

Summary
Interleukin-34 (IL-34) is an alternative ligand to colony-stimulating factor-1 (CSF-1) for the CSF-1 receptor that acts as a key regulator of monocyte/macrophage lineage. In this study, we show that tumor-derived IL-34 mediates resistance to immune checkpoint blockade regardless of CSF-1 existence in various murine cancer models. Consistent with its immunosuppressive characteristics, the expression of IL-34 in tumors correlates with decreased frequencies of cellular (such as CD8+ and CD4+ T cells and M1-biased macrophages) and molecular (including various cytokines and chemokines) effectors at the tumor microenvironment. Then, a neutralizing antibody against IL-34 improved the therapeutic effects of the immune checkpoint blockade in combinatorial therapeutic models, including a patient-derived xenograft model. Collectively, we revealed that tumor-derived IL-34 inhibits the efficacy of immune checkpoint blockade and proposed the utility of IL-34 blockade as a new strategy for cancer therapy.
Subject Areas: Immunology, Cancer
Graphical Abstract
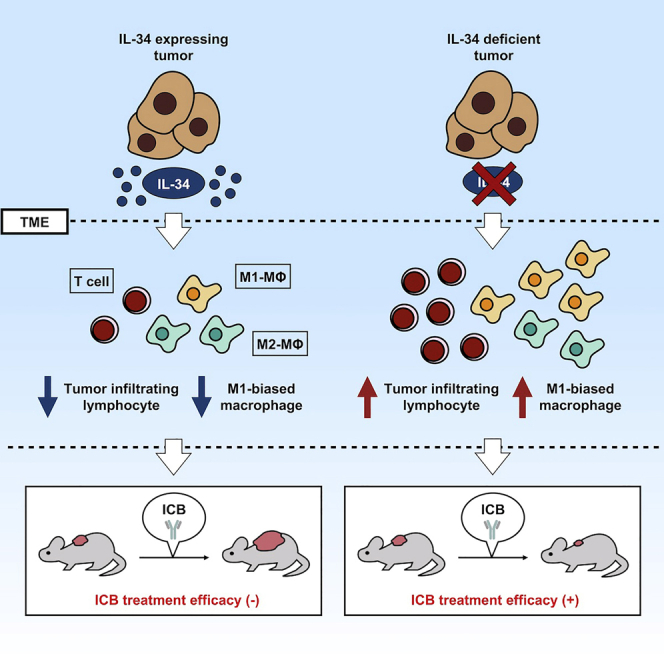
Highlights
-
•
IL-34-expressing tumors are resistant to ICB in several murine models
-
•
IL-34 upregulates the ratio of M2-biased to M1-biased macrophage in TME
-
•
Blockade of IL-34 with neutralizing antibody (Ab) enhances the efficacy of ICB
-
•
Anti-human IL-34 Ab shows the potential to enhance PD-1 blockade efficacy in PDX model
Immunology; Cancer
Introduction
The immune checkpoint blockade (ICB) is an attractive approach to activate therapeutic antitumor activity (Cheng et al., 2018; Garon et al., 2015; Hodi et al., 2010). However, tumors frequently develop immune resistance against T cells that are specific for tumor antigens, resulting in limited therapeutic benefits in the clinic (Herbst et al., 2014; Hugo et al., 2016; Wang et al., 2017). Several studies have suggested multiple mechanisms of immune resistance at the molecular and cellular levels, such as impaired infiltration and activation of T cells at the tumor microenvironment (TME), epigenetic changes in tumor cells that lead to impaired interferon-gamma (IFNγ) signaling, and immunosuppression at the local TME (O'Donnell et al., 2017; Pardoll, 2012; Ribas, 2015). In this context, the enrichment of the TME with immunosuppressive cells such as M2-biased tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) in addition to various metabolic and inflammatory mediators such as indoleamine 2,3-dioxygenase (IDO), arginase1 (ARG1), and prostaglandin E2 (PGE2) have been suggested to play critical roles in both innate and acquired resistance to immunotherapy (Kumar et al., 2016; Prima et al., 2017; Ugel et al., 2015). In many cases, the immunosuppressive TME is generated and continuously maintained by soluble factors secreted by the tumor cells (Binnewies et al., 2018). Accordingly, targeting these factors may help relieve immunosuppression and improve immunotherapeutic responses (Ghirelli and Hagemann, 2013; Pitt et al., 2016).
