

Abstract
The difficulty to synthesize and purify the Amyloid β (Aβ) peptide, combined with its high aggregation propensity and low solubility at physiological conditions, leads to a wide variety of experimental results from kinetic assays to biological activity. Thus, it becomes challenging to reproduce outcomes, which limits the ability to rely on reported results as the foundation for new research. This article examines variability of the Aβ peptide from different sources, comparing purity, and oligomer and fibril formation propensity side by side. The results highlight the importance of performing rigorous controls so meaningful biophysical, biochemical, and neurobiological results can be obtained to improve our understanding on Aβ.
Graphical Abstract

Know your source. Amyloid β (Aβ) has been the target of multiple (and failed) therapeutic approaches to prevent Alzheimer’s disease. Here, we summarize the challenges in data reproducibility of Aβ, and we evaluate the impact that different sources and methodologies can have in experimental results.
One of the main characteristic features in Alzheimer’s disease (AD) is the presence of amyloid plaques in the brain, as discovered by Dr. Alois Alzheimer in 1906 when examining the postmortem brain of Auguste Deter, a patient who suffered from dementia and memory loss.[1] However, it was not until 78 years later when the amyloid β (Aβ) peptide, the predominant component of the amyloid plaques, was first identified and characterized by Glenner and Wong in 1984.[2] Advances in the field have been made since then: In 1991, Cotman et al.[3] linked in vitro Aβ aggregation to neurotoxicity, and the amyloid cascade hypothesis was postulated as the driving force for AD model (Hardy & Higgins, Selkoe),[4,5] stating that the aggregation process of Aβ into fibrils was the seminal etiological agent of the disease. At the same time, it was observed that Aβ was produced by normal metabolic activity on cells,[6] showing that the production of Aβ is not only associated to AD patients exclusively. In fact, reports have shown that picomolar concentrations of Aβ have positive long-term potentiation (LTP) modulation effects.[7]
Since the amyloid cascade hypothesis was first postulated, the quantity of manuscripts published on Aβ has been exponentially increasing (Figure 1A). While initial research focused on amyloid fibrils and plaques as the main disease culprit, the field has evolved during the past two decades towards the consensus that soluble aggregation intermediates, commonly referred to as oligomers, represent the most neurotoxic agents.[8–10] This represented a shift towards a more complex mechanism, where a vast number of oligomers may aggregate in several pathways, generating an equilibrium of multiple transient conformations and different molecular sizes,[11] with each oligomerization state potentially contributing to neurodegeneration to a different degree.[12,13] Aβ oligomers have been reported to adopt ring,[14,15] annular,[16] spherical,[17] or β-barrel[18] structures, and the number of Aβ subunits in these metastable intermediates is also important. For example, Aβ dimers from AD brains are thought to be the minimal synaptotoxic oligomeric species,[19,20] and dodecameric units termed as Aβ*56 are associated to memory impairment in 3xTg-AD transgenic mice.[21] Aβ protofibrils, defined as short, curved fibrils with molecular weight >100,00 kDa and <200 nm length,[22] also seem to have an important role in patients carrying the “Arctic” familial mutation.[23] These oligomeric intermediates are thought to assemble in different pathways, and are often termed as “on-pathway” and “off-pathway” depending whether or not they lead to fibril formation.[24,25] The mechanism by which they aggregate into fibrils also depends on the nucleation type, with elongation, primary nucleation, and secondary nucleation as principal mechanisms of fibril formation,[26] being the latter considered to be the most relevant for the proliferation of neurotoxic species.[27] It is therefore not surprising to observe structural variability in published high-resolution Aβ40[28] and Aβ42 fibrils.[29–34] Recent findings even suggest that Aβ fibrils could even act as protective reservoirs against toxic Aβ oligomers[35,36] and enhancing Aβ aggregation was established as a strategy to reduce Aβ toxic effects.[37–39]
Figure 1.
