

Abstract
Most cancers harbor a small population of highly tumorigenic cells known as cancer stem cells (CSCs). Because of their stem cell‐like properties and resistance to conventional therapies, CSCs are considered to be a rational target for curable cancer treatment. However, despite recent advances in CSC research, CSC‐targeted therapies are not as successful as was initially hoped. The proliferative, invasive, and drug‐resistant properties of CSCs are regulated by the tumor microenvironment associated with them, the so‐called CSC niche. Thus, targeting tumor‐promoting cellular crosstalk between CSCs and their niches is an attractive avenue for developing durable therapies. Using mouse models of squamous cell carcinoma (SCC), we have demonstrated that tumor cells responding to transforming growth factor β (TGF‐β) function as drug‐resistant CSCs. The gene expression signature of TGF‐β–responding tumor cells has accelerated the identification of novel pathways that drive invasive tumor progression. Moreover, by focusing on the cytokine milieu and macrophages in the proximity of TGF‐β–responding tumor cells, we recently uncovered the molecular basis of a CSC–niche interaction that emerges during early tumor development. This review article summarizes the specialized tumor microenvironment associated with CSCs and discusses mechanisms by which malignant properties of CSCs are maintained and promoted.
Keywords: ADAP1, antioxidant responses, cancer invasion, cancer stem cells (CSCs), interleukin‐33 (IL‐33), macrophages, transforming growth factor β (TGF‐β)
Cancer stem cells (CSCs) are a rational target for curable cancer treatment, but the development of effective CSC‐targeted therapies is moving at a restricted pace. This review article summarizes the recent advances in the understanding of the mechanism of tumor‐promoting interactions between CSCs and their niche tumor microenvironment.

1. INTRODUCTION
During tumor development, neoplastic cells often become heterogeneous genetically and phenotypically. 1 Concurrently, functionally distinct nontumor cells, including fibroblasts, endothelial cells, and immune cells, increase the spatial complexity of the tumor microenvironment. 2 , 3 Extensive intratumor heterogeneity can be a cause of therapy resistance and a limiting factor in patient survival. 4 Thus, an improved understanding of the mechanisms that cause intratumor heterogeneity would yield better cancer diagnosis and treatment strategies.
Cancer stem cells (CSCs), also known as tumor‐initiating cells (TICs), are a major driver of intratumor heterogeneity. 5 CSCs are a long‐lived subset of tumor cells with properties resembling normal stem cells. 5 CSCs have been defined by their ability to propagate tumors in transplantation assays and to recapitulate phenotypic features of primary tumors in secondary tumors. 6 , 7 More recently, CSCs have been evaluated by their ability to maintain long‐term clonal growth using lineage tracing assays in mouse models of various cancers. 8 , 9 , 10 Based on cell surface markers and biochemical properties unique to CSC populations, CSCs have been purified from many cancer types in human and mouse, and their transcriptomic, phenotypic, and functional characteristics have been extensively profiled. CSCs typically activate gene expression programs relevant to stem cells and embryonic development. 11 , 12 Moreover, CSCs are known to have enhanced capacities for stress resistance, including DNA damage, 13 antioxidant, 14 and hypoxic responses, 15 and upregulated molecular programs involved in drug resistance, immune evasion, and metastasis. Thus, understanding the regulatory mechanisms and vulnerability of CSCs is a vital step to target CSCs therapeutically.
Normal stem cells are maintained in specialized microenvironments or the stem cell niche. 16 Similarly, CSCs are believed to reside in anatomically distinct regions within the tumor, the so‐called CSC niche. 17 External cues emanating from the niche regulate the stem‐like state of CSCs and malignant properties of their progeny. 3 , 18 Transforming growth factor β (TGF‐β) is a multifunctional cytokine that plays a critical role in the stem cell niche and tumor development. 19 , 20 , 21 , 22 TGF‐β binds to its receptor kinase complex on the cell surface that phosphorylates downstream signal transducers SMAD2 and SMAD3. Phosphorylated‐SMAD2/3 form protein complexes with SMAD4 and other transcriptional cofactors to regulate TGF‐β–responsive target genes. TGF‐β also induces a series of non‐SMAD signaling pathways. 23 It is well described in the literature that TGF‐β acts as a tumor suppressor or promoter in different stages of cancer development. For example, a conditional knockout (cKO) of the TGF‐β receptor in mouse epithelial tissues provided direct evidence of TGF‐β function in “tumor suppression.” 24 , 25 , 26 , 27 However, this approach (i.e., the lack of TGF‐β receptor throughout tumorigenesis) does not allow examination of the tumor cell‐intrinsic effects of TGF‐β in “tumor progression.” Because TGF‐β exerts a tumor‐supportive role by regulating nontumor cells in the stroma, such as the activation of cancer‐associated fibroblasts 28 and immunosuppression, 29 the functional significance of TGF‐β signaling activated in tumor cells has not been probed thoroughly in vivo. Given that TGF‐β is instrumental in orchestrating various stem cell niches, 20 TGF‐β signals activated in niche tumor microenvironments that participate in maintaining and promoting the malignant properties of CSCs could offer a novel therapeutic target.
