

Abstract
Brucellosis, caused by a number of Brucella species, remains the most prevalent zoonotic disease worldwide. Brucella establish chronic infections within host macrophages despite triggering cytosolic innate immune sensors, including Stimulator of Interferon Genes (STING), which potentially limit infection. In this study, STING was required for control of chronic Brucella infection in vivo. However, early during infection, Brucella down-regulated STING mRNA and protein. Down-regulation occurred post-transcriptionally, required live bacteria, the Brucella type IV secretion system, and was independent of host IRE1-RNase activity. STING suppression occurred in MyD88-/- macrophages and was not induced by Toll-like receptor agonists or purified Brucella lipopolysaccharide (LPS). Rather, Brucella induced a STING-targeting microRNA, miR-24-2, in a type IV secretion system-dependent manner. Furthermore, STING downregulation was inhibited by miR-24 anti-miRs and in Mirn23a locus-deficient macrophages. Failure to suppress STING expression in Mirn23a-/- macrophages correlated with diminished Brucella replication, and was rescued by exogenous miR-24. Mirn23a-/- mice were also more resistant to splenic colonization one week post infection. Anti-miR-24 potently suppressed replication in wild type, but much less in STING-/- macrophages, suggesting most of the impact of miR-24 induction on replication occurred via STING suppression. In summary, Brucella sabotages cytosolic surveillance by miR-24-dependent suppression of STING expression; post-STING activation “damage control” via targeted STING destruction may enable establishment of chronic infection.
Author summary
Cytosolic pattern recognition receptors, such as the nucleotide-activated STING molecule, play a critical role in the innate immune system by detecting the presence of intracellular invaders. Brucella bacterial species establish chronic infections in macrophages despite initially activating STING. STING participates in the control of Brucella infection, as mice or cells lacking STING show a higher burden of Brucella infection. However, we have found that early following infection, Brucella upregulates a microRNA, miR-24, that targets the STING messenger RNA, resulting in lower STING levels. Dead bacteria or bacteria lacking a functional type IV secretion system were defective at upregulating miR-24 and STING suppression, suggesting an active bacteria-driven process. Failure to upregulate miR-24 and suppress STING greatly compromised the capacity of Brucella to replicate inside macrophages and in mice. Thus, although Brucella initially activate STING during infection, the ensuing STING downregulation serves as a “damage control” mechanism, enabling intracellular infection. Viruses have long been known to target immune sensors such as STING. Our results indicate that intracellular bacterial pathogens also directly target innate immune receptors to enhance their infectious success.
Introduction
Brucella spp. are Gram-negative, facultative intracellular α-proteobacteria which cause the zoonotic disease brucellosis [1,2]. Human brucellosis is characterized by an acute undulating fever accompanied by flu-like myalgias before developing into a chronic disease, with long-term pathologies such as sacroiliitis, arthritis, liver damage, meningitis, and endocarditis [3]. Brucellosis in animals often causes orchitis and sterility in males and spontaneous abortions in females, leading to profound economic loss worldwide [4]. During chronic infection, Brucella live and replicate within macrophages and other phagocytes. This intracellular localization renders the organism refractory to even prolonged multiple antibiotic treatments, and relapses occur in 5–10% of cases [3]. In the U.S., brucellosis has been largely controlled through vaccination of livestock with live attenuated strains, though outbreaks still occur [5–7]. Currently, no safe and effective human vaccine exists. The mechanism(s) involved in supporting the intracellular persistence of Brucella remain unclear.
Innate immune responses form the first line of defense against bacterial pathogens. However, Brucella express multiple ‘atypical’ virulence factors, which stymie innate defenses. For example, Brucella spp. resist complement activation and express a weakly endotoxic “smooth” lipopolysaccharide that is a poor agonist for the innate immune sensor Toll-like receptor 4 [8]. Despite sequestration in membranous compartments, Brucella trigger cytosolic innate immune sensors including various inflammasomes and the Stimulator of Interferon Genes (STING) [9–12]. STING resides in the endoplasmic reticulum membrane and upon activation by bacterial cyclic-di-nucleotides or cyclic GMP-AMP (c-GAMP), STING translocates to peri-nuclear clusters where it co-localizes with and activates TANK binding kinase I (TBK1), which in turn phosphorylates the IFN-β regulatory transcription factor IRF3 [13,14]. In addition to Type I interferon induction, STING is essential for optimal induction of NF-κB-dependent pro-inflammatory cytokines and other host defense genes, and regulates autophagy [15]. Evidence from the cancer literature also suggests STING critically supports effective CD8+ T cell adaptive immune responses [16]. Previously, we have shown that STING is required for Type I interferon production in response to infection with Brucella abortus, and that STING contributes to control of B. abortus infection at 72 hours in vitro [9,17].
Here, we report that STING is critical for the control of acute and chronic Brucella infection in vivo. However, early during infection, Brucella down-regulate STING (Tmem173) mRNA expression and protein. Concurrently with STING suppression, Brucella induce a STING-targeting microRNA miR-24. Inhibition by anti-miR-24 or genetic deficiency of miR-24-2 leads to a significant increase in STING expression as well as augmented IFN-β production in macrophages. Inability to induce miR-24 and downregulate STING compromised Brucella survival in macrophages and in mice. These results suggest that Brucella mitigates the cost of innate immune activation by miR-24-dependent targeting of STING expression.
Results
STING is required for chronic control of Brucella in vivo
In previous studies, we showed that STING is required for control of Brucella replication in vitro from 24–72 hours [17,18]. We confirmed that by 24h, STING (Tmem173)-/- macrophages displayed significantly increased Brucella infection (Fig 1A). Recently, we had also shown that STING is required for control of Brucella infection in mice at 1 and 3 weeks [18]. To confirm these results and evaluate the role of STING in longer-term chronic Brucella infection [19], wild type C57BL/6 and STING-/- mice were infected with wild-type S2308 Brucella abortus for 1, 3 and 6 weeks. Splenic colony forming units (CFU) showed an approximately two-log difference between STING-/- mice and age-matched control C57BL/6 mice at 3 weeks and ~1.5 log difference at 6 weeks (Fig 1B). These data indicate that STING critically participates in the control of chronic Brucella infection in vivo.
Fig 1. STING is required for control of acute Brucella infection in vitro, and acute and chronic infection in vivo.
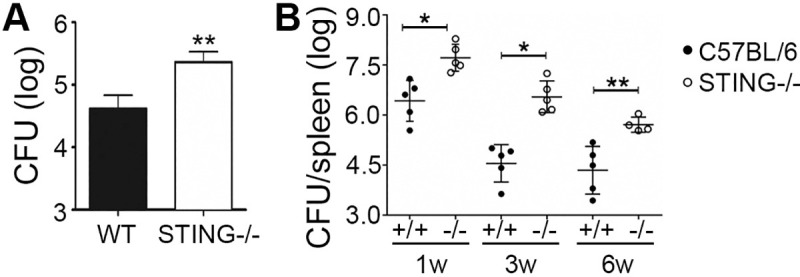
A) Bone marrow derived macrophages from wild type C57BL/6 control (WT) or STING-/- mice were infected with 10 multiplicity of infection (MOI) B. abortus for 24h prior to enumeration of colony forming units (CFU). Error bars denote triplicate determinations. B) Wild-type C57BL/6 (black circles, +/+) and STING-/- mice (open circles, -/-) were infected for 1, 3 or 6 weeks with 106 CFU Brucella abortus 2308 and splenocyte CFUs determined. Circles represent individual mice with 5 mice per group except the STING-/- from week 6 (4 mice). Bars denote median CFU/group. Results in (A) and (B) are representative of 3 independent experiments.
Brucella infection suppresses STING expression independently of IRE1 endonuclease activity and requires live bacteria and Type IV secretion
Given the requirement for STING in the control of chronic infection, it was surprising to note significant STING (Tmem173) mRNA down-regulation in bone marrow-derived macrophages (BMDMs) infected with wild type 16M Brucella melitensis at 24h (published RNAseq data set in [17]). To confirm the RNAseq data, and determine whether other Brucella species down-regulate STING, v-raf/v-myc immortalized murine bone marrow-derived macrophages [20] were uninfected or infected with different Brucella species for 24 hours and STING (Tmem173) mRNA levels assessed via RT-qPCR (Fig 2A). B. melitensis, B. abortus and B. suis are human pathogens and primarily infect ruminants, cattle and swine, respectively. B. neotomae has been isolated from wood rats and voles but also has been isolated in human neurobrucellosis [21]. The four species of Brucella significantly down-regulated STING mRNA compared to uninfected macrophages. STING protein levels also decreased in cells infected for 24 hours with Brucella (Fig 2B). In macrophages infected with B. melitensis, Tmem173 mRNA down-regulation was evident by 4h post-infection (Fig 2C).
Fig 2. Brucella suppresses STING expression.
