

Abstract
Non‐alcoholic fatty liver disease (NAFLD) is one of the most common causes of hepatocellular carcinoma (HCC), but the underlying mechanisms behind the correlation of NAFLD with HCC are unclear. We aimed to uncover the genes and potential mechanisms that drive this progression. This study uncovered the genes and potential mechanisms through a multiple ’omics integration approach. Quantitative proteomics combined with phenotype‐association analysis was performed. To investigate the potential mechanisms, a comprehensive transcriptome/lipidome/phenome‐wide association analysis was performed in genetic reference panel BXD mice strains. The quantitative proteomics combined with phenotype‐association results showed that VDAC1 was significantly increased in tumor tissues and correlated with NAFLD‐related traits. Gene co‐expression network analysis indicated that VDAC1 is involved in mitochondria dysfunction in the tumorigenic/tumor progression. The association between VDAC1 and mitochondria dysfunction can be explained by the fact that VDAC1 was associated with mitochondria membrane lipids cardiolipin (CL) composition shift. VDAC1 was correlated with the suppression of mature specie CL(LLLL) and elevation level of nascent CL species. Such profiling shift was supported by the significant positive correlation between VDAC1 and PTPMT1, as well as negative correlation with CL remodeling enzyme Tafazzin (TAZ). This study confirmed that the expression of VADC1 was dysregulated in NAFLD‐driven HCC and associated with NAFLD progression. The VDAC1‐driven mitochondria dysfunction is associated with cardiolipin composition shift, which causes alteration of mitochondria membrane properties.
Keywords: hepatocellular carcinoma, mitochondria dysfunction, non‐alcoholic fatty liver disease, system biology, voltage‐dependent anion channel
Abbreviations
- ACN
acetonitrile
- B6
C57BL/6J strain
- CL
lipids cardiolipin
- D2
DBA/2J strain
- eQTL
expression quantitative trait locus
- FDR
false discovery rate
- GO
Gene Ontology
- GRP
genetic reference population
- HCC
Hepatocellular carcinoma
- HCD
high collision dissociation
- IMM
the inner mitochondrial membrane
- IPA
Ingenuity Pathway Analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LRS
likelihood of the ratio statistics
- NAFLD
Non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- PG
phosphatidylglycerol
- PGP
phosphatidylglycerophosphate
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- TAZ
Tafazzin
- TCGA‐LIHC
Cancer Genome Atlas Liver Hepatocellular Carcinoma
- TFA
trifluoroacetic acid
- VDAC1
voltage‐dependent anion channel
- WB
western blot
- WGCNA
Weighted Gene Co‐expression Network Analysis
1. INTRODUCTION
NAFLD is characterized by the excessive abnormal accumulation of fatty acids and triglycerides within hepatocytes. 1 It encompasses a spectrum of diseases from non‐alcoholic fatty liver (NAFL) to non‐alcoholic steatohepatitis (NASH), fibrosis, and cirrhosis. 1 Even though NAFLD is implicated as a risk factor of hepatocellular carcinoma (HCC), the precise mechanism is still unclear. 2 As liver is the major organ in lipid metabolism, NAFLD‐driven HCC is in an extreme lipid‐enriched microenvironment. 3 This provides a unique scenario for us to understand how tumor cells adapt to and exploit such an environment for disease progression. 4 Thus, understanding the association between NAFLD and HCC will help us to reveal the mechanism of how malignant cells shift nutrition metabolic preference during the tumorigenesis process. We hypothesized that the genes that were associated with NAFLD progression and dysregulated in HCC would provide hints about HCC tumorigenesis mechanism induced by NAFLD.
Transcriptome‐wide association study is emerging as a novel approach for prioritizing casual genes and mechanisms for diseases progression. 5 , 6 Genes with significant correlation with disease markers or traits can be hypothesized for further validation, 7 Moreover, with the accumulation of diverse ’omics data, multiple ’omics‐wide association is achievable, such as gene‐protein and gene‐lipids correlation.
However, one of the drawbacks that limit the broad application of such association study is the need for a large sample size in the human population due to genetic and lifestyle diversity. 8 In this respect, the GRP from animal models provides an alternative opportunity. The well controlled genetic background could significantly reduce sample size and, even more, it was possible to integrate multiple ’omics studies. Therefore, system biology analysis on GRP has emerged as a potent approach to identify gene functions in biological questions and interpret the underlying molecular mechanisms. 9 Among various GRP animal models, the BXD mice family, which consists of more than 100 strains derived from a cross between the C57BL/6J strain (B6) and the DBA/2J strain (D2), contains comprehensive multiple ’omics data. With this unique GRP, it is possible to combine systematic genetics and multiple ’omics data to establish multi‐omics association network including genes, transcripts, metabolites, and phenotypes for diseases. 10 , 11
In this study, we collected 6 paired NAFLD‐driven HCC clinical samples and applied quantitative proteomics to reveal the differentially expressed proteins between tumor tissues and adjacent non‐tumor tissues. The association between candidate proteins and NAFLD‐related traits was further evaluated. The result indicated that VDAC1 was one of the genes that was significantly upregulated in tumor tissues and associated with NAFLD traits. A comprehensive analyzed on VDAC1 was performed to underlying the mechanisms of VDAC1 in NAFLD‐associated HCC, including quantitative trait locus (eQTL) mapping, and gene co‐expression network analysis. As VDAC1 is associated with mitochondria dysfunction, the association between VDAC1 and mitochondria membrane lipid profiling was investigated.
2. MATERIALS AND METHODS
2.1. Tissue samples
Six pairs of NAFLD‐driven HCC and adjacent non‐tumor liver tissues were collected and matched with normal tissues, and the patients were prospectively enrolled with written informed consent at Shandong Provincial Hospital (Table 1). This study was conducted in accordance with the Declaration of Helsinki of 1975, and the program was approved by the ethics committee of human research of Binzhou Medical University (approval number: 2016‐019). Participants were included if the following criteria were met: (a) Male, 18‐70 y old, HCC Grade III. (b) Previous diagnosis of fatty liver (laboratory tests combined with confirmation of medical imaging results). (c) Absence of regular or excessive use of alcohol (the norm is less than 140 g per wk for male). (d) Except viral hepatitis, drug‐induced liver disease, total parenteral nutrition, hepatolenticular degeneration, and other specific diseases that can lead to fatty liver. All samples were rinsed with Ringer solution (CAS No. 7647‐14‐5) to remove blood, immediately frozen in liquid nitrogen, and stored at −80°C until use. The diagnostic tissues were fixed in formalin and embedded in paraffin.
TABLE 1.
Clinical information of NAFLD‐related HCC patients selected for mass spectrometry‐based proteomics analysis
| Sample ID | Gender | Age | Pathological diagnosis | Non‐alcoholic fatty liver | Grade | TNM |
|---|---|---|---|---|---|---|
| 1N/ 1T | Male | 72 | HCC | + | III | T3N1M0 |
| 2N/ 2T | Male | 55 | HCC | + | III | T2N1M0 |
| 3N/3T | Male | 55 | HCC | + | III | T3N1M0 |
| 4N/ 4T | Male | 70 | HCC | + | III | T3N0M0 |
| 5N/ 5T | Male | 65 | HCC | + | III | T3N0M0 |
| 6N/ 6T | Male | 45 | HCC | + | III | T3N1M0 |
2.2. Protein digestion
The protocol was similar to that described in our previous work. 12 In short, tissues samples were suspended and homogenized in lysis buffer (9 mol/L urea, 20 m mol/L HEPES) containing proteinase inhibitor cocktail (Thermo, A32961). The samples were centrifuged at 4°C for 10 min at 12 000 × g after sonication, and the supernatant was collected. Then, protein concentrations were measured using the BCA protein assay kit. 20 µg of the total protein was diluted in 100 µL digestion buffer (6 mol/L urea, 100 mmol/L TEAB), 10 µL 50 mmol/L DTT reduction solution was added and the sample incubated at 50°C for 15 min, followed by addition of 10 µL 50 mmol/L IAA for alkylation in the dark for 15 min at room temperature. The samples were diluted 4 times with digestion buffer (50 mm NH4HCO3, pH 8.0), and digested with trypsin (Thermo, ADV5111) at a final concentration of 5% (w/w) overnight at 37°C. The reaction was stopped by diluting the sample 1:1 with trifluoroacetic acid (TFA) in acetonitrile (ACN) and Milli‐Q water (1/5/94 v/v). Finally, peptides were desalted using Pierce C18 Spin Columns (Thermo, SC2003) and dried completely in a vacuum centrifuge.
