

Abstract
Various clinical and experimental findings have revealed the causal relationship between autophagy failure and oncogenesis, and several mechanisms have been suggested to explain this relationship. We recently proposed two additional mechanisms: centrosome number dysregulation and the failure of autophagic cell death. Here, we detail the mechanical relationship between autophagy failure and oncogenesis.
Keywords: alternative autophagy, autophagic cell death, autophagy, centrosome, oncogenesis
Various clinical and experimental findings have revealed the causal relationship between autophagy failure and oncogenesis. Here, we detail their mechanical relationship.
![]()
1. INTRODUCTION
Autophagy is a cellular mechanism that degrades subcellular components, including proteins, lipids, and even organelles. Because autophagy is required for the maintenance of cellular homeostasis, its failure triggers various disease mechanisms, including oncogenesis. In this Review, we briefly summarize the molecular mechanisms of autophagy and then describe the relationship between autophagy and oncogenesis.
2. MOLECULAR MECHANISMS OF AUTOPHAGY
Autophagy is a catabolic process that degrades intracellular contents after enclosing them within autophagic membranes. 1 , 2 , 3 , 4 This constitutive process maintains cellular homeostasis by degradeing superfluous or damaged proteins and organelles. Autophagy is also activated to protect cells against a variety of cellular stressors, such as nutrient starvation and DNA damage. In some specific cases, the hyperactivation of autophagy has occasionally led to cell death. 5 , 6
Autophagy is driven by over 30 autophagy‐related proteins (Atgs), which are well conserved from yeasts to mammals. 1 , 2 , 3 , 4 Unc51‐like kinase 1 (Ulk1), a serine/threonine kinase, forms the Ulk1 complex together with Fip200, Atg13, and Atg101. In healthy conditions, Ulk1 is phosphorylated and inactivated by mammalian target of rapamycin complex 1 and AMP‐activated protein kinase. Upon starvation, Ulk1 is dephosphorylated by protein phosphatase 2A and subsequently translocates to pre‐autophagosomal membranes, which are the initial platforms of the phagophore membrane (Figure 1). Autophagy is also regulated by phosphatidylinositol 3‐kinase (PI3K) class III, which promotes the invagination of the membrane at domains rich in phosphatidylinositol‐3‐phosphate and generates phagophores. The phagophore membrane originates from the endoplasmic reticulum (ER) or mitochondria‐associated ER membranes (MAM). 7 , 8 The subsequent expansion and closure of phagophores generate autophagosomes via two ubiquitin‐like conjugation pathways, namely, the Atg5‐Atg12 pathway and the microtubule‐associated protein 1 light chain 3 (LC3)–phosphatidylethanolamine (PE) pathway. In the former pathway, Atg7 is required for the conjugation of Atg12 to Atg5 as an E1‐like enzyme. Conjugation of phosphatidylethanolamine to LC3 is mediated by the actions of Atg3 and the Atg5‐Atg12 complex, as E2‐like and E3‐like enzymes, respectively. Because autophagosome formation is largely disturbed in cells lacking Atg5 and Atg7, they are considered an essential molecule for autophagy (Figure 1). The modification of LC3 to LC3–PE and its translocation from the cytosol to the autophagic membrane are considered reliable autophagy markers.
FIGURE 1.