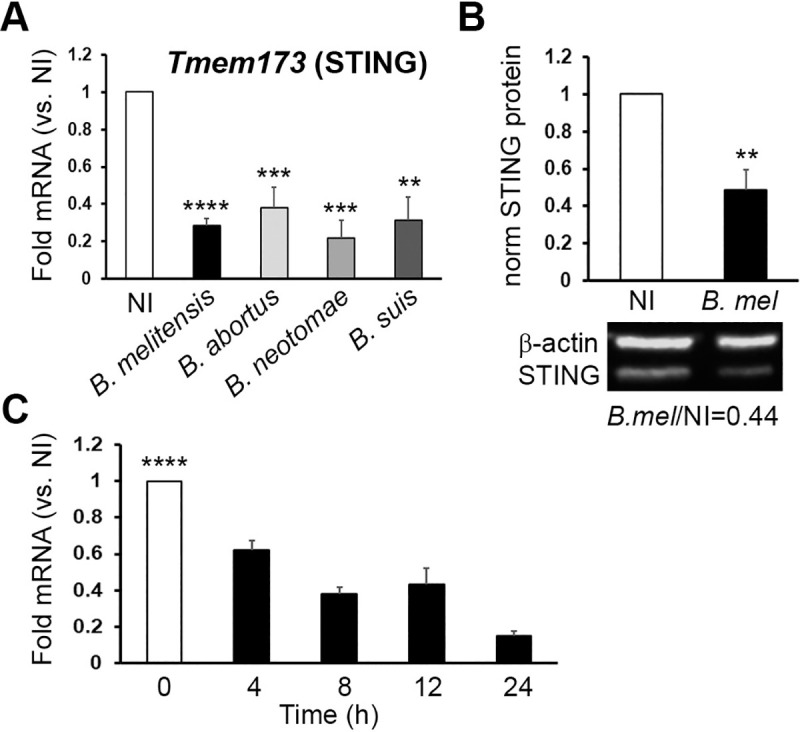
A) Immortalized murine bone marrow derived macrophages were not infected (NI) or infected with B. melitensis 16M (black bars), B. abortus (light gray), B. neotomae (medium gray) or B. suis (dark gray) as indicated at 100 MOI for 24h. Cells were lysed, RNA isolated and reverse transcribed, and relative Tmem173 (STING) mRNA levels determined by quantitative PCR (qPCR) with normalization to 18S rRNA and uninfected controls (NI, set = 1). Results are from 25, 7, 5, and 5 independent experiments respectively, with error bars denoting SEM. P-values are vs. NI control. B) Protein expression of STING: cells were infected with 100 MOI of B. melitensis for 24h, and lysates resolved using SDS PAGE. STING and β-actin proteins were detected by western blot. Band fluorescence was quantitated and results are means +/- SEM of 5 independent experiments. An example western blot is below the graph with the ratio of β-actin normalized STING fluorescence for B. melitensis vs. NI. C) Time course: Cells were infected with 100 MOI B. melitensis for the times indicated and processed for RNA quantitation as in (A). Results are means +/- SEM of 9 independent experiments and for all times tested, p<0.001 for NI vs. infected samples.
Tmem173 down-regulation required live bacteria, consistent with an active bacteria-driven process (Fig 3A). B. melitensis with mutations in the type IV secretion system (deletion of the critical VirB2 subunit [22]) displayed an intermediate phenotype with only modest downregulation of Tmem173, suggesting an intact type IV secretion system (T4SS) is required for full STING suppression (Fig 3B). Regarding the mechanism of suppression, one straightforward possibility was that Brucella infection suppresses the activity of transcription factors required for Tmem173 promoter activity. To address this possibility, we utilized a murine STING-promoter driven luciferase reporter (Fig 3C). Brucella infection increased the activity of the STING promoter-driven construct (at least 1.5-fold in 3 experiments and a 4-fold increase in one experiment). Heat-killed Brucella treatment also increased promoter-driven luciferase activity. This result suggested Brucella suppressed Tmem173 expression downstream of promoter activation.
Fig 3. Brucella down-regulation of STING requires live bacteria and Type IV secretion, and is RIDD-independent.
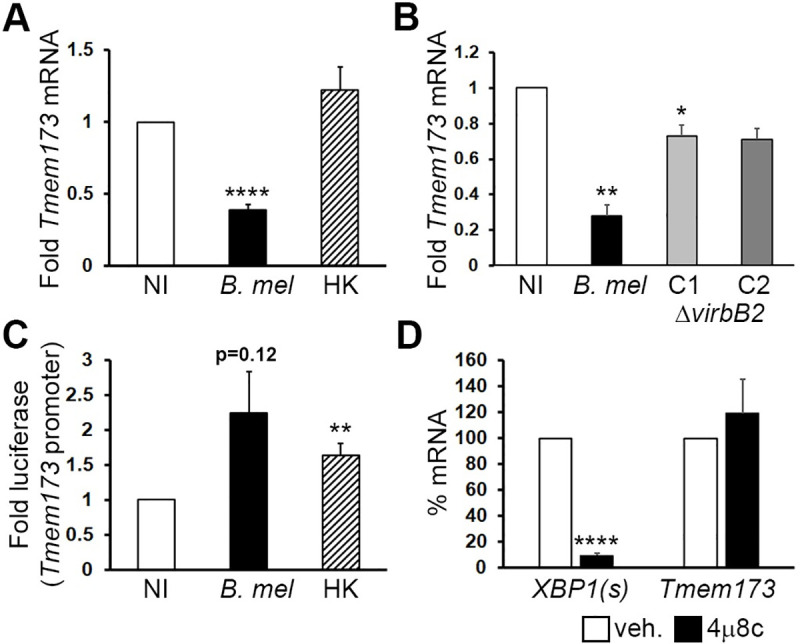
Macrophages were not infected (NI) or infected with 100 MOI B. melitensis 16M (B. mel), heat-killed B. melitensis (HK) in (A), or 2 clones of the ΔvirB2 mutant B. melitensis (C1 and C2) in (B) for 24 hours prior to harvesting for RNA processing. Relative Tmem173 mRNA levels were determined by qPCR with normalization to 18S rRNA and uninfected controls (NI set = 1); p-values are vs. NI. Results are from 21 experiments for HK and 3 independent experiments for the VirB2 mutants. C) Macrophages were transfected with a murine Tmem173 promoter luciferase reporter. Cells were then infected with 100 MOI live (B. mel) or heat-killed B. melitensis (HK). Lysates were analyzed by dual luciferase assay. Results are from 4 and 7 experiments respectively. P-values are vs. NI. D) Macrophages pre-treated with vehicle (veh.) or with the IRE1 endonuclease inhibitor 4μ8c one hour prior to infection with B. melitensis (N = 3 experiments). Levels of spliced XBP1 (XBP1(s)) or Tmem173 mRNA were determined by qPCR. Vehicle treated mRNA expression was set = 100%.
Our group and others have previously shown that Brucella infection induces the Unfolded Protein Response (UPR) in macrophages [23,24]. An important effector of the UPR is the transmembrane protein Inositol-requiring enzyme 1 (IRE1), which functions as both a kinase and an endonuclease. Both B. abortus and B. melitensis infections activate the IRE1 pathway [23,25]. Following activation and oligomerization, the IRE1 endonuclease cleaves 26bp from the XBP1 transcription factor mRNA, thus removing a premature stop codon in the “spliced” product [26]. With prolonged endoplasmic reticulum (ER) stress, the IRE1 endonuclease changes activity to a process termed RIDD (Regulated IRE1 Dependent Decay), whereby it non-specifically degrades ER-proximal mRNAs in the secretory pathways, thus decreasing ER client load [27]. To determine if RIDD degrades Tmem173 mRNA, macrophages were pre-treated with the IRE1 endonuclease inhibitor 4μ8c [28] before infection with B. melitensis. As a positive control for 4μ8c efficacy, we assessed inhibition of XBP1 splicing during B. melitensis infection via RT-qPCR (Fig 3D). Tmem173 levels were unaffected by 4μ8c pre-treatment, indicating that STING mRNA down-regulation does not occur via IRE1-dependent endonuclease activity.
Brucella infection upregulates miR-24, a STING-targeting microRNA
Another hypothesis for the reduction in STING mRNA is that its mRNA is a target of microRNA (miRNA). miRNA are endogenous, small non-coding RNAs 18–25 nucleotides in length that post-transcriptionally regulate gene expression via translational inhibition and mRNA destruction [29]. To search for possible miRNAs that target STING, we used the online tool TargetScanMouse to identify possible candidates. A top hit was a conserved micro-RNA miR-24, which has been shown to post-transcriptionally regulate endogenous STING in Rattus norvegicus epithelium cells [30]. More recently, a study of liver ischemia reperfusion injury reported critical downregulation of STING via miR-24-3p [31]. MiR-24-2 (encoded by the Mirn23a locus) was also increased in our RNAseq data set from Brucella-infected macrophages [17]. To confirm the effect of infection on miR-24 levels, macrophages were infected for 24 hours with B. melitensis (Fig 4A). Infected macrophages significantly and reliably increased miR-24 levels compared to uninfected cells, although the degree of induction was variable (50% up to 8-fold). MiR-24 induction was also observed in vivo, 24h post-infection in mouse spleen. To confirm the biologic relevance of this miR-24 increase, we examined expression of another predicted mRNA target BCL2-like 11 (Bim), an apoptosis facilitator [32]. Bcl2l11 mRNA levels were also significantly decreased in B. melitensis infected macrophages compared to uninfected cells (Fig 4B). Tmem173 levels decreased over time as miR-24 increased (Fig 4C). Fold induction of miR-24 significantly correlated with the extent of Tmem173 suppression (Fig 4D). Just as heat-killed Brucella failed to suppress STING, the killed Brucella did not induce miR-24 expression (Fig 4E). Brucella ΔvirB2 mutants also displayed a marked defect in miR-24 induction, consistent with the failure to fully suppress Tmem173 expression (Fig 4F). These defects in miR-24 induction and Tmem173 suppression were evident by 4 and 8h post infection, respectively (Fig 4G) and occurred in primary BMDM (S1 Fig). In the absence of VirB2, Tmem173 expression decreased slightly at 4h, but then did not diminish further. One explanation for the lesser effects on host gene expression could be impaired survival of the ΔvirB2 mutants. However, the defects in miR-24 induction and STING suppression were evident well before the ΔvirB2 mutant diverged from wild type in replication (S1 Fig). Host gene modulation defects at 8h were complemented with exogenous virB2 (S1 Fig), confirming the gene specificity of the phenotype.
