

Abstract
Myocardial infarction (MI) remains a major cause of mortality worldwide. Despite significant advances in MI treatment, many who survive the acute event are at high risk of chronic cardiac morbidity. Here we developed a cell-free therapeutic that capitalizes on the antifibrotic effects of micro(mi)RNA-101a and exploits the multi-faceted regenerative activity of mesenchymal stem cell (MSC) extracellular nanovesicles (eNVs). While the majority of MSC eNVs require local delivery via intramyocardial injection to exert therapeutic efficacy, we have developed MSC eNVs that can be administered in a minimally invasive manner, all while remaining therapeutically active. When loaded with miR-101a, MSC eNVs substantially decreased infarct size (12 ± 2.4% vs. 21.4 ± 5.7%) and increased ejection fraction (53.6 ± 7.6% vs. 40.3 ± 6.0%) and fractional shortening (23.6 ± 4.3% vs. 16.6 ± 3.0%) compared to control. These findings are significant as they represent an advance in the development of minimally invasive cardio-therapies.
Keywords: Extracellular vesicles, MSC, fibrosis, myocardial infarction, macrophage polarization
Background
Myocardial infarction (MI) remains a major cause of death in the US and beyond (1, 2). Even though advances in MI treatment by coronary reperfusion and/or systemic medication have significantly decreased mortality, many who survive the acute event continue to be at high risk of arrhythmias, congestive heart failure, and stroke (3). After MI, the myocardium undergoes extensive remodeling, with multiple processes affecting disease progression and healing of the damaged tissue.
Inflammatory monocytes and macrophages play important roles in disease progression and remodeling of the infarcted myocardium. Immediately after MI, macrophages infiltrate the infarcted myocardium, remove apoptotic cardiomyocytes, and degrade the extracellular matrix through the secretion of enzymes and cytokines (4). Subsequently, the injured tissue is replaced with non-contractile, fibrotic tissue and extracellular matrix, which increases myocardial stiffness and worsens cardiac function. Furthermore, fibrosis reduces blood flow through decreased capillary density and negatively affects mechanical strength (5). While established heart failure therapies including statins and aldosterone antagonists have minor anti-fibrotic effects, inhibiting fibrosis is not their main mode of action and anti-fibrotic mechanisms are poorly understood (6). Thus, no current therapy specifically and efficiently targets fibrotic tissue in the infarcted heart.
Transforming growth factor beta (TGF-β) signaling is extensively involved in cardiac fibrosis (7), so therapeutic agents capable of downregulating TGF-β would be highly desirable. MicroRNA (miR)-101a has been shown to target and modulate TGF-β and Wnt signaling, both of which are involved in fibroblast proliferation and activation and, therefore, fibrosis (8, 9). However, due to its anionic nature and large molecular weight, systemic delivery of naked miR-101a, as with other small RNAs, is ineffective and requires a delivery vehicle (10).
While macrophages are important for the removal of apoptotic cardiomyocytes in the initial phase post-MI, their excessive persistence can impair infarct healing. Aside from contributing to fibrosis, macrophages with a pro-inflammatory phenotype also secrete cytokines and chemokines that exacerbate myocardial inflammation (11). Since disease progression after MI is complex, a therapeutic system that simultaneously targets both cardiac remodeling and the inflammatory response after MI would be highly beneficial. Mesenchymal stem cell (MSC)-derived extracellular nanovesicles (eNVs) are one such promising therapeutic modality because they possess multifaceted regenerative effects. MSC eNVs possess intrinsic bioactivity and are anti-fibrotic, induce angiogenesis, and inhibit cardiomyocyte apoptosis (12, 13), all of which are pertinent to cardiac repair after MI.
The objective of this study was to exploit the antifibrotic effects of miR-101a and deliver it using a carrier with intrinsic tissue regenerative capacity, namely MSC eNVs (14). Carriers with intrinsic bioactivity are especially attractive in that they not only serve as drug vehicles but, unlike inert carriers, also exert therapeutic effects. One of the major drawbacks of current MSC eNV-based therapeutics is that they require direct intramyocardial injection to induce therapeutic effects. This is a highly invasive procedure that can lead to severe complications such as arrhythmia and tissue irritation. We showed that MSC eNVs mediate cardioprotective effects when they are loaded with anti-fibrotic miR-101a, even without direct myocardial injection. Intravenously administered miR-101a MSC eNVs improve cardiac function in a mouse model of MI, have antifibrotic effects, and also polarize macrophages to an anti-inflammatory phenotype. These findings are significant as they represent an advance in the development of minimally invasive cardio-therapies.
Methods
Methods and materials are detailed in the supplementary information.
Extracellular nanovesicle (eNVs) isolation
eNVs were isolated from human bone marrow-derived MSCs obtained from the American Type Culture Collection (ATCC PCS-500–012) and cultured with MesenPRO RS™ Medium (Gibco™, Gaithersburg, MD) (14–17). eNVs were isolated by differential centrifugation.
eNV characterization
eNVs were prepared as previously described and analyzed by TEM (14). The size distribution of eNVs was determined by nanoparticle tracking analysis (NTA) (NanoSight, Malvern) and the zeta potential of eNVs was measured by dynamic light scattering (DLS) using Nanobrook Omni (Brookhaven Instruments).
Enrichment of miRNAs into eNVs
miRNA enrichment was performed using the Neon Transfection System (Thermo Fisher Scientific). Briefly, 240,000 MSCs were electroporated with 30 μg of the double-stranded miR-101a-3p mimic (mature sequence 5’-UACAGUACUGUGAUAACUGAA-3’), which is also referred to as miR-101a. MSCs were washed thoroughly to remove free miRNA and cultured with EV-depleted medium. ENVs were collected 3 days after electroporation and analyzed for miR-101a-3p content by RT-PCR.
Animals
Male C57BL/6 mice were used (10–12 weeks) for the in vivo studies. All animal procedures were approved by the University at Buffalo - State University of New York Institutional Animal Care and Use Committee (IACUC) or by the University of North Carolina at Chapel Hill IACUC. All methods and experiments were performed in accordance with the U.S National Institutes of Health Guide for Care and Use of Laboratory Animals. Humane care and treatment of animals were ensured.
Mouse model of myocardial infarction
MI was induced as previously described (18). Briefly, mice were anesthetized with isoflurane. Body temperature was maintained with a heating pad. A small incision under the mandible was made to visualize the trachea. Mice were intubated with a 20-gauge blunt needle and connected to a ventilator (Harvard Rodent Ventilator; Harvard Apparatus, Holliston, MA). A left lateral thoracotomy was made to expose the heart. The left anterior descending (LAD) artery was occluded permanently with an 8–0 nylon suture. The thorax was closed in layers (ribs, muscles, and skin). Analgesic treatments were provided according to the protocol and mice were monitored carefully.
Biodistribution of MSC eNVs – Group I
For biodistribution studies (n = 3), 2 mg/kg of 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide (DiD)-labeled eNVs (n =3) or PBS (n=3) was administered via tail vein injection into C57BL/6 mice on day 2 after inducing MI. To fluorescently label the eNVs, 5 μl DiD was dissolved in ethanol and incubated with 1 ml eNVs at 37°C for 1 h. Ethanol and any unincorporated DiD were removed using the Pierce protein concentrator (3K). After 5 h of injection, blood was collected by cardiac puncture under isoflurane anesthesia followed by centrifugation at 2000 rpm for 10 min to obtain the serum. Organs including brain, lung, heart, liver, spleen, kidneys, and bones were collected and weighed. EV biodistribution was analyzed using the IVIS Spectrum in vivo imaging system (PerkinElmer, Waltham, MA), and average region of interest (ROI) signals were calculated using Living Image 4.5.2 software (PerkinElmer). A standard curve was generated to calculate the % of total injection dose (%ID) for each organ, and the data was presented as %ID divided by organ weight.
Assessment of cardiac function, immunofluorescent staining and infarct size - Group II
(1). Cardiac function
To assess therapeutic efficacy, 2 mg/kg of unmodified and miR-101a eNVs were administered to C57BL/6 mice via tail vein injection at day 2 and 3 after inducing MI (n=5 per group). Studies were performed in a blinded manner. Of the 15 animals randomized to treatment, 3 were excluded because of sudden death 2–3 days post MI [1 was PBS treated, 1 was treated with eNV, and 1 was treated with modified eNV]. PBS-treated mice served as controls. Heart function was evaluated by echocardiography on day 11 after surgery. Mice were anesthetized by isoflurane (1–3%). Echocardiograms were performed with the GE Healthcare Echocardiography Vivid 7 system (GE Healthcare, Chicago, IL) equipped with an i13L probe. Two-dimensional mode parasternal long and short axis views were recorded. The left ventricular (LV) dimensions and wall thicknesses were determined from M-mode images at the mid-papillary muscle level. The LV stroke volume (SV) was calculated as the difference between the end diastolic volume (EDV) and the end systolic volume (ESV). LV ejection fraction (EF) was calculated as (EF=[(EDV-ESV)/EDV]*100). All measurements were performed with investigators blinded to experimental groups.