Among several therapeutic candidates, colony-stimulating factor-1 (CSF-1) receptor (CSF-1R) has gained much attention as a key molecule that controls the survival, proliferation, and functions of M2-biased TAMs with enhanced immunosuppressive activities (Noy and Pollard, 2014). Importantly, CSF-1/CSF-1R axis has been involved in promoting resistance to programmed death-1 (PD-1)/PD-L1 blockade, including, but not limited to, melanoma, hepatocellular carcinoma, and pancreatic cancer (Cannarile et al., 2017; Gyori et al., 2018; Neubert et al., 2018; Quaranta et al., 2018; Zhu et al., 2019). In addition to CSF-1, CSF-1R can be alternatively activated by binding with its second ligand, IL-34 (Lin et al., 2008). Although both of CSF-1 and IL-34 share the same receptor and show comparable effects on myeloid cells cultured in vitro, the two cytokines surprisingly show neither similar sequences nor common motifs and bind to distinct pockets within the extracellular domain of CSF-1R, resulting in different activation patterns of CSF-1R (Kim et al., 2006). A major difference between IL-34 and CSF-1 is the selective expression of IL-34 in the brain and skin, in contrast to CSF-1 which is widely expressed in the body under physiological conditions (Wang et al., 2012). However, despite its selective expression, IL-34 can be expressed and secreted by tumor cells and plays important roles in tumor progression (Baghdadi et al., 2018, 2019; Franzè et al., 2017; Lin et al., 2008; Ségaliny et al., 2015; Wang et al., 2012; Zhou et al., 2016) and resistance against chemotherapy and molecular targeted therapy (Baghdadi et al., 2016; Giricz et al., 2018). Except for our previous report of the potential involvement of IL-34 in tumor resistance against PD-1 blockade in a clinical case of a refractory melanoma patient (Han et al., 2018), little is known regarding the real role of IL-34 in promoting immunotherapeutic resistance against ICB and the related mechanisms. In this study, we first explore the potential role of IL-34 in mediating immune resistance of tumors against ICB in various murine tumors in addition to patient-derived xenograft (PDX) model and examine its impact on the molecular and cellular components of the TME.
Results
IL-34-Expressing HM-1 Murine Ovarian Cancer Is Resistant to PD-1 Blockade Therapy
To evaluate the impact of IL-34 on the therapeutic effects of ICB, we first utilized a murine ovarian cancer cell line OV2944-HM-1 (HM-1), which secretes a substantial level of IL-34 (Figure 1A) and CSF-1 (Figure S1). We used CRISPR-Cas9 system to generate IL-34-deficient HM-1 cell line (named Il34KO HM-1), and knockout efficiency was confirmed by measuring the secreted levels of IL-34 by Enzyme-Linked Immunosorbent Assay (ELISA) (Figure 1A). We found that the defect of IL-34 expression did not affect the viability of HM-1 cells in vitro, as compared with mock-transfected (Mock) HM-1 cells (Figure 1B). These cell lines expressed comparably low level of surface PD-L1 molecule (Figure 1C), while other surface molecules, including CD115 (CSF-1R), CD80, CD86, and PD-L2, were undetectable (data not shown). Then, Mock or Il34KO HM-1 cells were subcutaneously (s.c.) injected into syngenic B6C3F1 mice to evaluate their response against PD-1 blockade (Figure 1D). Interestingly, only Il34KO HM-1 tumors responded to PD-1 blockade, showing smaller tumor volumes than control IgG treatment (Figures 1E and 1F). On the other hand, PD-1 blockade showed little effects in mice bearing Mock HM-1 tumors (Figures 1E and 1F), indicating that PD-1 blockade is obvious in the absence of IL-34 in this model.
Figure 1.

IL-34 Derived from HM-1 Cells Limits Therapeutic Efficacy of Anti-PD-1. Therapy with Inhibiting T Cell Accumulation
(A) IL-34 concentration in supernatants of HM-1 cell lines (n = 3/cell line).
(B) Mean cell viability of Mock and Il34KO HM-1 cells measured by MTT assay (n = 3, technical triplicates). Similar results were obtained in 2 independent experiments.
(C) Representative histograms of PD-L1 expression of HM-1 cell lines.
(D) Schematic of the anti-PD-1 treatment. The timeline shows the procedure of tumor inoculation and antibody treatment.
(E) Tumor growth in B6C3F1 mice inoculated with Mock or Il34KO HM-1 cells and treated with anti-PD-1 antibody (α-PD-1) or control IgG (n = 4–6/group). Similar results were obtained in 2 independent experiments.
(F) Tumor weight on day 19 after tumor inoculation (n = 4/group).
(G) Representative flow cytometry profiles showing CD8+ and CD4+ T cells within the tumor-infiltrating CD3+CD45 + cells on day 19. Bar graphs represent the frequency of each T cell subset (n = 4–6/group).
(H) Representative flow cytometry profiles showing CD11b+ and F4/80 + cells within the tumor-infiltrating CD45 + cells on day 19. Bar graphs represent the frequency of CD11b + F4/80 + cells within CD45 + cells (n = 4–6/group).
(I) qPCR analysis of Ifng, Tnfa, Cxcl9, Cxcl10, and Cxcl11 mRNA expression in HM-1 tumors on day 19 (n = 3–6/group).
(J) Schematic of the anti-PD-1 antibody (α-PD-1) treatment in combination with anti-IL-34 antibody (α-IL-34). The timeline shows the procedure of tumor inoculation and antibody treatment.
(K) Mock HM-1 tumor growth in B6C3F1 mice treated with the indicated antibodies (n = 3–4/group). Similar results were obtained in 2 independent experiments.
(L) Tumor weight on day 19 after tumor inoculation (n = 4/group).
(M) Bar graphs represent the frequency of CD8+ or CD4+ cells within the CD3+CD45 + cells and CD11b + F4/80 + cells within CD45 + cells infiltrated in the tumors described in Figure 2B (n = 3–7/group).
Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; two-tailed Student's t-test (E), Tukey's multiple comparison test (F-M). N.S., not significant.
See also Figures S1–S3.
