

Abstract
Cervical cancer (CC) remains one of the leading causes of mortality of female cancers worldwide, with more than 90% being cervical squamous cell carcinoma (CSCC). ΔNp63α is the predominant isoform expressed in cervical epithelial tissues and exerts its antitumor function in CSCC. In this study, we have identified 39 long noncoding RNAs as ΔNp63α targets in CSCC through RNA sequencing and chromatin immunoprecipitation sequencing, in which we further confirmed and focused on the two tumor‐related long noncoding RNAs, PART1 (lncPART1) and MIR17HG (lncMIR17HG). Experiments from stable overexpression/knockdown cell lines revealed that lncPART1 and lncMIR17HG regulated cell proliferation, migration, and invasion. In vivo experiments further showed that lncPART1 suppresses tumor growth in CSCC‐derived tumors. Examinations of clinical tissues indicated that the expression of lncPART1 was positively correlated with ΔNp63α expression, while lncMIR17HG was negatively correlated with ΔNp63α expression, suggesting that ΔNp63α plays a central role via regulating its direct targets in the progression of CSCC. These findings provide novel insights in targeted therapy of cervical cancers.
Keywords: cervical cancer, CSCC, lncMIR17HG, lncPART1, ΔNp63α
lncRNA PART1 and MIR17HG are ΔNp63α direct targets and regulate cell proliferation and metastasis of cervical squamous cell carcinoma (CSCC). ΔNp63α plays a central role via regulating its direct targets in the progression of CSCC.
1. INTRODUCTION
Cervical cancer is the fourth most common cancer among women and ranks 2 in mortality of female cancers. More than 90% of cervical cancers are cervical squamous cell carcinoma (CSCC) with abnormal regenerative proliferation and blocked differentiation. 1 , 2 , 3 , 4 Effective prophylactic vaccines against the most important carcinogenic human papillomavirus types are available, but the number of people receiving the vaccine remains low. 5 , 6 Clinically, surgery, radiotherapy, and chemotherapy have been widely used to improve the overall survival rate of CSCC; however, the 5‐year survival rate also remains low for advanced CSCC patients, especially for metastatic cervical cancer patients with a survival rate around 5%‐15%. 7 , 8 The underlying molecular mechanisms in tumorigenesis for CSCC need further clarification, which would promote our understanding of the clinical treatment of CSCC.
p63, a member of the p53 gene family, acts as a key transcription factor involved in cell growth, proliferation, apoptosis, and differentiation. p63 generates two different isoforms (TA isoform and ΔN isoform) from different transcription promoters. Both TA and ΔN isotypes can undergo alternative splicing to generate different carboxytermini, including α, β, γ, δ, and ε. 9 , 10 , 11 Among them, ΔNp63α is the predominant isoform expressed in cervical epithelial tissues. 12 , 13 , 14 As the major isotype controlling epithelium morphogenesis, aberrant expression of ΔNp63α leads to abnormal differentiation and epithelial‐mesenchymal transition behavior through various mechanisms. 15 , 16 , 17 We have previously demonstrated that ΔNp63α exerts its antitumor function in CSCC by regulation of its direct targets. 18 , 19 Further investigations of other ΔNp63α direct targets are needed to better understand the functional mechanisms of ΔNp63α in CSCC.
Aberrant activation of RNA regulatory networks is reported to promote changes in cell state that may be involved in tumorigenesis. Long noncoding RNAs (lncRNAs) are a class of RNA molecules with over 200 nucleotides in length. 20 , 21 , 22 With the development of RNA‐sequencing (RNA‐seq) techniques, more and more lncRNAs are reported to play important roles in epigenetic regulation, transcriptional regulation, and posttranscriptional regulation. 23 , 24 lncRNAs are widely involved in the progression of various tumor cells, such as bladder cancer, lung cancer, hepatocellular carcinoma, etc. 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 However, only a limited number of lncRNAs, such as MEG3, MALAT1, GAS5, HOTAIR, and EBIC, have been identified to relate to CSCC progression. 2 , 33 , 34 , 35 , 36 , 37
In this study, we identified that the lncRNAs lncPART1 and lncMIR17HG are the main direct transcriptional targets of ΔNp63α by overlapping chromatin immunoprecipitation sequencing (ChIP‐seq) and RNA‐seq. Overexpression of lncPART1 suppressed the proliferation, migration, and invasion of CSCC cells. Knockdown of lncPART1 promoted the proliferation, migration, and invasion of CSCC cells. In vivo experiments also showed that lncPART1 suppresses tumor growth in CSCC‐derived tumors. On the other hand, knockdown of lncMIR17HG suppressed the proliferation, migration, and invasion of CSCC cells, acting more like an oncogene. 38 , 39 Our work demonstrated that ΔNp63α plays a central role via regulating its direct targets in the progression of CSCC, either upregulating or downregulating, providing new insights for the diagnosis and treatment of CSCC.
2. MATERIALS AND METHODS
2.1. Cell lines
The cervical cancer cells SiHa, ME‐180, C‐33A, HeLa, HaCat, and 293T were obtained from the American Type Cancer Culture (ATCC). SiHa, C‐33A, HeLa, and 293T were inoculated with DMEM (HyClone), HaCat was inoculated with MEM (Gibco, Thermo Fisher Scientific), and ME‐180 was inoculated with Macoy's 5A medium (Gibco, Thermo Fisher Scientific) containing 10% fetal bovine serum (Gibco, Thermo Fisher Scientific), 100 units/mL penicillin, and 100 mg/mL streptomycin (HyClone) and cultured in an incubator at 37°C with 5% CO2 and saturated humidity. After cells had grown along the dish wall, the medium was changed every 1‐2 days and 0.25% trypsin (Sigma‐Company) was used for digestion and subculture. All cells were tested for mycoplasma by a PCR‐based method as well as DAPI staining to ensure the absence of contamination.
2.2. Construction of SiHa/PART1 and ME‐180/shPART1 stable cell lines
pLVX‐IRES‐mcherryy‐lnc‐PART1 plasmid was constructed by inserting a full length of human lnc‐PART1 cloned by RT‐PCR into the EcoRI and NotI site of pLVX‐IRES‐mcherryy. pLVX‐IRES‐mcherryy‐lncPART1 and the control plasmid with their packaging vectors pmd2g and pspax2 were cotransfected to the 293T cells. Then, virus supernatant was used to infect the SiHa cells. SiHa/PART1 stable cell lines and SiHa/Con cells were generated by selection with 100 μg/mL G418 for 2 weeks as described previously.
