

Summary
CDK6 is frequently overexpressed in various cancer types and functions as a positive regulator of the cell cycle and as a coregulator of gene transcription. We provide evidence that CDK6 is involved in the process of DNA methylation, at least in ALL. We observe a positive correlation of CDK6 and DNMT expression in a large number of ALL samples. ChIP-seq analysis reveals CDK6 binding to genomic regions associated with DNA methyltransferases (DNMTs). ATAC-seq shows a strong reduction in chromatin accessibility for DNMT3B in CDK6-deficient BCR-ABL+ Cdk6−/− cells, accompanied by lower levels of DNMT3B mRNA and less chromatin-bound DNMT3B, as shown by RNA-seq and chromatome analysis. Motif analysis suggests that ETS family members interact with CDK6 to regulate DNMT3B. Reduced representation bisulfite sequencing analysis uncovers reversible and cell line-specific changes in DNA methylation patterns upon CDK6 loss. The results reveal a function of CDK6 as a regulator of DNA methylation in transformed cells.
Subject Areas: Genetics, Genomics, Cancer
Graphical Abstract
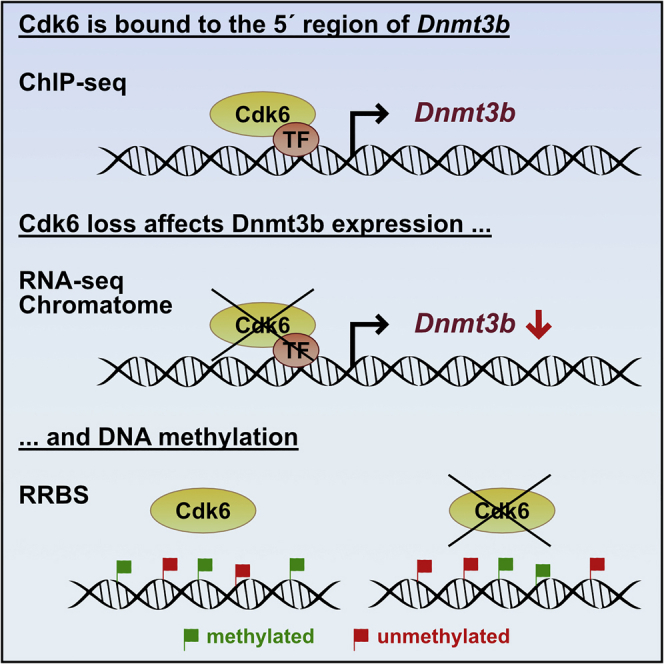
Highlights
-
•
CDK6 and DNMT/TET/HDAC expression correlate in ALL samples
-
•
CDK6 transcriptionally regulates DNMT3B in BCR-ABL+ cells
-
•
DNA methylation patterns change upon CDK6 loss in BCR-ABL+ cells
Genetics; Genomics; Cancer
Introduction
The transformation of normal to cancer cells is driven by a multitude of genetic and epigenetic alterations. There is recent evidence that overexpression of the cyclin-dependent kinase 6 (CDK6) is a frequent event in hematological malignancies and solid tumors (Tadesse et al., 2015; Nagel et al., 2008; Tigan et al., 2016). Functional characterization of CDK6 has revealed that it affects a variety of molecular processes (Bellutti et al., 2018; Scheicher et al., 2015; Uras et al., 2017, 2019; Kollmann et al., 2013; Wang et al., 2017). Besides binding D-type cyclins to drive the G1 phase of the cell cycle, it also regulates gene expression (Kollmann et al., 2013; Tigan et al., 2016). Independent of its kinase activity, it interacts with transcription factors to act as a chromatin-bound cofactor that induces or represses the expression of specific genes. We have shown that CDK6 interacts with STAT3 to induce the expression of p16INK4a and with the AP-1 transcription factor c-Jun to induce the expression of VEGF-A (Kollmann et al., 2013). It also cooperates with the NF-κB subunit p65 to induce an inflammatory gene response (Handschick et al., 2014). CDK6 can also repress transcription, as it does in hematopoietic stem cells by interacting with AP-1 to repress EGR1 expression and thereby to induce the exit from quiescence and the growth of leukemic stem cells (Scheicher et al., 2015). Together with NFYA and SP1, CDK6 induces a transcriptional program to block p53 in hematopoietic cells (Bellutti et al., 2018).
Gene transcription is frequently regulated by epigenetic mechanisms, such as DNA methylation, various chemical modifications of histone proteins and chromatin restructuring, which results in gene activation or gene silencing (Sandoval and Esteller, 2012). The main targets of methylation in the mammalian genome are cytosines within CpG dinucleotides. CpG dinucleotides are distributed throughout the genome but are concentrated in regions of about 0.5–2 kb in length, called CpG islands, that are associated with ∼60% of human gene promoters (Sandoval and Esteller, 2012). DNA methylation is considered important in the pathogenesis of many solid tumors and hematological malignancies (Heller et al., 2016; Guillamot et al., 2016; Kimura et al., 2019). The CpG islands of many cancer-associated genes are frequently methylated in cancer cells, resulting in the transcriptional silencing of these genes (Sandoval and Esteller, 2012; Heller et al., 2013). Changes in DNA methylation in regions outside CpG islands may be equally important in leukemogenesis, with hypomethylation as relevant as hypermethylation (Guillamot et al., 2016; Figueroa et al., 2010; Kimura et al., 2019; Irizarry et al., 2009; Qu et al., 2014). Surprisingly, DNA methylation aberrations in tumor cells of various cancer types are highly heterogeneous (Wenger et al., 2019; Sheffield et al., 2017; Brocks et al., 2014; Li et al., 2016; Landau et al., 2014; Nordlund et al., 2013).