Fig 4. Brucella induces a STING-targeting microRNA miR-24.

A) Left panel: Macrophages were not infected (NI) or infected with 100 MOI Brucella melitensis (B. mel) for 24 hours before harvesting for RNA. Right: Mice were infected with 106 B. abortus 2308 (B. abort) for 24 hours prior to processing of spleen for microRNA. Micro RNA levels were determined by qPCR with normalization toRNU6 and uninfected controls (NI set = 1). In vitro results are from 17 experiments, with error bars denoting SEM. In vivo, results are representative of 2 independent experiments. N = 3 uninfected and 4 infected mice, with SD error bars. B) Macrophages were infected as in (A) and processed for mRNA. Expression was normalized to 18S rRNA. Bcl2l11 (Bim) expression is from 8 experiments. C) Time course of quantitative expression of both miR24-3p and STING (Tmem173) mRNA. P<0.001 for changes over time (N = 6). D) Correlation is from 19 experiments performed and evaluated as in (A). R2 = 0.027, p = 0.022 E) Macrophages were infected as in (A) and processed for miRNA. Comparison of live and heat killed B. melitensis (B. mel vs. HK) is from N = 9. P<0.005 for B. mel vs. NI and HK. F) Macrophages were infected with wild type B. melitensis or VirB2 deletion mutant clones C1 and C2 and analyzed as in (A). Results are from 3 experiments. P<0.005 for B. mel vs. NI and vs. ΔVirb2 clones. G) Time course comparing effects of wild type B. melitensis (filled symbols) and Δ virB2 (clone 1, open symbols) on miR-24 (black circles) and Tmem173 mRNA (gray triangles). Gene expression changes were normalized to time 0 for each Brucella genotype infection (see methods) and error bars represent standard deviations of triplicate determinations. P-values compare Brucella genotypes at each time point: #p<0.005, ##p<0.001 for Tmem173 and *p<0.05, **p<0.01, ***p<0.005, ****p<0.001 for miR-24.
The requirement for live bacteria and the type IV secretion system to induce miR-24 and suppress STING expression suggested an active, bacterially driven process, rather than a passive host response to pathogen associated molecular patterns (PAMPs). Intriguingly, Ma et al. had reported that LPS suppressed STING expression via a MyD88-dependent pathway [33]. The mechanism was, and remains unknown. MyD88, a critical signaling intermediary downstream of multiple Toll-like receptors, is critical for control of Brucella infection in vivo [34,35]. To determine if MyD88 contributed to Tmem173 downregulation in Brucella-infected macrophages, we compared Tmem173 and miR-24 expression in MyD88-/- and wild type macrophages (Fig 5A and 5B). MyD88 was not required for Tmem173 mRNA suppression or for miR-24 induction, although both were less robust in MyD88-/- cells. Further, miR-24 induction in STING-/- macrophages is similar to wild type cells, indicating miR-24 induction does not require STING expression (Fig 5C). As another approach to examining whether Brucella PAMPS contribute to STING suppression, the regulation of Tmem173 expression by purified Toll-like receptor (TLR) agonists was examined. IL6 mRNA served as a control for stimulation. Brucella stimulates TLR2, TLR9 and TLR4, although Brucella LPS is 3–4 logs less endotoxic than E. coli LPS [35,36]. The ligands for TLR2 and TLR9, Pam3CSK4 and ODN 1585, respectively did not downregulate Tmem173, nor did purified Brucella LPS (Fig 5D).
Fig 5. MiR-24 induction and Tmem173 suppression do not require STING or TLR agonist PAMPs.

A) Wild type (WT) or Myd88-/- macrophages were infected with 100 MOI B. melitensis (B. mel) for 24h and then RNA levels assessed by qPCR as above. A) Tmem173 expression is from N = 20, with normalization to WT uninfected controls within each experiment (WT NI = 1). B) MiR-24 expression is from N = 10, normalized as in (A) (left panel) and normalized to uninfected MyD88-/- (right). C) WT or STING (Tmem173)-/- macrophages were infected with heat killed (HK) or live B. melitensis for 24h and analyzed for miR-24 expression by qPCR, with normalization to uninfected controls (NI = 1), N = 8. White bars: uninfected or HK-infected wild type; black bars: infected wild type; dotted bars: uninfected Myd88-/-; striped bars: infected Myd88-/-; light gray: HK infected STING-/-; dark gray: live Brucella infected STING-/-. D) Macrophages were treated for 24h with media (not treated, NT), 1 μM ODN 1585 (ODN, TLR9 agonist), 10ng/mL Pam3CSK4 (Pam3, TLR2 agonist), 100ng/mL E. coli LPS (E-LPS, TLR4 agonist) or 10μg/mL Brucella LPS (B LPS), and analyzed for Tmem173 and IL6 expression. E. coli LPS is from 3 independent experiments, and ODN 1585, Pam3CSK4, and Brucella LPS data from 2 experiments. P-values are vs. NT.
To confirm that miR-24 is required for the down-regulation of STING and Bim, we utilized anti-miR-24 miRNA inhibitors (S2 Fig). The restoration of Tmem173 and Bcl2l11 expression with anti-miR-24 treatment (Fig 6A) was consistent with the idea that miR-24 contributes to the down-regulation of these mRNAs during Brucella infection. STING is required for optimal Brucella-dependent IFN-β production in macrophages [9,18]. To determine if failure to suppress STING correlated with increased STING activity, we assessed the impact of the anti-miR-24 on IFN-β production. As shown in Fig 6B, IFN-β was significantly up-regulated in macrophages transfected with the miR-24 inhibitor compared to mock transfected control cells, consistent with increased STING activity. Together, these data support the idea that Brucella infection induces miR-24 to down-regulate STING.
Fig 6. STING suppression requires miR-24 induction.

A) Macrophage cells were transfected with an anti-miR24 inhibitor or control anti-miR, then infected with 100 MOI B. melitensis (B. mel) for 24h. Relative gene expression of Tmem173 (left) and Bcl2l11 (right) were determined via qPCR with normalization to 18S rRNA and non-infected control (NI set = 1). N = 5 (Tmem173) and 3 experiments (Bcl2l11). P-values are vs. NI (*) or vs. anti-miR24 (#). (A and B) White bars are NI; black B. mel; light gray anti-miR-24 + B. mel; and dark gray scrambled anti-miR control + B. mel. B) IFN-β production in culture supernatant after 24h of infection was determined by ELISA. Data are from 4 experiments. C) And (D) Wild type (WT) or Mirn23a-/- macrophage cells were infected with 100 MOI B. melitensis for 24h and expression of miR-24 (C) Tmem173 mRNA (D) determined as above. Results were normalized to uninfected wild type (NI = 1) within each experiment. Data are from 5 and 8 experiments respectively. E) WT or Mirn23a-/- macrophages were infected with B. melitensis for the times indicated prior to lysis for RNA extraction. Tmem173 levels were determined using qPCR with each genotype normalized to its own NI values (set = 1). 24h data is from 9 experiments with the other time points assessed in 5 experiments. STING protein 24h following infection was detected using western blot with normalization to β-actin and genotype-respective uninfected controls. N = 3. In E, p-values are for B. mel infected vs NI WT cells (*) and for WT vs Mirn23a-/- infected cells (#). F) Ifnb1 mRNA expression at 24h, normalized to uninfected control cells for each genotype. N = 3, #p-value is for WT vs Mirn23a-/-. For C-F, dotted bars are uninfected Mirn23a-/- and striped bars are infected Mirn23a-/- cells.
To further evaluate the requirement for miR-24, we utilized a genetic model of miR-24 deficiency. MiR-24-3p is 100% homologous between mouse, rat and human and is expressed from two genetic loci: Mirn23a encodes miR-23a, miR-24-2 and miR-27a and Mirn23b encodes miR-23b, miR-24-1 and miR-27b. Our previous RNAseq data suggested bone marrow macrophages induced miR-24-2 but not miR-24-1 [17]. Mirn23a is the predominant source of miR-24 in blood [37]. Mirn23a-/- macrophages showed decreased levels of miR-24 compared to wild type prior to infection and were deficient at miR-24 upregulation in response to Brucella infection (Fig 6C). As noted above, heat-killed Brucella did not induce miR-24 in either genotype. Mirn23a-/- macrophages were unable to suppress Tmem173 expression at 24h in relation to their uninfected state, although overall levels of Tmem173 mRNA were decreased compared to uninfected wild type macrophages, suggesting a balance between static miR-24 and Tmem173 levels (Fig 6D and 6E). STING protein suppression was also impaired. The defect in Tmem173 suppression in the Mirn23a-/- macrophages, more evident over time (Fig 6E), correlated with greatly increased Ifnb1 induction by 24h post-infection, consistent with increased STING activity (Fig 6F).
