

Summary
SMYD3 is frequently overexpressed in a wide variety of cancers. Indeed, its inactivation reduces tumor growth in preclinical in vivo animal models. However, extensive characterization in vitro failed to clarify SMYD3 function in cancer cells, although confirming its importance in carcinogenesis. Taking advantage of a SMYD3 mutant variant identified in a high-risk breast cancer family, here we show that SMYD3 phosphorylation by ATM enables the formation of a multiprotein complex including ATM, SMYD3, CHK2, and BRCA2, which is required for the final loading of RAD51 at DNA double-strand break sites and completion of homologous recombination (HR). Remarkably, SMYD3 pharmacological inhibition sensitizes HR-proficient cancer cells to PARP inhibitors, thereby extending the potential of the synthetic lethality approach in human tumors.
Subject Areas: Molecular Biology, Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
SMYD3 phosphorylation by ATM favors the formation of HR complexes during DSB response
-
•
SMYD3 mediates DSB repair by promoting RAD51 recruitment at DNA damage sites
-
•
SMYD3 inhibition triggers a compensatory PARP-dependent DNA damage response
-
•
Co-targeting SMYD3/PARP leads to synthetic lethality in HR-proficient cancer cells
Molecular Biology; Cell Biology; Cancer
Introduction
In recent years, the histone methyltransferase SET and MYND Domain containing 3 (SMYD3) gathered a lot of interest from researchers and pharmaceutical companies, and several SMYD3 chemical inhibitors were recently developed (Fabini et al., 2019a, 2019b; Bottino et al., 2020). Indeed, SMYD3 has been found overexpressed in colorectal cancer (CRC) as well as in other types of tumors, such as breast cancer (BC), ovarian cancer (OvCa), prostate cancer (PCa), pancreatic cancer (PC), gastric cancer, lung cancer, and hepatocellular carcinoma (Hamamoto et al., 2004, 2006; Tsuge et al., 2005; Mazur et al., 2014). SMYD3 has been initially identified as a member of the basal transcriptional machinery forming a complex with RNA polymerase II through the RNA helicase HELZ.3. This interaction, along with its ability to methylate histone tails at oncogene regulatory regions (Hamamoto et al., 2004, 2006; Zou et al., 2009; Sarris et al., 2016) and the increased susceptibility to some types of cancer conferred by the presence of tandem repeat polymorphisms in an E2F-binding element in its gene promoter (Tsuge et al., 2005), points to an oncogenic role for SMYD3. In normal cells, SMYD3 seems to be dispensable for development as well as for proliferation and survival. Indeed, SMYD3 homozygous conditional knockout (KO) mice, both male and female, did not show any significant abnormality after full phenotyping (http://www.informatics.jax.org/allele/key/571089; Mazur et al., 2014; Sarris et al., 2016). However, exogenous SMYD3 overexpression in normal cells is sufficient to accelerate cell growth and has a key role in the activation of genes acting downstream of pathways that are involved in tumor cell transformation and migration (Cock-Rada et al., 2012; Luo et al., 2014). It is worth noting that a number of sophisticated in vivo studies using SMYD3-KO mice models showed that this protein plays a key role in lung, pancreas, liver, and colon oncogenesis (Mazur et al., 2014; Sarris et al., 2016).
In a recently published work, we studied the expression and activity of SMYD3 in a CRC preclinical animal model and found that it is strongly upregulated throughout tumorigenesis both at the mRNA and protein levels. Our results also showed that RNAi-mediated SMYD3 ablation or its pharmacological blockade by a small-molecule inhibitor (BCI-121) induces a significant enrichment in the number of cancer cells in the S phase of the cell cycle (Peserico et al., 2015). Extended analysis revealed that SMYD3 is overexpressed in a wide variety of cancer cell lines, with cells expressing high levels of SMYD3 mRNA and protein (high SMYD3) being highly sensitive to its genetic depletion or pharmacological inhibition by BCI-121 (Peserico et al., 2015).
Several studies have been carried out to explore the mechanisms underlying SMYD3 oncogenic activity and suggest that, besides regulating gene expression-related processes, SMYD3 also interacts with and/or methylates non-histone proteins, through which it transactivates cancer-specific pathways. In the nucleus, SMYD3 interacts with heat shock protein 90 (HSP90), which modulates its binding to chromatin and activity (Hamamoto et al., 2004; Brown et al., 2015). SMYD3 also interacts with the PC4 coactivator, another component of the transcriptional machinery that promotes cell proliferation and invasion (Kim et al., 2015). Moreover, SMYD3 has been shown to interact with transcription factors involved in cancer, such as the estrogen receptor (ER), enhancing ER-mediated transcription (Kim et al., 2009). Additionally, it can methylate cytoplasmic proteins involved in signaling cascades that regulate cancer cell proliferation and survival, resulting in enhanced activation, as is the case for VEGFR1, AKT1, HER2, and the RAS/ERK signaling component MAP3K2 (Kunizaki et al., 2007; Yoshioka et al., 2016, 2017; Mazur et al., 2014). However, a recent work carried out by Thomenius and colleagues, who characterized in vitro hundreds of cancer cell lines by using several SMYD3 inhibitors (SMYD3is), SMYD3-specific siRNAs, and CRISPR/Cas9 KO cellular models, revealed that SMYD3’s main contribution in the regulation of tumorigenesis is not based on simply sustaining autonomous proliferation of cancer cells but is still largely unknown (Thomenius et al., 2018). Intriguingly, it has been recently suggested that SMYD3 might participate in the homologous recombination (HR) pathway by modulating the expression of certain HR genes (Chen et al., 2017). HR is a multistep process that is tightly linked to human cancer risk. It is activated by the DNA damage sensor ATM and, through the sequential involvement of BRCA1, CHK2, and BRCA2, finally leads to RAD51 recombinase loading on chromatin at double-strand break (DSB) sites to repair these DNA lesions (Sun et al., 2020; Falck et al., 2005).
To get insight into SMYD3 functions in cancer cells, we performed a proteomic screening to find novel SMYD3 direct interactors that could help clarify its role in tumorigenesis. Here we report that SMYD3 is a direct interactor of the key members of the HR pathway, ATM, CHK2, and BRCA2, and is required for DSB repair. SMYD3 phosphorylation by ATM induces the formation of HR complexes and promotes the recruitment of RAD51 at DSB sites in response to endogenous damage or administration of DNA-damaging agents in CRC and BC cells. Finally, we show that targeting SMYD3 could help extend synthetic lethality approaches based on PARP inhibitors (PARPis) to HR-proficient tumors originating from different tissues.
Results
SMYD3 Directly Interacts with ATM, CHK2, and BRCA2 In Vitro and In Cellulo
Based on the presence of a tetratricopeptide repeat module on SMYD3 C-terminal domain (Brown et al., 2015), we searched for new SMYD3-interacting proteins by screening tripeptides composed of rare amino acid residues, which are often found in specific interfaces for protein-protein interactions (Kanduc, 2010; Reiss and Schwikowski, 2004).