2. A SYSTEM TO STUDY TGF‐β SIGNALING IN TUMOR DEVELOPMENT AND DRUG RESISTANCE
Investigating a tumor cell‐intrinsic effect of TGF‐β signaling in cancer development and drug resistance requires specialized models. We previously generated a de novo squamous cell carcinoma (SCC) mouse model through the in utero injection of lentiviruses (Figure 1A). 30 In this model, a fluorescent reporter for the TGF‐β/SMAD signal allowed us to detect tumor cells responding to TGF‐β in HRAS‐driven primary SCCs in vivo. Interestingly, we observed that most TGF‐β–responding cells localized at the tumor‐stroma interfaces. Consistent with the cytostatic effects of TGF‐β, they were a slow‐cycling tumor cell population. When we purified fluorescent reporter‐positive, TGF‐β–responding cells and transplanted them into immunodeficient mice, TGF‐β–responding tumor cells formed secondary tumors more efficiently than did nonresponding cells. 30
FIGURE 1.
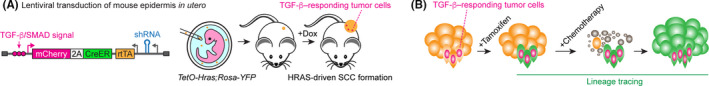
A, Lentiviral construct for in utero injection. Lentiviral particles were injected into amniotic fluid surrounding the TetO‐Hras,Rosa‐YFP transgenic mouse embryo to transduce the surface ectoderm. Postnatally, doxycycline administration induces oncogenic HRAS expression only in transduced cells (orange), which initiate the formation of squamous cell carcinoma (SCC). In the developing tumors, a fluorescent reporter driven by a SMAD‐dependent promoter enabled us to detect transforming growth factor β (TGF‐β)–responding tumor epithelial cells (pink). B, Lineage tracing of TGF‐β–responding tumor cells during chemotherapy and tumor recurrence. A tamoxifen‐activatable Cre (CreER) under the SMAD‐dependent promoter allowed investigation of the fate of TGF‐β–responding tumor cells.
This system also allowed us to examine how TGF‐β–responding tumor cells behaved in developing SCC and those under chemotherapy. Using a tamoxifen‐activatable Cre (CreER) explicitly expressed upon TGF‐β signaling, we revealed that TGF‐β–responding tumor cells gave rise to invasive, poorly differentiated, tumor cell progenies in vivo. These progenies tended to be more scattered in the tumor epithelial tissue and exhibited phenotypes resembling epithelial–mesenchymal transition (EMT). Moreover, under chemotherapy treatment, most TGF‐β–responding tumor cells evaded apoptosis and their progeny drove the recurrence of SCC, indicating that TGF‐β–responding tumor cells functioned as drug‐resistant CSCs (Figure 1B). 30
In addition to the slow‐cycling state and EMT, which are known contributors for drug resistance, TGF‐β–responding tumor cells activate other mechanisms to promote drug‐resistant properties. Transcriptomic analyses of TGF‐β–responding tumor cells revealed that they had enhanced glutathione metabolism. Glutathione is an antioxidant that helps to reduce oxidative stress and detoxify foreign substances, including anti‐cancer drugs. 31 Indeed, an inhibitor of glutathione transferases suppressed TGF‐β–mediated protection against cisplatin cytotoxicity. Transcription factor NRF2, the master regulator of antioxidant responses, 32 mediates the expression of glutathione metabolism genes in TGF‐β–responding tumor cells. When we upregulated or downregulated NRF2, tumor cells became resistant and sensitive to chemotherapy, respectively. 30
3. TGF‐β–RESPONDING TUMOR CELLS ACTIVATE THE ADAP1–ARF6‐MEDIATED ENDOCYTIC PATHWAY
Gene expression signatures of CSCs can be predictive of poor patient outcomes. 33 , 34 Therefore, we sought to identify clinically relevant genes from signature genes of the TGF‐β‐responding CSC population. Through this investigation, we recently identified ADAP1 (ArfGAP with dual PH domains 1, also known as centaurin‐α1), and our study suggested that ADAP1 could be a potential biomarker and therapeutic target for SCCs with the risk of metastatic progression.
