

Abstract
Tumor cells migrate through changing microenvironments of diseased and healthy tissue, making their migration particularly challenging to describe. To better understand this process, computational models have been developed for both the ameboid and mesenchymal modes of cell migration. Here, we review various approaches that have been used to account for the physical environment's effect on cell migration in computational models, with a focus on their application to understanding cancer metastasis and the related phenomenon of durotaxis. We then discuss how mesenchymal migration models typically simulate complex cell–extracellular matrix (ECM) interactions, while ameboid migration models use a cell-focused approach that largely ignores ECM when not acting as a physical barrier. This approach greatly simplifies or ignores the mechanosensing ability of ameboid migrating cells and should be reevaluated in future models. We conclude by describing future model elements that have not been included to date but would enhance model accuracy.
I. INTRODUCTION
Cell migration is an integral part of many biological functions and pathological conditions, from immune response and wound healing to organ development and cancer metastasis. A cell's ability to move through space and reach its destination is critically important for it to fulfill its intended function. Depending on the cell type and the circumstances it finds itself in, cells can adopt different modes of migration,1,2 but all modes of migration can be described with the same basic steps: membrane extension, attachment formation, contraction, and rear release.3 Mechanisms that control each step and the degree to which each step affects migration varies with cell migration mode. Although a continuum of possibilities exists between the extremes of migration modes, two main subsets of migration, ameboid and mesenchymal migration, are among the most described, especially in the context of—but not exclusive to—cancer metastasis.
Ameboid migration occurs both in single-celled organisms, such as the ameba Dictyostelium discoideum, and within specific cell types in multicellular organisms, such as neutrophils.4 Cells undergoing ameboid migration exhibit rounded protrusions, i.e., blebs, and show little spreading on their substrate. This mode of migration progresses through a three-step blebbing cycle: nucleation, growth, and contraction [Fig. 1(a)]. The formation and expansion of these blebs are driven by weaknesses in the actin cortex and cytoplasmic pressure differences that cause the cellular membrane to expand outward.5 During the nucleation and growth phases, it is not clear to what extent the actin cortex ruptures, but there is a clear separation between the two.6 As blebs transition from growth to contraction, myosin causes the bleb to retract back into the main body of the cell, which can result in an overall movement of the cell toward the direction of the bleb expansion.7 It should be noted that ameboid cells can exhibit other types of protrusions that are closer to the mesenchymal end of the migration spectrum, i.e., pseudopods.8 Pseudopods initiate as blebs; however, their expansion from the cell body is coupled with continuous active expansion of F-actin in the underlying cellular membrane. Both types of protrusions can form on the same ameboid cell, and blebs and pseudopods have been shown to operate cooperatively during chemotaxis.9 Regardless of the main protrusion type, ameboid cells exhibit clearly defined polarization with a leading and trailing side.10 They can also travel at relatively fast speeds compared to other modes of migration (∼10 μm/min)11 depending on their surroundings. Additionally, these cells do not form strong focal adhesions with the surrounding extracellular matrix (ECM)4 and tend to be more processive in their migration, especially in the dense matrix.12
FIG. 1.
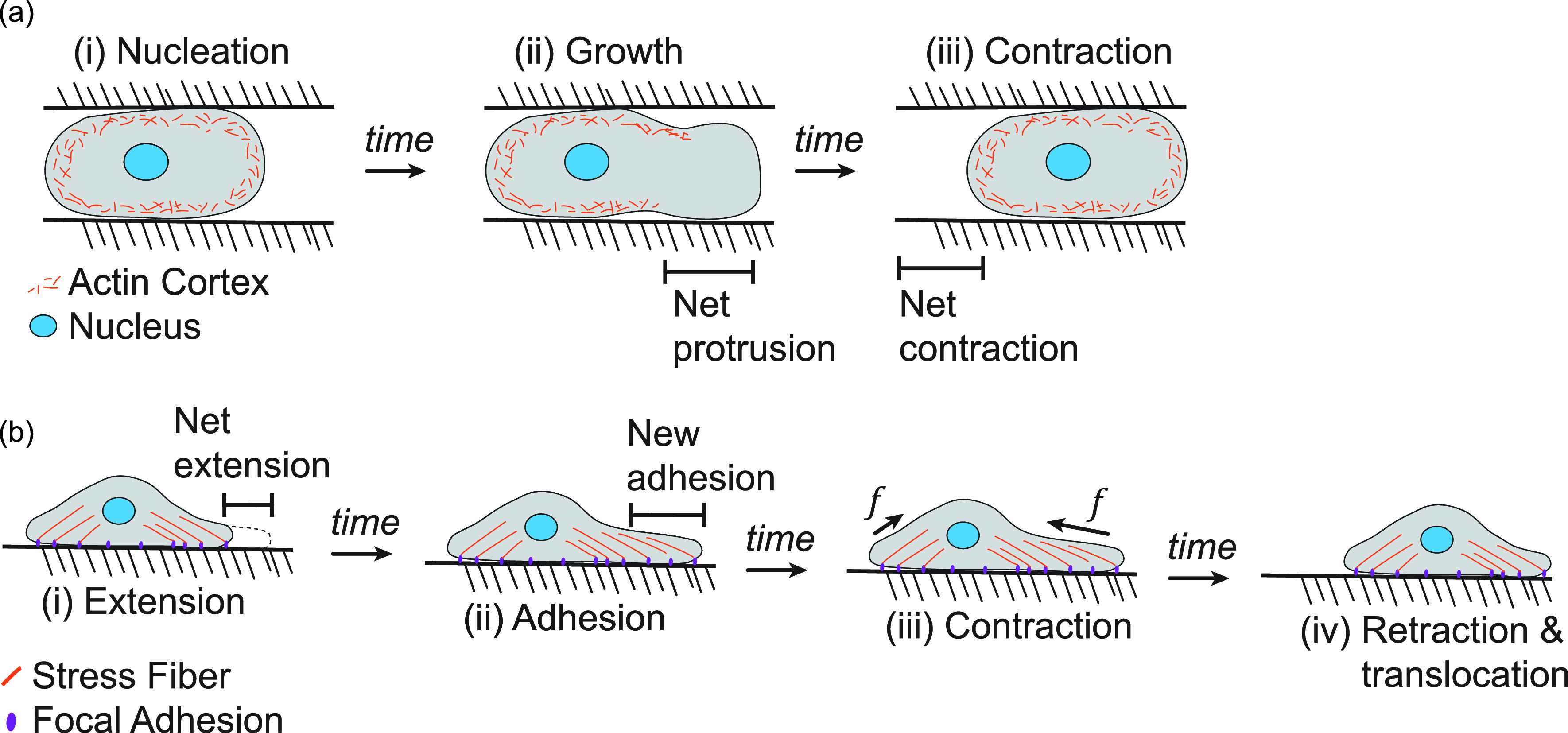
Processes of ameboid and mesenchymal migration in cancer. (a) Ameboid migration typically occurs as a three-step blebbing cycle with nucleation, growth, and contraction steps. (b) Mesenchymal migration typically involves a different process wherein cells extend their leading edge and adhere, contract, and release their trailing edge.
