

Abstract
B cells infiltrate pancreatic ductal adenocarcinoma (PDAC) and in preclinical cancer models, can suppress T cell immunosurveillance in cancer. Here, we conducted a pilot study to assess the safety and feasibility of administering lentiviral-transduced chimeric antigen receptor (CAR)-modified autologous T cells redirected against mesothelin to target tumor cells along with CART cells redirected against CD19 to deplete B cells. Both CARs contained 4-1BB and CD3ζ signaling domains. Three patients with chemotherapy-refractory PDAC received 1.5 g/m2 cyclophosphamide prior to separate infusions of lentiviral-transduced T cells engineered to express chimeric anti-mesothelin immunoreceptor SS1 (CART-Meso, 3 × 107/m2) and chimeric anti-CD19 immunoreceptor (CART-19, 3 × 107/m2). Treatment was well tolerated without dose-limiting toxicities. Best response was stable disease (1 of 3 patients). CART-19 (compared to CART-Meso) cells showed the greatest expansion in the blood, although persistence was transient. B cells were successfully depleted in all subjects, became undetectable by 7–10 days post-infusion, and remained undetectable for at least 28 days. Together, concomitant delivery of CART-Meso and CART-19 cells in patients with PDAC is safe. CART-19 cells deplete normal B cells but at the dose tested in these 3 subjects did not improve CART-Meso cell persistence.
Keywords: pancreatic cancer, chimeric antigen receptor, mesothelin, B cells, CD19
Graphical Abstract
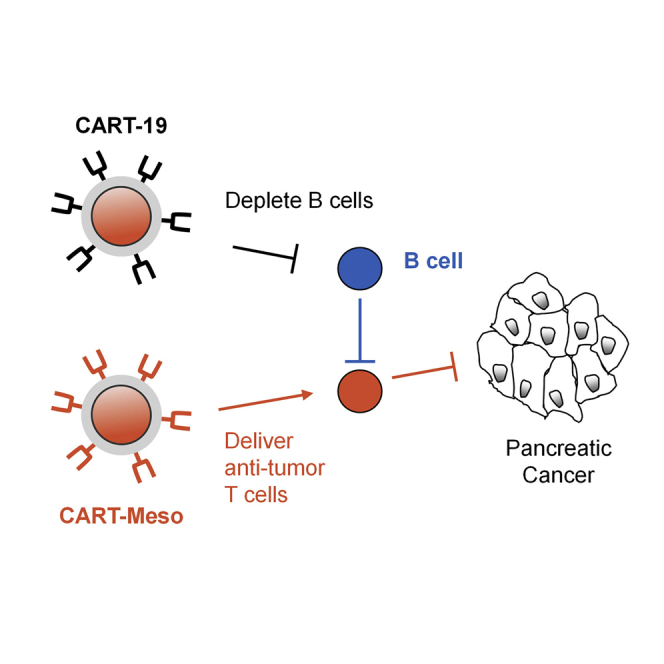
This study examined the safety/feasibility of administering two separate infusions of CART cells recognizing (1) mesothelin, a tumor-associated antigen, and (2) CD19 to deplete B cells. Treatment was well-tolerated in patients with pancreatic cancer and depleted peripheral B cells but did not improve CART cell anti-tumor activity or persistence.
Introduction
Enthusiasm generated by immunotherapy and its broad application to cancers has not yet translated to pancreatic ductal adenocarcinoma (PDAC).1 Over the past decade, efforts to leverage anti-tumor immunity for the treatment of PDAC (and many other solid malignancies) have included vaccines, immune checkpoint inhibitors, targeted therapies, and adoptive T cell therapy.1,2 However, these strategies do not produce meaningful clinical activity in the vast majority of patients.
One promising immunotherapy approach is the use of T cells engineered to express a chimeric-antigen receptor (CAR) that recognizes a tumor-associated antigen on the cell surface of tumor cells.3, 4, 5 However, unlike the significant clinical activity achieved with CART cells in hematological malignancies,6 success with CART cells in solid cancers, including PDAC, has been limited.7 Notably, this observation contrasts the capacity of CART cells to effectively lyse autologous human epithelial-derived cancer cells in vitro. To explain these incongruent findings, multiple mechanisms of immune resistance have been proposed, including poor CART cell persistence and expansion in vivo, immune rejection of CART cells, limited trafficking of CART cells to tumors, and an immunosuppressive tumor microenvironment that coerces tumor-infiltrating T cells into a hypofunctional state.7
Engineered T cells, such as CART cells, are an invaluable tool for tracking the fate of tumor-reactive T cells in vivo. In patients with advanced solid cancers, CART cells have been shown to rapidly expand in the peripheral blood after administration, but their persistence is transient.5,8, 9, 10 The mechanisms underlying this poor in vivo persistence of CART cells in patients with solid cancer remain ill-defined. Notably, antibody responses against CARs have been observed in patients,4,5,11 which raises the possibility that limited CART cell persistence in vivo may reflect anti-CAR humoral immunity mediated by B cells.
B cells can have fundamental roles in shaping T cell responses. For example, B cells can support anti-tumor immunity by acting as antigen-presenting cells for the priming of T cells and by producing tumor-specific antibodies.12 Recently, enrichment of memory B cells in tertiary lymphoid structures was found to associate with anti-tumor activity achieved with immunotherapy in patients with melanoma, renal cell carcinoma, and soft-tissue sarcoma.13, 14, 15 However, B cells have also been implicated as negative regulators of anti-tumor T cell immunity due to their capacity to promote immunosuppressive activity by macrophages and to dampen T cell-mediated anti-tumor immunity.16, 17, 18, 19, 20, 21 In this regard, mouse models of pancreatic cancer have shown that tumor outgrowth is slowed in B cell-deficient mice but can be rescued by the adoptive transfer of CD19+ B cells.22,23
Here, we sought to examine a potential role for B cells in regulating CART cell activity and biology in patients with PDAC. The primary objective was to assess the safety and feasibility of administering two separate intravenous (i.v.) infusions of lentiviral-transduced CART cells engineered to recognize (1) mesothelin as a tumor-associated antigen and (2) CD19 to deplete B cells. All patients received pre-conditioning with cyclophosphamide as a lymphodepleting chemotherapy. Secondary objectives were to evaluate CART cell persistence, B cell depletion, and clinical activity.