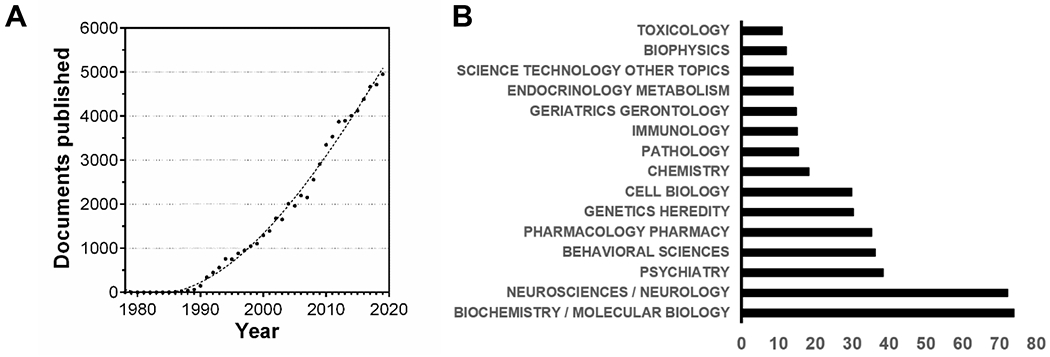
Document search using “amyloid beta” as keywords. Search did not exclude results related to amyloid precursor protein (APP). (A) Documents published per year using “amyloid beta” as keywords. Datapoints are fit with a cubic polynomial curve (R2 = 0.994) (B) % of 69,010 documents organized by research areas. Note that some areas may overlap. Source: Web of Science
As of February 29th, 2020, a search in Web of Science using the words “amyloid beta” finds 69,010 documents, “amyloid beta aggregation” results in 12,715 documents, and “amyloid beta toxicity” finds 6,684 documents. With such a vast amount of literature on a very diverse spectrum of research areas (Figure 1B), one should expect that the biophysical, biochemical, and biological properties of the Aβ peptide should be well-defined, and therefore adopted by new researchers to build new hypotheses and improve the understanding of Aβ aggregation and toxic actions. However, an insufficient level of rigor often impairs the possibilities of using this new knowledge to produce significant contributions and generate reproducible results that could be compared across different laboratories.[40,41] Although efforts have been made to classify the multiple aggregation intermediates and structures that Aβ can generate, i.e. by Glabe,[13] strikingly, as summarized by Teplow,[40] the Aβ field even suffers from not having a precise and systematic nomenclature, where usual terms such as oligomers, protofibrils, on-pathway species, or off-pathway species, among others, can have a different meaning depending on who writes about it.
Sources of Aβ
While Aβ is known for its problems with batch-to-batch reproducibility,[42,43] very little of this aspect is shown in published manuscripts, where sometimes, the employed Aβ material is ambiguously defined or insufficiently characterized. Obtaining Aβ from commercial sources might lead to the assumption that the peptide should have the expected physicochemical and biological properties, such as propensity to form oligomers and fibrils, and the ability to elicit toxic actions in cellular cultures and neuronal networks. However, because not all laboratories can check the ability of the peptide to form oligomers and fibrils before starting the experiments, assumptions are made about its properties based on reported literature. This fact is exemplified in Figure 2, where HPLC and mass spectrometry traces from two different commercial sources of Aβ42 are shown, and compared with peptide from our laboratory. The traces obtained for commercial Aβ -sold as >95% purity-, analyzed under the exact same conditions of our laboratory synthesized and characterized Aβ[38] is evidentially different, with Aβ from commercial source 1 displaying substantially lower purity by HPLC and mass spectrometry. Additionally, Thioflavin T (ThT) kinetic assays and photo-induced crosslinking experiments were performed on these samples to show how peptide purity and quality can also influence aggregation kinetics and oligomerization propensity (Fig. 3). While Aβ42 from commercial source 2 is comparable with our laboratory Aβ42 preparation, the Aβ42 sample obtained from commercial source 1 shows minimal aggregation propensity when monitored by ThT (Fig. 3A–C). This difference is also reflected in the ability of the peptides to form low-order oligomers when subjected to photo-induced crosslinking, where laboratory and commercial Aβ42 sources show oligomerization profiles similar to previously described by other laboratories,[10] but with commercial source 1 displaying almost no oligomer formation. It is therefore not surprising that different commercial sources of Aβ might lead to morphologically different Aβ structures,[14] and ultimately to distinct peptide properties and experiment conclusions that might be limited to a single Aβ preparation. Thus, it is advisable to check that Aβ samples pass baseline quality standards before starting to use it for experiments, such as ensuring peptide purity and its ability to aggregate into oligomers and fibrils. An additional factor to consider is the use of synthetic or recombinant sources of Aβ, where studies have shown that recombinant sources are more neurotoxic and aggregate faster,[44,45] a difference that could be arising from the distinct structures that the peptides might adopt or due to the nature of the contaminants and impurities obtained from each source.