Therefore, we performed a peptide screening and identified a set of 19 short amino acid motifs (P-tripeptides) that were able to bind to SMYD3 in vitro, thus being eligible to be used as a minimum probe to screen the human proteome in the search for new SMYD3 interactors. Of note, all P-tripeptides were shown to interact with recombinant SMYD3 in a surface plasmon resonance (SPR) assay (Figure S1). No significant difference in binding responses between S-adenosyl methionine (SAM)-free and SAM-saturated binding events emerged, indicating there was no allosteric effect related to the presence of the methyl donor (SAM). Various proteomic studies suggest that rare amino acids often represent functional and/or protein-protein interaction consensus motifs (Kanduc, 2010; Kusalik et al., 2009) and that trimeric peptide modules act as biological effectors in protein-protein interactions (Wong et al., 2016; Kataya et al., 2015; Nam et al., 2014; Nishimura and Linder, 2013; Sudnitsyna et al., 2012; Kieken et al., 2009; Jeon et al., 2007). Thus, we performed an in silico analysis to investigate the specific distribution of these P-tripeptides in the human proteome, with the aim of identifying proteins with the highest number of P-tripeptide occurrences at functional sites as potential SMYD3 interactors. Surprisingly, the occurrence of P-tripeptides in all human proteins proved much lower than the theoretically expected probability value, suggesting that their distribution in the human proteome is not stochastic. Indeed, our screening showed that among 169,671 reviewed human proteins (analysis performed in December 2018; www.uniprot.org, UniProt Consortium, 2014), only 8,650 (5.1%) contain at least one P-tripeptide. Intriguingly, we found ≥4 P-tripeptide occurrences in only 214 (0.12%) proteins, which represented our starting subset to identify new potential SMYD3 interactors. One of these 214 proteins was VEGFR1, a known SMYD3 interactor and substrate (Kunizaki et al., 2007). After clustering the selected proteins for their biological role, we observed an enrichment in the cluster involved in DNA repair and S-phase checkpoint (Figure S2). Then, we searched for members of the HR pathway (Reactome database, http://reactome.org.), as it has been proposed that SMYD3 might regulate the expression of certain HR genes (Chen et al., 2017). The best candidates identified by our in silico analysis were BRCA2 and ATM (with 6 P-tripeptide matches each). BRCA2 is a critical protein in the HR process for DSB repair (Krejci et al., 2012); it has been found mutated in various cancers (BC, OvCa, PC, CRC), and its germline mutations specifically predispose to breast and ovarian cancers (Petrucelli et al., 2010). First, we analyzed in vitro the interaction between a full-length HIS-tagged SMYD3 recombinant protein and a series of nine GST fusion proteins, designated B2-1 to B2-9 (Lee et al., 2004), which span the entire BRCA2 coding region (Figure 1A). Interestingly, HIS pull-down assay results showed that BRCA2 B2-4, B2-7, and B2-9 fragments interact with SMYD3 (Figure 1A). These fragments encompass BRC repeats and the C-terminal domain, which mediate RAD51 binding and regulation on resected DNA substrates (Carreira et al., 2009; Chatterjee et al., 2016). These results validated the predictions of our proteomic screening by showing that BRCA2-SMYD3 interaction occurs directly and specifically involves at least one region (B2-4) encompassing a P-tripeptide motif.
Figure 1.

SMYD3 Directly Interacts with ATM, CHK2, and BRCA2 In Vitro and In Cellulo
(A) Upper panel: Diagram showing the nine overlapping GST-BRCA2 (B2-1 to B2-9) fusion proteins used in this study. Lower panel: HIS-SMYD3 bound to histidine beads was incubated with GST-BRCA2 fusion proteins and washed. Bound proteins were visualized by immunoblotting using anti-GST and anti-HIS antibodies.
(B) Upper panel: Diagram showing the eight overlapping GST-ATM (A-1 to A-8) fusion proteins used in this study. Lower panel: HIS-SMYD3 bound to histidine beads was incubated with GST-ATM fusion proteins and washed. Bound proteins were visualized by immunoblotting using anti-GST and anti-HIS antibodies.
(C) Competition assay: HIS-SMYD3 bound to histidine beads was incubated with GST-BRCA2 B2-4 and GST-ATM A-8 fusion proteins in the presence of escalating doses of the purified P1 and P10 tripeptides, respectively. Bound proteins were visualized by immunoblotting using anti-GST and anti-HIS antibodies.
(D) Co-immunoprecipitation assay in MYC-SMYD3- and BRCA2-overexpressing HEK-293 cells using anti-MYC and anti-BRCA2 antibodies.
(E) Co-immunoprecipitation assay in MYC-SMYD3- and FLAG-ATM-overexpressing HEK-293 cells using anti-MYC and anti-FLAG antibodies.
(F) Co-immunoprecipitation of endogenous SMYD3 using anti-SMYD3 antibodies in MDA-MB-231 nuclei treated with MNase to limit indirect interaction through polynucleosomes.
(G) Upper panel: Domain structure of CHK2 protein. Lower panel: HIS-SMYD3 bound to histidine beads was incubated with recombinant CHK2 protein and washed. Bound proteins were visualized by immunoblotting using anti-GST and anti-CHK2 antibodies.
(H) Co-immunoprecipitation assay in MYC-SMYD3- and FLAG-CHK2-overexpressing HEK-293 cells using anti-MYC and anti-FLAG antibodies.
(A, B, C, and G) A 10% input of the purified fusion proteins was used as a loading control. (A, B, and G) GST-HSP90 C-terminal (616–736) was used as a positive control. (D, E, F, and H) Anti-IgGs were used as negative controls. P, P-tripeptide. Results are representative of at least three independent experiments. See also Figures S1 and S2.
ATM is the initiator kinase mediating DNA DSB response through activation of the HR cascade (You et al., 2007; Lee and Paull, 2007). We thus performed an ATM-SMYD3 in vitro pull-down assay by using HIS-tagged SMYD3 and eight GST-ATM fusion proteins, designated A-1 to A-8 (Takai et al., 2007), and found that A-7 and A-8 fragments interact with SMYD3 (Figure 1B). These two consecutive fragments encompass ATM phosphatidylinositol kinase (PIK) domain, which mediates its kinase activity (Bakkenist and Kastan, 2003) and contains one P-tripeptide.
To validate the involvement of the identified tripeptide motifs in SMYD3 interaction, we performed an in vitro competition assay for SMYD3 binding between the BRCA2/ATM fragments containing the relevant tripeptide sequences and the specific purified P-tripeptides. Specifically, we tested BRCA2 GST-B2-4 fragment (encompassing P1, NFF) with escalating doses of the purified P1 tripeptide and ATM GST-A-8 fragment (encompassing P10, NDF) with escalating doses of the purified P10 tripeptide. In both cases, the purified P-tripeptides interfered with the binding between HIS-SMYD3 and the indicated BRCA2/ATM fragments in a dose-dependent manner (Figure 1C).
Next, we assessed whether a direct interaction between SMYD3 and ATM or BRCA2 also occurs in cellulo. Co-immunoprecipitation (coIP) assays in HEK-293 cells transiently transfected with Myc-tagged SMYD3 and BRCA2 or FLAG-ATM confirmed the results obtained in vitro (Figures 1D and 1E). To validate these findings, we also performed coIP assays of the endogenous proteins in breast cancer MDA-MB-231 cells, which express high levels of SMYD3 (Figure S3A) and are wild-type for both ATM and BRCA2 (Figure S5F), in the presence or absence of MNase to rule out indirect interaction via chromatin. Our results showed similar signals in both experimental conditions, confirming that SMYD3 directly interacts with BRCA2 and ATM (Figure 1F).
These findings prompted us to further explore our screening list in the search for other key players of the HR pathway. Interestingly, we found that CHK2, a kinase that acts downstream of ATM and plays an effector role in the HR pathway by activating BRCA2 (Shiloh, 2003), also contains 2 P-tripeptides. Our data indicate that CHK2 can also interact with SMYD3 both in vitro and in cellulo (Figures 1G and 1H).