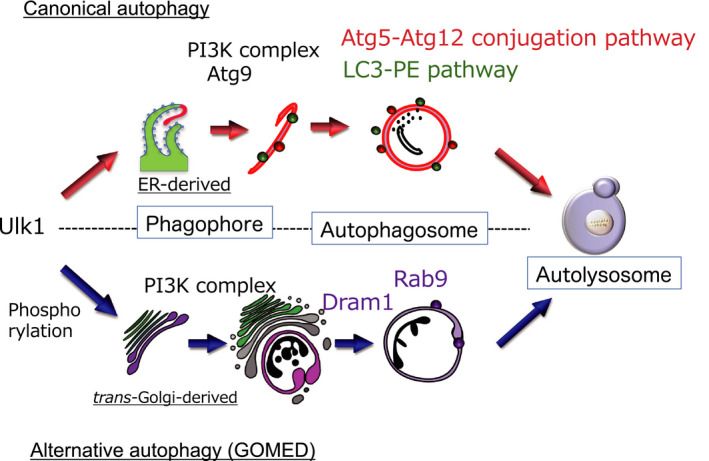
Hypothetical model of autophagy. There are at least two modes of autophagy, namely, canonical and alternative autophagy. Canonical autophagy requires autophagy‐related protein (Atg)‐5 and originates from the endoplasmic reticulum (ER) membrane. In contrast, alternative autophagy occurs independently of Atg5 and originates from the Golgi membrane. LC3, microtubule‐associated protein 1A/1B‐light chain 3; PI3K, phosphatidylinositol 3‐kinase; Rab9, Ras‐related protein 9; Ulk1, Unc51‐like kinase 1
3. NONCANONICAL AUTOPHAGY
The importance of Atg5 in autophagy has been widely accepted, as described. However, two types of Atg5‐independent autophagy have been reported. One is a residual canonical autophagy that is slowly driven via the standard autophagy mechanism and does not involve Atg5. 9 The other is an alternative type of autophagy, namely, alternative autophagy or Golgi membrane‐associated degradation (GOMED), which is driven via a different mechanism to canonical autophagy 10 , 11 , 12 , 13 (Figure 1). Alternative autophagy originates from the Golgi membrane instead of from the ER or MAM, 10 , 11 and with the exception of Ulk1 and PI3K, the molecules involved are different from those of canonical autophagy (Table 1). Ulk1 is an essential molecule for both types of autophagy, and we recently identified the mechanism whereby Ulk1 regulates each type 14 (Figure 2). Ulk1 function is largely dependent on its phosphorylation status, and its dephosphorylation at serine637 is required for the activation of both autophagy types. Subsequent phosphorylation at serine746 determines the type of autophagy; that is, serine746 phosphorylation induces alternative autophagy, but if this does not occur, canonical autophagy occurs. Importantly, phosphorylated Ulk1 at serine746 is entirely located on the Golgi membrane, where phagophores originate in alternative autophagy. p‐Ulk1746 induces the elongation of ministacked Golgi membranes. Then, Golgi membranes are elongated to become pahogophore membranes via PI3K‐dependent manner. The subsequent closure of phagophores generate autophagosomes via fusion between trans‐Golgi‐derived membranes and endosomal vesicular membranes. In these steps, Ras‐related protein 9 and Dram1 are utilized instead of Atg5, Atg7, and LC3 (Figure 1). Finally, Golgi membrane‐derived autophagosomes fuse with lysosomes to generate autolysosomes.
TABLE 1.
A comparison between canonical and alternative types of autophagy
| Canonical type | Alternative type | |
|---|---|---|
| Morphology | Autophagosome/Autolysosome | Autophagosome/Autolysosome |
| Phylogenetic conservation | Yeast to Mammals | Yeast to Mammals |
| Membrane source | ER, MAM | Golgi membrane |
| Inducible condition | Starvation, Rapamycin | Golgi stress |
| Specific substrate |
p62, LC3 Parkin‐dependent mitophagy |
Insulin granules, Erythrocyte mitophagy |
FIGURE 2.
