

Abstract
Folate metabolism enables cell growth by providing one-carbon (1C) units for nucleotide biosynthesis. The 1C units are carried by tetrahydrofolate (THF), whose production by the enzyme DHFR is targeted by the important anticancer drug methotrexate. 1C units come largely from serine catabolism by the enzyme SHMT, whose mitochondrial isoform is strongly upregulated in cancer. Here we report the SHMT inhibitor SHIN2 and demonstrate its in vivo target engagement with 13C-serine tracing. As methotrexate is standard treatment for T-cell acute lymphoblastic leukemia (T-ALL), we explored the utility of SHIN2 in this disease. SHIN2 increases survival in NOTCH1-driven mouse primary T-ALL in vivo. Low dose methotrexate sensitizes Molt4 human T-ALL cells to SHIN2, and cells rendered methotrexate resistant in vitro show enhanced sensitivity to SHIN2. Finally, SHIN2 and methotrexate synergize in mouse primary T-ALL and in a human patient-derived xenograft in vivo, increasing survival. Thus, SHMT inhibition offers a complementary strategy in the treatment of T-ALL.
Keywords: T-ALL, cancer, small molecule inhibitor, serine, folate
Introduction
One-carbon (1C) metabolism, mediated by the folate cofactor, enables cancer growth and proliferation by supporting purine and pyrimidine biosynthesis, as well as amino acid homeostasis (glycine, serine, and methionine), epigenetic maintenance, and redox defense (ref. 1–7). In rapidly proliferating cells including cancer cells, the amino acid serine is the main 1C donor (ref. 4,8–11). The enzyme serine hydroxymethyltransferase (SHMT), which has cytosolic (SHMT1) and mitochondrial (SHMT2) isoforms, catalyzes the conversion of serine and tetrahydrofolate (THF) into glycine and 5,10-methylene-THF (ref. 1,12–14). Consistent with their key role in providing 1C units for DNA synthesis, the 1C/folate metabolism enzymes SHMT2 and the immediately downstream mitochondrial enzyme 5,10-methylene-tetrahydrofolate dehydrogenase (MTHFD2) are among the most consistently overexpressed metabolic enzymes in cancer (ref. 15–17).
Due to the existence of functionally redundant mitochondrial and cytosolic 1C/folate metabolism branches (ref. 1,4), single deletion of key enzymes (e.g. SHMT1, SHMT2, MTHFD1L, MTHFD2) does not block cell proliferation under nutrient replete conditions (ref. 4,18,19). Double deletion of SHMT1/SHMT2 or SHMT1/MTHDF2, however, completely halts proliferation under standard culture conditions in vitro and tumor growth in vivo (ref. 4,18).
Motivated by this genetic evidence, we developed a dual SHMT1/2 inhibitor. Building from a pyrazolopyran scaffold that inhibits plant SHMT, we designed SHIN1, a folate-competitive cell-permeable inhibitor of human SHMT1/2 (ref. 18). SHIN1 demonstrates potent and specific on target activity against SHMT in HCT116 cells and inhibits proliferation across a wide range of cancer cell lines (ref. 18). Due to rapid clearance, however, SHIN1 is not suitable for in vivo studies.
Here we present SHIN2, the first in vivo active mammalian SHMT1/2 inhibitor. We validate the in vitro and in vivo on target activity against SHMT using metabolomics and isotope tracing, and apply SHIN2 in the context of T-cell acute lymphoblastic leukemia (T-ALL). The dihydrofolate reductase (DHFR) inhibitor methotrexate is a standard of care treatment in both pediatric and adult T-ALL (ref. 20,21). Despite advances in treatments, 20–50% of the T-ALL patients relapse. The prognosis of these T-ALL patients remains extremely poor, highlighting the need to discover novel therapeutic approaches (ref. 22,23).
SHIN2 inhibits proliferation in human T-ALL cell lines and has an antileukemic effect in vivo in a mouse model of NOTCH1-driven T-ALL and in a patient derived T-ALL xenograft. Interestingly, SHIN2 shows a synergistic activity when combined with methotrexate both in vitro and in vivo. Moreover, methotrexate-resistant human T-ALL cells showed increased sensitivity to SHIN2. Thus, here we present a novel drug suitable for dual SHMT1/2 inhibition in vivo and demonstrate that targeting SHMT is effective for the treatment of T-ALL, alone or in combination with DHFR inhibition, and might be useful in patients with methotrexate-resistant disease.
Materials and Methods
Cell lines, reagents, constructs and antibodies
Human colon cancer cell line HCT116 and human T-ALL cell lines Molt4, Molt3, Jurkat and KOPT-K1 were obtained from ATCC. HPBALL and DND41 human T-ALL cell lines were obtained from DSMZ (The Leibniz Institute). The CUTLL1 NOTCH1-dependent T-cell lymphoblastic cell line has been previously described (ref. 24). SHMT1 and SHMT2 were knocked out in HCT116 lines using CRISPR/Cas9 nickase method as previously described (ref. 4,18). Adherent cell lines were subcultured in 5% CO2 at 37 °C using DMEM (CellGro 10–017; Mediatech) supplemented with 10% FBS (F2442; Sigma-Aldrich); suspension cell lines were subcultured in 5% CO2 at 37 °C in RPMI-1640 (11875; Gibco) with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. For all experiments, media supplemented with 10% dialyzed FBS was used (F0392; Sigma-Aldrich). Cell lines were regularly tested for mycoplasma. Antibodies were used according to their manufacturers’ directions. Anti-SHMT1 (12612) and SHMT2 (12762) were obtained from Cell Signaling Technologies (1:1,000 dilution). Anti-β-ACTIN (A3854) was obtained from Sigma-Aldrich (1:50,000 dilution). Secondary antibody coupled to horseradish peroxidase (NA934) was obtained from Sigma-Aldrich. Signal was detected using enhanced chemiluminescence (34578, Thermo Scientific).
In vivo target engagement of SHIN2 in mice
Mouse studies followed protocols approved by the Princeton University Institutional Animal Care and Use Committee. Infusion was performed on single-housed 10 – 14 week old male C57BL/6 mice with a catheter surgically implanted on the right jugular vein (Charles River). U-13C-Serine was prepared at 30 mM in saline and infused at 0.1 μL/min/g. (+)SHIN2 was prepared at 20 mg/ml in a 20% 2-hydroxypropyl-β-cyclodextrin solution in water. Mice received either vehicle or a single dose of (+)SHIN2 (200 mg/kg) via an intraperitoneal (IP) injection at the beginning of the experiment. Blood (~10 μL) was collected by tail bleeding in blood collection tubes (Sarstedt 16.443.100), placed on ice for 20 min, and centrifuged at 16,000 g for 10 min at 4 °C to obtain serum and then kept at −80 °C until LC-MS analysis.
In vivo efficacy of SHIN2 in NOTCH1-driven mouse T-ALL and patient derived xenografts
Animals were maintained in specific pathogen-free facilities at the Rutgers Cancer Institute of New Jersey. The Rutgers Institutional Animal Care and Use Committee (IACUC) approved all animal procedures. Generation of NOTCH1-induced T-ALL tumors in mice has been previously described (ref. 25). Animals were randomly assigned to the different treatment groups, and investigators were not blinded to group allocation. Sample size was estimated based on previous reports (ref. 25). For survival studies, leukemia cells expressing a fusion protein consisting of the cherry fluorescent protein fused to luciferase (MigR1-mCherry-Luc) were transplanted from primary recipients into sublethally irradiated C57BL/6 (4.5 Gy) secondary recipients (Taconic Farms) (ref. 25). Animals were monitored for signs of illness, injury or abnormal behavior at least twice daily. Terminally ill leukemic animals were euthanized according to humane endpoints approved by Rutgers IACUC, including: body condition scoring (BCS) < 1,5, rough hair coat, hunched posture, lethargy, abnormal breathing, central nervous system signs (head tilt, spasticity or paralysis) and/or unresponsiveness to external stimuli. For in vivo drug dosing, (+)SHIN2 was dissolved at 20 mg/mL in a 20% 2-hydroxypropyl-β-cyclodextrin solution in water, and methotrexate at 1 mg/mL in PBS. Methotrexate was administered at 10 mg/kg (IP injection), (+)SHIN2 was administered at 200 mg/kg (IP injection). For the investigation of (+)SHIN2 as a single agent, mice were dosed BID with vehicle or (+)SHIN2 for 11 days. For the drug synergism studies in mouse primary T-ALL in vivo, we treated the mice with 4 cycles of intraperitoneal doses of day 1 methotrexate (10 mg/kg) and (+)SHIN2 or vehicle; days 2 to 4 two doses daily of (+)SHIN2 or vehicle, day 5 methotrexate (10 mg/kg) or vehicle, and 2 days off. We evaluated disease progression and therapy response by in vivo bioimaging with the In vivo Imaging System (IVIS, Xenogen). To investigate potential toxicity from the combination of methotrexate and (+)SHIN2 in vivo, healthy C57BL6 mice were treated with 2 cycles of methotrexate alone or in combination with (+)SHIN2 (BID). For the experiment using a human primary leukemia xenograft, we used a previously reported xenograft (PDTALL#10) expressing the cherry fluorescent protein and luciferase (FUW-mCherry-Puro-Luc) (ref. 25); cells were injected into male or female 8 – 10 week old NRG mice (the Jackson Laboratory). Mice were treated with two cycles of 11 days of treatment with (+)SHIN2 (BID) or vehicle. Mice were left off-treatment for 11 days between cycle 1 and cycle 2. Methotrexate (10 mg/kg) was injected the day before each (+)SHIN2 cycle started, and at day 6 of each cycle. We evaluated disease progression and therapy response by in vivo bioimaging with the In vivo Imaging System (IVIS, Xenogen).
