVs have an exclusive tropism for keratinocytes. Infection by beta-HPV is common and asymptomatic, except in patients with EV.
Typical or isolated EV is defined by a lack of other infections (which are seen in patients with T cell deficits underlying atypical or syndromic EV) (131). The first two genetic etiologies were discovered in 2002, with mutations in TMC6 and TMC8, encoding for EVER1 and EVER2, respectively (132). A third genetic etiology was discovered more recently, with mutations in CIB1 (133). The three proteins form a complex; in the absence of EVER1 or EVER2, the expression of CIB1 is dramatically reduced in keratinocytes. Moreover, this complex interacts with E5 and E8, suggesting that it behaves like a human restriction factor for HPVs. A plausible model is therefore that keratinocytes lacking EVER1, EVER2, or CIB1, become permissive to β-HPVs that lack the corresponding virulence factor E5 and E8, and cannot cause lesions in humans with a functional EVER-CIB1 complex. It has proven challenging to test this model experimentally, as it has not been yet possible to establish an in vitro model of the full HPV cycle across all layers of skin keratinocytes.
Another condition that also revealed the importance of cell-intrinsic immunity to viruses by resident, non-hematopoietic cells in specific tissues or organs is herpes simplex virus 1 (HSV-1) encephalitis (HSE) (134). In the course of primary infection by HSV-1, children rarely cannot prevent the virus from reaching the forebrain via the olfactory bulb or the brainstem via the trigeminal nerves. Until the advent of acyclovir, HSE was almost invariably lethal, and survivors continue to suffer from severe neurological sequelae. Despite its severity, HSE is restricted to the central nervous system: there are no lesions elsewhere and the virus cannot even be found in the bloodstream. A forward genetic approach of forebrain HSE found germline mutations in various components of the TLR3 signaling pathway (135). More recently, mutations were found also in SNORA31 gene, encoding a small nucleolar RNA, that do not affect the TLR3-responsive pathway (136). Finally, mutations in the only known RNA debranching enzyme DBR1 were found in patients with brainstem infection caused by HSV-1, influenza virus, or norovirus (137). Different anatomical territories of the central nervous system (CNS) therefore seem to rely on different mechanisms of host defense against viruses.
Leukocytes from patients with inborn errors of the TLR3 pathway do not display any detectable phenotype. In contrast, their fibroblasts and induced pluripotent stem cell (iPSC)-derived cortical neurons and oligodendrocytes show impaired immunity to HSV-1, which can be rescued by exogenous type I IFN (138). Consistent with this, the occurrence of HSE has been reported also in a patient with AR STAT1 deficiency, a condition that impairs cellular response to type I IFN. iPSC-derived trigeminal neurons lacking TLR3 are not more vulnerable than control cells, further suggesting that the cellular basis of disease involves an impairment of CNS-resident cortical neurons (139). The mechanisms by which SNORA31 haploinsufficiency impairs cortical neuron immunity remain to be deciphered. Likewise, neither brainstem neurons (nor any other CNS resident cell) from DBR1-deficient patients have been tested. Yet, their dermal fibroblasts showed enhanced RNA lariat accumulation and impaired cell-intrinsic immunity to the viruses tested, including HSV-1. The patients’ fibroblasts have only 1–3% residual activity, accounting for the massive accumulation of RNA lariats. The mechanism by which these lariats disrupt cell-intrinsic immunity is a topic of intensive study. Overall, the study of HSE and other types of viral encephalitis has revealed the essential role of human CNS-resident, non-hematopoietic cell-intrinsic immunity to viruses.
Advances in treatment (from supportive care to precision medicine)
The study of the cellular and molecular IEI has led to major contributions in the treatment of human diseases (Fig. 4). Upon characterization of the cellular phenotype of SCID, and shortly after the initial description of HLA in 1967 (140), allogeneic HSCT was successfully used for the first time in a baby with XR-SCID in 1968 (14). Initially, results were only successful when there was an HLA-identical sibling, as otherwise fatal graft-versus-host disease would develop. Development of methods that allow T-cell depletion from the bone marrow enabled the first successful application of an HLA-mismatched HSCT in a patient with SCID in 1983 (141), paving the way for the application of HSCT to a large number of severe hematological and metabolic disorders. Use of chemotherapy or irradiation is typically used to deplete the recipient’s hematopoietic stem and progenitor cells, but carries significant risks of toxicity. As an alternative approach to conditioning, injection of anti-CD117 mAb targeting hematopoietic stem cells (142) has recently been started (NCT02963064). Finally, some forms of severe T-cell lymphopenia are of extra-hematopoietic origin. Identification of the thymic nature of the immune deficiency has permitted development of thymic transplantation for complete DiGeorge syndrome and other congenital thymic defects (143).
Figure 4. Progress in the treatment of inborn errors of immunity.
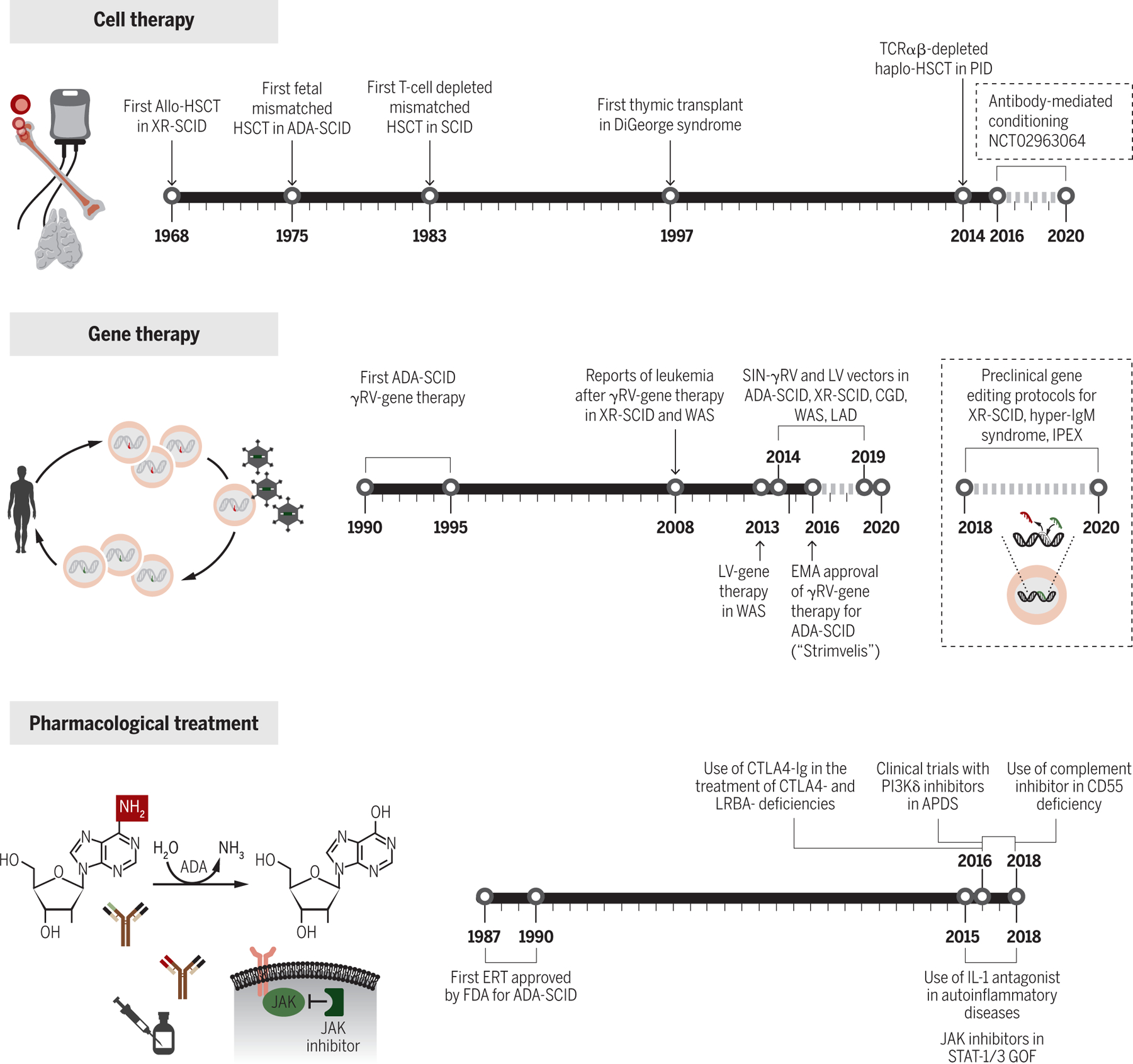
Three separate timelines display milestone events in the evolution of cell therapy, gene therapy and pharmacological treatment to address inborn errors of immunity.
