

Abstract
Background and methods:
Atrial fibrillation (AF) is the most common cardiac arrhythmia in clinical practice. The substrate of AF is composed of a complex interplay between structural and functional changes of the atrial myocardium often preceding the occurrence of persistent AF. However, there are only few animal models reproducing the slow progression of the AF substrate to the spontaneous occurrence of the arrhythmia. Transgenic mice (TG) with cardiomyocyte-directed expression of CREM-IbΔC-X, an isoform of transcription factor CREM, develop atrial dilatation and spontaneous-onset AF. Here we tested the hypothesis that TG mice develop an arrhythmogenic substrate preceding AF using physiological and biochemical techniques.
Results:
Overexpression of CREM-IbΔC-X in young TG mice (b8 weeks) led to atrial dilatation combined with distension of myocardium, elongated myocytes, little fibrosis, down-regulation of connexin 40, loss of excitability with a number of depolarized myocytes, atrial ectopies and inducibility of AF. These abnormalities continuously progressed with age resulting in interatrial conduction block, increased atrial conduction heterogeneity, leaky sarcoplasmic reticulum calcium stores and the spontaneous occurrence of paroxysmal and later persistent AF. This distinct atrial remodelling was associated with a pattern of non-regulated and up-regulated marker genes of myocardial hypertrophy and fibrosis.
Conclusions:
Expression of CREM-IbΔC-X in TG hearts evokes abnormal growth and development of the atria preceding conduction abnormalities and altered calcium homeostasis and the development of spontaneous and persistent AF. We conclude that transcription factor CREM is an important regulator of atrial growth implicated in the development of an arrhythmogenic substrate in TG mice.
Keywords: Atrial fibrillation, Transgenic mice, Substrate remodeling, cAMP-dependent gene regulation
1. Introduction
The structurally related transcription factors cAMP-response element protein (CREB) and cAMP-response element modulator (CREM) bind as dimers to the cAMP-response element (CRE) in various promoters and regulate transcriptional activation in response to cAMP and other signaling pathways [1]. CREB and CREM are expressed in the human heart [2, 3]; CREM is critical for β-adrenoceptor-mediated cardiac deterioration in mice [4]. Mice with cardiomyocyte-directed expression of CREM-IbΔC-X (TG), a CREM repressor isoform isolated from human myocardium [3], develop a complex cardiac phenotype dominated by spontaneous-onset of sustained atrial fibrillation (AF) associated with dilation of atria [5]. Atrial remodeling and AF dilation are not secondary to chronic hemodynamic overload as observed in other animal models of AF since left ventricular (LV) function is not impaired in TG mice [5]. Therefore, TG mice could constitute an interesting model to study the pathogenesis of the AF substrate associated with progressive atrial dilation.
Atrial fibrillation, the most common sustained cardiac arrhythmia in clinical practice, is often associated with progressive dilation and remodeling of the atria which constitute the substrate of the arrhythmia [6]. This atrial remodeling is characterized by complex structural and functional alterations of the atrial myocardium: short action potentials, heterogeneous refractory periods, dystrophic myocytes and interstitial fibrosis which act together to favor local conduction bloc, activation of ectopies and the formation of microreentries of the electrical excitation. However, only few experimental models reproduce these complex and chronic pathogenic processes, hence underlying molecular actors and signaling pathways are still poorly understood [7].
Here, we characterized the atrial remodeling associated with the occurrence of AF in CREM-IbΔC-X TG mice. The aim was to decipher the role of transcription factor CREM in structural and functional properties of the atria and to study the relevance of this TG model to the substrate of human AF. We found that atria of young mice displayed dilation of atria with little fibrosis and down-regulation of connexin 40 (Cx40). These structural alterations were the earliest changes associated with atrial ectopies and inducibility of AF. This suggests the development of an arrhythmogenic substrate in young TG atria which stands at the beginning of a chain of events, structural and electrical changes, continuously progressing to spontaneous-onset AF with age.
2. Methods
2.1. Animal model
Generation and breeding of TG mice were described previously [5]. Wild-type control mice (WT) and TG mice were from the same colony and were included in the experiments as sex-matched littermates where possible. The study was approved by the institutional review boards in Münster, Paris, Maastricht and Houston, and conformed to the local and US NIH guidelines for conduct of animal research. After an initial series of ECG monitoring experiments, we defined two time points for further characterization: One period in life (young mice, <8 weeks) when atrial fibrillation was not yet persistent, and another period (old mice, >12 weeks) when persistent AF started to develop.
2.2. Atrial electrical and hemodynamic function in vivo
6-lead electrocardiograms (EMKA technologies, Paris, France), cardiac 2D- and M-Mode echocardiography (VEVO 770 small animal ultrasound platform), and telemetric Holter ECGs (EMKA technologies) in freely roaming mice were used to assess atrial electrical and contractile function in vivo [8–11]. Telemetric ECG recordings were analyzed during 24 h of continuous recordings at predefined ages (6, 8, 11, and 14 weeks) for arrhythmias [8–11]. Some animals were followed by serial telemetric ECG recordings to determine arrhythmia patterns until the animals developed chronic atrial fibrillation.
2.3. Electrophysiological studies
2.3.1. Studies in isolated atria
For optical mapping of atrial activation, Langendorff-perfused (Tyrode’ s solution) atria were spread out by attaching 2 sutures per atrium to the side of the chamber to provide a view of left and right atrial free walls, Bachmann’s bundle and the pulmonary vein area. Hearts were stained with the voltage-sensitive dye di-4-ANEPPS and (−) blebbistatin (5 μM) was used as an excitation–contraction uncoupler. High-resolution optical mapping (SciMedia Ulitima-L CMOS camera, 100×100 pixels, 2 kHz) was used to record optical potentials during pacing from the left atrium with a unipolar platinum electrode (1.5× threshold; paced cycle lengths 150, 120, 90, and 60 ms). To reduce noise, six sweeps of optical potentials were averaged for each cycle length. Local activation was determined as the point of the fastest upstroke in the optical action potential. Conduction velocities were computed by custom software (OmaTool, Department of Physiology, University Maastricht) and activation maps were created. To quantify the degree of conduction heterogeneity, the maximal difference in activation time point between an electrode and any of its neighboring electrodes was divided by the interelectrode distance to yield the ‘phase difference’ for each electrode. Conduction heterogeneity was quantified as the difference between the 95th and 5th percentile (P95–P5) of the distribution of the phase differences.