Knockdown of PART1 was achieved using predesigned shRNA oligonucleotides. Specific shRNA has been annealed to connect to the pLKO.1 puro vector. Lentivirus packaging plasmid Gag/Pol, Rev, VSV‐G, and shNC, shPART1 were cotransfected to the 293T cells. Then, the virus supernatant infected the ME‐180 cells, respectively. Stable cell lines and were generated by selection with 2 μg/mL puromycin for 2 weeks.
2.3. Construction of SiHa/si Con and SiHa/si lncMIR17HG cell lines
Transfection of control plasmid and siRNA was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols.
2.4. RNA extraction and qRT‐PCR analyses
The total RNA samples of the cell line were isolated using TRIzol reagent according to the manufacturer's protocol (Life Technologies) for RNA extraction and quantitative real‐time polymerase chain reaction (qRT‐PCR) analyses. Then, 500 ng of the total RNA was reversely transcribed in a final volume of 20 μL, using random primers and standard conditions with the GoScript Reverse Transcription System (Promega). Subsequently, we performed qRT‐PCR using the SYBR Select Master Mix with 2 μL complementary DNA (cDNA) according to the manufacturer's instructions. The qRT‐PCR reaction included an initial denaturation step at 95°C for 30 seconds, which was then followed by 40 cycles at 95°C for 5 seconds and 60°C for 1 minute. All the primer sets are shown in Table S1. Note that the primers for ΔNp63α are specific for ΔNp63α, instead of TAp63α. GAPDH and specific transcript levels for each transfection condition were measured in triplicate. The ΔΔC T method was applied to quantify relative gene expression.
2.5. Chromatin immunoprecipitation (ChIP)
ChIP assay was performed as described previously. 40 , 41 Briefly, cells were cross‐linked in a UV cross‐linker (UVP) at 200 mJ. After rinsing with PBS twice, the cell pellets were lysed in 1 mL of SDS lysis buffer (1% (w/v), 10 mM EDTA, and 50 mM Tris‐HCl (pH 8.1) containing Complete protease inhibitor cocktail (Roche) and were incubated for 20 minutes on ice. Cell extracts were sonicated for 5 minutes with a Vibra‐Cell processor (Sonics & Materials, Inc.). A 100‐μL sample of the supernatant was saved as input. The remaining sample was diluted 1:10 in a ChIP dilution buffer (0.01% (w/v) SDS, 1.1% (v/v) Triton X‐100, 1.2 mM EDTA, 16.7 mM Tris‐HCl, pH 8.1, and 167 mM NaCl) containing protease inhibitors. The chromatin solution was precleared and immunoprecipitated with 2 μg of p63 antibody (CST, 13109S) or normal IgG control. The immunocomplexes were eluted in 1% (w/v) SDS and 50 mM NaHCO3, and crosslinks were reversed for 6 hours at 65°C. Samples were digested with proteinase K for 1 hour at 45°C, and the DNA was extracted with phenol/chloroform/isoamyl alcohol.
2.6. RNA‐seq preparation and sequencing
For lncRNA‐seq, 5 μg extraction of the total RNA was iron‐fragmented at 95°C and then subjected to end repair and 5′‐adaptor ligation. Then, reverse transcription was performed with random primers containing 3′ adaptor sequences and randomized hexamers. The cDNAs were purified, amplified, quantified, and stored at −80°C until sequencing. Differentially expressed lncRNAs were identified using a t‐test (P < 0.05) combined with fold change (FC) (log2(FC)>2 for upregulated lncRNAs, and log2(FC)<−2 for downregulated lncRNAs). These differentially expressed lncRNAs were visualized by a heatmap and volcano analyses using R program. All the primers are shown in Table S1.
2.7. Cell proliferation in vitro(ACEA)
6 × 104 cells (SiHa/Con, SiHa/lncPART1, ME‐180/Con, ME‐180/shlncPART1, SiHa/siCon, and SiHa/silncMIR17HG cells) were resuspended in 1 mL DMEM or Myco5A containing 10% FBS. 100 μL of the cell suspension was seeded into each well of the 16‐well plate (ACEA Biosciences Inc) and put into an xCELLigence RTCA DP measuring instrument (Roche). The number of cells per well was then counted at the indicated time points. Each type of cells was counted in triplicates. The cell growth plot was analyzed by xCELLigence (ACEA Biosciences Inc).
2.8. Colony formation assay
The number of all cells per hole was strictly counted, and the cells were kept in uniform distribution. For SiHa/Con, SiHa/lncPART1, ME‐180/Con, ME‐180/shlncPART1, SiHa/si Con, and SiHa/silncMIR17HG cells, 100 and 300 cells were distributed into 6‐well plates separately and were run in triplicate. Cells were allowed to grow for 2 weeks in 5% CO2 incubators before being stained with 0.5% crystal violet staining solution (Solarbio). The quantitation of the colony formation assays was described in a histogram. The results represent mean values of two duplicate experiments, and the error bars show SD.
2.9. Scratch, migration, and invasion assays
For scratch assays, 400 000 cells, including SiHa/Con, SiHa/lncPART1, ME‐180/Con, ME‐180/shlncPART1, SiHa/siCon, and SiHa/silncMIR17HG cells, were plated in 6‐well plates. After the cells were attached, cell monolayers were scraped by a middle pipet tip consistently and washed with PBS to gently remove cell debris. All cells were cultured in 1% FBS in DMEM or Myco5A. Photos were taken during the subsequent 12, 36, and 72 hours to monitor scratch closure. For the transwell migration assay, a cell suspension containing 4 × 105/mL cells was prepared in serum‐free media: 1 mL of media containing 10% fetal bovine serum was added to the lower chamber, and then 500 μL of the prepared cell suspension was added to each insert (Millipore# PIEP30R48, pore size: 8 μm). For the transwell invasion assay, 300 μL of warm serum‐free media was added to the interior of the inserts and allowed to rehydrate the ECM layer for 1 hour at room temperature. Then, 2 × 104 cells were plated into the transwell inserts (Chemicon #ECM550, pore size: 8 μm). All the steps were performed strictly following the instructions of the transwell migration and invasion assay kit. After 24 hours, cells that did not migrate were removed by scratching the upper side of the membrane with a cotton swab before fixation in 4% methanol for 5 minutes at room temperature. Cells were then stained with crystal violet staining solution for 5 minutes. The percentage of migration was determined by calculating the sum of the area of total migrated cells on the entire membrane by using ImageJ software.
2.10. Flow cytometry cell cycle assay
A total of 2 × 105 cells was plated in 6‐well plates per well and cultured overnight. Then, all cells were synchronized in serum‐free medium for 48 hours. After that, the cells were resuspended in complete medium for another 24 hours. The cells in each group were fixed with 75% ethanol overnight and washed with 1× phosphate buffer saline (PBS) twice. According to the protocol of the cell cycle detection kit (Multi Sciences), the cells were resuspended with 400 μL of 1× binding buffer and stained with 20 μL of propidium iodide (PI) for 20 minutes at room temperature in the dark. The cell cycle was analyzed immediately with a flow cytometer (Becton‐Dickinso, FACSCalibur). The percentage of cells at each phase of the cell cycle was obtained by Cell Quest software (Becton‐Dickinso, FACSCalibur).