SMYD3 Mediates DSB Repair in BC Cells
To assess SMYD3 direct involvement in the HR repair process, we analyzed the formation and clearance of nuclear foci of the damage sensor p53-binding protein 1 (53BP1), a DSB marker (Panier and Boulton, 2014). Since we found that SMYD3 is overexpressed in a large panel of BC cell lines (Figure S3A), including triple-negative breast cancer (TNBC) cells (a BC phenotype for which no standard therapy is currently recommended), we genetically silenced SMYD3 in the high-SMYD3 TNBC cell line MDA-MB-231 and evaluated 53BP1 foci in non-stressed asynchronous cells. Double-immunostaining experiments for foci containing 53BP1 and γH2AX, which is another DSB response component that co-localizes with 53BP1, revealed the presence of a higher number of 53BP1/γH2AX-positive foci in cells treated with SMYD3-specific siRNAs, indicating the presence of increased unrepaired endogenous DSB lesions in SMYD3-depleted cells (Figures 2A and S3B). An increase in 53BP1 foci was also observed when cells were treated with the SMYD3 inhibitors BCI-121 or EPZ031686 for 2.5 h (Figures 2B and 2C). Next, we determined the ability of SMYD3-interfered cells to repair neocarzinostatin (NCS)-induced DSBs (Dedon and Goldberg, 1992). Twenty-four hours after NCS exposure, cells that were genetically depleted for SMYD3 were incapable of completely repairing DNA compared with non-silenced cells (Figure 2A). This evidence indicates that SMYD3 genetic ablation prevents correct repair and resolution of DSBs at this time point (Figure 2A). In contrast, SMYD3i treatment did not affect the resolution of NCS-induced DNA lesions in MDA-MB-231 cells at the 24-h time point (Figures 2B and 2C). Similar results were obtained when 53BP1 foci were analyzed both in unstressed conditions and following NCS exposure in BCI-121-treated MCF7 cells, a different high-SMYD3 BC cell line (Figure 2D). Importantly, BCI-121 treatment did not affect the abundance of endogenous or NCS-induced 53BP1 foci in the MDA-MB-468 cell line, in which SMYD3 expression is barely detectable (low SMYD3) (Figure 2E). These findings confirmed that SMYD3 has a critical role in DSB repair and its protein expression is required for DNA restoration. However, although its function appears to be critical for endogenous damage, data related to residual damage after NCS exposure suggest that alternative DNA repair mechanisms might be involved when SMYD3 is pharmacologically impaired.
Figure 2.

SMYD3 Mediates DSB Repair in BC Cells
(A) High-SMYD3 MDA-MB-231 cells were transfected with control (siCTRL) or SMYD3-specific (siSMYD3) siRNAs, exposed to 0.8 nM neocarzinostatin (NCS) after 48 h, and fixed at the indicated time points. Upper panel: Number of 53BP1 foci/cell based on immunostaining for 53BP1. At least 100 cells were analyzed for each time point. Means and standard deviations obtained from at least three independent experiments are shown in the graph on the left. The graph in the middle shows the number of 53BP1 foci/cell before NCS addition (endogenous damage). The graph on the right shows the percentage of 53BP1 foci/cell induced by NCS and detectable 24 h after drug exposure (residual damage NCS 24h). Lower panel: double immunostaining for 53BP1 (red) and γH2AX (green). Nuclei were stained with DAPI (blue). Representative images are shown. The scale bar represents 5 μm.
(B) MDA-MB-231 cells were treated with 10 μM BCI-121 2.5 h before NCS addition. Immunostaining for 53BP1 and foci counting were performed and are graphically shown as in (A).
(C) Same as in (B) but cells were treated with 1 μM EPZ031686.
(D and E) Same as in (B), but experiments were performed on high-SMYD3 MCF7 cells (D) and low-SMYD3 MDA-MB-468 cells (E).
Data are presented as mean (SD), and significance was calculated using Student's t-test; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results are representative of at least three independent experiments.
See also Figure S3.
Of note, the results obtained upon 2.5 h pre-treatment with a SMYD3i (Figures 2B–2D) indicate that, at least in cancer cells, the main role of SMYD3 in DSB repair is not the epigenetic regulation of certain HR genes as previously suggested (Chen et al., 2017) but involves an important transcription-independent activity that positively mediates DSB repair in cancer cells expressing high levels of SMYD3.
SMYD3 Analysis in the Pan-Cancer Dataset and Identification of a Genetic Variant in a High-Risk BC Family
The involvement of SMYD3 in HR, together with its interaction with ATM, CHK2, and BRCA2, which are all encoded by genes that are highly mutated in sporadic cancers and are implicated in genetic predisposition to BC (Jerzak, et al., 2018; Apostolou and Papasotiriou, 2017; Tazzite, et al., 2020), prompted us to analyze SMYD3 genomic alterations and mRNA expression using publicly available human breast invasive carcinoma data from The Cancer Genome Atlas (TCGA) Pan-Cancer dataset. We included in this study a total of 981 primary tumors previously and systematically analyzed by integrating data on somatic truncating mutations, deep copy-number deletions, and epigenetic silencing events involving a curated list of 276 genes encompassing all major DNA repair pathways (Knijnenburg et al., 2018). We used the cBioPortal website (http://www.cbioportal.org) to assess somatic alterations involving the SMYD3 gene in 981 selected BC tumors. Tumors were stratified based on SMYD3 RNA-seq Z score, and the third quartile was identified as high SMYD3 (q3: SMYD3 Z score ≥ 1.06). This analysis revealed that the overall frequency of somatic alterations involving the SMYD3 gene was of about 33% in all BC tumors. Specifically, a low proportion of BC tumors (11/981; 1.12%) harbored missense (6/981), fusion (1/981), or deleterious (4/981) mutations involving the SMYD3 gene. Among the six SMYD3 missense mutations identified in the BC dataset, two (c.203C>T, p.Ala68Val; c.620G>T, p.Ser207Ile) were identified in diploid tumors with low/normal SMYD3 mRNA levels and were predicted to be probably deleterious by in silico analysis. Furthermore, in agreement with the observation that SMYD3 is overexpressed in several cancer types (Zhu and Huang, 2020; Bottino et al., 2020), increased SMYD3 mRNA levels were observed in 25% of BC tumors (245/981); of these, 32 also harbored copy number amplifications. The presence of copy number amplifications involving the SMYD3 gene was observed in 6.7% of all BC tumors (66/981). These data highlighted the role of SMYD3 overexpression in the pathogenesis of sporadic BC (Figures 3A and S4).
Figure 3.

SMYD3 Analysis in the PanCanAtlas BC Dataset and Identification of a Genetic Variant in a High-Risk BC Family
(A) Oncoprint of SMYD3 and HRD-associated genes in PanCanAtlas BC tumors. Overall profiling of 981 BC tumors (columns) carrying alterations involving the SMYD3 gene (mRNA overexpression, copy number alterations, and mutations) and deleterious mutations, deletions, and epigenetic silencing events for each HRD-associated gene (rows with gene names listed on the left) in more than 1% of cases. Gray boxes indicate the absence of alterations, and color/shape combinations corresponding to the various alteration types are indicated below the oncoprint. The overall frequency of each gene alteration in the oncoprint plot is indicated on the left.
(B) Pedigree of the BC family selected for whole-exome sequencing. The proband is indicated by an arrowhead. For each individual diagnosed with cancer, the age at diagnosis is reported. Tested family members are marked as +/− to indicate SMYD3 (C)794G>A (p.Arg265His) heterozygous mutation carriers or as −/− to indicate wild-type SMYD3 individuals.
(C) Partial electropherogram of SMYD3 exon 8 confirming the presence of the SMYD3 c.794G>A mutation (indicated by a black arrow) in both the germline and the tumor DNA of the proband.