2.3. LC‐MS/MS analysis
The peptides analyses were performed using an Q Exactive Plus Orbitrap™ mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), equipped with a nano‐electrospray ion source. Peptides were dissolved in 20 µL water/formic acid (0.1%, v/v) and separated by reversed phase liquid chromatography using the EASY‐nLC 1000 system (Thermo Fisher Scientific). In MS analysis, peptides were loaded onto a 2 cm EASY‐column pre‐column (ID 100 µm, 5 µm, C18) (Thermo Fisher Scientific), while the analytical column was a 10 cm EASY‐column (ID 75 µm, 3 µm, C18) (Thermo Fisher Scientific). Peptides were eluted with a 90 min linear gradient at 250 nL/min from 4% to 100% acetonitrile in the positive ion mode. The full range of MS spectra was performed at a resolution of 70 000, the 10 most abundant ions are obtained by HCD. 13
2.4. Proteomic data analysis
The proteomics raw data were analyzed using Maxquant (version 1.5.0.1) based on the Uniprot Homo sapiens database (release 2016‐06, with 20 199 protein entries). The searching parameters were set as follows: maximum 10 and 5 ppm error tolerance for the survey scan and MS/MS analysis, respectively; the specificity enzyme was trypsin; and a maximum of 2 missed cleavage sites was allowed; cysteine carbamidomethylation was set as static modification; Oxidation (M) was set as a dynamic modification. A maximum FDR was set to 1% for peptides and proteins identification. 14 The differentially expressed proteins (P < .05) and their log2‐transformed expression ratios were analyzed using the Ingenuity® Pathway Analysis (IPA) software (Qiagen, CA) and the top canonical pathways associated with the analyzed proteins were listed alongside the P‐values, which were calculated using right‐tailed Fisher exact test. We selected the top 10 pathways for further analysis.
2.5. Western blot (WB)
Prepared lysis buffer (mix RIPA and PMSF in the proportion of 100:1) and 30 mg tissues were homogenized in 150 µL of lysis buffer. Then the samples were collected after centrifugation at 4°C, 12 000 × g for 10 min. Total protein was quantified by the BCA protein assay kit and 20 µg protein samples were loaded and separated using 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE). Subsequently, proteins were transferred to a PVDF membrane and blocked with TBS (pH 7.4) containing 0.05% Tween 20 and 5% non‐fat dried milk. The following antibodies were used: rabbit VDAC1 antibody (CUSABIO, CSB‐RA025821A0HU, 1:1000), internal reference: RPLP0 (Proteintech, 11290‐2‐AP, Rabbit, 1:1000). After washing, membranes were incubated with goat anti‐rabbit antibody horseradish peroxidase conjugated (1:3000) at room temperature for 1 h. Enhanced chemiluminescence was used to detect protein expression.
2.6. BXD mice liver gene expression data
This data set was generated by our collaborator and uploaded in our Genenetwork website, which can be accessed publicly. 14 Briefly, 39 strains of the BXD family and both parental strains (B6 and D2) were maintained in a 12 h: 12 h light‐dark cycle. Animals were fasted overnight prior to sacrifice with isoflurane anesthesia at 29 wk of age. The livers were immediately removed, frozen, and stored in liquid nitrogen. RNA was extracted and pooled evenly (approximately 5 samples) into a single RNA sample for each strain. Quality‐passed samples (RIN values were >8.0) were run on the Affymetrix Mouse Gene 1.0 ST system at the University of Tennessee Health Science Center. Raw microarray data were normalized using the Robust Multichip Array (RMA) method. 15 The expression data were then re‐normalized using a modified Z‐score as described in a previous publication. 16 RMAs were first transformed into log2‐values; then the data of every single array was converted to a Z‐score, multiplied by 2, and a value of 8 was added.
2.7. Vdac1 genetic variants and expression QTL (eQTL) mapping
The genetic variants of the Vdac1 in parental strains B6 and D2 were determined using the mouse genomes variants querying site from the mouse genome project (https://www.sanger.ac.uk/science/data/mouse‐genomes‐project); and the variants included. 17 eQTL mapping of Vdac1 gene expression was performed with WebQTL (www.genenetwork.org). 16 Briefly, the expression of Vdac1 across BXD mice was treated as a quantitative trait, and the variation in Vdac1 mRNA levels was mapped using conventional QTL methods. In the current study, simple interval mapping was used to identify the potential genetic loci that regulated Vdac1 expression. An LRS was calculated and used to measure the association or linkage between differences in traits and differences in specific genotype markers. To evaluate the statistical significance (QTL significance), 2000 permutation tests were performed. The QTL confidence interval was estimated by a conventional 1.5 LOD drop‐off interval, in which LOD = LRS/4.61. 16
2.8. WGCNA
Probe sets were filtered in 2 steps: (a) probe sets with unannotated gene information were removed; (b) probe sets with a mean expression level < 7 across all the samples were filtered out. The samples underwent WGCNA using the R package version v.1.63. 18 Briefly, the absolute correlation coefficient between probes across the samples was first calculated. Soft threshold power β was selected based on the criterion of approximate scale‐free topology. Subsequently, an assigned adjacency matrix was obtained and used as the distance to generate a hierarchical cluster of genes. Genes were aggregated into modules by cutting the breaches using a high cut‐off of 0.99. The minimum module size (number of probes) was set to 30.
2.9. Gene set enrichment analysis
WEB‐based Gene SeT AnaLysis Toolkit (WebGestalt) was used to perform gene set enrichment analysis with the mouse genome reference gene set as background (http://www.webgestalt.org/). 19 The over‐representation of both GO and KEGG pathways was determined by the hypergeometric test. The adjusted P‐value was calculated with Benjamini and Hochberg methods. IPA software was used to analyze the pathways involved in the VDAC1 correlated genes.
2.10. Immunohistochemistry (IHC)
VDAC1 expression was detected by IHC in NAFLD‐driven HCC and adjacent non‐tumor liver tissues. The method was consistent with our previous report 12 ; in short, the paraffin section of liver tissues was heated at 60°C for 30 min, then deparaffinized with gradient ethanol, and incubated in boiling citrate buffer (0.01 mol/L, pH 6.0) for 10 min to accomplish the antigen retrieval. After treated with 3% hydrogen peroxide for 30 min, the samples were blocked with bovine serum albumin for 30 min at 37°C, and incubated in the VDAC1 antibody (CSB‐RA025821A0HU, CUSABIO, 1:100), then followed by the Histostain‐SP (streptavidin‐peroxidase) kit (SP‐0023, BIOSS) protocol. Finally, the DAB Horseradish Peroxidase Color Development Kit was deployed to detect the immunoreactivity.
2.11. Cardiolipin quantitation
The cardiolipin assay kit (ab241036, Abcam) was used to quantify the cardiolipin. HepG2 and Hep3B cells were cultured in the DMEM (sh30022.01, HyClone) supplemented with 10% fetal bovine serum (fbscn00519‐1, AusGeneX) and 1% penicillin/streptomycin (ma0110‐jan‐09f, Meilunbio). The cells were treated by knockdown, overexpression, or inhibition of VDAC1. The SiRNA (vdac1‐homo‐815, 19601) and plasmid (HG17755‐NF, Sino Biological) were transfected for knockdown and overexpression of VDAC1, respectively; 0.25 mmol/L DIDS (32029, MCE) was used to inhibit VDAC1. After 24 h, the transfection or inhibition efficiency was analyzed by western blot. The treated cells were suspended in CL Assay Buffer and lysed with brief sonication, then centrifuged at 10 000 g for 10 min at 4°C, and the supernatant was collected. The mixture of 50 μL sample and 50 μL probe was incubated at room temperature for 5‐10 min. Finally, fluorescence was measured at Ex/Em 340/480 nm.
