

Group B streptococcus (GBS) is the leading cause of neonatal sepsis, pneumonia, and meningitis. GBS strain CNCTC10/84 is a highly virulent blood isolate that has been used extensively to study GBS pathogenesis for over 20 years. Strain CNCTC10/84 has an unusually strong hemolytic activity, but the genetic basis is unknown. In this study, we discovered that a single-nucleotide insertion in an intergenic homopolymeric tract is responsible for the elevated hemolytic activity of CNCTC10/84.
KEYWORDS: CNCTC10/84, CovR-CovS two-component regulatory system, Streptococcus agalactiae, homopolymeric tract, hyperhemolytic phenotype, single-nucleotide insertion
ABSTRACT
Streptococcus agalactiae (group B streptococcus [GBS]) is a major cause of infections in newborns, pregnant women, and immunocompromised patients. GBS strain CNCTC10/84 is a clinical isolate that has high virulence in animal models of infection and has been used extensively to study GBS pathogenesis. Two unusual features of this strain are hyperhemolytic activity and hypo-CAMP factor activity. These two phenotypes are typical of GBS strains that are functionally deficient in the CovR-CovS two-component regulatory system. A previous whole-genome sequencing study found that strain CNCTC10/84 has intact covR and covS regulatory genes. We investigated CovR-CovS regulation in CNCTC10/84 and discovered that a single-nucleotide insertion in a homopolymeric tract in the covR promoter region underlies the strong hemolytic activity and weak CAMP activity of this strain. Using isogenic mutant strains, we demonstrate that this single-nucleotide insertion confers significantly decreased expression of covR and covS and altered expression of CovR-CovS-regulated genes, including that of genes encoding β-hemolysin and CAMP factor. This single-nucleotide insertion also confers significantly increased GBS survival in human whole blood ex vivo.
IMPORTANCE Group B streptococcus (GBS) is the leading cause of neonatal sepsis, pneumonia, and meningitis. GBS strain CNCTC10/84 is a highly virulent blood isolate that has been used extensively to study GBS pathogenesis for over 20 years. Strain CNCTC10/84 has an unusually strong hemolytic activity, but the genetic basis is unknown. In this study, we discovered that a single-nucleotide insertion in an intergenic homopolymeric tract is responsible for the elevated hemolytic activity of CNCTC10/84.
INTRODUCTION
Streptococcus agalactiae (group B streptococcus [GBS]) is a leading cause of neonatal sepsis, pneumonia, and meningitis (1–3). It also causes invasive infections in pregnant women and immunocompromised patients (1, 4–11). GBS strains are commonly epidemiologically classified into one of 10 known serotypes (Ia, Ib, and II to IX) based on the antigenicity of capsular polysaccharide variants (12–14). Among these variants, serotypes Ia, Ib, II, III, and V cause the majority of infant invasive GBS infections (15).
GBS strain CNCTC10/84 is a capsule serotype V strain that was isolated from the blood of a septic neonate (16). It is highly virulent in animal models and has been used extensively in many laboratories for molecular pathogenesis investigations (17–33). CNCTC10/84 has strong hemolytic activity due to the overproduction of β-hemolysin, a key virulence factor of GBS (16, 21, 34–41). The β-hemolysin made by GBS is a pigmented cytotoxic lipid synthesized and transported by proteins encoded by the cyl operon (cylX-cylK) (29, 41–44). Multiple studies have demonstrated that strain CNCTC10/84 is hyperhemolytic and highly pigmented due to β-hemolysin overproduction (16, 45, 46). However, the genetic basis underlying the overproduction of β-hemolysin by CNCTC10/84 is unknown.
In GBS, the CovR-CovS (control of virulence) two-component system is a major global regulator that controls the expression of many virulence factors and metabolic enzymes (47). CovR-CovS regulation suppresses GBS production of β-hemolysin and, conversely, promotes that of CAMP factor (47). Typically, as a consequence, GBS strains that are functionally deficient in CovR-CovS regulation have enhanced beta-hemolytic activity and diminished CAMP factor activity (47–49). Given that CNCTC10/84 is phenotypically similar to a CovR-CovS-deficient strain (i.e., hyperhemolytic and with hypo-CAMP activity) (16, 50), we hypothesized that CNCTC10/84 would likely have a loss of function (frequently a reading frameshift) mutation in either the covR or covS gene. However, a genome sequencing study done by Hooven et al. found that CNCTC10/84 has intact covR and covS genes (50), and therefore factors other than covR or covS gene disruption are responsible for the hyperhemolytic phenotype of CNCTC10/84.
In this study, we demonstrate that a single-nucleotide insertion in the covR promoter region is responsible for the unusual hyperhemolytic and hypo-CAMP factor activities of CNCTC10/84. We also show that this insertion is beneficial for GBS survival in human whole blood ex vivo.
RESULTS
A single-nucleotide insertion in the covRS promoter confers altered hemolytic activity and CAMP factor activity on GBS strain CNCTC10/84.
Given that strain CNCTC10/84 is phenotypically like GBS strains lacking CovR-CovS regulation but has intact covR-covS genes, we inspected the covRS upstream untranslated region for polymorphisms that potentially affect promoter function. Compared to GBS strains with the wild-type allele of the covRS promoter (such as serotype 1a strain A909), CNCTC10/84 has a single-nucleotide insertion in a homopolymeric nucleotide tract in the covRS promoter region (Fig. 1A). The wild-type covRS promoter has 9 consecutive “Ts” downstream of the CovR binding motif and upstream of the covR start codon (48), while the CNCTC10/84 covR promoter allele has 10 Ts (Fig. 1A). Because the CovR-CovS two-component system directly regulates its own expression along with that of β-hemolysin and CAMP factor (47), we hypothesized that this single-nucleotide insertion alters covR and covS expression and consequently causes aberrant regulation and production of β-hemolysin and CAMP factor in CNCTC10/84. To test this hypothesis, we generated an isogenic mutant strain of CNCTC10/84 (i.e., CNCTC10/84-9T) with 9 Ts in the homopolymeric tract (Fig. 1A). An isogenic covR-covS knockout strain (i.e., CNCTC10/84-ΔcovRS) was also generated for use as a CovR-CovS regulation-lacking control (Fig. 1). Our results show that both CNCTC10/84 and CNCTC10/84-ΔcovRS were strongly hemolytic on sheep blood agar and highly pigmented on Todd-Hewitt broth supplemented with yeast extract (THY) agar (Fig. 1B). In contrast, both CNCTC10/84-9T and A909 with the wild-type promoter (9 Ts), were weakly hemolytic and produced little pigment (Fig. 1B). Quantitative assay of hemolytic activity showed that strains CNCTC10/84 and CNCTC10/84-ΔcovRS caused significantly more hemolysis than strains CNCTC10/84-9T and A909 with the wild-type covR promoter (Fig. 1C). We also found that CNCTC10/84 and CNCTC10/84-ΔcovRS had diminished CAMP activity, while strains CNCTC10/84-9T and A909 with the wild-type covR promoter had robust CAMP activity (Fig. 1D). In summary, we demonstrate that deleting a single nucleotide from the poly(T) tract of the CNCTC10/84 covR promoter region restores what appears to be near-wild-type levels of hemolysin and CAMP activity.
