

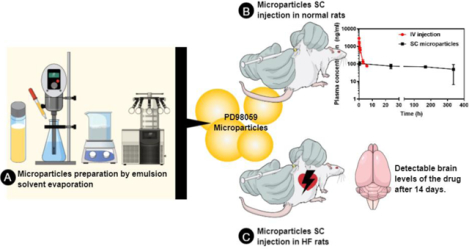
Abstract
PD98059 is a reversible MEK inhibitor that we are investigating as a potential treatment for neurochemical changes in the brain that drive neurohumoral excitation in heart failure. In preliminary studies in a rat model that closely resembles human heart failure, we found that PD98059 gains access to the brain to inhibit phosphorylation of ERK1/2 in the paraventricular nucleus of the hypothalamus, ultimately reducing sympathetic excitation which is a major contributor to clinical deterioration. Additional studies revealed that the pharmacokinetics of PD98059 matches a 2-compartment model with a short elimination half-life in plasma (approximately 73 minutes) that would severely limit its potential clinical usefulness. To increase its availability to tissues, we prepared a sustained-release PD98059-loaded PLGA microparticle formulation, using an emulsion solvent evaporation technique. The average particle size, yield percent, and encapsulation percent were found to be 16.73 μm, 76.6%, and 43%, respectively. In vitro drug release occurred over four weeks, with no noticeable burst release. Following subcutaneous injection of the microparticles in rats, steady plasma levels of PD98059 were detected by HPLC for up to two weeks. Furthermore, plasma and brain levels of PD98059 in rats with heart failure were detectable by LC/MS, despite expected erratic absorption. These findings suggest that PD98059-loaded microparticles hold promise as a novel therapeutic intervention countering sympathetic excitation in heart failure, and perhaps in other diseases processes, including cancers, in which activated MAPK signaling is a significant contributing factor.
Keywords: Microparticles, MEK1/2 inhibitor, sustained release, heart failure, LC/MS, PD98059
Graphical Abstract
Introduction
The mitogen-activated protein kinase (MAPK) cascade is a ubiquitous evolutionarily conserved serine/threonine protein kinase pathway essential for multiple cellular processes, including cell survival, proliferation, differentiation, development, apoptosis, metabolism, migration, and senescence [1–3]. This pathway transmits upstream signals from cell membrane receptors to the nucleus via a series of sequential phosphorylation processes [4]. Following external stimuli-related ligand binding to G-protein coupled receptors in the cell membrane, the signal transmission is initiated by the activation of the GDP/GTP binding protein Ras, which in turn activates (phosphorylates) Raf (also known as MAPK kinase kinase, MAPKKK, ERK kinase, or ERKK) [5, 6, 1]. Consequently, activation of MAPKKK activates MEK (also known as MAPKK or MAPK-ERK kinase), which finally activates one of four different effector protein kinases, namely ERK1/2 (extracellular signal-regulated kinase, also called p42/44), c-JNK (c-jun N-terminal kinase), p38 MAPK, and ERK5 [2, 7]. The most widely studied MAPK pathway is the Ras-Raf-MEK-ERK. Mutations in Ras (including the three highly conserved HRas, KRas, and NRas) predispose an individual to many types of cancer [6, 8–12]. The MEK-ERK pathway is also activated downstream of other tyrosine kinase receptors highly involved in cancer, such as epithelial growth factor receptor (EGFR) [13]. While targeting Ras mutations with small molecules may seem far from reach, a panel of inhibitors that target the downstream Raf-MEK-ERK kinases recently came into the focus as successful alternative approaches [14–16, 6, 17]. Most MEK1/2 inhibitors exhibit reversible kinase inhibitor activity and relatively short half-lives [4, 18]. Thus, in order to achieve long-term therapeutic effects, these agents would need to be administered frequently, or new molecules with longer half-lives would need to be developed [18].
Over the past decade, our laboratory has been investigating the role of the MAPK pathway in the hypothalamic paraventricular nucleus (PVN) in regulating the sympathetic excitation in a rat model of heart failure-induced by myocardial infarction [19–26]. Initial studies revealed that phosphorylated ERK1/2 (pERK1/2) was increased in the PVN of rats with chronic heart failure, along with PVN neuronal activitation, and that a 1-hour intracerebroventricular (ICV) infusion of the MEK1/2 inhibitor PD98059 decreased the pERK1/2 expression and neuronal excitation in PVN, and renal sympathetic nerve activity in rats with heart failure [23]. In subsequent work, chronic (4 week) ICV infusions of PD98059 in heart failure rats reduced plasma norepinephrine, an index of overall sympathetic nerve activity [19]. These powerful effects of ICV PD98059 likely reflect the central interactions between ERK1/2 and major neurochemical systems in brain that drive sympathetic activity, including the brain renin-angiotensin system, neuroinflammatory cytokines and chemokines, and endoplasmic reticulum stress [27, 28, 19–23, 29, 24, 25]. However, because of its short half-life and its reversible inhibition of MEK1/2,PD98059 requires persistent drug exposure to be effective. However, continuous drug administration is not practical and feasible clinically. In an effort to harness the therapeutic potential of PD98059 as an agent targeting central sympatho-excitatory mechanisms in heart failure, we sought to develop a novel pharmaceutical preparation that would deliver the drug to maintain a sustained plasma level sufficient to facilitate passage of effective levels of PD98059 into the brain.
In this research, we examined the pharmacokinetics of PD98059 dissolved in a U.S. FDA-approved vehicle (10% v/v Tween 80 in sterile phosphate buffer saline (PBS), pH 7.4) and injected intravenously (IV) in normal rats. To overcome its short plasma half-life and take advantage of its ability to cross the blood-brain barrier, we designed a poly lactide-co-glycolide (PLGA) microparticle formulation of PD98059. PLGA is a biocompatible and biodegradable polyester that has been utilized to prepare microparticles to provide sustained release of small molecules and macromolecules alike [30–35]. Slow drug release from the bulk-eroding polymer matrix not only provides sustained plasma concentrations at therapeutic levels, but also prevents sharp peaks and troughs in plasma levels that can result from multiple administrations and may result in toxicity or sub-therapeutic levels.
We hypothesized that this novel PLGA formulation would provide a sustained steady plasma drug level that is able to facilitate passage of PD98059 into the brain. We tested this hypothesis in normal rats and rats with heart failure, whose compromised circulation might adversely affect subcutaneous absorption, over a two-week interval following a single subcutaneous injection.
Experimental
Materials
PD98059 was purchased from Selleck Chemicals (Houston, TX). Poly (lactide-co-glycolide) (PLGA, Resomer RG 503 H) was purchased from Evonik (Parsippany, NJ). Poly vinyl alcohol (PVA, Mowiol 8–88, MW 67,000) and 7-hydroxyflavone were purchased from Sigma Aldrich (St Louis, MO). Tween 80 was purchased from Fisher Chemicals (Waltham, MA). All other chemicals and reagents were at least of analytical grade and were used as received without further purification.