Human primary xenografts
T-ALL samples were provided by the University of Padova. Written consent was obtained at study entry and samples were collected under the supervision of local Institutional Review Boards for participating institutions and analyzed under the supervision of Rutgers University.
Statistical Analyses
Statistical analyses were performed with Prism 7.0 (GraphPad) or R (ref. 26). Statistical significance between conditions was calculated using an unpaired two-tailed Student’s t test when comparing two groups or ANOVA followed by Tukey’s post hoc analysis when comparing more than two. Samples sizes, error bars, and p values are defined in each figure legend. Survival in mouse experiments was represented with Kaplan-Meier curves, and significance was estimated with the log-rank test. Synergy in vivo was estimated using a Cox regression introducing an interaction term using a Firth’s penalized maximum likelihood bias reduction method. Synergy in vitro was estimated by representing the IC50 isobolograms.
Additional methods (proliferation assays, cell cycle and apoptosis analyses, flow cytometry analysis of T-cell development, measurement of hematological parameters, cell metabolism studies, metabolite extraction, LC-MS-based untargeted metabolomics analysis, and chemical synthesis of SHIN2) are described in the supplementary information (Supplementary Materials and Methods).
Results
A small-molecule inhibitor of SHMT1/2 with in vivo target engagement.
We previously described SHIN1, a folate-competitive inhibitor of human SHMT1/2 (ref. 18). SHIN1 showed potent and specific cell-based target engagement and inhibited proliferation in a wide range of human cancer cell lines (ref. 18). SHIN1, however, lacked pharmacokinetic properties suitable for in vivo study of SHMT biology. To improve on SHIN1’s half-life, we synthesized a series of related molecules with different substitutions on the phenyl ring, leading to the discovery of an inhibitor with improved pharmacokinetic properties, SHIN2 (Fig. 1a).
Figure 1. SHIN2 inhibits SHMT in vitro and in vivo.
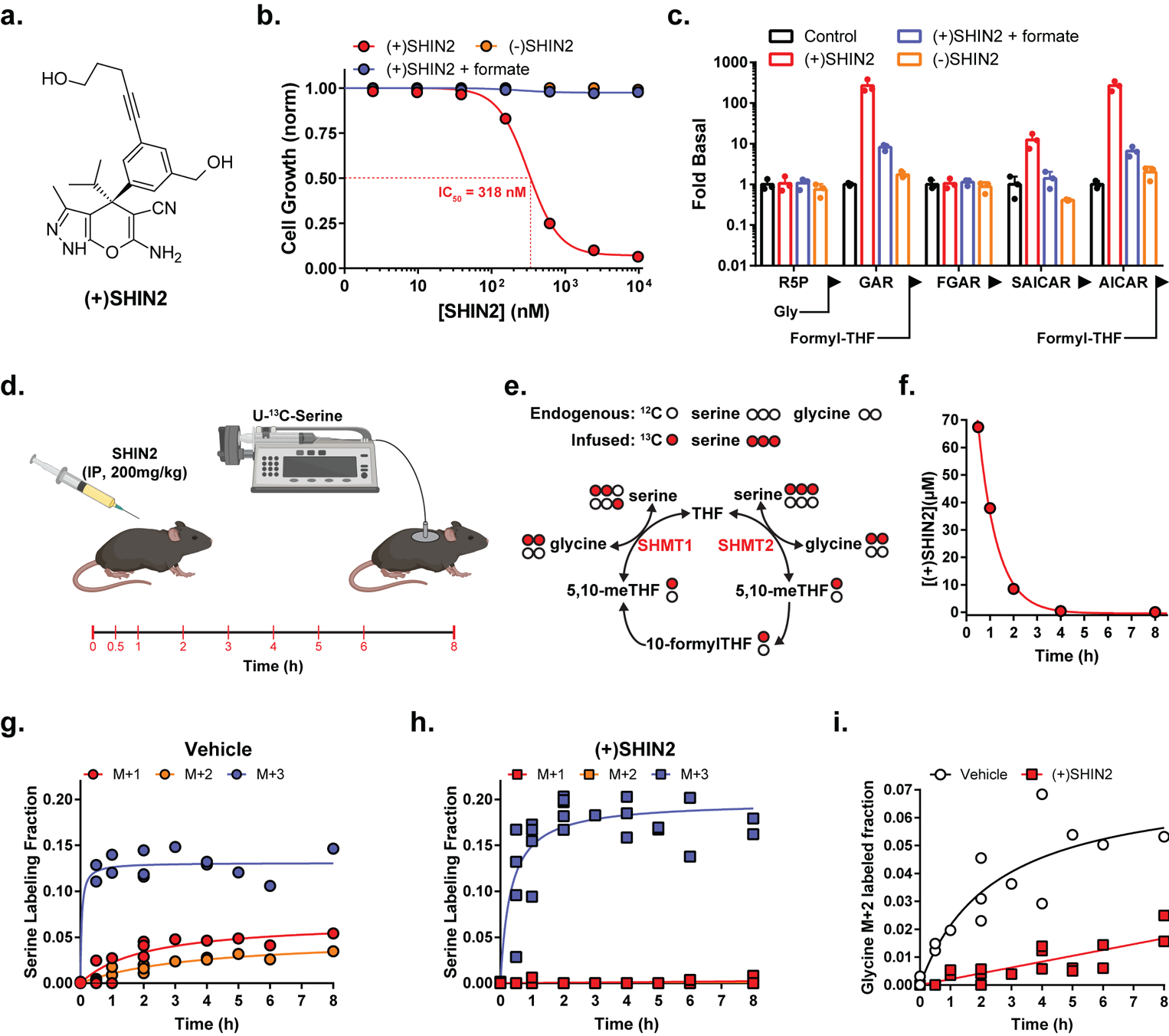
(a) Chemical structure. (b) Growth of HCT116 cells (n=3). (c) Normalized levels of purine biosynthetic pathway intermediates in HCT116 cells (24 h drug exposure at 2 μM) (mean ± SD, n=3). In b and c, formate concentration is 1 mM and (−)SHIN2 is the inactive enantiomer. (d) Experimental design for the analysis of in vivo target engagement using IP delivery of (+)SHIN2 followed by infusion of U-13C-serine. The times in red indicate blood collection. (e) Schematic showing labeling from infused U-13C-serine into glycine and serine via 1C/folate metabolism. (f) Plasma (+)SHIN2 concentration over time after a 200 mg/kg IP dose (mean, n = 2). (g,h) Circulating serine labeling pattern upon vehicle (g) or 200 mg/kg IP (+)SHIN2 (h) administration (n=2). (i) Circulating glycine M+2 fraction upon vehicle or (+)SHIN2 administration (n=2).
