

Abstract
Objective:
To improve our understanding of pulmonary arteriovenous malformations in univentricular congenital heart disease, our objective was to identify the effects of hepatic vein and superior vena cava constituents on lung microvascular endothelial cells independent of blood flow.
Methods:
Paired blood samples were collected from the hepatic vein and superior vena cava in children 0-10 years-old undergoing cardiac catheterization. Isolated serum was subsequently used for in vitro endothelial cell assays. Angiogenic activity was assessed using tube formation and scratch migration. Endothelial cell survival was assessed using proliferation (BrdU incorporation, cell cycle analysis) and apoptosis (caspase 3/7 activity, Annexin-V labeling). Data were analyzed using Wilcoxon signed-rank test and repeated measures analysis.
Results:
Upon incubating lung microvascular endothelial cells with 10% patient serum, hepatic vein serum increases angiogenic activity (tube formation, p=0.04, n=24; migration, p<0.001, n=18), increases proliferation (BrdU, p<0.001, n=32; S-phase, p=0.04, n=13), and decreases apoptosis (caspase 3/7, p<0.001, n=32; Annexin-V, p=0.04, n=12) compared to superior vena cava serum.
Conclusions:
Hepatic vein serum regulates lung microvascular endothelial cells by increasing angiogenesis and survival in vitro. Loss of hepatic vein serum signaling in the lung microvasculature may promote maladaptive lung microvascular remodeling and pulmonary arteriovenous malformations.
Introduction
Children born with functionally univentricular congenital heart disease (CHD) are increasingly surviving to adulthood (1–3). Despite improved survival, children with univentricular CHD are at risk for multiple complications. A common complication is the formation of pulmonary arteriovenous malformations (PAVMs) (4–5). PAVMs are abnormal vascular connections between arteries and veins in the lungs. Because PAVMs bypass the capillary bed, PAVMs cause hypoxia and can decrease quality of life. PAVMs develop as an unintended complication in 60-100% of patients who undergo staged surgical palliation for univentricular CHD (6–7). Despite the regular occurrence of CHD-associated PAVMs, little is known about them, and there are no effective medical therapies to treat CHD-associated PAVMs (8–10).
Clinical observations indicate that an unidentified factor in hepatic vein (HV) blood, called hepatic factor, is protective against developing PAVMs and can resolve existing PAVMs (11–12). Hepatic factor is excluded from pulmonary blood flow after surgical palliation with a superior cavopulmonary connection (SCPC), also known as Glenn palliation. In SCPC circulation, pulmonary blood flow is non-pulsatile and provided exclusively from superior vena cava (SVC) blood. PAVMs develop after SCPC palliation. After subsequent palliation with a total cavopulmonary connection (TCPC), also known as Fontan palliation, pulmonary blood flow remains non-pulsatile, but HV blood is re-directed to the pulmonary vasculature. PAVMs usually resolve over weeks to months after TCPC with re-incorporation of HV blood despite blood flow remaining non-pulsatile.
PAVM formation is a dynamic process of vascular remodeling. Animal models of SCPC palliation indicate that PAVMs may develop due to dysregulated angiogenesis, proliferation, and oxidative stress (13–15). The specific contributions of HV and SVC blood to regulate these fundamental cellular processes in the lung microvasculature is not well understood. Additionally, it remains unknown whether the cellular and molecular changes in these animal models of PAVMs are due to HV and SVC constituents or due to changes in blood flow from pulsatile to non-pulsatile. We hypothesized that HV and SVC constituents would differentially impact lung microvascular endothelial cells (ECs). In this study, we used blood samples from pediatric-age patients to investigate the effects of HV and SVC blood on lung microvascular ECs independent of flow.
Methods
Patient selection and Blood sample collection
Blood samples were collected from children 0-10 years old undergoing cardiac catheterization at Children’s Hospital of Wisconsin after obtaining parental written informed consent. Using fluoroscopic guidance, blood samples were specifically collected from two anatomic locations: (#1) the superior vena cava (SVC), and (#2) immediately cephalad to the hepatic vein (HV) insertion into the inferior vena cava (IVC). Site #2 will be referred to as HV. The volume of sample drawn was based on patient size. This study was approved by the Institutional Review Board (IRB).
After blood collection, samples were allowed to coagulate at room temperature for 30 minutes and centrifuged at 2000 x g for 15 minutes at 4°C. Serum was then aspirated, aliquoted, and stored at −80°C.
Data regarding patient diagnosis and clinical variables were obtained from the medical record. PAVMs were diagnosed using echocardiographic bubble studies during cardiac catheterization. PAVMs were suspected in patients with univentricular circulation without a bubble study if they had hypoxia (oxygen saturation [SpO2] < 80% for SCPC circulation or < 90% for TCPC circulation) without significant veno-venous collaterals, abnormal angiographic appearance of the pulmonary vasculature, or pulmonary vein desaturation (SpO2 < 95%).
Cell culture
Primary human pulmonary microvascular endothelial cells (HPMECs) isolated from the lung of a single donor were purchased and used for all experiments (Promocell, #C-12281). Mycoplasma-free cells were grown and maintained in Endothelial cell growth medium MV (Promocell, #C-22020) and studied at passage 4-6.