Conversely, a wide variety of migrating cells contract against focal adhesions in protrusions from the main cell body to move in a second method often termed mesenchymal migration [Fig. 1(b)]. Protrusions used in this mode are typically classified as filopodial or lamellipodial, with the former being thin spindle-like protrusions and the latter being sheet-like protrusions.3 Additionally, cells in the mesenchymal mode form strong focal adhesions to the ECM13–15 and, as a result, appear to spread over their substrate; this mode is typically slower than ameboid migration4 and is classically observed on two-dimensional substrates. In addition to morphological differences, there are mechanistic differences between the two modes of migration; cytoplasmic pressure gradients drive ameboid migration12 often in confined settings, whereas mesenchymal migration is driven by actin polymerization and the active maturation and turnover of focal adhesions coupled with actin-myosin contraction16 across a more spread cell. This different mechanism is not entirely distinct from the pressure-driven flowing actin networks of ameboid migration; mesenchymal migrating cells exhibit a retrograde actin flow away from the leading edge and toward the main cell body.17,18 However, computational models typically treat different migration modes as entirely distinct for the sake of simplicity and are used to answer specific questions. Contact guidance between these modes is also markedly different and results in proliferative differences that could underlie migration19 and hybrid cell formation.20 A summary of major differences in migration outcomes21–30 is shown in Table I.
TABLE I.
Major performance differences between migration modes for cancer cells.
| Ameboid | Mesenchymal | Reference | |
|---|---|---|---|
| Migration speed | 2–25 μm/min | 0.1–1 μm/min | 21–23 |
| Persistence | Low | High | 24, 25 |
| Morphology | Rounded | Elongated | 23, 26 |
| ECM attachment | Weak, short term, and lower integrin expression | Integrin clusters forming focal adhesions | 23, 27 |
| Migration in ECM | Squeezing or blebbing through ECM pores | Adhesion-mediated tractions and ECM degradation | 23, 28, 29 |
| CSK organization | Actin cortex | Actin meshwork, contractile stress fibers, and microtubules | 23, 29, 30 |
Although these two extreme modes represent a majority of cellular movements observed in vivo and in vitro (and models describing mesenchymal migration are significantly more common than ameboid migration), several other modes, both intermediate and distinct, have also been described but were omitted here for clarity.31 The use of these modes often depends on the environment's dimensionality (which can regulate adhesion assembly32), on the cell type, and on the receptor-ligand pairs as with selectins used in leukocyte migration.33 These modes often exhibit distinct features, making them easily identifiable, such as the crescent moon shape and gliding motion of keratocytes,34 but exist in a continuum between mesenchymal and ameboid modes.
II. MIGRATION AND CANCER METASTASIS
Cancer is the second leading cause of death in the United States, and the vast majority of its mortality is associated with secondary tumor formation.35 In order for cancer cells to metastasize and form secondary disease, they must migrate out of the primary tumor, intravasate into the bloodstream, and then extravasate into other tissues throughout the body.36 Cells within tumors are also very heterogeneous, making it difficult to separate indolent cancers from deadly ones, as only a subset of cells is able to disseminate from the main tumor and the others remain stationary and benign. Alongside migration mode, directionality is incredibly important for metastasis, yet remains poorly understood in certain contexts. For example, cancer cell chemotaxis (i.e., migration along a chemical concentration gradient) has been studied in-depth in ameboid cells but comparatively little for mesenchymal cells.4,37 More recently, effort has been made to understand the effect of cells' mechanosensing on migration. For example, the progression of metastatic breast cancer has been related to the levels of mechanosensing proteins in stiff ECM.38 Cells migrate at different speeds depending on substrate stiffness and oftentimes exhibit durotaxis, the ability to sense and migrate up a stiffness gradient.39–41 However, this seems counterintuitive for understanding cancer metastasis, as often times, the tumor microenvironment becomes much stiffer than the surrounding healthy stroma due to matrix secretion and cross-linking by cancer-associated fibroblasts.42,43 In these cases, the metastatic cells must exhibit adurotactic behavior in order to leave the primary tumor, which further complicates our current understanding of cancer cell migration and metastasis. Adding yet another level of complexity is the observation that tumor cells migrate in both the ameboid and mesenchymal modes and, depending on their environment, can switch between the two.1,2,44 They can also migrate individually or collectively,45 and their migration is highly dependent on the physical properties of their niche, such as stiffness, porosity, dimensionality, and toporgaphy,46 which can change as a result of clinical care.47 Despite these many influences, tumor migration models, thus far, largely focus on intracellular mechanisms governing mesenchymal and ameboid modes, and thus, we will describe the effects of additional modes and matrix properties in the context of model limitations later.
III. COMPUTATIONAL MODELING OF MIGRATION
Cancer cell interactions are often very complex; reductionist approaches using model systems, e.g., microfluidic bioreactors,48 explore many isolated variables, and more complex models may even include the vasculature to study extravasation.48,49 However, despite the simplicity of these model systems, fidelity with in vivo disease progression may be limited or at least require context and necessitate significant engineering to generate robust datasets. Computational models, however, may offer an alternative—where applicable—to create and test reasonably complex niches in silico to understand migration mechanisms prior to experimental studies, thus better informing the design of more effective and efficient experimental studies.
A key consideration for any computational model is the complexity of its physics; over- or under-determined systems can limit applicability and predictive value. With respect to cell migration, many models consider the following key concepts: force balance, mass conservation, biochemical activity, active forces, and passive forces50 (Fig. 2). Force balances are used in all models to determine the net force magnitude and direction, which governs a cell's movement. Mass conservation is especially prevalent in models with a focus on protrusion dynamics or morphology changes in migrating cells to determine cells' changing shapes with a constant mass. Biochemical activity connects intra, inter, and extracellular signaling to cellular and extracellular mechanics. Active forces include forces generated by cytoskeletal dynamics such as actin and microtubule polymerization and depolymerization and actomyosin contractility. On the other hand, passive forces include reaction forces arising within cells, between neighboring cells, and between cells and the surrounding environment elastic strain, viscous drag, and molecular friction.50 The integration of these components in the model, the degree to which they affect each other, and, more broadly, overall migration depends on a number of intracellular and extracellular parameters in the model's framework. A brief overview of some of the modeling approaches discussed below,51–68 and associated equations, is shown in Table II. Note that this is meant to introduce readers to the various ways that physical laws governing cell migration can be described mathematically and is meant to direct readers to specific examples where these methods are applied. As computational costs continue to go down, the equations and models can become more detailed and combine multiple approaches into hybrid models. For the remainder of this review, we will broadly discuss how the above described key concepts are modeled and affect cell migration mode citing specific examples.
FIG. 2.
