

Abstract
Suppressor of Ty 16 (Spt16) is a component of the facilitates chromatin transcription (FACT) complex, which is a histone chaperone and involved in gene transcription, DNA replication, and DNA repair. Previous studies showed that FACT is highly expressed in cancer, and cancer cells are more reliant on FACT than normal cells. However, the relationship between Spt16 and lung cancer remains unclear. In this study, we explored the functions of Spt16 in lung cancer cells. The effects of Spt16 on lung cancer cell proliferation, cell cycle progression, apoptosis, migration, and invasion were examined. We found that knockdown of Spt16 led to obvious decreases of both Rb and MCM7, and further activated the DNA damage response (DDR) pathway. In addition, a novel micro‐RNA, miR‐1227‐5p, directly targeted the 3′‐UTR of Spt16 and regulated the mRNA levels of Spt16. Furthermore, we found that CBL0137, the functional inhibitor of FACT, showed similar effects as loss of Spt16. Together, our data indicated that Spt16 is likely to be an essential regulator for lung cancer malignancy and is negatively regulated by miR‐1227‐5p.
Keywords: lung cancer, MCM7, MiR‐1227‐5p, Rb, Spt16
In this study, we examined the functions of suppressor of Ty 16 (Spt16) in lung cancer cells. We showed that Spt16 is upregulated in lung cancers and is correlated with the poor prognosis of lung cancer patients. Knockdown of Spt16 in lung cancer cells inhibited the proliferation and metastasis of lung cancer cells both in vitro and in vivo. We also determined a novel mechanism for the regulation of Spt16 expression by miRNA‐1227‐5p.

1. INTRODUCTION
Lung cancer is the second most common cancer, accounting for more than one‐quarter (26%) of all cancer deaths in 2019. 1 , 2 Currently, the 5‐year survival rate of patients remains quite low (15.6%). 3 Therefore, further exploring the mechanisms of lung cancer progression and identifying novel molecular targets for lung cancer therapy are required.
Suppressor of Ty 16 (Spt16) is a histone chaperon that regulates the organization of nucleosomes. 4 Spt16 binds structure‐specific recognition protein 1 (SSRP1), forming a heterodimeric complex, facilitates chromatin transcription (FACT). The FACT complex is involved in almost all chromatin remodeling‐related processes, including DNA replication, DNA repair, and gene transcription. 5 Spt16 usually exhibits its biological roles through forming the FACT complex by cooperating with SSRP1 to modulate chromatin. However, several studies show that Spt16 also possesses SSRP1‐independent roles. For instance, knockdown of Spt16 decreased the expression of the genes that were not influenced by loss of SSRP1. 6 Spt16, but not SSRP1, accelerated the exchange of H2A/H2B after UV‐irradiation. 7 Spt16 promoted the homologous recombination DNA repair through interacting with RNF20 in the early stages. 8
The protein levels of Spt16 are higher in cancerous tissues than in normal tissues in multiple types of cancers. 9 , 10 Moreover, the expression of Spt16 is further elevated in cancer cells with higher levels of stem cell markers compared with stem cell marker‐negative cancer cells. 11 , 12 , 13
Suppressor of Ty 16 regulates the re‐entry of G0 phase cells to the cell cycle in prostate and lung cancer cells. 14 Loss of Spt16 resulted in significant defects in tumor cell proliferation, while no obvious effect was observed in the growth of non–transformed cells. 10 In breast cancer, knockdown of Spt16 suppressed cell viability, and higher Spt16‐expressing cancer cells were more prone to Spt16 depletion‐induced cell death compared with lower Spt16‐expressing cancer cells. 13 Thus, loss of Spt16‐induced cell death closely depends on the levels of Spt16, suggesting that Spt16 might be used as a novel therapeutic target.
The relationship between Spt16 and lung cancer remains unclear. In the present study, we explore the functions of Spt16 in lung cancer cells. Our results suggest that Spt16 likely functions as an oncogene. Knockdown of Spt16 inhibited the proliferation and metastasis of lung cancer cells both in vitro and in vivo. In addition, miR‐1227‐5p directly targeted Spt16 and negatively regulated the expression of Spt16. Together, our results revealed that Spt16 promotes lung cancer progression and is regulated by miR‐1227‐5p, providing a potential target for lung cancer therapy.
2. MATERIALS AND METHODS
2.1. Cell culture and transfection
All cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences. BEAS‐2B was cultured in High Glucose DMEM (#SH30022.01, Hyclone) supplemented with 10% FBS (#SH30084.03, Hyclone). A549, NCI‐H1299, and NCI‐H460 were cultured in RPMI‐1640 medium (#SH30027.01, Hyclone) supplemented with 10% FBS. All cells were incubated in 5% CO2 at 37°C.
Two different siRNA specifically targeting Spt16 were designed and synthesized by GenePharma. miRNA mimic was synthesized by RIB‐BIO. Transfection of siRNA and miRNA mimic using Lipofectamine 2000 (#11668‐019, Invitrogene) were performed according to the manufacturer’s protocol. Spt16‐specific lentiviral shRNA were designed and constructed by GenePharma. Cells were infected in the presence of 5 μg/mL polybrene according to the manufacturer’s instructions. The sequences of siRNA and shRNA are shown in Table S1.
2.2. Patients and immunohistochemical staining
This work was approved by the Ethics Review Committee of the First Affiliated Hospital of Xi’an Jiaotong University. A total of 147 lung cancer specimens and adjacent normal tissues were obtained from the First Affiliated Hospital of Xi’an Jiaotong University. After antigen retrieval in sodium citrate buffer (PH = 9.0) in a microwave oven, the paraffin‐embedded tissues were stained with Spt16 (8D2, Biolegend), MCM7 (sc‐9966, Santa Cruz), and Rb (sc‐102, Santa Cruz) antibodies following the manufacturer’s protocol for the LSAB detection kit (ZSGB‐BIO). Each section was scored by the staining intensity (0, none; 1, weak; 2, moderate; and 3, strong) and the positive cell rate (0, no positive cells; 1, 10%–25%; 2, 25%–50%; 3, 50%–75%; 4, >75%). The total score is the product of the positive cell rate by staining intensity.
2.3. MTT assay
Equal amounts of cells were seeded into a 48‐well plate. At specific time points after seeding, 50 μL MTT (5 mg/mL) was added into the culture medium and incubated for 4 h at 37°C, and the medium was removed. The formazan crystals were dissolved in 375 μL DMSO, and the optical absorbance values were determined at 490 nm using a microplate reader (PerkinElmer).
2.4. Colony formation assay
Cells were plated in 6‐well plates with a density of 2 × 103 cells/well and incubated in 5% CO2 at 37°C. Colonies were stained with crystal violet dissolved in methanol. The colony numbers were counted.