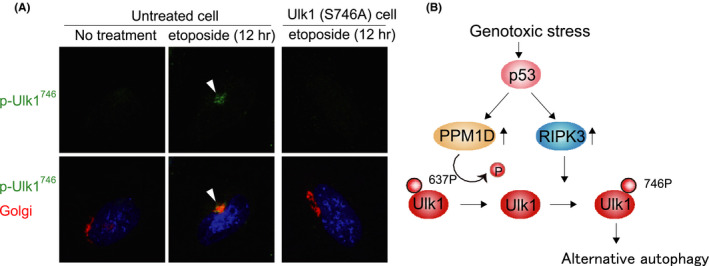
Mechanism of genotoxic stress‐induced autophagy (modified from ref. 14 ). (A) Genotoxic stress‐induced alternative autophagy is activated by phosphorylated Unc51‐like kinase 1 (p‐Ulk1)746, which is entirely localized on the Golgi membrane. The indicated mouse embryonic fibroblasts were treated with etoposide and immunostained with anti‐p‐Ulk1746 and anti‐GS28 antibodies. Representative images of p‐Ulk1746 (upper panels) and merged images (lower panels) are shown. Arrowheads indicate p‐Ulk1746 signals on the Golgi. (B) Schematic model of genotoxic stress‐induced autophagy. Genotoxic stress induces Ulk1 dephosphorylation at serine637 in a p53/protein phosphatase magnesium‐dependent 1D, delta isoform (PPM1D)‐dependent manner. This step is essential for both autophagy types. The subsequent phosphorylation of Ulk1 at serine746 by p53/RIPK3 is essential for the induction of alternative autophagy
Alternative autophagy is a common cell function that is phylogenetically conserved from yeasts to humans, 11 and it is not a compensatory mechanism for canonical autophagy. Almost all cells possess canonical and alternative autophagy mechanisms, and the type of autophagy depends on the substrate or type of cellular stress. For example, starvation stimulus mostly induces canonical autophagy, whereas genotoxic stress activates both autophagy types 10 (Table 1). Canonical autophagy selectively degrades p62 and LC3, but these molecules are not degraded by alternative autophagy. In contrast, mitochondrial elimination during erythrocyte maturation is mediated by alternative autophagy. 15 The presence of different degrading substrates indicates different biological functions (Table 1). Until now, alternative autophagy has been reported as involved in erythrocyte maturation, 15 the regulation of insulin secretion, 11 neuroprotection, 16 heart protection, 17 and the inhibition of inflammatory bowel diseases. 18
4. CANCER AND AUTOPHAGY
Various clinical and experimental findings regarding the relationship between cancer and autophagy have been reported. A deficiency of phosphatase and tensin homolog (PTEN), a protein inhibitor of PI3K, was found to cause cancer in mice. 19 Autophagy was considered to be involved in this mechanism of oncogenesis because PTEN inhibits autophagy‐inducing PI3K–protein kinase B–mammalian target of rapamycin (mTOR) signaling. 20 Several experimental studies have also suggested the involvement of autophagy in cancer. For example, liver‐specific Atg5 (or Atg7)‐deficient mice were found prone to benign tumors, 21 , 22 and the hetero‐knockout of Beclin 1, a key molecule for autophagy, resulted in cancer‐prone mice. In a xenograft cancer model, Beclin 1‐deficient cancer cells showed accelerated tumor growth that was suppressed by exogenously transfected Beclin 1. 23 , 24 Although Beclin 1 is involved in autophagy and in membrane trafficking, the involvement of autophagy in oncogenesis was proven by detailed analyses.
From a clinical aspect, a high frequency of Beclin 1 genomic mutations has been reported in ovarian (75%), breast (50%), and prostatic cancer (40%). 24 The involvement of gene mutations of Atg5, LC3, and FAK family kinase‐interacting protein of 200 kDa, have been reported in leukemia, glioblastoma, and breast cancer, respectively. 25 , 26 Excess accumulation of p62, which is induced by autophagy dysfunction, is also frequently found in hepatocellular carcinoma and glioma. 21 These findings imply the causal relationship between autophagy failure and oncogenesis.
In contrast with autophagy failure‐induced oncogenesis, autophagy acceleration is considered important for tumor growth. For example, the central region of solid tumors is hypoxic and undernourished. In such circumstances, autophagy‐mediated nutritional supplies are essential for cancer cell proliferation, 27 meaning that autophagy contributes to the survival and proliferation of cancer cells in suboptimal environments. The involvement of autophagy in cancer cell migration, metastasis, and dissemination has also been reported. 28 Thus, the role of autophagy in cancer development is dependent on the cell context.