We then collected tumors on day 19 and subjected them to flow cytometry analysis. Interestingly, we found that Il34KO HM-1 tumors were characterized by high infiltration of CD8+ and CD4+ T cells in the control IgG treatment group compared to Mock HM-1 tumors (Figure 1G), yet the enhancement of T cell infiltration in Il34KO HM-1 tumors was independent of PD-1 blockade. Additionally, we found that the frequencies of CD11b+F4/80+ cells were comparable among all groups and not affected by either IL-34 deficiency or PD-1 blockade (Figure 1H). Besides, quantitative PCR (qPCR) analysis showed enhanced expression of Ifng and Tnfa in Il34KO HM-1 tumors (Figure 1I). Together, these data suggest that IL-34 secreted by tumor cells, in itself, may disturb T cell-mediated antitumor immunity without affecting macrophage population, which apparently affected the efficacy of PD-1 blockade therapy.
The results shown above suggested that IL-34 derived from tumor cells controls T cell infiltration to tumor sites. Therefore, we sought to determine the factor that contributes to the chemotaxis of T cells. Cxcl9, Cxcl10, and Cxcl11 are known as T cell chemoattractants, and their expression is induced by IFNγ (Tokunaga et al., 2018). qPCR analysis revealed that, among these chemokines, Cxcl9, but not Cxcl10 and Cxcl11, expression was significantly upregulated in the anti-PD-1 treated Il34KO HM-1 tumors (Figure 1I). Cxcl9 has been shown to strongly stimulate T cell killer activity by binding to its receptor Cxcr3 (Chow et al., 2019), which may explain why the efficacy of PD-1 blockade was enhanced in Il34KO HM-1 tumors. Additionally, we performed flow cytometry analysis to identify the cell types expressing CSF-1R (CD115) and the populations of these cells. As a result, we revealed that CD115+ cell subset consisted of several immune cells, CD11c+MHC classⅡ+, CD11b+F4/80+, CD3ε+ cells, and others. Interestingly, these cell proportions were not affected by IL-34 knockout in HM-1, but the CD115+ cells population within tumor-infiltrating CD45+ cells was expanded (Figure S2). Our results suggested that tumor-derived IL-34 could suppress the differentiation, proliferation, or infiltration of CD115+ cells in TME.
Therapeutic Benefits of IL-34 Blockade with Neutralizing Antibody when Combined with anti-PD-1 Antibody In Vivo
Based on the above results, we aimed to evaluate the therapeutic benefits of IL-34 blockade when combined with immunotherapy. B6C3F1 mice were inoculated with Mock HM-1 cells (IL-34-expressing) and then treated with anti-PD-1 antibody alone or combined with IL-34-neutralizing antibody or control IgG (Figure 1J). Fourteen days after the onset of treatment, PD-1 blockade alone showed minimal effects on tumor size compared to the control group (Figures 1K and 1L). On the other hand, the combination of PD-1 blockade with anti-IL-34 antibody resulted in a significant suppression of tumor growth and tumor weight compared to anti-PD-1 monotherapy (Figures 1K and 1L). By analyzing the cellular components of the TME in each group, we found that IL-34 blockade resulted in enhanced infiltration of CD8+ T cells, but not CD4+ T cells (Figure 1M). In this case, in contrast to IL-34 knockout model, the frequency of CD11b+F4/80+ cells showed a slight reduction in combination therapy (Figure 1M). This observation may be explained by broad effect of neutralizing antibody which targets IL-34 produced not only by tumor cells but also by other cell types within TME. Previous studies demonstrated that CSF-1R blockade enhances antitumor response through anti-PD-1 treatment in several cancer models. To compare the antitumor efficacy of IL-34 blockade with CSF-1R blockade, we next performed in vivo experiments using anti-CSF-1R antibody under the same protocol used in anti-IL-34 antibody treatment. As a result, similar to anti-IL-34 mAb treatment, the anti-CSF-1R treatment showed a decrease in tumor growth when combined with anti-PD-1 mAb treatment (Figure S3). These data indicate that IL-34 blockade has the potential to enhance the efficacy of anti-PD-1 mAb as well as CSF-1R blockade.
IL-34 Impairs the Therapeutic Effects of PD-1 Blockade in Murine Colon and Breast Cancer Models
To extend our findings on the immunosuppressive role of tumor cell-derived IL-34 against PD-1 blockade, we utilized two more murine cancer cell lines: CT26 colon cancer that shows low-level expression of IL-34, and 4T1 breast cancer that secretes considerable level of IL-34 (Figure 2A). CRISPR-Cas9 system was used to generate CT26 and 4T1 cell lines deficient in IL-34; in addition, expression of IL-34 was enforced in Il34KO CT26 cell line to generate IL-34 overexpression cell line (Il34OE CT26). We note that, similar to HM-1 cells, these cell lines also secreted CSF-1 (Figure S1). Syngenic BALB/c mice were inoculated with these tumor cells and treated with anti-PD-1 antibody or control IgG (Figures 2B and 2K). As expected, Il34KO tumors exhibited a better response when treated with anti-PD-1 antibody, showing smaller tumor volumes than control IgG treatment, whereas the significant effect of PD-1 blockade was abrogated by the existence of IL-34 secreted by tumor cells (Figures 2C and 2L). To evaluate the gene set enhanced in the group showing the most effective antitumor efficacy, we performed next-generation sequencing (NGS) analysis and gene ontology (GO) analysis. The analyzed data suggested that the clusters associated with immune cell response, including "T cell receptor signaling pathway," "antigen processing and presentation," and “cytokine-cytokine receptor interaction,” were enriched in the group inoculated with Il34KO CT26 and treated with anti-PD-1 antibody (Figure S4). Moreover, we found that several genes associated with T cell accumulation (Cd3e, Cd4, Cd8a), inflammation (Tnf, Ifng, Cxcl9), and M1-macrophage subset (Cd86, Ciita, Nos2) were upregulated in Il34KO CT26 tumor treated with anti-PD-1 antibody (Figure 2D). On the other hand, gene expressions of M2-macrophage subset (Mrc1, Chi3l3, Arg1) in Il34KO CT26 tumor was lower than in Il34OE CT26 tumor treated with anti-PD-1 antibody (Figure 2D). We also performed qPCR to evaluate the expression of several genes presented in Figure 2D using CT26 and 4T1 tumor samples (Figure S5).