FIG 1.

A single-nucleotide insertion in the covR promoter confers altered hemolytic activity and CAMP activity on CNCTC10/84. (A) Schematic depiction of the covR upstream sequences of assayed strains. CovR binding sequence is underlined. (B) Hemolytic and pigmentation phenotypes of GBS strains. (C and D) Hemolytic activity (C) and CAMP activity (D) of GBS strains. OD, optical density. *, P < 0.05 versus 10/84 (n = 4, one-way analysis of variance with Dunnett’s multiple-comparison test).
A single-nucleotide insertion in the covR promoter significantly alters the expression of CovR-CovS-regulated genes in GBS strain CNCTC10/84.
We examined the effects of the covRS promoter region single-nucleotide insertion on CovR-CovS-regulated gene expression. Relative transcription was determined using quantitative reverse transcription PCR (qRT-PCR) for strains cultured in Todd-Hewitt broth supplemented with yeast extract (THY). CNCTC10/84 with the native 10-T covRS promoter allele had very low covR and covS transcript levels (Fig. 2A). The CNCTC10/84-9T isogenic mutant with one T deleted from the homopolymeric tract had significantly increased transcript levels of both covR and covS (Fig. 2A). Not surprisingly, A909 with the wild-type covR promoter (9 Ts) had significantly higher levels of covR and covS transcripts than those of CNCTC10/84. As expected, covR and covS expression was undetectable in the covR-covS deletion strain CNCTC10/84-ΔcovRS (Fig. 2A). These results show that the single-nucleotide insertion impairs the expression of covR and covS in CNCTC10/84.
FIG 2.

A single-nucleotide insertion in the covR promoter confers altered expression of CovR-CovS-regulated genes in CNCTC10/84. Data are expressed as means ± standard deviation (SD). *, P < 0.05 versus 10/84 (one-way analysis of variance with Dunnett’s multiple-comparison test). (E and F) A909 was not included because for bibA and fbsA, TaqMan primers and probes designed for strain CNCTC10/84 are not compatible with strain A909 due to sequence dissimilarity. (D) Locus number 0429 is relative to the genome of CNCTC10/84.
We next examined the expression of β-hemolysin synthesis genes. The cyl operon that encodes genes required for the production of β-hemolysin is negatively regulated by the CovR-CovS two-component system (47). As expected, the covR-covS deletion strain CNCTC10/84-ΔcovRS had high cylE and cylI transcript levels consistent with the lack of CovR-CovS repression (Fig. 2B). Similarly, CNCTC10/84 also had high cylE and cylI transcript levels (Fig. 2B), again consistent with a loss of CovR-CovS repression. In contrast, the isogenic mutant strain CNCTC10/84-9T had significantly lower cylE and cylI relative transcript levels (Fig. 2B, 10/84-ΔT), consistent with the single T deletion in the covR promoter restoring normal (wild-type-like) CovR-CovS repression/regulation. Strain A909 with the wild-type covR promoter had low transcript levels of the cyl genes, similar to those of CNCTC10/84-9T (Fig. 2B).
Inversely to the repression of the β-hemolysin synthesis genes, the CAMP factor-encoding cfb gene is positively regulated by CovR and CovS (47, 49). Consistent with the CAMP factor activity results (Fig. 1D), cfb expression was high in strains CNCTC10/84-9T and A909 with a wild-type 9-T covR promoter and normal covR-covS transcript levels but low in CNCTC10/84 and CNCTC10/84-ΔcovRS with significantly lower covR-covS transcript levels (Fig. 2B).
Lastly, to further investigate the effects of the single-nucleotide difference in the covR promoter poly(T) tract on GBS gene expression, we examined the three additional genes negatively regulated by CovR-CovS (47). These genes are 0429, encoding a putative secreted protein, bibA, encoding a cell wall-anchored adhesin (51), and fbsA, encoding a fibrinogen-binding protein (52). We found that compared to CNCTC10/84, strain CNCTC10/84-9T had significantly lower expression of these three genes (Fig. 2D to F). Consistent with these genes being under CovR-CovS system repression, the covR-covS deletion strain CNCTC10/84-ΔcovRS had relative transcript levels of these three genes that were significantly higher than those in strain CNCTC10/84 (Fig. 2D to F).
In summary, our gene expression data show that the single-nucleotide insertion in the covR promoter resulted in significantly decreased expression of covR and covS, as well as derepression of all CovR-CovS negatively regulated virulence factors examined.
A single-nucleotide insertion in covR promoter confers increased survival of CNCTC10/84 in human whole blood.
Because CNCTC10/84 was isolated from the blood of a septic neonate, we examined the effect of the single-nucleotide insertion on GBS growth in human whole blood collected from two healthy donors (Fig. 3). Our results show that the strongly hemolytic strains CNCTC10/84 and CNCTC10/84-ΔcovRS grew significantly better than the weakly hemolytic strain CNCTC10/84-9T in whole blood obtained from both donors (Fig. 3A and B). GBS causes hemolysis when interacting with human whole blood. After a 6-h incubation in the whole blood, strains CNCTC10/84 and CNCTC10/84-ΔcovRS caused significantly more hemolysis than strain CNCTC10/84-9T (Fig. 3C and D). Moreover, the extent of hemolysis caused by the covR-covS knockout mutant was significantly higher than that for CNCTC10/84 (Fig. 3C and D). None of the GBS strains grew significantly differently in THY (Fig. 3D), suggesting that the differences in hemolysis observed in human whole blood are likely not attributable to basic differences in capacity of the strains to grow in a nutrient-rich environment. In summary, our results show that the single-nucleotide insertion in the covR promoter confers significantly increased GBS survival in human whole blood ex vivo. Also, GBS strains with this single-nucleotide insertion caused significantly higher hemolysis when incubated with human whole blood.