Moreover, identification of the molecular bases of IEI has allowed development of targeted therapeutic approaches, based on replacement of the missing product, targeted pharmacological manipulation of the signaling pathway involved or correction of the gene defect. In 1987, Hershfield reported that enzyme replacement treatment (ERT) with polyethylene glycol-conjugated bovine ADA led to metabolic and immunological improvement in patients with ADA-SCID (144). In 1990, ADA-SCID became the first genetically-determined metabolic condition for which ERT was approved by the Food and Drug Administration.
Inborn errors of immune regulation have emerged as an important group of disorders in which newer therapeutic agents are being tested, often with promising initial results (see also Table 1). One example is the use of jakinibs to treat disease due to heterozygous gain-of-function (GOF) mutations in STAT1 or STAT3. These diseases cause combinations of infection susceptibility, autoimmunity, inappropriate inflammatory responses, and lymphoproliferation, which have been historically difficult to treat. STAT1 and STAT3 are transcription factors that normally become activated upon stimulation of various cytokine receptors, with IFN receptors activating STAT1, and IL-6, γc, IL-10, or IL-23 receptors activating STAT3. Since GOF mutations confer augmented activity upon cytokine receptor stimulation, which can exert complex downstream effects on immunity, disease should be amenable to inhibition of JAK adaptors that couple cytokine receptor activation to STAT activation. Indeed, efficacy of JAK inhibition in treating both STAT1 and STAT3 GOF patients has been recently demonstrated (145).
Table 1.
Targeted therapies for treatment of selected monogenic immune dysregulatory conditions.
| Immune dysregulatory condition | Molecular pathogenic pathway targeted | Treatment | References |
|---|---|---|---|
| STAT1 GOF | STAT1 hyperactivation | JAK inhibition (ruxolitinib) | (145) |
| STAT3 GOF | STAT3 hyperactivation | JAK inhibition (ruxolitinib, tofacitinib) | (145) |
| CANDLE syndrome (proteasome complex component LOF) | Type I IFN hyperactivation | JAK inhibition (baricitinib) | (93) |
| SAVI (STING GOF) | Type I IFN hyperactivation | JAK inhibition (baricitinib) | (93) |
| CTLA-4 haploinsufficiency | T cell costimulatory pathway hyperactivation | CTLA-4-Fc fusion protein (abatacept, belatacept) | (146) |
| LRBA deficiency | Increased CTLA-4 degradation leading to T cell costimulatory pathway hyperactivation | CTLA-4-Fc fusion protein (abatacept) | (84) |
| APDS (PASLI) (PIK3CD or PIK3R1 GOF) | PI3K pathway hyperactivation | PI3K inhibition (leniolisib) | (147) |
| CHAPLE (CD55 deficiency) | Complement hyperactivation | Eculizumab, pozelimab (in testing) | (148) |
| Inflammasomapathies (NLRP1, NLRP3, PSTPIP1 GOF; LPIN2, MEVK, WDR1, DIRA or IL1RN deficiencies, etc.) | IL-1 hyperactivation | IL-1 antagonists (anakinra, canakinumab, rilonacept) | (91) |
A second example where knowledge of the molecular etiology of PIDs has led to targeted therapies in immunodysregulatory disorders is CTLA-4-haploinsufficiency. While treatment with mTOR inhibitors in these patients can inhibit the increased costimulatory signals, CTLA-4-Fc fusion proteins such as abatacept can alternatively replace the missing CTLA-4 function (146). This new modality seems to be helpful in cases of refractory disease, and a clinical trial is planned for formal testing (NCT03733067). Additionally, CTLA-4-Fc fusion proteins may be useful in LRBA and DEF6 deficiencies which phenocopy CTLA4-haploinsufficiency by increasing CTLA-4 degradation within lysosomes (84).
A third example is the use of small molecule inhibitors of PI3K in activated phosphoinositide 3-kinase δ syndrome (APDS), a condition characterized by lymphoproliferation and immunodeficiency, with hypogammaglobulinemia with elevated serum IgM, accumulation of senescent T cells and increased proportion of transitional B cells. In this disease, treatment with leniolisib, a selective PI3Kδ inhibitor, was recently shown in a 12-week open-label dose-escalation safety/efficacy trial to suppress the hyperactive PI3Kδ pathway in patients, normalize abnormal immune biomarkers, and improve lymphoproliferation and autoimmune cytopenias (147). A randomized placebo-controlled study is currently in progress to validate these exciting findings (NCT02435173).
A final example of a disease where extrinsic targeted therapy is being tested is CD55 deficiency with early-onset protein-losing enteropathy and thrombosis. Loss of CD55 results in complement overactivation that is critical for disease pathogenesis, which is supported by anecdotal observations of improved disease upon treatment with the terminal complement inhibitor eculizumab (148). A randomized double-blind, placebo-controlled study is currently in progress to test the efficacy of a pozelimab, a fully human inhibitor of C5 (NCT04209634).
Ultimately, correction of the gene defect could provide definitive cure to patients with IEI (15). A first indication in this sense was provided by the observation of somatic mutations (reversions) that restore expression and function of the mutant protein in a clonal lineage, as first demonstrated in 1996 by Rochelle Hirschhorn for ADA deficiency (149). When the reversion event occurs in an early lymphoid progenitor, it confers a strong selective advantage to T-cell progenitors, and may even lead to apparent normalization of the immunological phenotype and clinical improvement, as observed in XR-SCID (150).
In the early 1990s gene therapy was attempted for ADA-SCID patients, initially using gene modified autologous T cells (151). In 1992, two ADA-SCID patients were treated with combined autologous T cells and hematopoietic stem cell precursors (HSPCs) carrying two distinct gamma-retroviral vectors expressing the wild-type ADA gene. This pioneering trial provided seminal demonstration of the selective advantage of cells with restored function, and also showed that while gene-modified T cells can be beneficial in the short term, long-term reconstitution is derived from gene-modified hematopoietic stem and progenitor cells (HSPCs) (152). Gamma-retroviral-mediated transfer of wild-type ADA cDNA under a constitutive promoter (to ensure persistent transgene expression) led to random but stable integration into the genome. The clinical success and safety of this approach, combined with ERT discontinuation and nonmyeloablative conditioning, was further confirmed in 10 patients (153). In 2016, this ex-vivo HSPC gene therapy designed to cure ADA-SCID (Strimvelis, Orchard Therapeutics), was approved by the European Medicines Agency, becoming the first gene therapy product approved for commercialization (154).
A similar therapeutic strategy, based on first-generation γ-retroviral vectors, led to successful and rapid reconstitution of T-cell immunity in infants with XR-SCID (155). However, six patients developed T cell acute lymphoblastic anemia due to insertion of the transgene into an oncogene (LMO2 in most cases), resulting in transactivation of the oncogene (156). Similar serious adverse events (SAEs) were reported also after initial attempts to treat Wiskott-Aldrich syndrome (WAS) by gene therapy (157). This prompted investigators to develop safer vector delivery systems. First, second-generation γ-retroviral vectors were generated, that were devoid of the potent transactivating long-term repeats of the viral vector and used a cellular promoter to drive expression of the transgene. Use of such self-inactivating (SIN) γ-retroviral vectors led to robust T-cell reconstitution, without occurrence of leukemic events, in patients with XR-SCID (158). Then, SIN-lentiviruses (SIN-LV) emerged as the most efficient and safest gene delivery vectors for both T cells and HSPCs. Gene therapy with SIN-LV vectors has now been used for numerous monogenic IEIs, including ADA-SCID, XR-SCID, WAS, CGD and LAD (reviewed in (15)). Furthermore, addition of reduced-toxicity conditioning regimens has allowed more robust engraftment of gene-corrected HSCs and multilineage immune reconstitution, overcoming the lack of B-cell and NK-cell reconstitution that has been observed after gene therapy for XR-SCID, when no chemotherapy had been used (159, 160).