Angiogenesis: Tube Fommtion
One day prior to tube formation assay, reduced growth factor Matrigel (VWR, #47743-718) was thawed overnight at 4°C and contact inhibited HPMECs were serum-starved overnight in 0.5% fetal bovine serum (FBS). Undiluted Matrigel was added (65 μL) to each well of a 96 well plate and allowed to solidify at 37°C for 1 hour. HPMECs were then trypsinized, counted, and 1 x 104 cells/well were added after mixing cells with 10% patient serum (triplicate). HPMECs were then incubated at 37°C for 4 hours. Tube formation was visualized using Keyence BZ-X700 bright-field/fluorescent microscope (Japan). Images were empirically obtained from the center of each well and analyzed with ImageJ using the “Angiogenesis Analyzer” function to quantify number of nodes, number of tubes, and tube length. The average of each sample (triplicate) was used for statistical analysis and is reported relative to control (HPMEC treated with complete growth medium).
Angiogenesis: Scratch migration
HPMECs were grown to confluence in a 24-well plate and then serum starved overnight in 0.5% FBS. The confluent monolayer was scratched with a sterile P200 pipette tip to create a zone free of HPMECs. The media was then gently aspirated and replaced with 10% patient serum in triplicate. Images from each well were obtained using Keyence BZ-X700 bright-field/fluorescent microscope immediately after replacing media (time (t) = 0 hours) and after 8-hour incubation at 37°C with 10% patient serum (t=8h). Migration was quantified using ImageJ where the denuded area at t=8h was subtracted from the denuded area at t=0h to calculate percent area closed. The average of each sample (triplicate) was used for analysis.
Cell Proliferation: BrdU incorporation
Contact inhibited HPMECs were seeded in a 96 well plate (5 x 103 cells/well), allowed to attach, serum-starved overnight in 0.5% FBS and then incubated with 10% patient serum. BrdU was added directly to each well and incubated for 24 hours. After a total of 48-hour incubation with patient serum, cells were fixed and immunostained according to the manufacturer’s recommendations (Roche, #11647229001). The average of each sample (triplicate) was used for statistical analysis and is reported relative to control (HPMEC treated with complete growth medium).
Cell Proliferation: Cell cycle analysis
Contact inhibited HPMECs were seeded in 6-well plates (2.5 x 105 cells/well), allowed to attach, serum-starved overnight in 0.5% fetal bovine serum (FBS), and then incubated with 10% patient serum for a total of 48 hours. Serum-treated cells were then trypsinized and incubated with LIVE/DEAD yellow dead cell stain kit (ThermoFisher, #L34968). After surface staining with viability dye, cells were washed, fixed and permeabilized with BD Pharmingen Transcription-Factor buffer set (BD Biosciences, #562574), and DNA was stained with propidium iodide (BD Biosciences, #550825). Samples were acquired on a BD Fortessa flow cytometer and data were analyzed with FlowJo software (version 10.5.3). Only viable cells (negative for yellow dead cell stain) were used for cell cycle quantification.
Apoptosis: Caspase 3/7 Activity
Contact inhibited HPMECs were seeded in a 96 well plate (5 x 103 cells/well), allowed to attach, serum-starved overnight in 0.5% fetal bovine serum (FBS), and then incubated with 10% patient serum for a total of 48 hours. Caspase 3/7 substrate was directly added to each well and incubated at room temperature for 6 hours. Fluorescent caspase 3/7 activity was determined according to the manufacturer’s recommendations (Promega, #G7790). The average of each sample (triplicate) was used for statistical analysis and is reported relative to control (HPMEC treated with complete growth medium).
Apoptosis: Annexin-Vlabeling
Contact inhibited HPMECs were seeded in 6-well plates (2.5 x 105 cells/well), allowed to attach, serum-starved overnight in 0.5% fetal bovine serum (FBS), and then incubated with 10% patient serum for a total of 48 hours. Serum-treated cells were then trypsinized and incubated with LIVE/DEAD yellow dead cell stain kit (ThermoFisher, #L34968). After surface staining with viability dye, cells were washed and incubated with Annexin V-PE conjugate (BD Biosciences, #563544). Samples were acquired on a BD Fortessa flow cytometer and data were analyzed with FlowJo software (version 10.5.3). Viable cells (negative for yellow dead cell stain) with positive Annexin-V staining specifically represent apoptotic cells and were used for apoptosis quantification.
Statistical Analysis
Patient demographics are summarized as median (range) for continuous data and n (%) for categorical data. Wilcoxon Signed Rank test, a non-parametric method, was used for comparisons between HV and SVC. Differences in medians with 95% confidence intervals for the observed differences in median levels were estimated using the Hodges-Lehmann method. For single experiments with multiple outcomes (tube formation assay and cell cycle analysis), we adjusted for multiple comparisons using false discovery rate (FDR) correction. Potential outliers were identified as outside of lower quartile − (1.5 x interquartile range (IQR)) and upper quartile + (1.5 x IQR). To investigate whether the results were impacted by patients’ demographics and clinical variables, we performed repeated measures analysis with the following covariates included separately or together: age, gender, SpO2, hypoxia (<94% vs ≥94%), PAVMs (yes vs no), univentricular (yes vs no), and biventricular circulation with left-to-right shunt (yes vs no). Statistical significance was accepted at the level of p<0.05 or FDR-adjusted p<0.05. All analyses were performed using SAS 9.4 (SAS Institute, Cary, NC), StatXact 8, and GraphPad Prism 8 (GraphPad Software, San Diego, CA).
Results
Patient characteristics
Blood samples from 32 patients at Children’s Hospital of Wisconsin were included. Patient age ranged from 3 months to 10 years with a median age of 29 months. Eighteen patients (56%) were female. SpO2 ranged 79-100% with a median SpO2 of 97%; twelve patients (38%) had SpO2 < 94%. Patient demographics, including primary cardiac diagnosis are summarized in Table 1. PAVMs were diagnosed or suspected in 7/32 patients (21.9%). Two patients with univentricular circulation were not suspected to have PAVMs due to normal SpO2 (95%) in one patient status post TCPC and one patient who underwent cardiac catheterization pre-SCPC. Experimental results from specific patient groups are reported in Supplemental Figs 1–4.