FIG 3.

A single-nucleotide insertion in the covR promoter contributes to CNCTC10/84 survival in human whole blood. (A and B) Growth of GBS strains in human whole blood. (C and D) Hemolysis caused by GBS strains after a 6-h incubation in human whole blood. (E) Growth of GBS strains in THY. (A to D) *, P < 0.05 versus 10/84 (n = 4, one-way analysis of variance with Dunnett’s multiple-comparison test).
Inserting a single T into the homopolymeric tract of the wild-type covR promoter is sufficient to confer hyperhemolysis.
We showed that deleting a single T from the CNCTC10/84 covR promoter abolished the hyperhemolytic phenotype of this strain (Fig. 1). To examine if this is a strain-specific phenomenon unique to CNCTC10/84 or a strain-independent mechanism that has evolved in GBS for altering virulence factor expression by changing covRS expression, we constructed isogenic mutant strain A909-10T by adding a T to the covR promoter of strain A909, a GBS isolate with the wild-type allele of the covR promoter (9 Ts) (Fig. 4A). Our results showed that compared to the wild-type parental strain A909, the isogenic mutant strain A909-10T had significantly decreased expression of covR, significantly increased expression of the hemolysin gene cylE, and significantly decreased expression of the CAMP factor gene cfb (Fig. 4B to D). Consistent with the gene expression profile, isogenic mutant strain A909-10T had markedly increased beta-hemolysis and pigment production, yet decreased CAMP factor activity, similarly to CNCTC10/84 (Fig. 4E and F). In summary, our results show that adding a T to the wild-type covR promoter is sufficient to confer hyperhemolysis in A909.
FIG 4.
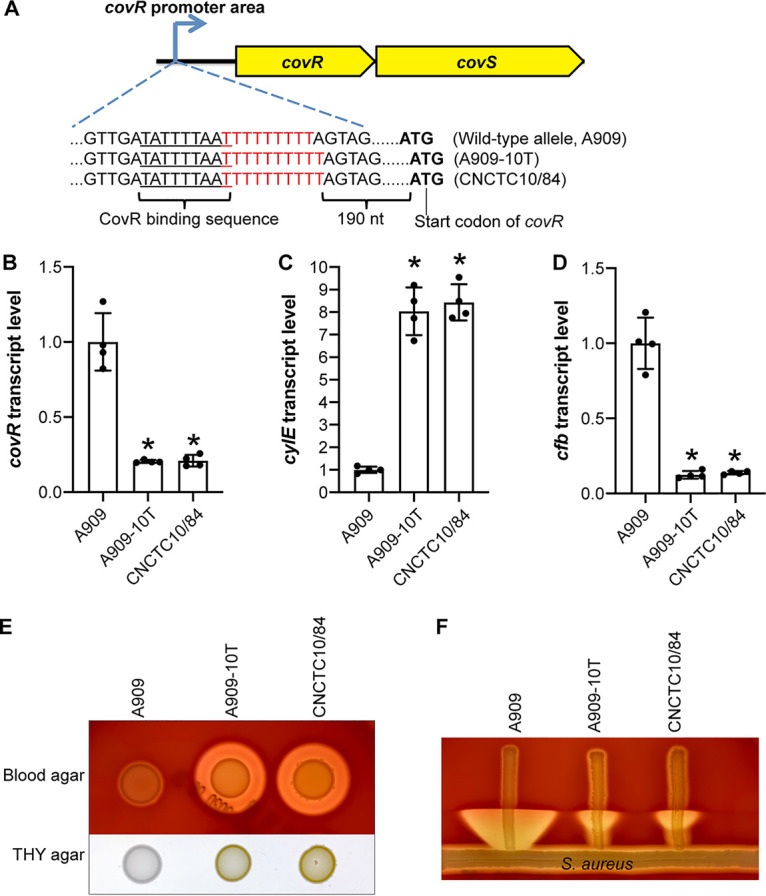
Inserting a single T into the wild-type covR promoter of strain A909 is sufficient to confer hyperhemolysis. (A) Schematic depiction of the covR upstream sequences of assayed strains. (B to D) Transcript levels of covR, the hemolysin gene cylE, and the CAMP factor gene cfb. *, P < 0.05 versus A909 (n = 4, one-way analysis of variance with Dunnett’s multiple-comparison test). (E) Hemolysis and pigment phenotype of GBS strains. (F) CAMP factor activity of GBS strains.
covR promoter region polymorphisms in a large collection of GBS clinical isolates causing invasive infections.
To study if the CNCTC10/84-like insertion in the covR promoter region is present in GBS clinical isolates, we investigated the extent of natural variation in the homopolymeric T-nucleotide tract in the covR promoter among 6,516 GBS invasive infection isolates using kmer counting as previously described for a homopolymeric nucleotide tract in Streptococcus pyogenes (53). The 6,516 isolates were collected and whole-genome sequenced as part of the Active Bacterial Core surveillance program of the Emerging Infections Program of the Centers for Disease Control and Prevention (54) (BioProject accession number PRJNA355303). The number of sequencing reads matching from 6 Ts to 13 Ts was determined for 6,509 of the 6,516 isolates (Fig. 4). Nearly all of the covRS promoters, 6,495 (99.8%), were wild type/A909 like and had 9 Ts. Eleven isolates had 8 Ts, and only 3 isolates were CNCTC10/84 like and had 10 Ts. None of the isolates were found to have 6, 7, 11, 12, or 13 Ts. Thus, the covRS promoter T-nucleotide tract was nearly invariant among the GBS invasive infection isolates examined.