(D) SMYD3 expression by immunohistochemistry in normal male breast tissue (left) and male breast tumors: cytoplasmic only localization (center) and nuclear and cytoplasmic localization (right). All the analyzed BC sections showed strong (3+) SMYD3 expression compared with normal breast tissue, regardless of SMYD3 mutational status. Magnification: 20×.
Results are representative of at least three independent experiments. See also Figure S4 and Table S1.
Next, we investigated whether SMYD3 could also be involved in genetic susceptibility to inherited BC. To this aim, we focused on a high-risk BC family who tested negative for BRCA1/2 germline mutations and comprised four patients with BC, two males and two females (Figure 3B), selected from the ongoing Italian Multicenter Study on Male Breast Cancer (MBC) (Rizzolo et al., 2019). Whole-exome sequencing was performed on the germline DNA of the two MBC patients. A rare SMYD3 missense variant (c.794G>A; p.Arg265His; rs61762672), which was predicted to be potentially pathogenic by in silico analysis, was found in both MBC patients (Figure 3C). Sequencing analysis of tumor DNA isolated from all four patients with BC showed that the two males and one of the females harbored this variant (Figure 3C). Further analysis of transcriptome data of one of the two BC males showed that the SMYD3 p.Arg265His variant and the wild-type allele were expressed in the tumor with similar percentages (the variant allele and the wild-type allele represented 43% and 57% of the total reads, respectively). SMYD3 protein immunohistochemical expression was evaluated in BC samples from all four affected family members. Strong (3+) SMYD3 expression was detected in tumor cells; by contrast, tumor-surrounding normal breast tissue exhibited weak/absent immunostaining (Figure 3D). Tumor samples displayed ≥70% SMYD3-positive cells: 95% in BC samples derived from family members with the germline SMYD3 p.Arg265His variant and 70% in the sample from the SMYD3 wild-type family member. SMYD3 immunostaining was detected both in the nucleus and in the cytoplasm, showing variable cellular localization across BC samples. Specifically, SMYD3 showed either cytoplasmic or cytoplasmic/nuclear localization in BC samples derived from family members with the germline SMYD3 p.Arg265His variant, whereas it displayed cytoplasmic only localization in the BC sample from the SMYD3 wild-type family member (Table S1 and Figure 3D). Overall, both RNA and protein expression data in BC samples suggest that the SMYD3 p.Arg265His variant could be a rare missense mutation acting with a dominant-negative effect on the wild-type protein.
SMYD3 Promotes the Formation of HR Complexes during DSB Response and Is a Substrate of ATM
Identification of the SMYD3 p.Arg265His variant prompted us to gain further molecular details on SMYD3 function in the HR pathway. To this end, we characterized this variant in our models. We first examined the role of wild-type SMYD3 (SMYD3-WT) in the formation of HR complexes by transfecting HEK-293 cells (Low SMYD3, Figure S3A) with a vector expressing FLAG-tagged SMYD3-WT. CoIP with anti-FLAG antibodies revealed that SMYD3-WT can bind to the HR complex members ATM, CHK2, BRCA2, and RAD51 following DNA damage induced by doxorubicin exposure, and ATM pharmacological inhibition affects this interaction (Figure 4A). These results showed that SMYD3 is part of the HR complex and may mediate its assembly and/or activity. The novel interaction with RAD51 emerging from this experiment, which is consistent with the observation that SMYD3 interacts with BRCA2 regions involved in RAD51 binding (Figure 1A), suggests that SMYD3 may recruit RAD51 on resected DNA ends as a final readout of DNA repair signals. Transfection of HEK-293 cells with a vector expressing the FLAG-tagged SMYD3 p.Arg265His variant (SMYD3-265H) showed that this variant binds to ATM, especially in its phosphorylated form, with a higher affinity than SMYD3-WT but loses the ability to bind other members of the complex (Figure 4A). These data suggest that the SMYD3 p.Arg265His variant might have a dominant-negative activity on ATM and may impair SMYD3 ability to modulate DNA repair. To test this hypothesis, we first generated a SMYD3-KO MDA-MB-231 cell line (MDA-MB-231 SMYD3-KO) using the CRISPR-Cas9 system for genome editing, along with a vector expressing both HA-tagged SMYD3-WT and FLAG-tagged SMYD3-R265H. Then, we transfected SMYD3-KO MDA-MB-231 cells with this vector and performed coIP assays with anti-ATM antibodies. After induction of DNA damage with doxorubicin, we found that ATM binds SMYD3-R265H with higher affinity than SMYD3-WT (Figure 4B). Moreover, analysis of 53BP1 foci in the high-SMYD3 cell line MDA-MB-231 transfected with FLAG-SMYD3-WT or FLAG-SMYD3-R265H revealed that increased endogenous DSBs are found in cells overexpressing the mutant form (Figure 4C). These latter results were further confirmed by a DR-GFP reporter assay, in which the I-SceI endonuclease is expressed in low-SMYD3 U2OS cells (Figure S3A) that are integrated with a direct repeat DR-GFP. I-SceI generates a DSB that restores GFP expression when repaired by HR (Gunn and Stark, 2012; Pierce et al., 1999). This assay showed that SMYD3-WT overexpression in the presence of I-SceI promoted HR repair efficiency and that this increase was not detectable when the SMYD3-R265H mutant was co-transfected (Figure 4D).
Figure 4.

SMYD3 Promotes the Formation of HR Complexes during DSB Response and Is a Substrate of ATM
(A) Co-immunoprecipitation assay with anti-FLAG antibodies in nuclear fractions from HEK-293 cells transfected with FLAG-SMYD3-WT or FLAG-SMYD3-R265H after treatment with doxorubicin (1 μM) and/or the ATM inhibitor KU60019 (1 μM) for 6 h.
(B) Upper panel: Schematic representation of the plasmid coding for both HA-SMYD3-WT and FLAG-SMYD3-R265H used in the subsequent assay. Lower panel: Co-immunoprecipitation assay with anti-ATM antibodies in nuclear fractions from SMYD3-KO MDA-MB-231 transfected cells after doxorubicin exposure (1 μM, 6 h). (A, B) Anti-IgGs were used as negative controls.
(C) After 48 h of transfection with the indicated constructs, high-SMYD3 MDA-MB-231 cells were fixed and immunostained with an anti-53BP1 antibody in combination with an anti-FLAG antibody. Cells positive for FLAG-SMYD3 nuclear staining were considered for 53BP1 counting. At least 50 cells were analyzed in each of three independent experiments. Data shown in the graph (endogenous damage) are presented as mean (SD).
(D) U2OS DR-GFP cells were transfected with the I-SceI and the indicated SMYD3 expression plasmids. After 24 h of transfection the percentage of GFP+ cells was determined for each condition by FACS and subsequently normalized to the control cells. Data are presented as the mean (SD) (n = 3). Significance was determined by one-way ANOVA followed by a Dunnett test. ∗p < 0.05.
(E) In vitro kinase assay showing SMYD3 phosphorylation by ATM, as measured by the luminescence signal resulting from ADP generation.
(F) Left panel: Schematic representation of the human SMYD3 protein highlighting the domains and residues located around the identified variant. The dotted square indicates the ATM phosphorylation site (Thr 268) based on in silico predictions by three different tools (http://phospho.elm.eu.org/; http://www.dabi.temple.edu/disphos/; http://www.cbs.dtu.dk/services/NetPhos/). Right panel: Co-immunoprecipitation assay with anti-FLAG antibodies in nuclear fractions from HEK-293 cells transfected with FLAG-SMYD3-WT or FLAG-SMYD3-R265H after treatment with doxorubicin (1 μM) and/or the ATM inhibitor KU60019 (1 μM) for 6 h.