Preparation of the microparticles
Microparticles were prepared using an emulsion-solvent evaporation method as previously reported [30]. Briefly, 200 mg of PLGA and 12 mg of PD98059 were dissolved in 1.5 ml of dichloromethane (DCM), and this organic solution was added into 30 ml of 1% PVA solution. The mixture was emulsified at 6500 rpm at room temperature for 5 min (Ultra-turrax T25 basic, Ika Works, Inc., Wilmington, NC). The emulsion was then magnetically stirred at room temperature at 600 rpm for 2 hours to evaporate DCM. The microparticle suspension was then collected by centrifugation at 1000 xg for 10 min (Eppendorf Centrifuge 5864 R, Eppendorf North America, Hauppauge, NY). The microparticles were resuspended in 45 ml of Nanopure water (Barnstead Thermolyne Nanopure water purification system, Thermo Fisher, Waltham, MA)., washed and centrifuged as mentioned earlier. This process was carried out twice to remove any remaining PVA and unencapsulated PD98059. Finally, the microparticles were resuspended in 1 ml of purified water and lyophilized overnight at 0.045 mbar and a collector temperature of −105° C (Labconco Free zone 4.5−105°C, Labconco, Kansas City, MO).
In vitro characterization of the prepared microparticles
Morphology of the microparticles
The morphology of the microparticles was investigated using scanning electron microscopy (SEM). Microparticles (lyophilized) were spread onto a carbon double-adhesive tape mounted on an aluminum stub, and then were sputter-coated with gold and palladium using an argon beam Emitech K550 sputter-coater. A Hitachi S-4800 scanning electron microscope (SEM) operated at 3 kV accelerating voltage (Hitachi High Technologies America Inc., Schaumburg, IL) was used to capture the images of the microparticles. The particle size was analyzed using ImageJ software (NIH, Bethesda, MA) after a minimum of 100 particles in SEM images were measured, and the data were plotted using Microsoft Excel
Determination of microparticles drug content
Microparticles were dissolved in DCM at 1 mg/ml, then 100 μl of this solution was added to 6.4 ml of methanol, and centrifuged (12,000 xg for 5 min). The supernatant was mixed with 3.5 ml of purified water, and the resultant solution was injected into the HPLC as mentioned below.
Drug content in the microparticles was calculated using equation 1, as follows:
| Equation 1 |
Yield percentage was calculated using equation 2, as follows:
| Equation 2 |
Finally, encapsulation efficiency percentage (EE%) was calculated using equation 3, as follows:
| Equation 3 |
The expected amount of PD98059 in 1 mg of microparticles, assuming 100% encapsulation, is 12 mg/212 mg (56.6 μg).
Differential scanning calorimetry
Weighed amounts of PD98059, PLGA, PLGA/PD98059 physical mixture (20:1 w/w), and PD98059-loaded PLGA microparticles were added into aluminum crimped pans and differential scanning calorimetric (DSC) thermograms were obtained using a TA Instruments model Q20 DSC (New Castle, DE, USA). A temperature ramp rate of 5 °C/min, within a range of 0 to 200 °C was used.
In vitro drug release
A weighed amount of the microparticles was suspended in 1X DPBS (Dulbecco’s phosphate buffered saline, Life Science, Waltham, MA) at 0.5 mg microparticles/ml. One ml of this suspension was transferred to a 1 ml screw-capped dialysis tube (Spectra/Por™ Float-A-Lyzer™ G2 MWCO 8–10 kDa, Sigma-Aldrich). The tube was submerged in 12 ml of 0.4% v/v solution of Tween 80 in 1X DPBS and placed in an orbital shaker (New Brunswick Scientific, Edison, NJ) at 300 rpm and 37° C. The solubility of PD98059 in this release medium was approximately 113 μg/ml at 37° C. At pre-determined time points, the whole volume of the release medium (12 ml) was removed and replaced completely with fresh medium. The concentration of PD98059 was measured in the samples using the HPLC method described below.
Experimental Protocols
Twenty three male Sprague-Dawley rats (6–8 weeks, 275–300 g, Harlan labs, Indianapolis, IN) were used in this experiment. Rats were kept under controlled temperature (23 ± 2° C) at the University of Iowa animal care facility. They were exposed to 12-hours of light and dark cycles, and food was provided ad libitum. All animal experiments performed were approved by the University of Iowa Institutional Animal Care and Use Committee.
Pharmacokinetics of PD98059. Rats were anaesthetized using urethane and a canula was inserted in the femoral vein. A solution of PD98059 dissolved in 10% Tween 80 in sterile 1X DPBS at a concentration of 0.5 mg/ml (total volume 2 ml) was administered slowly by intravenous infusion. After 5, 15, 30, 60, 180, and 360 minutes, blood samples were withdrawn from rats. Major organs (liver, kidney, brain, and heart) were collected from rats after 1 and 3 hours (n=3/time point).
Subcutaneous Administration of PD98059-loaded PLGA Microparticles - Normal Rats. PD98059-loaded microparticles suspended in sterile 1X DPBS were injected subcutaneously (SC) in healthy rats (n = 3) at a dose of 2.4 mg PD98059 in 1 ml per rat. At predetermined time intervals (30 min, 1 day, 7 days, and 14 days, 21 days, and 28 days), rats were anesthetized using isoflurane and blood samples were withdrawn from the tail vein. Plasma was collected and treated as mentioned previously, and drug levels were determined by HPLC.
Subcutaneous Administration of PD98059-loaded PLGA Microparticles - Heart Failure Rats. Under sterile conditions, male Sprague-Dawley rats were anesthetized with ketamine/xylazine and underwent left coronary artery ligation to induce heart failure, as previously described [22–24]. Twenty-four hours later, and after heart failure was confirmed by echocardiographic demonstration of reduced systolic function (left ventricular ejection fraction <40%), the rats were subcutaneously injected with the microparticle suspension at a dose of 3.6 mg in 1 ml sterile PBS. After 1, 7, and 14 days, rats were euthanized (n=2–3 per time point) and their plasma and brains were collected.
Plasma and Tissue Preparation.
All blood samples were collected into 4 ml BD Vacutainer® blood collection tubes (K2-EDTA, Becton, Dickinson, and Company, Franklin Lakes, NJ). Plasma samples were collected by centrifugation (3,300 xg, 15 min), and frozen at −80° C until analyzed. Plasma samples were thawed on ice, and a volume of 100 μl of plasma was transferred to a 15 ml tube, and spiked with 15 μl of the internal standard (IS) solution (10 μg/ml of 7-hydroxyflavone in methanol). Collected organs were rinsed in PBS, then frozen at −80° C For organs, a portion of each organ (200–400 mg) was accurately weighed and homogenized (Fisher Brand Bead Mill 4 Homogenizer, Hampton, NH) in 250 μl of 1X DPBS using 20–25 2.5 mm ceramic beads per sample, and spiked with 15 μl of the internal standard (IS) solution. One ml of cold acetonitrile was added to 100 μl of plasma samples or 400 μl of the tissue homogenate and vortexed for 1 min. The samples were kept on ice for 15 min to precipitate the proteins, then the tubes were centrifuged (4° C, 3,300 xg, 10 min) and the supernatant was collected, and evaporated under nitrogen stream.