2.11. Xenograft and orthotopic models of cancer in mice
Animal experiments were performed as described previously. Five‐week‐old female nude mice (Experiment Animal Center of Shanghai) (each group, n = 5) were subcutaneously injected with 6 × 106 SiHa/Con, SiHa/lncPART1, ME‐180/Con, or ME‐180/shlncPART1 cells in 0.1 mL PBS containing 20% matrigel, respectively. The growth of solid tumors of SiHa/Con, SiHa/lncPART1, ME‐180/Con, or ME‐180/shlncPART1 cells after injection were measured every 5 days for up to 30 days. All of the animals were sacrificed to take away the tumors for analysis. The use of mice was approved by the Animal Care and Use Committee of the USTC University (USTCACUC1801017).
2.12. Clinical samples
A total of 15 clinical samples were obtained from patients at Anhui Provincial Hospital, Hefei, China, including 5 cervical cancer patients and 10 uterine myoma patients. The detailed patients’ information is listed in Table S2. This study was reviewed and approved by the Ethics Review Board of Anhui Provincial Hospital. Written informed consent was obtained from each patient for this study.
2.13. Statistical evaluation
SPSS16.0 and GraphPad Prism 7 software were used for all statistical analyses. Data of all the experiments were presented as mean ± SD, which were replicated at least three times. Student's two‐tailed t‐test or analysis of variance (ANOVA) was used to assess the statistical significance of the difference. A P‐value <0.05 was considered statistically significant.
2.14. Data availability
The accession number for the RNA‐seq and ChIP‐seq data reported in this paper is GEO: GSE135257.
3. RESULTS
3.1. Identification of target genes of ΔNp63α in CSCC
We first established stable ME‐180 cells with ΔNp63α shRNA knockdown (ME‐180/shp63) as reported previously. 15 To investigate the transcriptional regulatory mechanisms of ΔNp63α in CSCC, we performed RNA‐seq in ME‐180/shp63 cell lines. Totally, 394 lncRNAs were significantly affected (cutoff of two FCs and P < 10−5) compared with the no‐knockdown control. To determine global direct targets of ΔNp63α, we performed ChIP‐seq of endogenous p63 in ME‐180 cells. Bioinformatics analyses of the ChIP‐seq data identified that 6505 genes were directly regulated by ΔNp63α. Among the 394 genes significantly affected by ΔNp63α in the RNA‐seq analyses, 39 possessed p63 ChIP‐seq binding sites, and thus should be direct targets of ΔNp63α (Figure 1A,B). These direct targets could be either activated or suppressed by ΔNp63α. All the 39 candidate lncRNAs were then analyzed by qRT‐PCR in ME‐180/shp63 cells (Figure S1).
FIGURE 1.
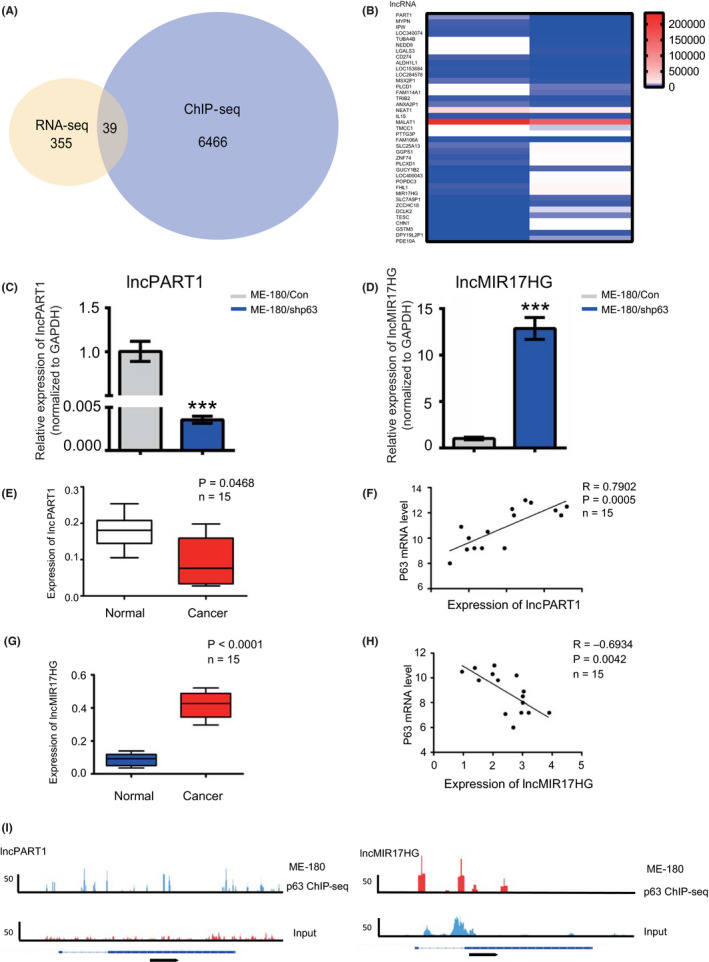
Identification of target long noncoding RNAs (lncRNAs) regulated by ΔNp63α in cervical squamous cancer cells. A, Analyses of RNA sequencing (RNA‐seq) and chromatin immunoprecipitation sequencing (ChIP‐seq); overlapped lncRNAs are shown. B, All the 39 candidate lncRNAs were analyzed by qRT‐PCR in ME‐180/Con and ME‐18 0/shp63 cells. C, qRT‐PCR analysis of the expression of PART1 in the ME180/shp63 transfected with ME180/Con cells group. D, qRT‐PCR analysis of the expression of lncMIR17HG in the ME180/shp63 transfected with ME180/Con cells group. E, The expression level of lncPART1 in normal tissues and cervical cancer tissues. F, The relationship between lncPART1 and P63 in cervical cancer tissues. G, The expression level of lncMIR17HG in normal tissues and cervical cancer tissues. H, The relationship between lncMIR17HG and P63 in cervical cancer tissues. I, ChIP‐seq peaks for both lncRNAs and ChIP efficiency. All the experiments were performed in triplicates. Error bars show SD; data are means ± SEM. n.s., not significant. *P < 0.05, **P < 0.01, ***P < 0.001, based on the Student's t‐test
Among the 39 directly regulated lncRNAs, previous studies indicated that lncPART1 and lncMIR17HG are associated to tumorigenesis. 42 , 43 , 44 , 45 , 46 , 47 Besides, lncPART1 and lncMIR17HG demonstrated significant differential expression in ME‐180/shp63 cells. LncPART1 showed significantly decreased expression levels, while lncMIR17HG showed significantly increased levels in ME‐180/shp63 cells (Figure 1C,D). We then detected the expression correlation of the two lncRNAs and ΔNp63α in clinical tissues. A total of 15 clinical samples were collected for analyses, including five cervical cancer patients and ten uterine myoma patients. The expression of lncPART1 was significantly lower than that in normal tissues (Figure 1E). The correlation analyses revealed that lncPART1 expression was positively correlated with ΔNp63α expression (Figure 1F). On the other hand, the expression of lncMIR17HG was significantly higher than that in normal tissues (Figure 1G). The correlation analyses revealed that MIR17HG expression was negatively correlated with ΔNp63α expression (Figure 1H). We hence focused on lncPART1 and lncMIR17HG for downstream examination. ChIP‐seq peaks for both lncRNAs and ChIP efficiency are shown in Figure 1I.