5. INVOLVEMENT OF AUTOPHAGY FAILURE IN ONCOGENESIS
How does autophagy failure contribute to oncogenesis? It is widely accepted that oncogenesis is primarily triggered by multiple gene alterations caused by point mutations, recombination, amplifications, and deletions, and hence, genotoxic stress plays an important role. Genotoxic insults include a wide variety of factors, such as DNA replication errors, spontaneous and UV‐induced mutations, toxic molecules, and reactive oxygen species. One of the most reasonable scenarios explaining autophagy failure‐induced oncogenesis is the hypergeneration of these genotoxic insults by autophagy failure. 29 In healthy cells, damaged or aged organelles, such as mitochondria and peroxisomes, are usually degraded by autophagy, which is evident because most cells contain abnormal organelles in autophagy‐deficient mice. Thus, in cells with reduced autophagic activity, damaged organelles are insufficiently degraded, resulting in the generation of toxic intra‐organellar molecules or oxygen radicals, which become genotoxic insults. Alternatively, autophagy failure increases necrotic cells due to the blockage of autophagy‐mediated nutrition supplies. 27 Necrotic cells are thought to release danger‐associated molecular patterns that trigger an inflammatory response, which may promote genotoxic stress.
Various studies have shown the involvement of p62 in cancer development. 21 , 30 p62 is a well‐known autophagy substrate that accumulates in cells showing decreased autophagy activity. p62 plays a role in protecting cells from cellular stress via several different signaling pathways. For example, p62 binds to kelch‐like ECH‐associated protein 1 to prevent it from trapping nuclear factor erythroid 2‐related factor 2 (Nrf2), the master transcription factor of the anti‐oxidative response, resulting in Nrf2 stabilization. 31 , 32 p62 also activates nuclear factor kappa‐light‐chain‐enhancer of activated B cells and mTOR via direct interaction with tumor necrosis factor receptor‐associated factor 6/receptor‐interacting serine/threonine‐protein kinase (RIPK)‐1 and mammalian mitogen‐activated protein kinase kinase kinase/regulatory‐associated protein of mTOR, respectively. 33 The former signal regulates multiple aspects of immune functions and inflammatory responses and also inhibits apoptosis signaling, whereas the latter signal regulates the nutrient response, both of which results in cell tolerance to various stressors. These mechanisms should contribute to cancer cell survival and promote tumor progression. The crucial role of p62 in cancer progression is evident in hepatocellular carcinoma because p62 accumulation is markedly observed in clinical liver tumors. 31 , 32 This is also supported by experimental findings, in which liver tumors that were observed in liver‐specific Atg5 (or Atg7)‐deficient mice were largely reduced by the concomitant loss of p62. 21
6. ALTERNATIVE MECHANISMS OF AUTOPHAGY FAILURE‐INDUCED ONCOGENESIS
In addition to the mechanisms described above, we recently identified two additional mechanisms that explain autophagy failure‐induced oncogenesis: centrosome number dysregulation and the failure of autophagic cell death.
The centrosome is an organelle that plays an essential role in the organization of the microtubule network during mitosis. During prophase, two centrosomes move to opposite poles of the cell to properly segregate chromosomes. If there are more than two centrosomes, mis‐segregation occurs, which leads to genomic instability and may trigger oncogenesis 34 (Figure 3). The centrosome number was considered to be regulated by the ubiquitin‐proteasome pathway. However, we recently found that autophagy also participates in the regulation of centrosome number. 35
FIGURE 3.
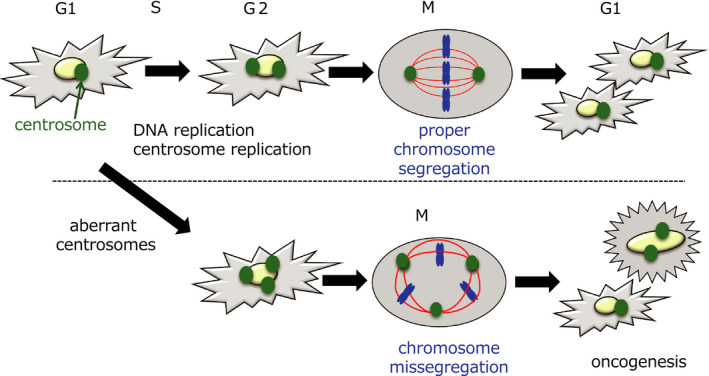
Mechanism of oncogenesis from an excess number of centrosomes. During prophase, two centrosomes move to opposite poles of the cell to properly segregate chromosomes. Excess centrosomes cause chromosome mis‐segregation, leading to genomic instability that may trigger oncogenesis
First, we noticed that many Atg5‐deficient cells contained three or more centrosomes (Figure 4A). This finding was confirmed using different Atg5‐ and Atg7‐deficient cells. Furthermore, chemical inhibition of autophagy also increased the number of centrosomes, confirming the involvement of autophagy in regulating the number of centrosomes. Therefore, autophagy failure generates multiple centrosomes, resulting in oncogenesis.