However, addition of wild-type copies of the relevant cDNAs would not cure disorders due to GOF and possibly dominant-negative mutations. Furthermore, use of constitutive cellular promoters driving expression of the transgene could be deleterious in the treatment of disease due to mutations of genes whose expression is tightly regulated, such as CD40LG and FOXP3. In this regard, a recent major advancement in the curative potential of gene therapy has been the development of “gene editing”. Here, a single nucleotide variant is replaced, or the entire gene coding sequence is inserted precisely at the endogenous locus, using a nuclease-mediated DNA break and subsequent repair by homologous recombination (161). Gene correction by editing preserves spatiotemporal regulated gene expression, which is important for genes that are differentially expressed during development and lineage differentiation, or that are only expressed upon cell activation. In addition, gene editing is particularly attractive for the correction of dominant-negative mutations. The use of different nucleases has been explored, with the CRISPR/Cas9 nuclease system combined with AAV-mediated delivery of the donor template, demonstrating the clearest translational efficacy. Although already in the clinic for the treatment of genetic blood disorders, CRISPR/Cas9 gene editing is still in pre-clinical phase for correction of IEI. However, feasibility and efficacy have been demonstrated both in vitro and in vivo in mice and in humanized mice models for XR-SCID (162), X-linked hyper-IgM (163), and IPEX (164). Collectively this work confirms that IEI offer an ideal model system for the exploration and advancement of innovative curative approaches, and supports the view that development of gene editing may lead to dramatic improvement in the life of affected patients. Yet, affordability of gene therapy remains a major financial and ethical challenge (165).
Conclusions
The last ten years have witnessed a gigantic progress in the field of IEI. The number of disorders being discovered is literally growing exponentially, including not only rare but also common genetic defects. The range of clinical phenotypes attributed to IEI is also diversifying at full speed, with an unsuspected diversity of infectious, malignant, autoimmune, autoinflammatory, and allergic phenotypes being caused by monogenic lesions. There is every reason to believe that we have only seen the tip of the iceberg, and that countless more IEI will be discovered. Vast areas of medicine, refractory to large population-based association studies, are being slowly but surely transformed by the family- and patient-based search for new types of IEI. The next ten years may very well see conditions as diverse as life-threatening COVID-19 and systemic lupus erythematosus each deciphered into a myriad of IEI. Noteworthy, an emerging feature of IEI is that their high level of genetic heterogeneity is typically associated with a high level of physiological homogeneity. Patients and their families have benefited the most from this avalanche of genetic and immunological studies. Indeed, these patient-based, basic studies ushered other types of studies, aimed at better diagnosing patients and better describing their clinical features, thereby improving genetic counseling. Progress in the molecular genetics and cellular immunology of IEI has also resulted in the development of innovative preventive and therapeutic approaches, in line with a long tradition. Indeed, enzyme replacement, hematopoietic transplantation, and gene therapy were also pioneered for IEI. Last, but not least, the field of IEI is also of great benefit to the scientific community for the basic knowledge it reveals. With the recent advent of many path-breaking genetic and immunological technologies, the extraordinary genetic and phenotypic diversity of the 7 billion living humans, which was previously seen as an insurmountable obstacle to scientific research, now provides a unique opportunity to delineate, for the first time, the essential and redundant functions of genes involved in one or another aspect of immunity.
Acknowledgments:
Funding: LDN and HCS are supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. JLC is supported by National Institutes of Health (NIH) (R01AI088364, R01NS072381, R37AI095983, P01AI061093, R01AI127564, R01AI143810); National Center for Advancing Translational Sciences (NCATS), NIH, Clinical and Translational Science Award (CTSA) program (UL1 TR001866), “Investments for the Future” program of the French National Research Agency (ANR) (ANR-10-IAHU-01), Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases (ANR-10-LABX-62-IBEID), French Foundation for Medical Research (FRM) (EQU201903007798), Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), and Paris University. RB is the Anne T. and Robert M. Bass Endowed Faculty Scholar in Pediatric Cancer and Blood Diseases, Stanford Maternal & Child Health Research Institute; she is supported by NIH (NIAID), California Institute for Regenerative Medicine, and the Stanford SPARK program.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: N/A.
References and Notes
- 1.Bruton OC, Agammaglobulinemia. Pediatrics 9, 722–728 (1952). [PubMed] [Google Scholar]
- 2.Glanzmann E, Riniker P, [Essential lymphocytophthisis; new clinical aspect of infant pathology]. Ann Paediatr 175, 1–32 (1950). [PubMed] [Google Scholar]
- 3.Hitzig WH, Biro Z, Bosch H, Huser HJ, [Agammaglobulinemia & alymphocytosis with atrophy of lymphatic tissue]. Helv Paediatr Acta 13, 551–585 (1958). [PubMed] [Google Scholar]
- 4.Hitzig WH, Barandun S, Cottier H, [The Swiss type of agammaglobulinemia]. Ergeb Inn Med Kinderheilkd 27, 79–154 (1968). [PubMed] [Google Scholar]
- 5.Cooper MD, A life of adventure in immunobiology. Annu Rev Immunol 28, 1–19 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Kostmann R, Hereditaer reticulos - en ny systemsjukdom (Hereditary reticulosis - a new systemic disease). Svenska Laekartidningen 47, 2861–2868 (1950). [Google Scholar]
- 7.Janeway CA, C. J; Davidson M; Downey W; Gitlin D; Sullivan JC, Hypergammaglobulinemia associated with severe, recurrent and chronic non-specific infection. American Journal of Diseases of Children 88, 388–392 (1954). [Google Scholar]
- 8.Berendes H, Bridges RA, Good RA, A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med 40, 309–312 (1957). [PubMed] [Google Scholar]
- 9.Seligmann M, Fudenberg HH, Good RA, A proposed classification of primary immunologic deficiencies. Am J Med 45, 817–825 (1968). [DOI] [PubMed] [Google Scholar]
- 10.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Picard C, Puck J, Torgerson TR, Casanova JL, Sullivan KE, Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casanova JL, Abel L, The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet 14, 215–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang SY, Jouanguy E, Zhang Q, Abel L, Puel A, Casanova JL, Human inborn errors of immunity to infection affecting cells other than leukocytes: from the immune system to the whole organism. Curr Opin Immunol 59, 88–100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, C. F. P. I. D. s. g. members of the, Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol 140, 1388–1393 e1388 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA, Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet 2, 1366–1369 (1968). [DOI] [PubMed] [Google Scholar]
- 15.Fischer A, Hacein-Bey-Abina S, Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Notarangelo LD, Fleisher TA, Targeted strategies directed at the molecular defect: Toward precision medicine for select primary immunodeficiency disorders. J Allergy Clin Immunol 139, 715–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gitlin D, Craig JM, The Thymus and Other Lymphoid Tissues in Congenital Agammaglobulinemia. I. Thymic Alymphoplasia and Lymphocytic Hypoplasia and Their Relation to Infection. Pediatrics 32, 517–530 (1963). [PubMed] [Google Scholar]
- 18.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ, Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet 2, 1067–1069 (1972). [DOI] [PubMed] [Google Scholar]
- 19.Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK, Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet 1, 1010–1013 (1975). [DOI] [PubMed] [Google Scholar]