2.5. Cell cycle analysis
Cells were trypsinized and fixed with cold 70% ethanol at 4°C overnight. Cells were incubated with 50 μg/mL sodium citrate and 10 μg/mL RNase A in the dark for 30 minutes. FACS Calibur flow cytometer (BD) was used to measure the DNA content. The data was analyzed using the MODFIT software program (Verity Software House).
2.6. EdU incorporation assay
Cells were cultured in 96‐well plates and incubated with 10 μM EdU for 2 hours. Incorporated EdU was detected using a kFluor555 Click‐iT EdU detection kit according to the manufacturer’s instructions (KGA337, KeyGEN). Briefly, cells were incubated for 30 minutes with kFluor555‐azide dye in TBS supplemented with CuSO4. Cells were then counterstained with DAPI and imaged with a laser scanning confocal microscope (Leica, Germany).
2.7. Apoptosis analysis
Cells were trypsinized and stained with 7‐AAD and FITC‐conjugated anti‐annexin V antibodies (R&D). Cells were resuspended in binding buffer for flow cytometry analysis using a FACSCalibur flow cytometer (BD).
2.8. Western blot analyses and antibodies
Cells were lyzed with RIPA buffer supplemented with protease and phosphatase inhibitors (A32959, Thermo). Cell extracts were separated in SDS‐PAGE and then transferred to PVDF membranes. The primary antibodies were incubated overnight at 4°C, and the HRP‐conjugated secondary antibodies were incubated for 1 h at room temperature. The chemiluminescent signals were detected with the ECL Plus Detection Reagents (WBULS0500, Millipore). Antibodies against Spt16 (8D2) was purchased from Biolegend. Antibodies against p‐ATR (2853), p‐ATM (5883), p‐BRCA1 (9009), p‐CHK2 (Thr1079, 8654), N‐cadherin (13116), E‐cadherin (3195), ZO‐1 (13663), vimentin (5741), Snail (3879), Bcl‐2 (4223), cleaved caspase 7 (8438), and cyclin D1 (55506) were purchased from Cell Signaling Technology. Antibodies against p‐Rb (sc‐377539), cyclin E (sc‐377100), MCM7 (sc‐9966), and Rb (sc‐102) were purchased from Santa Cruz Biotechnology. Antibodies against GAPDH (HRP‐6004) and β‐actin (HRP‐60008) were purchased from Proteintech.
2.9. Immunofluorescence analysis
Cells were seeded on coverslips and fixed in 4% paraformaldehyde for 10 minutes. Then, 0.2% Triton X‐100 was used for permeabilization. Cells were blocked for 1 hour, followed by incubation with primary antibodies overnight at 4°C. Alexa Fluor 488‐labeled secondary antibodies (Zhuangzhi Bio) were further incubated for another 2 hours. Cells were counterstained with DAPI and imaged with the laser scanning confocal microscope (Leica).
2.10. mRNA and miRNA expression analyses
For mRNA expression analyses (RT‐PCR), total RNA were isolated using the RNA Fast 200 isolation kit (220010, Feijie) and reversely transcribed using the cDNA Reverse Transcription Premix (11123, Yeasen) according to the manufacturer’s instructions. A total of 1 μL of cDNA was used for quantitative (qPCR) analyses. qPCR mixtures were prepared using the SYBR Green qPCR Mix (11201, Yeasen). qPCR analyses were performed with the Bio‐Rad CFX96TM Real‐Time PCR Detection System. mRNA expression was normalized to GAPDH and determined using the ΔΔCt method. The primers used in this study are shown in Table S2.
For miRNA expression analysis, miRNA were isolated with the miRcute miRNA Isolation Kit (DP501, TIANGEN). All miRNA were polyadenylated by poly(A) polymerase and converted into cDNA with the miDETECT A Track miRNA qRT‐PCR Starker Kit (C10712, RIB‐BIO). qPCR mixtures were prepared, and qPCR analyses were performed with the Bio‐Rad CFX96TM Real‐Time PCR Detection System. U6 was used as an internal reference to normalize miRNA expression. All primers used were obtained from RIB‐BIO. Two microDNA chips of lung cancer tissues (#micDNA‐HLugC030PT01 and #micDNA‐HLugA030PG01, each containing 15 pairs of lung cancer samples, respectively) used for analyzing miR‐1227‐5p expression were purchased from Shanghai OUTDO Biotech.
2.11. In vivo cell growth and migration studies
Female SCID/Beige mice were obtained from the Experimental Animal Center of Xi’an Jiaotong University. All animal experiments were conducted in accordance with the guidelines of Xi’an Jiaotong University Animal Care and Use Committee.
For tumorigenicity assay, 2 × 106 control or Spt16‐depleted A549 cells suspended in 100 μL PBS were injected subcutaneously into six mice. The tumor size was measured every 3 days, and the tumors were collected at the 40th day and weighted. The tumors were paraffin‐embedded and cut into 4‐μm‐thick sections.
For the metastasis assay, 1 × 106 control or Spt16‐depleted NCI‐H1299 cells were suspended in 100 μL PBS and injected into the tail vein of six mice. After 10 weeks, mice were killed, and the lungs and livers were collected. The tissues were paraffin‐embedded and cut into 4‐μm‐thick sections.
2.12. Wound healing assay
Cells were plated in 6‐well plates and grew to a sub‐confluence stage. A wound area was created using pipette tips. The wounded monolayer cells were cultured in serum‐free medium for 48 hours. Cell migration into the wounded area was monitored and photographed using the microscope (Leica).
2.13. Transwell assay
We used 8‐µm pore Transwell chambers (Corning) for transwell assays. The top insert chambers were coated with or without basement membrane growth factor‐reduced Matrigel (BD). Cells were suspended in serum‐free medium and added into top chambers. The lower chambers were filled with serum‐containing medium. After incubation at 37°C for specific times, the cells were stained with crystal violet. The transwell cells were photographed with the microscope (Leica).
2.14. Luciferase reporter analysis
The wild‐type or mutated 3′‐UTR of Spt16 were cloned into a firefly luciferase reporter construct. Cells were co–transfected with the firefly luciferase reporter and the Renilla luciferase reporter (Promega) for 48 hours using Lipofectamine 2000 according to the manufacturer’s instructions. The luciferase activity was measured using a dual luciferase reported gene assay kit (Promega) according to the manufacturer’s instructions. The relative activity of the reporter gene was calculated by dividing the signals from the firefly luciferase reporter by the signals from the Renilla luciferase reporter.
2.15. Data mining and statistical analysis
The LUAD and LUSC TCGA datasets were downloaded and re‐analyzed to obtain Spt16 mRNA expression data. The mRNA expression levels were log2‐transformed. Micro‐RNA and mRNA data of 155 surgically resected specimens of lung adenocarcinoma were downloaded from GEO database (GSE119269). The extracted data were processed by log2 transformation. Data are presented as mean ± SD. Statistical analyses were performed with a paired‐sample t test, an unpaired Student’s t test, or the Pearson correlation test using GraphPad Prism 5.0 (GraphPad Prism Software). P‐value < 0.05 was considered statistically significant.