Table 1:
Patient demographics
| Female patients, n (%) | 18 (56.3) |
| Age (months), median (range) | 29 (3-126) |
| SpO2 (%), median (range) | 97 (79-100) |
| Primary cardiac diagnosis, n (%) | Functionally univentricular, 9 (28.1) |
| Patent ductus arteriosus, 5 (15.6) | |
| Tetralogy of Fallot, 4 (12.5) | |
| Pulmonary valve stenosis, 4 (12.5) | |
| Atrial septal defect, 2 (6.3) | |
| Ventricular septal defect, 2 (6.3) | |
| Atrioventricular septal defect, 2 (6.3) | |
| Pulmonary hypertension, 2 (6.3) | |
| Double outlet right ventricle, 1 (3.1) | |
| Coarctation of aorta, 1 (3.1) | |
| Type of functionally univentricular, n (%) | Double inlet left ventricle, 1 (11) |
| Double outlet right ventricle, 1 (11) | |
| Hypoplastic left heart syndrome, 5 (56) | |
| Tricuspid atresia, 2 (22) |
SpO2= peripheral oxygen saturation
HV serum increased tube formation and cell migration of HPMECs in vitro
HV serum increased HPMEC tube formation and cell migration compared to SVC serum in vitro (Fig 1). HV serum increased multiple parameters of tube formation compared to SVC serum, including number of nodes (median difference (HV-SVC) 0.07, 95% CI for median difference 0.02, 0.17; p=0.02; FDR-adjusted p=0.04), number of tubes (median difference 0.10, 95% CI 0.01, 0.21; p=0.03; FDR-adjusted p=0.04), and tube length (median difference 0.08, 95% CI 0.004, 0.18; p=0.04; FDR-adjusted p=0.04) (n=24 for all parameters) (Fig 1A–E). HV serum also increased HPMEC wound closure compared to SVC serum using the scratch migration assay (median difference 0.03, 95% CI 0.02, 0.06; p<0.001; n=18) (Fig 1F–J). After adjusting for single and multiple patient variables, the differences in tube formation and scratch migration between HV and SVC remained statistically significant (Supp Table 1). Three potential outliers were identified in the tube formation assay. Patients included a 15-month-old male with patent ductus arteriosus (PDA) and normal SpO2 (98%), a 35-month-old female with PDA and normal SpO2 (98%), and a 57-month-old female with history of ventricular septal defect and pulmonary hypertension on pulmonary vasodilator therapy with normal SpO2 (95%). No outliers were identified in the scratch migration assay.
Figure 1:
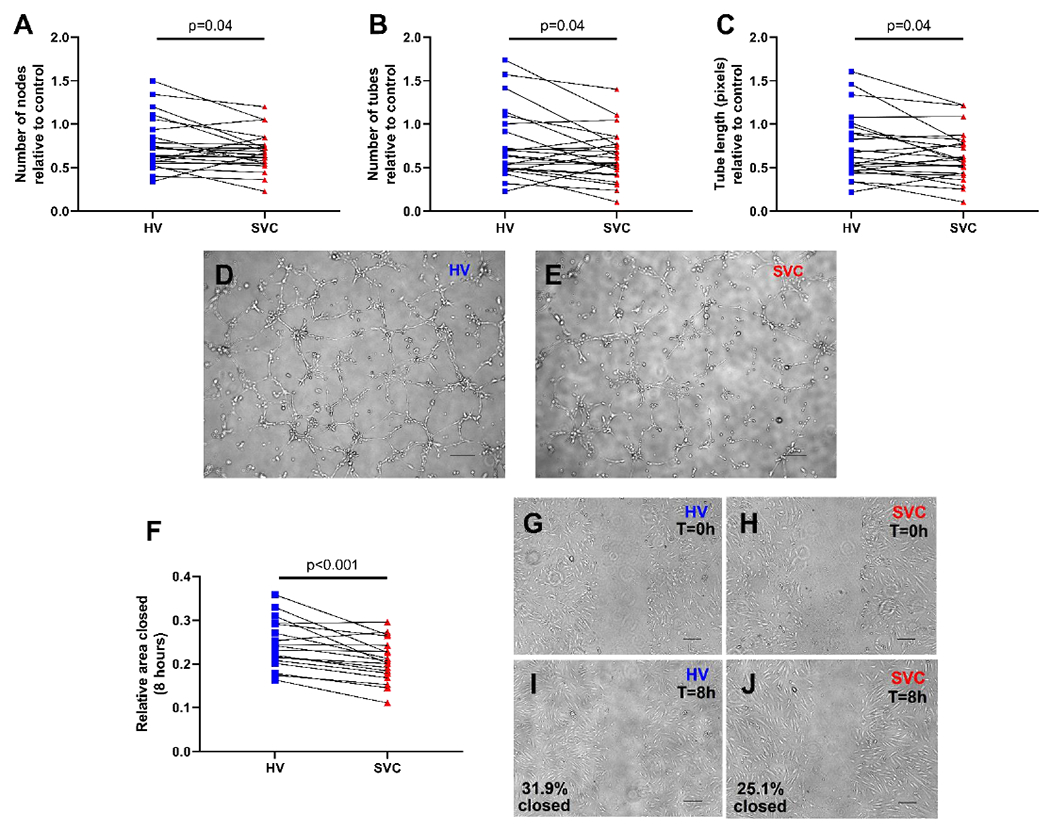
HV serum increases HPMEC angiogenic activity compared to SVC serum. (A-C) Quantification of HPMEC tube formation after 4-hour incubation with HV or SVC serum. N=24 per group. Before and after plots represent individual patient samples with lines connecting paired samples. (D-E) Representative images of tube formation. (F) Quantification of HPMEC migration 8-hours after scratch migration assay. N=18 per group. Before and after plot represents individual patient samples with lines connecting paired samples. (G-J) Representative images of scratch migration at 0 hours (T=0h) and 8 hours (T=8h). Scale bars represent 200 μm. HPMEC= human pulmonary microvascular endothelial cells, HV= hepatic vein, SVC= superior vena cava.