Figure 2.

Reversible Resistance against ICB of CT26 Colon Cancer and 4T1 Breast Cancer Cells by IL-34 Expression and Blockade
(A) IL-34 concentration in supernatants of WT, Il34KO, or Il34OE CT26 cells (n = 2/cell line, technical replicates) and Mock or Il34KO 4T1 cells (n = 2/cell line, technical replicates).
(B) Schematic of the anti-PD-1 antibody (α-PD-1) or control IgG treatment. The timeline shows the procedure of tumor inoculation and antibody treatment.
(C) Tumor growth in BALB/c mice inoculated with Il34KO or Il34OE CT26 cells and treated with anti-PD-1 antibody or control IgG (n = 3–4/group). Similar results were obtained in 2 independent experiments.
(D) Expression of selected genes is displayed by heatmap rendering of z-scores. Each column is the expression profile of a single tumor, and each row is a target gene, denoted on the left.
(E) Representative immunofluorescent stainings of F4/80, Nos2, and Arg1 of CT26 tumors. Green color indicates the expression of Nos2 (upper panel) or Arg1 (lower. panel) with F4/80 + macrophage infiltrating into tumor sites. Scale bars represent 20 μm.
(F) Proportions of Nos2+ or Arg1+ cells within the CT26 tumor-infiltrating CD11b + F4/80 + cells analyzed by flow cytometry on day 19. Bar graphs represent the frequency of each cell subset and the ratio of Arg1+ macrophage/Nos2+ macrophage (n = 8/group).
(G) Schematic of the anti-CTLA-4 or IL-34 antibody (α-CTLA-4, α-IL-34) or control IgG treatment. The timeline shows the procedure of tumor inoculation and antibody treatment.
(H) Tumor growth in BALB/c mice inoculated with Il34OE CT26 cells and treated with anti-CTLA-4 and/or IL-34 antibodies or control IgG (n = 3–4/group). Similar results were obtained in 2 independent experiments.
(I) Schematic of the anti-PD-1, CTLA-4 or IL-34 antibody, or control IgG treatment. The timeline shows the procedure of tumor inoculation and antibody treatment.
(J) Tumor growth in BALB/c mice inoculated with Il34OE CT26 cells and treated with anti-PD-1 and CTLA-4 antibodies with or without IL-34 antibody or control IgG (n = 5-7/group). Similar results were obtained in 2 independent experiments.
(K) Schematic of the anti-PD-1 antibody (α-PD-1) or control IgG treatment. The timeline shows the procedure of tumor inoculation and antibody treatment.
(L) Tumor growth in BALB/c mice inoculated with Mock or Il34KO 4T1 cells and treated with anti-PD-1 antibody or control IgG (n = 20/group). Similar results were obtained in 6 individual experiments.
(M) qPCR analysis of Ifng, Tnfa, and Nos2 mRNA expression in tumors on day 14 described in Figure 2J (n = 7/group).
(N) Representative immunofluorescent stainings of F4/80, Nos2, and Arg1 of 4T1 tumors. Green color indicates the expression of Nos2 (upper) or Arg1 (down) with F4/80 + macrophage infiltrating into tumor sites. Scale bars represent 20 μm.
(O) Proportions of Nos2+ or Arg1+ cells within the 4T1 tumor-infiltrating CD11b + F4/80 + cells analyzed by flow cytometry on day 14. Bar graphs represent the frequency of each cell subset and the ratio of Arg1+ macrophage/Nos2+ macrophage (n = 3/group).
Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01; Tukey's multiple comparison test (C, K, L) or Steel-Dwass nonparametric multiple comparison test (G, I), Student's t-test (E.M). N.S., not significant.
See also Figures S1, S4, and S5.
Next, we performed immunofluorescence staining and flow cytometry analysis to demonstrate whether tumor-derived IL-34 contributes to the populations of M1- and M2-biased macrophage in TME. As a result, the tumor-infiltrating Nos2+ M1-biased macrophage population was upregulated, and the ratio of Arg1+ M2-biased macrophage to Nos2+ M1-biased macrophage was decreased in Il34KO CT26 tumor (Figures 2E and 2F). These data suggest that tumor-derived IL-34 can interfere with the expansion of M1-biased macrophage in TME and form an anti-inflammatory microenvironment. 4T1 tumors, as well as CT26 tumors, showed significantly higher expression of inflammatory and pro-inflammatory cytokines when treated with PD-1 inhibitor (Figure 2M). Also, the population of tumor-infiltrating M1-biased macrophage was increased in Il34KO 4T1 tumor (Figures 2N and O).