Using in silico analysis, we found that the expression level of ADAP1 strongly correlated with poor survival of early‐stage head and neck SCC (HNSCC) patients. 35 ADAP1 is a GTPase‐activating protein (GAP) for the small GTPase ARF6 (Figure 2A), which is a critical regulator of endocytic vesicle transport between the cell surface and endosomal compartments (Figure 2B,C). ARF6 has been implicated in tumor development and metastasis. 36 , 37 ADAP1 was originally identified as a neuron‐specific phosphatidylinositol‐(3,4,5)‐trisphosphate (PIP3)‐binding protein, 38 , 39 but its role in cancer is largely unknown. Therefore, we explored the potential link between high ADAP1 expression and poor patient outcome. Using in vitro cell culture models of primary mouse keratinocytes and human SCC cell lines, we found that ADAP1 enhanced cell invasion through the Matrigel extracellular matrix (ECM) in a GAP activity‐dependent manner. Moreover, using our SCC mouse model, we showed that the overexpression of ADAP1 promoted SCC invasive progression with extensive disruption of the basement membrane (Figure 2D). In contrast, tumors overexpressing a GAP activity‐deficient ADAP1 better maintained the integrity of the basement membrane. Furthermore, Adap1 deletion in tumor epithelial cells prevented invasive tumor growth. 35
FIGURE 2.
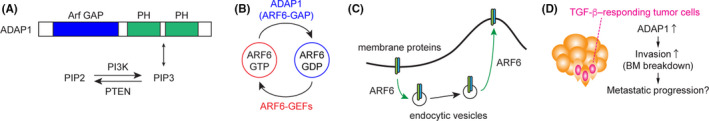
A, Domain structure of ADAP1. The N‐terminal Arf GAP domain functions as an ARF6‐directed GTPase‐activating protein (GAP). The C‐terminal dual PH domains bind to phosphatidylinositol 3,4,5‐trisphosphate (PIP3), a second messenger generated by PI3K activity. B, GTP‐GDP exchange cycle of ARF6 small GTPase. ADAP1 increases the GTPase activity of ARF6 to hydrolyze GTP to GDP, and guanine‐nucleotide exchange factors (GEFs) catalyze the exchange of GDP to GTP. C, The role of ARF6 in intracellular trafficking of cell surface proteins and cell membranes. ARF6‐associated endocytosis is a clathrin‐independent, plasma membrane‐endosomal recycling pathway. At the plasma membrane, ARF6‐GTP facilitates the internalization of various cargos using energy from GTP hydrolysis, and ARF6 also controls cargo recycling to coordinate cell adhesion and invasion. D, The role of ADAP1, which is highly expressed in transforming growth factor β (TGF‐β)–responding tumor cells, in invasive squamous cell carcinoma (SCC) progression.
The progression from in situ tumor to invasive tumor requires dissolution of cell–cell contacts, increased cell motility, and disruption of the basement membrane. Endocytic vesicle‐mediated membrane and protein transport underlies all of these processes, and aberrant expression of membrane trafficking regulators has been implicated in tumor progression. 40 However, it is mostly unknown whether CSCs activate a distinct endocytic transport to promote tumor progression. Potentially, ADAP1 might mediate this process. Through its C‐terminal dual PH domains, ADAP1 binds to PIP3‐containing membranes (Figure 2A), which are restricted to the basolateral surface of epithelial cells and the leading front of migrating cells. 41 Given that PI3K and PTEN, positive and negative regulators of PIP3 levels, are frequently mutated in a broad spectrum of cancers 42 and that PI3K activation promotes cell survival and invasion of CSCs of HNSCC, 43 PI3K‐induced aggressive phenotypes might be mediated by ADAP1. Moreover, an altered endocytic transport is known to reduce the uptake of cisplatin, suggesting the possible contribution to drug resistance. 44 There, the ADAP1–ARF6‐mediated endocytic mechanism might provide a novel insight into the vulnerability of CSCs.
4. CSCS AND THEIR NICHE MICROENVIRONMENTS
It is well established that the tumor microenvironment contributes to tumor progression. CSC niches are proposed as specific microenvironments to support the self‐renewal and survival of CSCs. Blood vessels, immune cells, fibroblasts, and other cell types with particular characteristics participate in the CSC niche. Cellular composition, signaling factors, and ECM proteins make the CSC niche molecularly distinct. Multiple studies have supported the idea that reciprocal interaction between CSCs and their putative niches is a crucial component of tumor growth and progression. 17 , 45 Cancer cells transform normal fibroblasts to cancer‐associated fibroblasts (CAFs) via IL‐6. In turn, CAFs produce matrix metalloproteinases MMP‐2 and MMP‐9, which promote EMT, stemness, and metastasis of cancer cells. 46 CSCs have elevated HIF‐1 activity, which promotes vascular endothelial growth factor (VEGF) expression and angiogenesis. 47 Then, endothelial cells promote stem cell‐like phenotypes in cancer cells through Notch signaling and nitric oxide signaling pathways. 48 Moreover, VEGF enhances the expression of PD‐1 and other inhibitory checkpoint molecules involved in CD8+ T cell exhaustion, establishing a perivascular, immunosuppressive niche microenvironment. The perivascular niches also serve as an entry site of circulating immune cells into the tumor. The spatial distribution of immune cells within tumors can be predictive of patient survival and metastasis. 49 , 50 In particular, tumor‐associated macrophages support CSC populations and promote EMT through cytokine signaling in multiple cancer types. However, it is still largely unknown how CSCs regulate the localization and function of immune cells in their spatial proximity. Using our mouse model, we recently unveiled the cellular and molecular basis for establishing a CSC niche that promoted the malignant progression and drug resistance of SCC. 51
4.1. Interleukin‐33 (IL‐33): a cytokine highly expressed in TGF‐β–responding tumor cells
Given that TGF‐β promotes functional properties of CSCs, understanding the cellular crosstalk that underlies the activation of TGF‐β signaling might provide a strategy for destabilizing CSCs. Our SCC mouse model offered a paradigm to investigate cellular crosstalk between the TGF‐β–responding CSCs and their neighboring stromal cells. We were particularly intrigued by the fact that the frequency of TGF‐β–responding tumor cells was strongly associated with locally enriched TGF‐β ligand in the adjacent stroma. Because abundant TGF‐β expression in the stroma is known to be associated with poor prognosis in human cancers, 52 , 53 “TGF‐β rich” stromal areas might serve as a niche microenvironment for CSCs. However, the mechanism by which the CSC niche emerges during early tumor development remains unclear.