FIGURE 4.
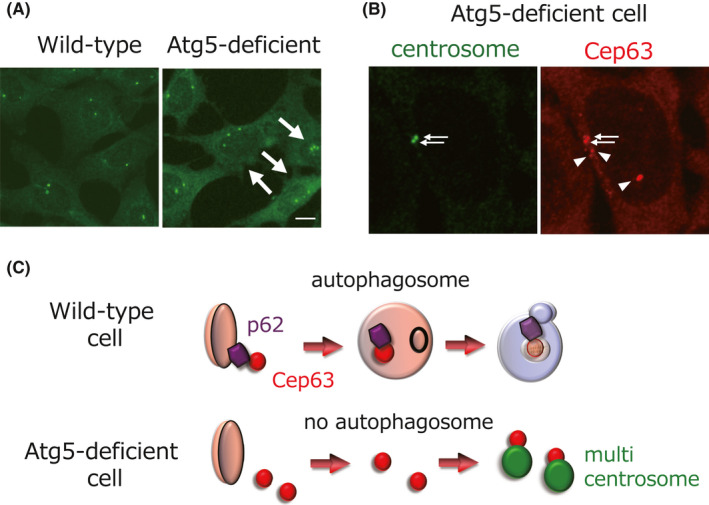
Autophagy regulates centrosome number (modified from ref. 15 ). (A) Autophagy‐related protein 5 (Atg5)‐deficient cells contain excess centrosomes. Centrosomes were immunostained with an anti‐γ‐tubulin antibody. Arrows indicate cells with excess centrosomes. (B) Atg5‐deficient cells contain many aberrant centrosomal protein 63 (Cep63) puncta. Atg5−/− mouse embryonic fibroblasts were immunostained with anti‐γ‐tubulin antibody (green; left) and anti‐Cep63 antibody (red; right) and were examined by fluorescence microscopy. Arrows and arrowheads indicate mature centrosomes and extra Cep63 dots, respectively. (C) Schematic model of autophagy failure‐induced centrosome overproduction. In healthy cells, the centrosome number remains normal (n = 2) because cytosolic Cep63 dots are degraded into autophagosomes via p62. In autophagy‐deficient cells, the centrosome number increases because many Cep63 dots exist in the cytosol
In order to elucidate the mechanism how autophagy failure generates multiple centrosomes, we first hypothesized that autophagy directly degraded excess centrosomes when aberrant centrosomes were generated. However, this proposal was disproved because we did not find any mature centrosomes in the autophagic vacuoles. After careful analysis, we identified centrosomal protein 63 (Cep63) as an autophagy substrate responsible for regulating the number of centrosomes. Cep63 usually localizes on the centrosome and plays an essential role for centrosome replication. However, this molecule is often abnormally generated in the cytosol and occasionally makes aberrant centrosomes (Figure 4B). But, autophagy degrades cytosolic Cep63 and hence prevents abnormal centrosome generation in healthy cells. In contrast, in autophagy‐deficient cells, abnormal centrosomes are generated from undigested cytosolic Cep63. This conclusion was obtained from the following findings: (a) multiple Cep63 dots appeared in autophagy‐deficient cells or in cells treated with autophagy inhibitors, (b) there were multiple Cep63 dots within autolysosomes when substrate degradation was blocked by E64d (lysosomal cysteine proteinase inhibitor) and pepstatin A (an inhibitor for lysosomal aspartic proteinases), (c) Cep63 interacted with p62 and was incorporated into autophagosomes, and (d) enforced expression of Cep63 generated aberrant centrosomes. Therefore, autophagy failure increases the expression level of cytosolic Cep63, resulting in the generation of aberrant centrosomes (Figure 4C), which may facilitate oncogenesis via chromosome mis‐segregation. It must be noted that Cep63 is degraded by canonical autophagy, not alternative autophagy, because Cep63 degradation occurs via its association with p62.