Despite a wealth of evidence, the mechanisms of aberrant methylation in cancer cells remain unclear. There is a preliminary indication that CDK6 may play a part, as inhibition of CDK kinase with palbociclib promotes the degradation of the DNA methyltransferase 1 (DNMT1) (Acevedo et al., 2016). We now confirm that CDK6 is involved in the regulation of DNA methylation, although its function does not depend on its kinase activity. In several independent gene expression microarray datasets from a large number of ALL samples CDK6 is co-expressed with DNMTs, TET1 and histone deacetylases (HDAC). ChIP-seq, ATAC-seq, RNA-seq, and mass spectrometry analysis of chromatin (referred to as chromatome) reveal that DNMT3B is a target for transcriptional regulation by CDK6 in BCR-ABL+ cells. Reduced representation bisulfite sequencing (RRBS) shows that loss of CDK6 causes changes in DNA methylation patterns in a cell line-specific manner, with both hypomethylation and hypermethylation prominent. Many of the changes in methylation can be reversed by re-expressing CDK6. The findings demonstrate that CDK6 has a function as a regulator of DNA methylation in transformed cells.
Results
CDK6 and DNMT/TET/HDAC Expression Correlate in ALL Samples
Epigenetic changes are frequent in leukemia and epigenetic regulators such as DNMTs and TETs are often mutated in hematopoietic malignancies (Yang et al., 2015, Delhommeau et al., 2009, Cancer Genome Atlas Research Network et al., 2013, Kimura et al., 2019, de Keersmaecker et al., 2013). Coexpression analysis using four independent gene expression microarray data sets from ALL patients showed a positive correlation between levels of CDK6 and of DNMT3A (mean R = 0.46 in 4 data sets; p < 0.0001), with an only slightly weaker positive correlation with levels of DNMT3B (mean R = 0.42 in 3 of 4 data sets; p < 0.0001) and a weaker correlation with levels of DNMT1 (mean R = 0.34 in 4 data sets; p < 0.0001; Figure 1). While CDK6 was positively correlated with TET1 (mean R = 0.49 in 4 data sets; p < 0.0001) and negatively with TET2 (mean R = −0.35 in 4 data sets; p < 0.0001), there was no correlation with TET3 in ALL patients (Figure 1).
Figure 1.

CDK6 and DNMT/TET/HDAC Expression Correlate in ALL Samples
Correlation of CDK6 expression and expression of DNMTs (red), TETs (green) and HDACs (yellow) in 4 independent cohorts of adult or pediatric acute lymphoblastic leukemia (ALL) patients. Spearman correlation coefficients are shown. Blue dots: positive correlation, orange dots: negative correlation.
HDACs are also affected in hematopoietic malignancies, although aberrant expression is more common than mutations (Moreno et al., 2010; Zhang et al., 2015). Of the large family of HDACs, we found HDAC1 and HDAC8 to be coexpressed with CDK6 in ALL patients (mean R = 0.55 and 0.42 in 4 data sets; p < 0.0001, respectively). The patterns of coexpression between CDK6 and DNMT/TET/HDACs are highly similar in the four gene expression data sets (Figure 1), showing that expression of CDK6 and several factors associated with DNA methylation are positively correlated in ALL. The data suggest that CDK6 might be involved in the changes in DNA methylation during leukemogenesis.
DNMT3B Is Transcriptionally Regulated by CDK6 in BCR-ABL+ Cells
The transcriptional regulator CDK6 is particularly significant in leukemia, where it is upregulated and contributes to leukemogenesis (Nagel et al., 2008; Tadesse et al., 2015; Tigan et al., 2016). The positive correlation between expression of CDK6 and DNMT3 prompted us to study whether DNMT1, DNMT3A and DNMT3B are transcriptionally regulated by CDK6. Murine (C57Bl/6J) Cdk6wt and Cdk6−/− cell lines obtained from single-cell bone marrow suspension were retrovirally transduced with a pMSCV-BCR-ABLp185-IRES-GFP vector as described (Bellutti et al., 2018). CDK6 ChIP-seq analysis of these cells revealed specific ChIP peaks in the 5′ regions of DNMT1 and DNMT3B (Figures 2A and S1A) and additional CDK6 peaks in the promoter and exonic/intronic regions of DNMT3A (Figure S1B). The chromatin accessibility of the genomic regions harboring the DNMT1 and DNMT3B genes also depended on the presence of CDK6. ATAC-seq analysis in BCR-ABL+ Cdk6wt and BCR-ABL+ Cdk6−/− cells showed that the chromatin accessibility in the DNMT3A region was comparable. However, the chromatin accessibility in the DNMT1 and DNMT3B regions was strongly reduced in CDK6-deficient BCR-ABL+ Cdk6−/− cells (Figures 2A and S1A). The significance of the differences in ATAC-seq were confirmed by RNA-seq analysis, which revealed reduced DNMT3B expression in BCR-ABL+ Cdk6−/− cells (Figure 2B), while expression of DNMT1 and DNMT3A remained unchanged.