Results
B Cell Infiltration in Pancreatic Ductal Adenocarcinoma
We first examined for the presence of CD19+ B cells in treatment-naive surgically resected human PDAC tissues using multiplex chromogenic immunohistochemistry (IHC). We found that B cells were focally distributed within tumors (Figure 1A). Most tissue regions examined showed a lack of CD19+ B cells. However, in all patient samples examined, B cells could be found in the stroma and detected adjacent to CD3+ T cells that surrounded CK19+ PDAC cells. B cells invariably co-localized with CD3+ T cells, sometimes forming structures consistent with tertiary lymphoid aggregates. These aggregates were found within the pancreatic parenchyma and differed significantly from lymph node structures that were detected within adjacent adipose tissue. Although CD3+ T cells dramatically outnumbered CD19+ B cells in PDAC (Figure 1B), their presence within tumor tissues was strongly correlated with CD19+ B cells (Figure 1C). Consistent with this observation, we also found that CD19 and CD3E transcripts were strongly correlated in human PDAC samples (n = 179) retrieved from The Cancer Genome Atlas (TCGA) human pancreatic adenocarcinoma (PAAD) dataset (Figure 1D). Collectively, these findings identify CD19+ B cells as a significant cellular component of the inflammatory reaction to human PDAC.
Figure 1.

B Cell Infiltration in Human Pancreatic Ductal Adenocarcinoma
In (A)–(C), human PDAC tissues were stained for CD3, CD19, and CK19 by multiplex immunohistochemistry. (A) Representative images showing four patterns of CD19+ B cell infiltration in PDAC. Scale bars, 100 μm. (B) Quantification of CD19 and CD3 expression in human PDAC tissues (n = 11). Statistical significance was evaluated using a Mann-Whitney test. (C) Correlation plot of number of CD3+ cells per mm2 versus number of CD19+ cells per mm2. Data points represent quantification of individual 5X fields captured across stained human PDAC tissues (n = 11). Spearman correlation was performed for r and p value (two-tailed). (D) Correlation plot showing gene expression of CD3E and CD19 in human pancreatic adenocarcinoma (PAAD) patient samples (n = 179) downloaded from The Cancer Genome Atlas (TCGA) dataset. Pearson correlation was performed for r and p value (two-tailed).
Expansion of B Cell Clones after CART-Meso Cellular Therapy
We previously showed that CART-Meso cellular therapy triggers a spreading of antibody responses against multiple proteins.4 We have also found an emergence of human anti-mouse antibodies (HAMAs) and human anti-chimeric (HACA) antibodies in some patients receiving CART-Meso cell therapy.4,5,11 Together, these findings suggest that B cell clones may undergo expansion after CART-Meso cell infusion and as such, we hypothesized that B cell expansion may limit the therapeutic potential of CART cells in vivo. To address this hypothesis, we performed high-throughput sequencing to compare the clonal structure of circulating B cells isolated from patients before and after treatment with CART-Meso cells. For this analysis, we examined previously collected blood samples from patients with PDAC treated with repeated infusions of CART-Meso cells engineered using RNA electroporation4 and from patients with PDAC, ovarian cancer, and mesothelioma treated with a single infusion of CART-Meso cells engineered using lentiviral transduction.5 We found that both RNA CART-Meso cells (Figure 2A) and Lentiviral CART-Meso cells (Figure 2B) triggered expansion of a subset of B cell clones detected 28 days after treatment in all patients analyzed.
Figure 2.

Peripheral Blood B Cell Clone Expansion after CART-Meso Cell Infusion in Patients with Advanced Solid Cancers
B cell clones were detected in the peripheral blood by DNA sequencing of the IgH CDR3. (A and B) Shown is the number and fold change of expanded B cell clones found in patients at day 28 after beginning treatment (compared to baseline) with (A) RNA CART-Meso or (B) lentiviral CART-Meso cells on prior clinical studies.
Patients and Treatment
To address the role of B cells in regulating CART-Meso cell therapy in patients with PDAC, we designed a pilot study in which patients received two separate infusions of (1) mesothelin-specific CART cells (CART-Meso) for targeting the tumor and (2) CD19-specific CART cells (CART-19) to deplete B cells. Between June 2015 and September 2015, four patients signed an informed consent. One patient died after pheresis due to progressive disease prior to receiving CART cell therapy. The full analysis set included 3 patients. Demographics and baseline characteristics are summarized in Table 1. Mesothelin expression was not an inclusion criterion for this study and was only available for patient 19214-03. In this patient, analysis of fine needle aspirates (FNAs) from a pancreatic body mass and liver mass showed 2+ membrane/cytoplasmic reactivity in 70% of rare atypical (tumor) cells in both specimens. For each patient, CART-Meso and CART-19 cells were successfully manufactured. All patients received a single dose of cyclophosphamide (1.5 g/m2 i.v.) administered 4 days prior to separate infusions of 3 × 107 CART-Meso cells/m2 and 3 × 107 CART-19 cells/m2. Cell product characteristics for CART-Meso and CART-19 cells are summarized in Tables S1 and S2, respectively. The average transduction efficiency for CART-Meso and CART-19 cells was 37% and 22%, respectively. The infused cell products were an average of 97% CD3+ (CART-Meso) and 98% CD3+ (CART-19) with an average CD4/CD8 ratio of 2.5 (CART-Meso) and 2.7 (CART-19). The average cell viability for CART-Meso was 98% (range of 95%–100%) and for CART-19 was 99% (range of 97%–100%).
Table 1.
Patient Characteristics
| Subject ID | Age/Sex | Performance Status (ECOG) | Disease | Prior Number of Therapies | Co-morbidities | Sites of Disease at Enrollment |
|---|---|---|---|---|---|---|
| 19214-01 | 64/F | 1 | pancreatic adenocarcinoma | 4 | GERD, IBS, hypoalbuminemia, anemia, percutaneous biliary drain, chronic back pain | lung, lymph nodes (mediastinal, intra/retro-peritoneal) |
| 19214-03 | 38/F | 0 | pancreatic adenocarcinoma | 2 | alopecia, anemia, depression | liver |
| 19214-04 | 50/M | 0 | pancreatic adenocarcinoma | 3 | DVT, peripheral sensory neuropathy | liver |
DVT, deep vein thrombosis; GERD, gastresophageal reflux disease.