- 20.Royer-Pokora B, Kunkel LM, Monaco AP, Goff SC, Newburger PE, Baehner RL, Cole FS, Curnutte JT, Orkin SH, Cloning the gene for an inherited human disorder--chronic granulomatous disease--on the basis of its chromosomal location. Nature 322, 32–38 (1986). [DOI] [PubMed] [Google Scholar]
- 21.Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, McBride OW, Leonard WJ, Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell 73, 147–157 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Leonard WJ, Lin JX, O’Shea JJ, The gammac Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 50, 832–850 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, et al. , Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 377, 65–68 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Puel A, Ziegler SF, Buckley RH, Leonard WJ, Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat Genet 20, 394–397 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Horev L, Unger S, Molho-Pessach V, Meir T, Maly A, Stepensky P, Zamir M, Keller B, Babay S, Warnatz K, Ramot Y, Zlotogorski A, Generalized verrucosis and HPV-3 susceptibility associated with CD4 T-cell lymphopenia caused by inherited human interleukin-7 deficiency. J Am Acad Dermatol 72, 1082–1084 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Kosumi H, Natsuga K, Takashima S, Miyauchi T, Huang YT, Nomura T, Yanagi T, Huang HY, Chiu FP, Chen PC, Hsu CK, Shimizu H, Two cases of interleukin-7 (IL-7)-deficient generalized verrucosis. Clin Infect Dis, (2020). [DOI] [PubMed] [Google Scholar]
- 27.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP, Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 105, 177–186 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Ahnesorg P, Smith P, Jackson SP, XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124, 301–313 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay JP, Revy P, Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124, 287–299 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Pannicke U, Honig M, Hess I, Friesen C, Holzmann K, Rump EM, Barth TF, Rojewski MT, Schulz A, Boehm T, Friedrich W, Schwarz K, Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet 41, 101–105 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, Demerens-de Chappedelaine C, Morillon E, Valensi F, Simon-Stoos KL, Mullikin JC, Noroski LM, Besse C, Wulffraat NM, Ferster A, Abecasis MM, Calvo F, Petit C, Candotti F, Abel L, Fischer A, Cavazzana-Calvo M, Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet 41, 106–111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steimle V, Otten LA, Zufferey M, Mach B, Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75, 135–146 (1993). [PubMed] [Google Scholar]
- 33.Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, Reith W, A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev 9, 1021–1032 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W, RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J 16, 1045–1055 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, Sanchez JC, Hochstrasser DF, Mach B, Reith W, A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet 20, 273–277 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Schatz DG, Oettinger MA, Baltimore D, The V(D)J recombination activating gene, RAG-1. Cell 59, 1035–1048 (1989). [DOI] [PubMed] [Google Scholar]
- 37.Oettinger MA, Schatz DG, Gorka C, Baltimore D, RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248, 1517–1523 (1990). [DOI] [PubMed] [Google Scholar]
- 38.Speckmann C, Doerken S, Aiuti A, Albert MH, Al-Herz W, Allende LM, Scarselli A, Avcin T, Perez-Becker R, Cancrini C, Cant A, Di Cesare S, Finocchi A, Fischer A, Gaspar HB, Ghosh S, Gennery A, Gilmour K, Gonzalez-Granado LI, Martinez-Gallo M, Hambleton S, Hauck F, Hoenig M, Moshous D, Neven B, Niehues T, Notarangelo L, Picard C, Rieber N, Schulz A, Schwarz K, Seidel MG, Soler-Palacin P, Stepensky P, Strahm B, Vraetz T, Warnatz K, Winterhalter C, Worth A, Fuchs S, Uhlmann A, Ehl S, P. C. s. o. t. I. E. W. P. o. t. EBMT, A prospective study on the natural history of patients with profound combined immunodeficiency: An interim analysis. J Allergy Clin Immunol 139, 1302–1310 e1304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A, A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, Fischer A, Feske S, STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 360, 1971–1980 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villa A, Notarangelo LD, RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev 287, 73–90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Notarangelo LD, Kim MS, Walter JE, Lee YN, Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 16, 234–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, Jamieson BD, Zack JA, Picker LJ, Koup RA, Changes in thymic function with age and during the treatment of HIV infection. Nature 396, 690–695 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Puck JM, Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol Rev 287, 241–252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, Kapoor N, Hanson IC, Filipovich AH, Jyonouchi S, Sullivan KE, Small TN, Burroughs L, Skoda-Smith S, Haight AE, Grizzle A, Pulsipher MA, Chan KW, Fuleihan RL, Haddad E, Loechelt B, Aquino VM, Gillio A, Davis J, Knutsen A, Smith AR, Moore TB, Schroeder ML, Goldman FD, Connelly JA, Porteus MH, Xiang Q, Shearer WT, Fleisher TA, Kohn DB, Puck JM, Notarangelo LD, Cowan MJ, O’Reilly RJ, Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 371, 434–446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haddad E, L. BR; Griffith LM; Buckley RH; Parrott RE; Prockop SE; et al. , SCID genotype and 6-month post-transplant CD4 count predict survival and immune recovery: a PIDTC Retrospective Study. Blood, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Constantine GM, Lionakis MS, Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev 287, 103–120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D, Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA, Aire Enforces Immune Tolerance by Directing Autoreactive T Cells into the Regulatory T Cell Lineage. Immunity 44, 1102–1113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D, Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, Rosen LB, Break TJ, Gu W, Hunsberger S, Browne SK, Hsu AP, Rampertaap S, Swamydas M, Collar AL, Kong HH, Lee CR, Chascsa D, Simcox T, Pham A, Bondici A, Natarajan M, Monsale J, Kleiner DE, Quezado M, Alevizos I, Moutsopoulos NM, Yockey L, Frein C, Soldatos A, Calvo KR, Adjemian J, Similuk MN, Lang DM, Stone KD, Uzel G, Kopp JB, Bishop RJ, Holland SM, Olivier KN, Fleisher TA, Heller T, Winer KK, Lionakis MS, Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devoss JJ, Shum AK, Johannes KP, Lu W, Krawisz AK, Wang P, Yang T, Leclair NP, Austin C, Strauss EC, Anderson MS, Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J Immunol 181, 4072–4079 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJ, Pura M, Schalke B, Strobel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, Meager A, Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 207, 299–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL, Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207, 291–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rieux-Laucat F, Magerus-Chatinet A, Neven B, The Autoimmune Lymphoproliferative Syndrome with Defective FAS or FAS-Ligand Functions. J Clin Immunol 38, 558–568 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Siegel RM, Chan FK, Chun HJ, Lenardo MJ, The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat Immunol 1, 469–474 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Lehle AS, Farin HF, Marquardt B, Michels BE, Magg T, Li Y, Liu Y, Ghalandary M, Lammens K, Hollizeck S, Rohlfs M, Hauck F, Conca R, Walz C, Weiss B, Lev A, Simon AJ, Gross O, Gaidt MM, Hornung V, Clevers H, Yazbeck N, Hanna-Wakim R, Shouval DS, Warner N, Somech R, Muise AM, Snapper SB, Bufler P, Koletzko S, Klein C, Kotlarz D, Intestinal Inflammation and Dysregulated Immunity in Patients With Inherited Caspase-8 Deficiency. Gastroenterology 156, 275–278 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP, Danner RL, Barb J, Munson PJ, Puck JM, Dale J, Straus SE, Fleisher TA, Lenardo MJ, NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A 104, 8953–8958 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takagi M, Shinoda K, Piao J, Mitsuiki N, Takagi M, Matsuda K, Muramatsu H, Doisaki S, Nagasawa M, Morio T, Kasahara Y, Koike K, Kojima S, Takao A, Mizutani S, Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood 117, 2887–2890 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG, Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol 142, 1679–1695 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Bacchetta R, Barzaghi F, Roncarolo MG, From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci 1417, 5–22 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr 100, 731–737 (1982). [DOI] [PubMed] [Google Scholar]