Figure 2.

DNMT3B Is Transcriptionally Regulated by CDK6 in BCR-ABL+ Cells
(A) Binding of CDK6 to the 5′ region of DNMT3B in BCR-ABL+ cells determined by ChIP-seq analysis (upper panel). Chromatin accessibility at the 5′ region of DNMT3B in BCR-ABL+ Cdk6wt (green) and BCR-ABL+ Cdk6−/− (red) cells determined by ATAC-seq analysis. See also Figure S1.
(B) Heatmap showing mRNA expression of DNMT1/3a/3b in BCR-ABL+ Cdk6wt (green, N = 3) and BCR-ABL+ Cdk6−/− (red, N = 3) cells analyzed by RNA-sequencing.
(C) Chromatin-bound DNMT1/3a/3b in BCR-ABL+ Cdk6wt (green) and BCR-ABL+ Cdk6−/− (red) cells analyzed by chromatome analysis. Mean values + SD are shown.
(D) ETS motifs found within the ATAC-seq peak at the 5′ UTR of DNMT3B.
(E) Binding of ETS1 to the 5′ region of DNMT3B in human K562 cells (left panel) and in murine spleen cells (right panel) determined by ChIP-seq analysis. See also Figure S1.
Analysis of the chromatin-bound proteome showed that the changes in transcription are reflected by alterations in protein abundance: less DNMT3B bound to chromatin in BCR-ABL+ Cdk6−/− cells than in BCR-ABL+ Cdk6wt cells (Figure 2C). Motif analysis of the genomic region underlying the CDK6 ChIP-seq/ATAC-seq peak (Figure 2A) identified ETS motifs overlapping with CDK6 binding sites in the 5′ region of DNMT3B (Figure 2D). To test if ETS1 binds to this genomic location, we analyzed ETS1 ChIP-seq data from human K562 cells and murine B-cells. We verified that an ETS1 peak overlaps with the CDK6 binding site in the promoter/exon 1 region of DNMT3B in both data sets (Figure 2E). These findings suggest that CDK6 directly regulates DNMT3B, possibly mediated by ETS factors.
DNA Methylation Patterns Change upon Loss of CDK6 in BCR-ABL+ Cells
To investigate whether the alterations in DNMT3 expression provoke changes of the methylome, we used a CRISPR-Cas9-based strategy to generate BCR-ABL+ cells deficient for CDK6 as confirmed by Western blotting (Figure S2). Our procedure (for details see Transparent Methods) maintains genetic stability and avoids altering p53: CDK6-deficient transformed cells consistently harbor p53 mutations that may affect the methylome (Tovy et al., 2017). Dnmt3b expression was downregulated upon Cdk6 loss in these cells (Figures 3A and S3). Further, Cdk6 reexpression strongly induced Dnmt3b mRNA expression (Figure 3A). We used 3 individually derived BCR-ABL+ cell lines for RRBS to study the methylome. Principal component analysis (PCA) based on CpG site methylation unequivocally separated BCR-ABL+ Cdk6−/− cells from BCR-ABL+ Cdk6wt cells (Figure 3B). The separation was also evident in the sample correlation plot, which showed weaker correlation between BCR-ABL+ Cdk6−/− and BCR-ABL+ Cdk6wt cells than within BCR-ABL+ Cdk6wt cells (Figure S4).
Figure 3.

Analysis of the Methylome of BCR-ABL+ Cdk6wt and BCR-ABL+ Cdk6−/− Cells By Reduced Representation Bisulfite Sequencing
(A) Quantification of Dnmt3b mRNA expression in BCR-ABL+ Cdk6wt, BCR-ABL+ Cdk6−/− and BCR-ABL+ Cdk6−/−+Cdk6 cells determined by RT-PCR. Mean fold changes +SEM are shown (N = 8), p values were calculated using one-sample t-tests.
(B) Principal component analysis of 3 BCR-ABL+ Cdk6wt (WT, red) and 3 BCR-ABL+ Cdk6−/− (KO, blue) cell lines based on CpG methylation. Each dot represents a unique sample. See also Figure S4.
(C) Volcano plots showing hypomethylated and hypermethylated CpG sites in 3 replicates of BCR-ABL+ Cdk6−/− cell lines compared to BCR-ABL+ Cdk6wt cells.
(D) Venn diagram illustrating the overlap of differentially methylated CpG sites in the 3 BCR-ABL+ Cdk6−/− cell lines.
(E) Density plot showing the location of hypermethylated (pink) and hypomethylated (blue) CpG sites relative to transcriptional start sites (TSS). See also Figure S4.
Unexpectedly, the BCR-ABL+ Cdk6−/− samples are not represented in a distinct cluster in the PCA but are separated from one another, indicating that loss of CDK6 is accompanied by different changes in DNA methylation in the different cell lines. To investigate the differences in more detail, we examined differential methylation between individual pairs of cell lines. We found 4,707 (2,028 hyper, 2,679 hypo), 4,565 (2,498 hyper, 2,067 hypo) and 5,528 (2,773 hyper, 2,756 hypo) CpG sites with a difference in methylation of at least 40% in each of the three pairwise comparisons (Figure 3C). Only ∼1.5% of differentially methylated CpG sites were shared in two of the cell lines and no single CpG site showed a consistent difference in methylation in all 3 cell lines (Figure 3D). Annotation of the differentially methylated CpG sites to genomic region revealed that both hyper- and hypomethylated CpG sites are closely associated with transcriptional start sites (TSSs) of genes (Figure 3E). The data confirm that loss of CDK6 has a dramatic effect on DNA methylation in BCR-ABL + cells and suggest that the precise nature of the effect depends on stochastic processes.