In the case of protocols 1 and 2, the residue in each tube was then dissolved in the mobile phase (described in the HPLC section below), centrifuged (12,000 xg, 5 min), and the supernatant was injected in the HPLC and analyzed by the method described below. A standard curve was prepared using plasma and tissues collected from naïve rats. These plasma or tissue homogenate samples were spiked with 15 μl of the IS solution as mentioned above, in addition to 15 μl of standard solutions of PD98059 in methanol at different concentrations. Pharmacokinetic parameters were calculated using PK Solver [36, 37].
In the case of protocol 3, residues were redissolved in the mobile phase and PD98059 levels in the plasma and brain were measured using LC/MS/MS with multiple reaction monitoring (MRM) using 7-hydroxyflavone as an IS. Standard curves were constructed in plasma and brain tissues collected from naïve rats.
HPLC and LC/MS/MS
An Agilent HPLC workstation was used for sample analysis (Agilent Infinity 1100, Santa Clara, CA) that consisted of an Agilent quaternary pump, automatic injection port, and Agilent diode array detector (Agilent Corporation, Santa Clara, CA). A RP-C18 column was used for analysis (Waters Symmetry, 5 μm pore size, 4.6 mm × 150 mm, Milford, MA). The mobile phase consisted of methanol: water 70:30 with 0.1% v/v trifluoroacetic acid, and the flow rate was 1 ml/min at room temperature. The detection wavelength was set to 275 nm.
The LC/MS/MS system consisted of a Waters Acquity TQD (Milliford, MA), which includes a triple quadruple mass spectrometer and Acquity H-Class UPLC. The same column, temperature, mobile phase, and flow rate stated above with the HPLC method were used. Quantitative analysis of PD98059 and IS was carried out using positive electrospray ionization via the highly sensitive and specific MRM mode. PD98059 was detected at 3 transition channels for brain samples (268.03→104.86, 268.03→121.01, and 268.03→133.06) and 5 transition channels for plasma samples (268.03→104.86, 268.03→121.01, 268.03→133.06, 268.03→148.08, and 268.03→236.07), while the IS was detected as 3 transition channels in both brain and plasma samples (239.03→77.04, 239.03→129.03, 239.03→136.97). The standard curves were linear over a range of 0.1–30 μg/ml for both plasma and brain.
Results
Preparation and in vitro characterization of the microparticles
The drug-loaded microparticles contained 24.3 μg PD98059/mg. Scanning electron microscopy showed that the microparticles were mostly spherical in shape, with smooth surfaces, while no unencapsulated drug was observed (Figure 1a), as observed when larger amounts of the drug were loaded. Particle size analysis revealed a normal distribution (Figure 1b). Average particle size was approximately 16.7 μm (Figure 1b). DSC thermograms (Figure 1c) showed that the drug exhibited a sharp endothermic peak at 171°C that indicates the drug melting point. The polymer had a brief endothermic peak at 48.33°C, which indicates the glass transition temperature (Tg) of the polymer. The physical mixture of the drug and polymer at a ratio of 1:20 showed a Tg of PLGA (at 49.1° C), while the drug melting point peak appeared as a broad endothermic incident at 162.3° C. The microparticles did not show any endothermic peaks, neither at the polymer Tg, nor at or around the melting point of PD98059. This may indicate physicochemical interaction between PD98059 and PLGA, which resulted from the drug being in an amorphous state, or may simply be due to the microparticle preparation processing. The average particle size, yield %, drug loading (μg drug/mg microparticles), and encapsulation efficiency % (EE %) are 16.73 ± 6.22 μm, 76.6 ± 2.35 %, 24.33 ± 3.1 μg/mg, and 43 ± 5.47 %, respectively. In vitro drug release from PLGA microparticles was slow, with less than 40% of loaded drug being released in the first week. Approximately 73% of the loaded drug was released within four weeks (Figure 1d). In general, the release followed a biphasic pattern with no burst release, where the initial release during the first two days was faster than the rest of the release period.
Figure 1:
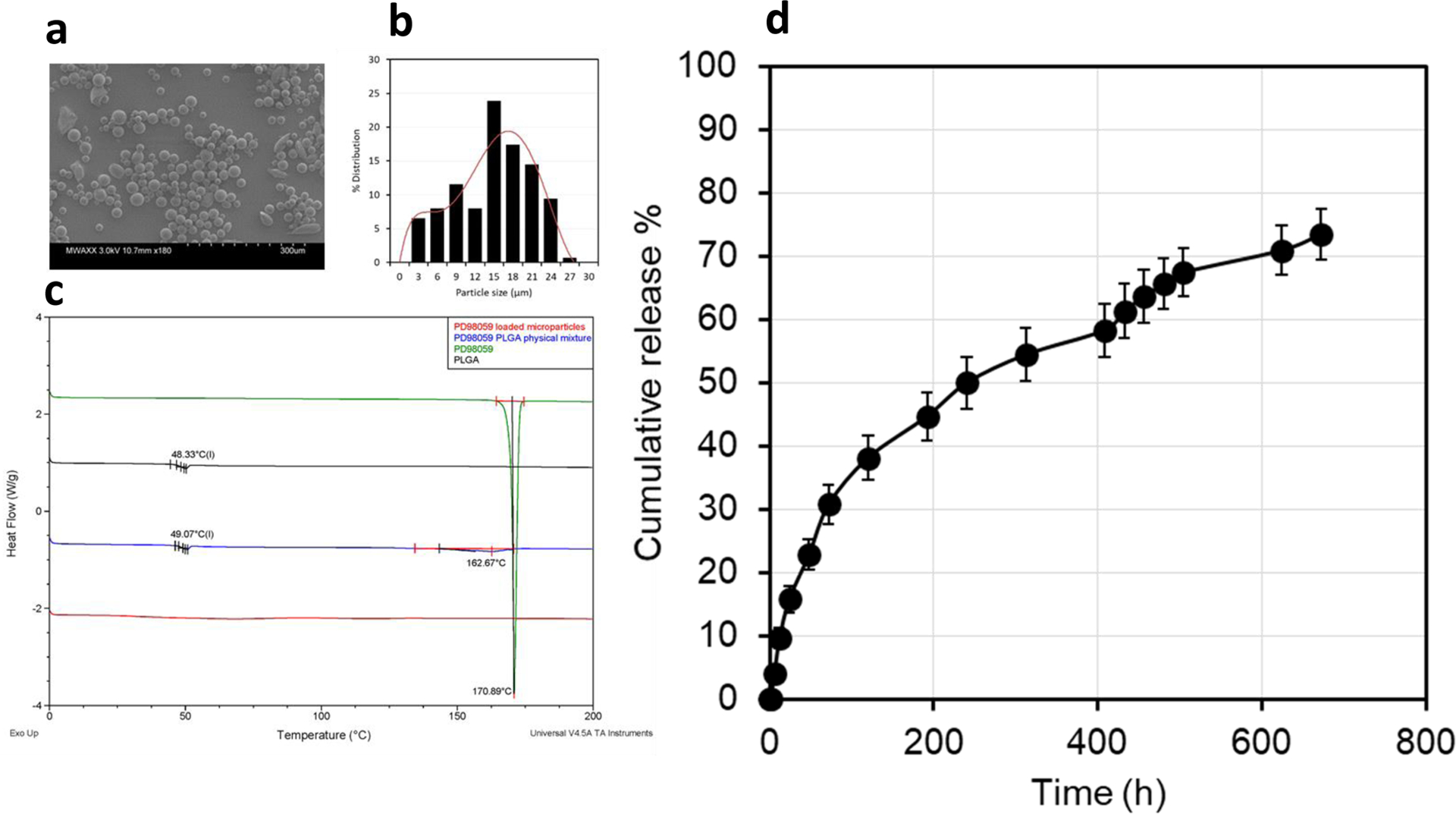
Characterization of the prepared PD98059-loaded PLGA microparticles. (a) Scanning electron microscopy (SEM) image of the drug-loaded microparticles. (b) Particle size distribution, measured by ImageJ analysis of 100 particles in SEM images. (c) Differential scanning calorimetry (DSC) thermograms of PD98059 (green), PLGA (black), a physical mixture of the two (blue), and PD98059-loaded microparticles (red). (d) In vitro release profile of PD98059 from PLGA microparticles (data represent mean ± SD, n=3).