2.12. NAO staining and laser confocal microscopy
The 1% NAO solution (75168‐11‐5, MAOKANGBIO) was prepared. HepG2 and Hep3B were cultured and treated to knockdown or overexpression of VDAC1 as mentioned above. After 24 h, the culture medium was discarded, and the cells were washed twice with PBS and serum‐free DMEM medium with the final concentration of 0.01% NAO was added. Then the cells were incubated at 37°C for 30‐60 min. After removal of the culture medium, the cells were washed with PBS and serum‐free DMEM medium was added. Finally, Laser scanning confocal microscope was performed to detect the cardiolipin content.
2.13. High‐resolution respirometry
Cell respiratory function was analyzed with high‐resolution respirometry in accordance with the manufacturer's instructions, and was performed in a two‐channel titration injection respirometer at 37°C (Oxygraph‐2k, Oroboros, Innsbruck, Austria). The cells were trypsinized and resuspended in FBS‐free medium to a final concentration of 106 cells/mL, and the respiration function was measured in the respirometry chamber.
2.14. Phenotypes information
As there are no specific and distinctive characteristics for NAFLD, the phenotype of body fat mass percentage (Record ID 12919) was screened to identify the NAFLD correlated gene 20 (Table S1). To further confirm the association between VDAC1 and NAFLD, we evaluated a cluster of fat liver‐related traits, among which, 7 phenotypes were selected, body fat mass (12920), 20 body weight (12909), 20 liver mass (17767), 14 white adipose mass (12957), 20 cholesterol in plasma (17801), 14 basal glucose in plasma under fasted state (17799) 21 and area under curve of glucose in oral glucose tolerance test (Glucose AUC in OGTT) (17661). 21 The correlations between Vdac1 and these phenotypes were analyzed (Table S2). To further analyze the association between Vdac1 and mitochondrial dysfunction, a correlation of Vdac1 and mitochondrial membrane cardiolipin was performed. To access and analyze the CL phenotypes, we first examined the Vdac1 gene, and computed the correlations with all BXD published phenotypes and selected the liver lipidomics CL phenotypes (Table S3), which were identified as detailed in a previous study. 11
2.15. Data analysis and online software
The raw mass spectrometry files were deposited in the ProteomeXchange Consortium with the online identifier PXD017072. Two‐tailed Student T test was performed on protein expression levels between the tumor tissue and adjacent non‐tumor tissue to identify proteins with significant statistical differences (P < .05).
The Cancer Genome Atlas (TCGA) LIHC cohort data analysis was performed using an online analysis tool (http://ualcan.path.uab.edu/analysis.html); and GEPIA analysis tools (http://gepia.cancer‐pku.cn/detail.php).
Correlation analysis was performed on the online server www.genenetwork.org. Pearson correlation coefficient and P‐value were used for assessment.
The liver gene expression data and the liver proteome data used in this study can be accessed via our online server (www.genenetwork.org). To access and analyze the Vdac1 gene expression, choose ‘Mouse (mm10)’ for Species, ‘BXD Family’ for Group, ‘Liver mRNA’ for Type, ‘EPFL/LISP BXD CD Liver Affy Mouse Gene 1.0 ST (Aug18) RMA’ for Data Set, and enter ‘VDAC1’ for Get Any. For phenotypes analysis, chose ‘Phenotypes’ for Type, ‘BXD Phenotypes’ for Data Set, and entered the phenotypes traits, such as ‘body fat’ for Combined. Peptide expression from VDAC1 was searched from the liver proteome data set ‘EPFL/ETHZ BXD Live Proteome CD (Nov19)’ (Table S4). All data in the study are provided in supplementary tables.
Heatmaps were plotted using the online software Morpheus (https://software.broadinstitute.org/morpheus). Graphs and analyses were made using GraphPad Prism software (8.0.1).
3. RESULTS
3.1. The proteome profiling differences between NAFLD‐driven HCC tumor tissues and adjacent non‐tumor tissues
In total, 1965 proteins were identified and quantified (Table S5). To avoid potential mis‐identification bias, we focused on 238 proteins that were identified in all samples. A volcano plot was generated to show the differential expression distribution (Figure 1A). Among the 238 proteins, 100 proteins presented significant differential expression between HCC tissues and adjacent non‐tumor tissues (P < .05) (Table S6). Here, 65 proteins were upregulated and 35 proteins were downregulated in the HCC. Pathway analysis indicated that the top enriched pathways included Ketogenesis, Mevalonate Pathway I and Isoleucine Degradation I (Figure 1B and Table S7). The significant altered proteins were analyzed by using the PANTHER classification system (http://pantherdb.org/). 22 The majority of proteins belonged to cell components (58.68%), possessed catalytic activity (45.33%), and were involved in metabolic processes (31.77%) (Figure S1). Pathway analysis was consistent with findings in a previous report of an increased glycolysis process and a decreased fatty acid beta‐oxidation process. Furthermore, our proteomics results were comparable with those of a previous study, indicating that a potential mechanism difference existed between NAFLD‐associated HCC and other types of HCC. 23 , 24 , 25 The results of early‐stage hepatocellular carcinoma proteomics showed that hepatocellular carcinoma was related to hepatitis B virus infection (HBV‐HCC) and was associated with disrupted cholesterol homeostasis, accompanied by high expression of sterol O‐acyltransferase 1 (SOAT1). 23 Nonetheless, our study suggested that the differences in protein expression between NAFLD‐driven HCC tissues and adjacent non‐tumor tissues were mainly focused on ketogenesis. This difference suggested that different diagnoses and therapeutic strategies should be applied to different types of HCC.
FIGURE 1.
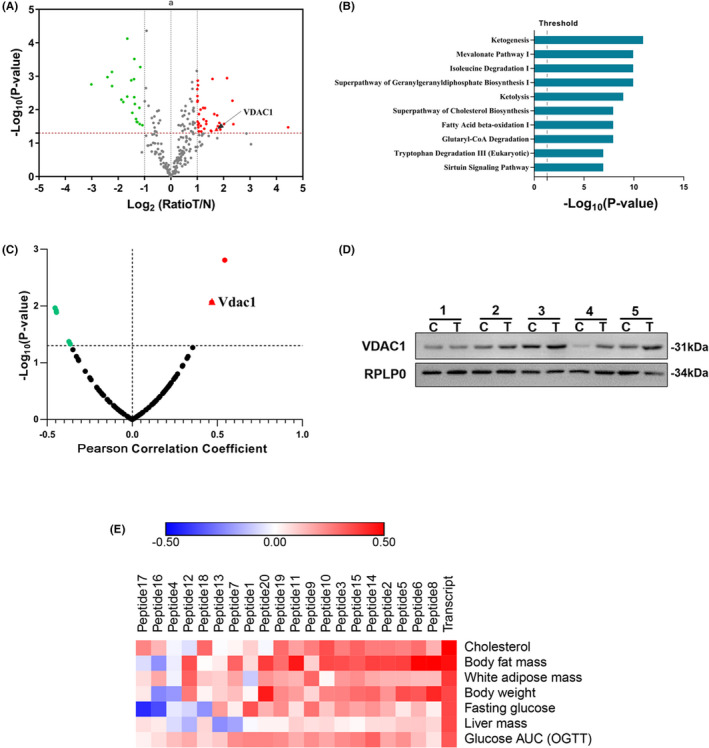
VDAC1 is dysregulated in NAFLD‐driven HCC and associated with NAFLD. A, Volcano plot graph showing the differential expressed proteins in the quantitative analysis. The −log10(P‐value) was plotted against the log2 (ratio Tumor/Normal). The upregulated proteins in HCC tissues were marked with red dots, and the downregulated proteins in HCC tissues with green dots. B, Pathways analysis of significantly altered proteins. The canonical pathways associated with the differential proteins were tested alongside the P‐values calculated using right‐tailed Fisher exact test. The top 10 pathways were listed. C, Transcriptome‐wide association study suggested that VDAC1 was associated with NAFLD. The correlation between differentially expressed protein and the phenotype of body fat mass percentage (Record ID 12 919) was shown. The red plots indicated the positive correlation, the green plots indicated the negative correlation, and the dark plots indicated no statistical significance. A significant correlation with the VDAC1 (R = 0.469, P = .008) was shown in the red triangle. D, Overexpression of VDAC1 in tumor tissues was validated with western blot. C represented the HCC tissues, and N represented the adjacent normal tissues. RPLP0 is used as internal reference. E, Heat map showed that VDAC1 was associated with a serial of fatty liver disease phenotypes in both gene and protein expression level. The VDAC1 transcript had significant correlations with the body fat mass (R = 0.472, P = .008), body weight (R = 0.361, P = .049), liver mass (R = 0.358, P = .021), white adipose mass (R = 0.391, P = .035), cholesterol in plasma (R = 0.525, P = .0003), Glucose AUC in OGTT (R = 0.331, P = .034) and fasting glucose level (R = 0.298, P = .058). Red represents positive correlations, and blue represents negative correlations. Transcript and peptide 1‐20 represent the gene and peptides expression level, respectively. A, B, D, obtained from the proteomic analysis of human samples; C, E were obtained from the transcriptomic and proteomic analysis of BXD family samples
3.2. VDAC1 is dysregulated in NAFLD‐driven HCC and associated with NAFLD
To prioritize the genes associated with NAFLD, a transcriptome‐wide association analysis was performed in BXD mice strains. The correlation between body fat mass percentage and the transcripts of 100 candidate proteins was calculated. Among all candidates, VDAC1 presented a significant correlation with body fat mass percentage (R = 0.469, P = .008) (Figure 1C and Table S1). To further confirm the association between VDAC1 and NAFLD, the correlation between VDAC1 and a series of fatty liver disease phenotypes was evaluated at both the transcript and peptide levels. The phenotypes included body fat mass, body weight, liver mass, white adipose mass, cholesterol in plasma, basal glucose in plasma under fasting and area under curve of glucose in an oral glucose tolerance test (Glucose AUC in OGTT) (Figure 1E and Table S2). The results suggested that both the gene and protein expression levels of VDAC1 were correlated with NAFLD traits.