Safety and Clinical Activity
Adverse events (AEs) at least possibly related to study treatment are summarized in Table 2. The most common AEs were fatigue and fever observed in all patients. The clinical course for each patient was as follows: (1) patient 19214-01 was a 64-year-old female diagnosed with metastatic PDAC involving the lung and lymph nodes. She had received multiple prior therapies including FOLFOX, gemcitabine/nab-paclitaxel, immunotherapy with single-agent CRS-207 (an attenuated mesothelin-expressing Listeria vaccine) as an investigational drug, and FOLFIRI. There was no clinical evidence of cytokine release syndrome associated with CART cell therapy. The patient experienced clinical decline within 2 months after treatment due to progression of pulmonary and nodal metastases and accumulation of new pelvic fluid suggestive of peritoneal carcinomatosis. (2) Patient 19214-02 was a 62-year-old male who had received neoadjuvant FOLFIRINOX and SBRT to his primary pancreatic tumor followed by Whipple resection, which revealed node-positive disease. The patient subsequently developed hepatic metastases that were treated with additional FOLFIRINOX, followed by gemcitabine/nab-paclitaxel before enrolling on study. At the time of study entry, pulmonary, hepatic, and peritoneal metastases were noted. The patient underwent leukapheresis, but due to disease progression died prior to receiving CART cell therapy. (3) Patient 19214-03 was a 38-year-old female diagnosed with metastatic PDAC involving the liver. She had previously received FOLFIRINOX and gemcitabine/nab-paclitaxel. Her treatment course after receiving CART cell therapy was notable for a fever reaching 104.3°F at 6 days post infusion. She was treated empirically with vancomycin and aztreonam during work-up, which revealed no evidence of underlying infection. Her fevers continued for 5 days before subsiding. At 2 months after CART cell infusion, imaging showed enlargement of the primary pancreatic tumor and modest growth in hepatic metastases. (4) Patient 19214-04 was a 50-year-old male with metastatic PDAC who had received multiple therapies over the course of 4 years, including several courses of FOLFIRINOX (one as part of a clinical trial in combination with a Hedgehog inhibitor), gemcitabine/nab-paclitaxel, and capecitabine plus temozolomide. At the time of study entry, the patient had multifocal hepatic metastases and received CART cell therapy without evidence of cytokine release syndrome. Early computed tomography (CT) imaging at 2 weeks after CART cell infusion showed slight disease progression, which was unchanged on follow-up scans at 2 weeks later. The patient opted to resume standard of care chemotherapy at 6 weeks.
Table 2.
Treatment-Related Adverse Events
| All Subjects (N = 3) | Grade 1 n | Grade 2 n | Grade 3 n | Total n |
|---|---|---|---|---|
| Clinical Events | ||||
| Fatigue | 2 | 1 | 3 | |
| Fever | 2 | 1 | 3 | |
| Dizziness | 2 | 2 | ||
| Night sweats | 2 | 2 | ||
| Nausea | 1 | 1 | ||
| Myalgia | 1 | 1 | ||
| Headache | 1 | 1 | ||
| Upper respiratory symptoms | 1 | 1 | ||
| Hypotension | 1 | 1 | ||
| Flu-like symptoms | 1 | 1 | ||
| Hematologic Events | ||||
| Lymphocyte count decreased | 1 | 2 | 3 | |
| Neutrophil count decreased | 2 | 2 | ||
| Platelet count decreased | 1 | 1 | ||
| Anemia | 1 | 1 | ||
| Total | 16 | 2 | 5 | 23 |
In summary, no dose-limiting toxicities were observed in the 3 treated patients. The best overall response (BOR) based on RECIST v1.1 was stable disease in 1 of 3 patients.
CART Cell Expansion, Persistence, and Biological Activity
The infusion of CART-19 cells is known to deplete normal B cells in patients. Thus, we used this endpoint as a measure of the in vivo effector activity of CART-19 cells. We found that CART cell therapy effectively depleted normal B cells in the blood for all patients (Figures 3A and 3B). Peripheral blood B cells became undetectable by day 10–14 after treatment and remained depleted through day 28 of follow-up. For patient 19214-01, analysis at month 2 showed continued depletion of B cells at a frequency of 0.7% compared to 23.3% at baseline. For patient 19214-02, analyses performed at months 6, 9, and 12 showed progressive re-population of B cells with frequencies of 3.2%, 6.3%, and 9.7%, respectively, compared to 8.3% at baseline. Follow-up data was not available for patient 19214-04. Depletion of B cells preceded the expansion of CART-19 cells in the peripheral blood for each patient. Notably, the peak expansion of CART-19 copies in the blood was a log higher than observed for CART-Meso (Figures 3C and 3D). The level of expansion for CART-19 was also at a similar magnitude as has been seen in patients with hematological malignancies.24 CART-19, but not CART-Meso copies, were also detected in on-treatment day 14 biopsies (details described in the Materials and Methods) of a lung mass for patient 19214-01 (0 and 1,567 copies/μg DNA for CART-Meso and CART-19, respectively) and liver mass for patient 19214-03 (0 and 325 copies/μg DNA for CART-Meso and CART-19, respectively), although in the latter patient there was no viable tumor identified in the liver biopsy specimen.
Figure 3.

CART Cell Expansion, Persistence and Activity
(A) Frequency of B cells detected in the peripheral blood of patients with PDAC at baseline (Pre) and at day 28 after receiving a 1:1 mixture of CART-19 and CART-Meso cells. (B) Frequency of CART-19 cells and B cells in blood over time for indicated patients. (C and D) Shown is the copy number of CART-Meso/μg DNA and CART-19/μg DNA detected in the blood at (C) peak expansion and (D) the indicated time points. Statistical significance in (A) and (C) was determined with unpaired Mann Whitney test (two-tailed). ∗p < 0.05.
The kinetics of expansion within the blood for CART-19 and CART-Meso were similar with peak copy number levels occurring within 7–14 days after infusion. The kinetics of CART19 cells detected in the blood by flow cytometry (Figure 3C) and by quantitative PCR (qPCR; Figure 3D) were also similar. We found no appreciable changes in the levels of HAMA responses in any of the patients after infusion with CART cells (Table S3). Analysis of serum cytokine levels reveals significant increases in interleukin-10 (IL-10) and IL-12 detected 10 days after CART cell infusion in all 3 patients (Figures 4A and 4B). In addition, 2 of 3 patients showed an increase in IL-6, and 1 of 3 patients showed an increase in interferon-γ (IFN-γ) detected after CART cell infusion (Figures 3C and 3D). All patients showed increases in the chemokine IP-10 (CXCL10) with peak levels detected between days 7 and 14, and 2 of 3 patients had an increase in serum MIG (CXCL9) levels. No changes in levels of cytokines associated with T cell activation including IL-2, IL-4, IL-5, IL-7, IL-17, IL-21, and tumor necrosis factor (TNF) were seen. However, despite evidence for CART cell biological activity seen with B cell depletion and cytokine release, neither CART-Meso nor CART-19 cells were found to persist in the blood beyond 28 days after infusion.
Figure 4.

Cytokine Release after CART Cell Infusion
(A–F) Serum cytokines, including (A) IL-10, (B) IL-12, (C) IL-6, (D) IFN-γ, (E) IP-10 (CXCL10), and (F) MIG (CXCL9) were detected in the peripheral blood at defined time points after CART cell infusion. Shown in each left panel is comparison of cytokine levels detected pre-infusion (Pre) and at day 10 post-infusion. In right panel, fold change in cytokine levels are depicted over time. Statistical significance was determined by paired t test. ∗p < 0.05.