HV serum promoted HPMEC survival through increased proliferation and decreased apoptosis
After incubating HPMECs with HV or SVC serum for 48 hours, HPMECs exposed to HV serum had more proliferation (Fig 2) and less apoptosis (Fig 3) than HPMECs exposed to paired SVC serum samples. HV serum increased BrdU incorporation (median difference 0.10, 95% CI 0.05, 0.15; p<0.001; n=32) relative to control (Fig 2A). HV serum also increased the percentage of HPMECs in S-phase of the cell cycle (median difference 0.4, 95% CI 0.02, 0.7; p=0.04; FDR-adjusted p=0.04; n=13), increased the percentage in G2-M phases (median difference 0.65, 95% CI 0.04, 1.74; p=0.02; FDR-adjusted p=0.03; n=13), and decreased the percentage in G1-G0 phases (median difference −1.16, 95% CI −2.34, −0.20; p=0.01; FDR-adjusted p=0.03; n=13) (Fig 2B–D). After adjusting for single and multiple patient variables, the differences in BrdU incorporation and percentage of cells in G1-G0 phases between HV and SVC remained statistically significant. Percentage of cells in S-phase remained significant after adjusting for single patient variables but not multiple patient variables. The difference in percentage of cells in G2-M phase of the cell cycle was not statistically significant when single and multiple covariates were included (Supp Table 2). No outliers were identified in the BrdU incorporation assay. One potential outlier was identified in the cell cycle analysis – a 28-month-old female with PDA and normal SpO2 (99%).
Figure 2:
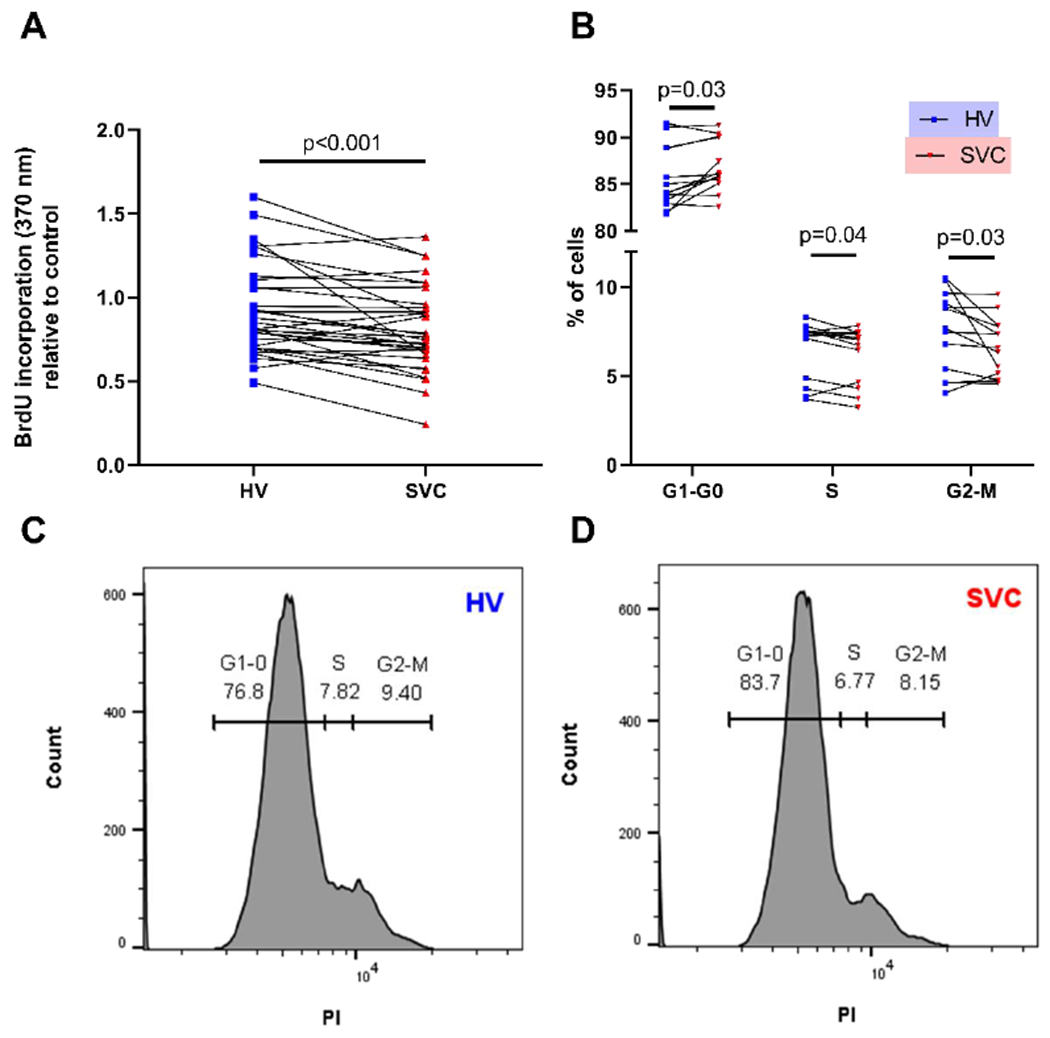
HV serum increases HPMEC proliferation compared to SVC serum. (A) Quantification of HPMEC proliferation using colorimetric absorbance of BrdU incorporation. N=32 per group. Before and after plot represents individual patient samples with lines connecting paired patient samples. (B) Quantification of HPMEC proliferation using cell cycle analysis with PI as detected by flow cytometry. N=13 per group. Before and after plots represent individual patient samples with lines connecting paired patient samples. (C-D) Representative histograms of cell cycle analysis. HPMEC= human pulmonary microvascular endothelial cells, HV= hepatic vein, PI=propidium iodine, SVC= superior vena cava.