We recorded standard glass microelectrode action potentials from isolated, superfused right or left atrial preparations. Rapidly excised atria were placed in an oxygenated (95% O2 and 5% CO2, 8 ml/min, 36.5 ±0.5 °C) pre-warmed Tyrode solution [12]. The atrial preparation was stimulated at 1 Hz using a unipolar electrode (5 ms impulse duration, twice diastolic pacing threshold). Transmembrane action potentials were recorded at different sites from the epicardial surface of the superfused left atrium by standard microelectrodes. At least 30 action potentials were obtained from each preparation. APs were digitized at 20 kHz using Acquis1 software (version 4.0).
2.3.2. Atrial electrophysiology in isolated beating Langendorff-perfused hearts
We recorded atrial monophasic action potentials and performed biatrial epicardial activation mapping in isolated Langendorff-perfused mouse hearts as described [9, 13]. We used the Langendorff-perfused preparation to perform high-density (spacing 300 μm) biatrial epicardial activation mapping using custom-designed 32 electrode grid multi-electrode arrays (Multichannelsystems, Stuttgart, Germany) placed simultaneously on the right and left atrial epicardium to record the right and left atrial epicardial activation. Unipolar electrograms were recorded using custom-made amplifiers (sampling rate 1 kHz, 0.1 Hz high-pass, 426 Hz low-pass filter, University of Maastricht Engineering Department, Maastricht, the Netherlands). Custom-made analysis software was used for offline analysis of activation times.
2.4. Quantitative real-time polymerase chain reaction
Total atrial RNA was isolated by using the ZR RNA MicroPrep™ Kit (Zymo Research, Orange, USA) and cDNAs were generated with the RevertAid First Strand cDNA Synthesis Kit (MBI Fermentas, St Leon-Rot, Germany). The quantitative real-time polymerase chain reaction (PCR) was performed with the LightCycler system following the manufacturer’s specifications with the use of LightCycler Fast Start DNA Master SYBR Green I Mix (Roche Diagnostics, Penzberg, Germany) and the following primers: Hprt, 5′-GGAGTCCTGTTGATGTTGCCAGTA-3′ (forward), 5′-GGGACG CAGCAACTGACATTTCTA-3′ (reverse); Ywhaz, 5′-AGCAGGCAGAGCGATATGATGACA-3′ (forward), 5′-TCCCTGCTCAGTGACAGACTTCAT-3′ (reverse); ANP, 5′-CCTGATGGATTTC AAGAATTTGCTGGA-3′ (forward), 5′-CTGCTTCCTCAGTCTGCTCACT-3′ (reverse); BNP, 5′-GTTACAGGAGCAGCGCAACCAT-3′ (forward), 5′-ATGCCGCCTCAGCACTTTGCA-3′ (reverse); α-skeletal-actin, 5′-GTGAGATTGTGCGCGACATC-3′ (forward), 5′-GGCAACGGA AACGCTCATT-3′ (reverse); cardiac-actin, 5′-CCAGCCCTCTTTCATTGGTA-3′ (forward), 5′-GCTGGAAGGTGGACAGAGAG-3′ (reverse); α-MHC, 5′-GATGCCCAGATGGCTGACTT-3′ (forward), 5′-GGTCAGCATGGCCATGTCCT-3′ (reverse); β-MHC, 5′-GCCAACACCAACCTG TCCAAGTTC-3′ (forward), 5′-TGCAAAGGCTCCAGGTCTGAGGGC-3′ (reverse); collagen type Ia, 5′-GTGTGATGGGATTCCCTGGACCTA-3′ (forward), 5′-CCGAGCTCCAGCTTCTC CATCTT-3′ (reverse); colIagen Typ IIIa, 5′-ACCCCCTGGTCCACAAGGATTA-3′ (forward), 5′-ACGTTCTCCAGGTGCACCAGAAT-3′ (reverse). Levels of mRNAs were determined using LightCycler software version 3.5 with appropriate calibration curves including different amounts of control cDNAs. Results are mean values of duplicate runs for each sample normalized to WT median value.
2.5. Protein expression
Western blot analysis was performed as described [4, 5]. To correct for potential differences in cardiomyocyte content in the homogenates, protein load was adjusted to the calsequestrin content in each sample. Connexin blots were performed using primary rabbit polyclonal antibodies against connexin 40 (1/500; Zymed Laboratories, South San Francisco, CA) or connexin 43 (1/500; Invitrogen, Carlsbad, CA) and 5% dried milk powder for blockade of non-specific binding.
2.6. Histology and immunohistochemistry
Indirect immunohistofluorescence was performed on 7 μm frozen sections. Primary antibodies were rabbit polyclonal antibodies against connexin 43 (1/50; Zymed) and connexin 40 (1/50; Zymed), and a mouse monoclonal antibody against sarcomeric α-actinin (1/400, Sigma, St-Louis, MO). Secondary antibodies were goat anti-rabbit IgG coupled to Alexa Fluor® 488 and goat anti-mouse IgG coupled to Alexa Fluor® 594 (1/100, Molecular Probes, Invitrogen, Eugene, OR). Images were collected with an Olympus IX50 inverted fluorescence microscope (Olympus, Center Valley, PA). In control experiments primary antibodies were omitted. Five-micrometre-thick LA sections were stained with Picrosirius Red F3BA, and fibrosis was quantified blindly on five sections per atrium (8–10 fields per section). The interstitial collagen volume fraction expressed as a percentage of the total atrial surface area was determined as described elsewhere [14]. To determine the length of atrial cardiomyocytes, atria were excised and digested using 0.9 mg/ml collagenase (Type II, 190 U/mg, Worthington), 0.57 mg/ml elastase (Type III, Sigma-Aldrich) and 0.35 U/ml protease (Type XIV, Sigma). After 10 min of digestion tissue parts were gently triturated to release single cells in Ca2 + free solution. For determination of cell length artial cells were transferred to a chamber and analysed by a Nikon Eclipse Ti microscope (Nikon Instruments Europe B.V., Germany).