Dysregulation of VDAC1 in HCC was further validated. Our proteomic results suggested a 3.3‐fold increase in VDAC1 in HCC tissues. This result was validated using a western blot (Figures 1D and S2), and further confirmed in an external clinical cohort. In The Cancer Genome Atlas (TCGA) LIHC cohort, Vdac1 expression levels were upregulated in HCC tissues compared with non‐tumor tissues (Normal N = 50, Tumor N = 371, P < .0001) (Figure 2A) and increased with increasing tumor grade (Figure 2B). Moreover, high‐level VDAC1 expression was correlated with poor clinical outcome (High, n = 91; Low, n = 91; P = .013) (Figure 2C). Collectively, these results indicated that VDAC1 was associated with NAFLD and dysregulated NAFLD‐driven HCC.
FIGURE 2.
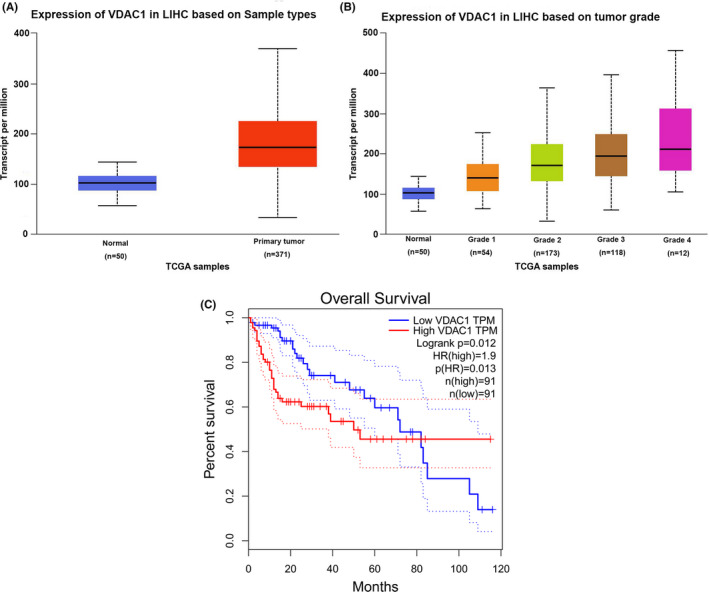
Identification of VDAC1 expression correlated with HCC in a human clinical cohort. A, Vdac1 upregulated in HCC samples compared with the normal samples was validated in the Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA‐LIHC) (n = 371), and Normal (n = 50) cohorts. B, Vdac1 is upregulated with HCC grade increase and was identified in TCGA‐LIHC Grade 1 (n = 54), Grade 2 (n = 173), Grade 3 (n = 118), Grade 4 (n = 12), and Normal (n = 50) cohorts. C, High‐level VDAC1 expression correlates with poor survival outcome in HCC and LIHC (High, n = 91; Low, n = 91) cohorts based on survival analysis
3.3. System genetics analysis of Vdac1 in BXD mice strains
System genetics analysis was then performed to reveal the expression regulation of VDAC1. Gene expression levels of VDAC1 in liver tissue 39 BXD mouse strains and their corresponding parental strains were examined. There was only 1 probe set (10 375 941) that targeted the first exon of the Vdac1 gene in the Affymetrix Mouse Gene 1.0 ST array. Vdac1 expression varied widely across BXD strains with a fold change of 1.41 (Figure 3A). The average expression of Vdac1 across all BXD strains was 12.12 ± 0.02 (log2 scale, mean ± standard error of the mean (SEM)). The strain with the highest level of expression was BXD45, while the strain with the lowest expression was BXD61. To identify sequence variants that affected the expression of Vdac1 in mouse liver, we performed simple interval mapping for Vdac1 across the mouse genome. One significant expression QTL (eQTL) was identified on chromosome 11 at 52.01 Mb with an LRS of 18.3, and which was close to the physical location of Vdac1 on chromosome 11 at 52.36 Mb (Figure 3B and Table S8). In addition, we also identified 4 suggestive eQTLs located on other chromosomes (Figure 3B and Table 2). These results indicated that the Vdac1 mRNA level was both cis‐ and trans‐regulated, simultaneously. For the other 4 trans‐eQTLs, we retrieved a total of 361 protein coding genes from their 1.5 LOD confidence interval (Table 2). Among them, 18 genes are significantly correlated with the Vdac1 expression level (Table 2). From the liver proteome data, there were 20 peptide‐targeted VDAC1 proteins. To evaluate the correlation between Vdac1 transcripts and peptides, a correlation analysis was performed. Among the 20 peptides, the top 5 peptides most highly correlated with the VDAC1 transcript were listed, Peptide 3 (R = 0.458, P = .005), Peptide 10 (R = 0.472, P = .004), Peptide 14 (R = 0.493, P = .002), Peptide 15 (R = 0.437, P = .008), and Peptide 19 (R = 0.496, P = .002) (Figure S3 and Table S4). Therefore, Peptides 3, 10, 14, 15, 19 were chosen to represent the VDAC1 protein for further analysis.
FIGURE 3.
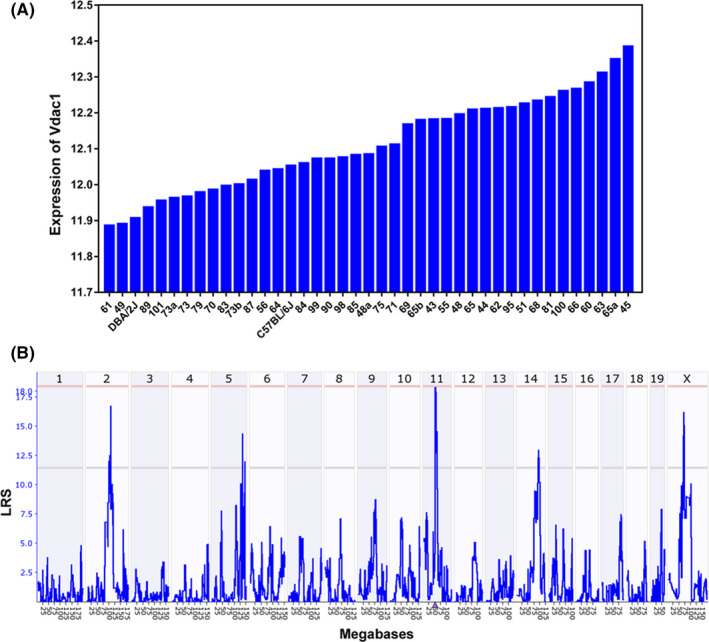
System genetics analysis of Vdac1 in BXD mice strains. A, Variable expression levels of Vdac1 in the liver tissue. Expression data for 39 BXD strains and their parental strains (B6 and D2). The x‐axis denotes the strain, while the y‐axis denotes mean expression given as log2. Each bar shows mean expression values ± standard error of the mean (SEM). B, Interval mapping of the Vdac1 gene in the liver. The upper x‐axis shows the chromosome, the lower x‐axis shows the location in megabases, the y‐axis provides the likelihood ratio statistics score (LRS). Blue lines indicate the LRS values at a given position, with a significant LRS at 18.35 and suggestive LRS at 11.14. Interval mapping identified a significant cis‐eQTL on chromosome 11 at 52.01 Mb and 4 suggestive trans‐eQTLs on chromosomes 2, 5, 14, and X
TABLE 2.