DISCUSSION
In this study, we discovered the genetic basis underlying the hyperhemolysis of GBS strain CNCTC10/84, a highly virulent strain that has been used extensively to study GBS pathogenesis. Our results show that a single-nucleotide insertion in the covR promoter area is responsible for the enhanced hemolytic activity of CNCTC10/84. This mutation is beneficial for GBS survival in human whole blood ex vivo.
The CovR-CovS two-component system is an important global regulator of virulence in GBS (47, 48). CovR positively autoregulates its own expression via binding to the covR promoter (Fig. 1) (48). In this study, we demonstrate that CNCTC10/84 carries a single-nucleotide insertion in the covR promoter that results in strongly reduced promoter activity (Fig. 2). This insertion increases the length of the homopolymeric tract; however, it does not alter the covR binding motif (48) (Fig. 1A). We speculate that the insertion does not affect CovR binding to the CNCTC10/84 covR promoter but instead affects interaction between CovR and RNA polymerase. Proper interaction between CovR and the RNA polymerase complex is essential for covR expression (55). In bacterial pathogens, it is not uncommon for intergenic homopolymeric tracts to play a role in gene regulation and virulence (56). For example, small insertions or deletions within an intergenic homopolymeric tract result in fimbrial phase variation in Bordetella pertussis (57). Similarly, length variation in a homopolymeric tract in the promoter region of nspA has been demonstrated to affect factor H-mediated serum resistance in Neisseria meningitidis (58). We recently discovered that a one-nucleotide indel in an intergenic homopolymeric tract significantly alters global transcript profiles and virulence of Streptococcus pyogenes (53, 59). In this study, we provide another example of intergenic homopolymeric tract polymorphism that affects promoter activity, specifically by altering hemolysin production and potentially virulence of GBS. Further study is needed to unravel the exact biological function of the homopolymeric tract in the covR promoter area.
Many studies have found that CNCTC10/84 is phenotypically similar to a covR-covS-deficient strain, due to its strong hemolytic activity and weak CAMP activity (16, 50, 60, 61). Here, we showed that CNCTC10/84 is a weak expresser of covR and covS, although it has intact covR and covS genes. We also show that CNCTC10/84 is phenotypically similar but not identical to an isogenic covR-covS knockout mutant strain (Fig. 2). For example, the expression of CovR-suppressed genes (cylE, cylI, 0429, bibA, and fbsA) is significantly higher in the covR-covS knockout strain than in CNCTC10/84 (Fig. 2). Also, the covR-covS knockout strain had significantly higher hemolytic activity than CNCTC10/84 (Fig. 1 and 3). These results suggest that the derepression of CovR-suppressed genes is incomplete in CNCTC10/84, presumably due to the residual expression of covR and covS (Fig. 2A). Finally, our results show that there are slight differences in the covR- and covS-regulated genes between A909 and isogenic mutant CNCTC10/84-9T (Fig. 2A and C). We speculate that the moderate difference in gene expression profiles is due to the distinct genetic backgrounds of A909 and CNCTC10/84.
In this study, we demonstrate that the single-nucleotide insertion in the covR promoter is beneficial for CNCTC10/84 survival in human whole blood (Fig. 3). Changing this promoter to the wild-type allele significantly impaired GBS growth in whole blood (Fig. 3). We speculate that CNCTC10/84 is more resistant to the neutrophils present in the whole blood due to the overproduction of the cytotoxic β-hemolysin. In support of this hypothesis, Liu et al. showed that β-hemolysin is essential for GBS survival in human whole blood and for resisting neutrophil killing by promoting cell death (34). We also showed that CNCTC10/84 overexpresses the virulence genes bibA and fbsA (Fig. 2), which encode surface-located adhesins known to protect GBS from opsonophagocytosis and which are required for GBS survival in human whole blood (51, 52). Therefore, the genetic bases underlying enhanced survival of CNCTC10/84 in whole blood could be multifactorial.
Although the insertion in the covRS promoter is beneficial for CNCTC10/84 growth in whole blood ex vivo (Fig. 3), we found that the mutation is rare among clinical isolates causing invasive infections. The large majority of the clinical isolates have the wild-type covR promoter. This indicates that proper expression of covR and covS is critical for GBS fitness and pathogenesis during human infections. That is, decreased expression or complete loss of CovR and CovS function may result in a fitness loss under certain intrahost conditions. Prior studies demonstrate that deletion of covR and covS in GBS can result in increased or decreased pathogenesis depending on the infection models used. For example, Lembo et al., showed that infection with CovR-deficient GBS strains resulted in increased sepsis in mice when injected intravenously (48). Also, CovR-deficient GBS strains were more proficient in induction of permeability and proinflammatory signaling pathways in brain endothelium and penetration of the blood-brain barrier (48). Conversely, there is compelling evidence showing that CovRS-deficient GBS strains are less fit in other infection models. For instance, a covR-covS knockout mutant was significantly impaired for persistence in human serum (47). Also, loss of CovS/CovR abrogates intracellular survival of a type III GBS in macrophages (62). Moreover, CovR-deficient GBS strains exhibit a decreased ability to invade brain microvascular endothelial cells and lung epithelial cells (48). Furthermore, a CovR-deficient serotype III GBS strain was significantly attenuated for colonization in mice and adhesion to uroepithelial cells (63). Lastly, CovR and CovS mutant strains were significantly attenuated for causing sepsis in mice when injected intraperitoneally (49). It is noteworthy that for intraperitoneal injection, GBS cells must invade the epithelium to gain access into the bloodstream and cause sepsis, and CovRS-deficient GBS strains have a defect in crossing epithelial barriers. Collectively, these results suggest that the CovRS two-component system is critical for colonization and epithelial cell invasion. Although CovRS-deficient strains (decreased covR-covS expression, or loss of CovRS function) may have advantages in the bloodstream due to the enhanced production of cytotoxin, these strains may have defects in transmission, colonization, and crossing epithelial barriers. Nevertheless, it is interesting that the GBS covR promoter has a homopolymeric tract that regulates covR-covS expression. The presence of this homopolymeric tract may favor GBS transition from a commensal to an invasive bacterial pathogen. Further investigations are needed to test this hypothesis.
MATERIALS AND METHODS
Construction of isogenic mutant strains.