In the next step, we asked if DNA methylation changes are also associated with CDK6 expression in patients. Thus, we stratified the TCGA LAML data set into CDK6low (lower quartile, N = 27) and CDK6high (upper quartile, N = 27) samples and tested for correlation between CDK6 and DNMT3B expression as well as for differences in DNA methylation. As for ALL samples (Figure 1), we found a positive correlation between CDK6 and DNMT3B expression in AML samples (R = 0.47, p < 0.005). Further, CDK6high samples showed a strong increase in methylation (β-difference >0.3) for 1,287 probes (Figure S5). Only 11 probes with decreased methylation (β-difference < -0.3) were found indicating that CDK6 expression is mainly associated with hypermethylation in patient samples.
Expression of CDK6 Reverses Changes in CpG Methylation Caused by Loss of CDK6
It is conceivable that the variable changes in DNA methylation are a result of different levels of CDK6. We tested this possibility by re-expressing CDK6 using retroviral transduction with a construct encoding HA-CDK6 (Figure S2) and RRBS analysis and compared methylation in the reconstituted BCR-ABL+ Cdk6−/−+Cdk6 cells and the maternal cells (BCR-ABL+ Cdk6wt and BCR-ABL+ Cdk6−/−). Re-expression of CDK6 did not result in the formation of a separate methylation cluster in the hierarchical cluster analysis but showed that BCR-ABL+ Cdk6−/−+Cdk6 cells clustered with BCR-ABL+ Cdk6−/− cells from the same background (Figure 4A). Thus, re-expression of CDK6 does not induce global changes of the methylome but regulates a distinct set of genes in each cell line. We then analyzed whether the methylation changes caused by loss of CDK6 are reverted by re-expression of CDK6. Hypomethylation of 79% (range: 70%–85%) of CpG sites was found to be reversible (>1.5-fold increase in methylation) in the three cell lines, while methylation of 34% of the hypermethylated CpG sites (mean: 34%; range: 26%–48%) was reverted when CDK6 is re-expressed (Figure 4B). The reversal of methylation patterns was independent of the location of CpG sites and was found consistently in CGIs, CGI shores, CGI shelves, inter CGI regions, promoters, introns, exons, and intergenic regions (Figure S6). The effect of re-expression of CDK6 on the methylation of 5,000 randomly selected CpG sites was weak (mean methylation changes: 6.6%; repeated 10 times), confirming that the changes in methylation mediated by CDK6 re-expression are specific (Figure S7).
Figure 4.

Reversal of CDK6 Loss Mediated CpG Site Methylation Changes by CDK6 Re-expression
(A) Dendrogram of hierarchical clustering of BCR-ABL+ Cdk6wt (WT, red), BCR-ABL+ Cdk6−/− (KO, blue) and BCR-ABL+ Cdk6−/−+Cdk6 (KO + CDK6, green) cells based on CpG site methylation.
(B) Heatmaps showing methylation values of differentially methylated CpG sites in 3 replicates (Rep 1–3) of BCR-ABL+ Cdk6wt, BCR-ABL+ Cdk6−/− and BCR-ABL+ Cdk6−/−+Cdk6 cell lines. Values are depicted as percentage of methylation and range from 0% (dark blue) to 100% (dark red). See also Figures S6 and S7.
As the re-expression of CDK6 produces varying amounts of the protein in the individual cells, the consistent nature of the changes in methylation caused by re-expression of CDK6 shows that the changes are independent of the level of CDK6. In other words, expression of CDK6 is associated with specific changes in CpG methylation, with the precise effects different for each individual clone. Re-expression of CDK6 largely reverts the hypomethylation mediated by loss of CDK6, although the hypermethylation in BCR-ABL+ Cdk6−/− cells is only partially revertable, strengthening the dominating hypermethylation seen in CDK6high patient samples (Figure S5).
CDK6-Dependent Differential Methylation Is Associated with Altered Gene Expression in BCR-ABL+ Cells
Loss of CDK6 is thus associated with changes in DNA methylation. As not all changes in DNA methylation result in differences in gene regulation, we performed RNA-seq analysis to examine the consequences of loss of CDK6 on gene expression. PCA based on all gene expression data revealed that BCR-ABL+ Cdk6−/− cells and BCR-ABL+ Cdk6wt cells cluster separately with lower heterogeneity than the RRBS clustering (Figure S8). The overlay of CDK6-dependent methylation and transcriptional regulation uncovered an inverse pattern of decreased methylation/increased gene expression or increased methylation/decreased gene expression for 12 (2 up, 10 down), 29 (16 up, 13 down) and 14 (8 up, 6 down) genes in the three cell lines (Figure 5A). In silico functional characterization of these genes identified them as involved in cell differentiation (e.g. Runx2, Shox2, Shb), cell proliferation (e.g. Fzd3, Satb1, TBX2), immune systems (e.g. Gfi1, Nfkbiz), MAPK signaling (e.g. Dusp4, Mlkl), Wnt signaling (e.g. Fzd3, Fzd6) or cell growth regulation (e.g. Lhx2, Bcl6) (Figure 5B). This shows that various genes involved in cancer-related pathways are transcriptionally deregulated by CDK6-mediated changes in DNA methylation.