We next investigated the activity of SHIN2 in cultured cells. SHIN2 blocked proliferation of HCT116 Ras-driven colon cancer cells, in a stereoselective manner, with a half-maximal inhibitory constant (IC50) of 300 nM for the (+) enantiomer. Proliferation was restored by the addition of 1 mM formate, which provides 1C units independently of SHMT activity (Fig. 1b). Knockout of SHMT2, which is the dominant SHMT isozyme in most cancer cells, but not SHMT1, sensitized HCT116 to (+)SHIN2 (Fig. S1a). Metabolomic analysis of HCT116 cells treated with (+)SHIN2, but not (–)SHIN2, revealed metabolic alterations consistent with SHMT inhibition, which were largely reversed by exogenous formate (Fig. S1b–d). These included dTTP and ATP depletion and serine and purine biosynthetic intermediates buildup, specifically the accumulation of purine intermediates directly preceding 1C-dependent reactions (AICAR, GAR) (Fig. 1c, Fig. S1b).
To evaluate (+)SHIN2 target engagement in vivo, we developed an assay based on continuous infusion of tracer amounts of [U-13C]-serine (which contains three 13C atoms and accordingly is M+3) and monitoring of circulating serine and glycine labeling by mass spectrometry (Fig. 1d). SHMT activity converts M+3 serine into M+2 glycine and M+1 methylene-THF. Due to SHMT reversibility, the M+2 glycine can recombine with an unlabeled 1C unit from methylene-THF to give rise to M+2 serine; similarly, unlabeled glycine can recombine with a labeled 1C unit to give rise to M+1 serine (Fig. 1e). IP administration of 200 mg/kg (+)SHIN2 resulted in micromolar plasma levels, which were sufficient to impair SHMT activity for 8 h, based on decreased circulating M+1 and M+2 serine and M+2 glycine (Fig. 1f–i). Thus, (+)SHIN2 blocks SHMT both in vitro and in vivo.
SHIN2 blocks T-ALL growth by inhibiting SHMT
We then sought tumor types with particular sensitivity to 1C/folate pathway inhibition. Analysis of publicly available data revealed that ALL cell lines show the highest sensitivity, of a wide range of cancer types, for both the DHFR inhibitor methotrexate (ref. 27) (Fig. S2a) and an earlier generation SHMT inhibitor (ref. 18) (Fig. S2b). Moreover, methotrexate is frequently used in the treatment of T-ALL (ref. 20,21). In addition, human T-ALL cell lines show increased expression of SHMT2 compared to normal hematological cells (i.e. human thymus, peripheral blood mononuclear cells and peripheral blood CD4+ T cells) (Fig. S2c). MYC has been previously shown to directly regulate SHMT1/2 (ref. 28–30). Moreover, the NOTCH1-MYC axis is critical for T-ALL growth, as over 60% of T-ALLs harbor activating mutations of NOTCH1 (ref. 31), and one of the critical functions of NOTCH1 in T-ALL generation and progression is to directly activate MYC through the distal N-Me enhancer (ref. 32–34). Thus, it is likely that SHMT2 is overexpressed in T-ALL, at least in part, via NOTCH1-MYC. Consistent with this hypothesis, NOTCH1 inhibition in T-ALL in vivo translates into reduced levels of Shmt1/2, independently of Pten mutational status (ref. 25) (Fig. S2d). Moreover, acute deletion of the N-Me enhancer in mouse T-ALLs in vivo leads to a drastic reduction in Myc levels (ref. 33), which translates into significant downregulation of both Shmt1/2 (Fig. S2e). Therefore, we decided to analyze the effect of (+)SHIN2 in the treatment of T-ALL.
(+)SHIN2 inhibited proliferation across various human T-ALL cell lines (Fig. S2f), and was particularly potent against the human T-ALL cell line Molt4 (IC50 ~ 90 nM) (Fig. 2a), a prototypical T-ALL cell line harboring activating mutations in NOTCH1 (ref. 31). Addition of formate to culture media rescued proliferation of cells treated with (+)SHIN2 (Fig. 2a). Next, analysis of cell proliferation and apoptosis in Molt4 and other T-ALL cell lines treated with (+)SHIN2 revealed that its antiproliferative effect is mainly cytostatic, characterized by a block in S phase of the cell cycle, with little induction of cytotoxicity (Fig. 2b and Fig. S2g–h).
Figure 2. SHIN2 blocks growth of the human T-ALL cell line Molt4 via SHMT inhibition.
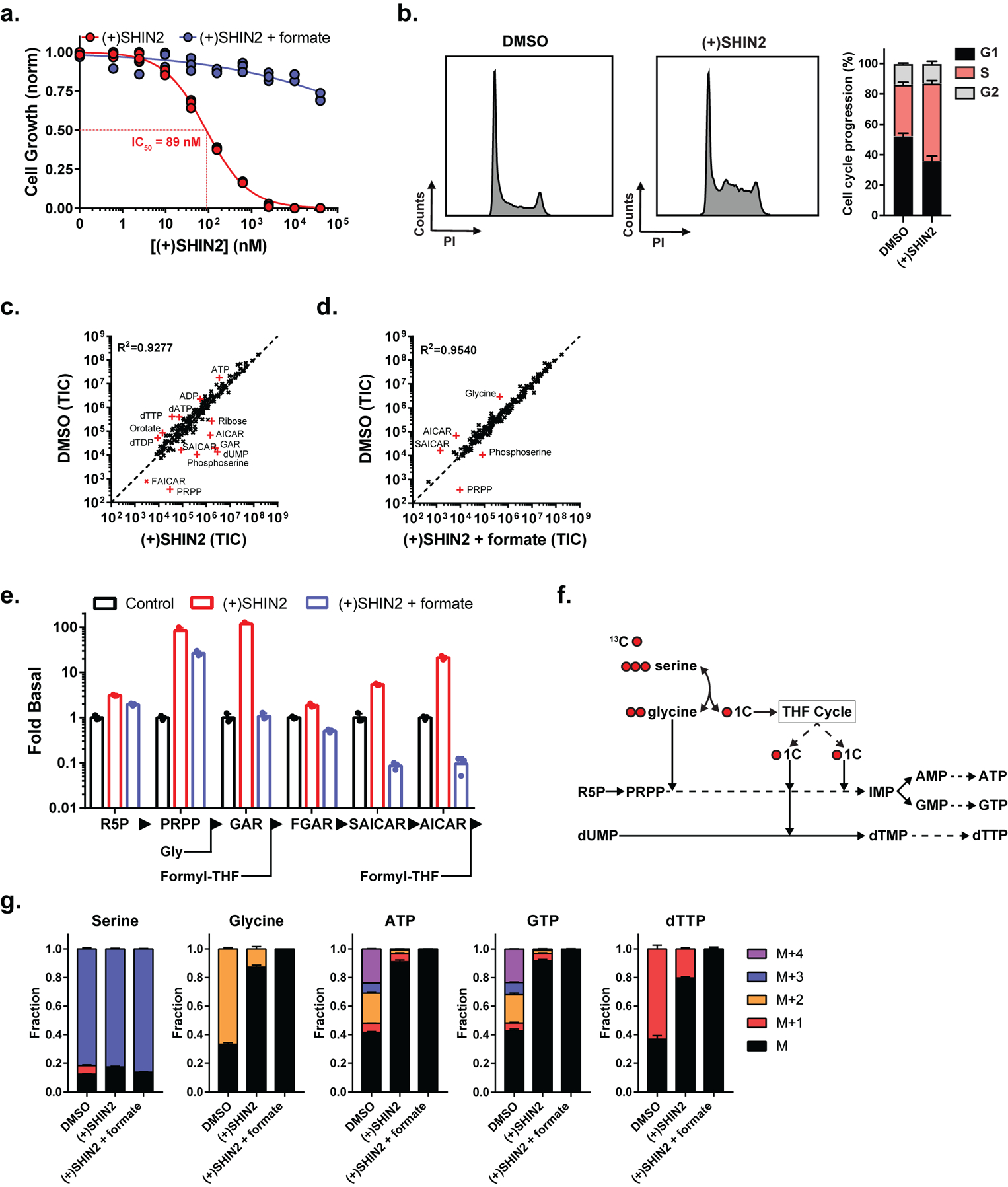
(a) Growth of Molt4 cells (n=3). (b) Analysis of (+)SHIN2 effects on cell cycle in Molt4 cells (48 h after treatment). Representative cell cycle histograms are shown on the panels on the left; quantification is shown on the panel on the right (mean ± SD, n=3). (c-e) Metabolite levels in Molt4 cells (24 h drug exposure) (mean, n=3). In c and d, metabolites displaying a fold-change > 4 are highlighted in red. (f) Schematic showing the incorporation of U-13C-serine-derived carbons into downstream products. (g) Metabolite labeling patterns in Molt4 cells after a 6 h incubation with U-13C-serine (mean ± SD, n=3). (+)SHIN2 concentration is 2 μM, formate concentration is 1 mM.