List of 4 suggested trans‐eQTLs for Vdac1
| Chr | Start | End | LRS | Marker | # of gene | Genes correlated with Vdac1 |
|---|---|---|---|---|---|---|
| 2 | 98 | 107 | 16.263 | rs33188980 | 87 | Tcp11l1 Apip Cd59a Cd59b Lmo2 Ldlrad3 Eif3m Qser1 |
| 5 | 132 | 136 | 14.215 | rs36653585 | 66 | Lat2 Limk1 Mdh2 |
| 14 | 73 | 100 | 12.941 | – | 151 | Klf5 Rcbtb2 Tpt1 Mzt1 Sucla2 Esd |
| X | 64 | 72 | 16.188 | rs33876026 | 57 | Prrg3 |
3.4. Gene co‐expression network and function enrichment analysis indicated VDAC1 is associated with mitochondria dysfunction
To explore the potential mechanism of VDAC1 involved in NAFLD‐driven HCC, the Vdac1 gene co‐expression network and functional enrichment analysis were implemented. With the aim to identify Vdac1 genes involved in gene modules, the filtered probes were submitted to the WGCNA 18 package for constructing gene co‐expression networks. Soft threshold power β (8) was chosen based on scale‐free topology criteria (Figure 4A,B). Using this soft‐thresholding power, we identified 35 distinct co‐expression modules that were assigned different colors; Vdac1 was located in the green module containing 589 genes (Figure 4C and Table S9). To verify the involvement of the green module in the biological processes, we performed GO and KEGG enrichment analysis using the WebGestalt online toolkit. We found that this module was significantly enriched in biological processes such as the oxidation‐reduction process, cell death, and the apoptotic signaling pathway (Table 3). This green module was also enriched in several KEGG pathways including oxidative phosphorylation, NAFLD, and the tricarboxylic acid cycle (TCA cycle) (Table 3). The analysis revealed that Vdac1 and its co‐expressed genes present in the same module were involved in biological functions such as energy metabolism and liver disease (NAFLD). Furthermore, pathway analysis using IPA showed that the mitochondrial dysfunction pathway was the top enriched pathway (Figure 5A and Table S10) and that 20 mitochondrial inner membrane proteins were involved in this pathway, which showed correlation with Vdac1 (Figure 5B and Table S11).
FIGURE 4.
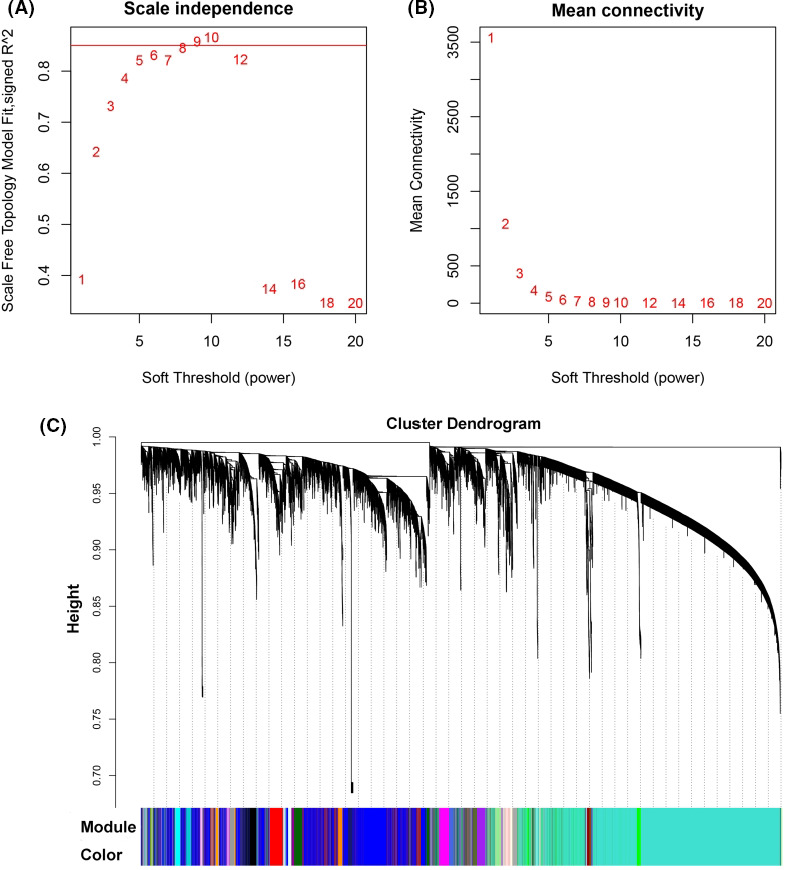
Weighted gene co‐expression network analysis. A, Soft‐thresholding index R 2 (y‐axis) as a function of different powers β (x‐axis). B, Mean connectivity (degree, y‐axis) as a function of the soft‐thresholding power (x‐axis). C, 35 co‐expression modules identified from mice liver transcriptome data by dendrogram branch cutting
TABLE 3.
Gene enriched terms for GO and KEGG pathways
| Term | Description | No. of Genes | P‐value | FDR |
|---|---|---|---|---|
| GO: Biological process | ||||
| GO:0055114 | Oxidation‐reduction process | 54 | 4.89E‐10 | 4.08E‐06 |
| GO:0045333 | Cellular respiration | 18 | 4.17E‐09 | 1.74E‐05 |
| GO:0006091 | Generation of precursor metabolites and energy | 24 | 6.68E‐08 | 1.85E‐04 |
| GO:0015980 | Energy derivation by oxidation of organic compounds | 20 | 2.06E‐07 | 2.86E‐04 |
| GO:0007005 | Mitochondrion organization | 29 | 4.36E‐06 | 2.45E‐03 |
| GO:0008219 | Cell death | 67 | 4.42E‐06 | 2.45E‐03 |
| GO:0097190 | Apoptotic signaling pathway | 30 | 5.72E‐06 | 2.75E‐03 |
| GO:0006915 | Apoptotic process | 62 | 1.04E‐05 | 3.61E‐03 |
| GO:0012501 | Programmed cell death | 62 | 1.63E‐05 | 4.84E‐03 |
| GO:0006099 | Tricarboxylic acid cycle | 6 | 3.27E‐05 | 7.79E‐03 |
| KEGG pathways | ||||
| mmu00190 | Oxidative phosphorylation | 22 | 2.17E‐12 | 6.49E‐10 |
| mmu05012 | Parkinson disease | 21 | 7.31E‐11 | 1.09E‐08 |
| mmu05010 | Alzheimer disease | 21 | 2.91E‐09 | 2.90E‐07 |
| mmu05016 | Huntington disease | 21 | 1.88E‐08 | 1.40E‐06 |
| mmu01100 | Metabolic pathways | 61 | 8.03E‐07 | 4.80E‐05 |
| mmu04932 | Non‐alcoholic fatty liver disease (NAFLD) | 16 | 1.60E‐06 | 7.98E‐05 |
| mmu01200 | Carbon metabolism | 13 | 8.85E‐06 | 3.78E‐04 |
| mmu00020 | Citrate cycle (TCA cycle) | 6 | 1.34E‐04 | 5.02E‐03 |
| mmu04142 | Lysosome | 10 | 1.23E‐03 | 4.09E‐02 |
| mmu00010 | Glycolysis/Gluconeogenesis | 7 | 1.39E‐03 | 4.16E‐02 |
FIGURE 5.