Figure 5.

Correlation of Differential Methylation and Gene Expression in BCR-ABL+ Cdk6−/− Cells Compared to BCR-ABL+ Cdk6wt Cells
(A) Heatmaps showing fold changes in DNA methylation (green panels) and gene expression (violet panels) determined by RNA-sequencing. Fold changes range from red (positive) to blue (negative). The 3 cell lines are presented separately.
(B) Association of hypermethylated/downregulated genes (red rectangles) and hypomethylated/upregulated genes (blue rectangles) with certain cancer-related molecular pathways. See also Figure S8.
Discussion
We show for the first time that CDK6 directly affects DNA methylation in a cell-specific manner. Global demethylation and de novo methylation of selected CpG islands are key events in the pathogenesis of malignant diseases (Michalak et al., 2019; Zhou et al., 2018). During the past decade, tremendous effort has been spent to identify the mechanisms of these epigenetic alterations in cancer cells and a variety of molecular changes have been associated with deregulated DNA methylation. Large-scale studies have shown that certain genes encoding enzymes involved in DNA methylation are affected by somatic mutations in certain types of cancer (Cancer Genome Atlas Research Network et al., 2013; Walter et al., 2011; Ley et al., 2010). Mutations of DNA methyltransferases (DNMTs), especially of DNMT3A, TET2, IDH1, and IDH2, have been found in AML and other hematopoietic malignancies (Yang et al., 2015; Spencer et al., 2017; Delhommeau et al., 2009; Paschka et al., 2010, Cancer Genome Atlas Research Network et al., 2013) and deregulated expression of DNMTs has been found in multiple tumor types, resulting in aberrant DNA methylation (Esteller, 2008; Peng et al., 2006; Saito et al., 2003).
We show that expression of DNMT3 and other epigenetic modifiers correlates with CDK6 expression in ALL patients. To investigate the mechanism we used a multi-omics approach in BCR-ABL+ cells. ChIP-seq analysis showed that CDK6 is located at multiple sites within the 5′ regions and/or intragenic regions of DNMTs. In the case of DNMT3B, ATAC-seq and RNA-seq analysis associated CDK6 binding with open chromatin and transcriptional regulation. The regulation of DNMT3B mediated by CDK6 is reflected on the protein level and chromatome analysis showed reduced levels of DNMT3B protein in CDK6-deficient cells. Of note, we observed a correlation between CDK6 and TET1 expression in the ALL datasets. However, because TET1 expression was not detected in our experimental system, this correlation was not further investigated.
While the frequency of DNMT3B mutations in cancer cells is generally low, deregulated expression of this gene is of potential clinical relevance in multiple cancer types (Hayette et al., 2012; Lamba et al., 2018; Niederwieser et al., 2015; Poole et al., 2017; Amara et al., 2010; Ibrahim et al., 2011). Overexpression of DNMT3B has been correlated with shorter event-free survival (EFS) and a trend toward poorer overall survival (OS) in a large panel of de novo AML patients (Hayette et al., 2012). Increased expression of DNMT3B has also been associated with poor clinical outcome, worse minimal residual disease, high rate of relapse or resistant disease and worse EFS in pediatric AML patients (Lamba et al., 2018) and older adults with primary, cytogenetically normal AML, and high levels of DNMT3B had fewer complete remissions, inferior disease-free survival, and shorter OS (Niederwieser et al., 2015). Prognostically relevant overexpression of DNMT3B is not confined to AML patients. It has also been described for patients with diffuse large B-cell lymphomas and DNMT3B overexpression was detected in clinical specimens from T-ALL and Burkitt's lymphoma patients (Poole et al., 2017; Amara et al., 2010). We observed heterogeneous expression of DNMT3A/B, with levels positively correlating with levels of CDK6 in adult or pediatric ALL samples (Figure 1). This supports the idea that the regulatory effect of CDK6 is not confined to our in vitro experiments but is also relevant in vivo.
CDK6 does not have a DNA-binding domain, so can only bind to DNA in conjunction with transcription factors. Such cooperations have been observed between CDK6 and STAT3, c-Jun, p65, AP-1, NFYA, and SP1 and result in the transcriptional regulation of specific target genes (Kollmann et al., 2013; Handschick et al., 2014; Scheicher et al., 2015; Bellutti et al., 2018). To determine which transcription factor interacts with CDK6 on the 5′ region of DNMT3B, we performed motif analysis of the region underlying the ChIP-seq/ATAC-seq peak as shown in Figure 2A. ETS motifs are the predominant binding sites and ETS1 ChIP-seq analysis identified ETS1 peaks in the DNMT3B 5′ region in human BCR-ABL+ K562 cells as well as murine B-cells. These data suggest that DNMT3B may be regulated by the binding of CDK6/ETS family to its promoter region. This idea will be addressed in further studies.