- 63.Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, Dupuis-Girod S, Cerf-Bensussan N, Cavazzana-Calvo M, Brousse N, Fischer A, Casanova JL, Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med 344, 1758–1762 (2001). [DOI] [PubMed] [Google Scholar]
- 64.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM, JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest 106, R75–81 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME, X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27, 18–20 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27, 20–21 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Fontenot JD, Gavin MA, Rudensky AY, Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4, 330–336 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Hori S, Nomura T, Sakaguchi S, Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Khattri R, Cox T, Yasayko SA, Ramsdell F, An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 4, 337–342 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, Dagna-Bricarelli F, Sartirana C, Matthes-Martin S, Lawitschka A, Azzari C, Ziegler SF, Levings MK, Roncarolo MG, Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 116, 1713–1722 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, Le DT, Liu W, Brusko TM, Anderson MS, Rudensky AY, Marson A, Chang HY, Bluestone JA, A Mutation in the Transcription Factor Foxp3 Drives T Helper 2 Effector Function in Regulatory T Cells. Immunity 50, 362–377 e366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, Nademi Z, Slatter MA, Ulloa ER, Shcherbina A, Roppelt A, Worth A, Silva J, Aiuti A, Murguia-Favela L, Speckmann C, Carneiro-Sampaio M, Fernandes JF, Baris S, Ozen A, Karakoc-Aydiner E, Kiykim A, Schulz A, Steinmann S, Notarangelo LD, Gambineri E, Lionetti P, Shearer WT, Forbes LR, Martinez C, Moshous D, Blanche S, Fisher A, Ruemmele FM, Tissandier C, Ouachee-Chardin M, Rieux-Laucat F, Cavazzana M, Qasim W, Lucarelli B, Albert MH, Kobayashi I, Alonso L, Diaz De Heredia C, Kanegane H, Lawitschka A, Seo JJ, Gonzalez-Vicent M, Diaz MA, Goyal RK, Sauer MG, Yesilipek A, Kim M, Yilmaz-Demirdag Y, Bhatia M, Khlevner J, Richmond Padilla EJ, Martino S, Montin D, Neth O, Molinos-Quintana A, Valverde-Fernandez J, Broides A, Pinsk V, Ballauf A, Haerynck F, Bordon V, Dhooge C, Garcia-Lloret ML, Bredius RG, Kalwak K, Haddad E, Seidel MG, Duckers G, Pai SY, Dvorak CC, Ehl S, Locatelli F, Goldman F, Gennery AR, Cowan MJ, Roncarolo MG, Bacchetta R, Primary C Immune Deficiency Treatment, B. the Inborn Errors Working Party of the European Society for, T. Marrow, Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol 141, 1036–1049 e1035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eriksson D, Bacchetta R, Gunnarsson HI, Chan A, Barzaghi F, Ehl S, Hallgren A, van Gool F, Sardh F, Lundqvist C, Laakso SM, Ronnblom A, Ekwall O, Makitie O, Bensing S, Husebye ES, Anderson M, Kampe O, Landegren N, The autoimmune targets in IPEX are dominated by gut epithelial proteins. J Allergy Clin Immunol 144, 327–330 e328 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J, DNA methylation controls Foxp3 gene expression. Eur J Immunol 38, 1654–1663 (2008). [DOI] [PubMed] [Google Scholar]
- 75.Baron U, Werner J, Schildknecht K, Schulze JJ, Mulu A, Liebert UG, Sack U, Speckmann C, Gossen M, Wong RJ, Stevenson DK, Babel N, Schurmann D, Baldinger T, Bacchetta R, Grutzkau A, Borte S, Olek S, Epigenetic immune cell counting in human blood samples for immunodiagnostics. Sci Transl Med 10, (2018). [DOI] [PubMed] [Google Scholar]
- 76.Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, Segundo GRS, Allenspach EJ, Favre C, Ochs HD, Torgerson TR, Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol 9, 2411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, Tobin V, Nguyen DN, Lee MR, Putnam AL, Ferris AL, Chen JW, Schickel JN, Pellerin L, Carmody D, Alkorta-Aranburu G, Del Gaudio D, Matsumoto H, Morell M, Mao Y, Cho M, Quadros RM, Gurumurthy CB, Smith B, Haugwitz M, Hughes SH, Weissman JS, Schumann K, Esensten JH, May AP, Ashworth A, Kupfer GM, Greeley SAW, Bacchetta R, Meffre E, Roncarolo MG, Romberg N, Herold KC, Ribas A, Leonetti MD, Marson A, Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, Modis Y, Alazami AM, Acres M, Haller W, Bowen C, Doffinger R, Sinclair J, Brothers S, Zhang Y, Matthews HF, Naudion S, Pelluard F, Alajlan H, Yamazaki Y, Notarangelo LD, Thaventhiran JE, Engelhardt KR, Al-Mousa H, Hambleton S, Rooryck C, Smith KGC, Lenardo MJ, Human interleukin-2 receptor beta mutations associated with defects in immunity and peripheral tolerance. J Exp Med 216, 1311–1327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandez IZ, Baxter RM, Garcia-Perez JE, Vendrame E, Ranganath T, Kong DS, Lundquist K, Nguyen T, Ogolla S, Black J, Galambos C, Gumbart JC, Dawany N, Kelsen JR, de Zoeten EF, Quinones R, Eissa H, Verneris MR, Sullivan KE, Rochford R, Blish CA, Kedl RM, Dutmer CM, Hsieh EWY, A novel human IL2RB mutation results in T and NK cell-driven immune dysregulation. J Exp Med 216, 1255–1267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L, Jasper H, Tepper A, Heinrich JJ, Rosenfeld RG. Growth hormine insensitivity associated with a STAT5b mutation. N Engl J Med 349, 1139–1147 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Kanai T, Jenks J, Nadeau KC. The STAT5b pathway defect and autoimmunity. Front Immunol 3, 234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, Frucht DM, Dumitriu B, Scheinberg P, Folio LR, Frein CA, Price S, Koh C, Heller T, Seroogy CM, Huttenlocher A, Rao VK, Su HC, Kleiner D, Notarangelo LD, Rampertaap Y, Olivier KN, McElwee J, Hughes J, Pittaluga S, Oliveira JB, Meffre E, Fleisher TA, Holland SM, Lenardo MJ, Tangye SG, Uzel G, Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schaffer AA, Gruning BA, Unger S, Frede N, Baumann U, Witte T, Schmidt RE, Dueckers G, Niehues T, Seneviratne S, Kanariou M, Speckmann C, Ehl S, Rensing-Ehl A, Warnatz K, Rakhmanov M, Thimme R, Hasselblatt P, Emmerich F, Cathomen T, Backofen R, Fisch P, Seidl M, May A, Schmitt-Graeff A, Ikemizu S, Salzer U, Franke A, Sakaguchi S, Walker LS, Sansom DM, Grimbacher B, Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 20, 1410–1416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, Husami A, Kissell D, Nortman S, Chaturvedi V, Haines H, Young LR, Mo J, Filipovich AH, Bleesing JJ, Mustillo P, Stephens M, Rueda CM, Chougnet CA, Hoebe K, McElwee J, Hughes JD, Karakoc-Aydiner E, Matthews HF, Price S, Su HC, Rao VK, Lenardo MJ, Jordan MB, AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349, 436–440 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed D, Hultenby K, Liu C, Baronio M, Vitali M, Philippet P, Dideberg V, Aghamohammadi A, Rezaei N, Enright V, Du L, Salzer U, Eibel H, Pfeifer D, Veelken H, Stauss H, Lougaris V, Plebani A, Gertz EM, Schaffer AA, Hammarstrom L, Grimbacher B, Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 90, 986–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Serwas NK, Hoeger B, Ardy RC, Stulz SV, Sui Z, Memaran N, Meeths M, Krolo A, Yuce Petronczki O, Pfajfer L, Hou TZ, Halliday N, Santos-Valente E, Kalinichenko A, Kennedy A, Mace EM, Mukherjee M, Tesi B, Schrempf A, Pickl WF, Loizou JI, Kain R, Bidmon-Fliegenschnee B, Schickel JN, Glauzy S, Huemer J, Garncarz W, Salzer E, Pierides I, Bilic I, Thiel J, Priftakis P, Banerjee PP, Forster-Waldl E, Medgyesi D, Huber WD, Orange JS, Meffre E, Sansom DM, Bryceson YT, Altman A, Boztug K, Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis. Nat Commun 10, 3106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, McDonald TJ, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy NP, Milenkovic T, Seppanen M, Lernmark A, Mustjoki S, Otonkoski T, Kere J, Morgan NG, Ellard S, Hattersley AT, Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet 46, 812–814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N, The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity 49, 1004–1019 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Glocker E-O, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, Hätscher N, Pfeifer D, Sykora K-W, Sauer M, Kreipe H, Lacher M, Nustede R, Woellner C, Baumann U, Salzer U, Koletzko S, Shah N, Segal AW, Sauerbrey A, Buderus S, Snapper SB, Grimbacher B, Klein C. Inflammatory bowel disease and mutatione affecting the interleukin-10 receptor. N Engl J Med 361, 2033–2045 (2009).