Discussion
In this study, we evaluated the safety and feasibility of administering two separate infusions of CART-Meso and CART-19 cells that recognize mesothelin and CD19, respectively. Our rationale for administering CART-19 cells was to deplete CD19+ B cells, which we hypothesized were involved in limiting the in vivo persistence of CART cells. In addition, preclinical data has shown a role for CD19+ B cells in restraining T cell immune responses against cancer.12,19,20 Consistent with this, we found in surgically resected specimens that CD19+ B cells actively infiltrate human PDAC and that their infiltration strongly correlates with CD3+ T cells. Thus, we also hypothesized that B cell depletion might enhance the activity of CART-Meso cells in patients. However, despite the capacity of CART-19 cells to deplete peripheral blood B cells in patients with PDAC, at the dose level tested we observed no clinical activity with combining CART-19 cells with CART-Meso cells in the three patients enrolled in this study. In addition, CART cell persistence detected in the blood was transient. Overall, our findings show that CART-19 cells can be safely combined with CART-Meso cells, but also indicate that mechanisms beyond CD19+ B cells regulate CART-Meso cell persistence and their anti-tumor activity in patients with PDAC.
The administration of a combination of CART cells recognizing distinct protein targets has recently been considered as a strategy to circumvent the emergence of tumor-antigen loss variants. For example, in hematologic malignancies, loss of CD19 expression on cancer cells is a recognized mechanism of resistance to CART-19 cell therapy.25,26 To circumvent this resistance, ongoing studies are combining CART-19 cells with CART cells recognizing other tumor-associated proteins, such as CD22 and BCMA.27,28 Similarly, in solid cancers, heterogeneous expression of tumor-associated proteins presents a significant challenge that may require the use of a combination of or dual-targeted CART cells recognizing multiple tumor antigens. Consistent with this, CART cell therapy targeting epidermal growth factor receptor variant III (EGFRvIII) was associated with the emergence of tumor antigen-loss variants in patients with glioblastoma.29 Here, we combined CART-Meso cells to target mesothelin-specific tumor cells with CART-19 cells as a strategy to deplete normal B cells. We hypothesized that B cells may limit the persistence and anti-tumor activity of CART cells; thus, our approach was to combine tumor-antigen-specific CART cells with CART cells capable of removing elements of immunosuppression. Notably, our study incorporates separate infusions of CART cells directed against two distinct self-antigens. In this respect, we observed on-target depletion of CD19+ B cells, but no evidence of off-tumor on-target toxicities related to CART-Meso cells. Mesothelin is expressed on the lining of the peritoneum, pericardium, and pleura. However, we did not find any evidence of serositis, which is consistent with our prior studies.3, 4, 5 Together, our findings demonstrate that CART cells designed to recognize distinct self-protein targets can be safely combined and administered to patients.
Preclinical models have identified B cells as key regulators of T cell immune responses in cancer. Through production of antibodies, B cells can promote the immunosuppressive activity of macrophages within tumors.21 B cells also produce cytokines, such as IL-10, which can impair T cell anti-tumor activity.30 In addition, we have previously shown that B cell-dependent humoral responses develop against CART cells.4,5 In this study, we depleted endogenous B cells using CART-19 cells. CART-19 cells have demonstrated the capacity to deplete B cells in patients with hematologic malignancies.31 CART-19 cells may also deplete plasma cells in the bone marrow resulting in a decline in serum immunoglobulin levels.31 However, it is also clear that at least some long-lived antibody-secreting plasma cells can persist in the setting of CART-19 cell therapy.32 In our study, we found that CART-19 cells successfully depleted peripheral blood B cells in each patient with PDAC. In addition, we did not detect any increase in human anti-mouse antibodies against the murine component of the SS1 mesothelin-specific CAR, which we have seen in previous studies.4,5,11 However, our findings also show that B cells in PDAC reside in multiple compartments including adjacent to tumor-infiltrating T cells, in lymphoid aggregates, and in tumor-draining lymph nodes. We were unable to address the capacity of CART-19 cells to eliminate B cells in these tissue compartments, where they may have roles in regulating the effector activity of CART-Meso cells.
The mechanisms that regulate the persistence of CAR-T cells after infusion into patients remain ill-defined. The incorporation of the 4-1BB signaling domain into the CAR has previously been shown to enhance the persistence of CART cells in preclinical models.33,34 Consistent with this, CART cells can persist for years in some patients.35, 36, 37 In contrast, we have shown that CART-Meso cells persist only transiently in the peripheral blood of patients with advanced solid cancers.5 We hypothesized that B cells, which are eliminated by CART-19 cells, may impair CART cell persistence in vivo, such as by antibody-dependent cellular toxicity due to B cell production of CAR-specific antibodies. However, we found that CART-19 cells also persisted transiently in patients with PDAC despite their capacity to eliminate peripheral blood B cells. This finding suggests that either the host receiving the CART cells, the quality of the CART cells, or both are fundamental in defining the capacity of CART cells to persist in vivo. The degree of host conditioning can also impact CD19 CART cells persistence,38 and in this respect our study used mild lymphodepletion with cyclophosphamide, while studies showing long-term B cell aplasia have used cyclophosphamide in combination with fludarabine. Accordingly, decreased persistence of CART-19 cells in patients with chronic lymphocytic leukemia has been associated with enrichment of genes in the transferred CART cells that are involved in effector differentiation, whereas sustained persistence associates with memory-related genes.37
Preclinical studies have also shown that the adoptive transfer of antigen-specific T cells derived from central memory T cells, but not effector memory T cells, persist long-term in vivo.39 We have recently shown that T cells from patients with advanced PDAC are transcriptionally altered with a terminally differentiated effector phenotype and display diminished capacity to proliferate ex vivo.40 Thus, the quality of T cells collected from patients with PDAC may be a determinant of the poor persistence seen with CART cells. A high CD4:CD8 ratio of infused cells has also been reported to associate with impaired survival of CART cells in vivo.41,42 In our studies, the CD4:CD8 ratio (range 1.86–3.74) of the infused CART cells was notably higher than reported for other studies where CART cell persistence is observed.37 Taken together, it may be necessary to enrich for distinct T cell populations with enhanced replicative capacity (e.g., central memory T cells) for generating CART cells capable of persisting and mediating effector activity in patients with PDAC.37
After infusion, CART cells expand in vivo with peak frequencies typically detected within 7–14 days. Consistent with observations in hematologic malignancies, we found that CART-19 cells expanded nearly 1,000-fold in patients with PDAC. In contrast, CART-Meso cells demonstrated a 10-fold less expansion. CART-Meso cell expansion in vivo can be enhanced with pre-conditioning lymphodepletion with cyclophosphamide,5 which we included in this study. However, our findings show that the expansion capacity of CART-19 and CART-Meso cells is distinct. This difference likely reflects the abundance and location of the CAR target. Specifically, CD19+ B cells are readily accessible in the blood, bone marrow, and lymph nodes to circulating CART-19 cells, whereas mesothelin-expressing tumor cells are mainly confined to solid tissues. We also observed serum cytokine changes in all patients examined in this study with notable increases seen in IL-10, IL-12, and IP-10 (CXCL10). This finding is in contrast to our observations with infusion of CART-Meso cells alone in patients with advanced solid cancers where appreciable changes in serum cytokines were not observed.5 Thus, it is likely that the cytokine changes detected in this study represent the biological activity of CART-19 cells. We also analyzed on-treatment tumor biopsies to evaluate the trafficking of CART cells to tumor tissues. For both biopsies, we detected CART-19 but not CART-Meso. The absence of CART-Meso in the biopsy specimens might reflect sampling error, differences in trafficking patterns for CART-19 and CART-Meso cells, or limited CART-Meso cell expansion within tissues. It is also important to recognize that mesothelin expression was not able to be determined in 2 of the 3 study subjects. Nonetheless, by including CART-19 cells in this study, we show that the expansion capacity of CART cells detected in the peripheral blood of patients with PDAC is comparable to findings seen in hematologic malignancies.