3. RESULTS
3.1. Suppressor of Ty 16 is associated with poor prognosis of lung cancer patients and is highly expressed in lung cancer
To explore the potential roles of Spt16 in lung cancer, we examined the relationship between Spt16 expression and the survival rate of lung cancer patients. Kaplan‐Meier plot analyses using the data from TCGA (http://kmplot.com) indicated that lung cancer patients with higher Spt16 mRNA expression showed poorer overall survival (OS) and disease‐free survival (DFS) than patients with lower Spt16 expression (Figure 1A). Next, we compared the expression levels of Spt16 in lung cancer tissues and normal lung tissues. A total of 147 pairs of lung cancer specimens and the relative normal adjacent tissues were collected and stained with Spt16 antibodies for immunohistochemistry assays. Lung cancer tissues (squamous cell carcinoma and adenocarcinoma) showed much higher Spt16 staining signals than adjacent tissues (Figure 1B and C). In accordance with the expression of Spt16 in lung cancer specimens (Figure 1B and C), we found that the protein levels of Spt16 were much higher in lung cancer cell lines than in non–transformed bronchial epithelial cells (Figure 1D). Furthermore, according to the TCGA database, significant increases of the Spt16 mRNA levels were observed in both squamous cell carcinoma (LUSC) and adenocarcinoma (LUAD) compared with the paired adjacent normal tissues (Figure 1E), which is consistent to the above experimental assays (Figure 1B‐D). Thus, these results provided a clue that Spt16 likely associates with the progression and prognosis of lung cancer.
Figure 1.
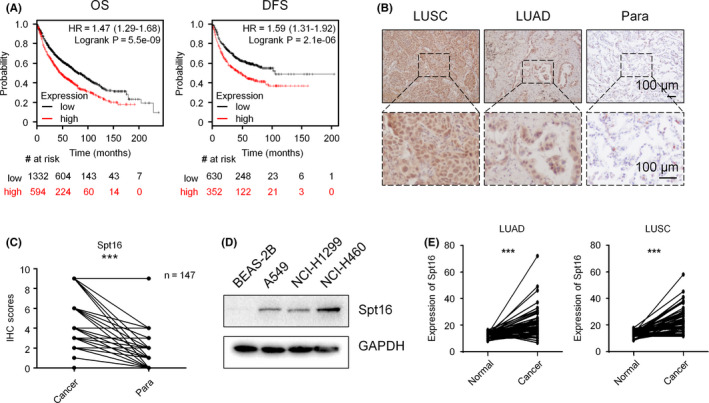
Suppressor of Ty 16 (Spt16) is elevated in human lung cancers and is associated with a poor prognosis of patients. A, Kaplan‐Meier Plotter analyses with a public database (http://kmplot.com/analysis/). B, Immunohistochemistry (IHC) for 147 pairs of human lung cancer specimens (LUSC and LUAD) and the relative normal adjacent tissues. C, Quantification of the data obtained in (B). D, Western blot analysis for cells as indicated. This experiment was repeated three times independently. E, Analysis of the mRNA levels of Spt16 in 53 pairs (LUAD) and 49 pairs (LUSC) of lung cancer tissues and para‐tissues from the TCGA database
3.2. Knockdown of suppressor of Ty 16 inhibits cell growth of lung cancer cells both in vitro and in vivo
To understand the biological impacts of Spt16 in the regulation of lung cancer, we knocked down the expression of Spt16 in A549, NCI‐H1299, and NCI‐H460 lung cancer cell lines, respectively, by two separate siRNA (Figure 2A). The effects of Spt16 on cell proliferation were determined by MTT and colony formation assays. Our results indicated that depletion of Spt16 resulted in cell proliferation impairment in all three lung cancer cell lines (Figure 2B and C). To further understand the roles of Spt16 in cell proliferation in vivo, xenograft tumor growth assays were performed by establishing a stable Spt16‐depleted A549 cell line. Consistently, we found that loss of Spt16 also led to an impaired xenograft tumor growth (Figure 2D). Together, these data suggested that knockdown of Spt16 inhibits lung cancer cell growth both in vitro and in vivo.
Figure 2.
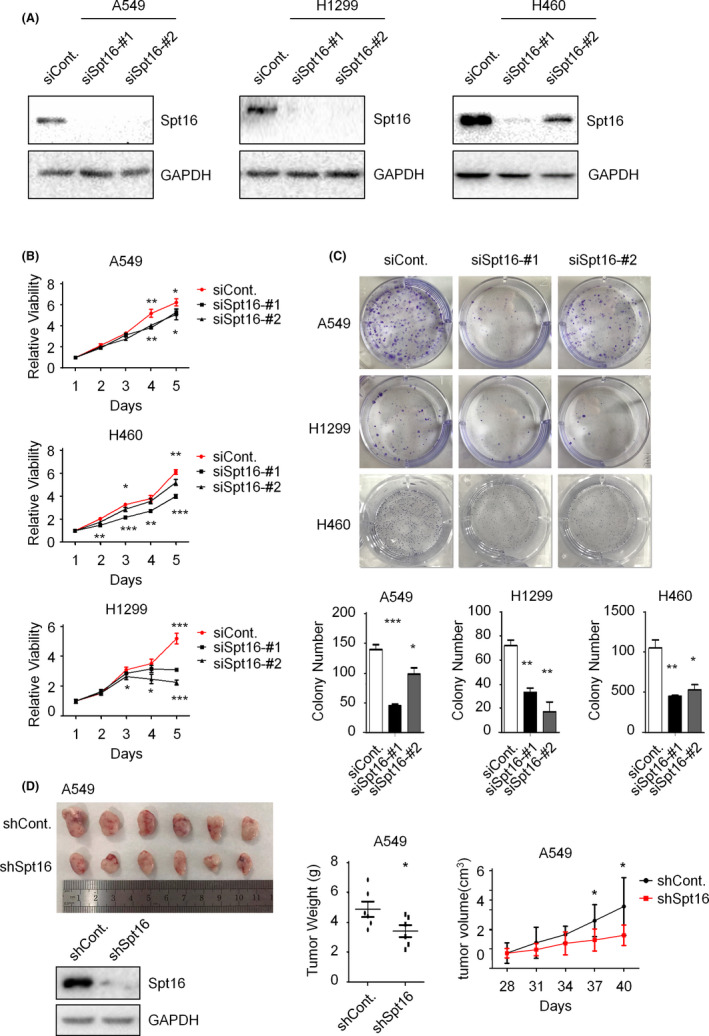
Suppressor of Ty 16 (Spt16) is essential for cell growth of lung cancer cells. A, A549, NCI‐H1299, and NCI‐H460 lung cancer cells were transfected with a control siRNA or two separate Spt16‐specific siRNA for 48 h. Cell extracts were then prepared and western blot analyses were performed with antibodies as indicated. This experiment was repeated three times independently. B, A549, NCI‐H1299, and NCI‐H460 lung cancer cells were transfected as in (A). MTT analyses were then performed. C, A549, NCI‐H1299, and NCI‐H460 lung cancer cells were transfected as in (A). Colony formation analyses were then performed. D, Spt16 stably depleted and control A549 cells were injected subcutaneously into six SCID/Beige mice, respectively. Tumor weight and volume were quantified. Bars and error bars are mean ± SD; n = 3 independent repeats. A two‐tailed unpaired Student’s t test was performed. *P < 0.05, **P < 0.01, ***P < 0.001
3.3. Knockdown of suppressor of Ty 16 impairs cell cycle progression and promotes apoptosis in lung cancer cells
To determine the underlying mechanisms of Spt16 regulating lung cancer cell growth (Figure 2), we tested the effects of Spt16 on cell cycle and apoptosis in lung cancer cells. Our data suggested that knockdown of Spt16 expression led to an accumulation of cells in the G0/G1‐phase and a decrease of cells in the S‐phase in both A549 and NCI‐H1299 cells (Figure 3A). Moreover, the changes in S‐phase cells were further verified by EdU incorporation assay. Consistently, the EdU positive cells (indicating S‐phase cells) decreased significantly in Spt16‐depleted cells (Figure 3B). The levels of a series of cell cycle regulators that are essential for G1/S transition are impaired after Spt16 depletion, including phosphorylated Rb (p‐Rb), cyclin D1, and cyclin E1 (Figure 3C). In addition, our apoptosis analyses revealed that an increase of cell apoptosis was observed after knockdown of Spt16 expression (Figure 3D). Moreover, the apoptosis inhibitor, Bcl‐2, was decreased in Spt16‐depleted lung cancer cells, while the apoptosis activator, cleaved caspase‐7, was increased (Figure 3E). Thus, the above data indicated that Spt16 depletion impairs cell cycle progression and promotes apoptosis in human lung cancer cells.