(2). Immunofluorescence staining for TGF- β1, collagen and LC3B
After heart function measurements, the same mice were sacrificed at day 13 post MI and the heart samples were collected for histological staining. Briefly, potassium chloride (30 mM) was injected into the LV to arrest the heart in diastole and harvested mouse hearts were embedded in OCT.
TGF- β1, collagen I and LC3B were analyzed by immunofluorescent staining. Briefly, heart cryosections were incubated with anti-TGF-β1 (Cat# NBP2–22114, Novus Biologicals), anti-collagen I (Cat# ab34710, Abcam) or anti-LC3B (Cat# NBP2–46892, Novus Biologicals) followed by Alexa Fluor 568-conjugated secondary antibodies (Invitrogen). For collagen staining, a total of 4 whole heart sections transversely from the apex to base at 900 μm intervals per heart were analyzed (n = 4 mice per group). For TGF-β1 and LC3B staining, 3 images were taken at the border zone of the heart with n=4 mice per group. Images were taken with a Zeiss AxioImager Microscope and analyzed using Image J.
(3). Immunofluorescence staining for CD68, iNOS, and CD206
Heart sections were incubated with the primary antibodies (CD68, Bio-Rad, Cat# MCA1957; iNOS, Abcam, Cat# ab15323; CD206, R&D Systems, Cat# AF2535). Samples were washed in TBST 3x and incubated with Alexa Fluor 646-, Alexa Fluor 568-, or Alexa Fluor 488-conjugated secondary antibodies (Invitrogen) for 45 min at RT. Samples were washed 3x with TBST, counter-stained with DAPI, washed 3x with TBST, mounted using FluorSave (Merck Millipore), and imaged using the Zeiss fluorescence microscope.
(4). Assessment of infarct size
To determine infarct size, hearts were sliced transversely from the apex to base at 900 μm intervals. Heart sections (10 μm) were fixed with 10% neutral formalin solution and stained with the Masson Trichrome Stain Kit (Cat# 87019; Richard-Allan Scientific Co, San Diego, CA). Whole heart images were taken using the Zeiss AxioImager Microscope (Karl Zeiss). Infarct size was measured by Image J and calculated as: (1) Area measurement: infarct size = (sum of infarct areas from all sections)/(sum of left ventricle areas from all sections) × 100, (2) Midline length measurement: midline infarct length was defined as midline length of infarct that included more than 50% of the whole thickness of myocardial wall, and infarct size = (sum of midline infarct length from all sections)/(sum of midline circumferences from all sections) × 100 (19).
Results
Characterization of MSC eNVs
The hydrodynamic diameter of MSC eNVs measured by NTA and TEM was approximately 81 nm (Figure 1A,B), and eNVs were positive for CD63 and CD9 as expected (20) (Figure 1C,D). The zeta potential of MSC eNVs was slightly negatively charged (−4.7 ± 2.0 mV). MSCs produced 4474.8 ± 468 exosomes per cell over three days of culturing as quantified by NTA.
Figure 1.
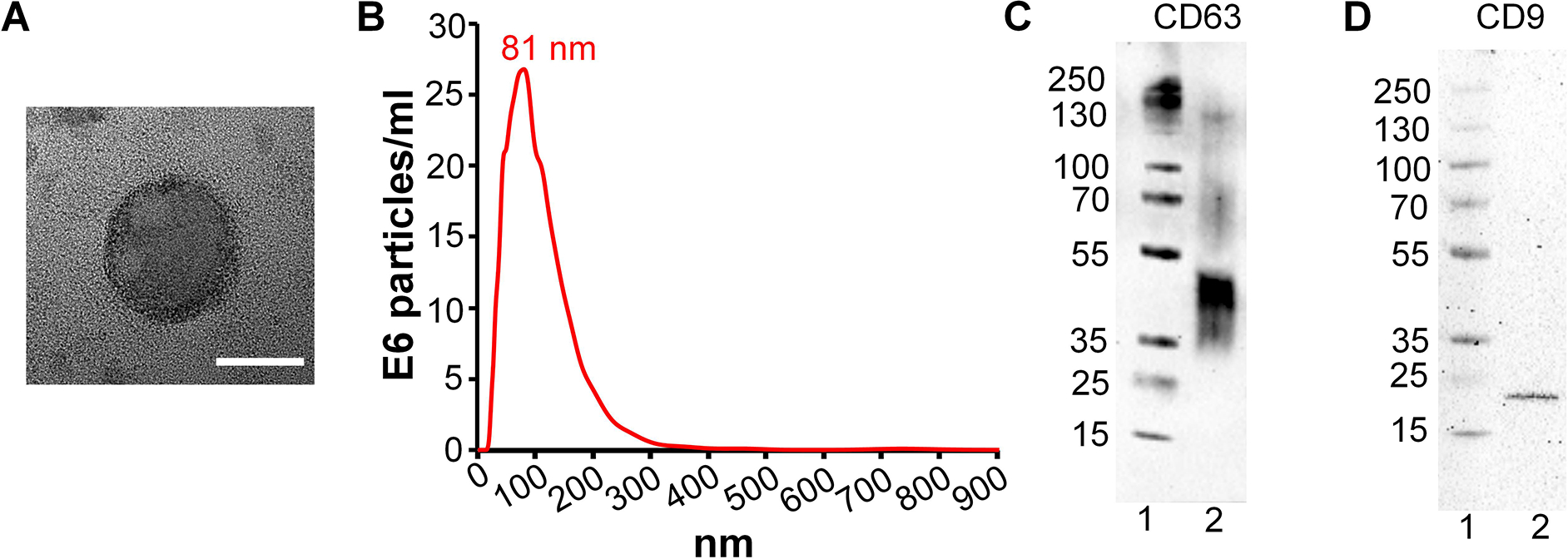
Characterization of MSC-derived eNVs. (A) TEM images of a representative MSC eNV, scale bar = 50 nm. (B) Size distribution of eNVs by NTA. (C) CD63 eNV surface marker protein expression by western blotting, protein ladder (lane 1) and eNV sample (lane 2) (D) CD9 MSC eNV surface marker expression by western blotting, protein ladder (lane 1) and eNV sample (lane 2).
miR-101a loaded MSC eNVs inhibit TGF-β and collagen production
To further improve the intrinsic anti-fibrotic effects of MSC eNVs, MSC eNVs were loaded with the anti-fibrotic miR-101a (Figure 2A,B). miR-101a was selected because it has been shown to inhibit fibrosis through targeting TGF-β in ischemic cardiac diseases (8, 9). MSC eNVs electroporated with miR-101a using the virus-free electroporation approach achieved 315-fold enrichment compared to eNVs derived from unmodified MSCs (p < 0.05, Figure 2B). Since AngII is a critical mediator of cardiac fibrosis by enhancing TGF-β secretion and has been shown to be increased after MI, we measured the effects of miR-101a loaded eNVs on AngII-stimulated TGF-β secretion (21). miR-101a eNVs significantly decreased AngII-induced TGF-β1, TGF-β2, and collagen type 1A1 expressions compared with unmodified eNVs (Figure 2C–E). Moreover, miR-101a eNVs significantly downregulated collagen type 1A1 in exogenously TGF-β-stimulated cardiac fibroblasts compared to non-treated cells (Figure 2F).
Figure 2.