To determine whether CDK6 affects the methylome we used the next-generation sequencing approach RRBS to perform extensive methylation analysis in BCR-ABL+ cells either expressing or lacking CDK6. We found both hypermethylation and hypomethylation. Although the numbers of differentially methylated CpG sites in different cell lines were comparable, we failed to detect a common pattern of differential methylation. It is attractive to speculate that this observation reflects the heterogeneity in tumor DNA methylation that has been described for various types of human cancer (Wenger et al., 2019; Sheffield et al., 2017; Brocks et al., 2014; Li et al., 2016; Landau et al., 2014; Nordlund et al., 2013). Our result is consistent with the report that aberrant DNA methylation is a signature for the heterogeneity of ALL patients of similar cytogenetic backgrounds (Nordlund et al., 2013). Importantly, many of the methylation changes mediated by CDK6 can be reverted by re-expressing CDK6. This indicates that CDK6 is directly involved in the process of DNA methylation rather than inducing secondary methylation changes. Our finding is consistent with indications that CDK6 is involved in epigenetic programming, such as the observation that DNMT1 is degraded by inhibition of CDK6 with the CDK4/6 inhibitor palbociclib (Acevedo et al., 2016) and that the CDK4/6 inhibitor abemaciclib causes cell cycle inhibition and changes DNA demethylation and immunogenicity (Dowless et al., 2018). However, we provide the first evidence of a direct effect of CDK6 on methylation.
The molecular consequences of the changes in DNA methylation mediated by CDK6 are unclear. It is possible that CDK6 does more than regulate gene transcription, conceivably also having effects on transcription factor binding and chromatin organization. DNA methylation can either mask consensus DNA-binding sites or create new binding sites for transcription factors and nuclear complexes (Bartke et al., 2010; Spruijt et al., 2013). The transcription factor CTCF, which is involved in large-scale chromatin organization, is known to be methylation-sensitive (Bell and Felsenfeld, 2000; Hashimoto et al., 2017; Ghirlando and Felsenfeld, 2016). DNA methylation prevents CTCF binding, thus, CDK6-mediated changes in DNA methylation may be associated with establishing and/or maintaining higher-order topological structures in the genome.
In summary, we show that loss of CDK6 contributes to DNA hypomethylation and hypermethylation. Importantly, the effect of CDK6 on DNA methylation is generally reversible, a point that should be considered when targeting CDK6 in anticancer therapy. We also identify DNMT3B as a transcriptional target of CDK6. This function of CDK6 was unknown so far but the extent to which CDK6-dependent changes in DNA methylation contribute to tumorigenesis and the maintenance of a malignant phenotype remains to be determined.
Limitations of the Study
In the present study, we demonstrated that CDK6 affects DNA methylation by transcriptional regulation of DNMT3B. However, because CDK6 does not have a DNA-binding domain, it can only bind to DNA in association with transcription factors. Our data suggest that ETS1 is one of these factors but the interaction between CDK6 and ETS1 remains to be investigated in more detail.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Veronika Sexl (Veronika.Sexl@vetmeduni.ac.at).
Materials Availability
This study did not generate new unique reagents.
All reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data Availability and Code Availability
The accession numbers for the data sets reported in this paper are: GEO: GSE145220, GEO: GSE156966, GEO: GSE145220, GEO: GSE113752.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Graham Tebb for scientific discussions and editing of the manuscript. This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program grant agreement No 694354 to VS and by the Austrian Science Fund, project P-31773 to KK.
Author Contributions
GH performed bioinformatic data analysis, data visualization, and wrote the manuscript; SN, FB, HÜ, LS, and KK performed wet lab experiments; MZ performed bioinformatic data analysis; VS and KK supervised the project team and wrote the manuscript; all authors carefully proof-read the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101602.
Supplemental Information
References
- Acevedo M., Vernier M., Mignacca L., Lessard F., Huot G., Moiseeva O., Bourdeau V., Ferbeyre G. A CDK4/6-dependent epigenetic mechanism protects cancer cells from PML-induced senescence. Cancer Res. 2016;76:3252–3264. doi: 10.1158/0008-5472.CAN-15-2347. [DOI] [PubMed] [Google Scholar]
- Amara K., Ziadi S., Hachana M., Soltani N., Korbi S., Trimeche M. DNA methyltransferase DNMT3b protein overexpression as a prognostic factor in patients with diffuse large B-cell lymphomas. Cancer Sci. 2010;101:1722–1730. doi: 10.1111/j.1349-7006.2010.01569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke T., Vermeulen M., Xhemalce B., Robson S.C., Mann M., Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A.C., Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Bellutti F., Tigan A.S., Nebenfuehr S., Dolezal M., Zojer M., Grausenburger R., Hartenberger S., Kollmann S., Doma E., Prchal-Murphy M. CDK6 antagonizes p53-induced responses during tumorigenesis. Cancer Discov. 2018;8:884–897. doi: 10.1158/2159-8290.CD-17-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocks D., Assenov Y., Minner S., Bogatyrova O., Simon R., Koop C., Oakes C., Zucknick M., Lipka D.B., Weischenfeldt J. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 2014;8:798–806. doi: 10.1016/j.celrep.2014.06.053. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A., Hoadley K., Triche T.J., Jr., Laird P.W. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Keersmaecker K., Atak Z.K., Li N., Vicente C., Patchett S., Girardi T., Gianfelici V., Geerdens E., Clappier E., Porcu M. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013;45:186–190. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F., Dupont S., Della Valle V., James C., Trannoy S., Masse A., Kosmider O., Le Couedic J.P., Robert F., Alberdi A. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Dowless M., Lowery C.D., Shackleford T., Renschler M., Stephens J., Flack R., Blosser W., Gupta S., Stewart J., Webster Y. Abemaciclib is active in preclinical models of ewing sarcoma via multipronged regulation of cell cycle, DNA methylation, and interferon pathway signaling. Clin. Cancer Res. 2018;24:6028–6039. doi: 10.1158/1078-0432.CCR-18-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N. Engl. J. Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Figueroa M.E., Lugthart S., Li Y., Erpelinck-Verschueren C., Deng X., Christos P.J., Schifano E., Booth J., Van Putten W., Skrabanek L. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R., Felsenfeld G. CTCF: making the right connections. Genes Dev. 2016;30:881–891. doi: 10.1101/gad.277863.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamot M., Cimmino L., Aifantis I. The impact of DNA methylation in hematopoietic malignancies. Trends Cancer. 2016;2:70–83. doi: 10.1016/j.trecan.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschick K., Beuerlein K., Jurida L., Bartkuhn M., Muller H., Soelch J., Weber A., Dittrich-Breiholz O., Schneider H., Scharfe M. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-kappaB-dependent gene expression. Mol. Cell. 2014;53:193–208. doi: 10.1016/j.molcel.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Hashimoto H., Wang D., Horton J.R., Zhang X., Corces V.G., Cheng X. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell. 2017;66:711–720 e3. doi: 10.1016/j.molcel.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayette S., Thomas X., Jallades L., Chabane K., Charlot C., Tigaud I., Gazzo S., Morisset S., Cornillet-Lefebvre P., Plesa A. High DNA methyltransferase DNMT3B levels: a poor prognostic marker in acute myeloid leukemia. PLoS One. 2012;7:e51527. doi: 10.1371/journal.pone.0051527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller G., Babinsky V.N., Ziegler B., Weinzierl M., Noll C., Altenberger C., Mullauer L., Dekan G., Grin Y., Lang G. Genome-wide CpG island methylation analyses in non-small cell lung cancer patients. Carcinogenesis. 2013;34:513–521. doi: 10.1093/carcin/bgs363. [DOI] [PubMed] [Google Scholar]
- Heller G., Topakian T., Altenberger C., Cerny-Reiterer S., Herndlhofer S., Ziegler B., Datlinger P., Byrgazov K., Bock C., Mannhalter C. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia. 2016;30:1861–1868. doi: 10.1038/leu.2016.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A.E., Arends M.J., Silva A.L., Wyllie A.H., Greger L., Ito Y., Vowler S.L., Huang T.H., Tavare S., Murrell A., Brenton J.D. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut. 2011;60:499–508. doi: 10.1136/gut.2010.223602. [DOI] [PubMed] [Google Scholar]
- Irizarry R.A., Ladd-Acosta C., Wen B., Wu Z., Montano C., Onyango P., Cui H., Gabo K., Rongione M., Webster M. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Seki M., Kawai T., Goto H., Yoshida K., Isobe T., Sekiguchi M., Watanabe K., Kubota Y., Nannya Y. DNA methylation-based classification reveals difference between pediatric T-cell acute lymphoblastic leukemia and normal thymocytes. Leukemia. 2019;34:1163–1168. doi: 10.1038/s41375-019-0626-2. [DOI] [PubMed] [Google Scholar]
- Kollmann K., Heller G., Schneckenleithner C., Warsch W., Scheicher R., Ott R.G., Schafer M., Fajmann S., Schlederer M., Schiefer A.I. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013;24:167–181. doi: 10.1016/j.ccr.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba J.K., Cao X., Raimondi S.C., Rafiee R., Downing J.R., Lei S., Gruber T., Ribeiro R.C., Rubnitz J.E., Pounds S.B. Integrated epigenetic and genetic analysis identifies markers of prognostic significance in pediatric acute myeloid leukemia. Oncotarget. 2018;9:26711–26723. doi: 10.18632/oncotarget.25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau D.A., Clement K., Ziller M.J., Boyle P., Fan J., Gu H., Stevenson K., Sougnez C., Wang L., Li S. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26:813–825. doi: 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley T.J., Ding L., Walter M.J., Mclellan M.D., Lamprecht T., Larson D.E., Kandoth C., Payton J.E., Baty J., Welch J. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Garrett-Bakelman F.E., Chung S.S., Sanders M.A., Hricik T., Rapaport F., Patel J., Dillon R., Vijay P., Brown A.L. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016;22:792–799. doi: 10.1038/nm.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak E.M., Burr M.L., Bannister A.J., Dawson M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019;20:573–589. doi: 10.1038/s41580-019-0143-1. [DOI] [PubMed] [Google Scholar]
- Moreno D.A., Scrideli C.A., Cortez M.A., De Paula Queiroz R., Valera E.T., Da Silva Silveira V., Yunes J.A., Brandalise S.R., Tone L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010;150:665–673. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]
- Nagel S., Leich E., Quentmeier H., Meyer C., Kaufmann M., Drexler H.G., Zettl A., Rosenwald A., Macleod R.A. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia. 2008;22:387–392. doi: 10.1038/sj.leu.2405028. [DOI] [PubMed] [Google Scholar]