Combination strategies will be necessary for unveiling the full therapeutic potential of CART cells in solid malignancies. For example, CART cells may need to be combined with treatments that improve their infiltration into tumors,43, 44, 45, 46 modulate immunosuppression imposed by tumors,47, 48, 49 or enhance the effector activity of CART cells by blocking immune checkpoints (e.g., PD-1)50,51 or stimulating immune activation (e.g., CD40 agonist).52 In this study, we demonstrate the safety and feasibility of administering two separate infusions of lentiviral CART cells recognizing mesothelin and CD19 to patients with PDAC. Our findings provide new insights into the biology of CART cells in solid cancer. We showed that CART-Meso and CART-19 cells display differential expansion capacity suggesting the importance of the CAR target and perhaps its tissue location as a determinant of in vivo expansion within the blood. However, unlike hematologic malignancies, CART-19 cells failed to persist indicating a role for either host determinants (e.g., cancer inflammation) or T cell intrinsic mechanisms. Finally, our study provides a framework for studies testing combinations of CART cells targeting distinct CAR targets, which can include both tumor and non-tumor associated proteins.
Materials and Methods
Patients
Patients with PDAC were enrolled in a phase I study (NCT02465983) at the Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco (San Francisco, CA, USA). Key inclusion criteria were age ≥18 years, histologically-confirmed pancreatic carcinoma, unresectable or metastatic disease, receipt of at least one prior standard chemotherapy, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, life expectancy >3 months, adequate organ, bone marrow, and clotting function (unless therapeutically anticoagulated for history of thrombosis with stable coagulation parameters), and agreement to use approved contraceptive methods and to abstain from other methods of contraception during the study and 6 months following study cell infusion or proof of sterility. Exclusion criteria included participation in a therapeutic study within 4 weeks prior to the screening visit or anticipating treatment with another investigational product while on study, anticipated need for systemic chemotherapy within 2 weeks before apheresis and 2 weeks before infusion of CART cell therapy, active invasive cancer other than PDAC, infection with HIV, HCV, or HBV or other ongoing or active infection, active autoimmune disease requiring immunosuppressive therapy within 4 weeks prior to screening visit planned concurrent treatment with systemic high-dose corticosteroids, requirement for supplemental oxygen therapy, prior therapy with gene modified cells, previous experimental therapy with SS1 moiety, murine or chimeric antibodies, history of allergy to murine proteins or study product excipients (human serum albumin, DMSO, and Dextran 40), clinically significant pericardial effusion, congestive heart failure (NY Heart Association Grade II–IV), or cardiovascular condition that would preclude assessment of pericarditis, and pregnancy or breast-feeding. Mesothelin expression on tumors was not a requirement for eligibility. All patients provided written informed consent and the study was approved by the local institutional review board of the University of California, San Francisco.
Clinical Samples
Patient samples were obtained after written informed consent and were de-identified. All studies were approved by the local institutional review boards of the University of Pennsylvania and University of California, San Francisco. Peripheral blood samples were collected in EDTA tubes for isolation of peripheral blood mononuclear cells. Research tubes were processed and stored as previously described.5 To examine the presence and distribution of B cells within pancreatic tumors, we obtained surgically resected tissue specimens from patients with PDAC from the Cooperative Human Tissue Network (CHTN).
Study Design and Treatment Plan
The primary objective of this pilot study was to determine the safety and feasibility of administering two separate infusions of lentiviral-transduced CART cells (CART-Meso and CART-19) with cyclophosphamide chemotherapy in patients with advanced or metastatic PDAC. Secondary objectives included assessment of clinical anti-tumor effect (as determined by RECIST v1.1) and measurement of progression-free survival and overall survival.
After screening for eligibility, patients underwent a large-volume leukapheresis for collection of peripheral blood mononuclear cells (PBMCs) to manufacture CART-Meso and CART-19 cells. Patients received cyclophosphamide (1.5 g/m2) administered i.v. 4 days prior to CART cell therapy. Two patient cohorts (N = 6) were initially planned to evaluate two dose levels of CART cells (cohort 1: 3 × 107/m2 and cohort 2: 3 × 108/m2). However, due to lack of clinical activity and lack of persistence of CART cells seen in the first three patients enrolled in cohort 1, the study was terminated early. For cohort 1, CART-Meso (3 × 107/m2) and CART-19 (3 × 107/m2) cells were administered i.v. in sequential fashion as separate infusions. Peripheral blood samples were obtained at defined time points to monitor for safety, B cell depletion, and CART cell persistence.
Safety Assessments
All patients who received CART-Meso and CART-19 cells were evaluated for safety. Safety assessments included monitoring and recording potential adverse effects of the treatment using Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03 at each study visit, including days +1, 3, 7, 10, 14, 21, and 28 days post-CART infusion. In the event of cytokine release syndrome (CRS), the Penn Grading Scale for Cytokine Release Syndrome (PGS-CRS53) was to be used since the CTCAE does not accurately capture CRS due to infusion therapy. AEs, including laboratory toxicities and clinical events, were defined as unrelated or possibly, probably, or definitely related to study participation. The study period for collection of AEs started on the day of cyclophosphamide administration and continued until a subject progressed or initiated another non-immune cancer-related therapy. A DLT was defined as grade 3 or higher hematologic or non-hematologic toxicity that developed within 4 weeks post infusion and was at least possibly related to CART cell therapy. DLTs included: (1) grade 3 or higher non-hematological toxicity with the exception of asymptomatic grade 3 electrolytes, grade 3 nausea, vomiting, diarrhea, or fatigue. Grade 3 toxicities associated with T cell therapy that are expected, manageable, and reversible within 3 days of care (e.g., high fevers, serositis, transient hypotension, and other sequelae of CRS) were not considered DLTs. (2) Grade 3 or higher hematologic toxicity reportable as an AE except asymptomatic lymphopenia or other blood counts that were pre-existing regardless of grading.
Tumor Response Assessment
Tumor response was evaluated by CT scans and CA19-9 levels. Radiographic responses were measured using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1.