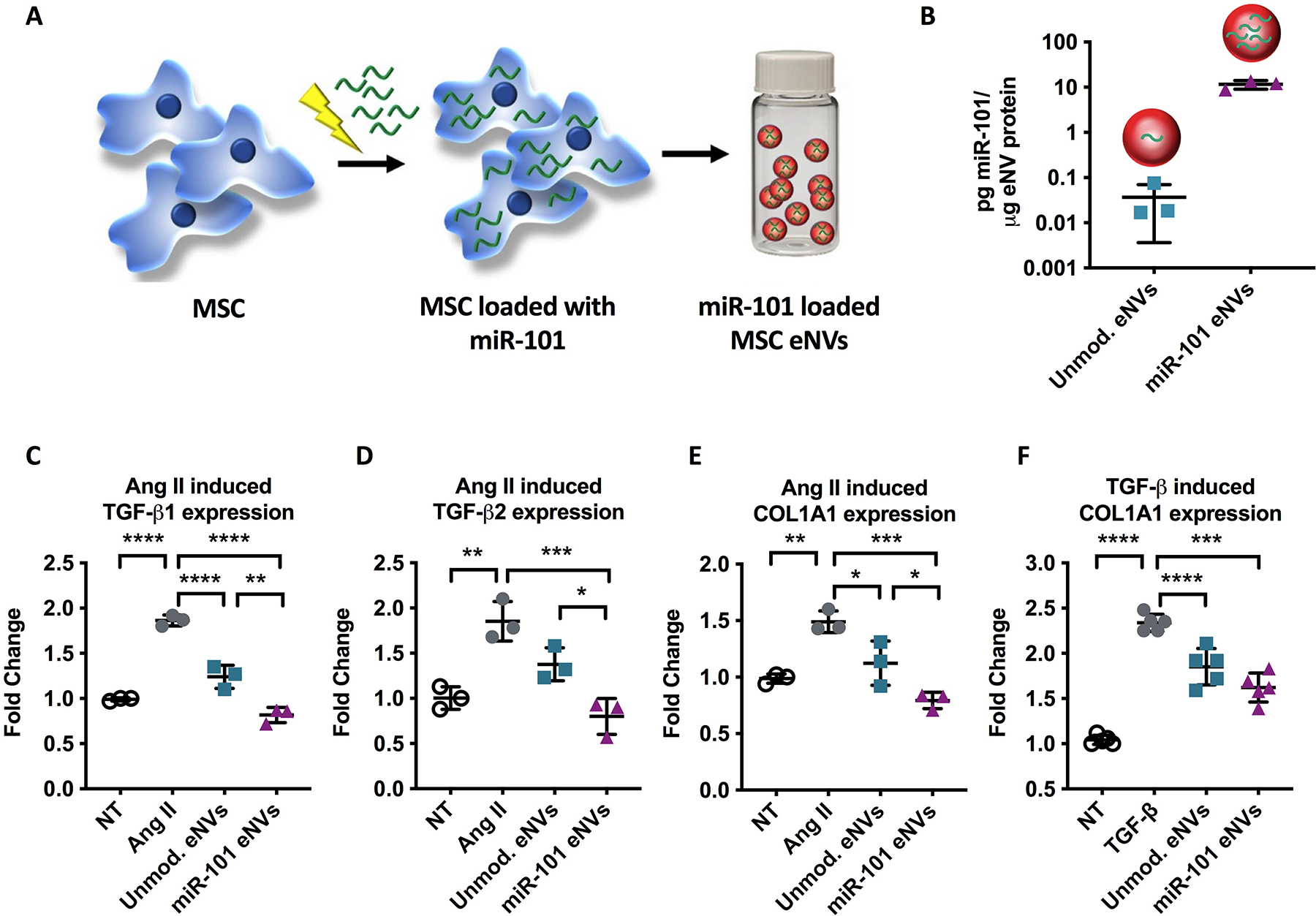
(A,B) eNVs derived from MSCs electroporated with miR-101a showed 315-fold enrichment with miR-101a than eNVs isolated from non-electroporated MSCs. (C-E) Angiotensin II (Ang II) induced TGF-β1, TGF-β2, and collagen expression in cardiac fibroblasts pre-treated with unmodified eNVs and miR-101a eNVs. (F) TGF-β induced collagen expression in cardiac fibroblast pretreated with unmodified eNVs and miR-101a eNVs. Data are presented as mean ± SD with **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=3–5.
Biodistribution of MSC eNVs and colocalization with CD68-positive macrophages in vivo
Next we assessed the biodistribution of DiD-labeled MSC eNVs after systemic administration via tail vein injection in a mouse model of MI. The majority of MSC eNVs were found in the spleen (64.8 ± 7.2%) and liver (56.7 ± 9.7%) followed by the lung (5.4 ± 1.3%), bone marrow (4.7 ± 1.3%), and kidneys (2.0 ± 0.7%) (Figure 3A–C). No brain deposition was detectable. About 3.8 ± 1% of MSC eNVs were found in the infarcted heart (Figure 3B), most likely as a result of the enhanced permeability of the leaky vasculature after MI (22). Importantly, MSC eNVs generally colocalized with macrophages present in the infarcted myocardium and spleen (Figure 3 D,E).
Figure 3.
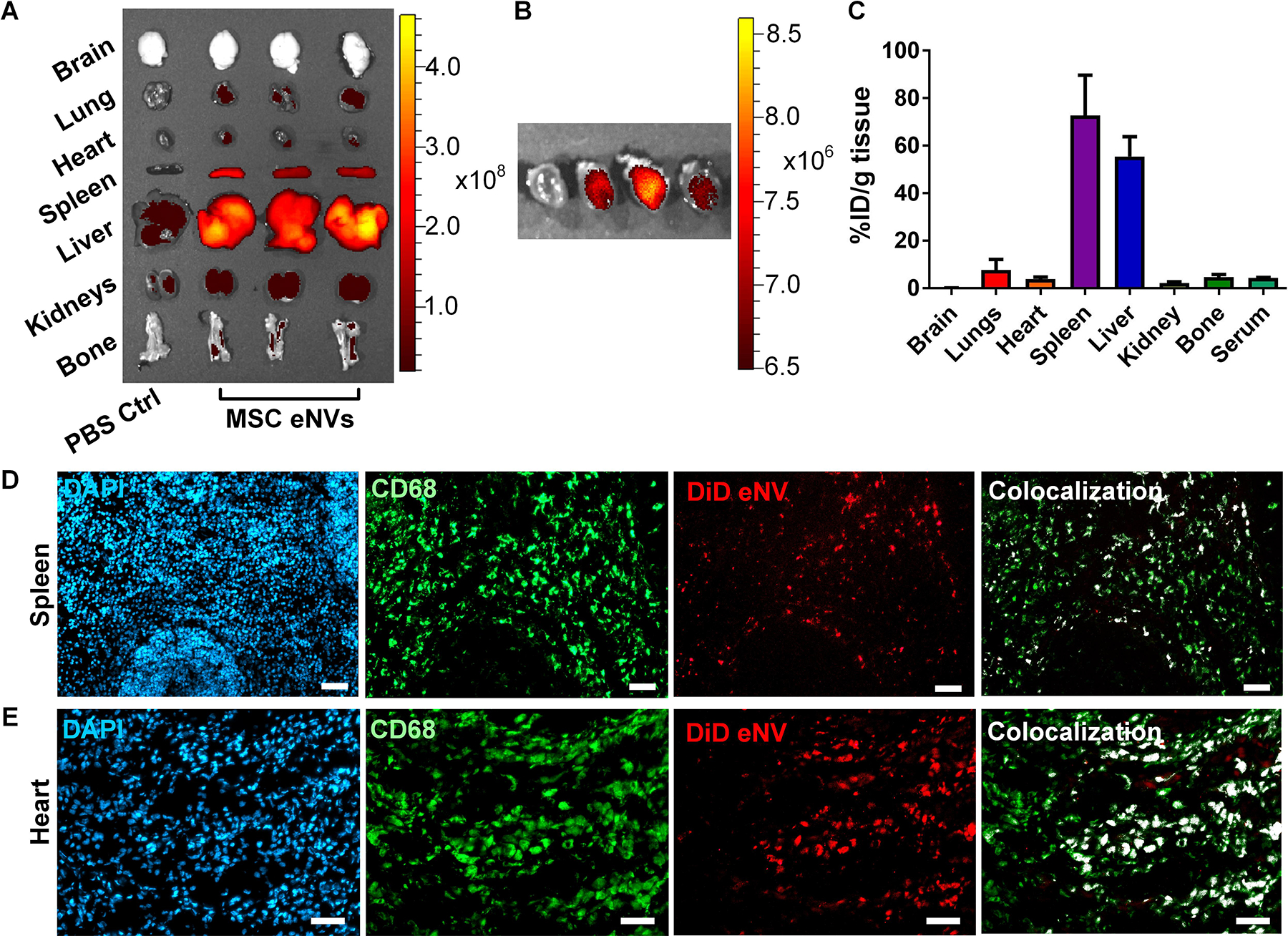
In vivo biodistribution of MSC-derived eNVs in mice with MI. (A) Representative images acquired using the IVIS Spectrum in vivo imaging system of DiD-labeled MSC eNVs in brain, lungs, heart, spleen, liver, kidneys, and bone. (B) IVIS imaging shows eNV deposition in the infarcted heart. (C) Quantification of % of full injection dose (% ID) in organs. Data are presented as mean ± SD, n=3. Colocalization studies from (D) splenic and (E) and cardiac tissues. Nuclei were stained with DAPI (blue), macrophages were stained with CD68 (green), eNV were stained with DiD (red), and colocalization of DiD-labeled eNV with CD68 are shown in white. Scale bar = 50 μm.