2.7. Calcium sparks measurements
Atrial myocytes were isolated, Fluo 4-AM loaded, and transferred to a chamber equipped with a pair of parallel platinum electrodes and placed on a LSM510 confocal microscope (Carl Zeiss, Thornwood, NY) as described [15]. Fluorescence images were recorded in line-scan mode with 1024 pixels per line at 500 Hz. Once steady state Ca2 + transient induced by 1 Hz-pacing (5 ms, 10 V) was observed, pacing was stopped for 45 s and Ca2 + sparks were counted. Steady state SR Ca2 + content was estimated by rapid application of 10 mM caffeine after pacing. SR Ca2 + leak in atrial myocytes was measured as described in detail previously [16]. Isolated cardiomyocytes were additionally measured for maximal length and diameter at the level of the nucleus.
2.8. Statistics
Nominal parameters were compared between genotypes using Fisher’s exact test and chi square test. Continuous parameters are indicated as mean±SEM and were compared using student’s T-test for normally distributed data except real-time PCR data which were analyzed using the REST software tool (http://www.gene-quantification.de/rest.html)[17]. Arrhythmia-free survival was compared by log-rank test. Two-sided p-values smaller than 0.05 were considered significant.
2.9. General
The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology.
3. Results
3.1. Development of atrial fibrillation in CREM-TG mice
Freely roaming TG mice showed atrial ectopies which increased exponentially after 7 weeks of age to several hundred atrial ectopic beats per hour (Fig. 1A) and preceded the development of AF (Fig. 1B,C). TG mice of 11 weeks (n=7) spent 15% of the monitored time in atrial ectopy (Fig. 1C, p<0.05 vs. 0.4% in 5 WT). In line with previous data [5], 40–50% of TG mice displayed at least short episodes of AF at an age around 14 weeks; the majority of TG mice older than 18 weeks showed long episodes of AF lasting more than a minute and often throughout the entire recording (chronic AF). Littermate WT controls did not show any AF (p<0.05, Fig. 1B). Moreover, 6 out of 18 isolated beating hearts from young TG mice (7 weeks) displayed spontaneous AF, and programmed right atrial stimulation with a single premature stimulus (S2) induced non-sustained episodes of AF in 14/17 young TG hearts vs. 5/17 WT hearts (p<0.05; Fig. 1D).
Fig. 1.

Development of AF. Freely roaming CREM-TG mice (TG) showed a spontaneous sequence of atrial ectopy, paroxysmal atrial fibrillation, and sustained atrial fibrillation (further description in text). A, Representative telemetric ECG showing atrial ectopy (arrow in inset) and number of ectopic beats per hour in individual TG mice plotted per week of life. B, Representative telemetric ECG of a short episode of atrial fibrillation (dotted line in inset) and Kaplan–Meier plot of survival without atrial fibrillation in 7 TG and 6 WT mice. C, Representative example of a hypothesis-free differentiation of cardiac rhythm clusters in the ECG by a self-learning computer algorithm [11] in one TG mouse. Ordinate indicates the percentage of time spent in each cluster, abscissa indicates the mouse age. All clusters were verified manually. D, Spontaneous and inducible atrial fibrillation in atria from young TG mice (age 7 weeks; monophasic action potential recording). Shown are a representative example of an episode of spontaneous atrial fibrillation (upper panel) and the numbers of isolated hearts with spontaneous (left) and inducible (right, single extra stimulus) atrial tachyarrhythmias in young TG and WT hearts (lower panel). *P<0.05 vs. WT.
3.2. Atrial dilatation precedes development of atrial ectopies and of AF in TG mice
TG mice developed a progressive atrial dilation detected at three to five weeks of age (Fig. 2A,B), resulting in more than tripling of atrial diameter at 10 weeks of age (Fig. 2B). In parallel, atrial weight was increased in TG mice at 7 weeks of age (WT: 4.9±0.9 mg, n=6; TG:8.5±0.9 mg, n=7, pb0.05). At 5–7 weeks of age, atrial contractile function was already reduced in vivo (Fig. 2C) and in electrically stimulated right atrial preparations (WT 3.0±0.5 mN, n=7; TG 0.7 ±0.2 mN, n=5, p<0.05). In contrast, there were no signs of LV dysfunction in echocardiography (n=9, data not shown) consistent with previous data obtained by LV catheterization [5].
Fig. 2.

Increase in atrial size. Age-dependent increase in atrial size in CREM-TG (TG) mice. A, Representative echocardiographic images of left atrial diameter in systole and diastole in a wild-type (WT) and a TG mouse (age 5 weeks). B, Echocardiographic analysis of age-dependent changes in atrial size. Each dot represents the left atrial size of an individual TG (●) or WT mouse (○) plotted against mouse age. Atrial enlargement preceded the development of sustained atrial arrhythmias. C, Statistical analysis of systolic and diastolic left atrial diameters and left atrial fractional shortening in 5 weeks old TG mice (filled bars, n=9) compared to WT mice (open bars, n=9). All parameters are expressed as percent of WT mean. *P<0.05 vs. WT.
3.3. Morphological substrate of AF in TG mice
In order to characterize the nature of the arrhythmogenic substrate of TG mice, we performed histological studies using Picro–Sirius stainings of atrial tissue slices. Both young and old TG mice showed dilated atria with an extremely thin atrial wall containing just a few trabeculae in contrast to the thick atrial wall of WT atria (Fig. 3A). At higher magnification, myocytes appeared elongated with some areas of interstitial fibrosis (9.5±2.2% vs 15.1±3.2% in WT and TG mice, respectively; n=6). Immunostaining of cryosections with an antibody directed against the sarcomeric α-actinin showed that atrial myocytes remained well striated without sign of dystrophic or myolytic sarcomeric apparatus in both TG and WT mice at an age below 10 weeks (3 mice, 75 sections). Isolated atrial myocytes from young and old TG mice were elongated as compared to WT controls (Fig. 3B,C,D). Old TG atrial cardiomyocytes were wider than WT atrial cardiomyocytes but the ratio of length to width was increased in TG mice compared to WT mice (Fig. 3D). The expression of gap junction channel protein Cx40 was reduced as illustrated by the faint staining detected in freeze sections of both left and right atria of young and old TG mice (Fig. 4A) and a 40% reduction of Cx40 protein levels in young TG vs. WT atria (Fig. 4B). Connexin 43 (Cx43) was delocalized from the intercalated disk to the lateral side of the plasma membrane while Cx43 protein levels were not different between groups (Fig. 4A,B). The increase in cardiomyocyte size was not accompanied by an up-regulation of the mRNAs encoding for the atrial natriuretic peptide (ANP), the brain-type natriuretic peptide (BNP), or the collagens type Ia or IIIa, all classical markers of myocardial hypertrophy and remodeling, in 10 old TG vs. 11 old WT atria (13–17 weeks of age; Table 1). All values were standardized to the mRNAs encoding housekeeping genes Hprt (hypoxanthine-guanine phosphoribosyltransferase) and Ywhaz (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide) which both were not different between genotypes. In contrast, mRNAs encoding other reported marker genes of hypertrophy, alpha skeletal actin and beta-myosin heavy chain, were up-regulated while alpha-myosin heavy chain mRNA was down-regulated in TG vs. WT atria (Table 1).