CART-Meso and CART-19 Design and Cell Manufacturing
Mesothelin-specific and CD19-specific CAR-modified T cells were manufactured in accordance with a US FDA accepted investigational new drug (IND) application. Leukocytes were collected from a large-volume (∼10–12 Liter) apheresis procedure performed at the UCSF apheresis center approximately 4–6 weeks prior to infusion. The apheresis product was shipped to the University of Pennsylvania Clinical Cell and Vaccine Production Facility (CVPF, https://www.med.upenn.edu/cvpf/) for CART cell manufacturing. Lymphocytes were isolated from the apheresis product by elutriation, enriched for T cells, and split in half for activation, transduction, and culture. The mesothelin-specific CAR was constructed using a chimeric anti-mesothelin immunoreceptor SS1 fused to the 4-1BB and CD3ζ signaling domains.5 The CD19-specific CAR was constructed using a chimeric anti-CD19 immunoreceptor fused to the 4-1BB and CD3ζ signaling domains.54 The separate lymphocyte cultures were transduced with a self-inactivating lentiviral vector expressing a CAR recognizing either mesothelin or CD19 and then continued expansion separately in vitro using bead-immobilized anti-CD3 and anti-CD28 antibodies. The CAR-transduced autologous T cell products were cryopreserved in separate cryobags in infusible cryoprotectant-supplemented solution. Quality control testing for cell purity, sterility, and identity per FDA-accepted release criteria was performed prior to release of cell product for infusion. Infusion bags were stored in a controlled and monitored freezer at the CVPF until needed and then shipped to UCSF several days prior to the scheduled infusion. The total T cell dose for infusion was based on the number for each patient required to achieve 3 × 107/m2 CAR-transduced cells. The pre-defined cell viability cut-off for infusion was ≥70%. CART cells were thawed at the bedside and infusion was administered as an inpatient, with overnight observation, at the UCSF Moffit-Long Hospital.
qPCR Analysis to Detect CART-19 and CART-Meso Transcripts
The persistence of CART-Meso and CART-19 cells in the peripheral blood was evaluated using total genomic DNA isolated from whole blood and measured using qPCR to detect a fragment unique to each CAR sequence as previously described.5,24 CART cell levels are reported as transgene copies per microgram of genomic DNA.
Flow Cytometry Detection of CART-19 Cells and Endogenous B Cells
Commercially available flow cytometry antibodies against CD3 and CD19 (BioLegend) were used in this study. CART-19 cells were detected using an Alexa Fluor 647-conjugated anti-idiotypic antibody, as previously described.55 Fluorescence-minus-one controls were used for the anti-idiotypic antibody to establish positive and negative gating strategies. Samples were acquired on an LSR Fortessa flow cytometer (BD Biosciences) and data were analyzed using FlowJo version 10 software. The frequency of CART-19+ CD3+ T cells among total CD3+ cells was reported. In addition, the frequency of CD19+ B cells in the blood was determined.
HAMAs and Serum Cytokine Analysis
HAMAs were assessed using an ELISA as previously described.3 Whole blood was collected in red top tubes without additive and processed to collect serum. Serum samples were analyzed as previously described4 to detect expression of panel of 30 cytokines including IL-1RA, fibroblast growth factor (FGF)-Basic, monocyte chemoattractant protein (MCP)-1, granulocyte colony-stimulating factor (G-CSF), IFN-γ, IL-12, IL-13, IL-7, granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor (TNF)-α, IL-1β, IL-2, IL-4, IL-5, IL-6, IFN-α, IL-15, IL-10, macrophage inflammatory protein (MIP)-1α, IL-17, IL-8, epidermal growth factor (EGF), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), monokine induced by gamma interferon (MIG), regulated on activation, normal T cell expressed and secreted (RANTES), Eotaxin, MIP-1β, interferon gamma-induced protein (IP)-10, and IL-2R.
Tumor Biopsy and Mesothelin Expression
Archival formalin fixed paraffin embedded cell blocks from FNAs at initial diagnosis were available for patients 19214-01 and 19214-03. Mesothelin expression was analyzed and scored as previously described.5 For patient 19214-01, a left lung FNAs showed inadequate sample present for analysis. For patient 19214-03, FNAs from pancreatic body mass and liver mass showed 2+ membrane/cytoplasmic reactivity for mesothelin in 70% of rare atypical (tumor) cells present in both specimens. On-treatment research biopsies were obtained at day 14 on patients 19214-01 (lung mass) and 19214-03 (liver mass) and evaluated by qPCR to detect for presence of CART-Meso and CART-19.
IHC
Automated IHC was performed on formalin-fixed paraffin embedded (FFPE) sections using a Ventana Discovery Ultra automated slide staining system (Roche). Primary antibodies against human antigens included rabbit anti-CD3 (Roche, clone 2GV6, cat# 790-4341), rabbit anti-CD19 (Cell Signaling, D4V4B, cat#90176S), and mouse anti-CK19 (Roche, clone A53-B/A2.26, cat#760-4281). Tissues were baked for 32 min at 60°C followed by deparaffinization. Slides were subjected to heat-induced epitope retrieval using Cell Conditioning Solution (CC1, Roche) for 64 min at 100°C. Slides were then incubated with discovery inhibitor (Roche, cat# 760-4840) for 8 min followed by S block (Roche, cat# 760-4212) for 8 min. Sequential IHC staining consisted of iterative cycles of staining with primary antibody, detection with a linking antibody and chromogen substrate, and denaturation of the applied antibodies. Anti-CD3 was incubated for 16 min at 37°C and detected using anti-rabbit NP (Roche) for 12 min at 37°C, anti-NP AP (Roche) for 12 min, and Discovery Yellow (Roche). Anti-CD19 was incubated for 32 min at 37°C and detected using anti-rabbit OMAP-HRP (Roche) for 16 min and Discovery Purple (Roche) for 60 min. Anti-CK19 was incubated for 16 min at 37°C and detected using anti-mouse HQ (Roche) for 12 min at 37°C, anti-HQ HRP (Roche) for 12 min and Discovery Teal (Roche) for 16 min. Antibody denaturation was performed at 91°C for 8 min in CC2 (Roche). Tissues were counterstained with hematoxylin and blue reagent for 4 min each. Slides were then prepared for mounting by washing in water and air drying at room temperature. Whole slide scanned images were acquired using an Aperio CS2 scanner system (Leica). Scanned images were analyzed using custom algorithms created using Visiopharm Integrator System (VIS) software (Version 2019.07). Tumor regions of interest were drawn on each image and a custom algorithm was developed to classify and detect cells based on colorimetric differences in yellow (CD3), purple (CD19), teal (CK19), and blue stains (nuclei). For correlational analyses, 1,042 μm by 1,042 μm square grids were superimposed upon scanned images. The number of CD3+ and CD19+ was then quantified and normalized to the area of each grid. Grids including CD19+ cells were included for analysis to assess relationship with CD3+ cell infiltrates.