With the indent to additionally evaluate IL-34 relevance to ICB resistance, we treated Il34OE CT26 tumors with anti-CTLA-4 antibody in combination with anti-IL-34 antibody (Figure 2G). Il34OE CT26 tumors treated with anti-CTLA-4 antibody exhibited a trend toward growth suppression (p = 0.06), and one of the tumors was completely eliminated (Figure 2H). Next, we tested the combination therapy of anti-PD-1 and CTLA-4 antibodies to treat Il34OE CT26 tumors with or without anti-IL-34 antibody (Figure 2I). While PD-1 and CTLA-4 combination therapy showed substantial tumor suppression, additional anti-IL-34 treatment resulted in a dramatical suppression. Among this treatment group, 2 out of 5 tumors were completely rejected (Figure 2J).
Collectively, these data indicate that IL-34 limits the efficacy of ICB not only by interfering T cell accumulation but also by reforming TME into the anti-inflammatory environment through the increase of M1-macrophage population. Also, IL-34 inhibition therapy with a neutralizing antibody efficiently reverses ICB resistance through recovering inflammatory circuit in the TME.
Therapeutic Potential of IL-34 Blockade in a PDX Model of Human Lung Adenocarcinoma
Finally, to translate these findings into clinical settings, we utilized a PDX model in which humanized-NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice, pre-injected with human hematopoietic stem cells (HSC), named HuNSG, were transplanted with human primary lung adenocarcinoma tissues (for detailed information, refer to Methods section). The tumor tissues exhibited considerable expression of both IL-34 and PD-L1 (Figures 3A–3C). HuNSG mice with established tumors were then treated with a monotherapy of anti-PD-1 or IL-34 antibody or a combination of both antibodies (Figure 3D). As a result, PD-1 blockade alone showed poor response when compared to the control baseline (Figure 3E). Anti-IL-34 monotherapy suppressed tumor growth in 1 out of 3 tumors, while the combination of anti-PD-1 and anti-IL-34 antibodies resulted in substantially suppressed tumor growth in 2 out of 3 tumors (Figure 3E). We observed complete necrosis in 1 responded tumor of the combination therapy group, and therefore, the tumor cannot be collected and not proceeded to further analyses (Figure 3F), suggesting that the combination therapy unleashed strong antitumor effect. Consistent with these data, strong immune cell infiltration was observed in the responded tumor in the combination therapy group (Figure 3G).
Figure 3.

Anti-IL-34 Treatment Enhances the Efficacy of Anti-PD-1 Treatment in PDX Model
(A) Pathological and gene expression profile of the tumor tissue used for the establishment of PDX model.
(B) Immunofluorescence staining of LU-TM-0007 sample for IL-34. Nuclei were counterstained with DAPI. Scale bars represent 20 μm.
(C) Representative immunohistochemistry staining of LU-TM-0007 sample with PD-L1 in the tumor. Scale bar represents 20 μm.
(D) Schematic of the anti-PD-1 (α-PD-1) and/or IL-34 (α-IL-34) antibody treatment. The timeline shows the procedure of human HSC injection, tumor inoculation, and antibody treatment.
(E) Tumor growth in HuNSG mice inoculated with LU-TM-0007 tumor cells and treated with the antibodies (n = 3/group). Dotted line indicates the size of nontreated tumor at the same time point. Dot plot shows the tumor size on day 28. Individual data points are shown with mean ± SEM.
(F) Macroscopic observation of s.c. injected xenografts in HuNSG mice sacrificed on day 28. Combination therapy caused strong necrosis in one of the treated tumors (∗) which could not be further analyzed.
(G) Representative hematoxylin and eosin staining of resected tumors in nontreated, anti-PD-1, or combination therapy groups on day 28. Arrowheads indicate infiltrating immune cells. Scale bars represent 100 μm.
Thus, these data suggest that IL-34 is involved in ICB resistance of human cancer and that its inhibition restores the therapeutic effect of ICB.
Discussion
In this study, we identify for the first time a potential involvement of IL-34 in immunotherapeutic resistance of cancer. By targeting IL-34 in the murine HM-1 ovarian cancer cells and 4T1 breast cancer cells using CRISPR-Cas9 system, we found that IL-34-deficient tumors exhibit a better response to therapeutic ICB than IL-34-expressing tumors. Nevertheless, CT26 murine colon cancer cells are known to be responsive to immune checkpoint inhibitors including anti-PD-1 and anti-CTLA-4 antibodies (Fu et al., 2019; Jure-Kunkel et al., 2013), overexpression of IL-34 in the cell line significantly impaired the outcome of ICB. More importantly, a neutralizing antibody against IL-34 showed therapeutic potential when combined with PD-1 blockade, which was further confirmed in a PDX model of lung adenocarcinoma.