7. THE ROLE OF AUTOPHAGIC CELL DEATH IN ONCOGENESIS
Autophagy is activated by most cellular stressors and plays a protective role against cellular stressors in most cases. However, autophagy can be utilized as a mechanism of cellular suicide, 5 , 36 which is referred to as autophagic cell death (Figure 5A). The simple association of autophagy with cell death is insufficient for autophagic cell death, and it is essential that the suppression of cell death by autophagy inhibitors (eg, 3‐methyl adenine) or by the genetic ablation of autophagy (eg, knockout or small interfering RNA silencing of essential autophagy genes) is also demonstrated. Thus, if autophagy inhibition does not prevent cell death, this process should not be referred to as autophagic cell death. The molecular mechanism of autophagic cell death has yet to be fully elucidated. However, it requires not only autophagy induction but also Jun amino‐terminal kinase (JNK) activation, because JNK inhibition largely suppresses autophagic cell death 37 (Figure 5B).
FIGURE 5.
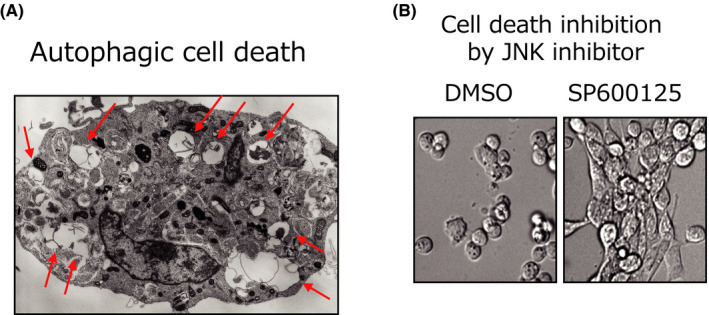
Involvement of Jun amino‐terminal kinase (JNK) in autophagic cell death. (A) Representative electron micrograph of autophagic cell death. Mouse embryonic fibroblasts (MEFs) were treated with etoposide for 24 h. There are many autophagic vacuoles (arrows) present in the cytosol, and the organelles are almost normal. (from ref. 5 ). (B) Reduction of etoposide‐induced death by the inhibition of JNK. MEFs were treated with etoposide in the presence of JNK inhibitor SP600125 for 24 h, and then examined by phase‐contrast microscopy (modified from ref. 37 )
A large body of evidence indicates that apoptosis inhibition is critical for tumorigenesis, but the elimination of cancer cells may also be mediated by autophagic cell death because this occurs in normal cells (eg, fibroblasts or thymocytes) but not in most cancer cells. 37 Furthermore, in some cancer cells, the magnitude of JNK activation, which is a crucial factor for autophagic cell death, is significantly lower compared with that in normal cells following exposure to cellular stress. 37 Thus, in these cancer cells, the JNK activity level may not attain the threshold level required to induce autophagic cell death. This conclusion is supported by evidence that the enforced expression of activated JNK in cancer cells induced autophagic cell death. 37 Taken together, it is likely that the failure of autophagic cell death by insufficient JNK activation enables the survival of precancerous cells, leading to malignant cancer.