Because normal stem cells coordinate their niches by sending short‐distant signals, 54 we hypothesized that CSCs might release unique signaling molecules to induce TGF‐β–rich microenvironments in the adjacent stroma for their maintenance and expansion. We searched candidate molecules from a list of signature genes of TGF‐β–responding tumor cells and identified IL‐33 as the most upregulated cytokine 51 (Figure 3A). IL‐33 is a cytokine that belongs to the IL‐1 family and has a vital role in tissue homeostasis and response to injury through its contribution to innate and adaptive immunity. 55 Interestingly, IL‐33 can play both pro‐ and anti‐tumorigenic roles in tumor growth and progression. In some cancer types IL‐33 can promote tumorigenesis by directly affecting tumor cells, 56 , 57 but in many cases IL‐33 regulates immune cells in the tumor microenvironment. 58 , 59
FIGURE 3.
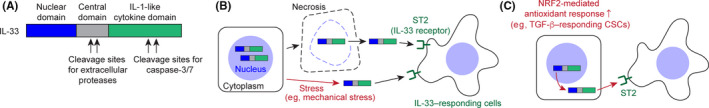
A, Domain structure of interleukin (IL)‐33. The N‐terminal domain has DNA/chromatin‐binding domains, and the C‐terminal domain functions as a cytokine molecule, which binds to its receptor known as ST2. The central domain contains cleavage sites for extracellular proteases, such as elastase and cathepsins, and truncated forms of IL‐33 are more bioactive. In contrast, caspase‐dependent cleavage in the C‐terminal cytokine domain prevents the IL‐33–dependent immune response. B, Regulation of the extracellular release of IL‐33. After necrotic cell death, full‐length IL‐33 is released from the nuclei of IL‐33–producing cells. Release of IL‐33 has also been reported after mechanical stress in the absence of cell death. Extracellular IL‐33 binds to the ST2 receptor expressed on IL‐33–responding cells, which leads to the activation of downstream signaling cascades, including ERK/JNK/p38 MAP kinases and NF‐κB pathways. C, Activity of NRF2‐mediated antioxidant responses in TGF‐β–responding tumor cells was correlated with nuclear‐to‐cytoplasmic translocation and extracellular release of IL‐33.
4.2. NRF2‐mediated antioxidant response facilitates the extracellular release of IL‐33
Under normal tissue homeostasis, IL‐33 is localized in the nucleus of epithelial and endothelial cells. The N‐terminal region of IL‐33 contains DNA‐binding domains, whereas its C‐terminal region harbors an IL‐1–like cytokine domain 60 (Figure 3A). The nuclear sequestration of IL‐33 is crucial because transgenic mice expressing IL‐33 whose nuclear targeting was abolished succumbed to lethal inflammation in multiple organs. 61 In contrast with the release of other IL‐1 family members, such as IL‐1β and IL‐18, which require the activation of caspase‐1/inflammasome, 62 inflammasome activation is dispensable for the extracellular release of IL‐33. Rather, caspase‐dependent cleavage in the cytokine domain of IL‐33 leads to its inactivation. Full‐length IL‐33 can be released from cells upon tissue injury mainly through necrosis (Figure 3B), and its cytokine activity is increased by proteolytic cleavage between the nuclear and cytokine domains in the extracellular space.
It remains unclear how IL‐33 can be released from living tumor cells, particularly CSCs. Interestingly, while IL‐33 was localized in the nucleus of normal skin epidermis, we detected IL‐33 in the cytoplasm of invasive SCC tumor cells. Moreover, a cytoplasmic IL‐33 signal was more frequently observed in TGF‐β–responding tumor cells compared with nonresponding cells, suggesting that IL‐33 may be preferentially released from this CSC population. It has been reported that nonlethal stress, such as mechanical stress, can induce the secretion of nuclear IL‐33 (Figure 3B). 63 Therefore, we addressed whether CSC‐intrinsic stress response properties, in particular the NRF2‐mediated antioxidant response, might be involved in the extracellular release of IL‐33.