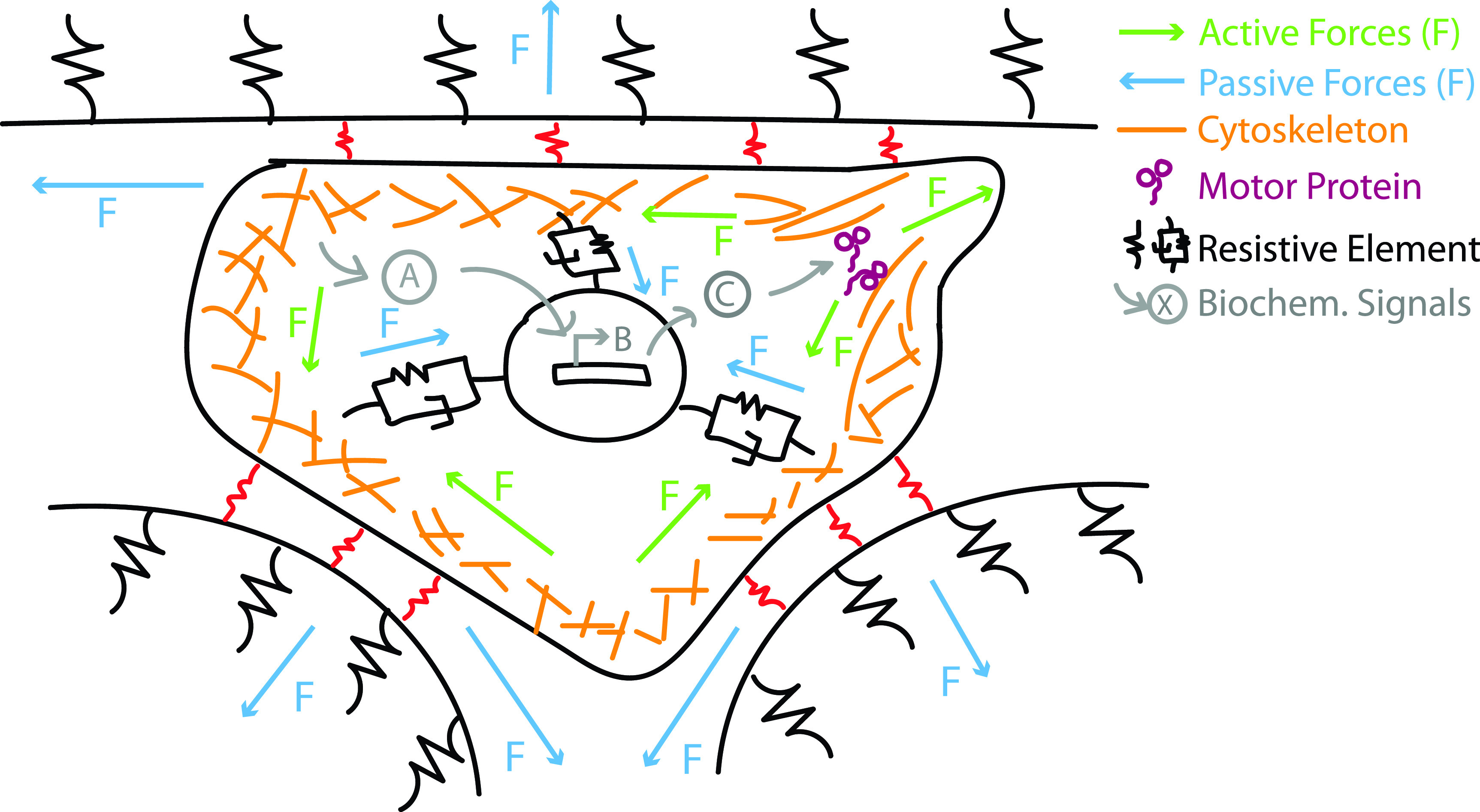
Conserved components of computational models of cancer migration. Four concepts are typically present to some degree in computational models of migration: force balance, mass conservation, active forces, and passive forces. Each is illustrated here and where active forces are those generated by motor proteins and polymerization and depolymerization of cytoskeletal filaments and passive forces are those from the viscoelastic parts of the cytoskeleton and ECM as well as from molecular friction.
TABLE II.
List of frequently physical frameworks used to model cancer cell migration and their applications.
| Common modeling approaches | Applications and examples |
|---|---|
| Chemo-mechanical models based on force-dependent reaction kinetics. | Used to model sub-cellular processes such as cell-substrate bond formation, filament polymerization and gliding and mechanosensing-based changes to predict resulting cell adhesion, traction, and migration (e.g., various spring/dashpot models,51–53 active matter models,54 and molecular clutch models55). |
| … Force balance between active forces driven by chemical potential, , and dissipative forces, . | |
| … stiffness, , and number, , and kinetics of molecular bonds, dictate drag coefficient, . | |
| Agent-based models focusing on force balance between individual cells and their environment. | Used to model cell populations interacting with each other and the environment. Coarse grained to implicitly include effects of various sub-cellular processes (e.g., force-based models,56–58 energy-based models,59,60 and lattice-based/cellular Potts models61–63). |
| … | |
| or an energy minimization approach | |
| Thermodynamic models based on equilibrium and non-equilibrium work-free energy change relationships | Used to model both cellular and sub-cellular processes and assess the energetic states that the system can occupy (e.g., free-energy-based models64,65). |
| …Free energy change of the system | |
| … work done by the system | |
| Equilibrium… minimize ( | |
| Non-equilibrium … | |
| Continuum phase-field models | Used to describe cell and surrounding free space as an evolving phase-field, with the moving boundary representing the cell membrane. Well suited to describe collective migration66,67 and migration of cell monolayers.68 |
| … Describes the dynamics of the cell shape in response to free energy changes. The free energy functional, , is chosen so that minima correspond to phases (i.e., intracellular and extracellular environment) of the system. |
IV. MESENCHYMAL MIGRATION MODELS: APPLICATIONS AND DIRECTIONS
Computational models describing mesenchymal cell migration primarily focus on intracellular active forces driving protrusion and retraction of lamellipodia and filopodia, balancing these forces against elastic, viscous, and friction forces within and outside the cell, and mass balance that defines the cell shape as the cell migrates under the action of these forces.50,69–71 At the nanoscale, these models can focus on the dynamics of actin polymerization and depolymerization, force generation by individual myosin motors, binding and unbinding of adhesion receptors to the extracellular matrix, clustering of adhesion receptors and maturation of adhesion sites, and binding of polymerized actin filaments to these receptors to form adhesion complexes.27,72,73 At this scale, models incorporate force generation and sensing aspects such as conformational changes in adhesion complex proteins, recruitment of additional actin-myosin fibers, and branching of actin fibers. At the mesoscale, models focus on cell spreading, filopodial and lamellipodial protrusion and contraction, coupling between the nucleus and the cytoskeleton, viscoelastic strains within the cytoskeleton and the nucleus under the influence of active and passive migration forces, and resulting cell shape changes.30,61,70,74,75 Models at this scale are particularly useful for predicting cell shape dynamics and interactions between two neighboring cells or a single cell and its environment. At the microscale, the focus of modeling is on overall cell migration dynamics under the influence of a driving force balanced by the drag forces from the environment.56–58,76,77 At this scale, the goal of the models is to predict cell migration velocity and path persistence as a function of the mechanistic interactions between the cell, its neighbors, and the surrounding extracellular matrix. Computational models may focus on a specific length scale or combine multiple length scales to predict migration dynamics. Models can also vary in their representation of the extracellular environment. For example, to describe cell migration on a 2D substrate, the ECM can be described as a continuous elastic material or discretized into a collection of binding sites connected to springs.61,63,78–80 In 3D, the ECM can be described as a viscous continuum, discretely as a collection of randomly or uniformly distributed binding sites, or as fibers distributed randomly or along the grid in 3D space.56,62,76,81,82 Depending on the choice of the ECM model, various aspects of cell–ECM interactions can be integrated such as ECM degradation, ECM remodeling, contact guidance along aligned fibers, and squeezing of cell through ECM pores. A common thread between all these models of mesenchymal cell migration is that the migration is driven by forces generated within long protrusions that grow along the surface in search of sufficient binding sites in 2D and along the length of fibers in 3D.
The primary goal of these computational models has been to predict how fast and persistently cells will migrate along or within a given substrate depending on their specific mechanical and chemical properties. Models have also been successful in predicting experimentally observed behavior of the biphasic dependence of migration speed on ECM density, adhesivity, and stiffness.41 Models can also recreate qualitative trends in migration persistence, which have been observed experimentally and clinically. More recently, modeling of experimentally observed emergent phenomena such as chemotaxis, durotaxis, haptotaxis, and contact guidance, which direct cells along specific directions, is gaining attention. How mechanobiology affects migration, i.e., how do changes in niche parameters direct processive migration and ultimately intravasation, is of extreme importance in understanding processes such as wound healing and cancer metastasis. Most adherent cells migrate toward a stiffer region of a substrate when presented with a gradient,83 i.e., “durotax;” yet, tumors are inherently stiff relative to adjacent, soft stroma in vivo84,85 created by cancer-associated fibroblasts. This creates a cancer cell “migration paradox” where tumor cells must migrate down ECM stiffness gradients that otherwise support migration in the opposing direction (Fig. 3). Here, we will focus on a few mesenchymal cell migration models that explain cancer cell durotaxis and their applicability toward answering the cancer cell migration paradox. That being said, it is important to note that we are restricting our discussion and this concept to the disseminating cells from the tumor. Many other cells must migrate toward the tumor, i.e., durotax, including inflammatory cells, among others.