As in the case of HCT116, untargeted LC-MS analysis of water-soluble metabolites showed that the changes induced by (+)SHIN2 were consistent with on-target SHMT inhibition, and most of them were reversed by formate (Fig. 2c–d, Fig. S2i–j). Interestingly, in T-ALL cell lines, (+)SHIN2 not only induced an increase in the purine biosynthesis intermediates GAR and AICAR, which require 1C units to be further metabolized, but also in phosphoribosyl pyrophosphate (PRPP), which is upstream of a step requiring glycine, which is also a product of the SHMT reaction (Fig. 2e, Fig. S2i–j). Consistent with (+)SHIN2 reducing functional glycine availability, formate addition normalized GAR and AICAR, but not PRPP and induced a more acute decrease in intracellular glycine (Fig. 2c–e, Fig. S2i–j).
Inhibition of cellular SHMT activity can be further monitored by isotope tracing using [U-13C]-serine as a tracer (Fig. 2f). (+)SHIN2 achieved a nearly complete blockade of SHMT activity as evidenced by the decrease in M+1 and M+2 serine, M+2 glycine, and the incorporation of serine-derived glycine and 1C units into ATP, GTP and dTTP (M+1 – M+4 ATP and GTP and M+1 dTTP) (Fig. 2g). Consistent with SHMT activity being the main route to intracellular glycine in T-ALL cell lines (as opposed to glycine uptake), M+2 glycine was predominant in Molt4 cells incubated with [U-13C]-serine (Fig. 2g). Thus, (+)SHIN2 exerts potent antiproliferative effect in human T-ALL cell lines through SHMT inhibition and associated depletion of both 1C units and intracellular glycine.
SHIN2 shows therapeutic activity in mouse primary T-ALL in vivo
SHIN2 showed potent antiproliferative effect against human T-ALL cell lines (Fig. 2 and S2) and in vivo SHMT target engagement over several hours from a single dose (Fig 1g–i) so we decided to test its antitumor properties in vivo. We first analyzed possible drug-induced toxicity in healthy mice treated with (+)SHIN2 (200 mg/kg BID, IP) for 11 consecutive days. SHIN2 administration did not impact total body weight (Fig. S3a). Similar to other anti-folates, it decreased hematological populations, including neutrophils, lymphocytes, monocytes and eosinophils, albeit to a lesser extent than methotrexate (Fig. S3b). All of these parameters returned to normal once the treatment was discontinued (Fig. S3b). We also analyzed T-cell development in these mice and, consistent with the effects of SHMT inhibition in T-cell proliferation (ref. 35), (+)SHIN2 treatment decreased thymus weight and cellularity, which normalized after treatment discontinuation (Fig. S4a). This phenotype reflected decreased thymocyte numbers across all populations (including double negative, CD4/CD8-double positive, mature CD4-single positive and mature CD8-single positive), rather than alteration of any specific thymocyte subpopulation (Fig.S4b–c). Thus, (+)SHIN2 is generally well tolerated with modest hematological toxicity.
NOTCH1 is the main oncogenic driver in T-ALL, as ~60% of patients show activating mutations in NOTCH1 (ref. 31). To model T-ALL in the mouse, we retrovirally transduced a GFP-expressing L1601P-ΔPEST-NOTCH1 (an oncogenic form of NOTCH1 that occurs in human T-ALL patients) into mouse bone marrow progenitor cells followed by transplantation in secondary recipients (ref. 25,36). This well-established model leads to T-ALL development in mice with high penetrance (ref. 37) and NOTCH1-induced T-ALL models have been previously used to uncover the role and the therapeutic effects of a wide variety of targets in this disease (ref. 25,38,39). Once the primary recipient mice developed leukemia, NOTCH1-driven leukemic cells were engineered to express luciferase (ref. 25) and transplanted into a secondary cohort of mice which were treated with vehicle or (+)SHIN2 (200 mg/kg BID, IP) for 11 consecutive days. Notably, (+)SHIN2 treatment significantly decreased tumor burden soon after treatment initiation, as assessed by either in vivo bioimaging (Fig. 3a–b) or FACS detection of leukemic GFP-positive cells in peripheral blood (Fig. 3c). Moreover, (+)SHIN2 treatment translated into extended survival, from median 16 days in control mice to 27 days in the (+)SHIN2 treated group (Fig. 3d). Thus, targeting of SHMT with (+)SHIN2 shows efficacy in primary mouse T-ALL.
Figure 3. SHIN2 has an antileukemic effect in T-ALL in vivo.
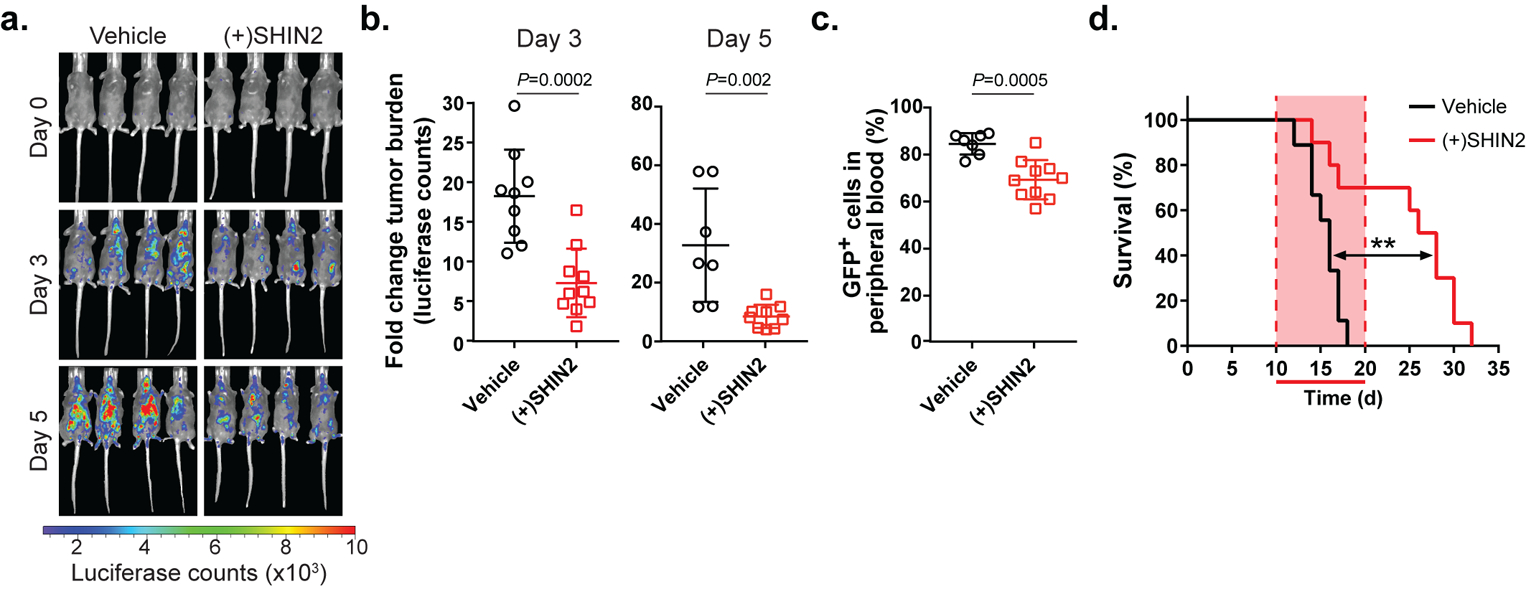
(a-b) Representative images from treated mice (a) and quantification (b) of changes in tumor burden at day 3 and day 5 post-treatment initiation with (+)SHIN2 (200 mg/kg, BID) as assessed by bioimaging in mice allografted with NOTCH1-induced mouse leukemia cells (n=9 for vehicle; n=10 for (+)SHIN2). P values were calculated using a two-tailed unpaired Student’s t-test. (c) Changes in leukemic burden at day 4 post treatment initiation with (+)SHIN2 (200 mg/kg, BID) as assessed by FACS detection of leukemic GFP-positive cells in peripheral blood (n=9 for vehicle; n=10 for (+)SHIN2). P value was calculated using a two-tailed unpaired Student’s t-test. (d) Kaplan-Meier survival curves of mice harboring NOTCH1-induced mouse T-ALL treated with vehicle or (+)SHIN2 (200 mg/kg) for 11 days (log-rank test; **P<0.01) (n=9 for vehicle; n=10 for (+)SHIN2).