Pharmacokinetics of PD98059 following IV injection
The PD98059 HPLC peak came after 3 min, while the retention time of the IS (7-hydroxyflavone) was 3.6 min. The pharmacokinetics of PD98059 were studied after the data were fit to 1-compartment or 2-compartments models using PK Solver add-in [37, 36]. It can be clearly seen that PD98059 pharmacokinetics follow the 2-compartment model (Figure 2a), which was confirmed by the R2 value (0.999 and 0.981 for 2- and 1-compartment models, respectively). Pharmacokinetics parameters are displayed in Table 1. Distribution and elimination half-lives of the drug were approximately 7 and 73 minutes, respectively. After 1 and 3 hours, levels of PD98059 in major organs were also measured (n=3) in order to gain information on tissue distribution and organ drug levels decline. It can be seen that the drug levels in the liver and kidney were comparatively higher than those in the brain and heart, and yet a significant portion of the drug crossed the blood brain barrier. Nevertheless, there was a quick decline in brain PD98059 levels from approximately 410 ng/g to 76 ng/g between 1 hour and 3 hours.
Figure 2:
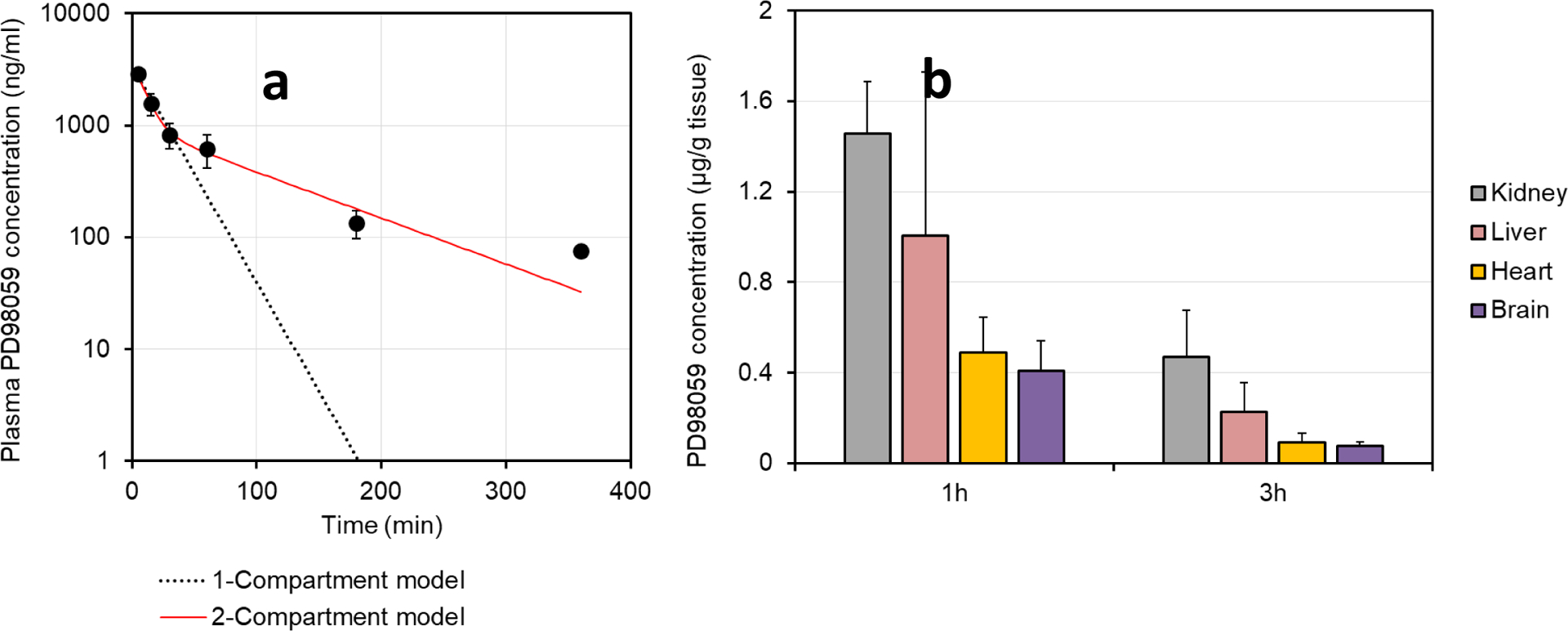
(a) Plasma PD98059 levels vs. time (n=3 for all time points, except 6 h, which has n=1) and (b) organ levels of PD98059 (data represent mean ± SD, n=3) following IV injection of 1 mg of PD98059 dissolved in 10% w/v Tween 80 aqueous solution in healthy rats.
Table 1:
Pharmacokinetics of PD98059 following IV injection.