Figure 3.
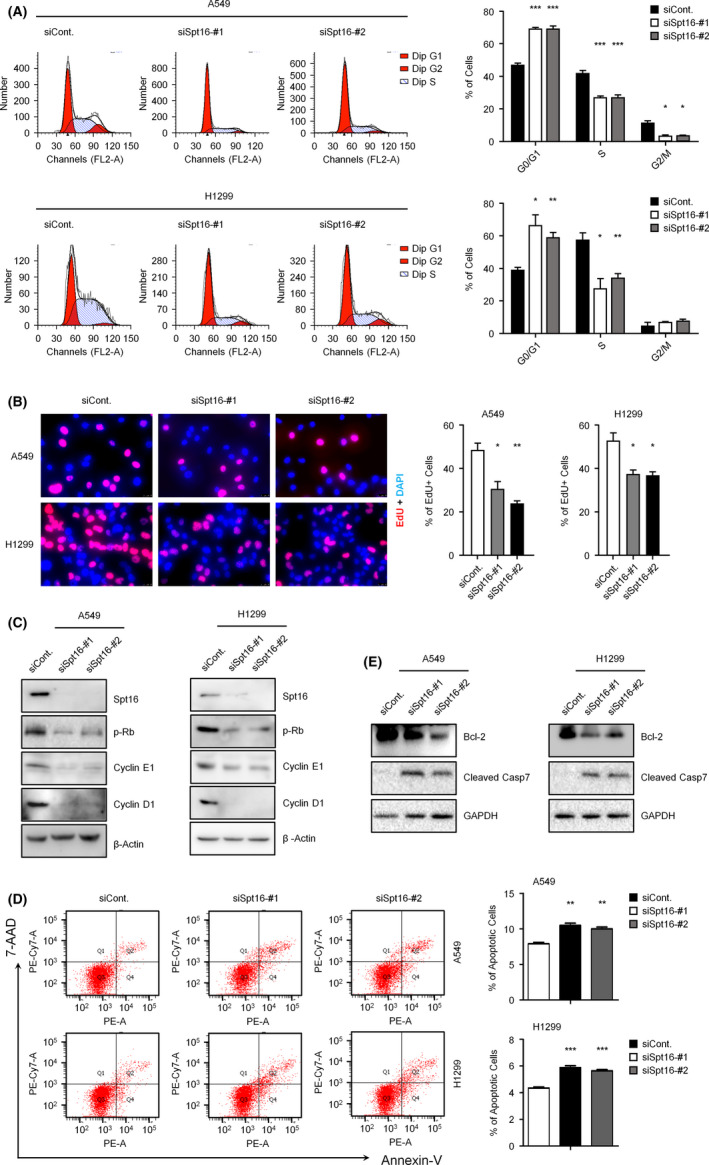
Loss of suppressor of Ty 16 (Spt16) impaired cell cycle progression and induced apoptosis in human lung cancer cells. A, A549 and NCI‐H1299 lung cancer cells were transfected with a control or two separate Spt16‐specific siRNA for 48 h. Flow cytometry (FACS) analyses with propidium iodide (PI) staining were performed to determine the effects of Spt16 on cell cycle progression. B, A549 and NCI‐H1299 lung cancer cells were transfected as in (A). EdU incorporation assays were performed. C, A549 and NCI‐H1299 lung cancer cells were transfected as in (A). Western blot analyses were performed with antibodies as indicated. This experiment was repeated three times independently. D, A549 and NCI‐H1299 lung cancer cells were transfected as in (A). Flow cytometry (FACS) analyses with Annexin‐V and 7‐AAD double staining were performed to determine the effects of Spt16 on cell apoptosis. E, A549 and NCI‐H1299 lung cancer cells were transfected as in (A). Western blot analyses were then performed with antibodies as indicated. This experiment was repeated three times independently. Bars and error bars are mean ± SD; n = 3 independent repeats. A two‐tailed unpaired Student’s t test was performed. *P < 0.05, **P < 0.01, ***P < 0.001
3.4. Knockdown of suppressor of Ty 16 activates DNA damage responses by negatively regulating both Rb and MCM7
We found that knockdown of Spt16 resulted in significant decreases in the protein levels of both Rb and MCM7 in A549, NCI‐H1299, and NCI‐H460 lung cancer cells (Figure 4A). The downregulation of Rb and MCM7 after Spt16 depletion is likely achieved via the transcriptional regulation, as the mRNA levels of both Rb and MCM7 were decreased in Spt16‐depleted cells (Figure 4B). Our previous work demonstrated that co–depletion of MCM7 and Rb caused severe DNA damage and further activated DDR. 15 , 16 Therefore, we tested the levels of multiple regulators of the DDR pathway (including p‐ATM, p‐ATR, p‐BRCA1, and p‐CHK2). In parallel with the changes of Rb and MCM7 after Spt16 depletion, our data confirmed that loss of Spt16 activated DDR (Figure 4C). Immunofluorescence analysis also supported this conclusion, as p‐BRCA1 foci, which are the typical patterns of p‐BRCA1 when DDR is activated, were increased in Spt16‐depleted lung cancer cells (Figure 4D). Next, we overexpressed PSM‐Rb in Spt16‐depleted cells to determine the involvement of Rb in Spt16‐activated DDR. PSM‐Rb is a constitutively active mutant of the Rb protein. However, this mutation resulted in the lack of epitopes that are recognized by most commercial Rb antibodies. We confirmed the overexpression of PSM‐Rb by checking the mRNA levels of PSM‐Rb (Figure 4E, right). Our results showed that overexpression of PSM‐Rb rescued the abnormal DDR activation observed in Spt16‐depleted lung cancer cells (Figure 4E, left). In addition, our immunochemistry staining assays of the xenograft tumors further supported the notion that the levels of MCM7 and Rb are downregulated in the Spt16‐depleted A549 cells (Figure 4F). Moreover, a positive correlation between the levels of Spt16 and Rb or MCM7 was observed in 147 clinical lung cancer specimens (Figure S1). Taken together, these data revealed that knockdown of Spt16 activates DDR likely by downregulating the expression of both Rb and MCM7.