The activity of NRF2 is tightly regulated by the KEAP1‐mediated ubiquitin‐proteasomal degradation pathway. 64 We previously found that TGF‐β–responding tumor cells often exhibited stabilized NRF2 in the nucleus. 30 Interestingly, in both human and mouse SCC, cytoplasmic IL‐33 localization was detected in tumor cells having stabilized NRF2 in the nucleus. 51 Moreover, using biochemical and genetic manipulation of the NRF2‐mediated antioxidant pathway, we found a correlation between NRF2 and cytoplasmic localization and the reduction of intracellular IL‐33 levels in vitro. Although the molecular mechanism is still largely unknown, our data suggested that the NRF2‐mediated antioxidant response activated in CSCs facilitated the extracellular release of IL‐33 (Figure 3C).
4.3. IL‐33 promotes paracrine TGF‐β signaling, tumor progression, and drug resistance
By comparing Il33 mRNA levels in tumor epithelial cells, fibroblasts, immune cells, and endothelial cells, we showed that tumor epithelial cells, particularly TGF‐β–responding tumor cells, are the primary source of IL‐33. So, what is the role of CSC‐derived IL‐33 in the development of SCC? Using Il33 shRNA in our lentiviral vector, we depleted IL‐33 in tumor cells, including the TGF‐β–responding CSC population. Il33 knockdown (KD) tumors showed a significantly smaller size and less malignant phenotypes, including smooth tumor–stroma boundaries and less invasive cells. Il33 KD tumor cells proliferated more frequently but maintained well differentiated states. 51
As described above, slow‐cycling, poorly differentiated, and invasive phenotypes are characteristics observed in TGF‐β–responding tumor cells and their progenies. 30 Therefore, we hypothesized that the stunted progression of Il33 KD tumors might be due to the reduction of TGF‐β–responding tumor cells. Indeed, by flow cytometry analysis, we found that Il33 KD tumors had significantly less TGF‐β–responding tumor cells. 51 Moreover, because TGF‐β exerts drug‐resistant effects in tumor cells, 30 we tested if Il33 KD tumors were more sensitive to chemotherapy treatment. Like the clinical problem in SCC patients, control tumors typically showed recurrence after cisplatin treatment. By contrast, most Il33 KD tumors shrunk dramatically and failed to regrow. These data supported the notion that CSC‐derived IL‐33 plays a crucial role in generating the TGF‐β–responding CSC population potentially through the formation of TGF‐β–rich stroma.
4.4. A subset of alternatively activated macrophages accumulated around CSCs
To address the potential crosstalk between CSCs and surrounding immune cells, using immunofluorescence microscopy we searched a variety of immune cells existing within a 50‐µm radius of the tumor edges. Consistent with the fact that TGF‐β promotes differentiation of regulatory T (Treg) cells, 65 FoxP3+ Treg cells existed more frequently around TGF‐β–responding tumor cells, suggesting the presence of a functional TGF‐β gradient in the stroma. Among the types of immune cells that we examined, high‐affinity immunoglobulin E (IgE) receptor FcεRIα‐expressing cells were most significantly accumulated in proximity to TGF‐β–responding tumor cells. 66 To identify the cell type expressing FcεRIα in SCC, we performed a comprehensive flow cytometry analysis using a panel of immune cell markers. The majority of FcεRIα+ cells was macrophages with low MHC‐II surface expression. 51
Macrophages play an essential role in innate and adaptive immune responses by detecting and ingesting damaged or diseased cells through phagocytosis. In the tumor microenvironment, macrophages are regulated by a variety of stimuli and cytokines, and their properties are dynamically changed between classically activated phenotypes (often associated with high MHC‐II expression) and alternatively activated phenotypes. The latter is known to promote angiogenesis, immunosuppression, tumor cell invasion, and metastasis. 67 , 68 The close spatial relationship between TGF‐β–responding tumor cells and FcεRIα+ macrophages might reflect their functional interactions and be involved in the formation of TGF‐β–rich stroma. To characterize this previously unappreciated macrophage population, we performed RNA‐seq analysis of FcεRIα+ macrophages isolated from SCC. FcεRIα+ macrophages upregulated alternatively activated macrophage markers, including Arg1, Cd206, Cd163, and Il10, among others. In contrast, they downregulated classically activated macrophage markers such as Tnf, Cd40, and MHC‐II. In multiple mouse models of SCC and human SCC patient samples, we confirmed that the FcεRIα+ population was CD206+ alternatively activated macrophages preferentially accumulated in peritumor areas (within a 50 µm radius from tumor edges). Importantly, these FcεRIα+ macrophages upregulated Tgfb1 expression and were overlapped with TGF‐β–rich stromal areas, suggesting that this macrophage population was a putative component of the CSC niche. 51
4.5. CSC‐derived IL‐33 promotes the formation of TGF‐β‐rich niche microenvironment
We sought to address the potential link between CSC‐derived IL‐33 and FcεRIα+ macrophages. We found that FcεRIα+ macrophages highly expressed the IL‐33 receptor ST2, 69 raising an intriguing possibility that CSCs directly induced FcεRIα+ macrophages by releasing IL‐33 in peritumor areas (Figure 4). We first tested this possibility using bone marrow‐derived cells in vitro. IL‐33 induced not only alternatively activated macrophage differentiation but also FcεRIα expression. Mechanistically, the nuclear factor κB (NF‐κB) signaling pathway activated downstream of ST2 was critical for FcεRIα expression. To evaluate the function of FcεRIα+ macrophages, we cocultured keratinocytes and macrophages in vitro. Compared with CSF1‐induced FcεRIαneg macrophages, IL‐33–induced FcεRIα+ macrophages activated paracrine TGF‐β signaling more effectively and promoted invasive phenotypes of keratinocytes. Moreover, Tgfb1 depletion in FcεRIα+ macrophages inhibited paracrine TGF‐β signaling and cell invasion. 51 Thus, IL‐33 released by CSCs may induce FcεRIα+ macrophage differentiation from immature myeloid cells recruited into the tumor and, by doing so, IL‐33–responding FcεRIα+ macrophages promoted invasive progression driven by CSCs.