By analyzing the cellular components of the TME in our therapeutic models, we found that IL-34 converts not only the cellular profiles within tumor site but also the inflammatory state of TME. It is already reported that CSF-1R blockade induces a dramatical decline of the macrophage population in tumor sites (MacDonald et al., 2010). However, this phenomenon was not fully recapitulated in IL-34 blockade treatment that macrophage population showed only a slight reduction (Figure 1M). These results led us to the hypothesis that IL-34 may affect the function rather than the frequencies of tumor-infiltrating myeloid cells. Consistent with this, the existence of tumor-derived IL-34 suppressed the expression of Cxcl9 responsible for T cell recruitment and activation (Figure 1I). Additionally, we found that the expression levels of inflammatory and pro-inflammatory factors such as Ifng, Tnfa, and Nos2 were higher in IL-34-deficient tumors (Figures 1I. 2D, and M), which may support the hypothesis that IL-34 modifies the function of tumor-infiltrating myeloid cells at the local TME. In accordance with this, immunofluorescence staining revealed the possible role of IL-34 to restrict the M1 polarization of tumor-infiltrating macrophage (Figures 2E, F, N, and O).
Although IL-34 shows similar biological activities with CSF-1 in vitro, several studies have reported differences in the response of myeloid cells toward IL-34 or CSF-1 stimulation, showing altered expression of pro-inflammatory cytokines and chemokines (Boulakirba et al., 2018; Nakamichi et al., 2013). The engagement of CSF-1R by IL-34 has been suggested to activate caspase and autophagy signaling pathways in monocytes, which results in an IL-34-induced macrophagic differentiation and polarization that differ from CSF-1 (Boulakirba et al., 2018). Differences in CSF-1R binding affinity, hydrophobic/hydrophilic binding feature, in addition to differences in the binding pockets have been suggested to explain the distinct signaling between IL-34 and CSF-1 (Boulakirba et al., 2018; Liu et al., 2012). However, the molecular mechanisms that explain such differences remain to be explored in future works (Boulakirba et al., 2018).
Accumulating evidence has indicated critical roles for tumor-derived cytokines in all aspects of the TME, including tumor growth, metastasis, angiogenesis, and therapeutic resistance (Chen et al., 2018; Eichbaum et al., 2011; Jones et al., 2016; Kim et al., 2006). Accordingly, many cytokines may serve as beneficial therapeutic targets in cancer (Berraondo et al., 2019; Lee and Margolin, 2011; Rossi et al., 2015; Setrerrahmane and Xu, 2017; Szebeni et al., 2016). As an alternative ligand to CSF-1 for CSF-1R, IL-34 is suggested to play important roles at the TME by direct effects on both tumor and immune cells (Baghdadi et al., 2016). Thus, neutralizing antibodies that target IL-34 or specific inhibitors that suppress IL-34 expression may help to control tumor progression and overcome the resistance problem. In this study, all murine cancer cell lines, including the genetically manipulated ones, expressed CSF-1 simultaneously with IL-34 (Figures 1A and S1). Despite the fact that tumor-derived CSF-1 may be released in TME, IL-34 blockade enhanced the efficacy of immune checkpoint blockade in vivo. It indicates that IL-34-expressing tumors may acquire resistance to immune checkpoint blockade independently of CSF-1 existence. Possible merit of targeting IL-34 in cancer is the fewer side effects that result from treatment, due to the limited expression of IL-34 under physiological conditions. On the other hand, it has been reported that IL-34 binds to other receptors, protein tyrosine phosphatase, receptor type Z, polypeptide 1 (PTPRZ1), and Syndecan-1. PTPRZ1 is primarily expressed on neuronal progenitors and glial cells. Via PTPRZ1, IL-34 can regulate intracellular signaling pathways that inhibit proliferation, clonogenicity, and motility of the cellular targets, indicating a CSF1-R-independent action (Nandi et al., 2013). Syndecan-1 is involved in cell proliferation, migration, and matrix interactions, expressed in a wide range of tissues. Syndecan-1 and IL-34 binding modulates IL-34-induced CSF-1R activation and affects a particular myeloid cell migration (Ségaliny et al., 2015). Altogether, it is possible that IL-34 exerts its biological activities through binding to PTPRZ1 and syndecan-1 in addition to interaction with CSF-1R in TME. However, whether blocking IL-34 binding to these receptors interferes with antitumor effect or causes adverse effects remains unclear. This is also supported by the phenotype of Il34 knockout mice that showed no remarkable effects in contrast to Csf1r or Csf1 knockout mice (Wang et al., 2012). Furthermore, our finding in this study that IL-34 blockade does not severely decrease the frequency but enhance the function of myeloid cells may be a great advance compared with the total CSF-1R blockade in which myeloid cell population is dramatically destroyed (MacDonald et al., 2010).
In conclusion, we added here new evidence that indicates a potential role for IL-34 in promoting therapeutic resistance against tumor immunotherapy. These results indicate that IL-34 could be a therapeutic target to enhance the efficacy of ICB in human cancer treatment.
Limitations of the Study
Here, we demonstrated tumor-derived IL-34 has potential as a target for therapy that enhances the efficacy of ICB, regardless of the expression of CSF-1 from tumor cells. However, the molecular mechanism that explains the difference between IL-34 and CSF-1 on the immune system in TME needs to be elucidated in further research.
Resource Availability
Lead Contact
Ken-ichiro Seino, Division of Immunobiology, Institute for Genetic Medicine, Hokkaido University, Kita-15 Nishi-7, Sapporo 060-0815, Japan (seino@igm.hokudai.ac.jp).
Materials Availability
Not applicable.
Data and Code Availability
The NGS data in this paper have been deposited in GEO repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157602).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Ms. Yui Umeyama for her helpful technical supports. This work was supported in part by the Japan Agency for Medical Research and Development (AMED; Practical Research for Innovative Cancer Control) (K.-S.).