Unmodified eNVs and miR-101a eNVs are non-toxic and show anti-inflammatory effects in vivo
While eNV biodistribution was highest in the spleen and liver, neither organ showed signs of toxicity after dosing with either unmodified or miR-101a eNVs (Figure S2). C57BL/6 mice were administered PBS, unmodified eNVs, or miR-101a eNVs and harvested 7 days after treatment. H&E staining of the mouse liver showed normal lobular tissue without any signs of atrophy, granulation, necrosis, or inflammatory cell infiltrate (Figure S2A). Masson’s trichrome staining of the same organ did not show dystrophic collagen staining and had minimal overall collagen staining (Figure S2A). Quantitative histological analysis of Masson’s trichrome stains showed that in liver tissue, miR-101a-eNV treated mice had slightly less overall collagen staining than animals treated with unmodified eNVs or PBS (Figure S2B). However, this decrease was not statistically significant. H&E staining of spleen tissue did not show signs of toxicity such as necrosis, granular tissue, apoptosis, or inflammation (Figure S2C). Masson’s trichrome staining of the spleen did not show any dystrophic collagen deposition, nor did it show any significant difference in collagen scoring between the PBS, unmodified eNV, or miR-101a eNV treated mice (Figure S2D).
Remarkably, both unmodified eNVs and miR101-eNVs robustly reduced a number of pro-inflammatory cytokines including IL-3, IL-4, IL-6, TNFα, and Trem-1 (Figure S3B). In vivo, MSC eNVs did not affect the majority of anti-inflammatory cytokines such as CCL1, IL-1ra, IL-13, IL-10, and CD54 (Figure S3A). Both eNV treated groups had higher expression of CXCL12, a chemokine associated with stem cell recruitment (23, 24). Stem cell recruitment to the heart has been shown to be beneficial for cardiac repair (Figure S3C) (25, 26). Unmodified and miR-101a eNVs also reduced the cleavage of C5 into C5a (Figure S3C). C5 cleavage inhibition has been shown to reduce MI in pre-clinical studies (27). Both MSC eNV treatment groups reduced chemokines such as IL-5, CCL17, CXCL1, CXCL2, CXCL10, CXCL11, and CXCL13 (Figure S3C & D). Elevated CCL17 is associated with increased cardiac fibrosis, and elevated CXCL1, CXCL2 are associated with poorer outcomes in MI patients (28–30). Elevated CXCL10 and CXCL11 have been associated with MI-related co-morbidities such as coronary artery disease, left ventricular dysfunction, atherosclerosis, and heart failure (31–34). It is interesting to note that both eNVs reduced G-CSF and GM-CSF, but did not reduce M-CSF, a macrophage-stimulating chemokine. Elevated G-CSF has been associated with acute MI in compromised clinical patients, and GM-CSF is associated with detrimental inflammation post-MI, but pre-clinical studies have shown that elevated M-CSF is beneficial post-MI (35–40).
MSC eNVs polarize macrophages to the anti-inflammatory phenotype
Because we discovered that MSC eNVs colocalized with macrophages, we examined the effects of MSC eNVs on macrophages. Depending on their microenvironment, macrophages can adopt different phenotypes, with the anti-inflammatory phenotype promoting wound healing and the inflammatory phenotype and contributing to disease progression (41–43). As shown in Figure 4 A, unmodified and miR-101a MSC eNVs increased the polarization of the bone marrow-derived macrophages to the anti-inflammatory phenotype by 1.5 to 2-fold as measured by Arginase 1 and CD206 expression. No significant effects on pro-inflammatory macrophage markers were observed (iNOS and IL-6). In line with the gene expression markers quantified by RT-PCR, immunofluorescence staining showed that BMDMs treated with both unmodified eNVs and miR-101a eNVs were CD206 positive (Figure 4B). Similar to the cells treated with IL-4, a positive control for anti-inflammatory BMDMs, BMDMs treated with unmodified eNVs and miR-101a eNVs adopted a more spindle-like shape. By contrast, pro-inflammatory BMDMs stimulated with LPS and IFN-ɣ stained positive for iNOS and were significantly larger and more spherical in morphology compared to IL-4-stimulated BMDMs (Figure 4B). No statistically significant differences between unmodified eNVs and miR-101a eNVs were observed (Figure 4A,B).
Figure 4.
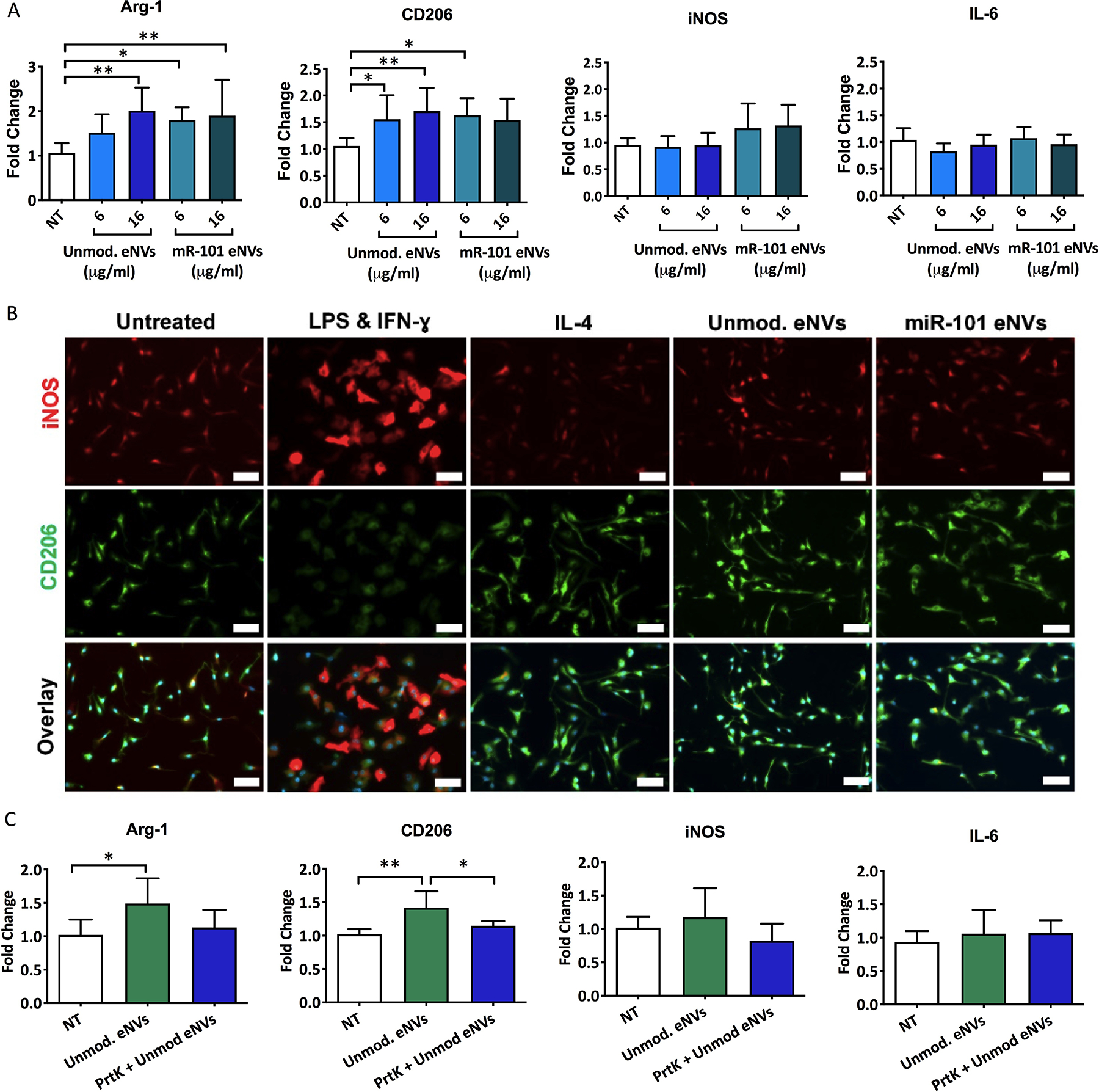
(A) Unmodified eNVs and miR-101a eNVs induce macrophage polarization to the anti-inflammatory phenotype as measured by Arginase 1 and CD206 expression. No statistically significant differences in pro-inflammatory markers iNOS and IL-6 were observed. (B) Immunofluorescent staining of BMDMs treated with LPS and IFN-ɣ, IL-4, unmodified MSC eNVs and miR-101a MSC eNVs. iNOS (red) served as a marker of the pro-inflammatory BMDM phenotype and CD206 served as a marker of the anti-inflammatory BMDM phenotype. Nuclei were stained with DAPI (blue). Scale bar = 50 μm. (C) Effect of proteinase K-treated BMDM eNVs on the gene expression of anti-inflammatory markers Arg-1 and CD206 and pro-inflammatory markers iNOS and IL-6. Data are presented as mean ± SD n=6–8 with *p<0.05, **p<0.01, and ****p<0.0001 with one-way ANOVA followed by Dunnett’s post-test.