All of the mutant strains used in this study were constructed using allelic exchange as described previously (64). Isogenic mutant strain CNCTC10/84-9T was derived from CNCTC10/84 by deleting one T in the homopolymeric tract in the covR promoter region. CNCTC10/84-ΔcovRS was constructed by deleting the covR and covS genes of CNCTC10/84. Isogenic mutant strain A909-10T was derived from A909 by inserting a T into the homopolymeric tract of the covR promoter. Primers used for mutant construction are listed in Table S1 in the supplemental material. All isogenic mutant strains were whole-genome sequenced as the final confirmation of the allelic replacement and to rule out the introduction of unwanted spurious mutations influencing covR-covS expression and GBS hemolytic activity. Briefly, the genomes of GBS strains were sequenced using an Illumina NextSeq 550 instrument. Sequence reads were quality filtered, adapter and artifact trimmed, and base call error corrected using Trimmomatic and Musket (65) and then mapped to the genome of CNCTC10/84 or A909 using SMALT (https://www.sanger.ac.uk/tool/smalt-0/). Single-nucleotide polymorphisms (SNPs) and insertions and deletions (indels) were identified using FreeBayes and Pilon (66).
Quantification of hemolytic activities of GBS strains.
A hemolytic activity assay was performed as described previously (16), with minor modifications. Briefly, GBS cells (108 CFU) were inoculated into 10 ml of sheep red blood cells (1%) suspended in phosphate-buffered saline (PBS) supplemented with 0.2% glucose and then incubated at 37°C. After an indicated period of incubation, the tubes were centrifuged to pellet intact red blood cells and GBS cells. An aliquot of 100 μl of the supernatant was transferred to a 96-well plate. Hemolysis was assessed by measuring hemoglobin released into the supernatant by absorbance at 420 nm.
Quantitative reverse transcription-PCR analysis of gene expression in GBS strains.
GBS strains were grown in THY broth (Todd-Hewitt broth supplemented with yeast extract) to an optical density (OD) at 600 nm of 0.5. RNA from GBS cultures was extracted with an RNeasy minikit (Qiagen) and converted into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative PCR was performed with the TaqMan Fast Universal PCR master mix (Applied Biosystems) and an ABI 7500 Fast System (Life Technologies) instrument. The sequences of the TaqMan primers and probes for the assayed genes are listed in Table S2 in the supplemental material. Each experiment was performed in quadruplicate (four biological replicates). A no-template control (NTC) was included to rule out false-positive signals generated by contamination or primer dimer formation. The statistical significance of relative expression differences between strains was evaluated using one-way analysis of variance with Dunnett’s multiple-comparison test.
GBS survival in human whole blood.
Heparinized human blood was collected from healthy volunteers under a Houston Methodist Research Institute Institutional Review Board human subject protocol (protocol number Pro00004933) and processed as described previously (67). To compare the ability of GBS strains to grow in human whole blood, GBS strains were grown to the mid-exponential phase in THY broth (OD of 0.5). GBS cells were washed three times with 10 ml of PBS and suspended with an equivalent volume of PBS. An aliquot of 20 μl of the GBS cell suspension (approximately 2 × 106 CFU) was inoculated into 2 ml of human whole blood and incubated at 37°C in 2-ml tubes rotated horizontally side over side. Aliquots (100 μl) of the seeded blood samples were recovered at 0, 3, and 6 h postinoculation, and GBS CFU were enumerated by serial dilution and growth on blood agar plates.
Supplementary Material
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Schuchat A. 1998. Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin Microbiol Rev 11:497–513. doi: 10.1128/CMR.11.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weston EJ, Pondo T, Lewis MM, Martell-Cleary P, Morin C, Jewell B, Daily P, Apostol M, Petit S, Farley M, Lynfield R, Reingold A, Hansen NI, Stoll BJ, Shane AL, Zell E, Schrag SJ. 2011. The burden of invasive early-onset neonatal sepsis in the United States, 2005–2008. Pediatr Infect Dis J 30:937–941. doi: 10.1097/INF.0b013e318223bad2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, Bizzarro MJ, Goldberg RN, Frantz ID III, Hale EC, Shankaran S, Kennedy K, Carlo WA, Watterberg KL, Bell EF, Walsh MC, Schibler K, Laptook AR, Shane AL, Schrag SJ, Das A, Higgins RD, Eunice Kennedy Shriver National Institute of Child Health, Human Development Neonatal Research Network. 2011. Early onset neonatal sepsis: the burden of group B streptococcal and E. coli disease continues. Pediatrics 127:817–826. doi: 10.1542/peds.2010-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francois Watkins LK, McGee L, Schrag SJ, Beall B, Jain JH, Pondo T, Farley MM, Harrison LH, Zansky SM, Baumbach J, Lynfield R, Snippes Vagnone P, Miller LA, Schaffner W, Thomas AR, Watt JP, Petit S, Langley GE. 2019. Epidemiology of invasive group B streptococcal infections among nonpregnant adults in the United States, 2008–2016. JAMA Intern Med 179:479–488. doi: 10.1001/jamainternmed.2018.7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jump RLP, Wilson BM, Baechle D, Briggs JM, Banks RE, Song S, Zappernick T, Perez F. 2019. Risk factors and mortality rates associated with invasive group B streptococcus infections among patients in the US Veterans Health Administration. JAMA Netw Open 2:e1918324. doi: 10.1001/jamanetworkopen.2019.18324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shelburne SA III, Tarrand J, Rolston KV. 2013. Review of streptococcal bloodstream infections at a comprehensive cancer care center, 2000–2011. J Infect 66:136–146. doi: 10.1016/j.jinf.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Jackson LA, Hilsdon R, Farley MM, Harrison LH, Reingold AL, Plikaytis BD, Wenger JD, Schuchat A. 1995. Risk factors for group B streptococcal disease in adults. Ann Intern Med 123:415–420. doi: 10.7326/0003-4819-123-6-199509150-00003. [DOI] [PubMed] [Google Scholar]