Fig. 3.
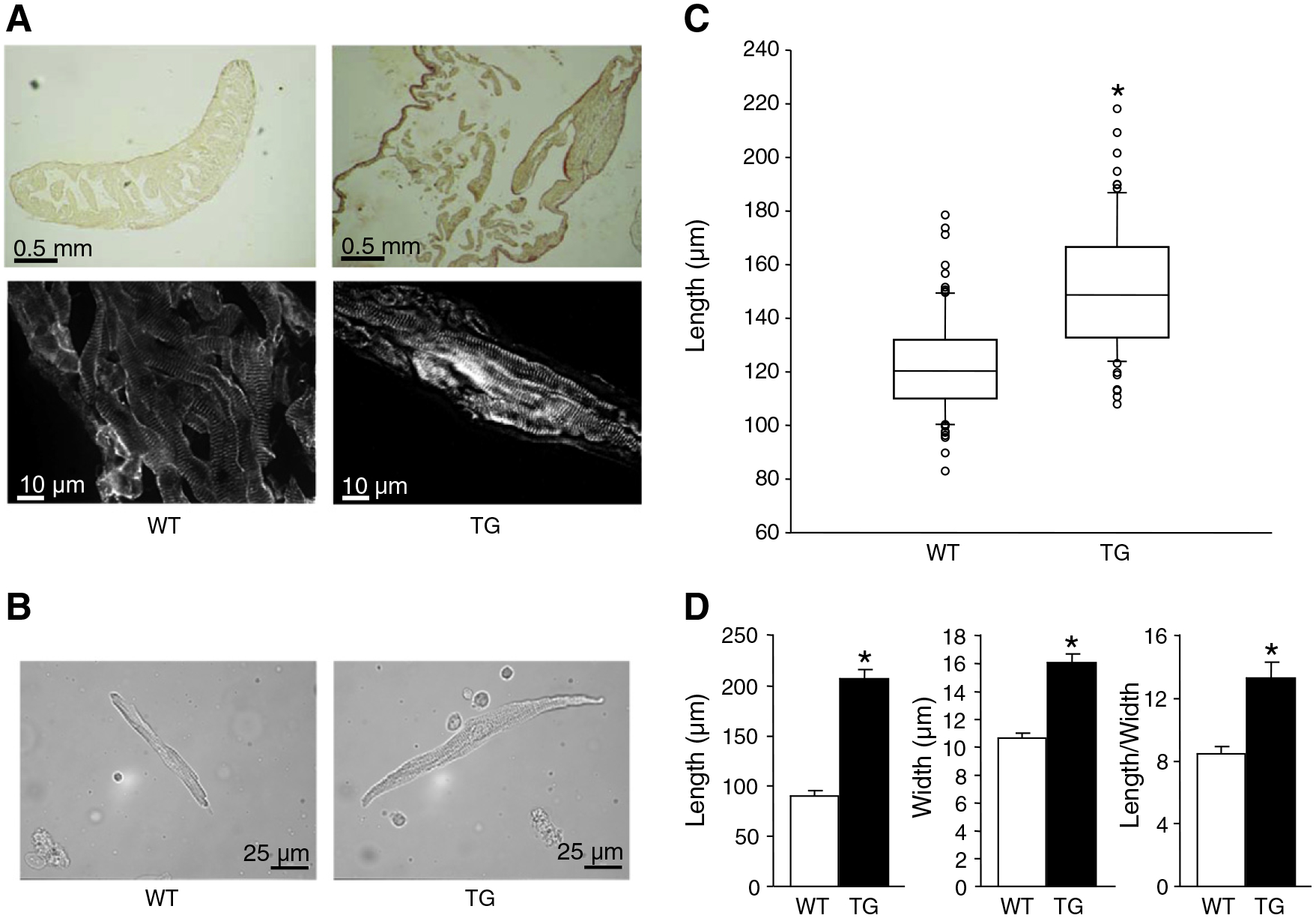
Abnormal growth of cardiomyocytes. Elongation of atrial myocytes in absence of atrial fibrosis in CREM-TG (TG) atria. A, Representative photographs of histological sections of wild-type (WT, 6 weeks) and TG atria (6 weeks) stained with Picro-Sirius Red (upper photos) or with an antibody directed against the sarcomeric α-actinin (lower photos), magnifications as indicated. B, Representative photomicrographs of isolated atrial myocytes from a WT and a TG mouse (7 weeks) used for measurements of cell size. C, Atrial myocyte length of young (7 weeks; WT: 91 cells/4 atria; TG: 77 cells/4 atria) mice. D, Mean length, width and ratio of length to width of 9 and 17 myocytes, respectively, from TG and WT atria at 18 weeks. *P<0.05 vs. WT.
Fig. 4.

Reduced expression of connexin 40. A, Immunohistochemical detection of α-actinin (upper panel), connexin 40 (Cx40) and connexin 43 (Cx43) in atrial slices from CREM-TG (TG) and WT mice, representative photographs of 3 hearts studied per group. B, Immunological detection of Cx40, Cx43 and calsequestrin in TG and WT atria, representative immunoblots. Statistical analysis of immunoblots revealed a reduced expression of Cx40 in TG atria as compared to WT controls (n=10; age 7 weeks) while protein levels of Cx43 were not changed (n=5). Values were normalized to calsequestrin and expressed in percent of WT. *P<0.05 vs. WT.
Table 1.