We next asked if proteins on the surface of the nanovesicles mediated some of the anti-inflammatory effects on BMDMs by treating MSC eNVs with proteinase K to digest all surface-bound proteins. Proteinase K-digested MSC eNVs did not upregulate anti-inflammatory-associated genes such as Arg-1 and CD206 (Figure 4C), indicating that surface proteins play a role in the anti-inflammatory effects of MSC eNVs.
MSC eNVs increase the number of anti-inflammatory macrophages in the infarcted heart
After demonstrating that MSC eNVs polarized macrophages to the anti-inflammatory M2 phenotype, we determined the number of infiltrating CD68-positive monocytes and the percentage of anti-inflammatory phenotype macrophages in the infarcted myocardium after treatment with MSC eNVs in vivo. To quantify the number of CD68-positive and anti-inflammatory macrophages, cardiac cryosections were stained with CD68 as a general macrophage marker, iNOS to identify pro-inflammatory macrophages, and CD206 to identify anti-inflammatory macrophages (Figure 5). Mice treated with unmodified MSC eNVs (56 ± 5% M2 macrophages) and miR-101a-loaded MSC eNVs (53 ± 6% M2 macrophages) showed a significant increase in infiltrating anti-inflammatory macrophages in the infarcted area compared to mice treated with PBS alone (36 ± 2% M2 macrophages). With respect to the CD206/iNOS (M2/M1) ratio, PBS-treated mice had a significantly lower ratio (0.67 ± 0.1) in the infarcted area than mice treated with unmodified MSC eNVs (1.08 ± 0.26) and miR-101a-loaded MSC eNVs (1.04 ± 0.09), indicating that unmodified MSC eNVs and miR-101a-loaded eNVs have anti-inflammatory effects. This is supported by the in vivo serum data, which showed that all MSC eNV treated mice had generally lower expression of pro-inflammatory cytokines (Figure S3). No differences were observed between the unmodified MSC eNVs and miR-101a-loaded eNVs. This is not unexpected since the miR-101a is not known to target inflammatory processes.
Figure 5.
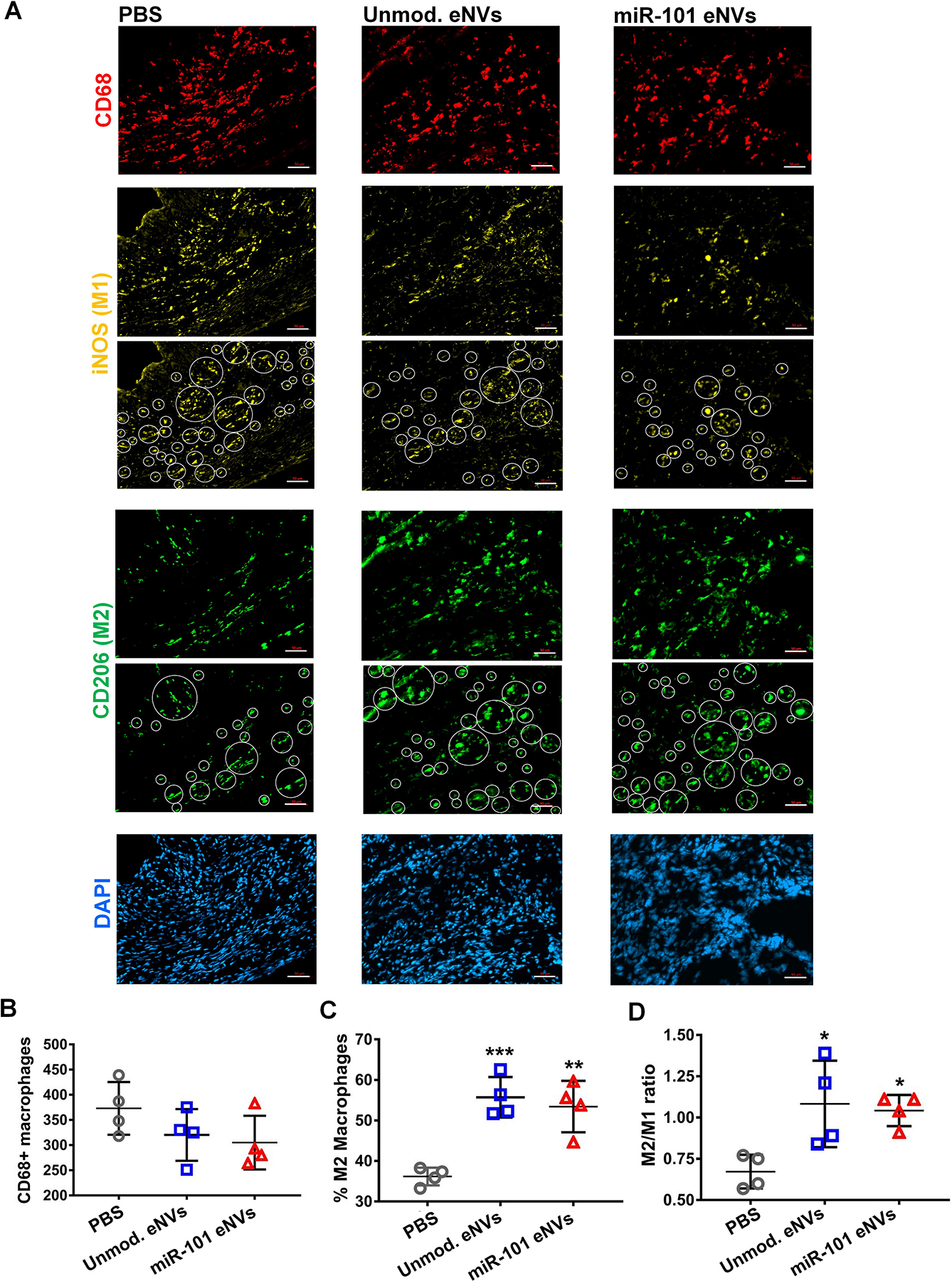
(A) Immunofluorescent staining of cryosections of the infarcted heart of mice treated with PBS, unmodified eNVs, and miR-101a-loaded eNVs. Cryosections were stained for CD68 (red), the pro-inflammatory marker iNOS (yellow), the anti-inflammatory marker CD206 (green), and nuclei (DAPI, blue). White circles indicate iNOS and CD206 positive cells that were detected in the infarcted area by ImageJ. Quantification of (B) CD68-positive macrophages in the infarcted area, (C) % anti-inflammatory macrophages, and (D) M2/M1 (CD206/iNOS) ratio. Data are presented as mean ± SD (n = 4 per group). Images were taken from the infarcted areas. Scale bar = 50 μm.
miR-101a MSC eNVs decrease TGF-β1 and collagen production in the infarcted myocardium
To assess the in vivo effects of miR-101a MSC eNVs and unmodified MSC eNVs on TGF-β and collagen production after MI, heart sections from mice treated with PBS, unmodified eNVs, and miR-101a-loaded MSC eNVs were stained with anti-TGF-β1 (Figure 6A) and anti-collagen antibody (Figure 6C) and assessed by immunofluorescence. Representative images taken from the border zone, which was defined as the 2 mm area encircling the infarct area were shown (Figure 6 A,C). miR-101a-loaded MSC eNVs treatment significantly decreased TGF-β1 expression (5.3 ± 1.3%) compared to the PBS group (13.5 ± 2.6%; P<0.01) or unmodified eNVs group (9.8 ± 3.3; P<0.05) (Figure 6B). Moreover, miR-101a-loaded MSC eNVs treated hearts had significantly less collagen (6.1 ± 0.3%) compared to mice treated with PBS (11.4 ± 1.5%; p<0.001) or unmodified eNVs (9.2 ± 1.0%; p<0.05) for the whole heart analysis (Figure 6D).
Figure 6.