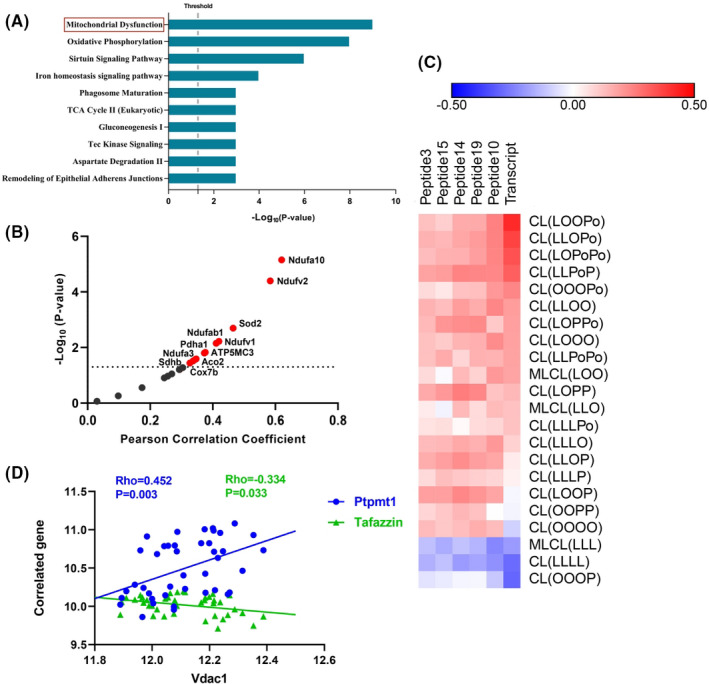
VDAC1 is associated with mitochondria dysfunction. A, Vdac1 co‐expression network is involved in mitochondrial dysfunction based on the mice liver transcriptome data. The Vdac1 co‐expression genes were analyzed by IPA. The top canonical pathways associated with the network genes were tested alongside the P‐values calculated using a right‐tailed Fisher exact test. Mitochondrial dysfunction was listed as the top pathway. B, Vdac1 shows correlation with 20 mitochondrial genes. The red plots indicate the significant positive correlation, and the dark plots indicate no statistical significance. C, Heatmap showing the correlation between VDAC1 and different species of CL based on the BXD family database. VDAC1 transcript had a negative relationship with the predominant CL species, CL (LLLL) and its precursor MLCL (LLL). VDAC1 had a positive relation with the nascent CL; and the VDAC1 correlated peptides showed the same trend. Red represents positive correlations, and blue represents negative correlations. D, Correlation between Vdac1 and cardiolipin synthetase gene Ptpmt1 and Tafazzin in a BXD mice cohort. Vdac1 has a positive correlation with Ptpmt1 (R = 0.452, P = .003); and negative correlation with Tafazzin (R=−0.334, P = .033) among 41 BXD strains. The blue line represents the correlation with Ptpmt1, and green with Tafazzin
3.5. VDAC1 is correlated with cardiolipin (CL) profiling shift
To further understand how VDAC1 was associated with mitochondrial dysfunction, we investigated the association between VDAC1 and mitochondrial membrane lipids. CL was the signature phospholipid of the IMM, which plays an important role in maintaining the structure and function of mitochondria. 26 , 27 A gene‐lipidome association analysis was performed between VDAC1 and 22 lipid species from the CL class. Expression levels of the VDAC1 transcript and peptides had a negative correlation with the mature CLs, CL (LLLL), and its precursor MLCL (LLL) (Figure 5C and Table S3). Conversely, VDAC1 had a positive correlation with the nascent CLs panel (Figure 5C and Table S3), thus we hypothesized that VDAC1 was associated with CL remodeling impairment. Therefore, we calculated the nascent CLs/mature CL species CL(LLLL) ratio for each nascent CL species that is the hallmark of CL remodeling. VDAC1 was positively correlated with 17 out 18 nascent/mature ratios (Table S12). Moreover, the significant correlation between VDAC1 and cardiolipin synthetase PTPMT1 and TAZ was confirmed in the BXD mice cohort with (R = 0.452, P = .003; R = 0.334, P = .033) (Figure 5D and Table S13). VDAC1 was correlated with CLs synthesis and CL remodeling, which might damage the stability of mitochondrial inner membrane structure by changing the CL components, and eventually cause mitochondrial dysfunction.
3.6. Mitochondrial function analysis with or without VDAC1 expression in Hep3B and HepG2
To further identify whether VDAC1 overexpression attenuates mitochondrial function or not, we performed pathological and biological experiments. The results of immunohistochemical analysis suggested that the expression of VDAC1 elevated in NAFLD‐driven HCC tissues compared with that in the adjacent non‐tumor tissues (Figure 6E). To illustrate the effects of VDAC1 on mitochondrial function, HCC cells (HepG2 and Hep3B) were applied to knockdown or overexpress VDAC1. Meanwhile, we also applied an inhibitor to reduce VDAC1 function. VDAC1 reduction and overexpression were determined by western blot (Figure 6A). After depletion of VDAC1 in HCC cells, the mitochondrial respiratory function enhanced significantly compared with the overexpression VDAC1 group (Figure 6D). Furthermore, overexpression of VDAC1 significantly affected mitochondrial respiration, with inhibition of oxygen consumption rates (OCRs) in basal respiration (Figure 6D). The IMM with abundant respiratory enzymes played an important role in mitochondrial respiration. Additionally, the inner membrane is rich in an unusual phospholipid, cardiolipin. Subsequently, cardiolipins were quantified by fluorometric probe kit and NAO staining. Cardiolipin content was decreased by overexpression of VDAC1 in both HepG2 and Hep3B cell lines. In contrast, decreased VDAC1 caused an increase in the cardiolipin content (Figure 6B,C). These data revealed an important role of VDAC1 overexpression in the decline in mitochondrial function.
FIGURE 6.
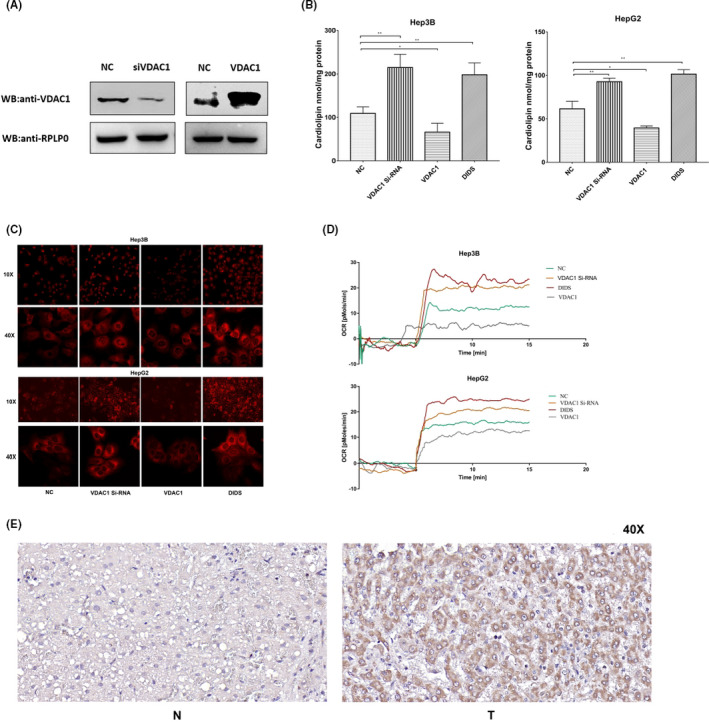
Mitochondrial function analysis with or without VDAC1 expression in Hep3B and HepG2. A, Western blot analysis of VDAC1 level in HepG2 cells after transfection of VDAC1 siRNA and VDAC1 expression plasmid. Compared with the negative control, the expression level of VDAC1 reduced in the knockdown group, and increased in overexpression group. RPLPO was the internal reference protein. B, Determination of cardiolipin in Hep3B and HepG2 cells. Cardiolipin was measured using the fluorometric probe assay kit (ab241036). The left panel shows the cardiolipin content in Hep3B. Compared with the NC group, the cardiolipin contents both in knockdown group and inhibitor group were significantly higher (P < .01), and the cardiolipin content in overexpression group was lower (P < .05). The right panel showed the cardiolipin content in HepG2. Compared with the NC group, the cardiolipin contents both in knockdown group and inhibitor group were significantly higher (P < .01), and the cardiolipin content in overexpression group was lower (P < .05). C, Detection of cardiolipin levels in HepG2 and Hep3B by NAO staining. Compared with negative control, the fluorescence intensity increased in the knockdown group and inhibitor group, and decreased in the overexpression group both in Hep3B and HepG2. The fluorescence intensity of the 2 cell lines was observed under a microscope at ×10 and ×40 magnification, respectively. D, Detection of mitochondrial respiratory function in Hep3B and HepG2 cells. High‐resolution respirometry performed in an Oroboros Oxygraph‐2k. The different color curves showed the mitochondrial oxygen consumption of the different groups. In Hep3B cells, the OCR of knockdown group and inhibitor group were higher than the NC group, and the OCR of overexpression group and inhibitor group was lower than the NC group. Also, the same trend was shown in HepG2 cells. E, Relative VDAC1 expression was detected by IHC between NAFLD‐driven HCC and adjacent non‐tumor liver tissues (×40). N represented the VDAC1 in adjacent non‐tumor liver tissues. T represented the VDAC1 in NAFLD‐driven HCC tissues. (NC represented the negative control group; VDAC1 siRNA represented VDAC1 knockdown group; VDAC1 represented the overexpression group and DIDS represented the inhibitor group)
3.7. VDAC1‐centralized transcript‐lipids‐phenotypes correlation network
To better illustrate the association of VDAC1 to NAFLD phenotypes through CL profiling, a VDAC1‐centralized transcript‐lipids phenotypes correlation network was generated to provide a reference for future studies (Figure 7A and Tables S2‐S4). In summary, our study indicated that VADC1 was associated with NAFLD and the dysregulation of VDAC1 in NAFLD‐driven HCC through mitochondrial dysfunction and by regulating mitochondrial membrane proteins and the CL acyl chain composition shift (Figure 7B).