Sequencing and Analysis of the B Cell Receptor (BCR) Immunoglobulin Heavy Chain
PBMC samples from patients who received either mRNA CART-Meso4 or Lentiviral CART-Meso cells5 on prior phase I studies were analyzed. Samples collected from patients at baseline prior to treatment and 28 days after treatment were analyzed. Genomic DNA was extracted from PBMC samples for DNA sequencing of the immunoglobulin heavy chain (IgH) complementarity-determining region 3 (CDR3), which was performed by Adaptive Biotechnologies (Seattle, WA). Sequencing data were uploaded to the Adaptive Biotech ImmunoSEQ Analyzer platform (Seattle, WA, USA) and then analyzed via the R programming language56 to identify unique IgH CDR3 sequences. Unique sequence frequencies were considered equivalent to specific B cell clone frequencies. The number of unique sequences was counted and compared between baseline and day 28 after treatment. B cell clone expansion was defined as a ≥2-fold expansion in the number of clones relative to baseline.
Database Analyses
Gene-expression profiles of CD3E and CD19 in PAAD patients were downloaded from TCGA dataset. Samples annotated as tumor were included (n = 179).
Statistics
Statistical analyses were performed using Prism (GraphPad Software, Version 8.4.0). For the analysis of B cell clone expansion, significance was tested using Fisher’s exact test. Unpaired group comparison testing was performed using two-tailed Mann-Whitney test. For the analysis of RNA-seq data from TCGA, correlation between selected genes was assessed using Pearson’s correlation. For the analysis of cell types detected by IHC, correlation was assessed using Spearman’s rank-order correlation. p values less than 0.05 were considered statistically significant.
Author Contributions
A.H.K., C.H.J., and G.L.B. conceived and designed the research. S.F.L., B.L.L., and J.J.M. performed the laboratory correlative experiments. A.H.K. conducted the clinical trial and clinical studies. All authors analyzed data. A.C.J., E.T., and G.L.B. wrote the manuscript, and all authors reviewed and edited the manuscript to its final version. The academic authors were fully responsible for the design, conduct, and analysis of the trial.
Conflicts of Interest
G.L.B. reports prior or active roles as consultant/advisory board member for Seattle Genetics, Aduro Biotech, AstraZeneca, Bristol-Myers Squibb, Cour Pharmaceuticals, Boehringer Ingelheim, Genmab, Merck, and BiolineRx and reports receiving commercial research grants from Incyte, Bristol-Myers Squibb, Verastem, Halozyme, Biothera, Newlink, Novartis, Arcus Biosciences, and Janssen. A.H.K. reports receiving commercial research grants from Celgene, Merrimack, Bristol-Myers Squibb, Roche/Genentech, Astra-Zeneca, Astellas, Aduro Biotech, Apexigen, Abgenomics, Halozyme, and Seattle Genetics (all paid directly to his institution) and prior or active roles as consultant/advisory board member for Erytech, Imugene, InCyte, ARMO Biosciences, Gilead, Genentech, Celgene, Seattle Genetics, and New Beta Innovation. L.F. reports receiving commercial grants from Abbvie, Bavarian Nordic, BMS, Dendreon, Janssen, Merck, and Roche/Genentech (all paid directly to his institution). C.H.J. reports receiving a commercial research grant from Tmunity Therapeutics. B.L.L., C.H.J., G.L.B., G.P., and J.J.M., and S.F.L. are inventors of intellectual property related to CART cells that is licensed by the University of Pennsylvania to Novartis and Tmunity Therapeutics. B.L.L., C.H.J., G.L.B., G.P., J.J.M., and S.F.L. are inventors of intellectual property related to CART cells that is licensed by the University of Pennsylvania to Novartis.
Acknowledgments
The authors thank the nurses of the UCSF Pancreas Center and Cancer Immunotherapy Program, Dr. Rebecca Kim for assistance in analysis of IgH sequencing, and Dr. Donald Siegel, Dr. Megan Davis, Dr. Andrew Fesnak, and the technical staff at the Clinical Cell and Vaccine Production Facility and the Translational and Correlative Studies Laboratory for outstanding trial support. This work was sponsored by the University of Pennsylvania(Philadelphia, PA) with funding provided by grants from National Institutes of Health grants R01 CA197916 (G.L.B.), Stand Up To Cancer, United States (C.H.J.), Lustgarten Foundation(C.H.J.), and from Novartis, United States. Research was also supported in part by the 2017 Pancreatic Cancer Action Network Precision Medicine Targeted Grant, grant number 17-85-BEAT (G.L.B.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.07.017.
Supplemental Information
References
- 1.Balachandran V.P., Beatty G.L., Dougan S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology. 2019;156:2056–2072. doi: 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beatty G.L., Eghbali S., Kim R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am. Soc. Clin. Oncol. Educ. Book. 2017;37:267–278. doi: 10.1200/EDBK_175232. [DOI] [PubMed] [Google Scholar]
- 3.Beatty G.L., Haas A.R., Maus M.V., Torigian D.A., Soulen M.C., Plesa G., Chew A., Zhao Y., Levine B.L., Albelda S.M. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beatty G.L., O’Hara M.H., Lacey S.F., Torigian D.A., Nazimuddin F., Chen F., Kulikovskaya I.M., Soulen M.C., McGarvey M., Nelson A.M. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology. 2018;155:29–32. doi: 10.1053/j.gastro.2018.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haas A.R., Tanyi J.L., O’Hara M.H., Gladney W.L., Lacey S.F., Torigian D.A., Soulen M.C., Tian L., McGarvey M., Nelson A.M. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019;27:1919–1929. doi: 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.June C.H., Sadelain M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beatty G.L., O’Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol. Ther. 2016;166:30–39. doi: 10.1016/j.pharmthera.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thistlethwaite F.C., Gilham D.E., Guest R.D., Rothwell D.G., Pillai M., Burt D.J., Byatte A.J., Kirillova N., Valle J.W., Sharma S.K. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maus M.V., Haas A.R., Beatty G.L., Albelda S.M., Levine B.L., Liu X. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fremd C., Schuetz F., Sohn C., Beckhove P., Domschke C. B cell-regulated immune responses in tumor models and cancer patients. OncoImmunology. 2013;2:e25443. doi: 10.4161/onci.25443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helmink B.A., Reddy S.M., Gao J., Zhang S., Basar R., Thakur R., Yizhak K., Sade-Feldman M., Blando J., Han G. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabrita R., Lauss M., Sanna A., Donia M., Skaarup Larsen M., Mitra S., Johansson I., Phung B., Harbst K., Vallon-Christersson J. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 15.Petitprez F., de Reyniès A., Keung E.Z., Chen T.W., Sun C.M., Calderaro J., Jeng Y.M., Hsiao L.P., Lacroix L., Bougoüin A. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 16.Andreu P., Johansson M., Affara N.I., Pucci F., Tan T., Junankar S., Korets L., Lam J., Tawfik D., DeNardo D.G. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17:121–134. doi: 10.1016/j.ccr.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schioppa T., Moore R., Thompson R.G., Rosser E.C., Kulbe H., Nedospasov S., Mauri C., Coussens L.M., Balkwill F.R. B regulatory cells and the tumor-promoting actions of TNF-α during squamous carcinogenesis. Proc. Natl. Acad. Sci. USA. 2011;108:10662–10667. doi: 10.1073/pnas.1100994108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Visser K.E., Korets L.V., Coussens L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Xiao X., Lao X.M., Chen M.M., Liu R.X., Wei Y., Ouyang F.Z., Chen D.P., Zhao X.Y., Zhao Q., Li X.F. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016;6:546–559. doi: 10.1158/2159-8290.CD-15-1408. [DOI] [PubMed] [Google Scholar]