Decreased Brucella replication in miR23 locus-/- macrophages
Although the data in Fig 1 suggested that STING regulates Brucella infection, the biologic consequences of miR-24 induction and STING suppression were not clear. To determine the role of the Mirn23a locus in infection, we compared replication (CFU) in wild type vs. Mirn23a-/- macrophages (Fig 7A). Initial uptake of Brucella was similar between genotypes, but diverged by 8h, with lower Brucella CFU recovered in the Mirn23a-/- macrophages. This divergence maintained or increased over the course of infection through 48–72 hours. These results were consistent with a role for the miRs encoded by this locus in supporting intracellular infection. We further confirmed that the observations in vitro were relevant in vivo by infecting Mirn23a-/- mice with Brucella. As predicted by the in vitro data, Mirn23a-/- mice were more resistant to B. neotomae, with greater than 1 log less splenic CFU one week post-infection. For gene expression in these splenocytes, see S3 Fig. To confirm the specificity of the Mirn23a-/- phenotype for miR-24, anti-miR24 and miR-24 mimics (Fig 7B) were introduced. Anti-miR24 greatly decreased the capacity of wild type but not Mirn23a-/- macrophages to control intracellular B. melitensis replication. In the converse experiment, addition of miR-24 mimics significantly enhanced B. melitensis replication in the Mirn23a-/- but not always in wild type macrophages. These data were consistent with the hypothesis that miR-24 is responsible for the decreased replication in Mirn23a-/- cells. Finally, to determine what proportion of the miR-24 effect was due to STING (vs. other miR-24 targets), STING-/- macrophages were transfected with anti-miR24 or mimics prior to infection. Whereas anti-miR24 suppressed Brucella replication in wild type macrophages, neither mimics nor anti-miRs exerted a significant magnitude of effect on replication in STING-/- cells (Fig 7C, anti-miRs: 5-40-fold in wild type vs <1.5-fold difference in STING-/-). These epistasis results suggested STING accounts for the majority of the miR-24 effect on replication during infection. Together, these data are consistent with the hypothesis that Brucella induction of miR-24 suppresses STING expression to increase infectious success.
Fig 7. Failure to induce miR-24 inhibits Brucella replication.

A) Wild type (WT, blue symbols) or Mirn23a-/- macrophages (orange symbols) were infected with 100 MOI B. melitensis for the times indicated, lysed, and then CFU were enumerated. Error bars are standard deviations of 8 replicates and results are representative of 3 independent experiments. B) C57BL/6 or Mirn23a-/- mice were infected with 106 B. neotomae (N = 7 mice per group). After 7 days, spleens were harvested for analysis of CFU. Bars are mean values. C) WT or Mirn23a-/- macrophages were transfected with anti-miR24 (left panel) or miR-24-3p mimic (right) or miR control then infected with B. melitensis for 24h. Cells were then lysed and CFU enumerated. Error bars are standard deviations of 8 replicates and representative of 4–5 experiments for the anti-miRs and miR-24-3p mimics respectively. Paler bars represent transfection of anti-miR24, whereas darker bars represent addition of the mimic. D) WT or STING-/- macrophages were transfected with anti-miR24 or miR-24-3p mimics and then infected with 100 MOI B. melitensis for 24h prior to enumeration of CFU. Results are representative of 4 experiments.
Discussion
The cytosolic DNA sensor STING plays a key role in innate immune defense via transcription of host defense genes including Type I interferons, induction of NF-κB-dependent responses and autophagy [15]. B. abortus DNA and cyclic-di-GMP activate STING, triggering IFN-β production [9,18]. In this report, at one, 3 and 6 weeks post infection, STING-/- mice had a 1-2-log higher burden of Brucella compared to age-matched wild-type counterparts, confirming that STING is ultimately required for control of Brucella infection. Although STING protects against Brucella, the striking suppression of STING mRNA expression early following infection suggests Brucella actively sabotages this innate immune sensor to gain a foothold inside macrophages.
STING suppression occurred post-transcriptionally, independently of UPR-mediated RNA decay, via upregulation of the microRNA miR-24. MiR-24 induction and STING suppression required live bacteria, and full suppression required the VirB-encoded Type IV secretion system, suggesting an active bacterial driven process rather than a simple host response to Brucella PAMPs. The independence of miR-24 upregulation from STING signaling and lack of STING suppression by purified TLR agonists supports this model. MiR-24 upregulation and STING downregulation were slightly less robust in the MyD88-/- macrophages, consistent with a minor role for MyD88 signaling. Divergence of the ΔvirB2 mutant and wild type Brucella <8h post-infection suggests the requirement for the type IV secretion system does not simply reflect its effect on replication. Rather, the type IV secretion system may contribute to miR-24 upregulation through secretion of a specific Brucella substrate or by enabling appropriate intracellular trafficking. The early divergence (4h) of STING expression between Mirn23a-/- and wild type macrophages supports the idea that the effect of miR-24 precedes intracellular Brucella replication.
Induction of the Mirn23a gene locus during infection comes at potential cost for Brucella infection. In NK cells, deletion of this locus (also known as Mirc11) resulted in decreased ability to contain Listeria infection, related to diminished IFN-γ and pro-inflammatory cytokine production [38]. Both IFN-γ and TNF-α have long been known to be critical for control of Brucella infection. However, effects of miR-24 on cytokine production may be cell-type specific. In CD4+ T cells, miR-24 was reported to target IFN-γ mRNA [39]. Over-expression of miR-24 in a Staphylococcus aureus infection model decreased “M1” inflammatory mediator production in macrophages and enhanced “M2” marker expression, which would benefit Brucella [40,41]. An earlier study had also suggested miR-24 modulates macrophage polarization towards an “alternative” M2 phenotype [42]. Manipulation of miR-24 levels had minimal effects on Brucella replication in STING-/- cells, suggesting that STING is the dominant or primary target of miR-24 induction during Brucella infection of macrophages that impacts intracellular replication.
Recently, the ability of another chronic intracellular pathogen, Mycobacterium tuberculosis, to manipulate host innate responses, autophagy, and apoptosis via host miRNA has garnered much interest [43–46]. In contrast, there is much less information regarding miRNA in the context of Brucella infection [47–49]. Budak et al. investigated the miRNA expression patterns in CD4+ and CD8+ T cells from patients, and reported discrete changes with acute vs. chronic brucellosis [50,51]. Another study reported the up-regulation of miR-1981 in RAW264.7 infected macrophages and showed the interaction of that microRNA with the 3’-UTR of Bcl-2, an apoptosis regulator [52]. Recently, Corsetti et al revealed several miRNA-dependent mechanisms of immune manipulation during B. abortus infection: upregulation of mmu-miR-181a-5p suppressed TNF-α and miR-21a-5p downregulation decreased IL10 and elevated GBP5 [49]. Here, we show that Brucella significantly induce miR-24. Additionally, another predicted target of miR-24, Bim, a key apoptosis-regulator induced by PERK signaling and C/EBP homologous protein (CHOP) transcriptional activity, was significantly down regulated during infection with B. melitensis. MiR-24 inhibition resulted in a significant recovery in both Bim and STING, indicating that miR-24 is targeting both these mRNAs during B. melitensis infection.
Our previous RNAseq data set [17] revealed upregulation of miR-24-2, encoded at the Mirn23a locus, but not miR-24-1 from the Mirn23b locus. Furthermore, although Mirn23a-/- cells expressed some miR-24, they were unable to upregulate its expression in response to infection. Throughout these experiments, fold-induction of miR-24 correlated with STING mRNA suppression. These results suggest that upsetting the balance between miR-24 and Tmem173 levels is the critical component. The strong effects of miR-24 manipulation through mimics and anti-miRs, as well as the defects in replication in the Mirn23a-/- macrophages together support the idea that upregulation of miR-24 is important for replication early during infection. In vivo, the inability to upregulate miR-24 correlated with decreased splenic CFU in the Mirn23a-/- mice one week post-infection, also suggesting that miR-24 supports acute infection. The greater replication in the Mirn23a-/- cells vs the anti-miR24 treated wild type macrophages (Fig 7) may reflect contributions from miR-23a and miR-27a encoded by that locus.
In addition to the increased STING mRNA, miR-24 inhibition or genetic deficiency resulted in a significantly increased IFN-β response compared to uninhibited macrophages. Although initially identified in its role in viral protection, Type I interferons have recently become a topic of interest in response to many bacterial pathogens [53]. During infections, the effect of Type I interferons can be protective or detrimental depending on the bacterial species. For example, Type I interferon protects mice against Salmonella typhimurium infection whereas Interferon-alpha/beta receptor (IFNAR)-mediated Type I interferon responses to Francisella tularensis and Listeria monocytogenes are harmful to the host [54–56]. The role of Type I interferon in response to Brucella is currently unclear; a study in 2007 showed no difference in splenic and liver CFUs in wild type versus IFNAR-/- mice [57]. However, a more recent study has shown a higher burden of Brucella in wild type mice compared to IFNAR-/- mice, indicating that Type I interferon response is detrimental to the host [9]. Resistance to B. abortus in the IFNAR-/- mice was accompanied by elevated production of IFN-γ and NO, and decreased apoptosis compared to wild-type mice. Although type I IFN served as a useful indicator for STING activity in our study, ultimately, the experience with the IFNAR-/- mice suggest STING is controlling infection through Type I IFN-independent mechanisms.