Synergism between SHIN2 and methotrexate in human T-ALL cell lines
DHFR and SHMT carry out sequential enzymatic reactions in the pathway from dihydrofolate to methylene-THF. Accordingly, we were curious if inhibition of both steps might have therapeutic benefits. In the human T-ALL cell line Molt4, the combination of methotrexate and (+)SHIN2 suppressed proliferation more strongly than either drug alone (Fig. 4a and Fig S5a), with methotrexate decreasing the IC50 for (+)SHIN2 (Fig. 4b) and the IC50 isobologram confirming synergy (Fig. 4c and Fig. S5a–c). Synergy between (+)SHIN2 and methotrexate was also observed in Molt3 and Jurkat human T-ALL cell lines (Fig S5d–e).
Figure 4. SHIN2 and methotrexate synergize in Molt4 cells.
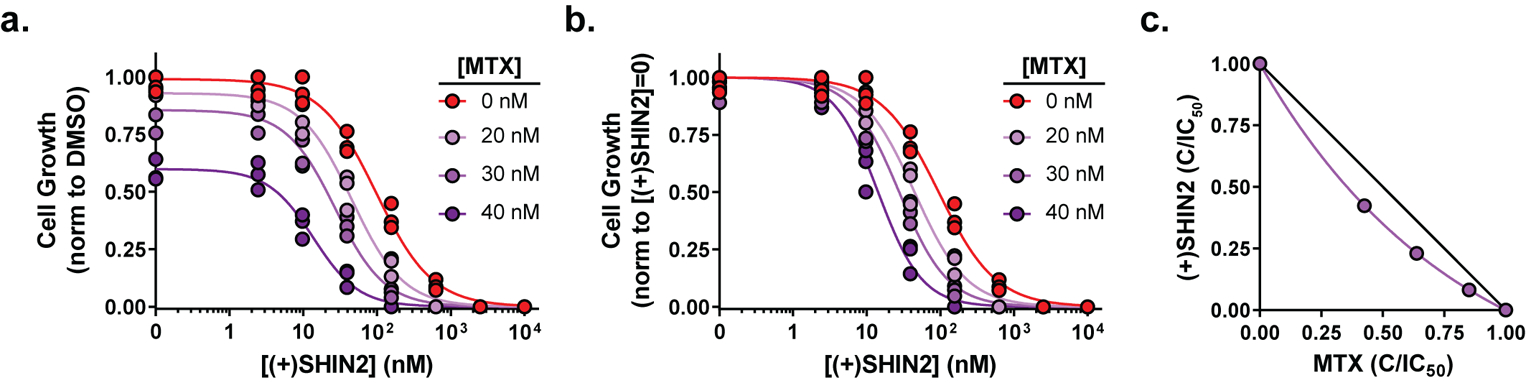
(a-b) Growth of Molt4 cells incubated with increasing concentrations of (+)SHIN2 in the presence of 0, 20, 30 and 40 nM methotrexate (MTX) normalized to DMSO control proliferation (a) or to proliferation for the same methotrexate dose in the absence of (+)SHIN2 (b) (n=3). (c) Isobologram for (+)SHIN2 and methotrexate showing the combinations of the drug that achieve a decrease in proliferation of > 50%; purple, actual values; black line, theoretical additive effect.
Synergism between SHIN2 and methotrexate in mouse T-ALL in vivo
We then decided to test the in vivo antitumor effect of the combination of (+)SHIN2 and methotrexate in our NOTCH1-driven murine T-ALL model. Here, we decided to implement a less aggressive dosage regimen to reduce the probability of potential toxicity observed in mice treated with both agents. Specifically, a 5-days ON and 2-days OFF treatment schedule with (+)SHIN2 dosed at 200 mg/kg and methotrexate at 10 mg/kg (as shown in Fig. 5a) did not lead to noticeable toxicity in healthy wild-type mice after two rounds of treatment, either in body weight or hematological parameters (Fig. S6a–b) and was therefore selected for the subsequent experiment in leukemic mice in vivo. Mice transplanted with NOTCH1-induced primary T-ALL were treated for 4 weeks with (+)SHIN2 and methotrexate, alone or in combination. This treatment significantly reduced tumor burden compared to vehicle treated mice, as measured by in vivo bioimaging after one cycle of treatment (Fig. 5b–c). Importantly, long-term treatment with the (+)SHIN2 and methotrexate combination significantly increased leukemic mice survival as compared to vehicle alone or either drug used in monotherapy (Fig. 5d). To evaluate the occurrence of a synergistic or an additive effect, we fit the data introducing an interaction term using a Firth’s penalized maximum likelihood bias reduction method for Cox regression, obtaining a significant non-zero coefficient for the interaction term (Fig. 5e). These results demonstrate that methotrexate and (+)SHIN2 synergize in prolonging survival in mouse T-ALL.
Figure 5. Synergistic in vivo antileukemic effect of SHIN2 and methotrexate in mouse primary leukemia.
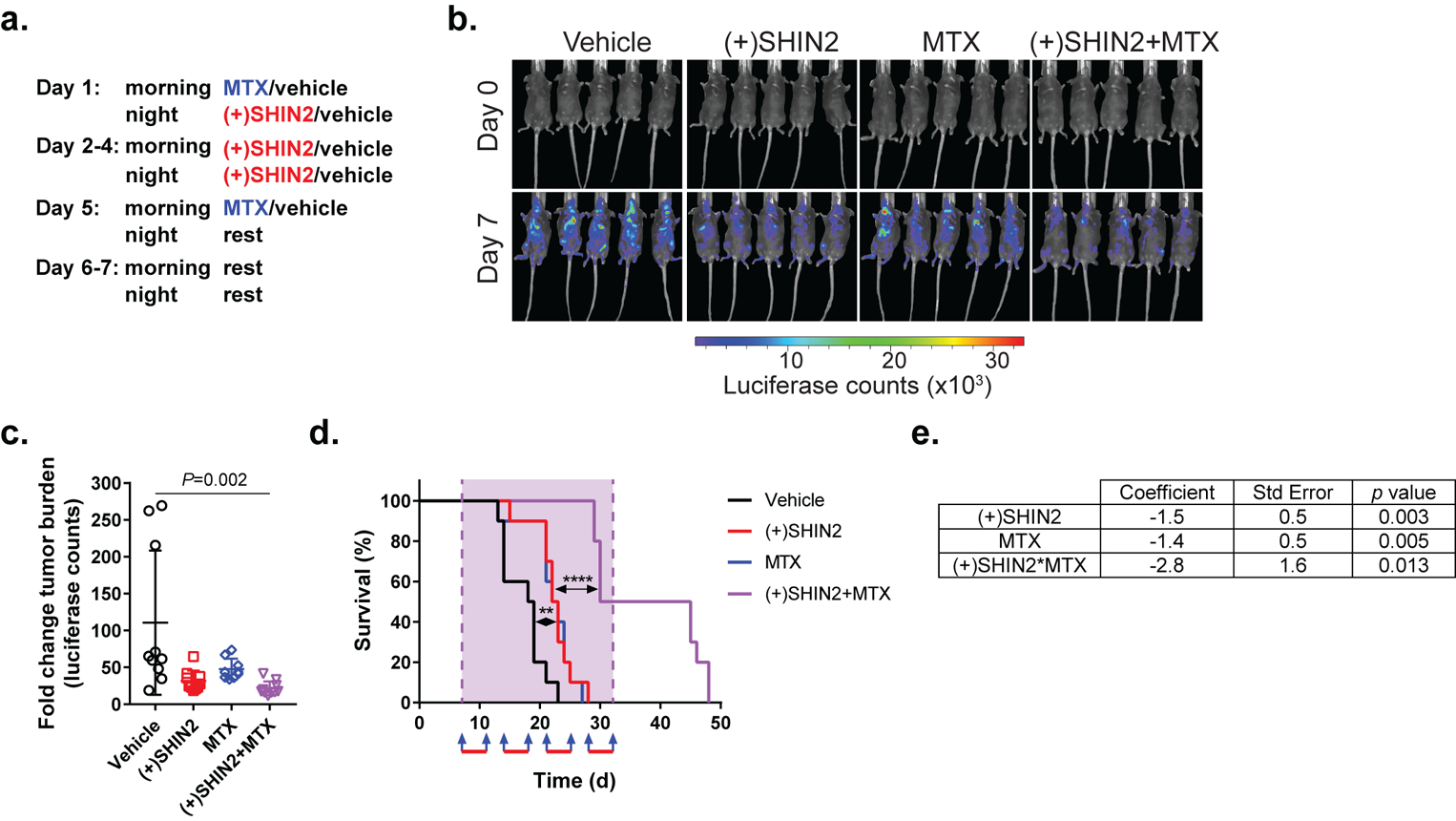
. (a) Dosage regimen for each of the 4 cycles of treatment. Methotrexate was administered at 10 mg/kg (IP injection), (+)SHIN2 was administered at 200 mg/kg (IP injection). (b-c) Representative images from five treated mice (b) and quantification (c) of changes in tumor burden as assessed by bioimaging in mice allografted with NOTCH1-induced mouse leukemia cells after one treatment cycle (n=10 for all groups). P value was calculated using one-way ANOVA testing. (d) Kaplan-Meier survival curves (n=10 for all groups, log-rank test; **P<0.01, ****P<0.001). Blue arrows represent methotrexate injection. Red bars represent days under (+)SHIN2 treatment. (e) Results of a Cox regression with Firth’s penalized likelihood including the terms for (+)SHIN2, methotrexate and the interaction between (+)SHIN2 and methotrexate. For each parameter the actual value of the coefficient, the standard error and the p value (null hypothesis coefficient = 0) are shown.