3.2. Overexpression of lncPART1 suppresses cell proliferation and migration
To investigate the functions of lncPART1 in cervical squamous tumorigenesis, we screened five cervical squamous cancer cell lines (HaCat and ME‐180 with high expression of ΔNp63α, and C‐33A, HeLa, and SiHa with low expression of ΔNp63α). Among them, SiHa cells showed the lowest expression levels of lncPART1 (Figure S2A,B). We hence selected SiHa (low expression of ΔNp63α) for the following in‐depth study. We first established stable SiHa cells with overexpression of lncPART1 (SiHa/lncPART1). The overexpression efficiency of lncPART1 was confirmed using qRT‐PCR analyses (Figure 2A). Compared with the control, cell proliferation and colony formation were significantly decreased in SiHa/ lncPART1 cells (Figure 2B,C). Overexpression of lncPART1 also decreased the proportion of cells in S phase and increased the proportion of cells in G1 phase in SiHa cells (Figure 2D). To investigate the role of lncPART1 in cell migration, we performed scratch assays and transwell assay. SiHa/lncPART1 cells migrated significantly slower compared with the control cells (Figure 2E). Moreover, SiHa/lncPART1 cells showed weaker migration and invasion capability in matrigel (Figure 2F).
FIGURE 2.
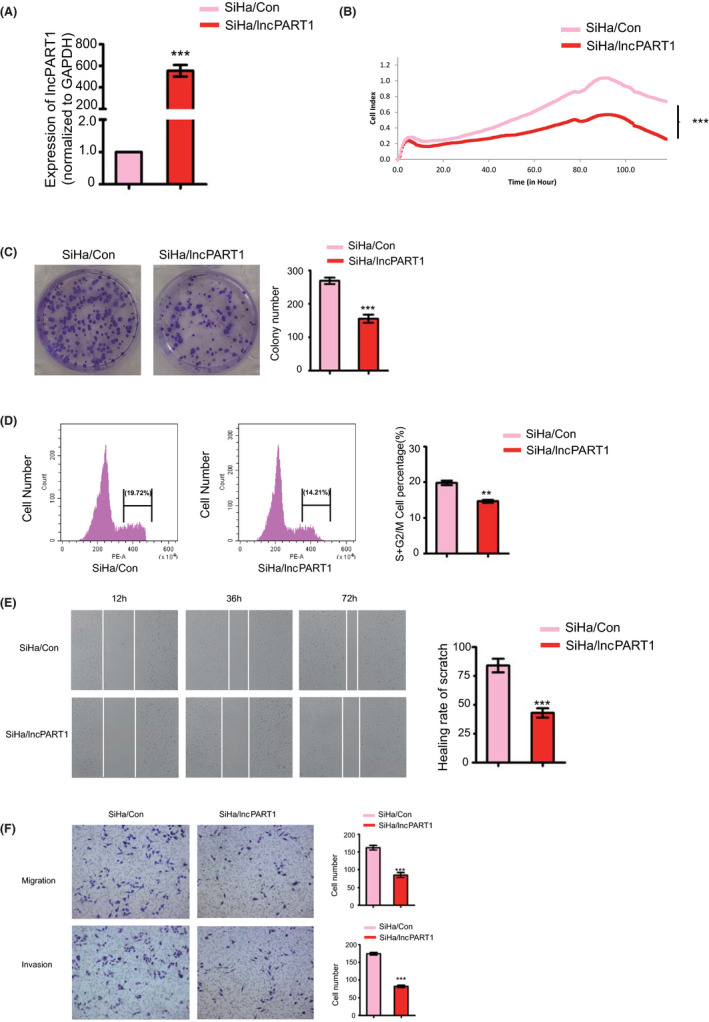
LncPART1 inhibits proliferation of cervical squamous cells in vitro. A, Expression levels of lncPART1 in SiHa/lncPART1 cells (SiHa cells with stable overexpression of lncPART1). B, Cell proliferation curves (as detected by RTCA assay) of SiHa cells in the two groups. C, Colony formation assays of SiHa/Con and SiHa/lncPART1 cells. Quantification of colony formation number is also shown. D, Cell cycle distribution analyses of SiHa/Con and SiHa/lncPART1 cells. Quantification of colony formation number is also shown. E, Representative images of wound healing in SiHa/Con and SiHa/lncPART1 cells. Quantification of healing rate is also shown. F, Representative images of transwell migration (up) and Matrigel invasion assays (down) of SiHa/Con and SiHa/lncPART1 cells. Quantification of migrating cells is also shown. All the experiments were performed in triplicates. Error bars show SD; data are means ± SEM. n.s., not significant. *P < 0.05, **P < 0.01, ***P < 0.001, based on the Student's t‐test
3.3. Knockdown of lncPART1 promotes cell proliferation and migration
ME‐180 cells harbor the highest expression level of lncPART1 of all five tested cell lines (Figure S2B); hence, we established stable ME‐180 cells with lncPART1 shRNA knockdown (ME‐180/shlncPART1‐1 and ME‐180/shlncPART1‐2). The knockdown efficiency of lncPART1 in the above two knockdown cell lines was confirmed using qRT‐PCR analyses (Figure 3A). Compared with the control, cell proliferation and colony formation were significantly increased in ME‐180/shlncPART1‐1 and ME‐180/shlncPART1‐2 cells (Figure 3B,C). Knockdown of lncPART1 increased the proportion of cells in S phase and decreased the proportion of cells in G1 phase in ME‐180 cells (Figure 3D). To investigate the role of lncPART1 in cell migration, we performed scratch assays and transwell assay. ME‐180/shlncPART1‐1 and ME‐180/shlncPART1‐2 cells migrated significantly faster compared with the control cells (Figure 3E). Moreover, ME‐180/shlncPART1‐1 and ME‐180/shlncPART1‐2 cells showed weaker migration and invasion capability in matrigel (Figure 3F).