| Non-compartmental analysis (NCA) | 1-Compartment model | 2-Compartment model | ||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Unit | Value | Parameter | Unit | Value | Parameter | Unit | Value |
| K | min−1 | 0.0074 | C0 | ng/ml | 3433.44 | A | ng/ml | 3236.5 |
| T1/2 | min | 93.93 | K | min−1 | 0.045 | α | min | 0.104 |
| C0 | ng/ml | 3863.5 | T1/2 | min | 15.5 | B | ng/ml | 980.76 |
| AUC0-t | ng.min/ml | 142723.37 | V | ml | 291.25 | β | min | 0.0094 |
| AUC0-inf | ng.min/ml | 152980.9 | CI | ml/min | 13.03 | K10 | min−1 | 0.031 |
| AUMC0-inf | ng.min2/ml | 14922241.77 | AUC0-t | ng.min/ml | 76774.82 | K12 | min−1 | 0.05 |
| MRT0-inf | min | 97.54 | AUC0-inf | ng.min/ml | 76774.83 | K21 | min−1 | 0.031 |
| V | ml | 885.82 | AUMC | ng.min2/ml | 1716754 | T1/2α | min | 6.7 |
| CI | ml/min | 6.54 | MRT | min | 22.36 | T1/2β | min | 73.36 |
| Vss | ml | 637.62 | Vss | ml | 291.25 | C0 | ng/ml | 4217.23 |
| R2 | 0.9807 | V | ml | 237.12 | ||||
| CI | ml/min | 7.4 | ||||||
| AUC0-t | ng.min/ml | 131605.22 | ||||||
| AUC0-inf | ng.min/ml | 135064 | ||||||
| AUMC | ng.min2/ml | 11287504.55 | ||||||
| MRT | min | 83.57 | ||||||
| Vss | ml | 618.76 | ||||||
| R2 | 0.999 | |||||||
K Elimination rate constant, T1/2 Plasma half-life, C0 Plasma concentration at 0 time., AUC Area under the plasma concentration-time curve, AUMC Area under the moment curve, MRT Mean residence time, V Volume of distribution, Cl Clearance, Vss Steady state volume of distribution
Pharmacokinetics and brain levels of PD98059 following subcutaneous injection of PD98059-loaded microparticles
In the normal rats treated subcutaneously with PD98059-loaded microparticles, there was a sustained level of PD98059 in the plasma of rats of 50–100 ng/ml over a period of 2 weeks (Figure 3). It was found that the AUC0-t values following the SC injection of 2.4 mg of PD98059 in PLGA microparticles was 22213.1 ng.h/ml, compared with 2378.7 ng.h/ml following IV injection of 1 mg of PD98059 in solution.
Figure 3:
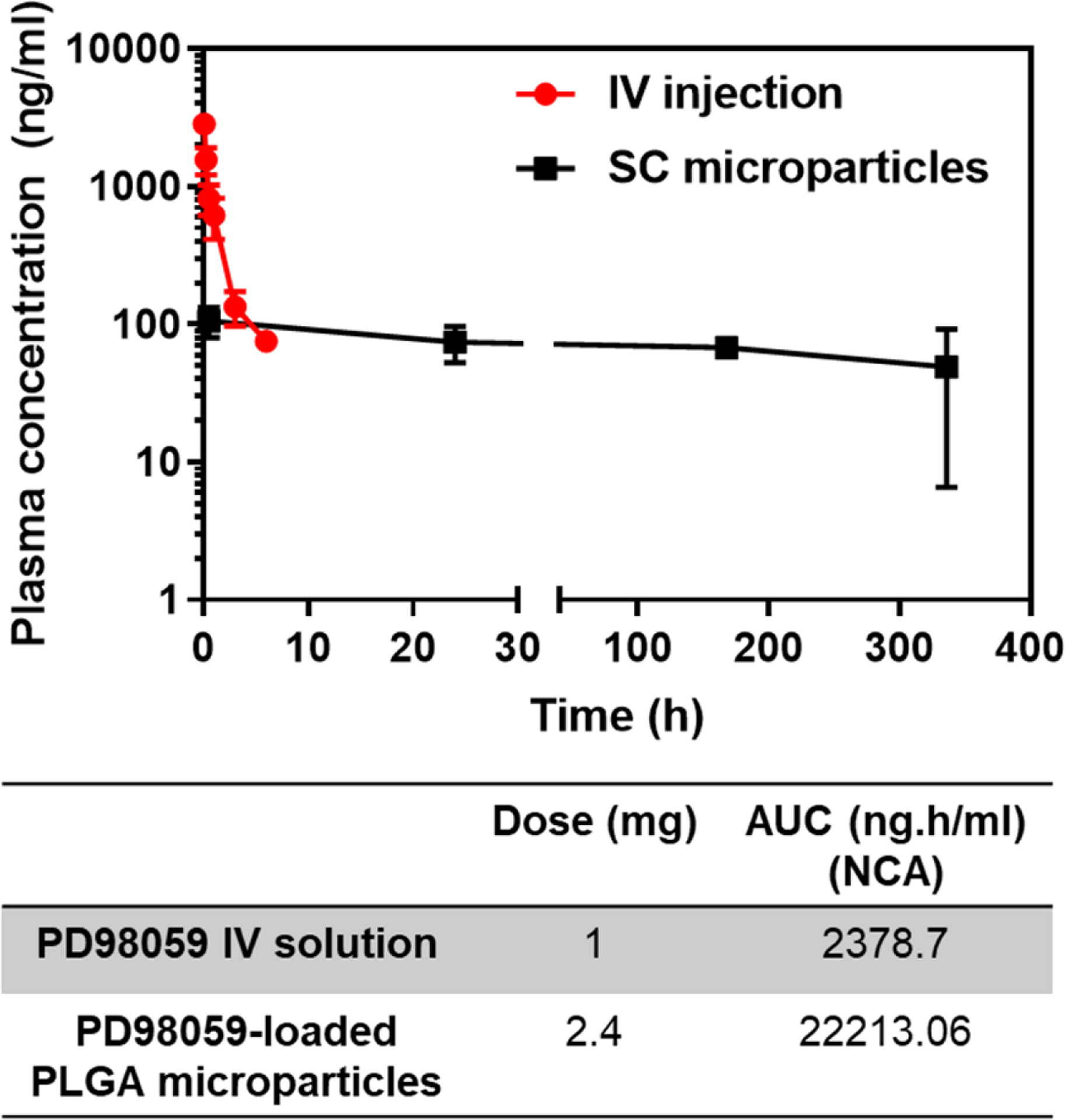
Comparison of Plasma PD98059 levels vs. time curves following IV injection of PD98059 and SC injection of PD98059-loaded PLGA microparticles (data represent mean ± SD, n=3). AUC0-t calculations were based on non-compartmental analysis (NCA) using PK Solver ad-in.
The brain levels of PD98059 in the heart failure rats declined gradually over the course of 2 weeks. The levels in the brain were found to be approximately 37, 16, and 8 ng/g after 1, 7, and 14 days, respectively. The plasma levels were found to be about 7.1, 3.5, and 8.6 ng/ml after 1, 7, and 14 days of injection of the microparticles.
Discussion
Our previous work highlighted the ability of centrally administered PD98059 to lower pERK1/2 levels in the PVN of rats with heart failure, with abrogation of the sympathetic excitation that contributes to further deterioration of cardiac function [21–23]. Such findings pave the way towards development of a new therapeutic modality in drug-based treatments of heart failure and introduces the new concept of using small molecules to target the central nervous system mechanisms driving sympathetic excitation in heart failure. Researchers in the cancer chemotherapy field have also successfully used inhibitors of the MEK-ERK pathway to potentiate existing cancer treatments [38, 39]. In both cases, the short half-life and reversibility of ERK1/2 inhibitory activity of most of these inhibitors have been major problems that have hindered their progression to the clinical applications. These two problems have necessitated either the repeated administration of these agents at high dosing frequencies [39], or the development of MEK-ERK inhibitors with long half-lives [18].