Relative expression of mRNAs encoding different markers of myocardial hypertrophy in atrial homogenates from 10 TG and 11 WT mice (age 13–17 weeks). The mRNAs encoding the housekeeping genes Hprt and Ywhaz were used for standardization. Levels of mRNAs in TG atria were expressed relative to levels in WT atria and were statistically analysed using the REST software with 2000 iterations.
| Gene | Expression relative to WT | 95% Confidence interval | Regulation |
|---|---|---|---|
| Hprt—reference | 0.963 | ||
| Ywhaz—reference | 1.039 | ||
| ANP | 1.031 | 0.536–1.752 | n.s. |
| BNP | 1.396 | 0.367–5.070 | n.s. |
| Cardiac-actin | 1.143 | 0.457–2.734 | n.s. |
| Skeletal-actin | 31.814 | 5.654–245.491 | Up-regulated |
| α-MHC | 0.769 | 0.460–1.629 | Down-regulated |
| β-MHC | 1.990 | 0.667–9.062 | Up-regulated |
| Collagen type Ia | 1.382 | 0.236–5.711 | n.s. |
| Collagen type IIIa | 1.208 | 0.090–4.620 | n.s. |
n.s. denotes not significant.
3.4. Alterations of the electrical properties of the atria of TG mice
We compared the conduction characteristics of right and left atria between young (>8 weeks) and old (>12 weeks) mice. Activation of the left atrium was delayed during right atrial pacing in young TG hearts (Fig. 5A), indicating interatrial conduction block and increased atrial size. Interatrial conduction block was further confirmed by epicardial activation mapping revealing an increase of interatrial conduction times in old TG hearts (Fig. 5B). Conduction heterogeneities were determined by high-density optical mapping in left and right atria from young and old mice (Fig. 5C). Atrial conduction heterogeneities were not significantly different in young mice; however, there was a trend to increased heterogeneity in TG right atria. In old mice, marked heterogeneity of conduction was observed in the two atria indicating aggravation of electrical alterations with time (Fig. 5C). In the vast majority of myocardial fibers of young WT mice using the glass microlectrode technique, it was possible to record well polarized action potentials, with a fast upstroke and a prominent plateau phase (Fig. 6A; Table 2). In contrast, only 25% of myocytes of TG mice had a normal resting membrane potential (<−70 mV) and generated a fast action potential (Fig. 6A,B). The majority of them were depolarized, and 25% were non-excitable or generated a slow-response AP (Fig. 6A,B; Table 2). There was a marked scattering of the distribution of resting membrane potentials (Fig. 6B) and action potential durations (table) between control and TG atria indicating electrical heterogeneity in TG atria. Moreover, frequent, repetitive, fast runs of atrial tachycardia were recorded in all isolated atrial trabeculae from old TG mice (n=6), in only one case of young TG mice and never in WT mice (Fig. 6C).
Fig. 5.

Impaired electrical conduction. A, Marked prolongation of activation times between right atrial pacing site and left atrial monophasic action potential recording in young (b8 weeks old) CREM-TG (TG) mice (filled bars, n=8) compared to wild-type (WT, open bars, n=7). The inset shows a scheme of the pacing and recording location. B, Representative recording of epicardial electrograms from two simultaneously recorded epicardial high-density 32-pole mapping electrodes (right and left atria) used for detailed assessment of conduction times in beating TG and WT hearts. Red lines indicate earliest and latest activation. There was marked interatrial conduction delay between sites A/B and C/D with an increased mean interatrial conduction time and a non-significant trend towards increased mean right (RA) and left atrial (LA) conduction times measured during sinus rhythm by multi-electrode epicardial mapping in old (>15 weeks, n=7) TG mice (filled bars) in comparison to 6 old WT controls (open bars); there was a non-significant trend towards an increased interatrial conduction time in young (< 8 weeks, n=15) TG atria as compared to 11 young WT controls. C, Representative atrial activation patterns recorded by optical mapping in young (<8 weeks) and old (>15 weeks) TG and WT atria. Activation patterns were homogeneous with clearly separated isochrones in young atria. In old TG atria, isochrones were more crowded with local conduction heterogeneity and mean local conduction heterogeneities in old TG (n=6) and WT (n=4) mouse atria. Phase differences between adjacent recording areas were analyzed. In young TG atria (n=5), left atrial conduction was normal, while right atrial conduction showed a trend towards increased heterogeneity compared to young WT (n=4). In atria from older mice, heterogeneity of conduction was markedly increased in both atria.
Fig. 6.

Prolongation of atrial action potentials and refractoriness, cellular depolarization and spontaneous arrhythmias. A, Typical recordings of action potentials by glass micro-electrodes in superfused right and left atrial preparations in wild-type (WT) and TG atria. B, Distribution of resting membrane potentials in TG (filled bars; 234 cells from 11 mice) and WT (open bars; 196 cells from 8 mice) atria. C, Fast spontaneous atrial tachycardia (left recording) in a right atrial TG trabeculum, representative recording.
Table 2.
Heterogeneity of diastolic potentials and prolongation of repolarization in atrial treabeculae from CREM-TG hearts. APD25, APD50 and APD90 (action potential duration at 20, 50 and 90 percent repolarization).
| WT (187 cells, 8 atria) | Young CREM-TG (<8 weeks old, 182 cells, 7 atria) | Old CREM-TG (>8 weeks old, 34 cells, 4 atria) | |
|---|---|---|---|
| Resting membrane potential (mV) | − 68.1 ± 0.3 | − 54.3 ± 1.8* | − 50.1 ± 2.4* |
| Partially depolarized cells | 2/187 | 56/182* | 14/34* |
| Overshoot, mV | 7.0 ± 0.5 | 2.8 ± 0.5* | 1.7 ± 0.8* |
| APD25, ms | 3.2 ± 0.1 | 6.1 ± 0.4* | 7.2 ± 0.6* |
| APD50, ms | 7.6 ± 0.3 | 11.0 ± 0.6* | 13.1 ± 0.8* |
| APD90, ms | 47.5 ± 1.4 | 57.3 ± 4.5* | 59.1 ± 7.1* |
| Maximal rate of depolarization (V/s) | 124.0 ± 3.0 (186 cells) | 84.2 ± 5.1* (154 cells) | 80.7 ± 11.7* (62 cells) |
P<0.05 vs. WT.