FIGURE 4.
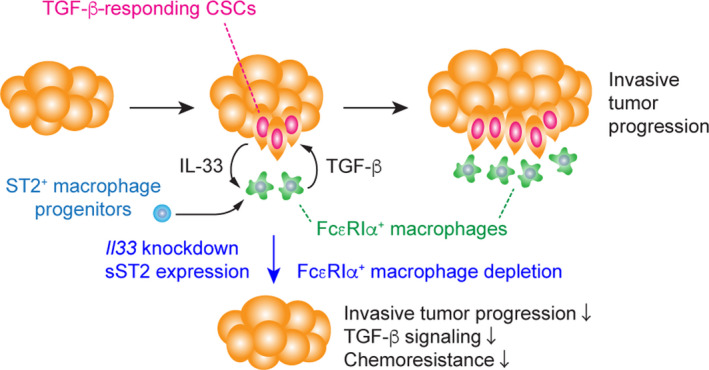
A model of cancer stem cell (CSC)‐niche interactions during invasive progression of squamous cell carcinoma (SCC). CSC cell populations release IL‐33 as a short‐distant signal, which promotes the differentiation of ST2+ hematopoietic progenitor cells recruited to the tumor tissue into FcεRIα+ alternatively activated macrophages. Those IL‐33–responding tumor cells produce “TGF‐β rich” niche microenvironments to activate paracrine transforming growth factor β (TGF‐β) signals, further promoting CSC‐relevant functions (eg, NRF2‐mediated antioxidant response) and IL‐33 production. In contrast, the suppression of IL‐33 expression in CSCs or FcεRIα+ macrophage depletion resulted in a reduction in TGF‐β signals and chemo‐resistance of SCC
Because IL‐33 serum levels are associated with several inflammatory diseases, 55 , 70 IL‐33 might have systemic effects in tumor‐bearing mice. However, FcεRIα+ macrophages were almost exclusively detected in the tumor microenvironment, but not in other hematopoietic organs such as the spleen and bone marrow. To block IL‐33–ST2 signals in the tumor microenvironment, we overexpressed soluble ST2 (sST2), a decoy receptor for IL‐33, 69 in tumor epithelial cells in vivo. Similar to Il33 KD tumors, sST2‐expressing tumors were significantly smaller and more differentiated compared with control tumors. Moreover, the number of FcεRIα+ macrophages was markedly lower in sST2‐expressing tumors, consistent with the reduction of IL‐33 in the tumor microenvironment. In contrast, the forced release of IL‐33 from tumor cells induced by NRF2 activation increased numbers of FcεRIα+ macrophages in the stroma. 51
To further evaluate the impact of IL‐33–ST2 signals on the differentiation of FcεRIα+ macrophages in the tumor microenvironment, we transplanted control and ST2 KD hematopoietic progenitor cells in the circulation of tumor‐bearing mice. ST2 depletion did not affect the recruitment of circulating progenitors into tumor tissues. However, the differentiation into FcεRIα+ macrophages was inhibited by ST2 KD. Finally, we depleted FcεRIα+ macrophages from tumors by injecting anti‐FcεRIα antibody and found that TGF‐β signaling in tumor cells was reduced. Moreover, tumor growth was perturbed by FcεRIα+ macrophage depletion, indicating that FcεRIα+ macrophages induced by IL‐33 are a crucial component of the niche that supports TGF‐β–responding CSCs (Figure 4). 51
5. CONCLUSIONS
The possible role of the niche as a tumor‐supportive microenvironment was suggested over 2 decades ago 71 , however, today, our incomplete understanding of CSC–niche interactions remains a significant barrier to improving therapeutic efficacy. Without filling this gap in knowledge, drug resistance is likely to remain a life‐threatening problem in cancer patients. To accommodate increasing numbers of CSCs during tumor development, neoplastic cells might transform the normal niche or create their niche de novo that would contribute to disease initiation, progression, and drug resistance. While the concept of exploring CSC–immune cell crosstalk is not novel, the specific tissue context and molecular underpinnings that underlie the emergence of CSC niches remain unclear. Previous studies have shown that IL‐33 induces macrophage recruitment and alters their phenotype to resemble alternatively activated macrophages. 56 , 72 Our recent study unveiled a novel CSC–niche signaling loop that emerged during the early stages of SCC development (Figure 4). 51 IL‐33 released by CSCs through their antioxidant activity directly created a spatially restricted, TGF‐β–rich niche microenvironment that was required for invasive progression and drug resistance of SCC. Because IL‐33–responding macrophages facilitated malignant properties of CSCs, identifying the pathways necessary for the maintenance of this macrophage subset and the development of reagent that disrupts CSC‐niche interactions, may offer durable cancer treatment.