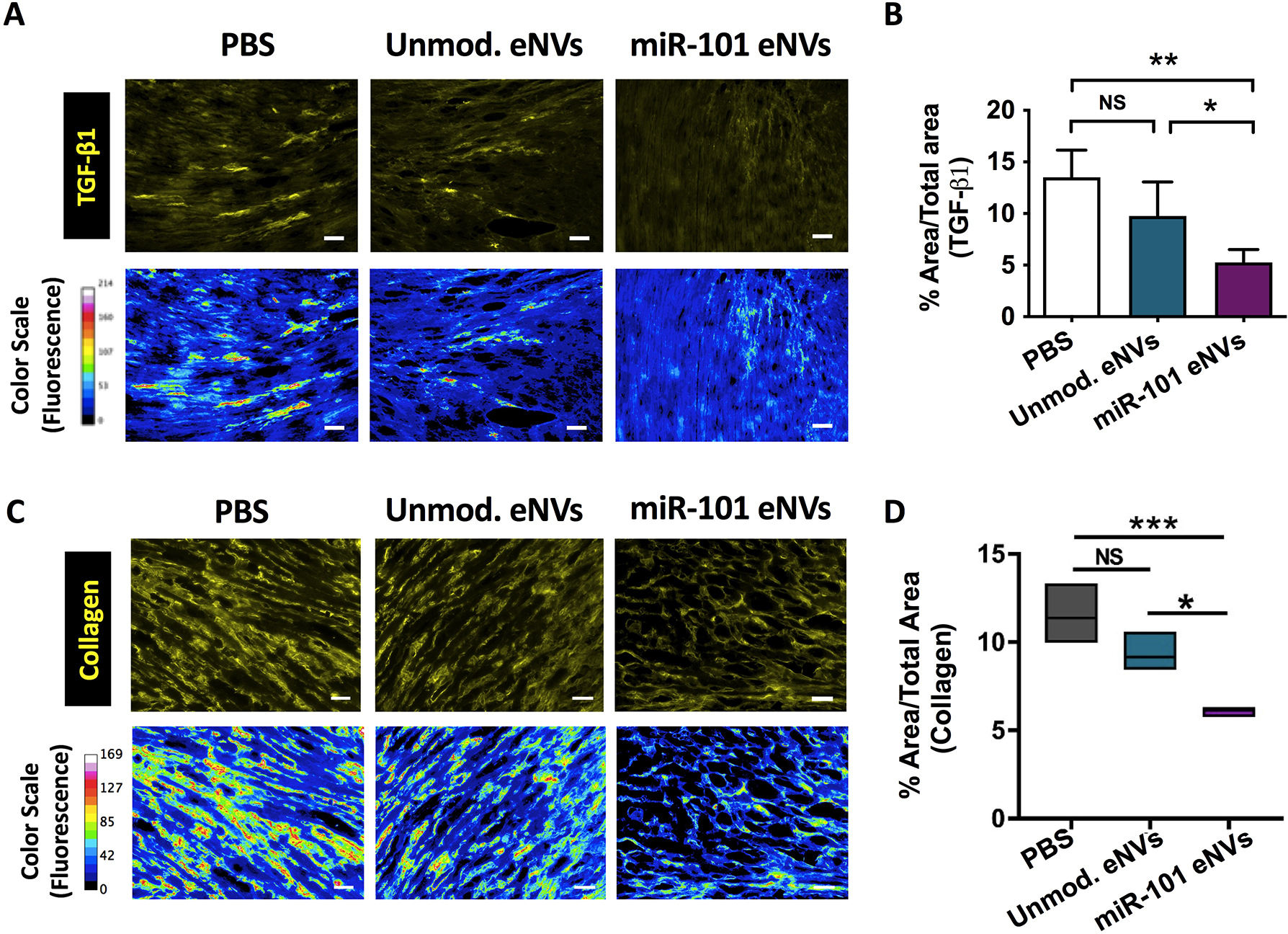
Effects of unmodified and miR-101a eNVs on TGF-β and collagen production. C57BL/6 with induced MI were treated with unmodified eNVs or miR-101a eNVs. (A) TGF-β1 and (C) collagen I were stained with anti-TGF-β1 antibody and anti-collagen I antibody with secondary visualization with Alexa Fluor 568 (yellow). Representative images from the border zones, which were defined as the 2 mm area encircling the infarcted area are shown. To better visualize TGF-β1 and collagen staining intensity, a 16-color range function in ImageJ was used to indicate negative staining (blue), yellow and orange indicate medium-intensity staining, and red and pink indicate high-intensity staining. (B) Quantification of TGF-β1 by image J. (D) Quantification of collagen of the entire cross-sectional area of the heart by Image J. Data are presented as mean ± SD (n =4) with *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA followed by Tukey’s post-test. Scale bar = 50 μm.
miR-101a-loaded MSC eNVs display cardioprotection after acute MI
Finally, we assessed the effect of miR-101a-enriched eNVs on myocardial function 13 days after MI. Unmodified eNVs showed a trend towards improving heart function by echocardiography and the improvements were not significant compared to PBS-treated mice (Figure 7D). However, miR-101a-loaded MSC eNVs significantly decreased infarct size compared to PBS-treated controls analyzed by area-based (12 ± 2.4% vs. 21.4 ± 5.7%; p<0.05; Figure 7B) and length-based measurement (14.55 ± 2.66% vs. 31.89 ± 7.06%; p<0.01; Figure 7B). Mice treated with miR-101a-loaded MSC eNVs displayed significantly higher EF (53.6 ± 7.6% vs. 40.3 ± 6.0% p<0.05; Figure 7D) and fractional shortening (23.6 ± 4.3% vs. 16.6 ± 3.0; p<0.05; Figure 7D) compared to mice treated with PBS. Other functional parameters assessed by echocardiography such as LVIDs and ESV (Figure 7D) were also significantly improved by miR-101a-loaded MSC eNVs.
Figure 7.
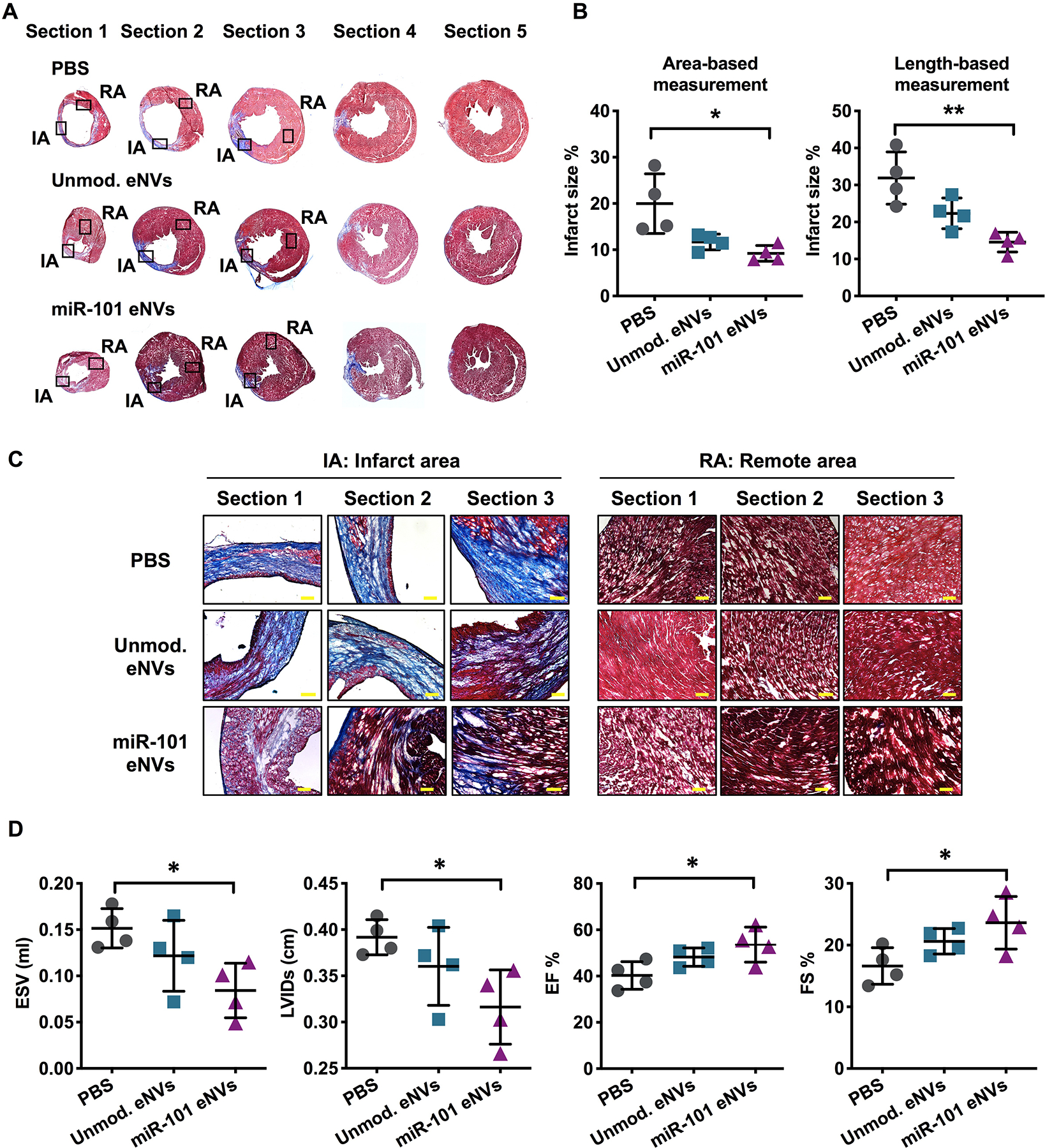
Heart function and infarct size after MI. (A,C) Masson trichrome staining of representative cryosections of infarcted hearts. (B) Infarct area expressed as % infarct size by area-based measurements (infarct area/LV (%)) and length-based measurements (midline infarct length/LV midline length (%)). (D) Analysis of ESV, LVIDs, ejection fraction (EF%), and fractional shortening (FS%) after MI by echocardiography. Data are presented as mean ± SD with *p < 0.05, **p < 0.01 by one-way ANOVA followed by Tukey’s post-test, n=4. Scale bar = 100 μm.