In this research, we successfully formulated a PLGA microparticle formulation capable of slow release of PD98059, which is a specific MEK inhibitor with a versatile application spectrum and high clinical potential in many conditions, including cancer and heart failure. Although there was significant variability in plasma and brain levels of PD98059 in this small number of animals, the findings suggest that PD98059 microparticles have a four-fold higher bioavailability, based on the dose-normalized AUC, compared to IV injection of soluble PD98059 (Figure 3). The rapid decline in drug plasma levels following IV injection (Figure 2) can be explained based on its relatively high volume of distribution, with a t1/2α as short as 6.7 min, and the quite short elimination half-life of 73 min (Table 1). Those two factors were compensated by the continuous slow release of PD98059 from the microparticles at the SC injection site, which guaranteed a steady plasma level in healthy rats (Figure 3). The pharmacokinetics of PD98059 were best fit to a 2-compartment model (Table 1). To maintain sustained levels in the brain of a drug that has a short half-life, it is crucial to maintain a continuous supply. PLGA-based microparticles are commonly used to provide sustained drug release in the body following intramuscular (IM) or SC injection. PLGA is a bulk-eroding polyester from which the drug release usually follows a biphasic pattern, comprising an initial diffusion-based release which extends for a few days to a few weeks (depending on the molecular weight of the polymer), and a subsequent constant release phase explained by erosion of the matrix combined with some contribution from the declining diffusion [40, 41].
The microparticles had an encapsulation efficiency of 43% and an average size of 16.7 μm in diameter. The drug release was monitored over 4 weeks, even though the plasma levels were detected for only two weeks. Plasma drug levels after 3 and 4 weeks could not be detected by HPLC. The in vitro release study showed that 55% of the drug was released within the first two weeks, compared to a further 19% over the next two weeks (between weeks 2 and 4). This marked reduction of drug release rate after the second week may explain the absence of measurable drug levels in the plasma during this time, taking into account the short half-life of the drug.
Ultimately, we hope to determine whether subcutaneous administration of this sustained-release microparticle preparation might be a novel therapeutic approach to modulate the excess ERK1/2 activity in cardiovascular regions of the brain that drive sympathetic excitation in a rat model of heart failure [21, 22]. Heart failure alters the pharmacokinetics of many drugs, mainly BCS class II and IV drugs (i.e. those with poor solubility and good permeation and those with poor solubility and poor permeability, respectively), and absorption of these drugs following oral administration was described as erratic, delayed, and poor [42–44]. Shammas and Dickstein reported that the reduced blood flow to the muscles in congestive heart failure adversely affects the absorption of poorly water soluble drugs and thus their intramuscular administration should be avoided [43]. More importantly, in a study on 46 patients, Ariza-Andraca reported that the rate and extent of absorption of subcutaneously injected insulin was significantly lowered in diabetic patients with generalized edema [45]. The insulin amount absorbed after 6 hours was three to four times less in diabetic patients with edema compared to patients without edema. The authors related this sharp decline in rate and extent to subcutaneous edema [45]. Congestive heart failure is one of the major reasons of peripheral edema [46].
In the present study, plasma levels in rats with established heart failure injected with PD98059-loaded microparticles were lower and more variable than those in healthy rats (Figure 4), which may be a result of decreased subcutaneous absorption in heart failure rats, the use of different rats at different time points, or both. Nevertheless, plasma levels, and of more significance brain levels, were still detectable for up to two weeks. Previously, we treated HF rats with PD98059 solution via ICV for 1 h and 4 weeks and obtained therapeutic effects in both cases. For the short-term experiment (1 h), the dose was 40 μl/h of 20 μM solution of PD98059 for 1 hour. This is equivalent to approximately 214 ng/h. In the long-term experiment (4 weeks), the dose was 0.25 μl/h of 0.6 mM solution of PD98059 for 4 weeks. This is equivalent to approximately 40 ng/h. Our data show that the brain levels are approximately 40 ng/g of brain tissue after 24 h, which makes the amount delivered to the brain (average weight 1.5–2 g) about 60–80 ng. The brain levels declined later, reaching about 8 ng/g after 2 weeks. We are currently optimizing the microparticle formulation to enable the delivery of even higher doses of PD98059, and to maintain constant brain levels for extended periods in HF rats.
Figure 4:
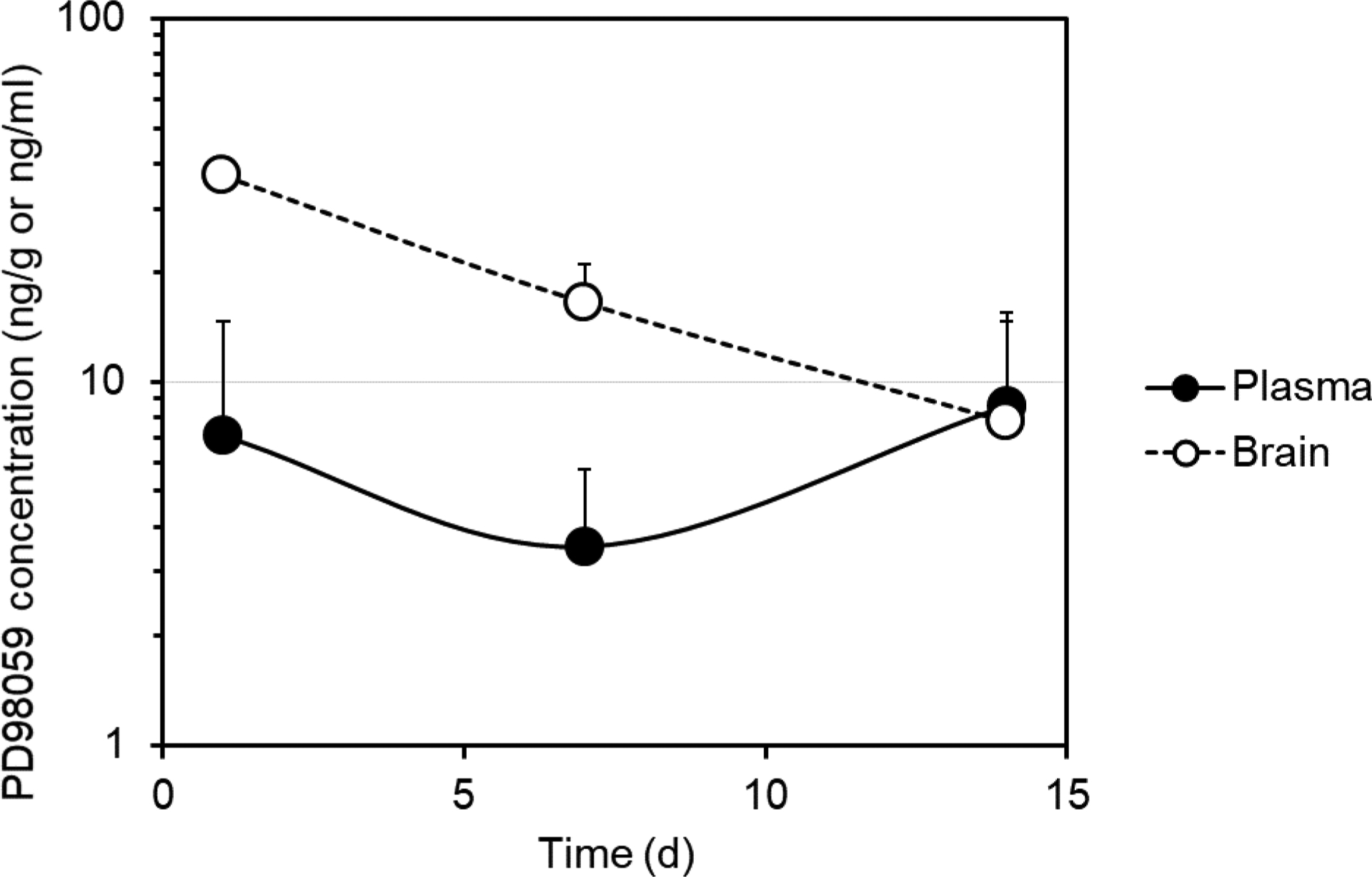
Plasma and brain PD98059 levels vs. time following the SC injection of 3.6 mg of PD98059 in PD98059-loaded PLGA microparticles in rats with heart failure (data represent mean ± SD, n=2–3).