Figure 4.
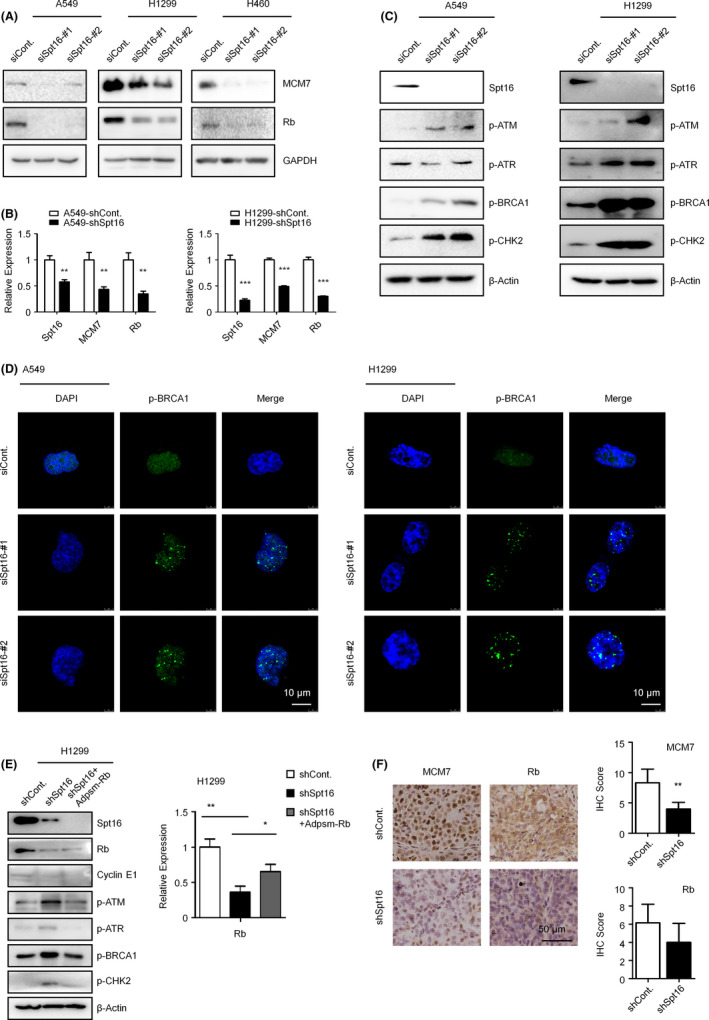
Loss of suppressor of Ty 16 (Spt16) decreases the levels of MCM7 and Rb and activates DNA damage responses (DDR). A, A549, NCI‐H1299, and NCI‐H460 human lung cancer cells were transfected with a control siRNA or two separate Spt16‐specific siRNA for 48 h. Cells were then harvested and western blot analyses were performed. This experiment was repeated three times independently. B, A549 and NCI‐H1299 cells were transfected with a control shRNA or an Spt16‐specific siRNA for 48 h. Quantitative RT‐PCR analyses were then performed. C, A549 and NCI‐H1299 human lung cancer cells were transfected as in (A). Cell extracts were then prepared and western blot analyses were performed. This experiment was repeated three times independently. D, A549 and NCI‐H1299 cells were transfected as in (A). Immunofluorescence (IF) analyses were then performed with phosphorylated BRCA1 (p‐BRCA1) antibodies. This experiment were was repeated three times independently. E, NCI‐H1299 cells were transfected with indicated constructs for 48 h. Western blot (left) and quantitative RT‐PCR (right) analyses were performed. F, Immunohistochemistry (IHC) in xenograft tumors generated as in Figure 2D. The levels of MCM7 and Rb were examined and quantified. Bars and error bars are mean ± SD; n = 3 independent repeats. A two‐tailed unpaired Student’s t test was performed. *P < 0.05, **P < 0.01, ***P < 0.001
3.5. Knockdown of suppressor of Ty 16 inhibits cell migration and invasion in lung cancer cells both in vitro and in vivo
Metastasis is the overwhelming cause of lung cancer mortality and results in a major hurdle for lung cancer therapy. 17 Therefore, it is essential to assess the effects of Spt16 on cancer metastasis. Wound healing and transwell assays were performed, and the loss of Spt16 significantly impaired the migration and invasion rates of both A549 and NCI‐H1299 cells (Figure 5A‐C). Epithelial‐mesenchymal transition is tightly involved in the regulation of tumor cell invasion and metastasis. We examined whether Spt16 affects the levels of epithelial‐mesenchymal transition (EMT) markers. Knockdown of Spt16 decreased the levels of mesenchymal markers (N‐cadherin and vimentin), while the levels of epithelial markers (E‐cadherin and ZO‐1) were increased (Figure 5D). Moreover, the levels of the EMT transcription factor, Snail, were also suppressed after Spt16 depletion (Figure 5D). To confirm the metastatic role of Spt16 in vivo, stable Spt16‐depleted and control NCI‐H1299 cells were injected into the tail veins of SCID‐mice. The mice bearing Spt16‐depleted cancer cells showed decreased lung and liver metastatic colonizations compared with the control group (Figure 5E andF). Together, these results suggested that knockdown of Spt16 inhibits EMT, invasion, and metastasis of lung cancer cells both in vitro and in vivo.
Figure 5.