Figure 3:
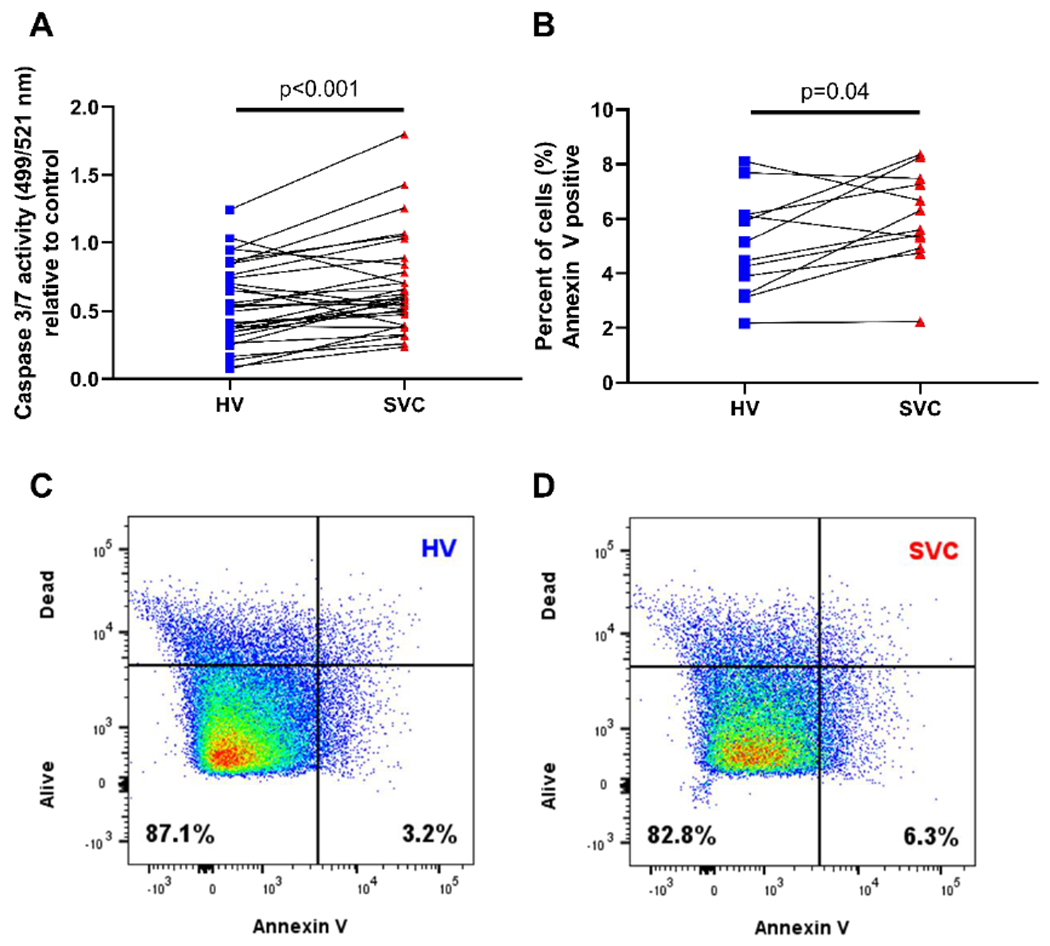
HV serum decreases HPMEC apoptosis compared to SVC serum. (A) Quantification of HPMEC apoptosis using fluorescence of caspase 3 and caspase 7 activity. N=32 per group. (B) Quantification of apoptosis using Annexin-V labeling of apoptotic cells as detected by flow cytometry. N=12 per group. Before and after plots represent individual patient samples with lines connecting paired patient samples. (C-D) Representative scatter plots of Annexin-V staining (x-axis) and cell viability staining (y-axis) to quantify apoptotic HPMECs. HPMEC= human pulmonary microvascular endothelial cells, HV= hepatic vein, SVC= superior vena cava.
HV serum decreased activity of caspase 3 and 7 (median difference −0.14, 95% CI −0.21, −0.07; p<0.001; n=32) relative to control (Fig 3A). HV serum also decreased the percentage of Annexin-V positive cells (median difference −1.11, 95% CI −2.11, −0.03; p=0.04; n=12) (Fig 3B–D). After adjusting for patient variables, the differences in caspase 3/7 activity and Annexin-V positive cells between HV and SVC remained statistically significant (Supp Table 3). No outliers were identified in the caspase activity assay or Annexin-V assay.