Synergism between SHIN2 and methotrexate in a patient-derived T-ALL xenograft in vivo
Next, we tested the efficacy of the combination therapy of (+)SHIN2 and methotrexate in a patient-derived T-ALL xenograft (PDX) in vivo. Mice transplanted with a luciferase-expressing T-ALL PDX (PDTALL#10; (ref. 25)) were treated on a schedule resulting from the combination of our previously used regimes (described in Materials and Methods). Consistent with our previous results in mouse primary leukemias (Fig. 5), (+)SHIN2 treatment alone or in combination with methotrexate led to decreased tumor burden as assessed by in vivo bioimaging (Fig. 6a–b). Moreover, treatment with (+)SHIN2 as single agent significantly increased survival of PDX-bearing mice, and combination of (+)SHIN2 with methotrexate led to a synergistic effect with further extended survival (Fig. 6c). These results demonstrate that methotrexate and (+)SHIN2 synergize in prolonging survival in mice harboring a patient-derived T-ALL xenograft in vivo.
Figure 6. Synergistic in vivo antileukemic effect of SHIN2 and methotrexate in a human patient-derived T-ALL xenograft.
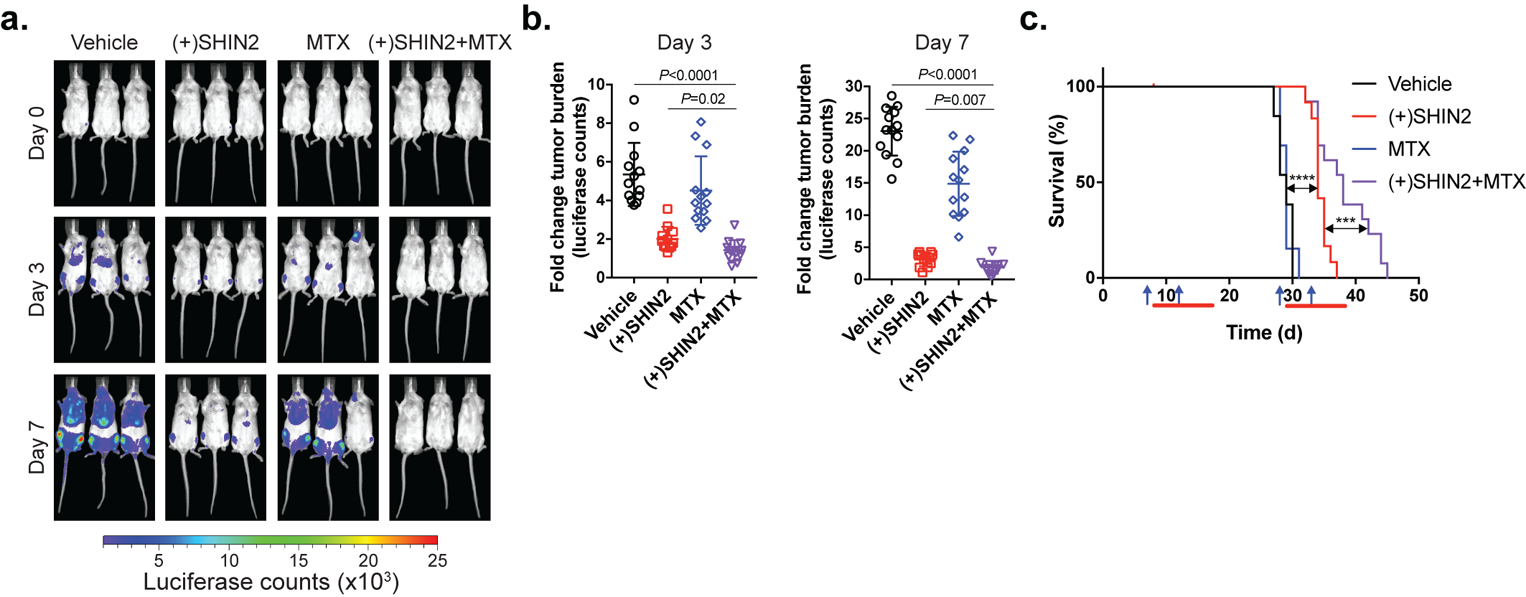
(a-b) Representative images from treated mice (a) and quantification (b) of changes in tumor burden as assessed by bioimaging in mice xenografted with a human patient-derived T-ALL xenograft at day 3 or day 7 after treatment initiation (n=12 for (+)SHIN2 and n=13 for the other groups). P value was calculated using one-way ANOVA testing, p value for (+)SHIN2 vs (+)SHIN2 + MTX comparison was calculated using Tukey’s multiple comparisons test. (c) Kaplan-Meier survival curves (n=12 for (+)SHIN2 and n=13 for the other groups, log-rank test; ***P<0.005, ****P<0.001). Blue arrows represent methotrexate injection. Red bars represent days under (+)SHIN2 treatment.
Methotrexate resistance sensitizes Molt4 cells to SHMT inhibition
Resistance to chemotherapy is an important clinical challenge in T-ALL (ref. 22,23). Given the favorable in vivo efficacy of the SHIN2-methotrexate combination, we decided to develop methotrexate-resistant Molt4 cells to test whether such cells would retain sensitivity to SHMT inhibition. To this end, Molt4 cells were cultured in increasing doses of methotrexate until we achieved a derived-cell line that showed a > 2-fold increase in IC50 for methotrexate (Fig. 7a). Surprisingly, methotrexate-resistant cells not only remained sensitive to SHMT inhibition, but actually showed enhanced sensitivity to (+)SHIN2 (4-fold decrease in IC50; Fig. 7b). Thus, SHMT inhibition represents a new metabolic vulnerability in methotrexate-resistant T-ALL cells.
Figure 7. Methotrexate resistance sensitizes Molt4 cells to SHIN2.