Figure 2.

(A) HPLC traces of different sources of Aβ42. Commercial Aβ42 source 1: AAPPTec. Commercial Aβ42 source 2: AnaSpec. (B) Linear trap quadrupole (LTQ) mass spectra of different sources of Aβ42, analyzed at the same sample concentration (1mg/mL). Star marks target Aβ42 peak.
Figure 3.

Aggregation and oligomerization propensity of distinct Aβ42 sources. Commercial Aβ42 source 1: AAPPTec. Commercial Aβ42 source 2: AnaSpec. (A-C) ThT-monitored aggregation kinetics at 37 ºC and continuous shaking of different Aβ42 sources. Peptide concentration and ThT concentration was 20 μM in all cases. (D) Oligomerization propensity analyzed by PICUP at 20 μM concentration of each Aβ42 source. After crosslinking, samples were subjected to SDS-PAGE and developed by silver staining.
Variability on aggregation and toxicity profiles.
Even at trace levels, impurities can have a profound effect on the aggregation and toxicity properties of Aβ.[45] These impurities may not only originate from synthesis or expression, but also from widely adopted Aβ pre-treatments. For example, acidic pretreatment with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) or trifluoroacetic acid (TFA), commonly employed to de-aggregate Aβ samples, can lead to increased toxicity of Aβ,[46] and it also can modify the aggregation propensity, stability, and morphology of the peptide.[47] In a similar way, high-pH pre-treatment like NaOH can lead to amino acid epimerization,[48] which can also drastically change the aggregation and biological activity of Aβ.[49–51] With such many variables, it is not surprising to find reports with fibril formation kinetic assays varying from minutes to days, and cytotoxicity values ranging from low-nanomolar to micromolar ranges in model cell lines. In vivo studies in rats show that Aβ oligomers can have negative hippocampal LTP modulation effects at nanomolar concentrations.[52] However, Aβ concentrations used in in vitro cytotoxicity experiments are typically within the micromolar range.[37,53,54] Furthermore, Aβ has different cytotoxic activity depending on the cell type,[55] cell confluence,[56] and cell differentiation.[57] Because Aβ binding to cellular membranes is thought to play a critical role in Aβ aggregation and toxicity,[58,59] these disparities could be a consequence of the distinct interaction of Aβ with cells having different membrane lipid composition. For example, while specific interactions between Aβ and phosphatidylethanolamine monolayers are limited,[60] incorporation of polar and negatively charged phospholipids leads to increased electrostatic interactions that can template Aβ aggregation by inducing β sheet conformation.[61,62] The nature of Aβ oligomers is also important, since non-fibrillary oligomers have been observed to cause macroscale membrane deformations in model lipid monolayers, while fibrillary oligomers induce membrane thinning and instability.[63] These are significant factors to consider since membrane poration and formation of calcium channels are thought to play a relevant role in Aβ toxicity.[64,65] Thus, Aβ reproducibility and variability issues do not only have a direct impact on the results itself, but it can also be one of the reasons why drugs against Aβ are not as efficient when tested in clinical trials.[66] As of July 2019, there were 38 amyloid-related ongoing trials,[67] with previous 23 amyloid-related drugs discontinued (source: Alzforum), and recent news of 2 new failures: Phase II/III study of solanezumab (Lilly) and gantenerumab (Roche/Genentech).