Discussion
In this study, we demonstrate that venous blood from different anatomic sources differentially impacts the pulmonary vasculature (Fig 4). Specifically, in static cell culture conditions, HV serum regulates HPMECs differently than SVC serum by increasing angiogenesis and cell survival. Our study reports a novel approach to investigate the etiology and pathogenesis of CHD-associated PAVMs. By specifically testing paired blood samples from the HV and SVC from children with CHD, we aim to improve our understanding of the precise roles of HV and SVC blood in CHD-associated PAVMs independent of blood flow. Greater understanding of HV blood and CHD-associated PAVMs may allow accurate identification of hepatic factor and subsequent development of targeted molecular-based therapies.
Figure 4:
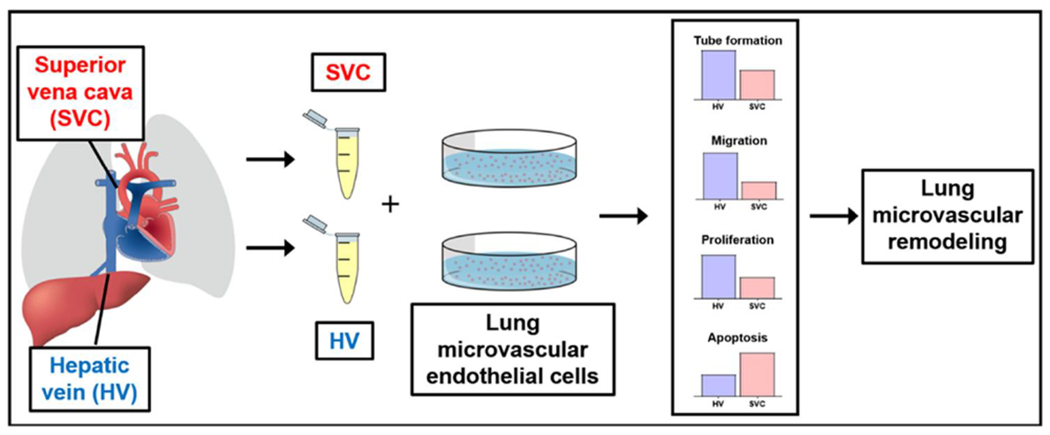
Patient-derived venous blood samples obtained from the HV and SVC differentially impacts the lung microvasculature in vitro. Differential regulation of microvascular endothelial cell angiogenesis (tube formation and migration), proliferation, and apoptosis can lead to lung microvascular remodeling and subsequent PAVM formation. HV= hepatic vein, PAVM= pulmonary arteriovenous malformation, SVC= superior vena cava.
There are several salient features of our study, which we will compare and contrast below with the published literature. Our data indicate that HV serum increases HPMEC angiogenesis in vitro compared to SVC serum, which differs from previous studies (14, 17–19). Angiogenesis is a dynamic, context-dependent, multi-staged process. Thus, differing experimental results from other studies are not surprising. Kavarana et al investigated angiogenic changes after SCPC using a pig model of SCPC physiology (“classic Glenn” – end-to-end SVC to right pulmonary artery anastomosis) (14). They harvested pulmonary artery ECs 6-8 weeks after SCPC by scraping the luminal surface of the branch pulmonary artery and culturing these macrovascular ECs. With the cultured ECs, they performed tube formation assays and reported increased number of tubes and tube length in ECs from the lung receiving exclusive SVC blood flow. Importantly, their study used ECs from a large branch pulmonary artery (macrovascular) rather than microvascular ECs, which are the affected ECs in PAVM pathophysiology. Additionally, as previously stated, it is unknown whether the increased tube formation is due to lack of HV blood (i.e., hepatic factor) perfusing the lungs or whether it is due to change from pulsatile to non-pulsatile blood flow.
Using patient lung biopsies after SCPC, Duncan et al reported initial findings from two patients, and Starnes et al subsequently reported follow-up findings from eight and thirteen patients (17–19). Their histological studies used immunostaining with von Willebrand factor to determine microvessel density (17–18). Microvessel density is often used as a surrogate marker for angiogenesis by quantifying microvascular ECs per surface area (20). Duncan and Starnes et al found that patients after SCPC had increased von Willebrand factor staining compared to controls in the initial and follow-up study. They concluded that lack of HV blood flow to the lungs increased microvessel density and thus increased angiogenesis, which is in contrast to our findings. Importantly, Starnes et al insightfully acknowledged that SCPC may cause PAVMs due to abnormal angiogenesis and vascular remodeling (18). This is supported by their data. In the initial study, Duncan et al reported diminished CD31 staining, and Starnes et al confirmed decreased CD31 staining in the follow-up study (17,19). Paradoxically, CD31 is also accepted as an immunostaining marker for microvessel density (20). Thus, two different EC markers for microvessel density give different results from the same histologic samples. These differences exemplify that angiogenesis is a dynamic multi-faceted process of remodeling that occurs both physiologically and pathologically. While our data do not fully reconcile with these previous studies, our findings are in agreement with the interpretation that vascular remodeling is likely altered in PAVM pathophysiology.
An important component of angiogenesis and vascular remodeling is the balance of EC proliferation and apoptosis. One of the initial studies investigating hepatic factor and CHD-associated PAVMs reported that hepatocyte-conditioned media inhibited pulmonary artery EC proliferation (16). This finding is consistent with the data reported by Kavarana et al (14). Kavarana et al reported that exclusive SVC blood flow in their pig model of SCPC physiology increased pulmonary artery EC proliferation compared to control ECs (14). These results are in contrast to our finding that HV serum increases HPMEC proliferation compared to SVC serum. Again, several major differences may account for these discrepancies: species (human versus pig), cell type (pulmonary artery macrovascular ECs versus lung microvascular EC), and flow (non-pulsatile blood flow versus static cell culture).