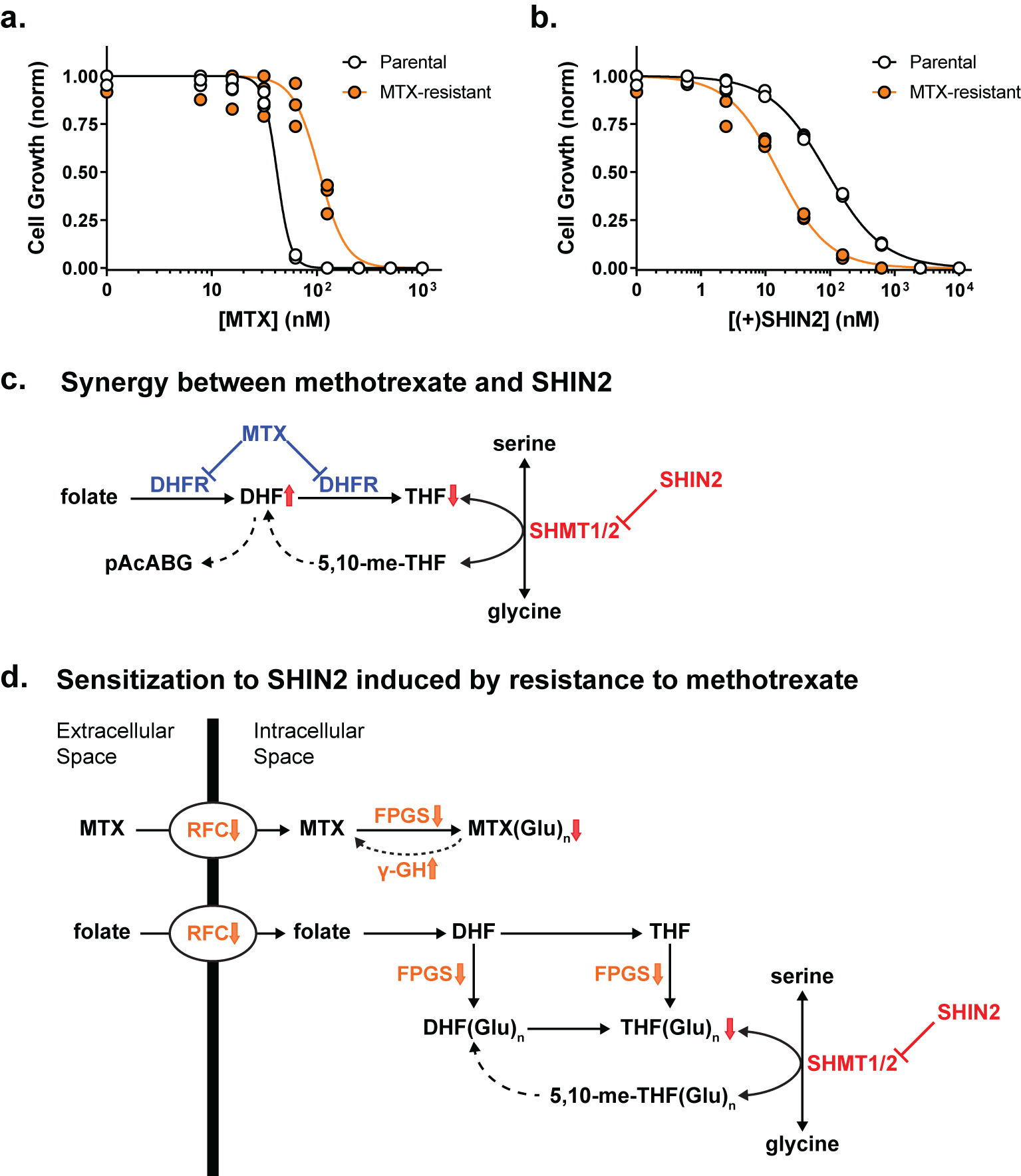
(a-b) Growth of parental or methotrexate (MTX)-resistant Molt4 cells incubated with increasing concentrations of methotrexate (a) or (+)SHIN2 (b) (n=3). (c) Proposed mechanism for the synergy between methotrexate and SHIN2 in T-ALL. DHFR inhibition by methotrexate decreases intracellular THF and thereby sensitizes cells to SHMT inhibition. (d) Proposed mechanism by which methotrexate resistance sensitizes to SHIN2. Decreases in folate import and polyglutamation (steps highlighted in orange) promote methotrexate resistance by decreasing intracellular polyglutamated-methotrexate (MTX(Glu)n), but also decrease polyglutamated-THF (THF(Glu)n), depleting the substrate of the SHMT reaction and thereby sensitizing the cells to SHIN2.
Discussion
Inhibition of folate metabolism and/or nucleotide biosynthesis is an important anticancer strategy. The main antifolates in current clinical practice are methotrexate, which primarily targets DHFR, and pemetrexed, which primarily targets thymidylate synthase (5-FU, acting as a dUMP analogue, also inhibits thymidylate synthase) (ref. 1,40). The product of DHFR is THF, while the substrate of thymidylate synthase is methylene-THF. The intervening chemical reaction, converting THF into methylene-THF, is carried out by SHMT, which uses serine as the one-carbon donor and makes glycine as an additional product. Given that SHMT sits in the folate pathway directly between these two valuable anticancer targets and that the mitochondrial isozyme SHMT2 is among the most consistently upregulated genes in cancer (ref. 16), there is strong rationale for exploring SHMT inhibition.
To this end, we developed the in vivo active SHMT inhibitor SHIN2. Three metabolic hallmarks of selective SHMT inhibition are (i) accumulation of purine intermediates immediately upstream of 10-formyl-THF requiring reactions (GAR, AICAR); (ii) blockade of passage of serine derived carbon into purines or dTTP as assayed by isotopic incorporation; and (iii) rescue of these effects by the soluble 1C-donor formate (ref. 4,18). SHIN2 induced these hallmarks of SHMT inhibition, suggesting on-target activity without significant off-target activity against other folate pathway enzymes.
To assess pharmacodynamic effects, it is convenient to monitor circulating metabolites, rather than relying on invasive tissue sampling. To this end, we took advantage of the fact that SHMT both synthesizes glycine and “scrambles” uniformly 13C-labeled serine into partially labeled serine (ref. 4). Serine and glycine are abundant in the circulation and rapidly exchange between the circulation and cells, which allowed us to monitor SHMT inhibition by infusing [U-13C]-serine and monitoring circulating serine and glycine labeling. In this manner, we were able to confirm prolonged (~ 8 h) in vivo SHMT inhibition by SHIN2.
Having proved the SHMT inhibitory effects of SHIN2 in vivo, we next decided to explore the potential therapeutic effect of this inhibition in T-ALL. These studies were motivated by (i) the sensitivity of ALL to folate/1C metabolism inhibition in vitro (ref. 18,27); (ii) the clinical use of methotrexate as standard of care for T-ALL therapy (ref. 20,41); (iii) resistance to methotrexate being a cause of treatment failure with limited alternative therapies available (ref. 20,21,41,42); and (iv) the possibility for methotrexate, by depleting one of the SHMT substrates (THF) to sensitize cancer cells to SHMT inhibition (Fig. 7c), much as dietary-induced 1C depletion can sensitize to 5-FU (ref. 43–45). Encouragingly, we observed single agent efficacy of SHIN2 as well as synergistic interaction of SHIN2 with methotrexate in decreasing tumor burden and extending survival in both mouse primary leukemias and human patient-derived T-ALL xenografts.
A striking feature of cells rendered genetically defective in mitochondrial folate metabolism is glycine auxotrophy. These cells can no longer produce sufficient glycine internally and rely on glycine uptake instead (ref. 4,14,19,46–50). While some solid tumor cell lines are very effective at glycine uptake (ref. 5,18), we have previously found that diffuse large B cell lymphoma are deficient in glycine uptake and thus sensitive to SHMT inhibition even in the presence of exogenous formate (ref. 18). In T-ALL cells in culture, isotope tracing shows that a majority of glycine is made by SHMT, but nevertheless formate rescues proliferation defects induced by SHMT inhibition. Thus, at least in cell culture, lack of 1C units seems to be the primary deficiency; however, glycine deficiency may also play a role in vivo.
Importantly, in addition to synergizing with methotrexate, SHIN2 is particularly effective in cells rendered resistant to methotrexate. Mechanisms of resistance to antifolates in general, and methotrexate in particular, include decreased transport mediated by the reduced folate carrier (RFC, SLC19A1), altered polyglutamylation due to decreased folylpolyglutamate synthetase (FPGS) activity or increased gamma glutamyl hydrolase activity (γ-GH), increased DHFR or TS activity, and mutations in DHFR (ref. 40,51). Unlike methotrexate and pemetrexed, SHIN2’s structure is distinct from folate and its activity does not depend on polyglutamation. A logical possibility is that decreases in folate transport or polyglutamation, which develop in response to methotrexate exposure, decrease the intracellular pool of THF and thereby render lower doses of SHIN2 effective in blocking SHMT (Fig. 7d). The cross-sensitization between methotrexate and SHMT inhibition renders the contemporaneous or sequential use of these agents a promising approach for leukemia treatment.
Supplementary Material
Acknowledgements.