Improving consistency and reproducibility.
Aβ research is frequently conducted in phenomenological fashion, describing observations of Aβ behavior without assessing robustness or reproducibility of the work; do the observed trends endure through different Aβ batches and show the same results by orthogonal techniques? In many cases of published work, the Aβ source either remains constant for the whole study (e.g. Aβ is always used from the same seller), or it is unspecified (e.g. if synthetic Aβ was used, did it all come from the same synthetic batch or from different batches?). While using a unique source could provide meaningful and valid results if the system is well-characterized, it might not ensure that other laboratories would be able to reproduce them. How does one then make sure that the obtained results are reproducible? From the commercial and the synthetic perspective, reproducing key results with Aβ from more than one seller, or with more than one synthetic batch, might ensure that results are reliable and reproducible by other laboratories. While there will always be some batch-to-batch variations, key and distinct findings must be consistent between batches.[43,51] This is not uncommon in other fields, for example, in medicinal chemistry, where “hits” obtained from screening assays are typically validated in a multifunctional process.[68]
Conclusions
To date, almost 70,000 documents have been published on Aβ research, with this number increasing year by year. However, the intricate properties of Aβ and the varied methodologies and sources commonly used to work with this peptide make it challenging to obtain reliable and reproducible results. Here, our goal was to emphasize the influence that different Aβ sources can have over experiments’ outcomes, which can ultimately lead to unreproducible or unrepresentative results. Thus, it becomes very important to ensure that experiments are performed with material that passes certain minimal quality controls. We propose that this baseline should include HPLC, mass spectrometry, and aggregation and oligomerization propensity assays, as well as validating the results by using multiple Aβ batches. Batch-to-batch variations should be regarded as an intrinsic property of Aβ rather than an inconsistency, and reporting these results as part of the manuscript should also become a common practice so researchers know what to expect when attempting to reproduce experiments. This increased diligence will provide with knowledge and results that other laboratories might find easier to adopt, and would help to increase the productivity and efficiency on Aβ research. We believe that more discussion is needed on this topic and hope this manuscript will stimulate such.
EXPERIMENTAL SECTION
Materials.
Laboratory Aβ42 was synthesized by solid-phase Fmoc chemistry on a Liberty Blue CEM peptide synthesizer assisted by a CEM discovery microwave, and purified by reverse-phase HPLC as previously described.[50] Commercial sources of Aβ42 were dissolved in 20 mM NH4OH aqueous solution, aliquoted, and lyophilized prior to the experiments.
Thioflavin T (ThT) assay.
Aβ42 samples were dissolved in 20 mM NaOH (not exceeding 3% volume of NaOH in final dilution amount) and diluted to 20 μM concentration with pH 7.4 1x PBS with 20 μM ThT. Aβ42 solutions were placed on a black, clear bottom 96-well plate and placed in a Molecular Device Gemini EM fluorescence plate reader to measure fluorescence (λem = 444 nm, λex = 485 nm). Aβ42-ThT solutions were 37 °C with continuous shaking and datapoints obtained every 5 minutes.
Photo-Induced crosslinking (PICUP) and SDS-PAGE.
PICUP experiments were performed as previously described.[50] Briefly, lyophilized Aβ42 samples were dissolved in 20 mM NaOH and diluted in pH 7.4 1x PBS. For the crosslinking reaction, 4 μL of 1 mM [Ru(bipy)3]2+ and 4 μL of 20 mM ammonium persulfate were added to 32 μL of the Aβ42 solutions, and irradiated with light for 1.2 seconds. The solutions were then quenched with 40 μL of loading buffer with 5% 2-mercaptoethanol. Finally, crosslinked samples were run on 12% tris-tricine polyacrylamide gels and developed by silver staining.