FIGURE 7.
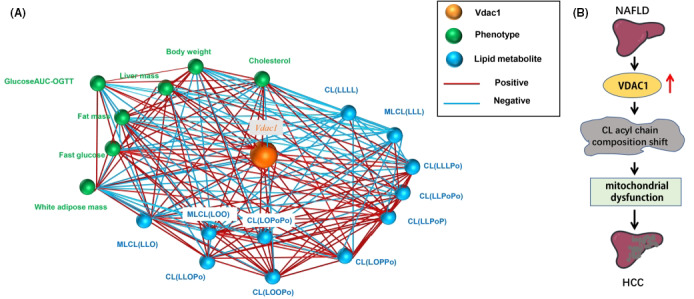
VDAC1‐centralized correlation network. A, Correlation network of VDAC1‐centralized transcript‐lipids phenotypes based on the BXD mice family database. The network was constructed including VDAC1 expression, NAFLD phenotypes and representative CL species. Red lines show positive correlations, and blue lines show negative correlations. The network illustrates the VDAC1 associated with NAFLD phenotypes through correlating with CL profiling. B, Schematic for VDAC1 involved in NAFLD‐related HCC. VADC1 is upregulated in NAFLD, and dysregulation of VDAC1 causes a CL acyl chain composition shift, which led the NAFLD‐driven HCC tumorigenesis through mitochondrial dysfunction
4. DISCUSSION
To investigate the potential mechanism linking NAFLD and liver cancer, we performed the first quantitative proteomic analysis on clinical biopsies from NAFLD‐related HCC patients. We quantified over 1900 proteins in hepatoblastoma tissues and compared them with non‐tumor adjacent liver tissues. For the significantly altered proteins, transcriptome‐wide association analysis suggested that VDAC1 was associated with NAFLD, suggesting that VDAC1 is an essential gene that may cause fatty liver disease progression to HCC. VDAC1 is the most abundant protein on the outer membrane of mitochondria, and plays an important role in regulating mitochondrial function. It is the gatekeeper for the passage of metabolites, nucleotides, and ions that control metabolism and apoptosis and interacts with many proteins also involved in lipid metabolism and cholesterol transport. As a hub protein, VDAC1 serves as an anchor point for mitochondrion‐interacting proteins such as hexokinase (HK), Bcl2, and Bcl‐xL, some of which are also overexpressed in cancer cells. In fact, multiple sources of evidence have confirmed the involvement of VDAC1 in several cancers. 28 Furthermore, we comprehensively analyzed the VDAC1 expression regulation network, which indicated mitochondria dysfunction as the major associated pathway. Further gene‐lipidome association analysis indicated that VDAC1 was associated with the CL composition profiling shift, which led to the mitochondrion membrane structure alteration and dysfunction. Mitochondrial dysfunction has been well accepted as a hallmark of various types of cancers. But an understanding of the mitochondrial membrane lipid in cancer is still limited. 26 We showed here the association between VDAC1 and mitochondrial dysfunction through regulating cardiolipin acyl chain composition.
Dysregulation of VDAC1 has been reported in both NAFLD and HCC. 27 , 29 However, the mechanism involved in VDAC1 mediation and NAFLD transition to HCC is still unclear. VDAC1 is considered an essential mitochondrial gatekeeper. 30 The significant increase in VDAC1 was associated with mitochondrial dysfunction. 31 This mechanism has not yet fully been revealed, we observed the association between VDAC1 and CL composition shift. CL is considered as the major negatively charged lipid in the inner mitochondria with high concentrations at the contact sites between inner and outer mitochondrial membranes, and co‐localization with VDAC1. 32 Among the lipid species, CL (LLLL), and its precursor MLCL (LLL) are 2 negative correlated lipids to VDAC1. In liver mitochondria, CL (LLLL) is the most predominate lipid species with tetra polyunsaturated acyl side chains (18:2) which provide a tight structure with high affinity binding of CL to proteins compared with saturated acyl side chains. 33 The loss of CL (LLLL) and shift of CL composition profiling will significantly change mitochondria dynamics and morphology. 34 Therefore, a proper composition for CL is critical for cells to maintain the structure and function of mitochondria. A loss of CL (LLLL) has been noted in a variety of metabolic disorders including NAFLD 14 and diabetes. 35
Such CL profiling shift related to VDAC1 can be explained by the negative correlation between VDAC1 and TAZ. The formation of CL (LLLL) did not occur during de novo biosynthesis because the enzymes of biosynthetic pathways lack the appropriate substrate, 36 a cardiolipin remodeling process is essential to achieve its appropriate acyl content. The main route is via CoA‐independent transacylation between different phospholipids in which TAZ plays a major part. Moreover, we also identified a significant positive correlation between VDAC1 and PTPMT1, which is an essential enzyme in CL biosynthesis. 37 It catalyzes the reaction of the transformation from phosphatidylglycerophosphate (PGP) to phosphatidylglycerol (PG), which is an essential intermediate in the biosynthetic pathway of cardiolipin. Taken together, our results suggested that VDAC1 association with CL profiling shift was through promoting CL synthesis and inhibiting the CL remodeling process, but the detailed mechanisms need to be further illustrated.
In this study, we integrated multiple ’omics data from BXD mice strains. Due to resource limitation, it was almost impossible to achieve all the ’omics analysis and phenotype measurements in one study, especially for a population level study. Thus, we took the advantage of BXD mice stains as a gene reference panel. Due to the well controlled breeding environment, the gene, protein, and lipid expression levels in each strain were stable enough for a cross‐study analysis. This made it possible to integrate diverse ’omics data from different studies. It was possible to establish a correlation network between genes, transcripts, proteins, metabolites, and phenotypes at a population level. With the unique GRP, we identified the association between VDAC1 and a series of fatty liver disease phenotypes and lipids components in mitochondria that identified the role of VDAC1 in NAFLD‐driven HCC from a systems point of view. Our study showed that this type of correlation analysis could generate and test novel association hypotheses, therefore this approach offered a novel alternative strategy to current research methods.
In conclusion, our study combined proteomics and system genetics analyses to reveal that VDAC1 was associated with NAFLD and dysregulated in NAFLD‐driven HCC. The dysregulation of VDAC1 was associated with mitochondria lipid composition shift and mitochondria dysfunction during tumorigenesis. Furthermore, the multiple ’omics‐wide association analysis provided a novel strategy to uncover the pathogenesis of diseases and discover new disease markers.