FIGURE 3.
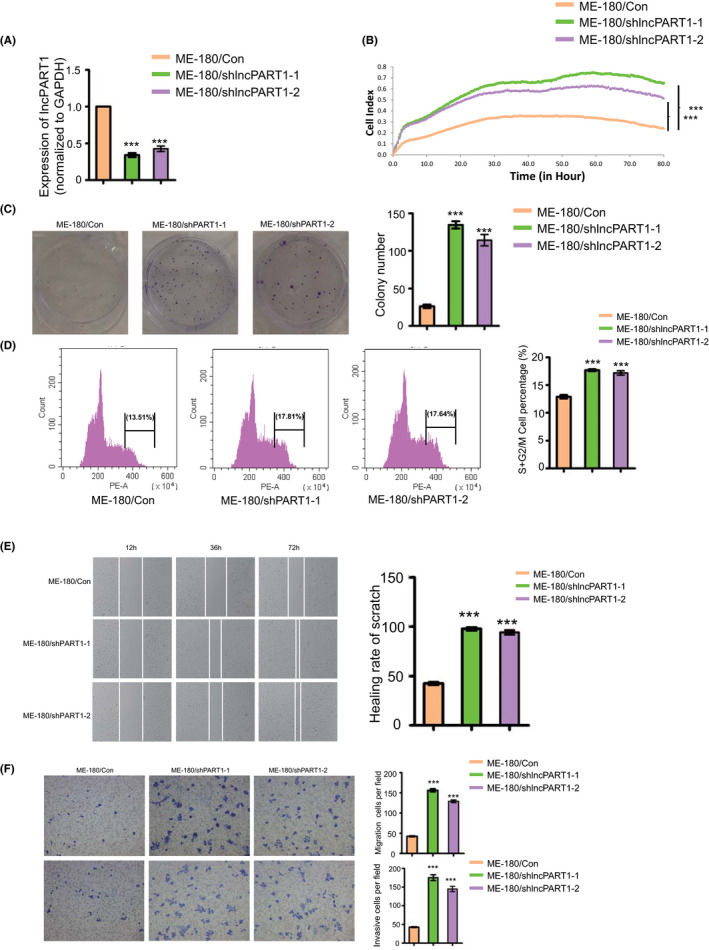
LncPART1 inhibits proliferation of cervical squamous cells in vitro. A, Expression levels of lncPART1 in ME180/shlncPART1‐1 and shlncPART1‐2 cells (ME‐180 cells with stable knockdown of lncPART1). B, Cell proliferation curves (as detected by RTCA assay) of SiHa cells in the three groups. C, Colony formation assays of ME‐180/Con, ME180/shlncPART1‐1, and shlncPART1‐2 cells. Quantification of colony formation number is also shown. D, Cell cycle distribution analyses of ME‐180/Con, ME180/shlncPART1‐1, and shlncPART1‐2 cells. Quantification of colony formation number is also shown. E, Representative images of wound healing in ME‐180/Con, ME180/shlncPART1‐1, and shlncPART1‐2 cells. Quantification of healing rate is also shown. F, Representative images of transwell migration (up) and Matrigel invasion assays (down) of ME‐180/Con, ME180/shlncPART1‐1. and shlncPART1‐2 cells. Quantification of migrating cells is also shown. All the experiments were performed in triplicates. Error bars show SD; data are means ± SEM. n.s., not significant. *P < 0.05, **P < 0.01, ***P < 0.001, based on the Student's t‐test
3.4. lncPART1 suppresses tumor growth in CSCC‐derived tumors
We then investigated the role of lncPART1 on tumorigenesis of cervical cancer cells in vivo. SiHa/lncPART1, ME‐180/shlncPART1‐1, and ME‐180/shlncPART1‐2 cells, along with their corresponding control cells, were subcutaneously injected into 5‐week‐old female athymic nude mice. The tumors from SiHa/lncPART1 cells grew significantly slower than those from control cells (Figure 4A‐C). On the other hand, tumors in the ME‐180/shlncPART1 and ME‐180/shlncPART1 groups grew significantly faster as compared with their control (Figure 4D‐F).
FIGURE 4.
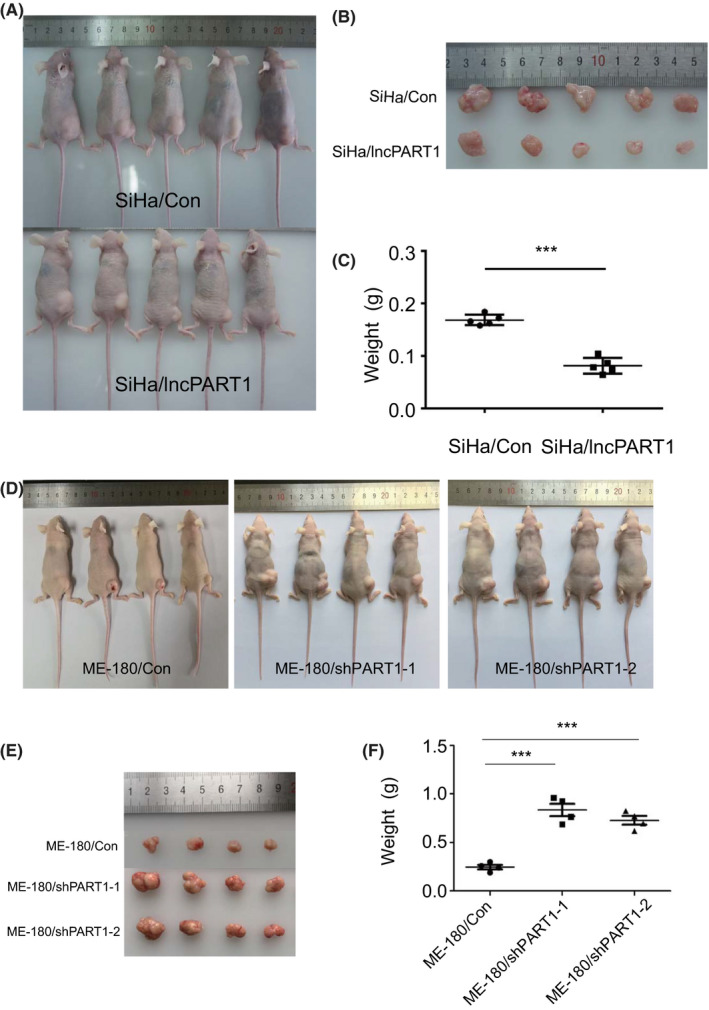
LncPART1 inhibits proliferation of cervical squamous cells in vivo. A, B, Images of the xenograft tumors in the SiHa/Con and SiHa/lncPART1 cells groups 30 days after injection. C, The tumor weight distribution in the SiHa/Con and SiHa/lncPART1 cells groups. D, E, Images of the xenograft tumors in the ME‐180/Con, ME180/shlncPART1‐1, and shlncPART1‐2 cells groups 30 days after injection. F, The tumor weight distribution in the ME‐180/Con, ME180/shlncPART1‐1, and shlncPART1‐2 cells groups. All the experiments were performed in triplicates. Error bars show SD; data are means ± SEM. n.s., not significant. *P < 0.05, **P < 0.01, ***P < 0.001, based on the Student's t‐test
3.5. Knockdown of lncMIR17HG suppresses cell proliferation and migration in CSCC
In investigation of the functions of lncMIR17HG in cervical squamous tumorigenesis, of all five tested CSCC cell lines, SiHa cells harbor the highest expression level of lncMIR17HG; hence, we selected SiHa (low expression of ΔNp63α) for the following in‐depth study (Figure S2C). We first established stable SiHa cells with the knockdown of lncMIR17HG (SiHa/shlncMIR17HG). The knockdown efficiency of lncMIR17HG was confirmed using qRT‐PCR analyses (Figure 5A). Compared with the control, cell proliferation and colony formation were significantly decreased in SiHa/lncMIR17HG cells (Figure 5B,C). To investigate the role of lncMIR17HG in cell migration, we performed scratch assays and transwell assay (Figure 5D,E). SiHa/shlncMIR17HG cells migrated significantly slower compared with the control cells. Moreover, SiHa/shlncMIR17HG cells showed weaker migration and invasion capability in matrigel (Figure 5E).