Brucella potently inhibits apoptosis, contributing to chronic infection; however, the mechanisms behind this process are unknown [34,58]. By down-regulating STING and subsequent IFN-β production, Brucella could be actively inhibiting apoptosis that is dependent upon Type I IFN signaling. Further, by up-regulating miR-24, which in turn down-regulates Bim, Brucella could be avoiding UPR-mediated apoptosis, which is partially dependent upon Bim in other experimental systems [59]. B. melitensis infection robustly induces CHOP, which is an upstream activator of Bim [23,60]. We did not detect a reliable effect of the anti-miR-24 on host cell apoptosis or cell death. One likely explanation is that there are unidentified miR-24-independent mechanisms that inhibit apoptosis independently of STING and Bim down-regulation. Indeed, our previous RNAseq data [17] suggested that Brucella suppresses the expression of multiple pro-apoptotic molecules.
In summary, our findings document the evasion of full STING activation during infection by an intracellular bacteria pathogen via miR-24-mediated suppression of STING expression. It is noteworthy that a single miRNA species should have such a profound impact on a major cytosolic innate immune sensor and consequent Brucella replication. Our data may have implications for other important pathogens. For instance, miR-24 was up-regulated and cited as one of 7 significantly altered microRNAs controlling the transcriptional response to M. tuberculosis in macrophages [61]. In a separate report, in transcriptomic data, Tmem173 was suppressed by more than 50% at 4 hours and >75% decreased 12 hours following M. tuberculosis infection [62]. Widely considered a “stealth” pathogen, Brucella can evade immune surveillance and persist chronically in macrophages [63]. In contrast to this idea of Brucella as “flying under the radar”, previous reports have described Brucella subversion of toll-like receptor signaling via Btp1/TcpB [64–66]. The data presented here elucidate a critical mechanism by which Brucella actively sabotages cytosolic surveillance by the innate immune sensor STING to establish its intracellular niche.
Methods
Ethics statement
Mouse care, handling and experimental procedures were approved by the Institutional Animal Care and Use Committees (IACUC) of the institutions involved in this project and performed with strict adherence.
Reagents, resources and associated sources and identifiers are listed below in Table 1 and primers in Table 2.
Table 1. Reagents and resources used in this study and their associated sources and identifiers.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-STING | Cell Signaling Technology | Cat# 13647 |
| Mouse monoclonal anti-β-actin | Santa Cruz | sc-47778 |
| Bacterial Strains | ||
| Brucella melitensis 16M | UW-Madison archive | N/A |
| Brucella abortus 2308 | UW-Madison archive | N/A |
| Brucella neotomae Stoenner and Lackman 1957 | ATC 23459 | N/A |
| Brucella suis 1330 | UW-Madison archive | N/A |
| Escherichia coli DH5a | UW-Madison archive | N/A |
| Chemicals | ||
| 4μ8c | EMD Millipore | Cat#412512-25MG |
| ODN 1585 | Invivogen | Tlrl-1585 |
| Pam3CSK4 | Invivogen | Tlrl-pms |
| Critical Commercial Assays | ||
| RNAzol RT | Molecular Research Center, Inc. | RN 190 |
| PowerUp SYBR Green Master Mix | AppliedBiosystems | Cat# A25742 |
| B-PER Bacterial Protein Extraction Reagent | ThermoFisher Scientific | Cat# 78248 |
| M-PER Mammalian Protein Extraction Reagent | ThermoFisher Scientific | Cat# 78501 |
| Halt Protease and Phosphatase Inhibitor | ThermoFisher Scientific | Cat# 78440 |
| Dual-Glo Luciferase assay system | Promega | Cat# E2490 |
| Cell Line Nucleofector kit V | Lonza | VVCA-1003 |
| Lipofectamine RNAiMAX Transfection Reagent | ThermoFisher Scientific | Cat# 13778030 |
| Opti-MEM | ThermoFisher Scientific | Cat# 31985070 |
| LEGEND MAX Mouse IFN-β ELISA Kit with Pre-coated Plates | BioLegend | Cat# 439407 |
| qScript microRNA cDNA Synthesis Kit | Quanta Biosciences | Cat# 95107–025 |
| SuperScript IV VILO Master Mix | ThermoFisher Scientific | Cat# 11756050 |
| LPS Extraction Kit | iNtRON Biotechnology | Cat# 17141 |
| Experimental Models: Cell Lines | ||
| iMACs (immortalized macrophages) | John-Demian Sauer | N/A |
| LADMAC (for CSF-1) | ATCC | CRL-2420 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus C57BL/6 | Federal University of Minas Gerais; IUSM South Bend | N/A |
| Mus musculus C57BL/6 STING-/- | Ishikawa and Barber, 2008 | N/A |
| Mus musculus mirn23a-/-(on C57BL/6 background) | Richard Dahl; IUSM South Bend | N/A |
| Mus musculus MyD88-/-(on C57BL/6 background) | Bruce Klein | N/A |
| Oligonucleotides and plasmids | ||
| mirVana miRNA inhibitor hsa-miR-24-3p | ThermoFisher Scientific | Cat# 4464084 |
| Anti-miR miRNA Inhibitor Negative Control #1 | ThermoFisher Scientific | Cat# AM17010 |
| miRNA Neg Control | SIGMA | MISSION miRNA Control |
| miRNA hsa-mir-24 | SIGMA | MISSION miRNA Mimic |
| miRNA inhibitor mmu-mir-24-3p | SIGMA | MISSION miRNA Inhibitor |
| VirB2 knock-out vector, pAV2.2 | Renee M. Tsolis | Den Hartigh et al., 2004 [22] |
| pBBR1-MCS4 | Lab Archive | N/A |
| pBBR1-VirB2 | This work | N/A |
| pSTING-254 murine STING promoter luciferase plasmid | Hua-Guo Xu | Xu et al., 2017 [67] |
| Software and Algorithms | ||
| GraphPad Prism 7 | Graphpad Software | N/A |
Table 2. Primers used in this study.
| Primers used in this work | ||
|---|---|---|
| Primer | Sequence | Source |
| 18S F | AGGGGAGAGCGGGTAAGAGA | IDT |
| 18S R | GGACAGGACTAGGCGGAACA | IDT |
| Bim F | TGTCTGACTCTGATTCTCGGA | IDT |
| Bim R | TGCAATTGTCCACCTTCTCTG | IDT |
| hsa-miR-24-3p | NA | SIGMA; MIRAP00056 |
| IFN-β F | GGCATCAACTGACAGGTCTT | IDT |
| IFN-β R | ACTCATGAAGTACAACAGCTACG | IDT |
| RNU6 | GCAAATTCGTGAAGCGTTCC | IDT |
| STING F | AAGTCTCTGCAGTCTGTGAAG | IDT |
| STING R | TGTAGCTGATTGAACATTCGGA | IDT |
| Xbp-1(t) F | TCCGCAGCACTCAGACTATGT | IDT |
| Xbp-1(t) R | ATGCCCAAAAGGATATCAGACTC | IDT |
| Xbp-1(s) F | GAGTCCGCAGCAGGTG | IDT |
| Xbp-1(s) R | GTGTCAGAGTCCATGGGA | IDT |
| virB2 F | CCAGACCGATAAGAGAACGATG | IDT |
| virB2 R | CCGATCAGGCACGCATATAA | IDT |
| virB2-988 F | CTCGAGGCTGCCCCAGTAAAAAAAACGAC | IDT |
| VIRB2-1562 R | ATCGATTCGGTCTGCTTGCTCAATGTCTAT | IDT |
| PerfeCTa Universal PCR Primer | NA | Quanta Biosciences; 95109–500 |
Primers used for qPCR determination of gene expression and their sources are listed.
Experimental model and method details
Mice
In vivo infections in mice were performed via intraperitoneal injection of 106 CFU/0.2ml of PBS diluent of Brucella abortus 2308 or Brucella neotomae Stoenner and Lackman 1957 or 0.2ml PBS (uninfected vehicle control). At indicated time points post infection, mice were euthanized following IACUC approved procedures. In Brazil, wild type C57BL/6 mice were purchased from the Federal University of Minas Gerais (UFMG). STING (Tmem173)-/-, mice were described previously [13]. Mice were maintained at UFMG and used at six weeks of age. To count Brucella colony forming units (CFU), individual spleens were macerated in 10 ml saline, serially diluted, and plated in duplicate on Brucella Broth agar. After 3 days of incubation at 37°C, the number of CFU was determined as described previously [34]. At the University of Notre Dame, seven wild-type C57/BL6 and Mirn23a-/- mice (mixed gender; 6 wks) were infected with B. neotomae, and one per genotype injected with PBS control, and euthanized at 7 days post-infection. Spleens were processed and assayed for CFU and RNA as follows: Single cell suspensions of mouse spleens were prepared using a gentleMACS dissociator (Miltenyi Biotec) following the manufacturer’s protocol. Splenocytes from each mouse were processed for either CFU determination (as described below) or RNA preparation.