As emptying the sarcoplasmic reticulum with the SERCA inhibitor thapsigargin (1 μM) completely suppressed atrial arrhythmias in 4/5 old TG hearts (25–33 weeks, Fig. 7A), we studied calcium homeostasis in isolated atrial myocytes using confocal microscopic imaging. We observed more spontaneous calcium release events (Fig. 7B) and an increased calcium leak from the SR associated with a decreased SR calcium load in TG atrial cardiomyocytes as compared to WT controls (Fig. 7C). This was likely not due to an altered expression of major calcium-regulatory SR proteins since protein levels of the Ca2+-ATPase of the sarcoplasmic reticulum (SERCA; n=6), phospholamban (PLB; n=3–4), troponin I (TnI; n=4) and the cardiac ryanodin receptor (RyR2; n=5–6) were not different in atrial homogenates from TG and WT atria aged 7 weeks (data not shown). Taken together, cardiomyocytes from dilated TG atria show severe electrical heterogeneity and display alterations of intracellular calcium homeostasis.
Fig. 7.
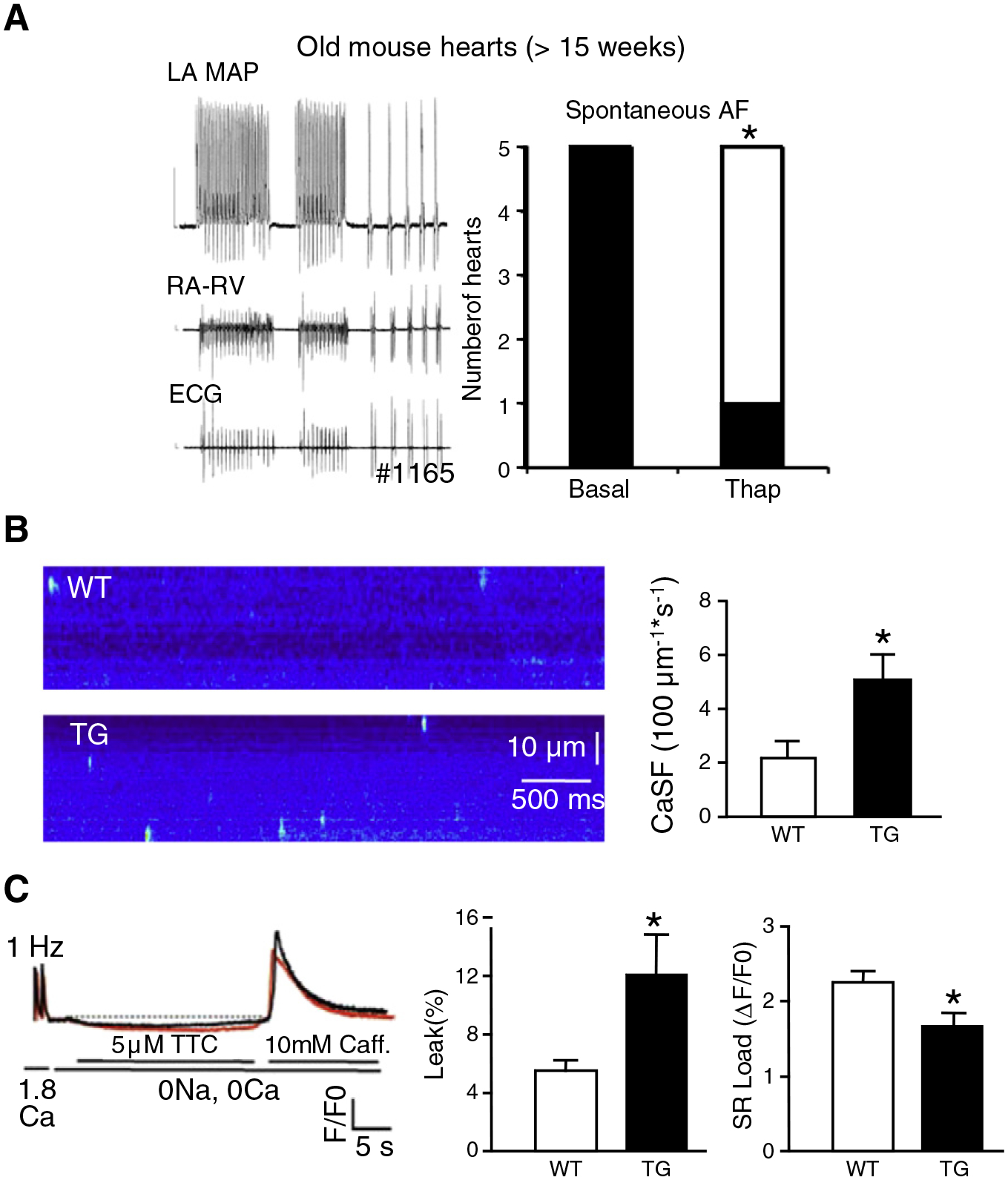
Calcium-triggered arrhythmogenesis. A, Thapsigargin (1 μM in perfusate) suppressed spontaneous atrial fibrillation in old CREM-TG (TG) mouse hearts (>15 weeks). Shown is a representative example of two short episodes of spontaneous atrial fibrillation (left) and the number of hearts with arrhythmias at baseline and with thapsigargin (right). *P<0.05 vs. WT. B, Confocal microscopic imaging of isolated atrial cardiomyocytes revealed more spontaneous calcium release events (“calcium sparks”) in 7 and 9 atrial cardiomyocytes, respectively, from TG atria as compared to wild-type (WT) atria (age >15 weeks). *P<0.05 vs. WT. C, Calcium transient recordings revealed a reduced SR calcium load and an increased diastolic calcium leak in TG vs. WT atrial cardiomyocytes (n=9, 15). *P<0.05 vs. WT.
4. Discussion
This study indicates the crucial role played by the transcriptional regulation mediated by the cAMP-response element (CRE) and transcription factors of the CREB/CREM family in the normal growth and structure of the atrial myocardium. One main feature of TG mice is an age-dependent continuous progression of profound structural alterations of the atrial myocardium which precedes spontaneous-onset transient and—later—sustained atrial arrhythmia. This is comparable to a clinically silent period of progressive atrial remodeling and susceptibility to arrhythmia foregoing the occurrence of human AF. As in human, in TG atria dilatation and sustained AF are associated with electrical heterogeneity, impaired conduction of atrial electrical activity and altered calcium homeostasis which are well-known factors promoting reentry mechanisms and triggered activities.