Discussion
The current management of acute MI involves restoring cardiac perfusion through mechanical and drug interventions. Even when blood flow is quickly restored, the ischemic myocardium suffers injury and more efficient therapeutic approaches are required to fully restore cardiac function. In the first few days after MI, pro-inflammatory macrophages infiltrate the injured myocardium where they predominate as they clear cellular debris and degrade the extracellular matrix (44, 45). Pro-inflammatory macrophages also release TGF-β, which stimulates fibroblast proliferation and collagen secretion from cardiac fibroblasts (46). Although extracellular production of collagen is essential for cardiac repair, excessive collagen production leads to maladaptive cardiac remodeling. Timely reprogramming of inflammatory macrophages to an anti-inflammatory phenotype would, therefore, be important to myocardial healing.
While MSC eNVs have been tested for their cardio-regenerative therapy, the majority of studies use the invasive approach of direct intramyocardial injection of MSC eNVs to achieve high local concentrations at the affected site. For example, miR-132-loaded eNVs preserved heart function after direct transplantation into the heart (47). Similarly, miR-19a-enriched eNVs isolated from MSCs with GATA-4 overexpression reduced infarct size when injected to the ischemic border (48). Since direct injection into the myocardium is (a) highly invasive, (b) can lead to severe complications such as arrhythmia and tissue irritation, and (c) is often limited to a few injections, having a non-invasive route of administration could represent a significant advance in the development of next-generation cardio-therapies.
To develop a minimally invasive cardio-therapeutic, we loaded MSC eNVs with anti-fibrotic miR-101a to further improve on the reparative effects of MSC eNVs. miR-101a is a key inhibitor of fibrosis through targeting TGF-β. To overexpress miR-101a, studies have utilized viral transfection systems such as adenoviral transfection (8). However, the risk of immunogenicity with adenoviral gene therapies limits its broad clinical application, and indeed the inflammatory response evoked by adenoviruses can be fatal (49). MSC eNVs are highly attractive as a nucleic acid carrier for several reasons. First, MSC eNVs display low immunogenicity due to a lack of MHCII molecules and thus could be used as an allogeneic therapy. Second, the lipid bilayer of eNVs protects encapsulated nucleic acids against degrading enzymes. Third, unlike many synthetic carriers that are inert, MSC eNVs display intrinsic regenerative properties as they are anti-fibrotic, anti-apoptotic, and mediate angiogenesis (14).
We discovered, that enriching MSC eNVs with the anti-fibrotic miRNA miR-101a enhanced the therapeutic effect of MSC eNVs. Both unmodified and miR-101a eNVs predominantly accumulated in the liver and spleen. However, neither organ showed signs of toxicity after dosing with either unmodified or miR101 eNVs. Instead, both eNV treatments reduced serum pro-inflammatory cytokines and several chemokines. The miR-101a eNVs significantly decreased AngII-induced TGF-β expression levels in cardiac fibroblast back to baseline level. They inhibited TGF-β deposition in the ischemic heart after MI (5.3% for miR-101a eNVs vs. 9.8% for unmodified eNVs, P<0.05). Remarkably, treatment with miR-101a-loaded eNVs significantly improved cardiac function and decreased infarct size compared to controls. The reparative effects of miR-101a loaded MSC eNVs compared to the unmodified eNVs appeared to be enhanced under in vivo conditions. Given that MSC eNVs have multifaceted effects, it is likely that additional, indirect mechanisms mediated by systemic exposure to MSC eNVs contribute to their overall function. Thus, we evaluated the effects of MSC eNVs on macrophage polarization. Pro-inflammatory macrophages are known to dominate in the early stage after MI and peak at 2–3 days after MI in humans and mice. A switch from the pro-inflammatory to the anti-inflammatory phenotype is required after MI to inhibit inflammation and promote wound healing (50). The persistence of pro-inflammatory macrophages has been shown to exacerbate heart injury due to persistent inflammation. When BMDMs were treated with unmodified eNVs and miR-101a loaded eNVs, anti-inflammatory markers such as CD206 and arginase-1 significantly increased whereas the effects on pro-inflammatory markers iNOS and IL-6 were negligible. Remarkably, mice treated with unmodified eNVs and miR-101a-loaded eNVs had increased numbers of anti-inflammatory macrophages (55% and 52%) compared to PBS treated control mice (38%) and significantly reduced circulating pro-inflammatory cytokines. This suggests that MSC eNVs possess anti-inflammatory effects. Furthermore, since 50% of monocytes and macrophages dispatched to the infarcted myocardium originate from the spleen (51), it is likely that high splenic deposition after systemic administration of MSC eNVs is advantageous for modulating macrophage and monocyte function and contributes to the overall cardio-regenerative effect. Further supporting this, we found high colocalization of MSC eNVs with CD68-positive macrophages, and Cho et al. demonstrated that MSCs have immuno-modulatory traits and accelerate cardiac repair by polarizing macrophages to the anti-inflammatory phenotype (52). MSC eNVs therefore share many features with their parent cells and are a promising alternative to cell-based therapeutics.
Additionally, miR-101a has been reported as a potent inhibitor of autophagy by suppressing ATG4D and blocking IL-3 processing (53, 54). While baseline autophagy is associated with protective effects, overly stimulated cardiac autophagy is known to promote cell death and worsen cardiac function (55). In contrast, inhibiting autophagy results in attenuated myocardial infarction (56). The immunofluorescent staining showed that miR-101a eNVs significantly decreased LC3B expression compared with unmodified eNVs (Figure S4). The results suggest that miR-101a-loaded eNVs not only regulated TGF-β mediated fibrosis, but also protected the infarcted myocardium from ischemic injury by regulating autophagy after MI.
In conclusion, MSC eNVs serve as both a small RNA carrier and, by virtue of their intrinsic bioactivity, a therapeutic, making them a highly promising platform for cardiac therapy. Importantly, miR-101a-loaded MSC eNVs significantly improved heart function, decreased infarct size, and decreased fibrosis after MI. Additionally, we revealed that MSC eNVs possess anti-inflammatory effects and polarized macrophages to the M2 phenotype. Notably, our studies showed that direct intramyocardial injection is not required for MSC eNVs to mediate cardio-protective effects when they are loaded with anti-fibrotic miR-101a. These results warrant further development of MSC eNV-based therapies for treating ischemic heart diseases.
Supplementary Material
Acknowledgements
We would like to thank Dr. Joseph Spernyak (Roswell Park Cancer Institute) for technical assistance in the use of IVIS imager and funding by the NIH (S10 OD 016450). We thank Dr. Maixian Liu for the TEM images. We thank Nana Nikolaishvili-Feinberg, Ph.D. in the Translational Pathology Laboratory (TPL) for expert technical assistance. The UNC TPL is supported in part by grants from NCI (5P30CA016080-42), NIH (U54-CA156733), NIEHS (3P30 EOS010126-17), UCRF and NCBT (2015-IDG-1007). Histological services for FFPE samples were provided by the Histology Research Core Facility in the Department of Cell Biology and Physiology at the University of North Carolina, Chapel Hill NC. Work in the Nguyen laboratory is supported by R01EB023262 (NIH), the Bruce Holm Technology award, and by the National Science Foundation (DMR 1751611). Work in the Canty laboratory is supported by HL-61610, the National Center for Advancing Translational Sciences UL1-TR-001412 and the Department of Veterans Affairs 1IO1BX002659.