FIG. 3.

The migration paradox. Most cells migrate toward stiffer regions of tissues in a process called durotaxis.59 However, tumor cells must migrate from stiff tumors through the progressively softer matrix to disseminate from a tumor core and intravasate into the blood stream. This metastatic migratory process is counter to conventional thought on stiffness gradient migration, and it is not clear which migration mode, if any based on available data, permits such migration. Note that for simplicity, additional cell types, e.g., cancer associated fibroblasts, have been omitted but play a key role in niche remodeling nonetheless.
To start, many models describe migrating cells as polarized with a higher probability of protrusion at the leading edge and retraction fibers at the trailing edge. Focal adhesions form preferentially along the leading edge and dissociate more frequently along the trailing edge, resulting in net motion in the direction of the leading edge.3 Some computational models replicate durotaxis on a surface using polarized cell filopodia, which are more likely to grow along elastic fibers aligned along an increasing stiffness gradient.81 This approach to simulating migration succeeds in predicting the general movement of durotactic cells and in creating a more realistic representation of ECM fiber networks rather than conventional models that treat the substrate as a continuum. Yet, such a rigid integration of durotaxis is not without disadvantages, e.g., durotactic behavior has to be built into the model, rather than durotactic behavior arising from it. Furthermore, while the elastic fibers can be deformed, the cell cannot remodel or degrade them. Thus, from such polarity-based models and those that take similar approaches, e.g., adhesions that bind more strongly on the stiffer region than softer regions forcing polarized shapes a priori,86 it may be difficult to infer mechanisms of durotaxis or even adurotaxis as observed in cancer in vivo. These models are, however, convenient to study the effect of inter-cellular heterogeneity in these parameters on overall population dynamics when predicting collective cell behavior. Indeed, within a tumor, not all cells metastasize, and thus, cancer migration models should highlight both cellular and temporal heterogeneity87 when describing gradients.
One of the early models that explained the origin of durotactic behavior rather than making it an intrinsic property of migrating cells was by Schwarz et al., which used a simple 2 spring attachment-detachment model between the actin myosin force generating elements, cell membrane attached adhesion protein, and the substrate.51 The model showed that as the substrate stiffness increases, the rate at which the force is generated within the actin–myosin filament increases, leading to larger overall traction forces within the lifetime of a cell–substrate adhesion bond. The increase in traction forces on stiffer substrates drives cell migration up the stiffness gradient explaining durotaxis. This phenomenon has since been integrated as an a priori mechanism of cell migration in a number of other models. A more recent addition to models explaining durotaxis is based on a rigidity sensing-based change in the biochemical activity of motor protein regulating units within the cell.72 Increased feedback from a stiffer substrate drives increased myosin activity and higher speeds for cells in stiffer regions, driving an accumulation of cells up a stiffness gradient. A third alternate model focuses on the mechanical response of the ECM fibers rather than that of the cell itself. The model is based on fiber mechanics that suggests that the deformation/extension of a fiber decreases as a cell gets closer to the point where the fiber is crosslinked to a stiffer environment. This generates a stronger passive force on the cell, pulling the cell toward the stiffer regions and driving durotaxis.81 There are many other models that explain durotaxis through variations or combinations of the above described themes that we are not discussing here. A more recent model by Heck et al.88 suggests yet another possible explanation for durotactic behavior. This model accounts for ECM as an obstacle that the cell must negotiate or degrade; this type of confined migration is often observed in dense tissue where degradation by the cancer cell proceeds filopodial extension89 and requires significant deformation (which can be measured in high throughput with fluidics90). The model predicts that migration is most affected by ECM stiffness, cell adhesion strength, and protrusion properties, e.g., number, lifetime, and length. The model shows the standard correlation between matrix stiffness and migration speed, consistent with past models and experimental observation using degradation and filopodial extension.91 They also show that adhesion turnover makes migration more processive. By incorporating force-dependent adhesion turnover rates and increased forces in stiffer ECM regions, they show an additional mechanism for durotactic behavior driven by increased persistence. A connection between processive migration and adhesion strength is also supported experimentally as seen when a weakly adherent phenotype results in a reduced disease-free interval.92 The implication that protrusion activity and matrix deformation in three-dimensions, not the development of robust focal adhesions as in two dimensions, as the origin of their processivity, may be consistent with some results in three dimensions.28
The models discussed so far cover most known aspects of mesenchymal cell migration and also can be used to describe a number of emergent phenomena. With regard to durotactic behavior, they all suggest using one argument or another that cells should durotax, and for the most part, adherent cells do so. However, the cancer cell migration paradox of metastatic cells migrating against a stiffness gradient to metastasize still remains unanswered. It may be necessary to develop mechanistic models that explain anti-, a-, and durotactic migration that focus not on the ECM but rather on the cytoskeletal elements that drive mesenchymal migration; here, we summarize two such models. To interrogate such cytoskeletal elements, the most common mesenchymal model used has been the molecular motor-clutch,93 which employs multiple molecular motors to pull actin filaments toward the cell body on a compliant, continuous substrate; conversely, the molecular clutch binds to actin stochastically and links it to the extracellular environment via a force-dependent Bell model connection.94 These models typically predict a stiffness-dependent relationship for migration where on compliant substrates, the motors undergo load-fail cycles, whereas on stiff substrates, the complex slips. This model predicts biphasic behaviors in force and migration that can reinforce adhesion,95 with an optimal stiffness region that cells will most likely migrate to. The model, thus, does not always predict durotaxis or antidurotaxis, but rather migration in the direction of the preferred stiffness for a given cell type. Optimal stiffness for a cell type is dependent on the actomyosin contractile force that the cells can generate and the number of clutches between the cell and the substrate. Overall, the clutch model may provide a possible reason for the adurotactic migration of metastatic cancer cells away from a stiff tumor ECM, but validation of experimentally observation of adurotaxis by highly metastatic cancer cells needs to be further explored. Another model in which the mechanical environment can be considered in migration is an equilibrium thermodynamics model. This model provides an alternative way to characterize the two-way feedback loop between cell contractility and matrix realignment. This model calculates the total change in free energy of the system, consisting of the energies of the cell, matrix, and adhesions to determine whether or not migration will occur as the system tries to move into a lower energy state.65 Similar to the clutch model, this thermodynamic migration model predicts a biphasic migration response to matrix stiffness and depends on the contractile force that a cell can generate and the strength of its adhesions. It is possible to envision that in the presence of gradients, cells could adurotactically migrate to an optimum away from the tumor, thus achieving goals similar to the clutch model.
Overall, due to important fluctuations in force,96 heterogeneous adhesion within a tumor,92 and stiffness gradients (vs changing but static substrates in these models) on the stroma,85 there is a need for new models where these or similar parameters are incorporated together, e.g., maximum force generated by a stress fiber, catch bond dynamics, etc. Changes to the force-bond lifetime relationship could then result in anti-, a-, and durotactic behavior depending on how each parameter varies with the others.