- 8.Seale AC, Bianchi-Jassir F, Russell NJ, Kohli-Lynch M, Tann CJ, Hall J, Madrid L, Blencowe H, Cousens S, Baker CJ, Bartlett L, Cutland C, Gravett MG, Heath PT, Ip M, Le Doare K, Madhi SA, Rubens CE, Saha SK, Schrag SJ, Sobanjo-Ter Meulen A, Vekemans J, Lawn JE. 2017. Estimates of the burden of group B streptococcal disease worldwide for pregnant women, stillbirths, and children. Clin Infect Dis 65:S200–S219. doi: 10.1093/cid/cix664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalin A, Acosta C, Kurinczuk JJ, Brocklehurst P, Knight M. 2015. Severe sepsis in women with group B streptococcus in pregnancy: an exploratory UK national case-control study. BMJ Open 5:e007976. doi: 10.1136/bmjopen-2015-007976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaleznik DF, Rench MA, Hillier S, Krohn MA, Platt R, Lee ML, Flores AE, Ferrieri P, Baker CJ. 2000. Invasive disease due to group B streptococcus in pregnant women and neonates from diverse population groups. Clin Infect Dis 30:276–281. doi: 10.1086/313665. [DOI] [PubMed] [Google Scholar]
- 11.Krohn MA, Hillier SL, Baker CJ. 1999. Maternal peripartum complications associated with vaginal group B streptococci colonization. J Infect Dis 179:1410–1415. doi: 10.1086/314756. [DOI] [PubMed] [Google Scholar]
- 12.Wessels MR, Kasper DL. 1993. The changing spectrum of group B streptococcal disease. N Engl J Med 328:1843–1844. doi: 10.1056/NEJM199306243282510. [DOI] [PubMed] [Google Scholar]
- 13.Slotved HC, Kong F, Lambertsen L, Sauer S, Gilbert GL. 2007. Serotype IX, a proposed new Streptococcus agalactiae serotype. J Clin Microbiol 45:2929–2936. doi: 10.1128/JCM.00117-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cieslewicz MJ, Chaffin D, Glusman G, Kasper D, Madan A, Rodrigues S, Fahey J, Wessels MR, Rubens CE. 2005. Structural and genetic diversity of group B streptococcus capsular polysaccharides. Infect Immun 73:3096–3103. doi: 10.1128/IAI.73.5.3096-3103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madrid L, Seale AC, Kohli-Lynch M, Edmond KM, Lawn JE, Heath PT, Madhi SA, Baker CJ, Bartlett L, Cutland C, Gravett MG, Ip M, Le Doare K, Rubens CE, Saha SK, Sobanjo-ter Meulen A, Vekemans J, Schrag S, Agarwal R, da Silva ARA, Bassat Q, Berkley JA, Dangor Z, Dhaded S, Giannoni E, Hammoud M, Kobayahsi M, O’Sullivan C, Sakata H, Sridhar S, Sigaúque B, Tyrrell G, Paul V, Infant GBS Disease Investigator Group. 2017. Infant group B streptococcal disease incidence and serotypes worldwide: systematic review and meta-analyses. Clin Infect Dis 65:S160–S172. doi: 10.1093/cid/cix656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nizet V, Gibson RL, Chi EY, Framson PE, Hulse M, Rubens CE. 1996. Group B streptococcal beta-hemolysin expression is associated with injury of lung epithelial cells. Infect Immun 64:3818–3826. doi: 10.1128/IAI.64.9.3818-3826.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Randis TM, Gelber SE, Hooven TA, Abellar RG, Akabas LH, Lewis EL, Walker LB, Byland LM, Nizet V, Ratner AJ. 2014. Group B streptococcus beta-hemolysin/cytolysin breaches maternal-fetal barriers to cause preterm birth and intrauterine fetal demise in vivo. J Infect Dis 210:265–273. doi: 10.1093/infdis/jiu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patras KA, Doran KS. 2016. A murine model of group B streptococcus vaginal colonization. J Vis Exp (117):54708. doi: 10.3791/54708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doran KS, Liu GY, Nizet V. 2003. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest 112:736–744. doi: 10.1172/JCI200317335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patras KA, Derieux J, Al-Bassam MM, Adiletta N, Vrbanac A, Lapek JD, Zengler K, Gonzalez DJ, Nizet V. 2018. Group B streptococcus biofilm regulatory protein A contributes to bacterial physiology and innate immune resistance. J Infect Dis 218:1641–1652. doi: 10.1093/infdis/jiy341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulkarni R, Randis TM, Antala S, Wang A, Amaral FE, Ratner AJ. 2013. beta-Hemolysin/cytolysin of group B streptococcus enhances host inflammation but is dispensable for establishment of urinary tract infection. PLoS One 8:e59091. doi: 10.1371/journal.pone.0059091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin SM, Jang AY, Zhi Y, Gao S, Lim S, Lim JH, Song JY, Sullam PM, Rhee JH, Seo HS. 2017. Vaccination with a latch peptide provides serotype-independent protection against group B streptococcus infection in mice. J Infect Dis 217:93–102. doi: 10.1093/infdis/jix565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gendrin C, Vornhagen J, Ngo L, Whidbey C, Boldenow E, Santana-Ufret V, Clauson M, Burnside K, Galloway DP, Adams Waldorf KM, Piliponsky AM, Rajagopal L. 2015. Mast cell degranulation by a hemolytic lipid toxin decreases GBS colonization and infection. Sci Adv 1:e1400225. doi: 10.1126/sciadv.1400225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee A, Kim BJ, Carmona EM, Cutting AS, Gurney MA, Carlos C, Feuer R, Prasadarao NV, Doran KS. 2011. Bacterial pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat Commun 2:462. doi: 10.1038/ncomms1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Sorge NM, Quach D, Gurney MA, Sullam PM, Nizet V, Doran KS. 2009. The group B streptococcal serine-rich repeat 1 glycoprotein mediates penetration of the blood-brain barrier. J Infect Dis 199:1479–1487. doi: 10.1086/598217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang NY, Patras KA, Seo HS, Cavaco CK, Rosler B, Neely MN, Sullam PM, Doran KS. 2014. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J Infect Dis 210:982–991. doi: 10.1093/infdis/jiu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alkuwaity K, Taylor A, Heckels JE, Doran KS, Christodoulides M. 2012. Group B streptococcus interactions with human meningeal cells and astrocytes in vitro. PLoS One 7:e42660. doi: 10.1371/journal.pone.0042660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cutting AS, Del Rosario Y, Mu R, Rodriguez A, Till A, Subramani S, Gottlieb RA, Doran KS. 2014. The role of autophagy during group B streptococcus infection of blood-brain barrier endothelium. J Biol Chem 289:35711–35723. doi: 10.1074/jbc.M114.588657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pritzlaff CA, Chang JC, Kuo SP, Tamura GS, Rubens CE, Nizet V. 2001. Genetic basis for the beta-haemolytic/cytolytic activity of group B streptococcus. Mol Microbiol 39:236–247. doi: 10.1046/j.1365-2958.2001.02211.x. [DOI] [PubMed] [Google Scholar]