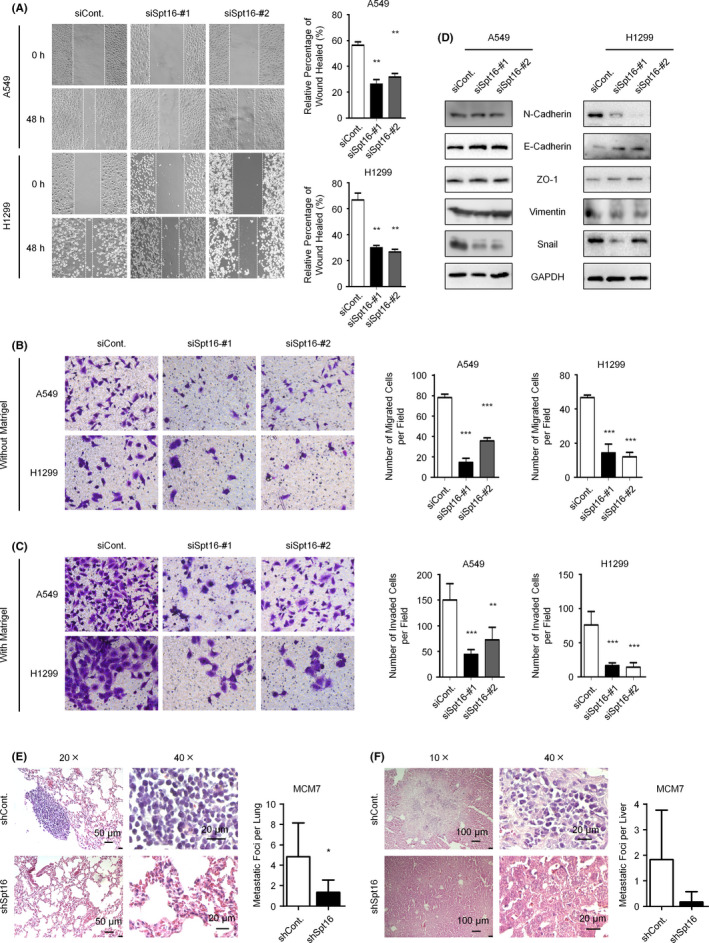
Loss of suppressor of Ty 16 (Spt16) inhibits cell migration and invasion in lung cancer cells both in vitro and in vivo. A, Wound healing assays were performed in A549 and NCI‐H1299 cells transfected as indicated. Cells were transfected with a control siRNA or two separate Spt16‐specific siRNA for 48 h. Cells were then subjected to wound healing assays, and the wound areas were photographed after another 48 h. B and C, Transwell assays without (B) or with (C) Matrigel were performed in A549 and NCI‐H1299 cells transfected with a control siRNA or two Spt16‐specific siRNA for 48 h. D, NCI‐H1299 cells were transfected with a control siRNA or two separate Spt16‐specific siRNA for 48 h. Cells were then harvested and subjected to western blot analyses with antibodies as indicated. This experiment was repeated three times independently. E and F, Control or Spt16‐depleted NCI‐H1299 cells were injected into the tail vein of six mice, and H&E staining and numbers of metastatic lung nodules in lung tissues (E) or in liver tissues (F) were analyzed. Bars and error bars are mean ± SD; n = 3 independent repeats. A two‐tailed unpaired Student’s t test was performed. *P < 0.05, **P < 0.01, ***P < 0.001
3.6. Suppressor of Ty 16 is negatively regulated by miR‐1227‐5p
The above data showed that Spt16 is highly expressed in lung cancer cells (Figure 1B‐E). However, how the expression of Spt16 is regulated remains unknown. Gain of copy numbers is the most frequent mechanism of oncogene overexpression in cancer cells. However, according to the data from cBioPortal (https://www.cbioportal.org/), the incidence of gene amplification in the Spt16 locus is quite low (<2%) in both lung adenocarcinoma and lung squamous cell carcinoma (Figure 6A). Therefore, other mechanisms may be involved in the upregulation of Spt16 expression in lung cancer. Micro‐RNA (miRNA) play significant roles in the control of protein levels. miRNA potentially targeting Spt16 were predicted by TargetScan (http://www.targetscan.org/), miRDB (http://mirdb.org/), and miRanda (http://www.microrna.org/). We found that miR‐1227‐5p ranked among the candidates of Spt16 regulatory miRNA, and miR‐1227‐5p is complementary to the 3ʹ‐UTR of Spt16 mRNA. We also found that, in 30 pairs of lung cancer and adjacent normal tissues (two microDNA chips were used, each containing 15 pairs of lung cancer samples, respectively), the levels of miR‐1227‐5p were significantly downregulated in cancer tissues (Figure 6B). After transfection with the miR‐1227‐5p mimic (Figure 6C), both the mRNA and protein levels of Spt16 were reduced in NCI‐H1299 and NCI‐H460 lung cancer cells (Figure 6D andE). The wild‐type or mutated 3′‐UTR of Spt16 were cloned into a luciferase reporter construct, and dual luciferase reporter assays were performed to determine the direct regulation of miR‐1227‐5p on the 3′‐UTR of Spt16 (Figure 6F). Consistently, miR‐1227‐5p transfection suppressed the activities of the reporter plasmids containing wild‐type Spt16 3′‐UTR, while there was no effect on the mutated Spt16 3′‐UTR containing plasmids (Figure 6G). Furthermore, a negative correlation between the miR‐1227‐5p levels and the Spt16 mRNA levels was observed in 155 surgically resected specimens of LUAD analyzed using GEO database (accession #: GSE119269) (Figure 6H). Together, these data indicated that Spt16 is negatively regulated by miR‐1227‐5p in lung cancer cells.
Figure 6.