Mammalian cell lines
V-raf/v-myc immortalized murine bone marrow derived macrophages (BMDM) were a generous gift from Dr. John-Demian Sauer at the University of Wisconsin-Madison. These macrophages (iMacs) were from C57BL/6 mice. V-raf/V-myc immortalized BMDM were generated in our lab from leg bones of MyD88-/-, STING-/-, and Mirn23a-/- mice obtained from researchers listed in Table 1 above. These mice were all on a C57BL/6 background. For the Mirn23a-/- experiments, wild type C57BL/6 bones were obtained from Dr. Dahl’s colony. All immortalized macrophage cell lines were cultured at 37°C with 5% CO2 in RPMI supplemented with 1mM Na pyruvate, 0.05mM 2-mercaptoethanol, and 10% FBS. Apart from Figs 1 and S1, immortalized macrophages were used for in vitro experiments.
Bone marrow derived macrophages (BMDM)
To generate CSF-1 containing media, LADMAC cells were grown to confluency and then pelleted. The supernatant was filtered (0.45 μm) prior to use. Mouse femur and tibia bones were cleaned of tissue. The ends of the bones were snipped aseptically using surgical scissors, and bone marrow was flushed using a PBS-filled syringe with a 27G needle. The flushed cells were then pulled and ejected through a syringe with an 18G needle to generate a single cell suspension. The suspension was filtered through a 70 μm cell strainer to remove solid fragments, pelleted (300 x g; 5 min), and resuspended in 35% LADMAC conditioned media. Bone marrow cells were differentiated into macrophages over 7 days.
Brucella strains
Brucella strains B. melitensis, B. abortus, B. suis, and B. neotomae were from archived stock of the University of Wisconsin-Madison and the University of Minas Gerais (B. abortus used in Figs 1 and 4). Brucella were cultured using Brain Heart Infusion broth or agar (Difco) at 37°C. All experiments with select agent Brucella strains were performed in a Biosafety Level 3 facility in compliance with the CDC Division of Select Agents and Toxins regulations according to standard operating procedures approved by the University of Wisconsin-Madison and UFMG Institutional Biosafety Committees. B. neotomae in vivo experiments were approved by the University of Notre Dame Institutional Biosafety Committee.
VirB2 deletion mutants of Brucella were derived through homologous recombination following the methods described by den Hartigh et. al., 2004 using the plasmid pAV2.2 (generous gift from R.M. Tsolis). Briefly, exponentially growing Brucella were made electrocompetent following standard microbiological methods. Electrocompetent Brucella were then electroporated with pAV2.2 and VirB2 deletion mutants were selected for kanamycin resistance and carbenicillin sensitivity. VirB2 deletion was confirmed in these clones using PCR. Where not otherwise specified, “clone 1” was used in experiments. The complementation plasmid for VirB2 was engineered through PCR amplification of the VirB2 ORF plus the ribosome binding site but lacking the promoter sequences. Primers virB2-988 F and virB2-1562 R also encoded appropriate restriction sites to aid cloning (Xho I/Cla I). VirB2 was then cloned into pBBR1-MCS4 containing a constitutive lac promoter shown in previous studies to be effective for complementation analysis in B. melitensis [68]. The PCR product was digested and ligated with similarly digested pBBR1-MCS4 to generate the complementation plasmid. The VirB2 amplified product was directionally cloned into pBBR1-MCS4 to ensure that these genes were transcribed from the lac promoter present in the plasmid. The resulting complementation plasmid, pBBR1-VirB2, was transformed into the VirB2 deletion mutant and selected for ampicillin resistance. In experiments using heat-killed (HK) Brucella as a control, inactivation of Brucella was as follows. After growing Brucella in culture, the sample was quantitated using spectrophotometry. Brucella was then aliquoted in microcentrifuge tubes and placed in a 56°C waterbath for 1 h. Brucella lipopolysaccharide (LPS) along with E. coli LPS was used as an agonist for TLR studies and was extracted as follows: Brucella melitensis was cultured in BHI broth for 3 days in a 37°C shaking incubator to an OD600 of approximately 1.2. Bacteria was pelleted and LPS extracted using an LPS extraction kit (iNtRon Biotechnology) following the manufacturer’s protocol. E. coli DH5a LPS was extracted using the same method after 1 day of culture.
Infections, treatments, transfections and CFU assays
Immortalized macrophage cell lines were plated on 6-well tissue culture plates at 0.4 x 106 cells per well in 2 ml culture media. Primary BMDM were plated on 6-well tissue culture plates at 1.2 x 106 cells per well in 2 ml culture media. The next day, the media was replaced, and cells were infected with 100 MOI Brucella determined by spectrophotometry (OD at 600 nm) through a formula established by a Brucella growth curve. Cells were then centrifuged (300 x g; 5 min) to synchronize infection and were incubated at 37°C with 5% CO2. One hour later, cells were washed 3x with 2 ml/well warm PBS and fresh media containing 10 μg/ml gentamycin was added. This one-hour point was considered “Time 0” post infection for the time courses. Cell treatments: 4μ8c (IRE1 endonuclease inhibitor) was dissolved at 10mM in DMSO and then diluted to 1mM (100x) in media. 4μ8c was added to the cultures 1 hour prior to infection. For TLR agonist treatment, cells were plated in 12 well plates at 0.5x106/well in 1 mL growth media. ODN 1585 and Pam3CSK4 were dissolved in media and used at concentrations of 1 μM and 10ng/mL respectively. Extracted E. coli LPS was used at 100ng/mL and Brucella LPS at 10μg/mL. Luciferase assay: 2 μg STING promoter driven luciferase reporter plasmid and 0.5μg Renilla TK were transfected into 1x106 cells suspended in solution V by AMAXA Nucleofection (Lonza), then cells were plated in 12 well plates. The following day, the cells were given fresh media and infected with Brucella melitensis (100 MOI) or stimulated with heat-killed Brucella melitensis (100 MOI). Twenty-four hours later, the cells were washed with 1ml PBS 3x, resuspended in 150 μL kit lysis buffer and assayed according to the manufacturer’s instructions. Assays were performed in triplicate using a Veritas microplate luminometer (Turner Biosystems). For microRNA transfections, immortalized macrophages were seeded on 6-well tissue culture plates at 0.4 x 106 cells/well. MiRNA mimics (miR-24-3p), miRNA control, and miRNA inhibitors for miR-24-3p were diluted to 0.28 μM using Opti-MEM and the cells were transfected using using RNAiMAX reagent following the manufacturer’s protocol. One day after transfection, cells were infected as described above, then processed for RNA or CFU. For CFU assays, cells were washed 3x in PBS to remove extracellular bacteria, then 1 ml of cell lysis buffer (dH2O + 0.1% Triton X-100) was added per well. CFU were determined by serial dilution plating with 8 replicates on BHI agar after 3–4 days.
Quantitative polymerase chain reaction (qPCR)
Total cellular RNA was processed using RNAzol RT reagent (Molecular Research Center, Inc.) following the manufacturer’s protocol. Then cDNA was prepared from either mRNA, using the Superscript IV VILO system (Invitrogen) or microRNA using the qScript system (Quanta biosciences). Samples for Quantitative PCRs were analyzed via SYBR Green and the delta-delta Ct method to calculate relative fold gene expression using a StepOnePlus thermocycler (ABI). Endogenous housekeeping genes used for comparative expression were either 18S (rRNA) or RNU6 (microRNA). The primers used in this study were designed using IDT’s online primer design tool or purchased.
Quantification of IFN-β by ELISA
Culture supernatants from cells were collected and frozen at -80°C until assayed. A mouse IFN-β ELISA was performed following the kit manufacturer’s protocol. Absorbance at 450 nm and 570 nm were determined utilizing a BioTek microplate reader. Mouse IFN-β was quantified by standard curve.
Western blot assays
Cell lines (infected or control) were washed with PBS, scraped off the well, then transferred to a microcentrofuge tube and pelleted (4k RPM, 5 min). The supernatant was removed and cells lysed with M-PER reagent (ThermoFisher Scientific) according to the manufacturer’s protocol. Whole-cell lysates were resolved by 12% SDS-PAGE. Samples were then transferred to polyvinyldene difluoride (PVD) membrane and immunoblotted with anti-STING primary antibody (ThermoFisher Scientific) and anti-β-actin primary antibody (Santa Cruz), followed by a fluorescence-conjugated secondary antibody (LI-COR). Proteins were visualized and quantitated with the Odyssey system (LI-COR).
Quantification and statistical analysis
CFU values and standardized mRNA expression levels were summarized in terms of means ± standard deviations and displayed in graphical format using bar charts, stratified by experimental conditions. Comparisons between experimental groups were conducted using two-sample t-test or analysis of variance (ANOVA). Pairwise comparisons between multiple groups were conducted using Tukey’s Honestly Significance Difference (HSD) method. Residual and normal probability plots were examined to verify the model assumptions. Linear regression and Pearson’s correlation analyses were conducted to evaluate bivariate associations. Statistical significance is indicated in the figures (* p<0.05, ** p<0.01, *** p<0.005, **** p<0.001, ns not significant). Statistical analyses were conducted using SAS (SAS Institute Inc., Cary NC), version 9.4.