FIGURE 5.
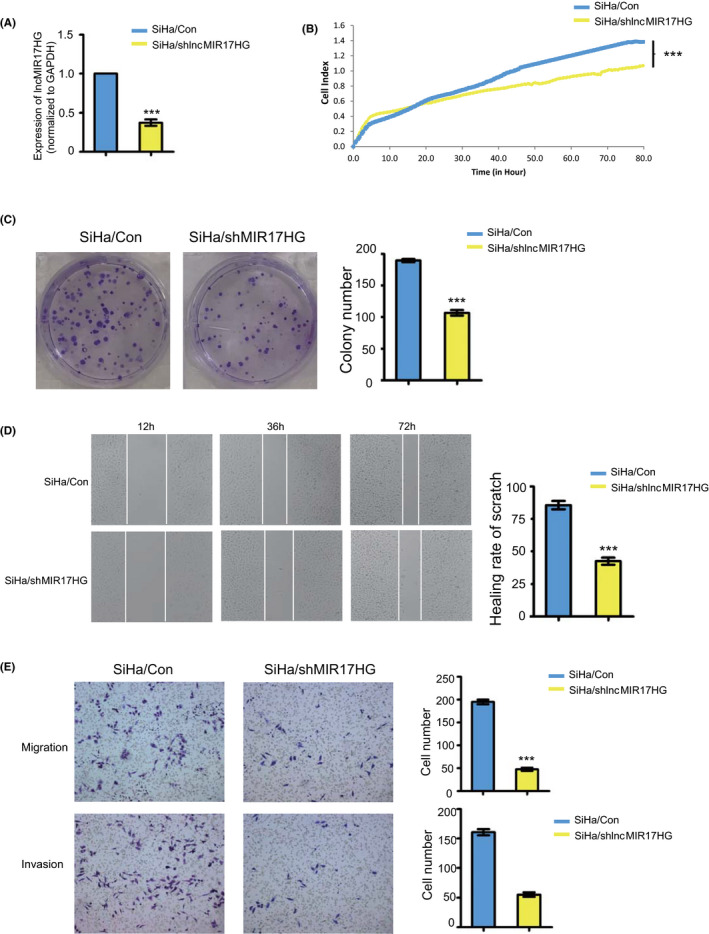
LncMIR17HG promotes proliferation of cervical squamous cells in vitro. A, Expression levels of lncMIR17HG in SiHa/shlncMIR17HG cells (SiHa cells with knockdown of lncMIR17HG). B, Proliferation curves (as detected by RTCA assay) of SiHa/shlncMIR17HG cells compared with the corresponding controls. C, Colony formation assays of SiHa/Con and SiHa/shlncMIR17HG cells. Quantification of colony formation number is also shown. D, Representative images of wound healing in SiHa/Con and SiHa/shlncMIR17HG cells. Quantification of healing rate is also shown. E, Representative images of transwell migration (up) and Matrigel invasion assays (down) of SiHa/Con and SiHa/shlncMIR17HG cells. Quantification of migrating cells is also shown. All the experiments were performed in triplicates. Error bars who SD; data are means ± SEM. n.s., not significant. *P < 0.05, **P < 0.01, ***P < 0.001, based on the Student's t‐test
4. DISCUSSION
Cervical cancer is the fourth most common cancer and the second leading cause of cancer deaths among women in the world. The discovery of critical diagnostic and therapeutic markers against CSCC would broaden our understanding on the molecular basis of CSCC. Besides, high‐throughput RNA‐seq has greatly enabled us to detect and quantify the entire transcriptome in CSCC. ΔNp63α plays a fundamental role in the regulation of cervical squamous tumorigenesis. We have previously identified that ΔNp63α acts as a tumor suppressor via regulating a cohort of downstream targets in CSCC. In this study, we found 39 potential ΔNp63α target lncRNAs by overlapping ChIP‐seq and RNA‐seq. ΔNp63α is the predominant isotype expressed in the cervix and ME‐180 cell line, while other isotypes (eg, TAp63α) are hardly detectable; thus, here we used p63α antibody for ChIP in ME‐180 cell line. 12 , 13 , 14 All the potential targets were verified by qRT‐PCR and the results were in accordance with those in the above sequencing data.
Among the 39 directly regulated lncRNAs, we found the expression level of lncPART1, a tumor‐associated lncRNA, was significantly decreased in ME‐180/shp63 cells, as compared with the control cells. Overexpression of lncPART1 suppressed, while knockdown of lncPART1 promoted proliferation, migration, and invasion of cervical cancer cell lines. We found that lncPART1 suppresses tumor growth in CSCC‐derived tumors. Correlation analysis of cervical cancer tissues revealed that lncPART1 expression was positively correlated with ΔNp63α expression. The above results indicated that lncPART1 might act as a tumor suppressor in CSCC. The detailed molecular mechanism of how lncPART1 functions remains for further investigation.