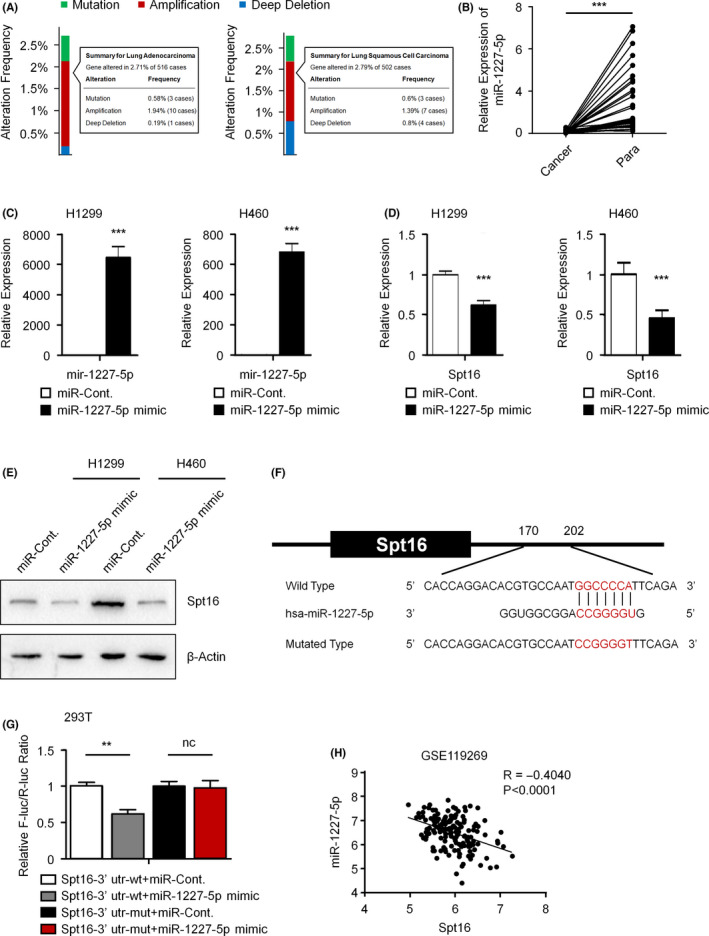
miR‐1227‐5p negatively regulates the expression of suppressor of Ty 16 (Spt16) in human lung cancer cells. A, Copy number variation (CNV) analysis with the cBioPortal online database (https://www.cbioportal.org/). B, The expressional levels of miR‐1227‐5p in 30 pairs of human lung cancer tissues and paracancerous tissues were examined with quantitative RT‐PCR analysis. C and D, Quantitative RT‐PCR analyses in NCI‐H1299 and NCI‐H460 lung cancer cells transfected as indicated. Cells were transfected with a control miRNA or a miR‐1227‐5p mimic as indicated for 48 h. Quantitative RT‐PCR analyses were performed. E, NCI‐H1299 and NCI‐H460 lung cancer cells were transfected with a control miRNA or a miR‐1227‐5p mimic for 48 h. Western blot analyses were performed. This experiment was repeated three times independently. F, Schematic diagram of the sequences used to construct the luciferase reporter plasmids. G, Dual luciferase reporter analyses in 293T cells transfected as indicated. 293T cells were transfected with indicated constructs for 48 h. Dual luciferase reporter analyses were performed. H, Pearson correlation analysis using GSE119269 data (containing 155 surgically resected specimens of LUAD) shows the reverse correlation between the miR‐1227‐5p levels and the Spt16 mRNA levels. Bars and error bars are mean ± SD; n = 3 independent repeats. A two‐tailed unpaired Student’s t test was performed. **P < 0.01, ***P < 0.001
3.7. Facilitates chromatin transcriptioninhibitor CBL0137 shows similar effects as loss of suppressor of Ty 16
To further confirm the effects of Spt16 on human lung cancer cells, CBL0317, an inhibitor of the FACT complex that can functionally inactivate the FACT complex by rearranging the distributions of FACT components including Spt16 to chromatin, was used. 18 Consistent with the effects of loss of Spt16, we found that treatment with CBL0317 also resulted in a significant reduction in cell proliferation in both A549 and NCI‐H1299 lung cancer cells (Figure S2A and B). FACS analyses showed that treatment with CBL0137 also resulted in an obvious increase in cell apoptosis in A549 and NCI‐H1299 lung cancer cells, which is similar to the effects of Spt16 depletion on cell apoptosis (Figure S2C). Finally, decreases in the protein levels of MCM7 and Rb (Figure S2D) and an activation of DDR (Figure S2E and F) were also detected in lung cancer cells treated with CBL0137. Thus, these findings further supported the critical roles of Spt16 in the regulation of cell growth, apoptosis, and DNA damage responses in human lung cancer cells. We also examined the combinational effects of CBL0137 treatment and miR‐1227‐5p mimic transfection. Our results indicated that both CBL0137 and miR‐1227‐5p mimic inhibited cell growth and promoted apoptosis in A549 lung cancer cells. However, no obvious differences were observed between CBL0137, miR‐1227‐5p single treatment and the combinational treatment (Figure S3). This might be because CBL0137 and miR‐1227‐5p modulate the same downstream target, Spt16, in the regulation of cell growth and apoptosis.
4. DISCUSSION
In this study, we examined the functions of Spt16 in lung cancer cells. We showed that Spt16 is upregulated in lung cancers and correlates with the poor prognosis of lung cancer patients. Knockdown of Spt16 in lung cancer cells inhibited the proliferation and metastasis of lung cancer cells both in vitro and in vivo. We also determined a novel mechanism for the regulation of Spt16 expression by miR‐1227‐5p.
Our results indicated that depletion of Spt16 inhibited the growth of lung cancer cells by disrupting the normal cell cycle progression and stimulating cell apoptosis (Figures 2 and 3). Cyclin D1, cyclin E1, and p‐Rb are well‐known cell cycle regulators, we found that the levels of these regulators were downregulated after Spt16 depletion. The changes of these regulators are consistent with the observed decreases in the population of S‐phase cells and increases in G0/G1 cells upon Spt16 depletion. 19 Thus, Spt16 likely functions as a negative regulator for cell proliferation and cell cycle progression in lung cancer cells.
Cleaved caspase‐7 plays a central role in the regulation of apoptosis. 20 Bcl‐2 is a well‐known negative regulator of apoptosis. 21 , 22 Here, consistent with the effects of cleaved caspase‐7 and Bcl‐2, we found that loss of Spt16 significantly increased the levels of the cleaved caspase‐7, while the levels of Bcl‐2 were downregulated (Figure 3E), supporting the observation that cell apoptosis was increased after Spt16 depletion (Figure 3D). Therefore, the elevated cell apoptosis may contribute to the defects of cell growth induced by Spt16‐depletion.
We demonstrated that depletion of Spt16 led to DDR activation likely due to the decreases in the protein levels of both MCM7 and Rb (Figure 4). MCM7 forms a hexametric complex by interacting with multiple MCM proteins, including MCM2‐6. 23 , 24 The MCM complex is a replicative DNA helicase involved in the assembly of pre–replication complex (pre–RC) and plays crucial roles in the initiation and elongation of DNA replication. 24 , 25 If the MCM complex was disrupted, cells were much more sensitive to replication stress. DNA damage was accumulated in the MCM‐depleted cells and eventually activated DDR. 24 , 26 Our previous studies have demonstrated that depletion of MCM7 in Rb‐negative cancer cells induced an obvious increase in the levels of γH2AX. Moreover, double depletion of Rb and MCM7 caused more severe DNA damage and DDR activation than individual depletion of Rb or MCM7. 15 , 16 Our results showed that the protein levels of both MCM7 and Rb were decreased significantly after Spt16 depletion (Figure 4A andB), and, in parallel with the decreases of MCM7 and Rb protein levels, an obvious DDR activation was observed in Spt16‐depleted lung cancer cells (Figure 4C andD). Accumulated DNA damage and activated DDR usually lead to cell cycle arrest and apoptosis. Thus, the activation of DDR after Spt16 depletion may also contribute to the observed cell growth inhibition of Spt16‐depleted lung cancer cells.