- 30.Hooven TA, Bonakdar M, Chamby AB, Ratner AJ. 2019. A counterselectable sucrose sensitivity marker permits efficient and flexible mutagenesis in Streptococcus agalactiae. Appl Environ Microbiol 85:e03009-18. doi: 10.1128/AEM.03009-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo HS, Mu R, Kim BJ, Doran KS, Sullam PM. 2012. Binding of glycoprotein Srr1 of Streptococcus agalactiae to fibrinogen promotes attachment to brain endothelium and the development of meningitis. PLoS Pathog 8:e1002947. doi: 10.1371/journal.ppat.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maisey HC, Hensler M, Nizet V, Doran KS. 2007. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol 189:1464–1467. doi: 10.1128/JB.01153-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim BJ, Bee OB, McDonagh MA, Stebbins MJ, Palecek SP, Doran KS, Shusta EV. 2017. Modeling group B streptococcus and blood-brain barrier interaction by using induced pluripotent stem cell-derived brain endothelial cells. mSphere 2:e00398-17. doi: 10.1128/mSphere.00398-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, Tissi L, Nizet V. 2004. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc Natl Acad Sci U S A 101:14491–14496. doi: 10.1073/pnas.0406143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosa-Fraile M, Dramsi S, Spellerberg B. 2014. Group B streptococcal haemolysin and pigment, a tale of twins. FEMS Microbiol Rev 38:932–946. doi: 10.1111/1574-6976.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forquin MP, Tazi A, Rosa-Fraile M, Poyart C, Trieu-Cuot P, Dramsi S. 2007. The putative glycosyltransferase-encoding gene cylJ and the group B streptococcus (GBS)-specific gene cylK modulate hemolysin production and virulence of GBS. Infect Immun 75:2063–2066. doi: 10.1128/IAI.01565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gibson RL, Nizet V, Rubens CE. 1999. Group B streptococcal beta-hemolysin promotes injury of lung microvascular endothelial cells. Pediatr Res 45:626–634. doi: 10.1203/00006450-199905010-00003. [DOI] [PubMed] [Google Scholar]
- 38.Hensler ME, Liu GY, Sobczak S, Benirschke K, Nizet V, Heldt GP. 2005. Virulence role of group B streptococcus beta-hemolysin/cytolysin in a neonatal rabbit model of early-onset pulmonary infection. J Infect Dis 191:1287–1291. doi: 10.1086/428946. [DOI] [PubMed] [Google Scholar]
- 39.Hensler ME, Miyamoto S, Nizet V. 2008. Group B streptococcal beta-hemolysin/cytolysin directly impairs cardiomyocyte viability and function. PLoS One 3:e2446. doi: 10.1371/journal.pone.0002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ring A, Braun JS, Pohl J, Nizet V, Stremmel W, Shenep JL. 2002. Group B streptococcal beta-hemolysin induces mortality and liver injury in experimental sepsis. J Infect Dis 185:1745–1753. doi: 10.1086/340818. [DOI] [PubMed] [Google Scholar]
- 41.Whidbey C, Harrell MI, Burnside K, Ngo L, Becraft AK, Iyer LM, Aravind L, Hitti J, Adams Waldorf KM, Rajagopal L. 2013. A hemolytic pigment of group B streptococcus allows bacterial penetration of human placenta. J Exp Med 210:1265–1281. doi: 10.1084/jem.20122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Armistead B, Whidbey C, Iyer LM, Herrero-Foncubierta P, Quach P, Haidour A, Aravind L, Cuerva JM, Jaspan HB, Rajagopal L. 2019. The cyl genes reveal the biosynthetic and evolutionary origins of the group B streptococcus hemolytic lipid granadaene. Front Microbiol 10:3123. doi: 10.3389/fmicb.2019.03123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spellerberg B, Martin S, Brandt C, Lutticken R. 2000. The cyl genes of Streptococcus agalactiae are involved in the production of pigment. FEMS Microbiol Lett 188:125–128. doi: 10.1111/j.1574-6968.2000.tb09182.x. [DOI] [PubMed] [Google Scholar]
- 44.Spellerberg B, Pohl B, Haase G, Martin S, Weber-Heynemann J, Lutticken R. 1999. Identification of genetic determinants for the hemolytic activity of Streptococcus agalactiae by ISS1 transposition. J Bacteriol 181:3212–3219. doi: 10.1128/JB.181.10.3212-3219.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burcham LR, Spencer BL, Keeler LR, Runft DL, Patras KA, Neely MN, Doran KS. 2019. Determinants of group B streptococcal virulence potential amongst vaginal clinical isolates from pregnant women. PLoS One 14:e0226699. doi: 10.1371/journal.pone.0226699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hooven TA, Catomeris AJ, Bonakdar M, Tallon LJ, Santana-Cruz I, Ott S, Daugherty SC, Tettelin H, Ratner AJ. 2017. The Streptococcus agalactiae stringent response enhances virulence and persistence in human blood. Infect Immun 86:e00612-17. doi: 10.1128/IAI.00612-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamy MC, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, Glaser P, Kunst F, Msadek T, Trieu-Cuot P, Poyart C. 2004. CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol 54:1250–1268. doi: 10.1111/j.1365-2958.2004.04365.x. [DOI] [PubMed] [Google Scholar]