Author Contribution
MB and KS designed the study. Naoki H, TK, Nanumi H, FK, NK, HW, HY, HL, and HR performed experiments. All authors analyzed data and discussed the results. Naoki H, TK, Nanumi H, RO, MB, and KS contributed to manuscript preparation. All authors approved the final version of this manuscript.
Declaration of Interests
The authors disclosed no potential conflicts of interest.
Published: October 23, 2020
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101584.
Supplemental Information
References
- Baghdadi M., Wada H., Nakanishi S., Abe H., Han N., Wira E.P., Endo D., Watari H., Sakuragi N., Hida Y. Chemotherapy-induced IL34 enhances immunosuppression by tumor-associated macrophages and mediates survival of chemoresistant lung cancer cells. Cancer Res. 2016;76:6030–6042. doi: 10.1158/0008-5472.CAN-16-1170. [DOI] [PubMed] [Google Scholar]
- Baghdadi M., Endo H., Takano A., Ishikawa K., Kameda Y., Wada H., Miyagi Y., Yokose T., Ito H., Nakayama H. High co-expression of IL-34 and M-CSF correlates with tumor progression and poor survival in lung cancers. Sci. Rep. 2018;8:1–10. doi: 10.1038/s41598-017-18796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdadi M., Ishikawa K., Nakanishi S., Murata T., Umeyama Y., Kobayashi T., Kameda Y., Endo H., Wada H., Bogen B. A role for IL-34 in osteolytic disease of multiple myeloma. Blood Adv. 2019;3:541–551. doi: 10.1182/bloodadvances.2018020008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berraondo P., Sanmamed M.F., Ochoa M.C., Etxeberria I., Aznar M.A., Pérez-Gracia J.L., Rodríguez-Ruiz M.E., Ponz-Sarvise M., Castañón E., Melero I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer. 2019;120:6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulakirba S., Pfeifer A., Mhaidly R., Obba S., Goulard M., Schmitt T., Chaintreuil P., Calleja A., Furstoss N., Orange F. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018;8:1–11. doi: 10.1038/s41598-017-18433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarile M.A., Weisser M., Jacob W., Jegg A.M., Ries C.H., Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer. 2017;5:1–13. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Tan W., Wang C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial–mesenchymal transition. Onco. Targets Ther. 2018;11:3817–3826. doi: 10.2147/OTT.S168317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W., Fu D., Xu F., Zhang Z. Unwrapping the genomic characteristics of urothelial bladder cancer and successes with immune checkpoint blockade therapy. Oncogenesis. 2018;7:2. doi: 10.1038/s41389-017-0013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow M.T., Ozga A.J., Servis R.L., Frederick D.T., Lo J.A., Fisher D.E., Freeman G.J., Boland G.M., Luster A.D. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50:1498–1512.e5. doi: 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichbaum C., Meyer A.S., Wang N., Bischofs E., Steinborn A., Bruckner T., Brodt P., Sohn C., Eichbaum M.H.R. Breast cancer cell-derived cytokines, macrophages and cell adhesion: implications for metastasis. Anticancer Res. 2011;31:3219–3227. [PubMed] [Google Scholar]
- Franzè E., De Simone V., Dinallo V., Rizzo A.M., Caprioli F., Colantoni A., Ortenzi A., Sica G., Monteleone G. OC.01.4: interleukin-34 sustains pro-tumorigenic signals in colon cancer tissue. Dig. Liver Dis. 2017;49:e78. doi: 10.18632/oncotarget.23289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C., Zhu X., Xu P., Li Y. Pharmacological inhibition of USP7 promotes antitumor immunity and contributes to colon cancer therapy. Onco. Targets Ther. 2019;12:609–617. doi: 10.2147/OTT.S182806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon E.B., Rizvi N.A., Hui R., Leighl N., Balmanoukian A.S., Eder J.P., Patnaik A., Aggarwal C., Gubens M., Horn L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- Ghirelli C., Hagemann T. Targeting immunosuppression for cancer therapy. J. Clin. Invest. 2013;123:2355–2357. doi: 10.1172/JCI69999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giricz O., Mo Y., Dahlman K.B., Cotto-Rios X.M., Vardabasso C., Nguyen H., Matusow B., Bartenstein M., Polishchuk V., Johnson D.B. The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma. JCI Insight. 2018;3:e120422. doi: 10.1172/jci.insight.120422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori D., Lim E.L., Grant F.M., Spensberger D., Roychoudhuri R., Shuttleworth S.J., Okkenhaug K., Stephens L.R., Hawkins P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight. 2018;3:e120631. doi: 10.1172/jci.insight.120631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han N., Baghdadi M., Ishikawa K., Endo H., Kobayashi T., Wada H., Imafuku K., Hata H., Seino K. Enhanced IL-34 expression in Nivolumab-resistant metastatic melanoma. Inflamm. Regen. 2018;38:2–6. doi: 10.1186/s41232-018-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi F.S., O’Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones V.S., Huang R.Y., Chen L.P., Chen Z.S., Fu L., Huang R.P. Cytokines in cancer drug resistance: cues to new therapeutic strategies. Biochim. Biophys. Acta. 2016;1865:255–265. doi: 10.1016/j.bbcan.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Jure-Kunkel M., Masters G., Girit E., Dito G., Lee F., Hunt J.T., Humphrey R. Synergy between chemotherapeutic agents and CTLA-4 blockade in preclinical tumor models. Cancer Immunol. Immunother. 