V. AMEBOID MIGRATION MODELS
Ameboid migration is dominated by propulsive membrane blebbing, i.e., the key concept of force balances, rather than spreading and forming strong focal adhesions to their substrate, i.e., the key concept of active contractile forces.4 It also relies less on modification or degradation of the adjacent ECM and more so on becoming highly deformable and pushing through matrix pores. Despite the differences between mesenchymal and ameboid migration, cancer cells display unique plasticity in their ability to switch between modes,24,97 making our understanding of ameboid migration even more critical. Unlike mesenchymal migration, ameboid migration models reviewed here tend to focus on intracellular parameters, e.g., the development of pressure gradients to form blebs or propel the cell forward,12 rather than on the cells' physical environment. Despite this, ameboid cells may indirectly mechano-sense, which may be necessary for tumor metastasis.
A common characteristic of ameboid models is that they highlight a specific aspect of migration based on the nature of their model. For example, cell membrane deformability—modeled as a system of springs—has been used to determine a cancer cell's migration speed through confined spaces,70 such as pores in a matrix. This model suggests that, but does not assess, stiffness gradients are able to drive a cells' direction of travel, but polarization in this model is simply defined to guide the cells through the obstacles and is not a result of it. The ECM playing only a passive role in influencing cell behavior is also observed in an earlier model from the study by Lim et al.7 In this model, the cell has a permeable actin cortex inside an impermeable outer membrane, with adhesion points connecting the two. Cell movement occurs when membrane-cortex adhesions rupture, the outer membrane expands, and the actin cortex is pulled toward the rupture by a cytoplasmic pressure gradient; again, the matrix is only an obstacle and polarization is built in to the model rather than a result of it. From such models, neither mechanosensing nor migration from gradients has been considered.
Ameboid-like migration has been frequently observed in 1-98 and three dimensions99,100 when the environment requires less adhesive, confined migration distinct from mesenchymal modes. Despite not engaging traditional machinery, cancer cells in this mode and confinement do recognize substrate stiffness unlike the models mentioned previously, and it is observed that stiffer, confined spaces support more ameboid migration.26 Thus, models that highlight this behavior often consider possible ways that gradient sensing could occur in ameboid migration, either de novo or as a switch between models. For example, probabilistic models highlight that migration mode switching occurs in heterogenous matrix conditions and that both migration modes and plasticity are advantageous in heterogenous tumors because cells can sense their niche and switch modes as needed. Modulating the degree of cytoskeletal polymerization can also induce transitions as observed by Niculescu et al.71 Simulated cells displayed ameboid blebbing or a spread lamellipodium and gliding behavior simply by changing maximum actin polymerization. Each mode fed back on itself, and so switching events were rare as in in vitro observations but were not dependent on local conditions, i.e., cell could not sense environmental changes.
All models discussed in this section portray a common trend and limitation of computational models used for ameboid migration: focus remains on processes within the cell rather than interactions between the cell and its environment. Even for the few models that incorporate interactions between migrating cells and their physical environment, they still simplified as they restrict the cell from taking some shape or prevent the membrane from expanding into the physical obstacle. Although an emerging part of the literature,100 mechanosensing in this mode should be validated so that we better understand the environmental conditions and gradients that could result in migrating cells switching between modes1,2 and if ameboid migration could support adurotaxis as the mechanisms still remain to be elucidated (Fig. 3). While these models are able to analyze how cells change the shape and what sized spaces they can fit through, thus providing information about the process of migration, they are limited in that they only account for one portion of complicated processes like tumor metastasis and adurotaxis. Thus, critical questions remain: Under what circumstances do ameboid migrating cells stop durotaxing and move out of the stiffer tumor region? Do ameboid migrating cells respond differently to stiffness gradients than mesenchymal migrating cells? What takes precedence if ameboid migrating cells are presented with competing durotactic and chemotactic signals?
VI. LIMITATIONS AND CONCLUSIONS
The models discussed in this review show varying degrees of importance given to cells' mechanosensing capabilities and the effects of the ECM on migration. This is especially important to understand in cancer, where cells' ability to sense and migrate against a stiffness gradient may contribute to their ability to metastasize.38,42,43 Due to the complex nature of cell migration and mechanosensing, computational models offer one of our best ways to understand and learn about migrating cell behavior. However, none of the models discussed consider all the variables involved in migration and to do so would likely be overcomplicated, computationally taxing, and, therefore, infeasible. Even the models that give extensive thought to the cells' physical environment do not consider cells' ability to switch between migration modes as other models do.26 Beyond this, few models explore the cooperative or inhibitory migration behaviors arising from interactions between multiple cell types such as metastatic cancer cells, cancer-associated fibroblasts, and senescent tumor cells, which can all occupy neighboring spaces within the tumor microenvironment. Multicellular interactions can be extremely complex and can assist or hinder durotactic behavior through short- and long-range mechanical and chemical coupling.101–104 No single modeling framework captures the complete breadth of these observations as far as we are aware. The current work on computational cell migration models has undoubtedly helped increase our knowledge of processes like cancer metastasis and migration in general to address the cancer cell migration paradox, but there are still many unanswered questions on how cell migration is guided by gradient sensing mechanisms illustrated in the paradox (Fig. 3).
Future models and experiments should examine the effects of stiffness gradients on ameboid migrating cells, despite their comparatively weak focal adhesions, and how the effects of the physical environment change as cells transition between the ameboid and mesenchymal migration modes. Additionally, the development of computational models and mechanistic explanations of migration are required in order to attain a balance between model accuracy, potential insight, and computational complexity. Mesenchymal migration models have shown significant inclusion of mechanosensing and the ECM's effects on migration, but they still omit other key parameters that could influence migration, such as fluid convection and flow,105 leaving room for growth in future models.
ACKNOWLEDGMENTS
The authors would like to thank Afsheen Banisadr for helpful discussions. A.J.E. acknowledges grant support from the National Institutes of Health (Nos. R01CA206880, R21CA217735, and R01NS116802) and the National Science Foundation (No. 1763139). P.K. acknowledges grant support from the National Science Foundation (No. 1763132) and the Army Research Office (No. W911NF-17–1-0413).
No potential conflicts of interest were disclosed.