The functions of lncPART1 were previously reported to be positively involved in cell migration and proliferation in multiple cancers. For example, lncPART1 promoted cell proliferation ability and apoptosis via the inhibition of Toll‐like receptor pathways in prostate cancer and promoted gefitinib resistance in esophageal squamous cell carcinoma by functioning as a competing endogenous RNA. 42 , 43 Our study demonstrates that, at least in CSCC cell lines examined and in the contexts of our experimental setup, lncPART1 is a tumor suppressor in CSCC, which broadens our insights into the function of lncPART1 in tumorigenesis.
Another core target of ΔNp63α is lncMIR17HG, a miR‐17‐92 cluster host gene lncRNA that is involved in cell proliferation and growth by modulating cell growth phenotype. However, the biological function of most lnc‐miRHGs in tumor progression is not well understood. In this study, we found that the level of lncMIR17HG was significantly increased in cultured cells and clinical tissues. Knockdown of lncMIR17HG markedly suppressed the proliferation, migration, and invasion of SiHa cells. The expression level of lncMIR17HG in cervical cancer tissues was significantly higher than in normal tissues. Correlation analysis of cervical cancer tissues revealed that lncMIR17HG expression was negatively correlated with ΔNp63α expression. The above results indicated that lncMIR17HG may be suppressed by ΔNp63α and functions as a protumor gene in CSCC. Previous studies indicated that lncMIR17HG normally functions through regulating its host miRNAs. For instance, lncMIR17HG promoted colorectal cancer progression via miR‐17‐5p, and downregulation of MIR17HG‐miR‐18a/miR‐19a axis expression and attenuating Wnt/β‐catenin signaling inhibited gastric cancer metastasis. 44 , 45 We have analyzed the association of lncMIR17HG and six lncMIR17HG cluster members (miR‐17, miR‐18a, miR‐19a, miR‐19b‐1, miR‐20a, and miR‐92a‐1). All of the members, except miR‐20a, showed positive correlation with lncMIR17HG (data not shown). The detailed molecular mechanism of how lncMIR17HG functions in CSCC needs further investigation.
Taken together, we have identified two direct transcriptional targets of ΔNp63α, lncPART1 and lncMIR17HG. We showed that lncPART1 suppressed the proliferation, migration, and invasion of CSCC cells and acted as a tumor suppressor gene in CSCC. On the other hand, lncMIR17HG is a protumor gene. Knockdown of lncMIR17HG suppressed the proliferation, migration, and invasion of CSCC cells. ΔNp63α, as a transcription factor, can both activate the expression of target genes (eg, lncPART1) and inhibit the expression of some other target genes (eg, lncMIR17HG), which reveals its combinatory effects of transcriptional regulators. Further identification and characterization of ΔNp63α targets in tumorigenesis are of fundamental significance in the understanding, prognosis, and treatment of CSCC.
DISCLOSURE
The authors have no conflict of interest to declare. All authors have read the journal's authorship agreement. The manuscript has been reviewed by and approved by all named authors.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (No. 81872110, 81902632, 31600657, and 8190101115), National Key Research and Development Program (2018YFC1003903), Anhui Provincial Key Research and Development Projects (1704a0802151), and the "Biological Basis of Aging and Therapeutic Strategies" of the Chinese Academy of Sciences (grant XDPB10).
Liu H, Zhu C, Xu Z, et al. lncRNA PART1 and MIR17HG as ΔNp63α direct targets regulate tumor progression of cervical squamous cell carcinoma. Cancer Sci. 2020;111:4129–4141. 10.1111/cas.14649
Hanyuan Liu and Chenchen Zhu contributed equally to this paper.
Contributor Information
Liang Chen, Email: anqingcl@ustc.edu.cn.
Ying Zhou, Email: caddie1234@gmail.com.
REFERENCES
- 1. Song W, Wang J, Liu H, et al. Effects of LncRNA Lnc‐LIF‐AS on cell proliferation, migration and invasion in a human cervical cancer cell line. Cytokine. 2019;120:165‐175. [DOI] [PubMed] [Google Scholar]
- 2. Aalijahan H, Ghorbian S. Long non‐coding RNAs and cervical cancer. Exp Mol Pathol. 2019;106:7‐16. [DOI] [PubMed] [Google Scholar]
- 3. Minion LE, Tewari KS. Cervical cancer – state of the science: from angiogenesis blockade to checkpoint inhibition. Gynecol Oncol. 2018;148:609‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7‐30. [DOI] [PubMed] [Google Scholar]
- 5. Rodriguez‐Carunchio L, Soveral I, Steenbergen RD, et al. HPV‐negative carcinoma of the uterine cervix: a distinct type of cervical cancer with poor prognosis. BJOG. 2015;122:119‐127. [DOI] [PubMed] [Google Scholar]
- 6. Rusan M, Li YY, Hammerman PS. Genomic landscape of human papillomavirus‐associated cancers. Clin Cancer Res. 2015;21:2009‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rose PG, Bundy BN, Watkins EB, et al. Concurrent cisplatin‐based radiotherapy and chemotherapy for locally advanced cervical cancer. N Engl J Med. 1999;340:1144‐1153. [DOI] [PubMed] [Google Scholar]
- 8. Sol ES, Lee TS, Koh SB, Oh HK, Ye GW, Choi YS. Comparison of concurrent chemoradiotherapy with cisplatin plus 5‐fluorouracil versus cisplatin plus paclitaxel in patients with locally advanced cervical carcinoma. J Gynecol Oncol. 2009;20:28‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mangiulli M, Valletti A, Caratozzolo MF, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009;37:6092‐6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714‐718. [DOI] [PubMed] [Google Scholar]
- 11. Candi E, Cipollone R, di Val R, et al. p63 in epithelial development. Cell Mol Life Sci. 2008;65:3126‐3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dohn M, Zhang S, Chen X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene. 2001;20:3193‐3205. [DOI] [PubMed] [Google Scholar]
- 13. Zhou Y, Xu Q, Ling B, Xiao W, Liu P. Reduced expression of ΔΝp63α in cervical squamous cell carcinoma. Clin Invest Med. 2011;34:E184‐E191. [DOI] [PubMed] [Google Scholar]
- 14. Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27‐29, encodes multiple products with transactivating, death‐inducing, and dominant‐negative activities. Mol Cell. 1998;2:305‐316. [DOI] [PubMed] [Google Scholar]