In contrast to Kavarana et al, Duncan and Starnes also investigated EC proliferation using lung biopsies obtained from children after SCPC (17, 19). They used immunostaining with proliferating cell nuclear antigen (PCNA) to assess proliferation. They reported no differences in PCNA staining between patients after SCPC and age-matched controls. These findings are also in contrast to our data because they reported no difference in proliferation, rather than decreased proliferation after SCPC. Without longitudinal assessments, we cannot determine if there are differences in EC proliferation at different stages of PAVM progression or resolution.
In terms of apoptosis, we demonstrate that HV serum decreases HPMEC apoptosis compared to SVC serum. To our knowledge, no previous studies of CHD-associated PAVMs directly investigated apoptosis. Malhotra et al reported an interesting finding of increased EC oxidative stress two and five weeks after SCPC (15). In their study, they compared lambs that underwent SCPC (“classic Glenn”), a unilateral branch PA band, or sham surgery. They reported that both SCPC and PA band increased angiogenic gene expression (VEGF) in whole lung lysates. On the other hand, SCPC increased EC stress response (heme-oxygenase 1, GLUT1, CD62), but PA band and sham did not. They concluded that endothelial stress response may be more relevant to PAVM formation than an angiogenic response. We speculate that our observed differences in apoptosis reflect a manifestation of SVC blood-induced EC stress.
Because PAVM formation is a dynamic process of vascular remodeling, direct comparison between our findings and previous animal models of SCPC should be interpreted cautiously. Our results represent the effects of patient serum on quiescent lung microvascular ECs after 48 hours of exposure, which differs from the longer-term exposure with animal models of SCPC (2-8 weeks). A snapshot of the lung microvasculature after PAVM formation in vivo may represent a different stage of vascular remodeling compared to our in vitro findings. Additionally, our patient blood samples were obtained from a heterogenous cohort at a single time-point. We cannot determine if the serum constituents in our patient samples primarily regulate the pulmonary microvasculature or if our patients’ serum contains factors that are upregulated or downregulated in compensation for remodeling pulmonary vasculature. Finally, we attempted to investigate how patient variables impacted our results using repeated measures analysis. These sub-analyses showed similar results with the exception of cell cycle analysis, which was performed with fewer patient samples. Ultimately, though, our analyses were limited to overall small patient numbers.
Despite previous data and our observations, there are many unknowns in the pathogenesis of CHD-associated PAVMs. We hypothesize that PAVMs develop following SCPC because (1) exclusive SVC blood flow causes increased cellular stress due to inflammatory and pro-apoptotic signals in SVC blood and loss of physiologic growth factors in HV blood. Chronically, decreased exposure of the pulmonary microvasculature to physiologic growth factors and increased EC stress leads to maladaptive microvascular remodeling and pathologic angiogenesis. Alternative etiologies are possible, though. For example, (2) HV blood may contain an inhibitory factor that maintains microvascular EC homeostasis by suppressing proliferation and angiogenesis. Exclusion of this factor from the pulmonary vascular bed (i.e., SCPC circulation) dysregulates the pulmonary microvasculature and leads to PAVM formation. (3) HV blood may contain a set of factors – both physiologic growth factors and inhibitory factors that regulate pulmonary microvascular homeostasis. Redundant factors may help explain the variability in PAVM severity. (4) HV and SVC blood may not directly regulate pulmonary microvascular ECs. Rather, HV blood may regulate vascular support cells (smooth muscle cells and pericytes) and the extracellular matrix, which provide the necessary scaffold for ECs. Loss of the vascular scaffold leads to vascular remodeling and PAVM formation. (5) Finally, multiple studies indicate the flow plays a role in vascular homeostasis (21–23). Resolution of PAVMs after TCPC palliation suggests that hepatic factor functions independent of flow; however, we cannot exclude that flow has a role in pulmonary vascular remodeling and PAVMs. Thus, our study in the context of the literature implicates that vascular remodeling is a key event in PAVM pathogenesis. Further ongoing studies will clarify the underlying mechanisms.
Conclusion
In static cell culture conditions, HV serum regulates HPMECs differently than SVC serum by increasing angiogenesis and cell survival. Loss of HV serum signaling in the lung microvasculature, which occurs with SCPC circulation, may promote lung microvascular remodeling and PAVM formation. The specific constituents of HV and SVC serum that mediate these effects, and whether these changes occur in vivo, requires further investigation.
Supplementary Material
Central Message.
Hepatic vein serum differentially regulates the lung microvasculature in vitro compared to paired samples from the superior vena cava. These effects may prevent pulmonary arteriovenous malformations.
Perspective statement.
Pulmonary arteriovenous malformations commonly form in children during surgical palliation for univentricular congenital heart disease, yet they remain poorly understand. The un-identified hepatic factor in hepatic vein blood prevents this complication. Here we report a novel approach to characterize the effects of hepatic vein blood on the lung microvasculature and potentially identify hepatic factor.
Acknowledgements:
We thank members of the Developmental Vascular Biology lab, Pediatric Cardiology Division, Herma Heart Institute, and MCW Cardiovascular Center for their input. We sincerely acknowledge the generous contribution of patients and families to this study, as well as the staff of the Cardiac Catheterization Lab who assisted in sample collection.
Disclosure of grants or other funding: AS: Funding was provided by Department of Pediatrics and Mend A Heart Foundation (Clarendon Hills, IL). RR: Supported by Department of Pediatrics, Children’s Research Institute, HL123338, and Department of Obstetrics and Gynecology.
Glossary of Abbreviations
- CHD
congenital heart disease
- EC
endothelial cell
- HPMEC
human pulmonary microvascular endothelial cell
- HV
hepatic vein
- PAVMs
pulmonary arteriovenous malformations
- SCPC
superior cavopulmonary connection
- SpO2
peripheral oxygen saturation
- SVC
superior vena cava
- TCPC
total cavopulmonary connection
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare that they have no conflict of interest and no relationship with industry.