ACKNOWLEDGMENT
Authors thank Mr. David B. Teplow, Ms. Ariel Kuhn and Mr. Ka Chan for helpful discussions during manuscript preparation stage. Authors acknowledge AnaSpec Inc. for providing Aβ samples (commercial Aβ42 source 2). J.A.R. thanks NIH for funding (R21AG058074). A.R.F. thanks NIH for funding (2R25GM058903-20 IMSD).
Biographies

Alejandro Rodriguez Foley obtained his B.S. in Chemistry at University of Sevilla (Spain). As an undergraduate and predoctoral student, he received training in organic synthesis from Prof. Dr. Alain Burger (Université de Nice Sophia Antipolis) Prof. Dr. J. Enrique Oltra (University of Granada), and Dr. Kirk R. Gustafson (NCI-NIH). Alejandro is currently a PhD candidate in Organic Chemistry at the University of California Santa Cruz in the laboratory of Prof. Dr. Jevgenij A. Raskatov. His research lies at the interface of chemistry and biology, specifically focused on studying amyloid β aggregation and neurotoxicity through biochemical, biophysical, and biological approaches. His scientific interests include peptide and protein chemistry and the application of fundamental chemistry concepts to biomedical research.

Prof. Jevgenij Raskatov received his undergraduate degree at the Heidelberg University (2001-2006), where he performed DFT-based studies of organometallic catalysis under guidance of Prof. Guenter Helmchen and was elected Fellow of the “Studienstiftung des Deutschen Volkes” in recognition of his academic achievements. He subsequently moved to Oxford as a Kekulé fellow to conduct experimental studies of chiral ion pairing in transition metal catalysis with Prof. John Brown, FRS (2006-2009). He subsequently received a Feodor von Lynen postdoctoral fellowship from the Alexander von Humboldt foundation to study DNA-binding Py-Im polyamides with Prof. Peter Dervan (2009-2014). The Raskatov lab at UCSC (2014-) uses a Chemical Neuroscience approach that combines peptide synthesis with an array of biological techniques, using chirality as a unique probe of structure and function of Alzheimer’s Amyloid β. We recently developed Aβ Chiral Inactivation (Aβ-CI) as a molecular approach to convert toxic natural (“L”; “all-L”) Aβ oligomers to non-toxic fibrils using mirror-image (“D”; “all-D”) Aβ as the structural converter. The experiments using mirror-image Aβ were recognized
REFERENCES
- [1].Alzheimer A, Neurol. Zentralblatt 1906, 25, 1134 [Google Scholar]
- [2].Glenner GG, Wong CW, Biochem. Biophys. Res. Commun 1984, 120, 885–890. [DOI] [PubMed] [Google Scholar]
- [3].Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW, Brain Res. 1991, 563, 311–314. [DOI] [PubMed] [Google Scholar]
- [4].Hardy JA, Higgins GA, Science 1992, 256, 184–185. [DOI] [PubMed] [Google Scholar]
- [5].Selkoe DJ, Neuron 1991, 6, 487–498 [DOI] [PubMed] [Google Scholar]
- [6].Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ, Nature 1992, 359, 322–325. [DOI] [PubMed] [Google Scholar]
- [7].Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O, J. Neurosci 2008, 28, 14537–14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG, Science 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- [9].Haass C, Selkoe DJ, Nat. Rev. Mol. Cell Biol 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- [10].Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB, Proc. Natl. Acad. Sci. U. S. A 2003, 100, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Raskatov JA, Teplow DB, Sci. Rep 2017, 7, 12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bucciantini M, Giannoni E, Chiti F, Baroni F, Taddei N, Ramponi G, Dobson CM, Stefani M, Nature 2002, 416, 507–511. [DOI] [PubMed] [Google Scholar]