DISCLOSURE
The author has no conflicts of interest to declare.
ACKNOWLEDGMENTS
Thanks to Weston Anderson for organizational edits of the manuscript. This work was supported by in part by the following grants and foundations: the NIH K99‐R00 Pathway to independence award (4R00CA178197‐03), the Medical Foundation of Oregon, the Collins Medical Trust, the Oregon Health & Science University (OHSU) Knight Cancer Institute (KCI) Pilot Grant (2018‐CCSG‐22), and the start‐up support from the KCI/OHSU to NO.
Oshimori N. Cancer stem cells and their niche in the progression of squamous cell carcinoma. Cancer Sci. 2020;111:3985–3992. 10.1111/cas.14639
REFERENCES
- 1. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Junttila MR, de Sauvage FJ. Influence of tumour micro‐environment heterogeneity on therapeutic response. Nature. 2013;501:346‐354. [DOI] [PubMed] [Google Scholar]
- 3. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275‐291. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 5. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. 2016;11:47‐76. [DOI] [PubMed] [Google Scholar]
- 6. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645‐648. [DOI] [PubMed] [Google Scholar]
- 7. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J, Li Y, Yu T‐S, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schepers AG, Snippert HJ, Stange DE, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730‐735. [DOI] [PubMed] [Google Scholar]
- 11. Ben‐Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell‐like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong DJ, Liu H, Ridky TW, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L. DNA Damage in Stem Cells. Mol Cell. 2017;66:306‐319. [DOI] [PubMed] [Google Scholar]
- 14. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keith B, Simon MC. Hypoxia‐inducible factors, stem cells, and cancer. Cell. 2007;129:465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880‐1885. [DOI] [PubMed] [Google Scholar]
- 17. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16:225‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui W, Fowlis DJ, Bryson S, et al. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531‐542. [DOI] [PubMed] [Google Scholar]
- 20. Oshimori N, Fuchs E. The harmonies played by TGF‐β in stem cell biology. Cell Stem Cell. 2012;11:751‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013;13:788‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. David CJ, Massagué J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat Rev Mol Cell Biol. 2018;19:419‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Derynck R, Budi EH. Specificity, versatility, and control of TGF‐β family signaling. Sci Signal. 2019;12:eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu S‐L, Herrington H, Reh D, et al. Loss of transforming growth factor‐β type II receptor promotes metastatic head‐and‐neck squamous cell carcinoma. Genes Dev. 2006;20:1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas‐specific blockade of transforming growth factor‐beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147‐3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muñoz NM, Upton M, Rojas A, et al. Transforming growth factor β receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837‐9844. [DOI] [PubMed] [Google Scholar]
- 27. Guasch G, Schober M, Pasolli HA, et al. Loss of TGFβ signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer. 2020;20:174‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batlle E, Massagué J. Transforming growth factor‐β signaling in immunity and cancer. Immunity. 2019;50:924‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oshimori N, Oristian D, Fuchs E. TGF‐β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217:2291‐2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931‐947. [DOI] [PubMed] [Google Scholar]
- 33. Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011;17:1086‐1093. [DOI] [PubMed] [Google Scholar]
- 34. Liu R, Wang X, Chen GY, et al. The prognostic role of a gene signature from tumorigenic breast‐cancer cells. N Engl J Med. 2007;356:217‐226. [DOI] [PubMed] [Google Scholar]
- 35. Van Duzer A, Taniguchi S, Elhance A, Tsujikawa T, Oshimori N. ADAP1 promotes invasive squamous cell carcinoma progression and predicts patient survival. Life Sci Alliance. 2019;2:e201900582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hashimoto S, Onodera Y, Hashimoto A, et al. Requirement for Arf6 in breast cancer invasive activities. Proc Natl Acad Sci USA. 2004;101:6647‐6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. D'Souza‐Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347‐358. [DOI] [PubMed] [Google Scholar]
- 38. Hammonds‐Odie LP, Jackson TR, Profit AA, et al. Identification and cloning of centaurin‐alpha. A novel phosphatidylinositol 3,4,5‐trisphosphate‐binding protein from rat brain. J Biol Chem. 1996;271:18859‐18868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kreutz MR, Böckers TM, Sabel BA, et al. Expression and subcellular localization of p42IP4/centaurin‐alpha, a brain‐specific, high‐affinity receptor for inositol 1,3,4,5‐tetrakisphosphate and phosphatidylinositol 3,4,5‐trisphosphate in rat brain. Eur J Neurosci. 1997;9:2110‐2124. [DOI] [PubMed] [Google Scholar]