- Niederwieser C., Kohlschmidt J., Volinia S., Whitman S.P., Metzeler K.H., Eisfeld A.K., Maharry K., Yan P., Frankhouser D., Becker H. Prognostic and biologic significance of DNMT3B expression in older patients with cytogenetically normal primary acute myeloid leukemia. Leukemia. 2015;29:567–575. doi: 10.1038/leu.2014.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund J., Backlin C.L., Wahlberg P., Busche S., Berglund E.C., Eloranta M.L., Flaegstad T., Forestier E., Frost B.M., Harila-Saari A. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013;14:r105. doi: 10.1186/gb-2013-14-9-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschka P., Schlenk R.F., Gaidzik V.I., Habdank M., Kronke J., Bullinger L., Spath D., Kayser S., Zucknick M., Gotze K. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010;28:3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- Peng D.F., Kanai Y., Sawada M., Ushijima S., Hiraoka N., Kitazawa S., Hirohashi S. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis. 2006;27:1160–1168. doi: 10.1093/carcin/bgi361. [DOI] [PubMed] [Google Scholar]
- Poole C.J., Zheng W., Lodh A., Yevtodiyenko A., Liefwalker D., Li H., Felsher D.W., Van Riggelen J. DNMT3B overexpression contributes to aberrant DNA methylation and MYC-driven tumor maintenance in T-ALL and Burkitt's lymphoma. Oncotarget. 2017;8:76898–76920. doi: 10.18632/oncotarget.20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y., Lennartsson A., Gaidzik V.I., Deneberg S., Karimi M., Bengtzen S., Hoglund M., Bullinger L., Dohner K., Lehmann S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 2014;9:1108–1119. doi: 10.4161/epi.29315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y., Kanai Y., Nakagawa T., Sakamoto M., Saito H., Ishii H., Hirohashi S. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer. 2003;105:527–532. doi: 10.1002/ijc.11127. [DOI] [PubMed] [Google Scholar]
- Sandoval J., Esteller M. Cancer epigenomics: beyond genomics. Curr. Opin. Genet. Dev. 2012;22:50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Scheicher R., Hoelbl-Kovacic A., Bellutti F., Tigan A.S., Prchal-Murphy M., Heller G., Schneckenleithner C., Salazar-Roa M., Zochbauer-Muller S., Zuber J. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015;125:90–101. doi: 10.1182/blood-2014-06-584417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield N.C., Pierron G., Klughammer J., Datlinger P., Schonegger A., Schuster M., Hadler J., Surdez D., Guillemot D., Lapouble E. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017;23:386–395. doi: 10.1038/nm.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer D.H., Russler-Germain D.A., Ketkar S., Helton N.M., Lamprecht T.L., Fulton R.S., Fronick C.C., O'laughlin M., Heath S.E., Shinawi M. CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell. 2017;168:801–816 e13. doi: 10.1016/j.cell.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W., Bauer C., Munzel M., Wagner M., Muller M., Khan F. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Tadesse S., Yu M., Kumarasiri M., Le B.T., Wang S. Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle. 2015;14:3220–3230. doi: 10.1080/15384101.2015.1084445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigan A.S., Bellutti F., Kollmann K., Tebb G., Sexl V. CDK6-a review of the past and a glimpse into the future: from cell-cycle control to transcriptional regulation. Oncogene. 2016;35:3083–3091. doi: 10.1038/onc.2015.407. [DOI] [PubMed] [Google Scholar]
- Tovy A., Spiro A., Mccarthy R., Shipony Z., Aylon Y., Allton K., Ainbinder E., Furth N., Tanay A., Barton M., Oren M. p53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017;31:959–972. doi: 10.1101/gad.299198.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uras I.Z., Maurer B., Nivarthi H., Jodl P., Kollmann K., Prchal-Murphy M., Milosevic Feenstra J.D., Zojer M., Lagger S., Grausenburger R. CDK6 coordinates JAK2 (V617F) mutant MPN via NF-kappaB and apoptotic networks. Blood. 2019;133:1677–1690. doi: 10.1182/blood-2018-08-872648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uras I.Z., Scheicher R.M., Kollmann K., Glosmann M., Prchal-Murphy M., Tigan A.S., Fux D.A., Altamura S., Neves J., Muckenthaler M.U. Cdk6 contributes to cytoskeletal stability in erythroid cells. Haematologica. 2017;102:995–1005. doi: 10.3324/haematol.2016.159947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M.J., Ding L., Shen D., Shao J., Grillot M., Mclellan M., Fulton R., Schmidt H., Kalicki-Veizer J., O'laughlin M. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–1158. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Nicolay B.N., Chick J.M., Gao X., Geng Y., Ren H., Gao H., Yang G., Williams J.A., Suski J.M. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature. 2017;546:426–430. doi: 10.1038/nature22797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger A., Ferreyra Vega S., Kling T., Bontell T.O., Jakola A.S., Caren H. Intratumor DNA methylation heterogeneity in glioblastoma: implications for DNA methylation-based classification. Neuro Oncol. 2019;21:616–627. doi: 10.1093/neuonc/noz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Rau R., Goodell M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer. 2015;15:152–165. doi: 10.1038/nrc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Zhong J.F., Stucky A., Chen X.L., Press M.F., Zhang X. Histone acetylation: novel target for the treatment of acute lymphoblastic leukemia. Clin. Epigenetics. 2015;7:117. doi: 10.1186/s13148-015-0151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Dinh H.Q., Ramjan Z., Weisenberger D.J., Nicolet C.M., Shen H., Laird P.W., Berman B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018;50:591–602. doi: 10.1038/s41588-018-0073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the data sets reported in this paper are: GEO: GSE145220, GEO: GSE156966, GEO: GSE145220, GEO: GSE113752.