2013;62:1533–1545. doi: 10.1007/s00262-013-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R., Emi M., Tanabe K., Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–5536. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- Kumar V., Patel S., Tcyganov E., Gabrilovich D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011;3:3856–3893. doi: 10.3390/cancers3043856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Lee E., Hestir K., Leo C., Huang M., Bosch E., Halenbeck R., Wu G., Zhou A., Behrens D. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- Liu H., Leo C., Chen X., Wong B.R., Williams L.T., Lin H., He X. The mechanism of shared but distinct CSF-1R signaling by the non-homologous cytokines IL-34 and CSF-1. Biochim. Biophys. Acta. 2012;1824:938–945. doi: 10.1016/j.bbapap.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald K.P.A., Olver S., Raffelt N.C., Kuns R., Hill G.R., Palmer J.S., Cronau S., Seppanen E., Wainwright B., Pettit A.R. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–3963. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
- Nakamichi Y., Udagawa N., Takahashi N. IL-34 and CSF-1: similarities and differences. J. Bone Miner. Metab. 2013;31:486–495. doi: 10.1007/s00774-013-0476-3. [DOI] [PubMed] [Google Scholar]
- Nandi S., Ciose M., Yeung Y.G., Nieves E., Tesfa L., Lin H., Hsu A.W., Halenbeck R., Cheng H.Y., Gokhan S. Receptor-type protein-tyrosine phosphatase ζ is a functional receptor for interleukin-34. J. Biol. Chem. 2013;288:21972–21986. doi: 10.1074/jbc.M112.442731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert N.J., Bordry N., Wald N., Martignier C., Tillé L., Homicsko K., Maby-El Hajjami H., Ioannidou K., Coukos G., Fuertes Marraco S.A. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018;10:eaan3311. doi: 10.1126/scitranslmed.aan3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R., Pollard J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell J.S., Long G.V., Scolyer R.A., Teng M.W.L., Smyth M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt J.M., Marabelle A., Eggermont A., Soria J.C., Kroemer G., Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016;27:1482–1492. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- Prima V., Kaliberova L.N., Kaliberov S., Curiel D.T., Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. U S A. 2017;114:1117–1122. doi: 10.1073/pnas.1612920114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaranta V., Rainer C., Nielsen S.R., Raymant M.L., Ahmed M.S., Engle D.D., Taylor A., Murray T., Campbell F., Palmer D.H. Macrophage-derived granulin drives resistance to immune checkpoint inhibition in metastatic pancreatic cancer. Cancer Res. 2018;78:4253–4269. doi: 10.1158/0008-5472.CAN-17-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 2015;5:915–919. doi: 10.1158/2159-8290.CD-15-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J.F., Lu Z.Y., Jourdan M., Klein B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015;21:1248–1257. doi: 10.1158/1078-0432.CCR-14-2291. [DOI] [PubMed] [Google Scholar]
- Ségaliny A.I., Mohamadi A., Dizier B., Lokajczyk A., Brion R., Lanel R., Amiaud J., Charrier C., Boisson-Vidal C., Heymann D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer. 2015;137:73–85. doi: 10.1002/ijc.29376. [DOI] [PubMed] [Google Scholar]
- Setrerrahmane S., Xu H. Tumor-related interleukins: old validated targets for new anti-cancer drug development. Mol. Cancer. 2017;16:1–17. doi: 10.1186/s12943-017-0721-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebeni G.J., Vizler C., Nagy L.I., Kitajka K., Puskas L.G. Pro-tumoral inflammatory myeloid cells as emerging therapeutic targets. Int. J. Mol. Sci. 2016;17:1958. doi: 10.3390/ijms17111958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga R., Zhang W., Naseem M., Puccini A., Berger M.D., Soni S., McSkane M., Baba H., Lenz H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation – a target for novel cancer therapy. Cancer Treat. Rev. 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S., De Sanctis F., Mandruzzato S., Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-Associated macrophages. J. Clin. Invest. 2015;125:3365–3376. doi: 10.1172/JCI80006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.Y., Eroglu Z., Ozgun A., Leger P.D., Zhao S., Ye F., Luke J.J., Joseph R.W., Haq R., Ott P.A. Clinical features of acquired resistance to anti–PD-1 therapy in advanced melanoma. Cancer Immunol. Res. 2017;5:357–362. doi: 10.1158/2326-6066.CIR-16-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Szretter K.J., Vermi W., Gilfillan S., Rossini C., Cella M., Barrow A.D., Diamond M.S., Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.L., Hu Z.Q., Zhou Z.J., Dai Z., Wang Z., Cao Y., Fan J., Huang X.W., Zhou J. miR-28-5p-IL-34-macrophage feedback loop modulates hepatocellular carcinoma metastasis. Hepatology. 2016;63:1560–1575. doi: 10.1002/hep.28445. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Yang J., Xu D., Gao X.M., Zhang Z., Hsu J.L., Li C.W., Lim S.O., Sheng Y.Y., Zhang Y. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68:1653–1666. doi: 10.1136/gutjnl-2019-318419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NGS data in this paper have been deposited in GEO repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157602).