- 40. Lanzetti L, Di Fiore PP. Behind the scenes: endo/exocytosis in the acquisition of metastatic traits. Cancer Res. 2017;77:1813‐1817. [DOI] [PubMed] [Google Scholar]
- 41. Shewan A, Eastburn DJ, Mostov K. Phosphoinositides in cell architecture. Cold Spring Harb Perspect Biol. 2011;3:a004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wong K‐K, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keysar SB, Le PN, Miller B, et al. Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J Natl Cancer Inst. 2017;109:djw189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang X‐J, Mukherjee S, Shen D‐W, Maxfield FR, Gottesman MM. Endocytic recycling compartments altered in cisplatin‐resistant cancer cells. Cancer Res. 2006;66:2346‐2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. 2019;24:41‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Giannoni E, Bianchini F, Masieri L, et al. Reciprocal activation of prostate cancer cells and cancer‐associated fibroblasts stimulates epithelial‐mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945‐6956. [DOI] [PubMed] [Google Scholar]
- 47. Maxwell PH, Bianchini F, Masieri L, et al. Hypoxia‐inducible factor‐1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1997;94:8104‐8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem‐like character in PDGF‐induced glioma cells. Cell Stem Cell. 2010;6:141‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Galon J. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960‐1964. [DOI] [PubMed] [Google Scholar]
- 50. DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taniguchi S, Elhance A, Van Duzer A, et al. Tumor‐initiating cells establish an IL‐33‐TGF‐β niche signaling loop to promote cancer progression. Science. 2020;369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Calon A, Espinet E, Palomo‐Ponce S, et al. Dependency of colorectal cancer on a TGF‐β‐driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Richardsen E, Uglehus RD, Johnsen SH, Busund L‐T. Immunohistochemical expression of epithelial and stromal immunomodulatory signalling molecules is a prognostic indicator in breast cancer. BMC Res Notes. 2012;5:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baghdadi MB, Castel D, Machado L, et al. Reciprocal signalling by Notch‐Collagen V‐CALCR retains muscle stem cells in their niche. Nature. 2018;557:714‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liew FY, Girard J‐P, Turnquist HR. Interleukin‐33 in health and disease. Nat Rev Immunol. 2016;16:676‐689. [DOI] [PubMed] [Google Scholar]
- 56. Fang M, Li Y, Huang K, et al. IL33 promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer Res. 2017;77:2735‐2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang J‐F, Wang P, Yan Y‐J, et al. IL‐33 enhances glioma cell migration and invasion by upregulation of MMP2 and MMP9 via the ST2‐NF‐κB pathway. Oncol Rep. 2017;38:2033‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wasmer M‐H, Krebs P. The Role of IL‐33‐Dependent Inflammation in the Tumor Microenvironment. Front Immunol. 2016;7:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fournié J‐J, Poupot M. The Pro‐tumorigenic IL‐33 Involved in Antitumor Immunity: A Yin and Yang Cytokine. Front Immunol. 2018;9:2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carriere V, Roussel L, Ortega N, et al. IL‐33, the IL‐1‐like cytokine ligand for ST2 receptor, is a chromatin‐associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bessa J, Meyer CA, de Vera Mudry MC, et al. Altered subcellular localization of IL‐33 leads to non‐resolving lethal inflammation. J. Autoimmun. 2014;55:33‐41. [DOI] [PubMed] [Google Scholar]
- 62. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LAB. Inflammasome activation and IL‐1β and IL‐18 processing during infection. Trends Immunol. 2011;32:110‐116. [DOI] [PubMed] [Google Scholar]
- 63. Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 2012;287:6941‐6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018;34:21‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF‐β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18:693‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309‐322. [DOI] [PubMed] [Google Scholar]
- 68. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kakkar R, Lee RT. The IL‐33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mitsui A, Tada Y, Takahashi T, et al. Serum IL‐33 levels are increased in patients with psoriasis. Clin Exp Dermatol. 2016;41:183‐189. [DOI] [PubMed] [Google Scholar]
- 71. Dührsen U, Hossfeld DK. Stromal abnormalities in neoplastic bone marrow diseases. Ann Hematol. 1996;73:53‐70. [DOI] [PubMed] [Google Scholar]
- 72. He Z, Chen L, Souto FO, et al. Epithelial‐derived IL‐33 promotes intestinal tumorigenesis in Apc Min/+ mice. Sci Rep. 2017;7:5520‐5614. [DOI] [PMC free article] [PubMed] [Google Scholar]