Significance was calculated using Student's t-test; ∗p < 0.05. DOXO, doxorubicin. Results are representative of at least three independent experiments.
See also Figure S3.
These data indicate that the SMYD3 p.Arg265His variant exerts a dominant-negative activity resulting in the accumulation of unrepaired DNA, an effect possibly mediated by enhanced ATM binding. These findings suggest that SMYD3 could be a substrate of ATM. To verify this hypothesis, we performed an in vitro kinase assay using the purified proteins. Our results showed that active ATM can efficiently phosphorylate SMYD3 (Figure 4E). Intriguingly, an in silico analysis suggested that the best candidate site for ATM phosphorylation is T268, a residue that is very close to the mutation site (R265H) we identified in the high-risk BC family (Figure 4F). To ascertain whether ATM can also phosphorylate SMYD3-R265H in cellulo, we transfected HEK-293 cells with a vector expressing FLAG-tagged SMYD3-WT or FLAG-tagged SMYD3-R265H and performed coIP assays with anti-FLAG antibodies. Phosphorylation of SMYD3-WT following doxorubicin exposure was observed with phospho-specific SQ/TQ antibodies, which recognize proteins phosphorylated on these motifs. This site was specifically targeted by ATM, as confirmed by the loss of phosphorylation observed after ATM inhibition. Remarkably, no phosphorylation was detected in cells overexpressing the mutant form SMYD3-R265H (Figure 4F).
SMYD3 Localizes at DSBs and Its R265H Mutation Prevents RAD51 Recruitment
Because of SMYD3 ability to associate to chromatin (Peserico et al., 2015; Sarris et al., 2016; Fenizia, et al., 2019; Proserpio et al., 2013), we wondered whether it is recruited at DSBs. First, we performed chromatin immunoprecipitation (ChIP) assays in low-SMYD3 DR-GFP U2OS cells (Figure S3A) transfected with FLAG-SMYD3-WT or empty vectors and we found that FLAG-SMYD3-WT associates to chromatin in close proximity to the DSB (Figure 5A). Of note, ATM pharmacological inhibition prevented the recruitment of SMYD3 at the DSB site (Figure 5A), thus confirming the role of ATM-dependent SMYD3 phosphoactivation in the assembly of the HR multiprotein complex (Figures 4A and 4E).
Figure 5.

SMYD3 Localizes at DSBs and Its R265H Mutation Prevents RAD51 Recruitment
(A) SMYD3 association to DSB regions was assayed by chromatin immunoprecipitation (ChIP) qPCR in DR-GFP U2OS cells transiently co-transfected with a plasmid carrying the I-SceI coding sequence and either an empty vector or the FLAG-SMYD3-WT plasmid and treated or not with KU60019 (1 μM). ChIP assays were performed using antibodies against SMYD3, and the region +1300 bp from the cut site was analyzed by qPCR.
(B) MDA-MB-231 cells were pre-treated for 4 h with BCI-121 (30 μM) or EPZ031686 (1 μM) and then subjected to DNA damage with NCS (1 nM) for 6 h. The graph reflects the quantification of RAD51 foci analyzed by immunofluorescence. ★p ≤ 0.05, all treatments compared with control (-NCS); #p ≤ 0.05, SMYD3i treatment compared with its respective control (±NCS).
(C) DR-GFP U2OS cells were transiently co-transfected with a plasmid coding for I-SceI and either FLAG-SMYD3-WT or the FLAG-SMYD3-R265H variant. ChIP assays were performed using antibodies against SMYD3 and RAD51, and the region +1300 bp from the cut site was analyzed by qPCR.
(D) MDA-MB-231 cells were transfected with FLAG-SMYD3-WT or FLAG-SMYD3-R265H and then treated with NCS (1 μM) for 6 h. The graph reflects the quantification of RAD51 foci analyzed by immunofluorescence.
(A and C) The ubiquitin B promoter was used as a negative control. Data are expressed as fold enrichment compared with IgGs and represent means (SD).
Statistical analysis was performed using Student's t-test: ∗p ≤ 0.05 was considered statistically significant. Results are representative of at least three independent experiments. See also Figures S3 and S5.
Based on these findings, we tested the hypothesis that SMYD3 might be involved in RAD51 loading on DSBs by analyzing RAD51 foci in MDA-MB-231 cells treated or not with BCI-121 or EPZ031686. We found that SMYD3 inhibition impairs the formation of RAD51 foci on DSBs after NCS exposure (Figures 5B and S5A).
Since the SMYD3-R265H mutant strongly binds to (phospho-)ATM but failed to interact with CHK2, BRCA2, and RAD51 in doxorubicin-treated cancer cells (Figure 4A), we assessed whether it could be recruited at DSB sites. To this end, we performed a ChIP assay in low-SMYD3 DR-GFP U2OS cells overexpressing FLAG-SMYD3-WT or FLAG-SMYD3-R265H and observed that both proteins occupy the DNA break region. However, in SMYD3-WT-overexpressing cells RAD51 co-occupied the break site, whereas overexpression of FLAG-SMYD3-R265H dramatically impaired RAD51 recruitment in proximity to the damage (Figure 5C). Consistent with the dominant-negative effect of FLAG-SMYD3-R265H overexpression observed in MDA-MB-231 cells (Figure 4C), our results showed that the mutant variant impairs RAD51 loading at DSBs after damage (Figures 5D and S5B).
Overall, these data suggest that SMYD3 associates with DSBs and the R265H variant does not affect SMYD3 localization at these sites but prevents proper HR complex formation and RAD51 recruitment at DSBs.
SMYD3 Inhibition Triggers a Compensatory PARP-Dependent DNA Damage Response
We observed a difference between the effect of SMYD3 genetic ablation or pharmacological inhibition on the residual damage of BC cells exposed to a DNA damaging agent, as reported in Figure 2. These findings prompted us to analyze in depth the possibility that other DNA repair signals may be activated to restore efficient DSB religation in the presence of a SMYD3i. PARP1 is an important player in DNA damage response: it acts upstream of various pathways and is implicated both in single-strand break repair mechanisms and in the regulation of DSB repair (Pascal, 2018). Thus, in order to investigate the potential compensatory effect of DNA repair mechanisms, other than HR, acting in BC cells treated with NCS and a SMYD3i, we inhibited PARP in these cells by using olaparib, an FDA-approved PARPi for BRCA1/2-mutated ovarian and pancreatic tumors (Buchtel et al., 2018). We first analyzed 53BP1 foci in MDA-MB-231 cells pre-treated with a SMYD3i alone, olaparib alone, or a combination of both, followed by 24 h NCS exposure in order to induce DSBs (Figure 6A, right panel, and S5C). Of note, cells pre-treated with both a SMYD3i and olaparib were found to be incapable of completely repairing DNA when compared with single drug-treated cells, confirming that combined inhibition impairs DSB repair. Interestingly, the activity of both SMYD3 and PARP1 was also required to efficiently repair endogenous damage (Figure 6A, left panel, and S5C). These results prompted us to evaluate the effect of PARP1 inhibition in wild-type and SMYD3-KO MDA-MB-231 cells. Our data showed that in the absence of SMYD3, BC cells became more sensitive to olaparib (Figure 6B). Moreover, SMYD3-KO cancer cells appeared to have higher levels of basal apoptosis compared with their wild-type counterpart (Figure 6B). To clarify this observation, we analyzed wild-type and SMYD3-KO MDA-MB-231 cells throughout several passages in culture. Our results showed that the SMYD3-KO cells we generated have a limited number of passages; indeed, we observed decreased cell growth and increased cell death after 10–15 passages (Figure S5D). Of note, exogenous re-expression of wild-type SMYD3 in SMYD3-KO MDA-MB-231 cells was able to rescue their phenotype by lowering apoptotic levels (Figure S5E). Finally, to confirm the effect of SMYD3 pharmacological inhibition or genetic ablation observed in BC cells, we analyzed the impact of the SMYD3-R265H mutant on olaparib sensitivity in HEK-293 cells (Low SMYD3, Figure S3A). Although overexpression of wild-type SMYD3 reduced HEK-293 sensitivity to olaparib, the R265H mutant significantly increased apoptosis induction (Figure 6C).