DISCLOSURE
The authors declare that they have no competing interests. All authors have agreed to publish this manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was conducted in accordance with the Declaration of Helsinki of 1975, and the program was approved by the ethics committee of human research of Binzhou Medical University (approval number: 2016‐019), and the patients were prospectively enrolled with written informed consent at Shandong Provincial Hospital.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
We thank Professor Robert W. Williams from the University of Tennessee Health Science Center for valuable scientific advices to the project and Dr. Zelu Zhang for technical assistance. This work was supported by Taishan Scholars Construction Engineering, The Shandong Excellent Young Scientist Award (ZR2016JL026), National Natural Science Foundation of China (31671139, 31771284), Shandong provincial natural science foundation (2017GSF18103, 2018GSF118131, 2018GSF118183, 2018GSF118230), BZMC Scientific Research Foundation (BY2017KYQD08), Swedish Research Grant 2015‐4870.
Zhu Y, Zhang C, Xu F, et al. System biology analysis reveals the role of voltage‐dependent anion channel in mitochondrial dysfunction during non‐alcoholic fatty liver disease progression into hepatocellular carcinoma. Cancer Sci. 2020;111:4288–4302. 10.1111/cas.14651
Yanping Zhu and Chao Zhang contributed equally to this work.
Funding information
Swedish Research Grant, (Grant/Award Number: 2015‐4870) BZMC Scientific Research Foundation, (Grant/Award Number: BY2017KYQD08) Natural Science Foundation of Shandong Province, (Grant/Award Numbers: 2017GSF18103, 2018GSF118131, 2018GSF118183, 2018GS) National Natural Science Foundation of China, (Grant/Award Number: 31671139, 31771284) Taishan Scholars Construction Engineering, The Shandong Excellent Young Scientist Award, (Grant/Award Number: ZR2016JL026).
Contributor Information
Lu Lu, Email: llu@uthsc.edu.
Jia Mi, Email: jia.mi@bzmc.edu.cn.
Geng Tian, Email: tiangeng@bzmc.edu.cn.
DATA AVAILABILITY STATEMENT
All data and materials are available from the authors upon request.
REFERENCES
- 1. Manne V, Handa P, Kowdley KV. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis. 2018;22(1):23‐37. [DOI] [PubMed] [Google Scholar]
- 2. Coilly A, Desterke C, Guettier C, Samuel D, Chiappini F. Fabp4 and Mmp9 levels identified as predictive factors for poor prognosis in patients with nonalcoholic fatty liver using data mining approaches and gene expression analysis. Sci Rep. 2019;9(1):19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fujiwara N, Nakagawa H, Enooku K, et al. Cpt2 Downregulation adapts Hcc to lipid‐rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018;67(8):1493‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakagawa H, Hayata Y, Kawamura S, Yamada T, Fujiwara N, Koike K. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers (Basel). 2018;10(11):447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wainberg M, Sinnott‐Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome‐wide association studies. Nat Genet. 2019;51(4):592‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nicholson JK, Holmes E, Elliott P. The METABOLOME‐WIDE ASSOCIATION STUDY: A NEW LOOK AT HUMAN DISEASE RISK FACTORS. J Proteome Res. 2008;7(9):3637‐3638. [DOI] [PubMed] [Google Scholar]
- 7. Ryaboshapkina M, Hammar M. Human hepatic gene expression signature of non‐alcoholic fatty liver disease progression, a meta‐analysis. Sci Rep. 2017;7(1):12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong EP, Park JW. Sample size and statistical power calculation in genetic association studies. Genomics Inform. 2012;10(2):117‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kitano H. Computational systems biology. Nature. 2002;420(6912):206‐210. [DOI] [PubMed] [Google Scholar]
- 10. Chella Krishnan K, Kurt Z, Barrere‐Cain R, et al. Integration of multi‐omics data from mouse diversity panel highlights mitochondrial dysfunction in non‐alcoholic fatty liver disease. Cell Syst. 2018;6(1):103‐15.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jha P, McDevitt MT, Gupta R, et al. Systems analyses reveal physiological roles and genetic regulators of liver lipid species. Cell Syst. 2018;6(6):722‐33.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Y, Qi X, Yu C, et al. Identification of prothymosin alpha (Ptma) as a biomarker for esophageal squamous cell carcinoma (Escc) by label‐free quantitative proteomics and quantitative dot blot (Qdb). Clin Proteomics. 2019;16(12):1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Wang D, Li M, et al. Quantitative proteomics of tramp mice combined with bioinformatics analysis reveals that Pdgf‐B regulatory network plays a key role in prostate cancer progression. J Proteome Res. 2018;17(7):2401‐2411. [DOI] [PubMed] [Google Scholar]
- 14. Wu Y, Williams EG, Dubuis S, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158(6):1415‐1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185‐193. [DOI] [PubMed] [Google Scholar]
- 16. Chesler EJ, Lu L, Shou S, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet. 2005;37(3):233‐242. [DOI] [PubMed] [Google Scholar]
- 17. Keane TM, Goodstadt L, Danecek P, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Langfelder P, Wgcna HS. An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(559):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. Webgestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45(W1):W130‐W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andreux PA, Williams EG, Koutnikova H, et al. Systems genetics of metabolism: the use of the bxd murine reference panel for multiscalar integration of traits. Cell. 2012;150(6):1287‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Williams EG, Wu Y, Jha P, et al. Systems proteomics of liver mitochondria function. Science. 2016;352(6291):aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mi H, Muruganujan A, Ebert D, et al. PANTHER version 14: more genomes, a new PANTHER GO‐slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419‐D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early‐stage hepatocellular carcinoma. Nature. 2019;567(7747):257‐261. [DOI] [PubMed] [Google Scholar]
- 24. Megger DA, Bracht T, Kohl M, et al. Proteomic differences between hepatocellular carcinoma and nontumorous liver tissue investigated by a combined gel‐based and label‐free quantitative proteomics study. Mol Cell Proteomics. 2013;12(7):2006‐2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buczak K, Ori A, Kirkpatrick JM, et al. Spatial tissue proteomics quantifies inter‐ and intratumor heterogeneity in hepatocellular carcinoma (Hcc). Mol Cell Proteomics. 2018;17(4):810‐825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, Yu W, Ryu SW, et al. Cardiolipins are biomarkers of mitochondria‐rich thyroid oncocytic tumors. Cancer Res. 2016;76(22):6588‐6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pittala S, Krelin Y, Kuperman Y, Shoshan‐Barmatz V. A Mitochondrial Vdac1‐based peptide greatly suppresses steatosis and Nash‐associated pathologies in a mouse model. Mol Ther. 2019;27(10):1848‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shoshan‐Barmatz V, Pittala S, Mizrachi D. VDAC1 and the TSPO: expression, interactions, and associated functions in health and disease states. Int J Mol Sci. 2019;20(13):3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pittala S, Krelin Y, Shoshan‐Barmatz V. Targeting liver cancer and associated pathologies in mice with a mitochondrial Vdac1‐based peptide. Neoplasia. 2018;20(6):594‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arif T, Stern O, Pittala S, Chalifa‐Caspi V, Shoshan‐Barmatz V. Rewiring of Cancer Cell Metabolism By Mitochondrial Vdac1 depletion results in time‐dependent tumor reprogramming: glioblastoma as a proof of concept. Cells. 2019;8(11):1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lemasters JJ, Holmuhamedov EL, Czerny C, Zhong Z, Maldonado EN. Regulation of mitochondrial function by voltage dependent anion channels in ethanol metabolism and the warburg effect. Biochim Biophys Acta. 2012;1818(6):1536‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shoshan‐Barmatz V, Zalk R, Gincel D, Vardi N. Subcellular localization of Vdac in mitochondria and Er in the cerebellum. Biochim Biophys Acta. 2004;1657(2–3):105‐114. [DOI] [PubMed] [Google Scholar]
- 33. Schlame M, Horvath L, Vigh L. Relationship between lipid saturation and lipid‐protein interaction in liver mitochondria modified by catalytic hydrogenation with reference to cardiolipin molecular species. Biochem J. 1990;265(1):79‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cells. 2019;8(7):728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46(21):6417‐6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J Biomed Res. 2010;24(1):6‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J, Guan Z, Murphy AN, et al. Mitochondrial phosphatase Ptpmt1 Is essential for cardiolipin biosynthesis. Cell Metab. 2011;13(6):690‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data Availability Statement
All data and materials are available from the authors upon request.