- 48.Lembo A, Gurney MA, Burnside K, Banerjee A, de los Reyes M, Connelly JE, Lin WJ, Jewell KA, Vo A, Renken CW, Doran KS, Rajagopal L. 2010. Regulation of CovR expression in group B streptococcus impacts blood-brain barrier penetration. Mol Microbiol 77:431–443. doi: 10.1111/j.1365-2958.2010.07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang SM, Cieslewicz MJ, Kasper DL, Wessels MR. 2005. Regulation of virulence by a two-component system in group B streptococcus. J Bacteriol 187:1105–1113. doi: 10.1128/JB.187.3.1105-1113.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hooven TA, Randis TM, Daugherty SC, Narechania A, Planet PJ, Tettelin H, Ratner AJ. 2014. Complete genome sequence of Streptococcus agalactiae CNCTC 10/84, a hypervirulent sequence type 26 strain. Genome Announc 2:e01338-14. doi: 10.1128/genomeA.01338-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santi I, Scarselli M, Mariani M, Pezzicoli A, Masignani V, Taddei A, Grandi G, Telford JL, Soriani M. 2007. BibA: a novel immunogenic bacterial adhesin contributing to group B streptococcus survival in human blood. Mol Microbiol 63:754–767. doi: 10.1111/j.1365-2958.2006.05555.x. [DOI] [PubMed] [Google Scholar]
- 52.Schubert A, Zakikhany K, Schreiner M, Frank R, Spellerberg B, Eikmanns BJ, Reinscheid DJ. 2002. A fibrinogen receptor from group B streptococcus interacts with fibrinogen by repetitive units with novel ligand binding sites. Mol Microbiol 46:557–569. doi: 10.1046/j.1365-2958.2002.03177.x. [DOI] [PubMed] [Google Scholar]
- 53.Eraso JM, Kachroo P, Olsen RJ, Beres SB, Zhu L, Badu T, Shannon S, Cantu CC, Saavedra MO, Kubiak SL, Porter AR, DeLeo FR, Musser JM. 2020. Genetic heterogeneity of the Spy1336/R28-Spy1337 virulence axis in Streptococcus pyogenes and effect on gene transcript levels and pathogenesis. PLoS One 15:e0229064. doi: 10.1371/journal.pone.0229064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Metcalf BJ, Chochua S, Gertz RE Jr, Hawkins PA, Ricaldi J, Li Z, Walker H, Tran T, Rivers J, Mathis S, Jackson D, Glennen A, Lynfield R, McGee L, Beall B, Active Bacterial Core surveillance t. 2017. Short-read whole genome sequencing for determination of antimicrobial resistance mechanisms and capsular serotypes of current invasive Streptococcus agalactiae recovered in the USA. Clin Microbiol Infect 23:574.e7–574.e14. doi: 10.1016/j.cmi.2017.02.021. [DOI] [PubMed] [Google Scholar]
- 55.Gusa AA, Gao J, Stringer V, Churchward G, Scott JR. 2006. Phosphorylation of the group A streptococcal CovR response regulator causes dimerization and promoter-specific recruitment by RNA polymerase. J Bacteriol 188:4620–4626. doi: 10.1128/JB.00198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orsi RH, Bowen BM, Wiedmann M. 2010. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genomics 11:102. doi: 10.1186/1471-2164-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willems R, Paul A, van der Heide HG, ter Avest AR, Mooi FR. 1990. Fimbrial phase variation in Bordetella pertussis: a novel mechanism for transcriptional regulation. EMBO J 9:2803–2809. doi: 10.1002/j.1460-2075.1990.tb07468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Claus H, Hubert K, Becher D, Otto A, Pawlik MC, Lappann I, Strobel L, Vogel U, Johswich K. 2019. A homopolymeric adenosine tract in the promoter region of nspA influences factor H-mediated serum resistance in Neisseria meningitidis. Sci Rep 9:2736. doi: 10.1038/s41598-019-39231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kachroo P, Eraso JM, Beres SB, Olsen RJ, Zhu L, Nasser W, Bernard PE, Cantu CC, Saavedra MO, Arredondo MJ, Strope B, Do H, Kumaraswami M, Vuopio J, Grondahl-Yli-Hannuksela K, Kristinsson KG, Gottfredsson M, Pesonen M, Pensar J, Davenport ER, Clark AG, Corander J, Caugant DA, Gaini S, Magnussen MD, Kubiak SL, Nguyen HAT, Long SW, Porter AR, DeLeo FR, Musser JM. 2019. Integrated analysis of population genomics, transcriptomics and virulence provides novel insights into Streptococcus pyogenes pathogenesis. Nat Genet 51:548–559. doi: 10.1038/s41588-018-0343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rajagopal L, Vo A, Silvestroni A, Rubens CE. 2006. Regulation of cytotoxin expression by converging eukaryotic-type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol 62:941–957. doi: 10.1111/j.1365-2958.2006.05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dramsi S, Morello E, Poyart C, Trieu-Cuot P. 2012. Epidemiologically and clinically relevant group B streptococcus isolates do not bind collagen but display enhanced binding to human fibrinogen. Microbes Infect 14:1044–1048. doi: 10.1016/j.micinf.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 62.Cumley NJ, Smith LM, Anthony M, May RC. 2012. The CovS/CovR acid response regulator is required for intracellular survival of group B streptococcus in macrophages. Infect Immun 80:1650–1661. doi: 10.1128/IAI.05443-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sullivan MJ, Leclercq SY, Ipe DS, Carey AJ, Smith JP, Voller N, Cripps AW, Ulett GC. 2017. Effect of the Streptococcus agalactiae virulence regulator CovR on the pathogenesis of urinary tract infection. J Infect Dis 215:475–483. doi: 10.1093/infdis/jiw589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu L, Olsen RJ, Nasser W, Beres SB, Vuopio J, Kristinsson KG, Gottfredsson M, Porter AR, DeLeo FR, Musser JM. 2015. A molecular trigger for intercontinental epidemics of group A streptococcus. J Clin Invest 125:3545–3559. doi: 10.1172/JCI82478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Y, Schroder J, Schmidt B. 2013. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 29:308–315. doi: 10.1093/bioinformatics/bts690. [DOI] [PubMed] [Google Scholar]
- 66.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thavasu PW, Longhurst S, Joel SP, Slevin ML, Balkwill FR. 1992. Measuring cytokine levels in blood: importance of anticoagulants, processing, and storage conditions. J Immunol Methods 153:115–124. doi: 10.1016/0022-1759(92)90313-i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.