8. P53, AUTOPHAGY, AND ONCOGENESIS
p53 is a tumor suppressor gene. It is the most frequently mutated gene in cancers, and its mutations cause oncogenesis. The p53 gene encodes a transcription factor that mediates various cellular stresses, particularly genotoxic stress. Because a wide variety of factors cause genotoxic stress, the ratio of DNA damage occurs at a rate of 10 000‐1 000 000 molecular lesions per cell per day. 38 p53 plays a role in surveying and correcting gene mutations, and the lack of p53 increases DNA damage and triggers oncogenesis. p53 also regulates a wide variety of cellular responses against genotoxic stress, and most of them contribute to the avoidance of oncogenesis. For example, cells with severe DNA damage are killed by apoptosis in a p53‐dependent manner. This mechanism is driven by the transcriptionally upregulated Puma, Noxa, Bim, and Bax pro‐apoptotic proteins in a p53‐dependent manner, 39 and is important to eliminate cancer‐prone cells with severe DNA damage. p53 is also involved in autophagy in response to genotoxic stress. Genotoxic stress almost equivalently activates canonical and alternative autophagy in a p53‐dependent manner. We recently identified how p53 activates genotoxic stress‐induced autophagy. One mechanism is the transcriptional upregulation of Ulk1. 10 As described, this molecule functions in the initial step of both types of autophagy (Figure 1), but its upregulation alone is insufficient to induce autophagy, and an additional factor, Ulk1 dephosphorylation at serine637, is required (Figure 2). The responsible phosphatase is protein phosphatase magnesium‐dependent 1D, delta isoform, which is also transcriptionally upregulated by p53. 40 Accordingly, autophagy is activated by the concomitant upregulation and dephosphorylation of Ulk1. Thus, genotoxic stress‐induced autophagy mediated via the p53‐Ulk1 axis should be useful to prevent oncogenesis.
9. CLOSING REMARK
As described, autophagy is an important cellular function against oncogenesis. However, the relationship between autophagy activity and oncogenesis or cancer development from a clinical aspect has not been fully elucidated. Such an approach appears to require an understanding of the precise role of autophagy in cancer. Furthermore, autophagy‐based cancer chemotherapeutics could be promising.
DISCLOSURE
The authors declare no conflicts of interest associated with this manuscript.
ETHICAL APPROVAL
The Tokyo Medical and Dental University Ethics Committee for Animal Experiments approved all experiments in this study, and all experiments were performed according to their regulations.
ACKNOWLEDGMENTS
This study was supported in part by Grant‐in‐Aid for Scientific Research (A) (17H01533, 20H00467), Grant‐in‐Aid for Scientific Research on Innovative Areas (17H06413, 17H06414) from the MEXT of Japan, by the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) (JP18cm0106109), by the Project for Psychiatric and Neurological Disorders (JP18dm0107136), by the Project for Practical Research Project for Rare/Intractable Diseases (JP19ek0109407) from the Japan Agency for Medical Research and Development. This study was also supported by the Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University.
Torii S, Honda S, Murohashi M, Yamaguchi H, Shimizu S. Autophagy involvement in oncogenesis. Cancer Sci. 2020;111:3993–3999. 10.1111/cas.14646
REFERENCES
- 1. Ohsumi Y. Molecular dissection of autophagy: two ubiquitin‐like systems. Nat Rev Mol Cell Biol. 2001;2:211‐216. [DOI] [PubMed] [Google Scholar]
- 2. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451:1069‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107‐132. [DOI] [PubMed] [Google Scholar]
- 4. Shimizu S. Biological roles of alternative autophagy. Mol Cells. 2018;41:50‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl‐2 family proteins in a non‐apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221‐1228. [DOI] [PubMed] [Google Scholar]
- 6. Bialik S, Dasari SK, Kimchi A. Autophagy‐dependent cell death – where, how and why a cell eats itself to death. J Cell Sci. 2018;131(18):jcs215152. [DOI] [PubMed] [Google Scholar]
- 7. Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER‐mitochondria contact sites. Nature. 2013;495:389‐393. [DOI] [PubMed] [Google Scholar]
- 8. Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831‐835. [DOI] [PubMed] [Google Scholar]
- 9. Tsuboyama K, Koyama‐Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036‐1041. [DOI] [PubMed] [Google Scholar]