Figure 6.

Targeting SMYD3 to Extend the Synthetic Lethality Approach to HR-Proficient Tumors
(A) Upper panel: Treatment scheme. MDA-MB-231 cells were pre-treated for 4 h with BCI-121 (30 μM) and/or olaparib (10 μM), then exposed to NCS (1 nM) for 24 h. Lower panel: Immunostaining with anti-53BP1 antibodies was performed to count nuclear foci. At least 100 cells were analyzed for each time point. The graphs show the number of 53BP1 foci/cell before NCS addition (endogenous damage, left) and the percentage of 53BP1 foci/cell induced by NCS and detectable 24 h after drug exposure (residual damage NCS 24 h, right). Data are presented as mean (SD).
(B) Cell death analysis by annexin V staining. Wild-type and SMYD3-KO MDA-MB-231 cells were treated with olaparib (10 μM) for 72 h and analyzed by flow cytometry for annexin V staining. The indicated percentages of total apoptotic cells include early and late apoptotic and dead cells. Statistical analysis was performed using Student's t-test; ★p ≤ 0.05, all treatments compared with control; Δp ≤ 0.05, SMYD3-KO MDA-MB-231 versus wild-type MDA-MB-231.
(C) Immunoblot analysis showing cleaved PARP levels in HEK-293 cells transfected with FLAG-SMYD3-WT or FLAG-SMYD3-R265H and treated with olaparib for 48 h. FLAG was analyzed as an overexpression control and actin was used as a loading control.
(D) Colony formation assay of MDA-MB-231 and HCT116 cells pre-treated with a SMYD3i (100 μM BCI-121 or 10 μM EPZ031686) for 6 h and then treated with olaparib (10 μM) in the presence of the SMYD3i for a total of 72 h.
(E and F) BC and CRC cell lines were treated as indicated in (D) and cell survival and cell death were assessed by colony formation assay (E) and by immunoblot analysis of cleaved PARP (F), respectively. Actin was used as a loading control.
(A and E) ★p ≤ 0.05, all treatments compared with control; #p ≤ 0.05, combined treatments compared with the respective single treatments.
Results are representative of at least three independent experiments. See also Figures S3 and S5.
Targeting SMYD3 to Extend the Synthetic Lethality Approach to HR-Proficient Tumors
Based on the above evidence, we hypothesized that combined inhibition of SMYD3 and PARP could represent a valid strategy to induce cancer cell death as a synthetic lethality approach. Specifically, high-SMYD3 and HR-proficient cancer cells might become sensitive to PARP inhibition when combined with SMYD3 inhibition, which impairs HR repair response. Tumors with HR defects are more dependent on PARP to preserve genome integrity (Telli and Ford, 2010). Indeed, inhibition of PARP enzymatic activity directly activates the HR pathway as a way to compensate for the dysfunction; as a consequence, cells with impaired key HR proteins fail to repair DNA damage and restore replication, resulting in cell death (Wang and Weaver, 2011). To get insight into this potential treatment strategy, we treated CRC and BC cells with a SMYD3i and/or olaparib for 72 h and assessed cell death response in single and dual treatment conditions. We found that the combined treatment has a synergistic cytotoxic effect both in MDA-MB-231 and HCT116 cell lines (Figure 6D). We thus extended our analysis to a large panel of BC and CRC cell lines with different mutation profiles and SMYD3 levels (Figure S3A and S5F). Our results confirmed the efficacy of combined SMYD3 and PARP inhibition both in CRC and BC cells with high SMYD3 levels, whereas no significant difference was observed in low-SMYD3 cancer cells (Figure 6E). We further analyzed the biological impact of the combined treatment by characterizing changes in cell fate. Our data revealed that treatment with both a SMYD3i and olaparib promotes apoptosis (Figure 6F). As PARPis are used in the clinics for ovarian and pancreatic BRCA1/2-mutant tumors (Gupta et al., 2019; Zhu et al., 2020; Penson et al., 2020), we further investigated the apoptotic efficacy of the combined treatment in our panel of BC and CRC cells and in OvCa and PC cells with different mutation profiles and SMYD3 levels (Figures S3A and S5F) by annexin V staining (Figures 7A and 7B). Our results confirmed that the combined treatment has a synergistic cytotoxic effect; in particular, PARPi-resistant high-SMYD3 cancer cells are sensitized by SMYD3 inhibition and undergo apoptosis. The high-SMYD3 CAPAN-1 PC cell line was used as a control since it has already been shown to be sensitive to PARP inhibition due to BRCA2 deficiency (McCabe et al., 2005). These data revealed that SMYD3 is synthetic lethal with olaparib in HR-proficient cancer cells, thus extending the potential of the synthetic lethality approach in human tumors.
Figure 7.

Apoptotic Efficacy of SMYD3i and PARPi Combined Treatment in a Panel of BC, CRC, PC, and OvCa Cell Lines
(A and B) Cell death analysis by annexin V staining. OVCAR-3 (A), BC (red), CRC (blue), PC (green), and OvCa (orange) cell lines (B) were pre-treated with a SMYD3i (100 μM BCI-121 or 10 μM EPZ031686) for 6 h, then treated with olaparib (10 μM) in the presence of the SMYD3i for a total of 72 h and analyzed by flow cytometry for annexin V staining. The indicated percentages of total apoptotic cells include early and late apoptotic and dead cells. Statistical analysis was performed using Student's t-test; ★p ≤ 0.05, all treatments compared with control; #p ≤ 0.05, combined treatments compared with the respective single treatments. Results are representative of at least three independent experiments. See also Figure S3.
To evaluate the potential clinical application of SMYD3i/PARPi combined therapy, we determined the fraction of BCs that could be eligible for this therapeutic strategy. To this end, we considered the HR deficiency (HRD) score—which is a biomarker that defines the HRD status and was calculated by Knijnenburg and colleagues by combining scores related to HRD-loss of heterozygosity, large-scale state transitions, and the number of telomeric allelic imbalances—in BCs with high SMYD3 mRNA expression (Knijnenburg et al., 2018). This analysis revealed that 62.4% (153/245) of BC tumors with high SMYD3 mRNA levels have a low HRD score (i.e., equal or below the median within the BC dataset) (Figure S4), thus suggesting that 15.6% (153/981) of all analyzed BCs could be eligible for SMYD3i/PARPi combined therapy (high SMYD3/low HRD). Moreover, we determined in the BC dataset the prevalence (Figure S4) and mutual exclusivity of alterations in genes that were previously shown to be significantly associated with a higher HRD score, which means a higher correlation with a homologous recombination deficiency (Figure 8A, Table S2). Alterations with a frequency above 1% were identified in genes (TP53, BRCA1, NEIL2, HERC2, ATM, RAD51C, BRCA2, NSMCE3, NUDT15, MLH1, POLE, CHEK1, PER1, RFC3, NEIL3, FAAP20) involved in DDR pathways, including homology-dependent recombination, nucleotide excision repair, and mismatch repair (Knijnenburg et al., 2018) (Figures 3A and S4). Importantly, SMYD3 mRNA overexpression is mutually exclusive with loss-of-function alterations in several genes associated with HRD in cancer, i.e., TP53, BRCA1, NEIL2, HERC2, ATM, RAD51C, and BRCA2 (Figure 8A and Table S2), supporting the potential of the therapeutic protocol relying on combined SMYD3 and PARP inhibition for HR-proficient tumors with high levels of SMYD3.