The atrial remodeling very likely represents a key event at the beginning of the chain of events leading to spontaneously occurring sustained AF at about 18 weeks of age in TG mice: (i) it is detectable as early as at three to four weeks of age and is the earliest alteration observed in this model so far; (ii) it is not secondary to ventricular dysfunction since LV function is increased in this model [5]; (iii) it is associated with a high incidence of atrial ectopies and the triggering of transient episodes of AF by programmed stimulation in young TG mice, suggesting that an arrhythmogenic substrate is present before the occurrence of chronic AF; (iv) the aggravation of atrial dilatation goes along with other structural alterations similar to the atrial remodeling and the development of a substrate of AF pathogenesis in human. These alterations are paralleled by the progression from frequent atrial ectopies to sustained AF and therefore suggest a continuum in the progression of the disease in TG mice from atrial dilation with sinus rhythm and ectopies to chronic or persistent AF.
The atrial remodeling in TG mice is characterized by a huge distension of atria combined with thin atrial walls containing just a few layers of disorganized and elongated myocytes and with little interstitial fibrosis. This is reminiscent of thin atrial walls with reduction in the number of myocytes which are disorganized as observed in volume-overloaded and dilated atria [18]. As in TG mice, the atrial myocardial remodelling in patients or experimental models is associated with depolarized and poorly excitable myocytes, heterogeneous AP morphology and abnormal conduction of the depolarization wave[18]. However, TG atrial myocytes are rather elongated than hypertrophic or dystrophic as indicated by a marked increase in length/width ratio, and a normal microscopical appearance of the sarcomeric apparatus. These histological characteristics together with the extremely thin atrial wall, suggest an abnormal myocardial growth in TG mice resulting in marked elongation of the cardiomyocytes and subsequently of the atrial myocardium. The pattern of up-regulated and non-regulated hypertrophy marker genes further suggests that this atrial remodeling is distinct from the classical hypertrophic and fibrotic remodelling process of the myocardium. Taken together these data indicate a default of growth and development of the atria of TG mice.
The structural alterations further include a down-regulation of Cx40, another feature of human AF, which could contribute to the conduction abnormalities in TG atria [23]. In addition to the disorganized conduction and electrical heterogeneity, calcium-dependent mechanisms (“triggered activity”) probably contribute to the occurrence of AF in old TG mice as indicated by the AF-suppressing effect of thapsigargin, by the increased incidence of calcium sparks and by an increased calcium leak from the sarcoplasmic reticulum. The latter events go along with an increased calcium/calmodulin-dependent protein kinase II (CaMKII) mediated phosphorylation at S2814 of the ryanodine receptor 2 [26], which—together with similar findings in other mouse models and in patients with AF—suggests an important role of CaMKII in the progression of AF. This altered calcium homeostasis might also be favored by the depolarization and elongation of atrial myocytes [27,28].
Our data suggest that overexpression of CREM leads to atrial structural and functional changes via transcriptional repression of CRE-controlled target genes. It may be speculated that Cx40 is a direct target gene of repressor proteins translated from CREM-IbΔC-X mRNA. In line with this, five half-site CREs were identified in the murine Cx40 gene promoter of which two are located between −3 kb and −300 bp of the transcriptional start site, one of them in direct vicinity to a TATA box [29]. The hypothesis that CRE-binding factors of the CREB/CREM family contribute to the regulation of atrial growth is further underscored by data about the cardiac role of c-Jun dimerization protein 2 (JDP2), a repressor of CRE-mediated transcription structurally related to CREM-IbΔC-X [3,30]. Cardiomyocyte-directed expression of JDP2 in mice causes atrioventricular conduction defects combined with atrial dilatation, down-regulation of Cx40, and other changes [31], similar to our data from TG mice and consistent with the function of CREM as a repressor of CRE-mediated transcription and a role of this mechanism in the regulation of atrial growth. CREs were identified in the promoters of multiple genes in the mouse genome [29], including genes with known functions in cardiomyocytes. While the majority of TG develops spontaneous-onset AF, there was only anecdotal evidence of spontaneous AF in JDP2 mice, likely due to different sets of target genes regulated by CREM and JDP2 [32]. The genes repressed by CREM or JDP2 in mouse atria are not known; it is unlikely that there is a single target gene of CREM at the beginning of the chain of events leading to AF in this model, and the identification of CREM-regulated genes in mouse atrium will be necessary to identify target genes functionally relevant in regard to AF. The hypothesis of multigenetic mechanisms is in good agreement with transcriptomic studies showing profound modifications of gene expression in human hearts with AF [33].
In conclusion, we suggest CREM as an important regulator of atrial growth and development associated with the development of an arrhythmogenic substrate in mouse atrium. It is generally believed that dilated atria from large animals with extensive structural alterations are required for sustained AF to develop, while “focal”, i.e. calcium-dependent mechanisms may cause atrial ectopy and paroxysmal AF in smaller atria. Our study challenges this dichotomic view by providing evidences that small areas of disorganized and elongated atrial myocardium with little fibrosis are sufficient for the activation of sustained arrhythmogenicity. TG mice further represent a unique mouse model of a structural substrate of AF consisting of abnormal growth of atrial cardiomyocytes and structural disorganisation of atrial myocardium and reflect important functional alterations observed in human AF. Therefore, the TG mouse model should help to identify therapeutic targets to prevent the progression of the atrial remodelling and to decipher signalling pathways involved in some aspects of the substrate of sustained AF.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (MU 1376/10-3 to FUM,MU 1376/11-1, and FA 413-3/1 to LF); Interdisciplinary Centre for Clinical Research IZKF Münster (Kih1/020/07 to LF, core unit CarTel to PK); Fondation Leducq (ENAFRA, to PK and US); ‘Alliance for Calmodulin Kinase Signaling in Heart Disease’ (to XHTW); Śtructural Alterations in the Myocardium and the Substrate for Cardiac Fibrillatioń (to EM, NM, GM, SH); the National Institute of Health (R01-HL089598 and R01-HL091947 to XHTW); Fédération Française de Cardiologie (to EM); University of Münster, Ínnovative Medical Research´ (IMF FA120431 to LF and KA620805 to SK); and the European Union (EUTRAF, to FUM, PK, SH, US). We thank Nina Kreienkamp, Nina Nordsiek, Meike Rolfing, Marcel Tekook, and Daniela Volkery for expert technical assistance, and Thomas Waldeyer and Kai Rothaus for the algorithm for hypothesis-free rhythm analysis. The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology.
Footnotes
Conflict of interest
None declared.