Footnotes
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol. 2013;168(2):934–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naghavi M, Abajobir AA, Abbafati C, Abbas KM, Abd-Allah F, Abera SF, et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet. 2017;390(10100):1151–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59(11):969–78. [DOI] [PubMed] [Google Scholar]
- 4.Liehn EA, Postea O, Curaj A, Marx N. Repair After Myocardial Infarction, Between Fantasy and Reality: The Role of Chemokines. J Am Coll Cardiol. 2011;58(23):2357–62. [DOI] [PubMed] [Google Scholar]
- 5.Travers Joshua G, Kamal Fadia A, Robbins J, Yutzey Katherine E, Blaxall Burns C. Cardiac Fibrosis. Circ Res. 2016;118(6):1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang L, Murphy AJ, Dart AM. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front Pharmacol. 2017;8:186–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51(4):600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, et al. MicroRNA-101 Inhibited Postinfarct Cardiac Fibrosis and Improved Left Ventricular Compliance via the FBJ Osteosarcoma Oncogene/Transforming Growth Factor-β1 Pathway. Circulation. 2012;126(7):840–50. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Wang K, Liao Y, Zeng Q, Li Y, Hu F, et al. MicroRNA-101a Inhibits Cardiac Fibrosis Induced by Hypoxia via Targeting TGFβRI on Cardiac Fibroblasts. Cell Physiol Biochem. 2015;35(1):213–26. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen J, Szoka FC. Nucleic acid delivery: the missing pieces of the puzzle? Acc Chem Res. 2012;45(7):1153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nature reviews Cardiology. 2014;11(5):255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barile L, Lionetti V, Cervio E, Matteucci M, Gherghiceanu M, Popescu LM, et al. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res. 2014;103(4):530–41. [DOI] [PubMed] [Google Scholar]
- 13.Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem cell research. 2010;4(3):214–22. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson SW, Wang J, Lee CJ, Liu M, Neelamegham S, Canty JM, et al. The microRNA regulatory landscape of MSC-derived exosomes: a systems view. Sci Rep. 2018;8(1):1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson S, Kim S, Lee C, Deci M, Nguyen J. The Phenotypic Effects of Exosomes Secreted from Distinct Cellular Sources: a Comparative Study Based on miRNA Composition. AAPS J. 2018;20(4):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferguson SW, Megna JS, Nguyen J. 3 - Composition, Physicochemical and Biological Properties of Exosomes Secreted From Cancer Cells In: Amiji M, Ramesh R, editors. Diagnostic and Therapeutic Applications of Exosomes in Cancer: Academic Press; 2018. p. 27–57. [Google Scholar]
- 17.Ferguson SW, Nguyen J. Exosomes as therapeutics: The implications of molecular composition and exosomal heterogeneity. J Control Release. 2016;228:179–90. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Seo MJ, Deci MB, Weil BR, Canty JM, Nguyen J. Effect of CCR2 inhibitor-loaded lipid micelles on inflammatory cell migration and cardiac function after myocardial infarction. INTERNATIONAL JOURNAL OF NANOMEDICINE. 2018;13:6441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, et al. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol (1985). 2007;102(6):2104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences. 2016;113(8):E968–E77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenkranz S TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovascular research. 2004;63(3):423–32. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen J, Sievers R, Motion JPM, Kivimae S, Fang QZ, Lee RJ. Delivery of Lipid Micelles into Infarcted Myocardium Using a Lipid-Linked Matrix Metalloproteinase Targeting Peptide. Mol Pharm. 2015;12(4):1150–7. [DOI] [PubMed] [Google Scholar]
- 23.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell–derived factor-1α plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110(21):3300–5. [DOI] [PubMed] [Google Scholar]
- 24.Ghadge SK, Mühlstedt S, Özcelik C, Bader M. SDF-1α as a therapeutic stem cell homing factor in myocardial infarction. Pharmacology & therapeutics. 2011;129(1):97–108. [DOI] [PubMed] [Google Scholar]
- 25.Krishna KA, Krishna KS, Berrocal R, Rao K, Rao KS. Myocardial infarction and stem cells. Journal of Pharmacy and Bioallied Sciences. 2011;3(2):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madigan M, Atoui R. Therapeutic use of stem cells for myocardial infarction. Bioengineering. 2018;5(2):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pischke SE, Gustavsen A, Orrem HL, Egge KH, Courivaud F, Fontenelle H, et al. Complement factor 5 blockade reduces porcine myocardial infarction size and improves immediate cardiac function. Basic research in cardiology. 2017;112(3):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Ye Y, Yang X, Yang X, Zhang S. GW29-e1858 Chemokine CCL17 acts as a new biomarker and therapeutic target in pathological cardiac hypertrophy. 2018. [Google Scholar]
- 29.Pordel S, Sajedi Khanian M, Karimi MH, Nikoo H, Doroudchi M. Plasma CXCL 1 levels and TRAF 3 IP 2 variants in patients with myocardial infarction. Journal of clinical laboratory analysis. 2018;32(6):e22402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Y, Bai J, Tang L, Zhang P, Pu J. Anti-CCL21 antibody attenuates infarct size and improves cardiac remodeling after myocardial infarction. Cellular Physiology and Biochemistry. 2015;37(3):979–90. [DOI] [PubMed] [Google Scholar]
- 31.Altara R, Gu Y-M, Struijker-Boudier HA, Thijs L, Staessen JA, Blankesteijn WM. Left ventricular dysfunction and CXCR3 ligands in hypertension: from animal experiments to a population-based pilot study. PloS one. 2015;10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altara R, Gu Y, Struijker-Boudier H, Staessen J, Blankesteijn WM. Circulating CXCL-9,−10 and-11 levels improve the discrimination of risk prediction models for left ventricular dysfunction. The FASEB Journal. 2015;29(1_supplement):46.2. [Google Scholar]
- 33.Altara R, Manca M, Hessel MH, Gu Y, van Vark LC, Akkerhuis KM, et al. CXCL10 is a circulating inflammatory marker in patients with advanced heart failure: a pilot study. Journal of cardiovascular translational research. 2016;9(4):302–14. [DOI] [PubMed] [Google Scholar]
- 34.van der Vorst EP, Daissormont I, Aslani M, Seijkens T, Wijnands E, Lutgens E, et al. Interruption of the CXCL13/CXCR5 chemokine axis enhances plasma IgM levels and attenuates atherosclerosis development. Thrombosis and Haemostasis. 2020;120(02):344–7. [DOI] [PubMed] [Google Scholar]
- 35.Bilir C, Engin H, Temi YB, Toka B, Karabağ T. Acute Myocardial Infarction Caused by Filgrastim: A Case Report. Case reports in oncological medicine. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. Journal of Experimental Medicine. 2017;214(11):3293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okazaki T, Ebihara S, Asada M, Yamanda S, Saijo Y, Shiraishi Y, et al. Macrophage colony-stimulating factor improves cardiac function after ischemic injury by inducing vascular endothelial growth factor production and survival of cardiomyocytes. The American journal of pathology. 2007;171(4):1093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frangogiannis NG, Mendoza LH, Ren G, Akrivakis S, Jackson PL, Michael LH, et al. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. American Journal of Physiology-Heart and Circulatory Physiology. 2003. [DOI] [PubMed] [Google Scholar]
- 39.Zidar DA. B Lymphocytes: Adding Insult to Injury After Myocardial Infarction. Science Translational Medicine. 2013;5(205):205ec164–205ec164. [Google Scholar]
- 40.Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circulation research. 2015;116(2):354–67. [DOI] [PubMed] [Google Scholar]
- 41.Deci MB, Ferguson SW, Scatigno SL, Nguyen J. Modulating Macrophage Polarization through CCR2 Inhibition and Multivalent Engagement. Mol Pharm. 2018;15(7):2721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deci MB, Liu M, Gonya J, Lee CJ, Li T, Ferguson SW, et al. Carrier-Free CXCR4-Targeted Nanoplexes Designed for Polarizing Macrophages to Suppress Tumor Growth. Cell Mol Bioeng. 2019;12(5):375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int J Mol Sci. 2017;18(7):1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Wang H, Li J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin Med Insights Cardiol. 2016;10:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. 2018;186:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dean RG, Balding LC, Candido R, Burns WC, Cao Z, Twigg SM, et al. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53(10):1245–56. [DOI] [PubMed] [Google Scholar]
- 47.Ma T, Chen Y, Chen Y, Meng Q, Sun J, Shao L, et al. MicroRNA-132, Delivered by Mesenchymal Stem Cell-Derived Exosomes, Promote Angiogenesis in Myocardial Infarction. Stem Cells Int. 2018;2018:3290372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu B, Kim HW, Gong M, Wang J, Millard RW, Wang Y, et al. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int J Cardiol. 2015;182:349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ritter T, Lehmann M, Volk H-D. Improvements in Gene Therapy. Biodrugs. 2002;16(1):3–10. [DOI] [PubMed] [Google Scholar]
- 50.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science (New York, NY). 2009;325(5940):612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho DI, Kim MR, Jeong HY, Jeong HC, Jeong MH, Yoon SH, et al. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp Mol Med. 2014;46:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frankel LB, Wen J, Lees M, Høyer‐Hansen M, Farkas T, Krogh A, et al. microRNA‐101 is a potent inhibitor of autophagy. The EMBO journal. 2011;30(22):4628–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen C, Ponnusamy M, Liu C, Gao J, Wang K, Li P. MicroRNA as a therapeutic target in cardiac remodeling. BioMed research international. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du Wu KZ, Hu P. The Role of Autophagy in Acute Myocardial Infarction. Frontiers in Pharmacology. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C-Y, Zhang Y-H, Li R-B, Zhou L-Y, An T, Zhang R-C, et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nature communications. 2018;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.