Figure 8.

Analysis of Genomic Alterations and Mutual Exclusivity of SMYD3 and HRD-Associated Genes in PanCanAtlas BC and COAD-READ Datasets
(A) Oncoprint of mutual exclusivity between SMYD3 mRNA overexpression and deleterious alterations of HRD-associated genes (TP53, BRCA1, NEIL2, HERC2, ATM, RAD51C, and BRCA2) identified using DISCOVER algorithm at maximum false discover rate (FDR) of 1% across 981 PanCanAtlas BC tumors. BC tumors with somatic alterations in mutually exclusive genes are depicted in black; BC tumors without somatic alterations in mutually exclusive genes are depicted in gray. Number of BC tumors is indicated below the oncoprint graph (x axis).
(B) Oncoprint of SMYD3 and HRD-associated genes in PanCanAtlas colon and rectum adenocarcinomas (COAD-READ). Overall profiling of 459 COAD-READ tumors (columns) carrying alterations involving the SMYD3 gene mRNA overexpression and deleterious mutations, deletions, and epigenetic silencing events for each HRD-associated gene (rows with gene names listed on the left). The association between COAD-READ tumors and the HRD score, low (≤12) and high (>12), is represented as yellow and green bars, respectively. Gray boxes indicate the absence of alterations, and color/shape combinations corresponding to the various alteration types are indicated below the oncoprint. The overall frequency of each gene alteration in the oncoprint plot is indicated on the left.
(C) Oncoprint of mutual exclusivity between SMYD3 mRNA overexpression and deleterious alterations of HRD-associated genes (MLH1, ATM, EXO5, and HERC2) identified using DISCOVER algorithm at maximum false discover rate (FDR) of 1% across 459 PanCanAtlas COAD-READ tumors. COAD-READ tumors with somatic alterations in mutually exclusive genes are depicted in black; COAD-READ tumors without somatic alterations in mutually exclusive genes are depicted in gray. The number of COAD-READ tumors is indicated below the oncoprint graph (x axis).
See also Figures S4 and S6 and Table S2.
These results prompted us to evaluate the prevalence and mutual exclusivity of SMYD3 mRNA overexpression in association with genomic alterations of 43 HRD-associated genes (Knijnenburg et al., 2018), across 459 PanCanAtlas colon and rectum adenocarcinomas (COAD-READ). These tumors were previously analyzed by integrating data on somatic deleterious mutations, deep copy number deletions, and epigenetic silencing events involving a curated list of 276 genes encompassing all major DNA repair pathways (Knijnenburg et al., 2018). This analysis revealed that 126/459 COAD-READ tumors (27%) show SMYD3 mRNA overexpression and that a low HRD score (i.e., equal or below the median within the COAD-READ dataset) is found in 41.2% of tumors (52/126) with high SMYD3 mRNA levels (Figure 8B). Furthermore 83% of COAD-READ tumors (383/459) display one or more deleterious alterations in a DNA damage repair gene associated with HRD in cancer, including MLH1, ATM, EXO5, and HERC2, which are mutually exclusive with SMYD3 mRNA overexpression (Figure 8C; Table S2). This analysis suggests that 11% of total CRCs could benefit from SMYD3i/PARPi combined therapy (high SMYD3/low HRD).
Since PARPi therapy has been approved for the treatment of patients with germline or somatic BRCA1/BRCA2-mutant ovarian and pancreatic cancers, we used TCGA Pan-Cancer ovarian cancer (OV) and pancreatic adenocarcinoma (PAAD) data to evaluate the fraction of patients who could be eligible for SMYD3i/PARPi combined therapy. We included in this analysis a total of 177 OV and 152 PAAD tumors, which were previously characterized for deleterious mutations in DDR genes, to assess the prevalence of HR-proficient and SMYD3 mRNA-overexpressing tumors. This analysis revealed that 24 of the 51/177 OV tumors with high SMYD3 mRNA levels and 16 of the 41/152 PAAD tumors with high SMYD3 mRNA levels have a low HRD score (i.e., equal or below the median within OV and PAAD datasets). Moreover, at least one mutation from a list of 43 HRD-associated genes was detected in 170/177 OV tumors (96%) and 116/152 PAAD tumors (65.3%) (Figure S6). Thus, 13.5% of total OvCas and 10.5% of total PCs could be eligible for SMYD3i/PARPi combined therapy.
Altogether, these results support the potential benefit of combined SMYD3 and PARP inhibition in the management of several solid tumors with HR proficiency and high levels of SMYD3.
Discussion
Personalized medicine is revolutionizing cancer therapy by targeting genes and pathways that are mutated in specific patients' tumors. At present, two different approaches are moving from bench to bedside: oncogene addiction and synthetic lethality. The oncogene addiction approach relies on the observation that tumor cells bearing an activating mutation in a cancer gene are addicted to the signal generated by the encoded gene product and are thus hypersensitive to drugs that specifically target the activated cancer pathway (Weinstein and Joe, 2006). The HER2-targeted antibody trastuzumab used in BC and the VEGF-targeted antibody bevacizumab used in CRC are just a couple of examples (Nahta, 2012; Rosen et al., 2017). Synthetic lethality approaches are based on the assumption that the presence of a mutation in a cancer gene is often associated with a novel vulnerability that can be targeted therapeutically. The genetic principle is that the combination of two genetic perturbations is lethal, whereas each of them individually is not, because the function of the targeted genes is compensatory or partially redundant. Thus, the clinical effect of single drugs individually targeting one of the genes is limited, but their impact is greatly potentiated when they are used in combination (Shen and Ideker, 2018). At present, the only synthetic lethality approach approved in the clinics is based on the use of PARP inhibitors (i.e., olaparib, rucaparib, niraparib, and talazoparib) in BRCA1/2-mutated tumors, as breast, ovarian, and pancreatic cancers.
SMYD3 is a methyltransferase of particular interest for pharmaceutical companies. Indeed, novel inhibitors of this enzyme were recently developed (Fabini et al., 2019a, 2019b; Bottino et al., 2020). However, although its involvement in tumorigenesis is well established—supported by the fact that it is highly overexpressed in several cancers and is required for the development of some types of tumors even in advanced mice models (Sarris et al., 2016)—its role in cancer formation has recently been debated following the publication of a report showing that its genetic ablation or inhibition of its activity does not impair cancer cell autonomous proliferation in vitro (Thomenius et al., 2018). This evidence prompted us to undertake an in-depth study of the functional role of SMYD3. A better understanding of SMYD3 function might indeed provide new therapeutic avenues to treat cancer.
As SMYD3 mediates cancer progression by interacting with and regulating key cancer-associated non-histone proteins (Mazur et al., 2014), we performed an in silico peptide screening that identified novel SMYD3 interactors, including ATM, CHK2, and BRCA2. These proteins are strictly linked and play important initiator and effector roles in the HR cascade responding to DSBs (Holloman, 2011; Maréchal and Zou, 2013). These data are in agreement with a preliminary study suggesting that SMYD3 is linked to HR repair—although the authors only focused on its gene expression-modulating activity and its effects on long recovery time (Chen et al., 2017)—and with another report hypothesizing that the absence of SMYD3 could arrest cells in the G2/M phase via the ATM-CHK2/p53-Cdc25C pathway (Wang et al., 2017). However, our results clearly support a direct interaction between SMYD3 and ATM. This interaction enables the propagation of a signal cascade through CHK2, which also interacts with SMYD3, by modulating the activity of the third SMYD3 interactor BRCA2, thereby promoting its ability to recruit RAD51 on resected DNA ends. Of note, we found that SMYD3 is a substrate of ATM, and an in silico analysis predicted that the targeted amino acid could be residue T268.