References
- [1].Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2001;2:599–609. [DOI] [PubMed] [Google Scholar]
- [2].Müller FU, Bokník P, Horst A, et al. cAMP response element binding protein is expressed and phosphorylated in the human heart. Circulation 1995;92:2041–3. [DOI] [PubMed] [Google Scholar]
- [3].Müller FU, Bokník P, Knapp J, et al. Identification and expression of a novel isoform of cAMP response element modulator in the human heart. FASEB J 1998;12:1191–9. [DOI] [PubMed] [Google Scholar]
- [4].Lewin G, Matus M, Basu A, et al. Critical role of transcription factor cyclic AMP response element modulator in beta1-adrenoceptor-mediated cardiac dysfunction. Circulation 2009;119:79–88. [DOI] [PubMed] [Google Scholar]
- [5].Müller FU, Lewin G, Baba HA, et al. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem 2005;280:6906–14. [DOI] [PubMed] [Google Scholar]
- [6].Camm AJ, Kirchhof P, Lip GY, et al. Guidelines for the management of atrial fibrillation: the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC). Eur Heart J 2010;31:2369–429. [DOI] [PubMed] [Google Scholar]
- [7].Nattel S, Duker G, Carlsson L. Model systems for the discovery and development of antiarrhythmic drugs. Prog Biophys Mol Biol 2008;98:328–39. [DOI] [PubMed] [Google Scholar]
- [8].Kirchhof P, Fabritz L, Zwiener M, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006;114:1799–806. [DOI] [PubMed] [Google Scholar]
- [9].Fabritz L, Kirchhof P, Fortmüller L, et al. Gene dose-dependent atrial arrhythmias, heart block and atrial brady-cardiomyopathy in mice overexpressing the A3-adenosine receptor. Cardiovasc Res 2004;62:500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wittköpper K, Fabritz L, Neef S, et al. Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest 2010;120:617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rothaus K, Jiang X, Waldeyer T, Fabritz L, Vogel M, Kirchhof P. Data mining for detecting disturbances in heart rhythm. International Conference on Machine Learning and Cybernetics, Kumming, China; 2008. p. 3211–6. [Google Scholar]
- [12].Abi-Char J, Maguy A, Coulombe A, et al. Membrane cholesterol modulates Kv1.5 potassium channel distribution and function in rat cardiomyocytes. J Physiol 2007;582:1205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Blana A, Kaese S, Fortmüller L, et al. A knock-in gain-of-function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm 2010;7:1862–9. [DOI] [PubMed] [Google Scholar]
- [14].Milliez P, Deangelis N, Rucker-Martin C, et al. Spironolactone reduces fibrosis of dilated atria during heart failure in rats with myocardial infarction. Eur Heart J 2005;26:2193–9. [DOI] [PubMed] [Google Scholar]
- [15].Sood S, Chelu MG, van Oort RJ, et al. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 2008;5:1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG, Wehrens XH. Genetic inhibition of PKA phosphorylation of Ryr2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A 2010;107:13165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pfaffl MW, Horgan GW, Dempfle L. Relative Expression Software Tool (Rest) for group wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 2002;30:E36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Boyden PA, Tilley LP, Pham TD, Liu SK, Fenoglic JJ Jr, Wit AL. Effects of left atrial enlargement on atrial transmembrane potentials and structure in dogs with mitral valve fibrosis. Am J Cardiol 1982;49:1896–908. [DOI] [PubMed] [Google Scholar]
- [19].Boyden PA, Tilley LP, Albala A, Liu S-K, Fenoglio JJ Jr, Wit AL. Mechanisms for atrial arrhythmias associated with cardiomyopathy: a study of feline hearts with primary myocardial disease. Circulation 1984;69:1036–47. [DOI] [PubMed] [Google Scholar]
- [20].Mary-Rabine L, Albert A, Pham TD, et al. The relationship of human atrial cellular electrophysiology to clinical function and ultrastructure. Circ Res 1983;52:188–99. [DOI] [PubMed] [Google Scholar]
- [21].Deroubaix E, Folliguet T, Rucker-Martin C, et al. Moderate and chronic hemodynamic overload of sheep atria induces reversible cellular electrophysiologic abnormalities and atrial vulnerability. J Am Coll Cardiol 2004;44:1918–26. [DOI] [PubMed] [Google Scholar]
- [22].Boutjdir M, Le Heuzey JY, Lavergne T, et al. Inhomogeneity of cellular refractoriness in human atrium: factor of arrhythmia? Pacing Clin Electrophysiol 1986;9:1095–100. [DOI] [PubMed] [Google Scholar]
- [23].Hagendorff A, Schumacher B, Kirchhoff S, Lüderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 1999;99:1508–15. [DOI] [PubMed] [Google Scholar]
- [24].Verheule S, van Batenburg CA, Coenjaerts FE, Kirchhoff S, Willecke K, Jongsma HJ. Cardiac conduction abnormalities in mice lacking the gap junction protein connexin40. J Cardiovasc Electrophysiol 1999;10:1380–9. [DOI] [PubMed] [Google Scholar]
- [25].Bagwe S, Berenfeld O, Vaidya D, Morley GE, Jalife J. Altered right atrial excitation and propagation in connexin 40 knockout mice. Circulation 2005;112:2245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chelu MG, Satyam S, Sood S, et al. Calmodulin kinase II-mediated sarcoplasmatic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 2009;119:1940–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Iribe G, Ward CW, Camelliti P, et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2 + spark rate. Circ Res 2009;104:787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kong CR, Bursac N, Tung L. Mechanoelectrical excitation by fluid jets in monolayers of cultured cardiac myocytes. J Appl Physiol 2005;98:2328–36. [DOI] [PubMed] [Google Scholar]
- [29].Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A 2005;102:4459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jin C, Ugai H, Song J, et al. Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett 2001;489:34–41. [DOI] [PubMed] [Google Scholar]
- [31].Kehat I, Heinrich R, Ben-Izhak O, Miyazaki H, Gutkind JS, Aronheim A. Inhibition of basic leucine zipper transcription is a major mediator of atrial dilatation. Cardiovasc Res 2006;70:543–54. [DOI] [PubMed] [Google Scholar]
- [32].Donald JE, Shakhnovich EI. Predicting specificity-determining residues in two large eukaryotic transcription factor families. Nucleic Acids Res 2005;33:4455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Barth AS, Merk S, Arnoldi E, et al. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res 2005;96:1022–9. [DOI] [PubMed] [Google Scholar]