Micro‐RNA have been reported to regulate development and diseases by targeting complementary mRNA for degradation or translational inhibition. 27 We found that Spt16 is directly targeted by miR‐1227‐5p. The roles of miR‐1227‐5p in cancers are poorly studied. A previous report indicated that miR‐1227‐5p is downregulated in the early stages of gastric cancer. 28 In addition, it has been reported that cigarette smoking also influences the expressional levels of miR‐1227‐5p in colorectal cancer. 29 Here, we found that miR‐1227‐5p is downregulated in lung cancers, negatively regulates the expression of Spt16, and likely contributes to the elevation of Spt16 in lung cancer cells (Figure 6). However, further studies are needed to comprehensively dissect the roles of miR‐1227‐5p in cancers.
Small molecular inhibitor CBL0137 binds chromatin, changes the topology of the DNA helix, and further causes chromatin trapping of FACT. 30 , 31 Such chromatin trapping of FACT complex results in a depletion of the functional FACT complex. We found that treatment with CBL0137 showed similar effects on lung cancer cells as the effects of Spt16 depletion (Figure S2), supporting the function of Spt16 in lung cancer cells. Furthermore, our results showed that CBL0137 significantly inhibited lung cancer proliferation and induced apoptosis, suggesting that CBL0137 may be used as a novel drug candidate for lung cancer therapy.
DISCLOSURE
The authors report no conflict of interest.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported by grants from the National Natural Science Foundation of China (No. 81872272).
Yang L, Wang X, Jiao X, et al. Suppressor of Ty 16 promotes lung cancer malignancy and is negatively regulated by miR‐1227‐5p. Cancer Sci. 2020;111:4075–4087. 10.1111/cas.14627
Contributor Information
Guanjun Zhang, Email: zgjdoc@163.com.
Peijun Liu, Email: liupeijun@mail.xjtu.edu.cn.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 2. Blandin Knight S, et al. Progress and prospects of early detection in lung cancer. Open Biol. 2017;7:170070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dela Cruz CS, Tanoue LT, Matthay RA. Lung cancer: epidemiology, etiology, and prevention. Clin Chest Med. 2011;32:605‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013;20:14‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gurova K, Chang H‐W, Valieva ME, Sandlesh P, Studitsky VM. Structure and function of the histone chaperone FACT – Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech. 2018;1861:892‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Y, Zeng SX, Landais I, Lu H. Human SSRP1 has Spt16‐dependent and ‐independent roles in gene transcription. J Biol Chem. 2007;282:6936‐6945. [DOI] [PubMed] [Google Scholar]
- 7. Dinant C, Ampatziadis‐Michailidis G, Lans H, et al. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV‐induced DNA damage. Mol Cell. 2013;51:469‐479. [DOI] [PubMed] [Google Scholar]
- 8. Oliveira DV, Kato A, Nakamura K, et al. Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J Cell Sci. 2014;127:763‐772. [DOI] [PubMed] [Google Scholar]
- 9. Koman IE, Commane M, Paszkiewicz G, et al. Targeting FACT complex suppresses mammary tumorigenesis in Her2/neu transgenic mice. Cancer Prev Res (Phila). 2012;5:1025‐1035. [DOI] [PubMed] [Google Scholar]
- 10. Garcia H, Miecznikowski J, Safina A, et al. Facilitates chromatin transcription complex is an "accelerator" of tumor transformation and potential marker and target of aggressive cancers. Cell Rep. 2013;4:159‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dermawan JKT, Hitomi M, Silver DJ, et al. Pharmacological targeting of the histone chaperone complex FACT preferentially eliminates glioblastoma stem cells and prolongs survival in preclinical models. Cancer Res. 2016;76:2432‐2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burkhart C, Fleyshman D, Kohrn R, et al. Curaxin CBL0137 eradicates drug resistant cancer stem cells and potentiates efficacy of gemcitabine in preclinical models of pancreatic cancer. Oncotarget. 2014;5:11038‐11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fleyshman D, Prendergast L, Safina A, et al. Level of FACT defines the transcriptional landscape and aggressive phenotype of breast cancer cells. Oncotarget. 2017;8:20525‐20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bi L, Xie C, Yao M, et al. The histone chaperone complex FACT promotes proliferative switch of G0 cancer cells. Int J Cancer. 2019;145:164‐178. [DOI] [PubMed] [Google Scholar]
- 15. Li J, Liu J, Liang Z, et al. Simvastatin and Atorvastatin inhibit DNA replication licensing factor MCM7 and effectively suppress RB‐deficient tumors growth. Cell Death Dis. 2017;8:e2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liang Z, Li W, Liu J, et al. Simvastatin suppresses the DNA replication licensing factor MCM7 and inhibits the growth of tamoxifen‐resistant breast cancer cells. Sci Rep. 2017;7:41776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Inamura K, Ishikawa Y. Lung cancer progression and metastasis from the prognostic point of view. Clin Exp Metastasis. 2010;27:389‐397. [DOI] [PubMed] [Google Scholar]
- 18. Gasparian AV, Burkhart CA, Purmal AA, et al. Curaxins: anticancer compounds that simultaneously suppress NF‐kappaB and activate p53 by targeting FACT. Sci Transl Med. 2011;3:95ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220‐5227. [DOI] [PubMed] [Google Scholar]
- 20. Slee EA, Harte MT, Kluck RM, et al. Ordering the cytochrome c‐initiated caspase cascade: hierarchical activation of caspases‐2, ‐3, ‐6, ‐7, ‐8, and ‐10 in a caspase‐9‐dependent manner. J Cell Biol. 1999;144:281‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kale J, Osterlund EJ, Andrews DW. BCL‐2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hardwick JM, Soane L. Multiple functions of BCL‐2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drissi R, Chauvin A, McKenna A, et al. Destabilization of the MiniChromosome Maintenance (MCM) complex modulates the cellular response to DNA double strand breaks. Cell Cycle. 2018;17:2593‐2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci USA. 2008;105:8956‐8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Labib K, Tercero JA, Diffley JF. Uninterrupted MCM2‐7 function required for DNA replication fork progression. Science. 2000;288:1643‐1647. [DOI] [PubMed] [Google Scholar]
- 26. Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci USA. 2004;101:10078‐10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281‐297. [DOI] [PubMed] [Google Scholar]
- 28. Bibi F, Naseer MI, Alvi SA, et al. microRNA analysis of gastric cancer patients from Saudi Arabian population. BMC Genom. 2016;17(Suppl 9):751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mullany LE, Herrick JS, Wolff RK, Stevens JR, Slattery ML. Association of cigarette smoking and microRNA expression in rectal cancer: Insight into tumor phenotype. Cancer Epidemiol. 2016;45:98‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin M‐Z, Xia B‐R, Xu Y, Jin W‐L. Curaxin CBL0137 exerts anticancer activity via diverse mechanisms. Front Oncol. 2018;8:598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Safina A, Cheney P, Pal M, et al. FACT is a sensor of DNA torsional stress in eukaryotic cells. Nucleic Acids Res. 2017;45:1925‐1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material