Current heart failure therapy has little impact on central nervous system mechanisms contributing to sympathetic excitation. The prospect of targeting a central pathway regulating sympathetic outflow in heart failure with a systemically administered long-acting drug preparation has clear translational potential.
Despite being widely used in in vitro testing, very few in vivo applications, and no clinical trials have been reported for PD98059. Sufficient pharmacokinetics information are difficult to find/unavailable in the literature. To our knowledge, this is the first report that describes the pharmacokinetics of this drug, and also gives a hint about its biodistribution in major organs. Also, it is worth mentioning that this is one of the very few reports to shed some light on the effect of heart failure on subcutaneous absorption of drug molecules from a slow release microparticles. Further research to optimize the microparticle preparation to provide higher steady-state plasma and brain levels of PD98059 for prolonged periods (1–2 months) and to determine whether these microparticles are capable of producing long-term inhibition of PVN pERK1/2 levels and reducing sympathetic activation in heart failure rats without inducing adverse systemic side effects, is currently ongoing.
Conclusion
PD98059, a potent but reversible MEK inhibitor, has a short elimination half-life, barely above 1 hour, in rats. When combined with a reversible MEK inhibitory activity, a continuous supply of the drug needs to be provided in order to achieve long-term therapeutic goals. PD98059-loaded PLGA microparticles were successfully prepared and characterized using emulsion solvent evaporation technique. The prepared microparticles produced steady plasma levels of PD98059 in rats following SC injection. Detectable levels of PD98059 in the brain were also present for up to two weeks in rats with heart failure, encouraging the further development of this formulation for long-term inhibition of pERK1/2 in brain regions like PVN that contributes to the increased sympathetic nerve activity in heart failure and for use in cancers in which ERK1/2 activity may contribute to progression.
Acknowledgements
This work was supported by the National Institute of Health grants R01-HL-136149 to R.B.F. and S10-OD-019941 to R.M.W., and by the Lyle and Sharon Bighley Professorship (A.K.S.). B.E.G. was supported by the National GEM Consortium, the Alfred P. Sloan Minority Ph.D. Scholarship and the University of Iowa Graduate College Dean’s Fellowship. Graphical abstract was designed using mindthegraph.com.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine reviews. 2001;22(2):153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 3.Cseh B, Doma E, Baccarini M. “RAF” neighborhood: protein-protein interaction in the Raf/Mek/Erk pathway. FEBS letters. 2014;588(15):2398–406. doi: 10.1016/j.febslet.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu PK, Park JI. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Seminars in oncology. 2015;42(6):849–62. doi: 10.1053/j.seminoncol.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaukat A, Barac A, Cross MJ, Offermanns S, Dikic I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Galpha(q) and Galpha(i) signals. Molecular and cellular biology. 2000;20(18):6837–48. doi: 10.1128/mcb.20.18.6837-6848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacology & therapeutics. 2014;141(2):160–71. doi: 10.1016/j.pharmthera.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–93. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncology letters. 2016;12(5):3045–50. doi: 10.3892/ol.2016.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costigan DC, Dong F. The extended spectrum of RAS-MAPK pathway mutations in colorectal cancer. Genes, chromosomes & cancer. 2019. doi: 10.1002/gcc.22813. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nature reviews Cancer. 2018;18(12):767–77. doi: 10.1038/s41568-018-0076-6. [DOI] [PubMed] [Google Scholar]
- 11.Savoia P, Fava P, Casoni F, Cremona O. Targeting the ERK Signaling Pathway in Melanoma. International journal of molecular sciences. 2019;20(6). doi: 10.3390/ijms20061483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang AX, Qi XY. Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB life. 2013;65(9):748–58. doi: 10.1002/iub.1193. [DOI] [PubMed] [Google Scholar]
- 13.Basu S, Harfouche R, Soni S, Chimote G, Mashelkar RA, Sengupta S. Nanoparticle-mediated targeting of MAPK signaling predisposes tumor to chemotherapy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):7957–61. doi: 10.1073/pnas.0902857106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Y, Tian H. Current Development Status of MEK Inhibitors. Molecules. 2017;22(10). doi: 10.3390/molecules22101551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao L, Tian H. Development of ERK1/2 inhibitors as a therapeutic strategy for tumour with MAPK upstream target mutations. Journal of drug targeting. 2019:1–12. doi: 10.1080/1061186X.2019.1648477. [DOI] [PubMed] [Google Scholar]
- 16.Broman KK, Dossett LA, Sun J, Eroglu Z, Zager JS. Update on BRAF and MEK inhibition for treatment of melanoma in metastatic, unresectable, and adjuvant settings. Expert opinion on drug safety. 2019;18(5):381–92. doi: 10.1080/14740338.2019.1607289. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Liu S, Deng J, Akbay EA, Hai J, Ambrogio C et al. Assessing Therapeutic Efficacy of MEK Inhibition in a KRAS(G12C)-Driven Mouse Model of Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(19):4854–64. doi: 10.1158/1078-0432.CCR-17-3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(5):989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 19.Wei SG, Yu Y, Weiss RM, Felder RB. Inhibition of Brain Mitogen-Activated Protein Kinase Signaling Reduces Central Endoplasmic Reticulum Stress and Inflammation and Sympathetic Nerve Activity in Heart Failure Rats. Hypertension. 2016;67(1):229–36. doi: 10.1161/HYPERTENSIONAHA.115.06329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei SG, Yu Y, Weiss RM, Felder RB. Endoplasmic reticulum stress increases brain MAPK signaling, inflammation and renin-angiotensin system activity and sympathetic nerve activity in heart failure. American journal of physiology Heart and circulatory physiology. 2016;311(4):H871–H80. doi: 10.1152/ajpheart.00362.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. American journal of physiology Heart and circulatory physiology. 2009;296(5):H1425–33. doi: 10.1152/ajpheart.00942.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Mitogen-activated protein kinases mediate upregulation of hypothalamic angiotensin II type 1 receptors in heart failure rats. Hypertension. 2008;52(4):679–86. doi: 10.1161/HYPERTENSIONAHA.108.