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Glocker E-O, Frede N, Perro M, Sebire N, Elawad M, Shah N, Grimbacher B. Infant colitis – It’s in the genes. Lancet 376, 1272 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Manthiram K, Zhou Q, Aksentijevich I, Kastner DL, The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 18, 832–842 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, Lee YS, Hernandez N, Drutman S, de Jong YP, Vivier E, Bruneau J, Beziat V, Boisson B, Lorenzo-Diaz L, Boucherit S, Sebagh M, Jacquemin E, Emile JF, Abel L, Rice CM, Jouanguy E, Casanova JL, Inherited IL-18BP deficiency in human fulminant viral hepatitis. J Exp Med 216, 1777–1790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rodero MP, Crow YJ, Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med 213, 2527–2538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, Rosain J, Buta S, Bousfiha A, Casanova JL, Bustamante J, Bogunovic D, Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alsohime F, Martin-Fernandez M, Temsah MH, Alabdulhafid M, Le Voyer T, Alghamdi M, Qiu X, Alotaibi N, Alkahtani A, Buta S, Jouanguy E, Al-Eyadhy A, Gruber C, Hasan GM, Bashiri FA, Halwani R, Hassan HH, Al-Muhsen S, Alkhamis N, Alsum Z, Casanova JL, Bustamante J, Bogunovic D, Alangari AA, JAK Inhibitor Therapy in a Child with Inherited USP18 Deficiency. N Engl J Med 382, 256–265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, Schalm S, Murias S, Dare JA, Brown D, Stone DL, Gao L, Klausmeier T, Foell D, de Jesus AA, Chapelle DC, Kim H, Dill S, Colbert RA, Failla L, Kost B, O’Brien M, Reynolds JC, Folio LR, Calvo KR, Paul SM, Weir N, Brofferio A, Soldatos A, Biancotto A, Cowen EW, Digiovanna JJ, Gadina M, Lipton AJ, Hadigan C, Holland SM, Fontana J, Alawad AS, Brown RJ, Rother KI, Heller T, Brooks KM, Kumar P, Brooks SR, Waldman M, Singh HK, Nickeleit V, Silk M, Prakash A, Janes JM, Ozen S, Wakim PG, Brogan PA, Macias WL, Goldbach-Mansky R, JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 128, 3041–3052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aksentijevich I, Zhou Q, NF-kappaB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front Immunol 8, 399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, Liu L, Stoffels M, Kratina T, Lawlor KE, Zaal KJM, Hoffmann PM, Etemadi N, Shield-Artin K, Biben C, Tsai WL, Blake MD, Kuehn HS, Yang D, Anderton H, Silke N, Wachsmuth L, Zheng L, Moura NS, Beck DB, Gutierrez-Cruz G, Ombrello AK, Pinto-Patarroyo GP, Kueh AJ, Herold MJ, Hall C, Wang H, Chae JJ, Dmitrieva NI, McKenzie M, Light A, Barham BK, Jones A, Romeo TM, Zhou Q, Aksentjevich I, Mullikin JC, Gross AJ, Shum AK, Hawkins ED, L Masters S, Lenardo MJ, Boehm M, Rosenzweig SD, Pasparakis M, Voss AK, Gadina M, Kastner DL, J Silke, Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Davis SD, Schaller J, Wedgwood RJ, Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet 1, 1013–1015 (1966). [DOI] [PubMed] [Google Scholar]
- 100.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O’Connell AC, Puck JM, Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med 340, 692–702 (1999). [DOI] [PubMed] [Google Scholar]
- 101.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H, Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448, 1058–1062 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, Pellier I, Zoghi S, Baris S, Keles S, Gray P, Du N, Wang Y, Zerbib Y, Levy R, Leclercq T, About F, Lim AI, Rao G, Payne K, Pelham SJ, Avery DT, Deenick EK, Pillay B, Chou J, Guery R, Belkadi A, Guerin A, Migaud M, Rattina V, Ailal F, Benhsaien I, Bouaziz M, Habib T, Chaussabel D, Marr N, El-Benna J, Grimbacher B, Wargon O, Bustamante J, Boisson B, Muller-Fleckenstein I, Fleckenstein B, Chandesris MO, Titeux M, Fraitag S, Alyanakian MA, Leruez-Ville M, Picard C, Meyts I, Di Santo JP, Hovnanian A, Somer A, Ozen A, Rezaei N, Chatila TA, Abel L, Leonard WJ, Tangye SG, Puel A, Casanova JL, A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, Bulashevska A, Proietti M, Frobel P, Noltner C, Yang L, Rojas-Restrepo J, Langer N, Winzer S, Engelhardt KR, Glocker C, Pfeifer D, Klein A, Schaffer AA, Lagovsky I, Lachover-Roth I, Beziat V, Puel A, Casanova JL, Fleckenstein B, Weidinger S, Kilic SS, Garty BZ, Etzioni A, Grimbacher B, ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, Capitani M, McGowan SJ, Sweeney E, Weber A, Chen L, Bowness P, Riordan A, Cant A, Freeman AF, Milner JD, Holland SM, Frede N, Muller M, Schmidt-Arras D, Grimbacher B, Wall SA, Jones EY, Wilkie AOM, Uhlig HH, A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med 214, 2547–2562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen YH, Grigelioniene G, Newton PT, Gullander J, Elfving M, Hammarsjo A, Batkovskyte D, Alsaif HS, Kurdi WIY, Abdulwahab F, Shanmugasundaram V, Devey L, Bacrot S, Brodszki J, Huber C, Hamel B, Gisselsson D, Papadogiannakis N, Jedrycha K, Gurtl-Lackner B, Chagin AS, Nishimura G, Aschenbrenner D, Alkuraya FS, Laurence A, Cormier-Daire V, Uhlig HH, Absence of GP130 cytokine receptor signaling causes extended Stuve-Wiedemann syndrome. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, Staal J, Aschenbrenner D, Roels L, Worley L, Claes K, Gartner L, Kohn LA, De Bruyne M, Schmitz-Abe K, Charbonnier LM, Keles S, Nammour J, Vladikine N, Maglorius Renkilaraj MRL, Seeleuthner Y, Migaud M, Rosain J, Jeljeli M, Boisson B, Van Braeckel E, Rosenfeld JA, Dai H, Burrage LC, Murdock DR, Lambrecht BN, Avettand-Fenoel V, Vogel TP, Undiagnosed Diseases N., Esther CR, Haskologlu S, Dogu F, Ciznar P, Boutboul D, Ouachee-Chardin M, Amourette J, Lebras MN, Gauvain C, Tcherakian C, Ikinciogullari A, Beyaert R, Abel L, Milner JD, Grimbacher B, Couderc LJ, Butte MJ, Freeman AF, Catherinot E, Fieschi C, Chatila TA, Tangye SG, Uhlig HH, Haerynck F, Casanova JL, Puel A, Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Spencer S, Kostel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, Gurel M, Zhang Y, Sun G, Sabroe RA, Greene D, Rae W, Shahin T, Kania K, Ardy RC, Thian M, Staples E, Pecchia-Bekkum A, Worrall WPM, Stephens J, Brown M, Tuna S, York M, Shackley F, Kerrin D, Sargur R, Condliffe A, Tipu HN, Kuehn HS, Rosenzweig SD, Turro E, Tavare S, Thrasher AJ, Jodrell DI, Smith KGC, Boztug K, Milner JD, Thaventhiran JED, Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med 216, 1986–1998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nieminen P, Morgan NV, Fenwick AL, Parmanen S, Veistinen L, Mikkola ML, van der Spek PJ, Giraud A, Judd L, Arte S, Brueton LA, Wall SA, Mathijssen IM, Maher ER, Wilkie AO, Kreiborg S, Thesleff I, Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am J Hum Genet 89, 67–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kotlarz D, Zietara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, Krawitz PM, Robinson PN, Hecht J, Puchalka J, Gertz EM, Schaffer AA, Lawrence MG, Kardava L, Pfeifer D, Baumann U, Pfister ED, Hanson EP, Schambach A, Jacobs R, Kreipe H, Moir S, Milner JD, Schwille P, Mundlos S, Klein C, Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med 210, 433–443 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, Mele F, Latorre D, Mahdaviani SA, Aytekin C, Mansouri D, Bryant VL, Jabot-Hanin F, Deswarte C, Nieto-Patlan A, Surace L, Kerner G, Itan Y, Jovic S, Avery DT, Wong N, Rao G, Patin E, Okada S, Bigio B, Boisson B, Rapaport F, Seeleuthner Y, Schmidt M, Ikinciogullari A, Dogu F, Tanir G, Tabarsi P, Bloursaz MR, Joseph JK, Heer A, Kong XF, Migaud M, Lazarov T, Geissmann F, Fleckenstein B, Arlehamn CL, Sette A, Puel A, Emile JF, van de Vosse E, Quintana-Murci L, Di Santo JP, Abel L, Boisson-Dupuis S, Bustamante J, Tangye SG, Sallusto F, Casanova JL, Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kebudi R, Kiykim A, Sahin MK, Primary Immunodeficiency and Cancer in Children; A Review of the Literature. Curr Pediatr Rev 15, 245–250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cohen JI, Epstein-Barr virus infection. N Engl J Med 343, 481–492 (2000). [DOI] [PubMed] [Google Scholar]
- 113.Tangye SG, Latour S, Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood, (2020). [DOI] [PubMed] [Google Scholar]