- 15. Ying H, Chang DL, Zheng H, McKeon F, Xiao ZX. DNA‐binding and transactivation activities are essential for TAp63 protein degradation. Mol Cell Biol. 2005;25:6154‐6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koster MI, Roop DR. p63 and epithelial appendage development. Differentiation. 2004;72:364‐370. [DOI] [PubMed] [Google Scholar]
- 17. Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene. 2007;26:5169‐5183. [DOI] [PubMed] [Google Scholar]
- 18. Zhou Y, Liu H, Wang J, et al. DeltaNp63alpha exerts antitumor functions in cervical squamous cell carcinoma. Oncogene. 2019;39:905‐921. [DOI] [PubMed] [Google Scholar]
- 19. Qian L, Xu F, Wang X, et al. LncRNA expression profile of DeltaNp63alpha in cervical squamous cancers and its suppressive effects on LIF expression. Cytokine. 2017;96:114‐122. [DOI] [PubMed] [Google Scholar]
- 20. Chan JJ, Tay Y. Noncoding RNA:RNA regulatory networks in cancer. Int J Mol Sci. 2018;19:1310–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cipolla GA, de Oliveira JC, Salviano‐Silva A, et al. Long non‐coding RNAs in multifactorial diseases: another layer of complexity. Noncoding RNA. 2018;4:13–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun T. Long noncoding RNAs act as regulators of autophagy in cancer. Pharmacol Res. 2018;129:151‐155. [DOI] [PubMed] [Google Scholar]
- 23. Li X, Fu XD. Chromatin‐associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet. 2019;20:503‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engreitz JM, Haines JE, Perez EM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gutschner T, Hammerle M, Eissmann M, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J, Piao HL, Kim BJ, et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat Genet. 2018;50:1705‐1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitra R, Chen X, Greenawalt EJ, et al. Decoding critical long non‐coding RNA in ovarian cancer epithelial‐to‐mesenchymal transition. Nat Commun. 2017;8:1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duguang L, Jin H, Xiaowei Q, et al. The involvement of lncRNAs in the development and progression of pancreatic cancer. Cancer Biol Ther. 2017;18:927‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsumura K, Kawasaki Y, Miyamoto M, et al. The novel G‐quadruplex‐containing long non‐coding RNA GSEC antagonizes DHX36 and modulates colon cancer cell migration. Oncogene. 2017;36:1191‐1199. [DOI] [PubMed] [Google Scholar]
- 30. Deguchi S, Katsushima K, Hatanaka A, et al. Oncogenic effects of evolutionarily conserved noncoding RNA ECONEXIN on gliomagenesis. Oncogene. 2017;36:4629‐4640. [DOI] [PubMed] [Google Scholar]
- 31. He Y, Meng XM, Huang C, et al. Long noncoding RNAs: novel insights into hepatocelluar carcinoma. Cancer Lett. 2014;344:20‐27. [DOI] [PubMed] [Google Scholar]
- 32. Li MM, Liu XH, Zhao YC, et al. Long noncoding RNA KCNQ1OT1 promotes apoptosis in neuroblastoma cells by regulating miR‐296‐5p/Bax axis. FEBS J. 2020;287:561‐577. [DOI] [PubMed] [Google Scholar]
- 33. Al‐Rugeebah A, Alanazi M, Parine NR. MEG3: an oncogenic long non‐coding RNA in different cancers. Pathol Oncol Res. 2019;25:859‐874. [DOI] [PubMed] [Google Scholar]
- 34. Wang N, Hou MS, Zhan Y, Shen XB, Xue HY. MALAT1 promotes cisplatin resistance in cervical cancer by activating the PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2018;22:7653‐7659. [DOI] [PubMed] [Google Scholar]
- 35. Yang W, Xu X, Hong L, Wang Q, Huang J, Jiang L. Upregulation of lncRNA GAS5 inhibits the growth and metastasis of cervical cancer cells. J Cell Physiol. 2019;234:23571‐23580. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y, Cheng X, Liang H, Jin Z. Long non‐coding RNA HOTAIR and STAT3 synergistically regulate the cervical cancer cell migration and invasion. Chem Biol Interact. 2018;286:106‐110. [DOI] [PubMed] [Google Scholar]
- 37. Sun NX, Ye C, Zhao Q, et al. Long noncoding RNA‐EBIC promotes tumor cell invasion by binding to EZH2 and repressing E‐cadherin in cervical cancer. PLoS One. 2014;9:e100340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Molinari C, Salvi S, Foca F, et al. miR‐17‐92a‐1 cluster host gene (MIR17HG) evaluation and response to neoadjuvant chemoradiotherapy in rectal cancer. Onco Targets Ther. 2016;9:2735‐2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu J, Meng Q, Li X, et al. Long non‐coding RNA MIR17HG promotes colorectal cancer progression via miR‐17‐5p. Cancer Res. 2019;79:4882‐4895. [DOI] [PubMed] [Google Scholar]
- 40. Hu S, Wang X, Shan G. Insertion of an Alu element in a lncRNA leads to primate‐specific modulation of alternative splicing. Nat Struct Mol Biol. 2016;23:1011‐1019. [DOI] [PubMed] [Google Scholar]
- 41. Chen L, Rashid F, Shah A, et al. The isolation of an RNA aptamer targeting to p53 protein with single amino acid mutation. Proc Natl Acad Sci USA. 2015;112:10002‐10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kang M, Ren M, Li Y, Fu Y, Deng M, Li C. Exosome‐mediated transfer of lncRNA PART1 induces gefitinib resistance in esophageal squamous cell carcinoma via functioning as a competing endogenous RNA. J Exp Clin Cancer Res. 2018;37:171. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Sun M, Geng D, Li S, Chen Z, Zhao W. LncRNA PART1 modulates toll‐like receptor pathways to influence cell proliferation and apoptosis in prostate cancer cells. Biol Chem. 2018;399:387‐395. [DOI] [PubMed] [Google Scholar]
- 44. Xu J, Meng Q, Li X, et al. Long noncoding RNA MIR17HG promotes colorectal cancer progression via miR‐17‐5p. Cancer Res. 2019;79:4882‐4895. [DOI] [PubMed] [Google Scholar]
- 45. Yuan J, Tan L, Yin Z, et al. MIR17HG‐miR‐18a/19a axis, regulated by interferon regulatory factor‐1, promotes gastric cancer metastasis via Wnt/β‐catenin signalling. Cell Death Dis. 2019;10:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhu D, Yu Y, Wang W, et al. Long noncoding RNA PART1 promotes progression of non‐small cell lung cancer cells via JAK‐STAT signaling pathway. Cancer Med. 2019;8:6064‐6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li M, Zhang W, Zhang S, Wang C, Lin Y. PART1 expression is associated with poor prognosis and tumor recurrence in stage I‐III non‐small cell lung cancer. J Cancer. 2017;8:1795‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Data Availability Statement
The accession number for the RNA‐seq and ChIP‐seq data reported in this paper is GEO: GSE135257.