DATA AVAILABILITY
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. Taddei M. L., Giannoni E., Comito G., and Chiarugi P., “ Microenvironment and tumor cell plasticity: An easy way out,” Cancer Lett. 341, 80–96 (2013). 10.1016/j.canlet.2013.01.042 [DOI] [PubMed] [Google Scholar]
- 2. Brabek J., Mierke C. T., Rosel D., Vesely P., and Fabry B., “ The role of the tissue microenvironment in the regulation of cancer cell motility and invasion,” Cell Commun. Signal 8, 22 (2010). 10.1186/1478-811X-8-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lauffenburger D. A. and Horwitz A. F., “ Cell migration: A physically integrated molecular process,” Cell 84, 359–369 (1996). 10.1016/S0092-8674(00)81280-5 [DOI] [PubMed] [Google Scholar]
- 4. Bear J. E. and Haugh J. M., “ Directed migration of mesenchymal cells: Where signaling and the cytoskeleton meet,” Curr. Opin. Cell Biol. 30, 74–82 (2014). 10.1016/j.ceb.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charras G. T., Coughlin M., Mitchison T. J., and Mahadevan L., “ Life and times of a cellular bleb,” Biophys. J. 94, 1836–1853 (2008). 10.1529/biophysj.107.113605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cox C. D. et al. , “ Removal of the mechanoprotective influence of the cytoskeleton reveals PIEZO1 is gated by bilayer tension,” Nat. Commun. 7, 10366 (2016). 10.1038/ncomms10366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lim F. Y., Koon Y. L., and Chiam K. H., “ A computational model of amoeboid cell migration,” Comput. Methods Biomech. Biomed. Eng. 16, 1085–1095 (2013). 10.1080/10255842.2012.757598 [DOI] [PubMed] [Google Scholar]
- 8. Bravo-Cordero J. J., Hodgson L., and Condeelis J., “ Directed cell invasion and migration during metastasis,” Curr. Opin. Cell Biol. 24, 277–283 (2012). 10.1016/j.ceb.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tyson R. A., Zatulovskiy E., Kay R. R., and Bretschneider T., “ How blebs and pseudopods cooperate during chemotaxis,” Proc. Natl. Acad. Sci. U. S. A. 111, 11703–11708 (2014). 10.1073/pnas.1322291111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bosgraaf L. and Van Haastert P. J., “ Navigation of chemotactic cells by parallel signaling to pseudopod persistence and orientation,” PLoS One 4, e6842 (2009). 10.1371/journal.pone.0006842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lammermann T. et al. , “ Rapid leukocyte migration by integrin-independent flowing and squeezing,” Nature 453, 51–55 (2008). 10.1038/nature06887 [DOI] [PubMed] [Google Scholar]
- 12. Petrie R. J., Koo H., and Yamada K. M., “ Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix,” Science 345, 1062–1065 (2014). 10.1126/science.1256965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gkretsi V. and Stylianopoulos T., “ Cell adhesion and matrix stiffness: Coordinating cancer cell invasion and metastasis,” Front. Oncol. 8, 145 (2018). 10.3389/fonc.2018.00145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ray A. et al. , “ Anisotropic forces from spatially constrained focal adhesions mediate contact guidance directed cell migration,” Nature Commun. 8, 14923 (2017). 10.1038/ncomms14923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jacquemet G., Hamidi H., and Ivaska J., “ Filopodia in cell adhesion, 3D migration and cancer cell invasion,” Curr. Opin. Cell Biol. 36, 23–31 (2015). 10.1016/j.ceb.2015.06.007 [DOI] [PubMed] [Google Scholar]
- 16. O'Neill G. M., “ The coordination between actin filaments and adhesion in mesenchymal migration,” Cell Adhes. Migr. 3, 355–357 (2009). 10.4161/cam.3.4.9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexandrova A. Y. et al. , “ Comparative dynamics of retrograde actin flow and focal adhesions: Formation of nascent adhesions triggers transition from fast to slow flow,” PLoS One 3, e3234 (2008). 10.1371/journal.pone.0003234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cramer L. P., “ Molecular mechanism of actin-dependent retrograde flow in lamellipodia of motile cells,” Front. Biosci. 2, d260–d270 (1997). 10.2741/A189 [DOI] [PubMed] [Google Scholar]
- 19. Pourfarhangi K. E., De La Hoz E. C., Cohen A. R., and Gligorijevic B., “ Contact guidance is cell cycle-dependent,” APL Bioeng. 2, 031904 (2018). 10.1063/1.5026419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chitwood C. A. et al. , “ Breast tumor cell hybrids form spontaneously in vivo and contribute to breast tumor metastases,” APL Bioeng. 2, 031907 (2018). 10.1063/1.5024744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krakhmal N. V., Zavyalova M. V., Denisov E. V., Vtorushin S. V., and Perelmuter V. M., “ Cancer invasion: Patterns and mechanisms,” Acta Nat. 7, 17–28 (2015). 10.32607/20758251-2015-7-2-17-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Zijl F., Krupitza G., and Mikulits W., “ Initial steps of metastasis: Cell invasion and endothelial transmigration,” Mutat. Res. 728, 23–34 (2011). 10.1016/j.mrrev.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pankova K., Rosel D., Novotny M., and Brabek J., “ The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells,” Cell Mol. Life Sci. 67, 63–71 (2010). 10.1007/s00018-009-0132-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friedl P. and Wolf K., “ Plasticity of cell migration: A multiscale tuning model,” J. Cell Biol. 188, 11–19 (2010). 10.1083/jcb.200909003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ly D. L. and Lumsden C. J., “ 3D amoeboid migration of a eukaryotic cell in a fiber matrix,” Artif. Life Rob. 14, 1–6 (2009). 10.1007/s10015-008-0708-0 [DOI] [Google Scholar]
- 26. Talkenberger K., Cavalcanti-Adam E. A., Voss-Bohme A., and Deutsch A., “ Amoeboid-mesenchymal migration plasticity promotes invasion only in complex heterogeneous microenvironments,” Sci. Rep. 7, 9237 (2017). 10.1038/s41598-017-09300-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakamoto Y., Prudhomme S., and Zaman M. H., “ Modeling of adhesion, protrusion, and contraction coordination for cell migration simulations,” J. Math. Biol. 68, 267–302 (2014). 10.1007/s00285-012-0634-6 [DOI] [PubMed] [Google Scholar]
- 28. Fraley S. I. et al. , “ A distinctive role for focal adhesion proteins in three-dimensional cell motility,” Nat. Cell Biol. 12, 598–604 (2010). 10.1038/ncb2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diz-Munoz A. et al. , “ Steering cell migration by alternating blebs and actin-rich protrusions,” BMC Biol. 14, 74 (2016). 10.1186/s12915-016-0294-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paul R., Heil P., Spatz J. P., and Schwarz U. S., “ Propagation of mechanical stress through the actin cytoskeleton toward focal adhesions: Model and experiment,” Biophys. J. 94, 1470–1482 (2008). 10.1529/biophysj.107.108688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamada K. M. and Sixt M., “ Mechanisms of 3D cell migration,” Nat. Rev. Mol. Cell Biol. 20, 738–752 (2019). 10.1038/s41580-019-0172-9 [DOI] [PubMed] [Google Scholar]
- 32. Cukierman E., Pankov R., Stevens D. R., and Yamada K. M., “ Taking cell-matrix adhesions to the third dimension,” Science 294, 1708–1712 (2001). 10.1126/science.1064829 [DOI] [PubMed] [Google Scholar]
- 33. Ivetic A., Hoskins Green H. L., and Hart S. J., “ L-selectin: A major regulator of leukocyte adhesion, migration and signaling,” Front. Immunol. 10, 1068 (2019). 10.3389/fimmu.2019.01068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verkhovsky A. B., Svitkina T. M., and Borisy G. G., “ Self-polarization and directional motility of cytoplasm,” Curr. Biol. 9, 11–20 (1999). 10.1016/S0960-9822(99)80042-6 [DOI] [PubMed] [Google Scholar]