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Angiotensin II-triggered p44/42 mitogen-activated protein kinase mediates sympathetic excitation in heart failure rats. Hypertension. 2008;52(2):342–50. doi: 10.1161/HYPERTENSIONAHA.108.110445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y, Wei SG, Zhang ZH, Weiss RM, Felder RB. ERK1/2 MAPK signaling in hypothalamic paraventricular nucleus contributes to sympathetic excitation in rats with heart failure after myocardial infarction. American journal of physiology Heart and circulatory physiology. 2016;310(6):H732–9. doi: 10.1152/ajpheart.00703.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu Y, Xue BJ, Zhang ZH, Wei SG, Beltz TG, Guo F et al. Early interference with p44/42 mitogen-activated protein kinase signaling in hypothalamic paraventricular nucleus attenuates angiotensin II-induced hypertension. Hypertension. 2013;61(4):842–9. doi: 10.1161/HYPERTENSIONAHA.111.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang ZH, Yu Y, Wei SG, Felder RB. Aldosterone-induced brain MAPK signaling and sympathetic excitation are angiotensin II type-1 receptor dependent. American journal of physiology Heart and circulatory physiology. 2012;302(3):H742–51. doi: 10.1152/ajpheart.00856.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felder RB, Yu Y, Zhang ZH, Wei SG. Pharmacological treatment for heart failure: a view from the brain. Clinical pharmacology and therapeutics. 2009;86(2):216–20. doi: 10.1038/clpt.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang YM, Zhang ZH, Xue B, Weiss RM, Felder RB. Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia-induced heart failure. American journal of physiology Heart and circulatory physiology. 2008;295(1):H227–36. doi: 10.1152/ajpheart.01157.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei SG, Zhang ZH, Yu Y, Weiss RM, Felder RB. Central actions of the chemokine stromal cell-derived factor 1 contribute to neurohumoral excitation in heart failure rats. Hypertension. 2012;59(5):991–8. doi: 10.1161/HYPERTENSIONAHA.111.188086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khaled KA, Sarhan HA, Ibrahim MA, Ali AH, Naguib YW. Prednisolone-loaded PLGA microspheres. in vitro characterization and in vivo application in adjuvant-induced arthritis in mice. AAPS PharmSciTech. 2010;11(2):859–69. doi: 10.1208/s12249-010-9445-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geary SM, Hu Q, Joshi VB, Bowden NB, Salem AK. Diaminosulfide based polymer microparticles as cancer vaccine delivery systems. Journal of controlled release : official journal of the Controlled Release Society. 2015;220(Pt B):682–90. doi: 10.1016/j.jconrel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Intra J, Salem AK. Fabrication, characterization and in vitro evaluation of poly(D,L-lactide-co-glycolide) microparticles loaded with polyamidoamine-plasmid DNA dendriplexes for applications in nonviral gene delivery. Journal of pharmaceutical sciences. 2010;99(1):368–84. doi: 10.1002/jps.21840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lew B, Kim IY, Choi H, Kim KK. Sustained exenatide delivery via intracapsular microspheres for improved survival and function of microencapsulated porcine islets. Drug delivery and translational research. 2018;8(3):857–62. doi: 10.1007/s13346-018-0484-x. [DOI] [PubMed] [Google Scholar]
- 34.Ansary RH, Rahman MM, Awang MB, Katas H, Hadi H, Doolaanea AA. Preparation, characterization, and in vitro release studies of insulin-loaded double-walled poly(lactide-co-glycolide) microspheres. Drug delivery and translational research. 2016;6(3):308–18. doi: 10.1007/s13346-016-0278-y. [DOI] [PubMed] [Google Scholar]
- 35.Andhariya JV, Shen J, Choi S, Wang Y, Zou Y, Burgess DJ. Development of in vitro-in vivo correlation of parenteral naltrexone loaded polymeric microspheres. Journal of controlled release : official journal of the Controlled Release Society. 2017;255:27–35. doi: 10.1016/j.jconrel.2017.03.396. [DOI] [PubMed] [Google Scholar]
- 36.Naguib YW, Lansakara PD, Lashinger LM, Rodriguez BL, Valdes S, Niu M et al. Synthesis, Characterization, and In Vitro and In Vivo Evaluations of 4-(N)-Docosahexaenoyl 2′, 2′Difluorodeoxycytidine with Potent and Broad-Spectrum Antitumor Activity. Neoplasia. 2016;18(1):33–48. doi: 10.1016/j.neo.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Computer methods and programs in biomedicine. 2010;99(3):306–14. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Yao J, Qian C, Shu T, Zhang X, Zhao Z, Liang Y. Combination treatment of PD98059 and DAPT in gastric cancer through induction of apoptosis and downregulation of WNT/beta-catenin. Cancer biology & therapy. 2013;14(9):833–9. doi: 10.4161/cbt.25332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Awasthi N, Monahan S, Stefaniak A, Schwarz MA, Schwarz RE. Inhibition of the MEK/ERK pathway augments nab-paclitaxel-based chemotherapy effects in preclinical models of pancreatic cancer. Oncotarget. 2018;9(4):5274–86. doi: 10.18632/oncotarget.23684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cilurzo F, Selmin F, Minghetti P, Montanari L. Design of methylprednisolone biodegradable microspheres intended for intra-articular administration. AAPS PharmSciTech. 2008;9(4):1136–42. doi: 10.1208/s12249-008-9158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Gao S. Temozolomide/PLGA microparticles and antitumor activity against glioma C6 cancer cells in vitro. International journal of pharmaceutics. 2007;329(1–2):122–8. doi: 10.1016/j.ijpharm.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 42.Ogawa R, Stachnik JM, Echizen H. Clinical pharmacokinetics of drugs in patients with heart failure: an update (part 2, drugs administered orally). Clinical pharmacokinetics. 2014;53(12):1083–114. doi: 10.1007/s40262-014-0189-3. [DOI] [PubMed] [Google Scholar]
- 43.Shammas FV, Dickstein K. Clinical pharmacokinetics in heart failure. An updated review. Clinical pharmacokinetics. 1988;15(2):94–113. doi: 10.2165/00003088-198815020-00002. [DOI] [PubMed] [Google Scholar]
- 44.Nies AS. Clinical Pharmacokinetics in Congestive Heart Failure In: Hosenpud JD, Greenberg BH, editors. Congestive Heart Failure: Pathophysiology, Diagnosis, and Comprehensive Approach to Management. New York, NY: Springer New York; 1994. p. 323–40. [Google Scholar]
- 45.Ariza-Andraca CR, Altamirano-Bustamante E, Frati-Munari AC, Altamirano-Bustamante P, Graef-Sanchez A. Delayed insulin absorption due to subcutaneous edema. Archivos de investigacion medica. 1991;22(2):229–33. [PubMed] [Google Scholar]
- 46.Navas JP, Martinez-Maldonado M. Pathophysiology of edema in congestive heart failure. Heart disease and stroke : a journal for primary care physicians. 1993;2(4):325–9. [PubMed] [Google Scholar]