- [13].Glabe CG, J. Biol. Chem 2008, 283, 29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Suvorina MY, Selivanova OM, Grigorashvili EI, Nikulin AD, Marchenkov VV, Surin AK, Galzitskaya OV, J. Alzheimer’s Dis 2015, 47, 583–593. [DOI] [PubMed] [Google Scholar]
- [15].Galzitskaya OV, Surin AK, Glyakina AV, Rogachevsky VV, Selivanova OM, Alzheimer’s Dis J. Reports 2018, 2, 181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT, Nature 2002, 418, 291. [DOI] [PubMed] [Google Scholar]
- [17].Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K, Proc. Natl. Acad. Sci. U. S. A 2003, 100, 6370–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe CG, Science 2012, 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shankar GM, Li S. Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ, Nat. Med. 2008, 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ, Proc. Natl. Acad. Sci. U. S. A 2011, 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lesné S, Ming TK, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH, Nature 2006, 440, 352–357. [DOI] [PubMed] [Google Scholar]
- [22].Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB, J. Biol. Chem 1999, 274, 25945–25952. [DOI] [PubMed] [Google Scholar]
- [23].Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG,Naslund J, Lanfelt L, Nat. Neurosci 2001, 4, 887–893. [DOI] [PubMed] [Google Scholar]
- [24].Soto C, Pritzkow S, Nat. Neurosci 2018, 21, 1332–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schnabel J, Nature 2011, 475, S12–S14 [DOI] [PubMed] [Google Scholar]
- [26].Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SIA, Dobson CM, Linse S, Knowles TPJ, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].White DA, Hellstrand E, Cohen SIA, Linse S, Luheshi LM, Rajah L, Knowles TPJ, Vendruscolo M, Otzen DE, Dobson CM, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 9768–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tycko R, Neuron 2015, 86, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 17342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y, Nat. Struct. Mol. Biol 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG, J. Am. Chem. Soc 2016, 138, 9663–9674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wälti MA, Böckmann A, Meier BH, Arai H, Ravotti F, Glabe CG, Güntert P, Wall JS, Riek R, Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, Tusche M, Lopez-Iglesias C, Hoyer W, Heise H, Willbold D, Schroder GF, Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ, Jucker M, Fändrich M, Nat. Commun 2019, 10, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Treusch S, Cyr DM, Lindquist S, Cell Cycle 2009, 8, 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Selkoe DJ, Hardy J, EMBO Mol. Med 2016, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez Del Amo JM, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Gunther S, Walsh DM, Wanker Nat EE. Chem. Biol 2012, 8, 93–101. [DOI] [PubMed] [Google Scholar]
- [38].Dutta S, Foley AR, Warner CJA, Zhang X, Rolandi M, Abrams B, Raskatov JA, Angew. Chemie - Int. Ed 2017, 56, 11506–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Limbocker R, Chia S, Ruggeri FS, Perni M, Cascella R, Heller GT, Meisl G, Mannini B, Habchi J, Michaels TCT, Challa PK, Ahn M, Casford ST, Fernando N, Xu CK, Kloss ND, Cohen SIA, Kumita JR, Cecchi C, Zasloff M, Linse S, Knowles TPJ, Chiti F, Vendruscolo M, Dobson CM, Nat. Commun 2019, 10, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Teplow DB, Alzheimer’s Res. Ther 2013, 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nat. Neurosci 2011, 14, 399. [DOI] [PubMed] [Google Scholar]