We thank members of the Rabinowitz and Herranz laboratories for helpful discussions. We also thank Adolfo A. Ferrando (Columbia University) for sharing with us the human normal thymus samples used in this study. Work in the laboratory of J. D. Rabinowitz is supported by US National Institutes of Health (1DP1DK113643 and R01CA163591, to J. D. Rabinowitz and R00CA215307 to G. S. Ducker) and the Rutgers Cancer Institute of New Jersey (P30CA072720). J. C. Garcia-Cañaveras is supported by funding from the European Union’s Horizon 2020 research and innovation program (Marie Sklodowska-Curie grant agreement No 751423). Work in the laboratory of D. Herranz is supported by the US National Institutes of Health (R00CA197869 and R01CA236936), a Research Scholar Grant from the American Cancer Society (RSG-19161-01-TBE), the Alex’s Lemonade Stand Foundation, the Leukemia Research Foundation, the Children’s Leukemia Research Association, the Gabrielle’s Angel Foundation for Cancer Research and the Rutgers Cancer Institute of New Jersey (P30CA072720). V.da Silva-Diz is funded by the New Jersey Commission on Cancer Research (DCHS19PPC008).
Footnotes
Conflict of Interest Disclosures. G. S. Ducker, J. M. Ghergurovich, H. Kim, and J. D. Rabinowitz are inventors on a Princeton University patent covering serine hydroxymethyltransferase inhibitors and their use in cancer. J. D. Rabinowitz is a co-founder of Raze Therapeutics and advisor and stock owner in Kadmon, Agios, L.E.A.F., and Rafael Pharmaceuticals. No competing interests were disclosed by the other authors.
References
- 1.Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab 2017; 25: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014; 510: 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maddocks ODK, Labuschagne CF, Adams PD, Vousden KH. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell 2016; 61: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, Teng X et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab 2016; 23: 1140–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL et al. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science (80-) 2012; 336: 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab 2015; 22: 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer 2016; 16: 650–662. [DOI] [PubMed] [Google Scholar]
- 8.Labuschagne CF, van den Broek NJF, Mackay GM, Vousden KH, Maddocks ODK. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 2014; 7: 1248–1258. [DOI] [PubMed] [Google Scholar]
- 9.Fu TF, Schirch V, Rife JP. The role of serine hydroxymethyltransferase isozymes in one-carbon metabolism in MCF-7 cells as determined by 13C NMR. Arch Biochem Biophys 2001; 393: 42–50. [DOI] [PubMed] [Google Scholar]
- 10.Herbig K, Chiang EP, Lee LR, Hills J, Shane B, Stover PJ. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem 2002; 277: 38381–38389. [DOI] [PubMed] [Google Scholar]
- 11.Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013; 493: 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrow TA, Brenner AA, Whitehead VM, Chen XN, Duncan RG, Korenberg JR et al. Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J Biol Chem 1993; 268: 11910–11916. [PubMed] [Google Scholar]
- 13.Girgis S, Nasrallah IM, Suh JR, Oppenheim E, Zanetti KA, Mastri MG et al. Molecular cloning, characterization and alternative splicing of the human cytoplasmic serine hydroxymethyltransferase gene. Gene 1998; 210: 315–324. [DOI] [PubMed] [Google Scholar]
- 14.Stover PJ, Chen LH, Suh JR, Stover DM, Keyomarsi K, Shane B. Molecular cloning, characterization, and regulation of the human mitochondrial serine hydroxymethyltransferase gene. J Biol Chem 1997; 272: 1842–1848. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science (80-) 2016; 351: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee GY, Haverty PM, Li L, Kljavin NM, Bourgon R, Lee J et al. Comparative oncogenomics identifies psmb4 and shmt2 as potential cancer driver genes. Cancer Res 2014; 74: 3114–3126. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun 2014; 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ducker GS, Ghergurovich JM, Mainolfi N, Suri V, Jeong SK, Hsin-Jung Li S et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc Natl Acad Sci 2017; 114: 11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel H, Di Pietro E, MacKenzie RE. Mammalian fibroblasts lacking mitochondrial NAD+-dependent methylenetetrahydrofolate dehydrogenase-cyclohydrolase are glycine auxotrophs. J Biol Chem 2003; 278: 19436–19441. [DOI] [PubMed] [Google Scholar]
- 20.Litzow MR, Ferrando AA. How I treat T-cell acute lymphoblastic leukemia in adults. Blood 2015; 126: 833–841. [DOI] [PubMed] [Google Scholar]
- 21.Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood 2017; 129: 1134–1142. [DOI] [PubMed] [Google Scholar]
- 22.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 2012; 122: 3398–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet 2013; 381: 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palomero T, Barnes KC, Real PJ, Glade Bender JL, Sulis ML, Murty VV. et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to γ-secretase inhibitors. Leukemia 2006; 20: 1279–1287. [DOI] [PubMed] [Google Scholar]
- 25.Herranz D, Ambesi-Impiombato A, Sudderth J, Sánchez-Martín M, Belver L, Tosello V et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 2015; 21: 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Core Team. R: A Language and Environment for Statistical Computing. 2019https://www.r-project.org/.
- 27.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013; 41: 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikiforov MA, Chandriani S, O’Connell B, Petrenko O, Kotenko I, Beavis A et al. A Functional Screen for Myc-Responsive Genes Reveals Serine Hydroxymethyltransferase, a Major Source of the One-Carbon Unit for Cell Metabolism. Mol Cell Biol 2002; 22: 5793–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov 2014; 4: 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pikman Y, Puissant A, Alexe G, Furman A, Chen LM, Frumm SM et al. Targeting MTHFD2 in acute myeloid leukemia. J Exp Med 2016; 213: 1285–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weng AP, Ferrando AA, Lee W, Morris IV JP, Silverman LB, Sanchez-Irizarry C et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (80-) 2004; 306: 269–271. [DOI] [PubMed] [Google Scholar]
- 32.Palomero T, Wei KL, Odom DT, Sulis ML, Real PJ, Margolin A et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A 2006; 103: 18261–18266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014; 20: 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017; 129: 1124–1133. [DOI] [PubMed] [Google Scholar]
- 35.Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 2017; 25: 345–357. [DOI] [PubMed] [Google Scholar]
- 36.Chiang MY, Xu L, Shestova O, Histen G, L’Heureux S, Romany C et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest 2008; 118: 3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med 1996; 183: 2283–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kourtis N, Lazaris C, Hockemeyer K, Balandrán JC, Jimenez AR, Mullenders J et al. Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. 2018. doi: 10.1038/s41591-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014; 514: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao R, Goldman ID. Resistance to antifolates. Oncogene 2003; 22: 7431–7457. [DOI] [PubMed] [Google Scholar]
- 41.Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J 2017; 7: e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016; 16: 494–507. [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Ducker GS, Lu W, Teng X, Rabinowitz JD. An LC-MS chemical derivatization method for the measurement of five different one-carbon states of cellular tetrahydrofolate. Anal Bioanal Chem 2017; 409: 5955–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng Y, Lin TY, Lee G, Paddock MN, Momb J, Cheng Z et al. Mitochondrial One-Carbon Pathway Supports Cytosolic Folate Integrity in Cancer Cells. Cell 2018; 175: 1546–1560.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019; 572: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tibbetts AS, Appling DR. Compartmentalization of Mammalian Folate-Mediated One-Carbon Metabolism. Annu Rev Nutr 2010; 30: 57–81. [DOI] [PubMed] [Google Scholar]
- 47.Taylor RT, Hanna ML. Folate-dependent enzymes in cultured Chinese hamster ovary cells: Impaired mitochondrial serine hydroxymethyltransferase activity in two additional glycine - auxotroph complementation classes. Arch Biochem Biophys 1982; 217: 609–623. [DOI] [PubMed] [Google Scholar]
- 48.Chasin LA, Feldman A, Konstam M, Urlaub G. Reversion of a Chinese Hamster Cell Auxotrophic Mutant. Proc Natl Acad Sci 1974; 71: 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin BF, Shane B. Expression of Escherichia coli folylpolyglutamate synthetase in the Chinese hamster ovary cell mitochondrion. J Biol Chem 1994; 269: 9705–9713. [PubMed] [Google Scholar]
- 50.Titus SA, Moran RG. Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J Biol Chem 2000; 275: 36811–36817. [DOI] [PubMed] [Google Scholar]
- 51.Bertino Göker, Gorlick Li, Banerjee. Resistance Mechanisms to Methotrexate in Tumors. Oncologist 1996; 1: 223–226. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.