Supporting information
A) Bone marrow derived macrophages were infected with 100 MOI B. melitensis or ΔvirB2 mutant and harvested for RNA at 12h and 24h post infection. Tmem173 and miR-24 expression was normalized to Time 0 for each genotype (set = 1). B) Immortalized macrophages were infected as above and CFU (8 replicates) determined over time. By 24h, the ΔvirB2 mutant was at a significant disadvantage for replication (p<0.001) vs. wild type B. mel and the complemented mutant. C) Macrophages were infected with 100 MOI of B. melitensis, ΔvirB2 mutant, or the mutant transfected with the virB2 gene for 8h prior to harvest for RNA. Error bars are triplicate standard deviations.
(TIF)
A) Macrophages were not transfected (white and black bars) or transfected with anti-miR-24 inhibitor (pale gray), scrambled control nucleotide (dark gray), or miR-24 mimic (striped bars). Cells were subsequently uninfected (-) or infected with 100 MOI B. melitensis (+). After 24h, cells were harvested for RNA and miR-24 levels determined by qPCR with normalization to RNU6. N = 3 experiments. Error bars are SEM. *p<0.05, ****p<0.001. B) Macrophages were transfected as in (A). 24h following infection with Brucella, cells were lysed and whole cell lysates resolved by SDS-PAGE. STING and β-actin were detected using western blot and immunofluorescence. Blot is representative of 2 experiments. Graph bars are actin-normalized optical densities of the STING bands.
(TIF)
C57BL/6 (+/+) and Mirn23a-/- (-/-) mice were infected intra-peritoneally with 106 B. neotomae (7 mice per group). After 7 days, mice were sacrificed and spleens harvested for CFU (Fig 7) and RNA. Gene expression was determined by qPCR. Results from individual infected mice were normalized to an uninfected control (set = 1) for each genotype. ****p<0.001 and **p<0.01 vs. infected C57BL/6.
(TIF)
Acknowledgments
We would like to thank Dr. John-Demian Sauer for providing us with the v-raf/v-myc immortalized murine bone marrow-derived macrophages and Glen Barber for providing the STING-/- mice. Bruce Klein supplied MyD88-/- femurs and Richard Dahl the Mirn23a -/- mouse bones.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
M.K. received the F31 AI115931 National Institutes of Health training award. S.C.O. received the NIH R01 AI116453 National Institutes of Health award. G.A.S. received the NIH R01 AI073558 National Institutes of Health award. R.D. received NIH R01 DK109051 National Institutes of Health award. J.A.S. was a multi-PI on NIH R01 AI073558. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med. 2005;352(22):2325–36. Epub 2005/06/03. 352/22/2325 [pii] 10.1056/NEJMra050570 . [DOI] [PubMed] [Google Scholar]
- 2.Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect Dis. 2006;6(2):91–9. Epub 2006/01/28. S1473-3099(06)70382-6 [pii] 10.1016/S1473-3099(06)70382-6 . [DOI] [PubMed] [Google Scholar]
- 3.Franco MP, Mulder M, Gilman RH, Smits HL. Human brucellosis. Lancet Infect Dis. 2007;7(12):775–86. Epub 2007/11/30. S1473-3099(07)70286-4 [pii] 10.1016/S1473-3099(07)70286-4 . [DOI] [PubMed] [Google Scholar]
- 4.Rossetti CA, Arenas-Gamboa AM, Maurizio E. Caprine brucellosis: A historically neglected disease with significant impact on public health. PLoS Negl Trop Dis. 2017;11(8):e0005692 10.1371/journal.pntd.0005692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serpa JA, Knights S, Farmakiotis D, Campbell J. Brucellosis in Adults and Children: A 10-Year Case Series at Two Large Academic Hospitals in Houston, Texas. South Med J. 2018;111(6):324–7. 10.14423/SMJ.0000000000000810 . [DOI] [PubMed] [Google Scholar]
- 6.Joseph R, Crotty MP, Cho J, Wilson MH, Tran J, Pribble J, et al. A single-institution experience with a brucellosis outbreak in the United States. Am J Infect Control. 2018;46(10):1195–7. 10.1016/j.ajic.2018.03.022 . [DOI] [PubMed] [Google Scholar]
- 7.Sfeir MM. Raw milk intake: beware of emerging brucellosis. J Med Microbiol. 2018;67(5):681–2. 10.1099/jmm.0.000722 . [DOI] [PubMed] [Google Scholar]
- 8.Byndloss MX, Tsolis RM. Brucella spp. Virulence Factors and Immunity. Annu Rev Anim Biosci. 2016;4:111–27. 10.1146/annurev-animal-021815-111326 . [DOI] [PubMed] [Google Scholar]
- 9.de Almeida LA, Carvalho NB, Oliveira FS, Lacerda TL, Vasconcelos AC, Nogueira L, et al. MyD88 and STING signaling pathways are required for IRF3-mediated IFN-beta induction in response to Brucella abortus infection. PLoS One. 2011;6(8):e23135 10.1371/journal.pone.0023135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, et al. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity. 2015;43(3):451–62. 10.1016/j.immuni.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, et al. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J Immunol. 2013;190(7):3629–38. 10.4049/jimmunol.1202817 . [DOI] [PubMed] [Google Scholar]
- 12.Guimaraes ES, Gomes MTR, Campos PC, Mansur DS, Dos Santos AA, Harms J, et al. Brucella abortus Cyclic Dinucleotides Trigger STING-Dependent Unfolded Protein Response That Favors Bacterial Replication. J Immunol. 2019;202(9):2671–81. 10.4049/jimmunol.1801233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–8. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–92. 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–70. 10.1038/nri3921 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38. 10.1111/imr.12765 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan M, Harms JS, Marim FM, Armon L, Hall CL, Liu YP, et al. The Bacterial Second Messenger Cyclic di-GMP Regulates Brucella Pathogenesis and Leads to Altered Host Immune Response. Infect Immun. 2016;84(12):3458–70. 10.1128/IAI.00531-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costa Franco MM, Marim F, Guimaraes ES, Assis NRG, Cerqueira DM, Alves-Silva J, et al. Brucella abortus Triggers a cGAS-Independent STING Pathway To Induce Host Protection That Involves Guanylate-Binding Proteins and Inflammasome Activation. J Immunol. 2018;200(2):607–22. 10.4049/jimmunol.1700725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grillo MJ, Blasco JM, Gorvel JP, Moriyon I, Moreno E. What have we learned from brucellosis in the mouse model? Vet Res. 2012;43:29 10.1186/1297-9716-43-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blasi E, Mathieson BJ, Varesio L, Cleveland JL, Borchert PA, Rapp UR. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318(6047):667–70. 10.1038/318667a0 . [DOI] [PubMed] [Google Scholar]
- 21.Villalobos-Vindas JM, Amuy E, Barquero-Calvo E, Rojas N, Chacon-Diaz C, Chaves-Olarte E, et al. Brucellosis caused by the wood rat pathogen Brucella neotomae: two case reports. J Med Case Rep. 2017;11(1):352 10.1186/s13256-017-1496-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.den Hartigh AB, Sun YH, Sondervan D, Heuvelmans N, Reinders MO, Ficht TA, et al. Differential requirements for VirB1 and VirB2 during Brucella abortus infection. Infect Immun. 2004;72(9):5143–9. 10.1128/IAI.72.9.5143-5149.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith JA, Khan M, Magnani DD, Harms JS, Durward M, Radhakrishnan GK, et al. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog. 2013;9(12):e1003785 10.1371/journal.ppat.1003785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Jong MF, Starr T, Winter MG, den Hartigh AB, Child R, Knodler LA, et al. Sensing of bacterial type IV secretion via the unfolded protein response. MBio. 2013;4(1):e00418–12. 10.1128/mBio.00418-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Jong MF, Starr T, Winter MG, den Hartigh AB, Child R, Knodler LA, et al. Sensing of Bacterial Type IV Secretion via the Unfolded Protein Response. mBio. 2013;4(1). Epub 2013/02/21. 10.1128/mBio.00418-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. Epub 2005/06/15. 10.1146/annurev.biochem.73.011303.074134 . [DOI] [PubMed] [Google Scholar]
- 27.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–31. 10.1083/jcb.200903014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109(15):E869–78. 10.1073/pnas.1115623109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. 10.1016/s0092-8674(04)00045-5 . [DOI] [PubMed] [Google Scholar]
- 30.Huang Z, Chen X, Yu B, Chen D. Cloning and functional characterization of rat stimulator of interferon genes (STING) regulated by miR-24. Dev Comp Immunol. 2012;37(3–4):414–20. 10.1016/j.dci.2012.02.010 . [DOI] [PubMed] [Google Scholar]
- 31.Shen A, Zheng D, Luo Y, Mou T, Chen Q, Huang Z, et al. MicroRNA-24-3p alleviates hepatic ischemia and reperfusion injury in mice through the repression of STING signaling. Biochem Biophys Res Commun. 2020;522(1):47–52. 10.1016/j.bbrc.2019.10.182 . [DOI] [PubMed] [Google Scholar]