- [42].Soto C, Castaño EM, Asok Kumar R, Beavis RC, Frangione B, Neurosci. Lett 1995, 200, 105–108. [DOI] [PubMed] [Google Scholar]
- [43].Teplow DB, Methods Enzymol. 2006, 413, 20–33. [DOI] [PubMed] [Google Scholar]
- [44].Finder VH, Vodopivec I, Nitsch RM, Glockshuber R, J. Mol. Biol 2010, 396, 9–18. [DOI] [PubMed] [Google Scholar]
- [45].Adams DJ, Nemkov TG, Mayer JP, Old WM, Stowell MHB, PLoS One 2017, 12, e0182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lioudyno MI, Broccio M, Sokolov Y, Rasool S, Wu J, Alkire MT, Liu V, Kozak JA, Dennison PR, Glabe CG, Losche M, Hall JE, PLoS One 2012, 7, e35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nichols MR, Moss MA, Reed DK, Cratic-McDaniel S, Hoh JH, Rosenberry TL, J. Biol. Chem 2005, 280, 2471–2480. [DOI] [PubMed] [Google Scholar]
- [48].Liardon R, Hurrell RF, Agric J. Food Chem. 1983, 31, 432–437. [Google Scholar]
- [49].Tomiyama T, Asano S, Furiya Y, Shirasawa T, Endo N, Mori H, J. Biol. Chem 1994, 269, 10205–10208. [PubMed] [Google Scholar]
- [50].Warner CJA, Dutta S, Foley AR, Raskatov JA, Chem. - A Eur. J 2016, 22, 11967–11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Foley AR, Finn TS, Kung T, Hatami A, Lee H-W, Jia M, Rolandi M, Raskatov JA, ACS Chem. Neurosci 2019, 10, 3880–3887. [DOI] [PubMed] [Google Scholar]
- [52].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ, Nature 2002, 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [53].Ono K, Condron MM, Teplow DB, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Breaken D, Callewaert G, Bartic C, D’Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B, EMBO J. 2010, 29, 3408–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang Y, McLaughlin R, Goodyer C, LeBlanc A, J. Cell Biol. 2002, 156, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Balcells M, Wallins JS, Edelman ER, Neurosci. Lett 2008, 441, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Krishtal J, Bragina O, Metsla K, Palumaa P, Tõugu V, PLoS One 2017, 12, e0186636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yamaguchi H, Maat-Schieman MLC, Van Duinen SG, Prins FA, Neeskens P, Natté R, Roos RAC, J. Neuropathol. Exp. Neurol 2000, 59, 723–732. [DOI] [PubMed] [Google Scholar]
- [59].Jin S, Kedia N, Illes-Toth E, Haralampiev I, Prisner S, Herrmann A, Wanker EE, Bieschke J, J. Biol. Chem 2016, 291, 19590–19606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Maltseva E, Brezesinski G, ChemPhysChem 2004, 5, 1185–1190 [DOI] [PubMed] [Google Scholar]
- [61].Maltseva E, Kerth A, Blume A, Möhwald H, Brezesinski G, ChemBioChem 2005, 6, 1817–1824. [DOI] [PubMed] [Google Scholar]
- [62].Chi EY, Ege C, Winans A, Majewski J, Wu G, Kjaer K, Lee KYC, Proteins Struct. Funct. Genet 2008, 72, 1–24. [DOI] [PubMed] [Google Scholar]
- [63].Vander Zanden CM, Wampler L, Bowers I, Watkins EB, Majewski J, Chi EY, Langmuir 2019, 35, 16024–16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Arispe N, Rojas E, Pollard HB, Proc. Natl. Acad. Sci. U. S. A 1993, 90, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG, J. Biol. Chem 2004, 279, 46363–46366. [DOI] [PubMed] [Google Scholar]
- [66].Doig AJ, Del Castillo-Frias MP, Berthoumieu O, Tarus B, Nasica-Labouze J, Sterpone F, Nguyen PH, Hooper NM, Faller P, Derreumaux P, ACS Chem. Neurosci 2017, 8, 1435–1437. [DOI] [PubMed] [Google Scholar]
- [67].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K, Alzheimer’s Dement. Transl. Res. Clin. Interv 2019, 5, 272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hughes JP, Rees SS, Kalindjian SB, Philpott KL, Br. J. Pharmacol 2011, 162, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]