- 10. Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7‐ independent alternative macroautophagy. Nature. 2009;461:654‐658. [DOI] [PubMed] [Google Scholar]
- 11. Yamaguchi H, Arakawa S, Kanaseki T, et al. Golgi membrane‐associated degradation pathway in yeast and mammals. EMBO J. 2016;35:1991‐2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Codogno P, Mehrpour M, Proikas‐Cezanne T. Canonical and non‐canonical autophagy: variations on a common theme of self‐eating? Nat Rev Mol Cell Biol. 2011;13:7‐12. [DOI] [PubMed] [Google Scholar]
- 13. Galluzzi L, Green DR. Autophagy‐independent functions of the autophagy machinery. Cell. 2019;177:1682‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Torii S, Yamaguchi H, Nakanishi A, et al. Identification of a phosphorylation site on Ulk1 required for genotoxic stress‐induced alternative autophagy. Nat Commun. 2020;11:1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. Ulk1‐mediated Atg5‐independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun. 2014;5:4004. [DOI] [PubMed] [Google Scholar]
- 16. Baron O, Boudi A, Dias C, et al. Stall in canonical autophagy‐lysosome pathways prompts nucleophagy‐based nuclear breakdown in neurodegeneration. Curr Biol. 2017;27:3626‐3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saito T, Nah J, Oka SI, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019;129:802‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ra EA, Lee TA, Kim SW, et al. TRIM31 promotes Atg5/Atg7‐independent autophagy in intestinal cells. Nat Commun. 2016;7:11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue‐by‐tissue survey. Oncogene. 2008;27:5398‐5415. [DOI] [PubMed] [Google Scholar]
- 20. Arico S, Petiot A, Bauvy C, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3‐kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243‐35246. [DOI] [PubMed] [Google Scholar]
- 21. Takamura A, Komatsu M, Hara T, et al. Autophagy‐deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077‐15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winardi D, Tsai HP, Chai CY, et al. Correlation of altered expression of the autophagy marker LC3B with poor prognosis in astrocytoma. Biomed Res Int. 2014;2014:723176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iqbal J, Kucuk C, deLeeuw RJ, et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer‐cell malignancies. Leukemia. 2009;23:1139‐1151. [DOI] [PubMed] [Google Scholar]
- 27. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshida T, Tsujioka M, Honda S, Tanaka M, Shimizu S. Autophagy suppresses cell migration by degrading GEF‐H1, a RhoA GEF. OncoTarget. 2016;7:34420‐34429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu EY, Ryan KM. Autophagy and cancer – issues we need to digest. J Cell Sci. 2012;125:2349‐2358. [DOI] [PubMed] [Google Scholar]
- 30. Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1‐Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618‐631. [DOI] [PubMed] [Google Scholar]
- 32. Saito T, Ichimura Y, Taguchi K, et al. p62/Sqstm1 promotes malignancy of HCV‐positive hepatocellular carcinoma through Nrf2‐dependent metabolic reprogramming. Nat Commun. 2016;7:12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moscat J, Karin M, Diaz‐Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell. 2016;167:606‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Godinho SA, Kwon M, Pellman D. Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009;28:85‐98. [DOI] [PubMed] [Google Scholar]
- 35. Watanabe Y, Honda S, Konishi A, et al. Autophagy controls centrosome number by degrading Cep63. Nat Commun. 2016;7:13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arakawa S, Tsujioka M, Yoshida T, et al. Role of Atg5‐dependent cell death in the embryonic development of Bax/Bak double‐knockout mice. Cell Death Differ. 2017;24:1598‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shimizu S, Konishi A, Nishida Y, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29:2070‐2082. [DOI] [PubMed] [Google Scholar]
- 38. Lodish H, Berk A, Matsudaira P, et al. Molecular Cell Biology, 5th ed New York, NY: WH Freeman; 2004:963. [Google Scholar]
- 39. Shimizu S, Tsujimoto Y. Proapoptotic BH3‐only Bcl‐2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage‐dependent anion channel activity. Proc Natl Acad Sci USA. 2000;97:577‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Torii S, Yoshida T, Arakawa S, Honda S, Nakanishi A, Shimizu S. Identification of PPM1D as an essential Ulk1 phosphatase for genotoxic stress‐induced autophagy. EMBO R. 2016;17:1552‐1564. [DOI] [PMC free article] [PubMed] [Google Scholar]