- 114.Kanellopoulou C, George AB, Masutani E, Cannons JL, Ravell JC, Yamamoto TN, Smelkinson MG, Jiang PD, Matsuda-Lennikov M, Reilley J, Handon R, Lee PH, Miller JR, Restifo NP, Zheng L, Schwartzberg PL, Young M, Lenardo MJ, Mg(2+) regulation of kinase signaling and immune function. J Exp Med 216, 1828–1842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Matsuda-Lennikov M, Biancalana M, Zou J, Ravell JC, Zheng L, Kanellopoulou C, Jiang P, Notarangelo G, Jing H, Masutani E, Oler AJ, Olano LR, Schulz BL, Lenardo MJ, Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes. J Biol Chem 294, 13638–13656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ravell JC, Matsuda-Lennikov M, Chauvin SD, Zou J, Biancalana M, Deeb SJ, Price S, Su HC, Notarangelo G, Jiang P, Morawski A, Kanellopoulou C, Binder K, Mukherjee R, Anibal JT, Sellers B, Zheng L, He T, George AB, Pittaluga S, Powers A, Kleiner DE, Kapuria D, Ghany M, Hunsberger S, Cohen JI, Uzel G, Bergerson J, Wolfe L, Toro C, Gahl W, Folio LR, Matthews H, Angelus P, Chinn IK, Orange JS, Trujillo-Vargas CM, Franco JL, Orrego-Arango J, Gutierrez-Hincapie S, Patel NC, Raymond K, Patiroglu T, Unal E, Karakukcu M, Day AG, Mehta P, Masutani E, De Ravin SS, Malech HL, Altan-Bonnet G, Rao VK, Mann M, Lenardo MJ, Defective glycosylation and multisystem abnormalities characterize the primary immunodeficiency XMEN disease. J Clin Invest 130, 507–522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rodriguez R, Fournier B, Cordeiro DJ, Winter S, Izawa K, Martin E, Boutboul D, Lenoir C, Fraitag S, Kracker S, Watts TH, Picard C, Bruneau J, Callebaut I, Fischer A, Neven B, Latour S, Concomitant PIK3CD and TNFRSF9 deficiencies cause chronic active Epstein-Barr virus infection of T cells. J Exp Med 216, 2800–2818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bustamante J, Mendelian susceptibility to mycobacterial disease: recent discoveries. Human genetics, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptove immune responses. Adv Immunol 96, 41–101 (2007). [DOI] [PubMed] [Google Scholar]
- 120.Kerner G, Rosain J, Guerin A, AlKhabaz A, Oleaga-Quintas C, Rapaport F, Massaad MJ, Ding JY, Khan T, Al Ali F, Rahman M, Deswarte C, Martinez-Barricarte R, Geha RS, Jeanne-Julien V, Garcia DST, Chi CY, Yang R, Roynard M, Fleckenstein B, Rozenberg F, Boisson-Dupuis S, Ku CL, Seeleuthner Y, Beziat V, Marr N, Abel L, Al-Herz W, Casanova JL, Bustamante J, Inherited human IFNgamma deficiency underlies mycobacterial disease. J Clin Invest, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL, Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, Lim CK, Krementsov DN, Hernandez N, Ma CS, Zhang Q, Markle J, Martinez-Barricarte R, Payne K, Fisch R, Deswarte C, Halpern J, Bouaziz M, Mulwa J, Sivanesan D, Lazarov T, Naves R, Garcia P, Itan Y, Boisson B, Checchi A, Jabot-Hanin F, Cobat A, Guennoun A, Jackson CC, Pekcan S, Caliskaner Z, Inostroza J, Costa-Carvalho BT, de Albuquerque JAT, Garcia-Ortiz H, Orozco L, Ozcelik T, Abid A, Rhorfi IA, Souhi H, Amrani HN, Zegmout A, Geissmann F, Michnick SW, Muller-Fleckenstein I, Fleckenstein B, Puel A, Ciancanelli MJ, Marr N, Abolhassani H, Balcells ME, Condino-Neto A, Strickler A, Abarca K, Teuscher C, Ochs HD, Reisli I, Sayar EH, El-Baghdadi J, Bustamante J, Hammarstrom L, Tangye SG, Pellegrini S, Quintana-Murci L, Abel L, Casanova JL, Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kerner G, Ramirez-Alejo N, Seeleuthner Y, Yang R, Ogishi M, Cobat A, Patin E, Quintana-Murci L, Boisson-Dupuis S, Casanova JL, Abel L, Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc Natl Acad Sci U S A 116, 10430–10434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, Wong N, Soudais C, Henderson LA, Marzouqa H, Shamma J, Gonzalez M, Martinez-Barricarte R, Okada C, Avery DT, Latorre D, Deswarte C, Jabot-Hanin F, Torrado E, Fountain J, Belkadi A, Itan Y, Boisson B, Migaud M, Arlehamn CS, Sette A, Breton S, McCluskey J, Rossjohn J, de Villartay JP, Moshous D, Hambleton S, Latour S, Arkwright PD, Picard C, Lantz O, Engelhard D, Kobayashi M, Abel L, Cooper AM, Notarangelo LD, Boisson-Dupuis S, Puel A, Sallusto F, Bustamante J, Tangye SG, Casanova JL, IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349, 606–613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL, Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL, Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208, 1635–1648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, Migaud M, Boisson B, Bolze A, Itan Y, Goudin N, Cottineau J, Picard C, Abel L, Bustamante J, Casanova JL, Puel A, Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med 212, 619–631 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, Bougarn S, Cypowyj S, Grodecka L, Levy R, Beziat V, Shang L, Payne K, Avery DT, Migaud M, Boucherit S, Boughorbel S, Guennoun A, Chrabieh M, Rapaport F, Bigio B, Itan Y, Boisson B, Cormier-Daire V, Syx D, Malfait F, Zoppi N, Abel L, Freiberger T, Dietz HC, Marr N, Tangye SG, Colombi M, Casanova JL, Puel A, Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, Dobbs AK, Masneri S, Joshi A, Otaizo-Carrasquero F, Myers TG, Ganesan S, Bondioni MP, Ho ML, Marks C, Alajlan H, Mohammed RW, Zou F, Valencia CA, Filipovich AH, Facchetti F, Boisson B, Azzari C, Al-Saud BK, Al-Mousa H, Casanova JL, Abraham RS, Notarangelo LD, PAX1 is essential for development and function of the human thymus. Sci Immunol 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Orth G, Host defenses against human papillomaviruses: lessons from epidermodysplasia verruciformis. Curr Top Microbiol Immunol 321, 59–83 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Crequer A, Troeger A, Patin E, Ma CS, Picard C, Pedergnana V, Fieschi C, Lim A, Abhyankar A, Gineau L, Mueller-Fleckenstein I, Schmidt M, Taieb A, Krueger J, Abel L, Tangye SG, Orth G, Williams DA, Casanova JL, Jouanguy E, Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J Clin Invest 122, 3239–3247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M, Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet 32, 579–581 (2002). [DOI] [PubMed] [Google Scholar]
- 133.de Jong SJ, Crequer A, Matos I, Hum D, Gunasekharan V, Lorenzo L, Jabot-Hanin F, Imahorn E, Arias AA, Vahidnezhad H, Youssefian L, Markle JG, Patin E, D’Amico A, Wang CQF, Full F, Ensser A, Leisner TM, Parise LV, Bouaziz M, Maya NP, Cadena XR, Saka B, Saeidian AH, Aghazadeh N, Zeinali S, Itin P, Krueger JG, Laimins L, Abel L, Fuchs E, Uitto J, Franco JL, Burger B, Orth G, Jouanguy E, Casanova JL, The human CIB1-EVER1-EVER2 complex governs keratinocyte-intrinsic immunity to beta-papillomaviruses. J Exp Med 215, 2289–2310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang SY, Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity. Human genetics, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL, Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314, 308–312 (2006). [DOI] [PubMed] [Google Scholar]
- 136.Lafaille FG, Harschnitz O, Lee YS, Zhang P, Hasek ML, Kerner G, Itan Y, Ewaleifoh O, Rapaport F, Carlile TM, Carter-Timofte ME, Paquet D, Dobbs K, Zimmer B, Gao D, Rojas-Duran MF, Kwart D, Rattina V, Ciancanelli MJ, McAlpine JL, Lorenzo L, Boucherit S, Rozenberg F, Halwani R, Henry B, Amenzoui N, Alsum Z, Marques L, Church JA, Al-Muhsen S, Tardieu M, Bousfiha AA, Paludan SR, Mogensen TH, Quintana-Murci L, Tessier-Lavigne M, Smith GA, Notarangelo LD, Studer L, Gilbert W, Abel L, Casanova JL, Zhang SY, Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat Med 25, 1873–1884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang SY, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, Mandel H, Garcia P, Ciancanelli MJ, Biran A, Lafaille FG, Tsumura M, Cobat A, Luo J, Volpi S, Zimmer B, Sakata S, Dinis A, Ohara O, Garcia Reino EJ, Dobbs K, Hasek M, Holloway SP, McCammon K, Hussong SA, DeRosa N, Van Skike CE, Katolik A, Lorenzo L, Hyodo M, Faria E, Halwani R, Fukuhara R, Smith GA, Galvan V, Damha MJ, Al-Muhsen S, Itan Y, Boeke JD, Notarangelo LD, Studer L, Kobayashi M, Diogo L, Fairbrother WG, Abel L, Rosenberg BR, Hart PJ, Etzioni A, Casanova JL, Inborn Errors of RNA Lariat Metabolism in Humans with Brainstem Viral Infection. Cell 172, 952–965 e918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova JL, Studer L, Notarangelo LD, Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491, 769–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zimmer B, Ewaleifoh O, Harschnitz O, Lee YS, Peneau C, McAlpine JL, Liu B, Tchieu J, Steinbeck JA, Lafaille F, Volpi S, Notarangelo LD, Casanova JL, Zhang SY, Smith GA, Studer L, Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci U S A 115, E8775–E8782 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bach FH, Amos DB, Hu-1: Major histocompatibility locus in man. Science 156, 1506–1508 (1967). [DOI] [PubMed] [Google Scholar]