- 35. Siegel R. L., Miller K. D., and Jemal A., “ Cancer statistics, 2017,” CA Cancer J. Clin. 67, 7–30 (2017). 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- 36. Wirtz D., Konstantopoulos K., and Searson P. C., “ The physics of cancer: The role of physical interactions and mechanical forces in metastasis,” Nat. Rev. Cancer 11, 512–522 (2011). 10.1038/nrc3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roussos E. T., Condeelis J. S., and Patsialou A., “ Chemotaxis in cancer,” Nat. Rev. Cancer 11, 573–587 (2011). 10.1038/nrc3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rubashkin M. G. et al. , “ Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate,” Cancer Res. 74, 4597–4611 (2014). 10.1158/0008-5472.CAN-13-3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong J. Y., Velasco A., Rajagopalan P., and Pham Q., “ Directed movement of vascular smooth muscle cells on gradient-compliant hydrogels,” Langmuir 19, 1908–1913 (2003). 10.1021/la026403p [DOI] [Google Scholar]
- 40. Pelham R. J. and Wang Y.-L., “ Cell locomotion and focal adhesions are regulated by substrate flexibility,” Proc. Natl. Acad. Sci. U. S. A. 94, 13661–13665 (1997). 10.1073/pnas.94.25.13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peyton S. R. and Putnam A. J., “ Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion,” J. Cell. Physiol. 204, 198–209 (2005). 10.1002/jcp.20274 [DOI] [PubMed] [Google Scholar]
- 42. McKenzie A. J. et al. , “ The mechanical microenvironment regulates ovarian cancer cell morphology, migration, and spheroid disaggregation,” Sci. Rep. 8, 7228 (2018). 10.1038/s41598-018-25589-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fusco S., Panzetta V., and Netti P. A., “ Mechanosensing of substrate stiffness regulates focal adhesions dynamics in cell,” Meccanica 52, 3389–3398 (2017). 10.1007/s11012-017-0676-3 [DOI] [Google Scholar]
- 44. Zhao Y. et al. , “ Acetylcholinesterase, a key prognostic predictor for hepatocellular carcinoma, suppresses cell growth and induces chemosensitization,” Hepatology 53, 493–503 (2011). 10.1002/hep.24079 [DOI] [PubMed] [Google Scholar]
- 45. Wolf K. et al. , “ Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force,” J. Cell Biol. 201, 1069–1084 (2013). 10.1083/jcb.201210152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Helvert S., Storm C., and Friedl P., “ Mechanoreciprocity in cell migration,” Nat. Cell Biol. 20, 8–20 (2018). 10.1038/s41556-017-0012-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miller J. P. et al. , “ Clinical doses of radiation reduce collagen matrix stiffness,” APL Bioeng. 2, 031901 (2018). 10.1063/1.5018327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thrivikraman G. et al. , “ Rapid fabrication of vascularized and innervated cell-laden bone models with biomimetic intrafibrillar collagen mineralization,” Nat. Commun. 10, 3520 (2019). 10.1038/s41467-019-11455-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramamoorthy P. et al. , “ Metastatic tumor-in-a-dish, a novel multicellular organoid to study lung colonization and predict therapeutic response,” Cancer Res. 79, 1681–1695 (2019). 10.1158/0008-5472.CAN-18-2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Danuser G., Allard J., and Mogilner A., “ Mathematical modeling of eukaryotic cell migration: Insights beyond experiments,” Annu. Rev. Cell Dev. Biol. 29, 501–528 (2013). 10.1146/annurev-cellbio-101512-122308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwarz U. S., Erdmann T., and Bischofs I. B., “ Focal adhesions as mechanosensors: The two-spring model,” Bio Syst. 83, 225–232 (2006). 10.1016/j.biosystems.2005.05.019 [DOI] [PubMed] [Google Scholar]
- 52. Moreo P., Garcia-Aznar J. M., and Doblare M., “ Modeling mechanosensing and its effect on the migration and proliferation of adherent cells,” Acta Biomater. 4, 613–621 (2008). 10.1016/j.actbio.2007.10.014 [DOI] [PubMed] [Google Scholar]
- 53. Crow A. et al. , “ Contractile equilibration of single cells to step changes in extracellular stiffness,” Biophys. J. 102, 443–451 (2012). 10.1016/j.bpj.2011.11.4020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marcq P., Yoshinaga N., and Prost J., “ Rigidity sensing explained by active matter theory,” Biophys. J. 101, L33–L35 (2011). 10.1016/j.bpj.2011.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chan C. E. and Odde D. J., “ Traction dynamics of filopodia on compliant substrates,” Science 322, 1687–1691 (2008). 10.1126/science.1163595 [DOI] [PubMed] [Google Scholar]
- 56. Zaman M. H., Kamm R. D., Matsudaira P., and Lauffenburger D. A., “ Computational model for cell migration in three-dimensional matrices,” Biophys. J. 89, 1389–1397 (2005). 10.1529/biophysj.105.060723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ribeiro F. O., Gomez-Benito M. J., Folgado J., Fernandes P. R., and Garcia-Aznar J. M., “ Computational model of mesenchymal migration in 3D under chemotaxis,” Comput. Methods Biomech. Biomed. Eng. 20, 59–74 (2017). 10.1080/10255842.2016.1198784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schluter D. K., Ramis-Conde I., and Chaplain M. A., “ Computational modeling of single-cell migration: The leading role of extracellular matrix fibers,” Biophys. J. 103, 1141–1151 (2012). 10.1016/j.bpj.2012.07.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun M., Zaman M. H., “ Modeling, signaling and cytoskeleton dynamics: Integrated modeling-experimental frameworks in cell migration,” Wiley Interdiscip. Rev. Syst. Biol. Med. 9, e1365 (2017). 10.1002/wsbm.1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rens E. G. and Edelstein-Keshet L., “ From energy to cellular forces in the Cellular Potts Model: An algorithmic approach,” PLoS Comput. Biol. 15, e1007459 (2019). 10.1371/journal.pcbi.1007459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Enderling H. et al. , “ Dependence of invadopodia function on collagen fiber spacing and cross-linking: Computational modeling and experimental evidence,” Biophys. J. 95, 2203–2218 (2008). 10.1529/biophysj.108.133199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maffei J. S., Srivastava J., Fallica B., and Zaman M. H., “ Combinative in vitro studies and computational model to predict 3D cell migration response to drug insult,” Integr. Biol. 6, 957–972 (2014). 10.1039/C4IB00167B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scianna M., Preziosi L., and Wolf K., “ A Cellular Potts Model simulating cell migration on and in matrix environments,” Math. Biosci. Eng. 10, 235–261 (2013). 10.3934/mbe.2013.10.235 [DOI] [PubMed] [Google Scholar]
- 64. Shenoy V. B., Wang H., and Wang X., “ A chemo-mechanical free-energy-based approach to model durotaxis and extracellular stiffness-dependent contraction and polarization of cells,” Interface Focus 6, 20150067 (2016). 10.1098/rsfs.2015.0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ahmadzadeh H. et al. , “ Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion,” Proc. Natl. Acad. Sci. U. S. A. 114, E1617–E1626 (2017). 10.1073/pnas.1617037114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alert R. and Trepat X., “ Physical models of collective cell migration,” Annu. Rev. Condens. Matter Phys. 11, 77–101 (2020). 10.1146/annurev-conmatphys-031218-013516 [DOI] [Google Scholar]
- 67. Ziebert F. and Aranson I. S., “ Computational approaches to substrate-based cell motility,” npj Comput. Mater. 2, 16019 (2016). 10.1038/npjcompumats.2016.19 [DOI] [Google Scholar]