- 32.Gogada R, Yadav N, Liu J, Tang S, Zhang D, Schneider A, et al. Bim, a proapoptotic protein, up-regulated via transcription factor E2F1-dependent mechanism, functions as a prosurvival molecule in cancer. J Biol Chem. 2013;288(1):368–81. 10.1074/jbc.M112.386102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma F, Li B, Yu Y, Iyer SS, Sun M, Cheng G. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. 2015;16(2):202–12. 10.15252/embr.201439366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macedo GC, Magnani DM, Carvalho NB, Bruna-Romero O, Gazzinelli RT, Oliveira SC. Central role of MyD88-dependent dendritic cell maturation and proinflammatory cytokine production to control Brucella abortus infection. J Immunol. 2008;180(2):1080–7. 10.4049/jimmunol.180.2.1080 . [DOI] [PubMed] [Google Scholar]
- 35.Oliveira SC, de Almeida LA, Carvalho NB, Oliveira FS, Lacerda TL. Update on the role of innate immune receptors during Brucella abortus infection. Vet Immunol Immunopathol. 2011. Epub 2011/06/28. 10.1016/j.vetimm.2011.05.036 . [DOI] [PubMed] [Google Scholar]
- 36.Goldstein J, Hoffman T, Frasch C, Lizzio EF, Beining PR, Hochstein D, et al. Lipopolysaccharide (LPS) from Brucella abortus is less toxic than that from Escherichia coli, suggesting the possible use of B. abortus or LPS from B. abortus as a carrier in vaccines. Infect Immun. 1992;60(4):1385–9. 10.1128/IAI.60.4.1385-1389.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurkewich JL, Boucher A, Klopfenstein N, Baskar R, Kapur R, Dahl R. The mirn23a and mirn23b microrna clusters are necessary for proper hematopoietic progenitor cell production and differentiation. Exp Hematol. 2018;59:14–29. 10.1016/j.exphem.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nanbakhsh A, Srinivasamani A, Holzhauer S, Riese MJ, Zheng Y, Wang D, et al. Mirc11 Disrupts Inflammatory but Not Cytotoxic Responses of NK Cells. Cancer Immunol Res. 2019;7(10):1647–62. 10.1158/2326-6066.CIR-18-0934 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fayyad-Kazan H, Hamade E, Rouas R, Najar M, Fayyad-Kazan M, El Zein N, et al. Downregulation of microRNA-24 and -181 parallels the upregulation of IFN-gamma secreted by activated human CD4 lymphocytes. Hum Immunol. 2014;75(7):677–85. 10.1016/j.humimm.2014.01.007 . [DOI] [PubMed] [Google Scholar]
- 40.Jingjing Z, Nan Z, Wei W, Qinghe G, Weijuan W, Peng W, et al. MicroRNA-24 Modulates Staphylococcus aureus-Induced Macrophage Polarization by Suppressing CHI3L1. Inflammation. 2017;40(3):995–1005. 10.1007/s10753-017-0543-3 . [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Li Y, Li H, Song H, Zhai N, Lou L, et al. Brucella Dysregulates Monocytes and Inhibits Macrophage Polarization through LC3-Dependent Autophagy. Front Immunol. 2017;8:691 10.3389/fimmu.2017.00691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fordham JB, Naqvi AR, Nares S. miR-24 Regulates Macrophage Polarization and Plasticity. J Clin Cell Immunol. 2015;6(5). 10.4172/2155-9899.1000362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JK, Kim TS, Basu J, Jo EK. MicroRNA in innate immunity and autophagy during mycobacterial infection. Cell Microbiol. 2017;19(1). 10.1111/cmi.12687 . [DOI] [PubMed] [Google Scholar]
- 44.Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 2016;17(6):677–86. 10.1038/ni.3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahu SK, Kumar M, Chakraborty S, Banerjee SK, Kumar R, Gupta P, et al. MicroRNA 26a (miR-26a)/KLF4 and CREB-C/EBPbeta regulate innate immune signaling, the polarization of macrophages and the trafficking of Mycobacterium tuberculosis to lysosomes during infection. PLoS Pathog. 2017;13(5):e1006410 10.1371/journal.ppat.1006410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdalla AE, Duan X, Deng W, Zeng J, Xie J. MicroRNAs play big roles in modulating macrophages response toward mycobacteria infection. Infect Genet Evol. 2016;45:378–82. 10.1016/j.meegid.2016.09.023 . [DOI] [PubMed] [Google Scholar]
- 47.Rong H, Jiao H, Hao Y, Pang F, Li G, Peng D, et al. CD14 gene silencing alters the microRNA expression profile of RAW264.7 cells stimulated by Brucella melitensis infection. Innate Immun. 2017;23(5):424–31. 10.1177/1753425917707025 . [DOI] [PubMed] [Google Scholar]
- 48.Budak F, Bal SH, Tezcan G, Akalin H, Goral G, Oral HB. Altered Expressions of miR-1238-3p, miR-494, miR-6069, and miR-139-3p in the Formation of Chronic Brucellosis. J Immunol Res. 2016;2016:4591468 10.1155/2016/4591468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corsetti PP, de Almeida LA, Goncalves ANA, Gomes MTR, Guimaraes ES, Marques JT, et al. miR-181a-5p Regulates TNF-alpha and miR-21a-5p Influences Gualynate-Binding Protein 5 and IL-10 Expression in Macrophages Affecting Host Control of Brucella abortus Infection. Front Immunol. 2018;9:1331 10.3389/fimmu.2018.01331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Budak F, Bal SH, Tezcan G, Guvenc F, Akalin EH, Goral G, et al. MicroRNA Expression Patterns of CD8+ T Cells in Acute and Chronic Brucellosis. PLoS One. 2016;11(11):e0165138 10.1371/journal.pone.0165138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Budak F, Bal SH, Tezcan G, Akalin EH, Yilmaz A, Hiz P, et al. The microRNA expression signature of CD4+ T cells in the transition of brucellosis into chronicity. PLoS One. 2018;13(6):e0198659 10.1371/journal.pone.0198659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng K, Chen DS, Wu YQ, Xu XJ, Zhang H, Chen CF, et al. MicroRNA expression profile in RAW264.7 cells in response to Brucella melitensis infection. Int J Biol Sci. 2012;8(7):1013–22. 10.7150/ijbs.3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marinho FV, Benmerzoug S, Oliveira SC, Ryffel B, Quesniaux VFJ. The Emerging Roles of STING in Bacterial Infections. Trends Microbiol. 2017;25(11):906–18. 10.1016/j.tim.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol. 2002;169(4):1665–8. 10.4049/jimmunol.169.4.1665 . [DOI] [PubMed] [Google Scholar]
- 55.Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200(4):527–33. 10.1084/jem.20040976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol. 2010;184(7):3755–67. 10.4049/jimmunol.0902065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roux CM, Rolan HG, Santos RL, Beremand PD, Thomas TL, Adams LG, et al. Brucella requires a functional Type IV secretion system to elicit innate immune responses in mice. Cell Microbiol. 2007;9(7):1851–69. 10.1111/j.1462-5822.2007.00922.x . [DOI] [PubMed] [Google Scholar]
- 58.Gross A, Terraza A, Ouahrani-Bettache S, Liautard JP, Dornand J. In vitro Brucella suis infection prevents the programmed cell death of human monocytic cells. Infect Immun. 2000;68(1):342–51. Epub 1999/12/22. 10.1128/iai.68.1.342-351.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–49. 10.1016/j.cell.2007.04.027 . [DOI] [PubMed] [Google Scholar]
- 60.Altman BJ, Wofford JA, Zhao Y, Coloff JL, Ferguson EC, Wieman HL, et al. Autophagy provides nutrients but can lead to Chop-dependent induction of Bim to sensitize growth factor-deprived cells to apoptosis. Mol Biol Cell. 2009;20(4):1180–91. 10.1091/mbc.e08-08-0829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothchild AC, Sissons JR, Shafiani S, Plaisier C, Min D, Mai D, et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2016;113(41):E6172–E81. 10.1073/pnas.1608255113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roy S, Schmeier S, Kaczkowski B, Arner E, Alam T, Ozturk M, et al. Transcriptional landscape of Mycobacterium tuberculosis infection in macrophages. Scientific reports. 2018;8(1):6758 10.1038/s41598-018-24509-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahmed W, Zheng K, Liu ZF. Establishment of Chronic Infection: Brucella's Stealth Strategy. Front Cell Infect Microbiol. 2016;6:30 10.3389/fcimb.2016.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR Domain-containing Protein Mimics Properties of the Toll-like Receptor Adaptor Protein TIRAP. J Biol Chem. 2009;284(15):9892–8. Epub 2009/02/07. M805458200 [pii] 10.1074/jbc.M805458200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, et al. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008;4(2):e21 Epub 2008/02/13. 10.1371/journal.ppat.0040021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W, Ke Y, Wang Y, Yang M, Gao J, Zhan S, et al. Brucella TIR-like protein TcpB/Btp1 specifically targets the host adaptor protein MAL/TIRAP to promote infection. Biochem Biophys Res Commun. 2016;477(3):509–14. 10.1016/j.bbrc.2016.06.064 . [DOI] [PubMed] [Google Scholar]
- 67.Xu YY, Jin R, Zhou GP, Xu HG. Involvement of GATA1 and Sp3 in the activation of the murine STING gene promoter in NIH3T3 cells. Scientific reports. 2017;7(1):2090 10.1038/s41598-017-02242-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajashekara G, Glover DA, Banai M, O'Callaghan D, Splitter GA. Attenuated bioluminescent Brucella melitensis mutants GR019 (virB4), GR024 (galE), and GR026 (BMEI1090-BMEI1091) confer protection in mice. Infect Immun. 2006;74(5):2925–36. Epub 2006/04/20. 10.1128/IAI.74.5.2925-2936.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]