- 141.Reisner Y, Kapoor N, Kirkpatrick D, Pollack MS, Cunningham-Rundles S, Dupont B, Hodes MZ, Good RA, O’Reilly RJ, Transplantation for severe combined immunodeficiency with HLA-A,B,D,DR incompatible parental marrow cells fractionated by soybean agglutinin and sheep red blood cells. Blood 61, 341–348 (1983). [PubMed] [Google Scholar]
- 142.Kwon HS, Logan AC, Chhabra A, Pang WW, Czechowicz A, Tate K, Le A, Poyser J, Hollis R, Kelly BV, Kohn DB, Weissman IL, Prohaska SS, Shizuru JA, Anti-human CD117 antibody-mediated bone marrow niche clearance in nonhuman primates and humanized NSG mice. Blood 133, 2104–2108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Markert ML, Devlin BH, McCarthy EA, Thymus transplantation. Clin Immunol 135, 236–246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hershfield MS, Buckley RH, Greenberg ML, Melton AL, Schiff R, Hatem C, Kurtzberg J, Markert ML, Kobayashi RH, Kobayashi AL, et al. , Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N Engl J Med 316, 589–596 (1987). [DOI] [PubMed] [Google Scholar]
- 145.Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, Plant AS, Su HC, Allenspach EJ, Slatter M, Abinun M, Lilic D, Cunningham-Rundles C, Eckstein O, Olbrich P, Guillerman RP, Patel NC, Demirdag YY, Zerbe C, Freeman AF, Holland SM, Szabolcs P, Gennery A, Torgerson TR, Milner JD, Leiding JW, Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol 142, 1665–1669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, Hoshino A, Kobayashi M, Imai K, Takagi M, Dybedal I, Haddock JA, Sansom DM, Lucena JM, Seidl M, Schmitt-Graeff A, Reiser V, Emmerich F, Frede N, Bulashevska A, Salzer U, Schubert D, Hayakawa S, Okada S, Kanariou M, Kucuk ZY, Chapdelaine H, Petruzelkova L, Sumnik Z, Sediva A, Slatter M, Arkwright PD, Cant A, Lorenz HM, Giese T, Lougaris V, Plebani A, Price C, Sullivan KE, Moutschen M, Litzman J, Freiberger T, van de Veerdonk FL, Recher M, Albert MH, Hauck F, Seneviratne S, Pachlopnik Schmid J, Kolios A, Unglik G, Klemann C, Speckmann C, Ehl S, Leichtner A, Blumberg R, Franke A, Snapper S, Zeissig S, Cunningham-Rundles C, Giulino-Roth L, Elemento O, Duckers G, Niehues T, Fronkova E, Kanderova V, Platt CD, Chou J, Chatila TA, Geha R, McDermott E, Bunn S, Kurzai M, Schulz A, Alsina L, Casals F, Deya-Martinez A, Hambleton S, Kanegane H, Tasken K, Neth O, Grimbacher B, Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 142, 1932–1946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rao VK, Webster S, Dalm V, Sediva A, van Hagen PM, Holland S, Rosenzweig SD, Christ AD, Sloth B, Cabanski M, Joshi AD, de Buck S, Doucet J, Guerini D, Kalis C, Pylvaenaeinen I, Soldermann N, Kashyap A, Uzel G, Lenardo MJ, Patel DD, Lucas CL, Burkhart C, Effective “activated PI3Kdelta syndrome”-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood 130, 2307–2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kurolap A, Eshach-Adiv O, Hershkovitz T, Paperna T, Mory A, Oz-Levi D, Zohar Y, Mandel H, Chezar J, Azoulay D, Peleg S, Half EE, Yahalom V, Finkel L, Weissbrod O, Geiger D, Tabib A, Shaoul R, Magen D, Bonstein L, Mevorach D, Baris HN, Loss of CD55 in Eculizumab-Responsive Protein-Losing Enteropathy. N Engl J Med 377, 87–89 (2017). [DOI] [PubMed] [Google Scholar]
- 149.Hirschhorn R, Yang DR, Puck JM, Huie ML, Jiang CK, Kurlandsky LE, Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet 13, 290–295 (1996). [DOI] [PubMed] [Google Scholar]
- 150.Bousso P, Wahn V, Douagi I, Horneff G, Pannetier C, Le Deist F, Zepp F, Niehues T, Kourilsky P, Fischer A, de Saint Basile G, Diversity, functionality, and stability of the T cell repertoire derived in vivo from a single human T cell precursor. Proc Natl Acad Sci U S A 97, 274–278 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klein H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF, T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 270, 475–480 (1995). [DOI] [PubMed] [Google Scholar]
- 152.Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, Mazzolari E, Maggioni D, Rossi C, Servida P, Ugazio AG, Mavilio F, Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science 270, 470–475 (1995). [DOI] [PubMed] [Google Scholar]
- 153.Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, Tabucchi A, Carlucci F, Eibl M, Aker M, Slavin S, Al-Mousa H, Al Ghonaium A, Ferster A, Duppenthaler A, Notarangelo L, Wintergerst U, Buckley RH, Bregni M, Marktel S, Valsecchi MG, Rossi P, Ciceri F, Miniero R, Bordignon C, Roncarolo MG, Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 360, 447–458 (2009). [DOI] [PubMed] [Google Scholar]
- 154.Aiuti A, Roncarolo MG, Naldini L, Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med 9, 737–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, Bousso P, Deist FL, Fischer A, Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288, 669–672 (2000). [DOI] [PubMed] [Google Scholar]
- 156.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M, Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118, 3132–3142 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, Modlich U, Beier R, Gohring G, Steinemann D, Fronza R, Ball CR, Haemmerle R, Naundorf S, Kuhlcke K, Rose M, Fraser C, Mathias L, Ferrari R, Abboud MR, Al-Herz W, Kondratenko I, Marodi L, Glimm H, Schlegelberger B, Schambach A, Albert MH, Schmidt M, von Kalle C, Klein C, Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Sci Transl Med 6, 227ra233 (2014). [DOI] [PubMed] [Google Scholar]
- 158.Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, Bleesing J, Blondeau J, de Boer H, Buckland KF, Caccavelli L, Cros G, De Oliveira S, Fernandez KS, Guo D, Harris CE, Hopkins G, Lehmann LE, Lim A, London WB, van der Loo JC, Malani N, Male F, Malik P, Marinovic MA, McNicol AM, Moshous D, Neven B, Oleastro M, Picard C, Ritz J, Rivat C, Schambach A, Shaw KL, Sherman EA, Silberstein LE, Six E, Touzot F, Tsytsykova A, Xu-Bayford J, Baum C, Bushman FD, Fischer A, Kohn DB, Filipovich AH, Notarangelo LD, Cavazzana M, Williams DA, Thrasher AJ, A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 371, 1407–1417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.De Ravin SS, Wu X, Moir S, Anaya-O’Brien S, Kwatemaa N, Littel P, Theobald N, Choi U, Su L, Marquesen M, Hilligoss D, Lee J, Buckner CM, Zarember KA, O’Connor G, McVicar D, Kuhns D, Throm RE, Zhou S, Notarangelo LD, Hanson IC, Cowan MJ, Kang E, Hadigan C, Meagher M, Gray JT, Sorrentino BP, Malech HL, Kardava L, Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med 8, 335ra357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, Ma Z, Condori J, Dowdy J, Triplett B, Li C, Maron G, Aldave Becerra JC, Church JA, Dokmeci E, Love JT, da Matta Ain AC, van der Watt H, Tang X, Janssen W, Ryu BY, De Ravin SS, Weiss MJ, Youngblood B, Long-Boyle JR, Gottschalk S, Meagher MM, Malech HL, Puck JM, Cowan MJ, Sorrentino BP, Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N Engl J Med 380, 1525–1534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Porteus MH, A New Class of Medicines through DNA Editing. N Engl J Med 380, 947–959 (2019). [DOI] [PubMed] [Google Scholar]
- 162.Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE, Lee C, Bao G, Kildebeck EJ, Punjya N, Sindhu C, Inlay MA, Saxena N, DeRavin SS, Malech H, Roncarolo MG, Weinberg KI, Porteus MH, Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun 10, 1634 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kuo CY, Long JD, Campo-Fernandez B, de Oliveira S, Cooper AR, Romero Z, Hoban MD, Joglekar AV, Lill GR, Kaufman ML, Fitz-Gibbon S, Wang X, Hollis RP, Kohn DB, Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cell Rep 23, 2606–2616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, Passerini L, Narula M, Sheikali A, Lee CM, Bao G, Bauer CS, Miller HK, Garcia-Lloret M, Butte MJ, Bertaina A, Shah A, Pavel-Dinu M, Hendel A, Porteus M, Roncarolo MG, Bacchetta R. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv 6, eaaz0571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fischer A, Dewatripont M, Goldman M, Benefit Corporation: a path to affordable gene therapies? Nat Med 25, 1813–1814 (2019). [DOI] [PubMed] [Google Scholar]