- 68. Palmieri B., Bresler Y., Wirtz D., and Grant M., “ Multiple scale model for cell migration in monolayers: Elastic mismatch between cells enhances motility,” Sci. Rep. 5, 11745 (2015). 10.1038/srep11745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim M. C., Whisler J., Silberberg Y. R., Kamm R. D., and Asada H. H., “ Cell invasion dynamics into a three dimensional extracellular matrix fibre network,” PLoS Comput. Biol. 11, e1004535 (2015). 10.1371/journal.pcbi.1004535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen J., Weihs D., Van Dijk M., and Vermolen F. J., “ A phenomenological model for cell and nucleus deformation during cancer metastasis,” Biomech. Model. Mechanobiol. 17, 1429–1450 (2018). 10.1007/s10237-018-1036-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Niculescu I., Textor J., and de Boer R. J., “ Crawling and gliding: A computational model for shape-driven cell migration,” PLoS Comput. Biol. 11, e1004280 (2015). 10.1371/journal.pcbi.1004280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bangasser B. L. and Odde D. J., “ Master equation-based analysis of a motor-clutch model for cell traction force,” Cell Mol. Bioeng. 6, 449–459 (2013). 10.1007/s12195-013-0296-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Partridge M. A. and Marcantonio E. E., “ Initiation of attachment and generation of mature focal adhesions by integrin-containing filopodia in cell spreading,” Mol. Biol. Cell 17, 4237–4248 (2006). 10.1091/mbc.e06-06-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim M. C., Neal D. M., Kamm R. D., and Asada H. H., “ Dynamic modeling of cell migration and spreading behaviors on fibronectin coated planar substrates and micropatterned geometries,” PLoS Comput. Biol. 9, e1002926 (2013). 10.1371/journal.pcbi.1002926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cuvelier D. et al. , “ The universal dynamics of cell spreading,” Curr. Biol. 17, 694–699 (2007). 10.1016/j.cub.2007.02.058 [DOI] [PubMed] [Google Scholar]
- 76. Yeoman B. M. and Katira P., “ A stochastic algorithm for accurately predicting path persistence of cells migrating in 3D matrix environments,” PLoS One 13, e0207216 (2018). 10.1371/journal.pone.0207216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Collins T. A., Yeoman B. M., and Katira P., “ To lead or to herd: Optimal strategies for 3D collective migration of cell clusters,” Biomech. Model. Mechanobiol. 19, 1551–1564 (2020). 10.1007/s10237-020-01290-y [DOI] [PubMed] [Google Scholar]
- 78. Stokes C. L. and Lauffenburger D. A., “ Analysis of the roles of microvessel endothelial cell random motility and chemotaxis in angiogenesis,” J. Theor. Biol. 152, 377–403 (1991). 10.1016/S0022-5193(05)80201-2 [DOI] [PubMed] [Google Scholar]
- 79. Tranquillo R. T. and Lauffenburger D. A., “ Stochastic model of leukocyte chemosensory movement,” J. Math. Biol. 25, 229–262 (1987). 10.1007/BF00276435 [DOI] [PubMed] [Google Scholar]
- 80. Dickinson R. B. and Tranquillo R. T., “ A stochastic model for adhesion-mediated cell random motility and haptotaxis,” J. Math. Biol. 31, 563–600 (1993). 10.1007/BF00161199 [DOI] [PubMed] [Google Scholar]
- 81. Kim M. C., Silberberg Y. R., Abeyaratne R., Kamm R. D., and Asada H. H., “ Computational modeling of three-dimensional ECM-rigidity sensing to guide directed cell migration,” Proc. Natl. Acad. Sci. U. S. A. 115, E390–E399 (2018). 10.1073/pnas.1717230115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zaman M. H., Matsudaira P., and Lauffenburger D. A., “ Understanding effects of matrix protease and matrix organization on directional persistence and translational speed in three-dimensional cell migration,” Ann. Biomed. Eng. 35, 91–100 (2006). 10.1007/s10439-006-9205-6 [DOI] [PubMed] [Google Scholar]
- 83. Lo C. M., Wang H. B., Dembo M., and Wang Y. L., “ Cell movement is guided by the rigidity of the substrate,” Biophys. J. 79, 144–152 (2000). 10.1016/S0006-3495(00)76279-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Levental K. R. et al. , “ Matrix crosslinking forces tumor progression by enhancing integrin signaling,” Cell 139, 891–906 (2009). 10.1016/j.cell.2009.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Paszek M. J. et al. , “ Tensional homeostasis and the malignant phenotype,” Cancer Cell 8, 241–254 (2005). 10.1016/j.ccr.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 86. Malik A. A., Wennberg B., and Gerlee P., “ The impact of elastic deformations of the extracellular matrix on cell migration,” Bull. Math. Biol. 82, 49 (2020). 10.1007/s11538-020-00721-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kwon T., Kwon O. S., Cha H. J., and Sung B. J., “ Stochastic and heterogeneous cancer cell migration: Experiment and theory,” Sci. Rep. 9, 16297 (2019). 10.1038/s41598-019-52480-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Heck T. et al. , “ The role of actin protrusion dynamics in cell migration through a degradable viscoelastic extracellular matrix: Insights from a computational model,” PLoS Comput. Biol. 16, e1007250 (2020). 10.1371/journal.pcbi.1007250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wisdom K. M. et al. , “ Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments,” Nat. Commun. 9, 4144 (2018). 10.1038/s41467-018-06641-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ahmmed S. M. et al. , “ Multi-sample deformability cytometry of cancer cells,” APL Bioeng. 2, 032002 (2018). 10.1063/1.5020992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ehrbar M. et al. , “ Elucidating the role of matrix stiffness in 3D cell migration and remodeling,” Biophys. J. 100, 284–293 (2011). 10.1016/j.bpj.2010.11.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Beri P. et al. , “ Cell adhesiveness serves as a biophysical marker for metastatic potential,” Cancer Res. 80, 901–911 (2020). 10.1158/0008-5472.CAN-19-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bangasser B. L. et al. , “ Shifting the optimal stiffness for cell migration,” Nat. Commun. 8, 15313 (2017). 10.1038/ncomms15313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bell G. I., “ Models for the specific adhesion of cells to cells,” Science 200, 618–627 (1978). 10.1126/science.347575 [DOI] [PubMed] [Google Scholar]
- 95. Elosegui-Artola A. et al. , “ Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity,” Nat. Cell. Biol. 18, 540–548 (2016). 10.1038/ncb3336 [DOI] [PubMed] [Google Scholar]
- 96. Plotnikov S. V., Pasapera A. M., Sabass B., and Waterman C. M., “ Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration,” Cell 151, 1513–1527 (2012). 10.1016/j.cell.2012.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hecht I. et al. , “ Tumor invasion optimization by mesenchymal-amoeboid heterogeneity,” Sci. Rep. 5, 10622 (2015). 10.1038/srep10622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Doyle A. D., Wang F. W., Matsumoto K., and Yamada K. M., “ One-dimensional topography underlies three-dimensional fibrillar cell migration,” J. Cell Biol. 184, 481–490 (2009). 10.1083/jcb.200810041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Holle A. W. et al. , “ Cancer cells invade confined microchannels via a self-directed mesenchymal-to-amoeboid transition,” Nano Lett. 19, 2280–2290 (2019). 10.1021/acs.nanolett.8b04720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang M. et al. , “ Microchannel stiffness and confinement jointly induce the mesenchymal-amoeboid transition of cancer cell migration,” Nano Lett. 19, 5949–5958 (2019). 10.1021/acs.nanolett.9b01597 [DOI] [PubMed] [Google Scholar]
- 101. Sunyer R. et al. , “ Collective cell durotaxis emerges from long-range intercellular force transmission,” Science 353, 1157–1161 (2016). 10.1126/science.aaf7119 [DOI] [PubMed] [Google Scholar]
- 102. Escribano J. et al. , “ A hybrid computational model for collective cell durotaxis,” Biomech. Model. Mech. 17, 1037–1052 (2018). 10.1007/s10237-018-1010-2 [DOI] [PubMed] [Google Scholar]
- 103. Ladoux B. and Mege R. M., “ Mechanobiology of collective cell behaviours,” Nat. Rev. Mol. Cell Biol. 18, 743–757 (2017). 10.1038/nrm.2017.98 [DOI] [PubMed] [Google Scholar]
- 104. Boocock D., Hino N., Ruzickova N., Hirashima T., and Hannezo E., “ Theory of mechanochemical patterning and optimal migration in cell monolayers,” Nat. Phys. (published online) (2020). 10.1038/s41567-020-01037-7 [DOI] [Google Scholar]
- 105. Kingsmore K. M. et al. , “ MRI analysis to map interstitial flow in the brain tumor microenvironment,” APL Bioeng. 2, 031905 (2018). 10.1063/1.5023503 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.