References:
- 1.Hirsch JC, Goldberg C, Bove EL, et al. Fontan operation in the current era: a 15-year single institution experience. Ann Surg. 2008;248:402–410. [DOI] [PubMed] [Google Scholar]
- 2.Rogers LS, Glatz AC, Ravishankar C, et al. 18 years of the Fontan operation at a single institution: results from 771 consecutive patients. J Am Coll Cardiol. 2012;60:1018–1025. [DOI] [PubMed] [Google Scholar]
- 3.d’Udekem Y, Iyengar AJ, Galati JC, et al. Redefining expectations of long-term survival after the Fontan procedure: twenty-five years of follow-up from the entire population of Australia and New Zealand. Circulation. 2014;130:S32–38. [DOI] [PubMed] [Google Scholar]
- 4.Kopf GS, Faks H, Stansel HC, Hellenbrand WE, Kleinman CS, Talner NS. Thirty-year follow-up of superior vena cava-pulmonary artery (Glenn) shunts. J Thorac Cardiovasc Surg. 1990;100:662–671. [PubMed] [Google Scholar]
- 5.Kim SJ, Bae EJ, Cho DJ, et al. Development of pulmonary arteriovenous fistulas after bidirectional cavopulmonary shunt. Ann Thorac Surg.2000;70:1918–1922. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein HS, Brook MM, Silverman NH, Bristow J. Development of pulmonary arteriovenous fistulae in children after cavopulmonary shunt. Circulation. 1995;92:11309–314. [DOI] [PubMed] [Google Scholar]
- 7.Vettukattil JJ, Slavik Z, Monro JL, et al. Intrapulmonary arteriovenous shunting may be a universal phenomenon in patients with the superior cavopulmonary anastomosis: a radionuclide study. Heart. 2000;83:425–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spearman AD, Kindel SJ, Woods RK, Ginde S. Arteriovenous fistula creation for hypoxia after single ventricle palliation: A single-institution experience and literature review. Congenit Heart Dis. 201900:1–8 [DOI] [PubMed] [Google Scholar]
- 9.McElhinney DB, Marx GR, Marshall AC, Mayer JE, del Nido PJ. Cavopulmonary pathway modification in patients with heterotaxy and newly diagnosed or persistent pulmonary arteriovenous malformations after a modified Fontan operation. J Thorac Cardiovasc Surg. 2011;141(6):1362–1370 [DOI] [PubMed] [Google Scholar]
- 10.Hickey EJ, Alghamdi AA, Elmi M, et al. Systemic arteriovenous fistulae for end-stage cyanosis after cavopulmonary connection: a useful bridge to transplantation. J Thorac Cardiovasc Surg. 2010;139:128–134. [DOI] [PubMed] [Google Scholar]
- 11.Mathur M, Glenn WW. Long-term evaluation of cava-pulmonary artery anastomosis. Surgery. 1973;74:899–916. [PubMed] [Google Scholar]
- 12.Jonas RA. Invited letter concerning: The importance of pulsatile flow when systemic venous return is connected directly to the pulmonary arteries. J Thorac Cardiovasc Surg. 1993;105:173–176. [PubMed] [Google Scholar]
- 13.Tipps RS, Mumtaz M, Leahy P, Duncan BW. Gene array analysis of a rat model of pulmonary arteriovenous malformations after superior cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2008;136:283–289. [DOI] [PubMed] [Google Scholar]
- 14.Kavarana MN, Mukherjee R, Eckhouse SR, et al. Pulmonary artery endothelial cell phenotypic alterations in a large animal model of pulmonary arteriovenous malformations following the glenn shunt. Ann Thorac Surg. 2013;96(4):1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malhotra SP, Reddy VM, Thelitz S, et al. The role of oxidative stress in the development of pulmonary arteriovenous malformations after cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2002;124:479–485. [DOI] [PubMed] [Google Scholar]
- 16.Marshall B, Duncan BW, Jonas RA. The role of angiogenesis in the development of pulmonary arteriovenous malformations in children after cavopulmonary anastomosis. Cardiol Young. 1997;7:370–374. [Google Scholar]
- 17.Duncan BW, Kneebone JM, Chi EY, et al. A detailed histologic analysis of pulmonary arteriovenous malformations in children with cyanotic congenital heart disease. J Thorac Cardiovasc Surg.1999;117:931–938. [DOI] [PubMed] [Google Scholar]
- 18.Starnes SL, Duncan BW, Kneebone JM, et al. Pulmonary microvessel density is a marker of angiogenesis in children after cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2000;120:902–908. [DOI] [PubMed] [Google Scholar]
- 19.Starnes SL, Duncan BW, Kneebone JM, et al. Angiogenic proteins in the lungs of children after cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2001;122:518–523. [DOI] [PubMed] [Google Scholar]
- 20.Nowak-Sliwinska P, Alitalo K, Allen E, et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis. 2018;21:425–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baeyens N, Larrivee B, Ola R, et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J Cell Biol. 2016;214:807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabit CE, Chen P, Kim GH, et al. Elevated angiopoietin-2 level in patients with continuous flow left ventricular assist devices leads to altered angiogenesis and is associated with higher non-surgical bleeding. Circulation. 2016;134:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodel CJ, Otten C, Donat S, et al. Blood flow suppresses vascular anomalies in a zebrafish model of cerebral cavernous malformations. Circ Res. 2019;125:e43–e54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.