- 20.Ren Z., Peng H., Fu Y.X. PD-1 Shapes B Cells as Evildoers in the Tumor Microenvironment. Cancer Discov. 2016;6:477–478. doi: 10.1158/2159-8290.CD-16-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Affara N.I., Ruffell B., Medler T.R., Gunderson A.J., Johansson M., Bornstein S., Bergsland E., Steinhoff M., Li Y., Gong Q. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014;25:809–821. doi: 10.1016/j.ccr.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunderson A.J., Kaneda M.M., Tsujikawa T., Nguyen A.V., Affara N.I., Ruffell B., Gorjestani S., Liudahl S.M., Truitt M., Olson P. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270–285. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pylayeva-Gupta Y., Das S., Handler J.S., Hajdu C.H., Coffre M., Koralov S.B., Bar-Sagi D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016;6:247–255. doi: 10.1158/2159-8290.CD-15-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sotillo E., Barrett D.M., Black K.L., Bagashev A., Oldridge D., Wu G., Sussman R., Lanauze C., Ruella M., Gazzara M.R. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orlando E.J., Han X., Tribouley C., Wood P.A., Leary R.J., Riester M., Levine J.E., Qayed M., Grupp S.A., Boyer M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018;24:1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 27.Yan Z., Cao J., Cheng H., Qiao J., Zhang H., Wang Y., Shi M., Lan J., Fei X., Jin L. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol. 2019;6:e521–e529. doi: 10.1016/S2352-3026(19)30115-2. [DOI] [PubMed] [Google Scholar]
- 28.Pan J., Zuo S., Deng B., Xu X., Li C., Zheng Q., Ling Z., Song W., Xu J., Duan J. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood. 2020;135:387–391. doi: 10.1182/blood.2019003293. [DOI] [PubMed] [Google Scholar]
- 29.O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalos M., June C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhoj V.G., Arhontoulis D., Wertheim G., Capobianchi J., Callahan C.A., Ellebrecht C.T., Obstfeld A.E., Lacey S.F., Melenhorst J.J., Nazimuddin F. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. 2016;128:360–370. doi: 10.1182/blood-2016-01-694356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milone M.C., Fish J.D., Carpenito C., Carroll R.G., Binder G.K., Teachey D., Samanta M., Lakhal M., Gloss B., Danet-Desnoyers G. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carpenito C., Milone M.C., Hassan R., Simonet J.C., Lakhal M., Suhoski M.M., Varela-Rohena A., Haines K.M., Heitjan D.F., Albelda S.M. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O’Connor R.S., Hwang W.T. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hay K.A., Gauthier J., Hirayama A.V., Voutsinas J.M., Wu Q., Li D., Gooley T.A., Cherian S., Chen X., Pender B.S. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–1663. doi: 10.1182/blood-2018-11-883710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berger C., Jensen M.C., Lansdorp P.M., Gough M., Elliott C., Riddell S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J., Sai H., Li Y., Jordan A.C., McGettigan S.E., Chen J.H., Bedoya F., Fraietta J.A., Gladney W.L., Melenhorst J.J., Beatty G.L. Peripheral Blood T-Cell Fitness Is Diminished in Patients With Pancreatic Carcinoma but Can Be Improved With Homeostatic Cytokines. Cell. Mol. Gastroenterol. Hepatol. 2019;8:656–658.e6. doi: 10.1016/j.jcmgh.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radvanyi L.G., Bernatchez C., Zhang M., Fox P.S., Miller P., Chacon J., Wu R., Lizee G., Mahoney S., Alvarado G. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012;18:6758–6770. doi: 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon E.K., Carpenito C., Sun J., Wang L.C., Kapoor V., Predina J., Powell D.J., Jr., Riley J.L., June C.H., Albelda S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Craddock J.A., Lu A., Bear A., Pule M., Brenner M.K., Rooney C.M., Foster A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moon E.K., Wang L.S., Bekdache K., Lynn R.C., Lo A., Thorne S.H., Albelda S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology. 2018;7:e1395997. doi: 10.1080/2162402X.2017.1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caruana I., Savoldo B., Hoyos V., Weber G., Liu H., Kim E.S., Ittmann M.M., Marchetti D., Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moon E.K., Wang L.C., Dolfi D.V., Wilson C.B., Ranganathan R., Sun J., Kapoor V., Scholler J., Puré E., Milone M.C. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wattenberg M.M., Beatty G.L. Overcoming immunotherapeutic resistance by targeting the cancer inflammation cycle. Semin. Cancer Biol. 2020 doi: 10.1016/j.semcancer.2020.01.002. Published online January 15, 2020. 10.1016/j.semcancer.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burga R.A., Thorn M., Point G.R., Guha P., Nguyen C.T., Licata L.A., DeMatteo R.P., Ayala A., Joseph Espat N., Junghans R.P., Katz S.C. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015;64:817–829. doi: 10.1007/s00262-015-1692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maude S.L., Hucks G.E., Seif A.E., Talekar M.K., Teachey D.T., Baniewicz D. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL) J. Clin. Oncol. 2017;35:103. [Google Scholar]
- 51.Li S., Siriwon N., Zhang X., Yang S., Jin T., He F., Kim Y.J., Mac J., Lu Z., Wang S. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017;23:6982–6992. doi: 10.1158/1078-0432.CCR-17-0867. [DOI] [PubMed] [Google Scholar]
- 52.Liu C., Lewis C.M., Lou Y., Xu C., Peng W., Yang Y., Gelbard A.H., Lizée G., Zhou D., Overwijk W.W., Hwu P. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J. Immunother. 2012;35:276–282. doi: 10.1097/CJI.0b013e31824e7f43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee D.W., Gardner R., Porter D.L., Louis C.U., Ahmed N., Jensen M., Grupp S.A., Mackall C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jena B., Maiti S., Huls H., Singh H., Lee D.A., Champlin R.E., Cooper L.J. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS ONE. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.R Core Team . 2018. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna.https://www.R-project.org [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.