This molecular evidence prompted us to study a group of 981 sporadic BCs from the TCGA BC Pan-Cancer dataset. This study revealed that SMYD3 is altered in 1.2% of all BCs, whereas its mRNA is overexpressed in 25% of them. These data are in agreement with previous reports on cancer tissues (Mazur et al., 2014; Fei et al., 2017) and with an extensive characterization performed on cancer cell lines including BCs and in particular the TNBC phenotype (Peserico et al., 2015 and Figure S3A). Moreover, we found a BC high-risk family carrying a SMYD3 genetic variant predicted to be deleterious. Intriguingly, the mutation occurs at residue 265, which is located very close to threonine 268, the potential ATM phosphorylation site. This variant correlates with a male and female BC phenotype and was also found in a dataset of patients with CRC (Cerami et al., 2012; Gao et al., 2013). The identification of this variant allowed us to gain further molecular clues about SMYD3 protein function in tumors. Our results showed that the R265H mutant protein acts as a dominant negative on ATM, confirming the data obtained by SMYD3 pharmacological inhibition and genetic ablation and unveiling the mechanisms underlying SMYD3 recruitment on DSBs and its active involvement in the repair of DNA lesions.
At the molecular level, we propose that SMYD3 is phosphorylated by ATM and recruited to the site of damaged DNA. At DSB sites, SMYD3 favors the recruitment of various key factors that drive DNA repair through its direct interaction with ATM, BRCA2, and CHK2. Remarkably, the R265H variant identified in the family case study is still capable of associating to DNA break sites but prevents the assembly of a proper DNA repair complex. This suggests that it plays a dominant-negative role, as it displays a very strong interaction with ATM (and phospho-ATM) but loses the ability to interact with BRCA2 and CHK2 to recruit RAD51 to DNA damage sites.
Our findings suggest that SMYD3 plays a role in HR DNA repair also in normal cells. Indeed, the SMYD3 R265H germline mutation was identified in the affected members (two males and one female) of a high-risk BC family. This mutant variant may exert a dominant-negative effect on the wild-type allele by sequestering ATM and preventing proper HR protein complex formation. The analysis of SMYD3 alterations in tumors shows that deleterious mutations have a low frequency, and SMYD3 overexpression, which accounts for around one-third of all cases, is mutually exclusive with mutations occurring in major HR genes. Thus, it seems that these molecular alterations (SMYD3 overexpression and HR gene mutations) may identify two different tumor subsets: one that is HR competent and addicted to SMYD3 overexpression and the other that “classically” displays impairment of major HR genes. Consistently, in our SMYD3-overexpressing cancer cell models, we found that SMYD3 genetic ablation or pharmacological inhibition could trigger a compensatory DNA repair mechanism mediated by PARP1 (Figure 9).
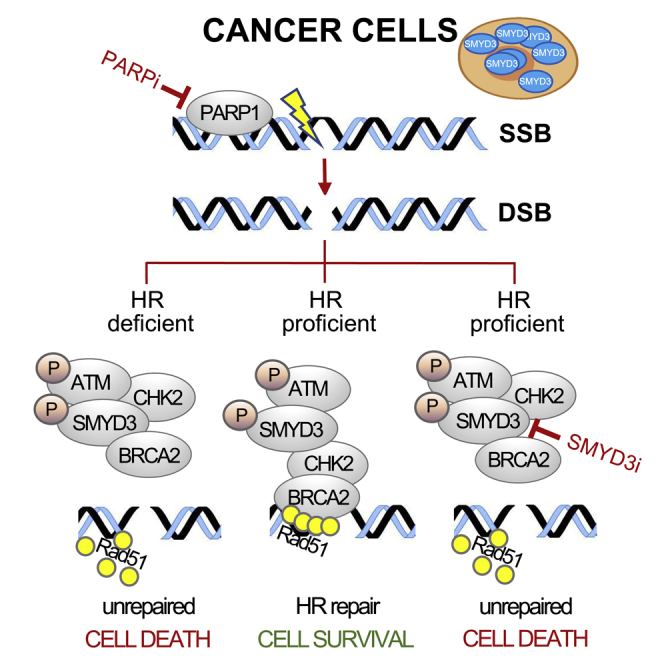
Figure 9.

DSB Repair in Depleted/Mutated and Overexpressing SMYD3 Cells
Based on our findings, SMYD3 might play a role in HR DNA repair response in normal cells. Indeed, the presence of the SMYD3 R265H germline mutation may exert a dominant-negative effect on the wild-type allele by sequestering ATM and preventing proper HR protein complex formation. In high SMYD3-expressing cancer cells, phosphorylated SMYD3 by ATM interacts with CHK2 and BRCA2 forming the HR complex required for the final loading of RAD51 at DBS sites. Molecular alterations as SMYD3 overexpression and HR gene mutations may identify two different tumor subsets: one that is HR competent and addicted to SMYD3 overexpression and the other that is influenced by HR gene mutations. In SMYD3-overexpressing cancer cell models, SMYD3 genetic ablation or pharmacological inhibition could trigger a compensatory DNA repair mechanism mediated by PARP1.
Consistent with a role for SMYD3 in DNA repair, SMYD3 depletion, SMYD3 inhibition, or expression of the R265H variant were all capable, by themselves, to induce endogenous DNA damage accumulation in SMYD3-expressing cancer cells. However, only SMYD3-depleted cells showed a significant defect in rejoining DNA breaks induced by a damaging agent like NCS. Therefore, we hypothesized that activation of one or more compensatory DNA repair signals may occur upon SMYD3 pre-inhibition. Indeed, our experiments highlighted the crucial role of PARP1 in mediating DSB repair in DNA-damaged cells with impaired SMYD3 activity. Thus, we tested the combined treatment with a SMYD3i and a PARPi in order to inhibit both signals, which resulted in cancer cells being incapable of repairing DNA damage. Based on these data, we assessed the efficacy of this combined treatment in BC, CRC, OvCa, and PC cell lines and found that it has a cytotoxic effect and induces apoptotic cell death.
Targeted therapy is currently considered the best approach for fighting cancer. It is based on the use of drugs that specifically interfere with selected molecules involved in cancer development. The increased specificity of this approach allows one to achieve better outcomes and to decrease toxic side effects (Sawyers, 2004). The identification of small-molecule inhibitors that can be associated with PARPis paves the way to new therapeutic protocols and opens up the possibility to extend PARPi/SMYD3i-based treatments even to cancers that are HR proficient and overexpress SMYD3. These represent a significant proportion not only of BC (15%) but also of CRC (11%), OvCa (15%), and PC (10%) cases, according to data retrieved from the TCGA Pan-Cancer Atlas.
We believe that our findings, which should be further tested in preclinical cancer models, may provide the basis for future clinical trials to assess the potential of SMYD3 inhibitors in novel therapeutic protocols.
Limitations of the Study
Our results showed that ATM can efficiently phosphoactivate SMYD3 in response to DNA damage, which promotes HR repair; however, we cannot rule out that additional post-translational modifications mediated by and/or affecting SMYD3 activity may be involved in the modulation of HR response. Thus, further investigations are warranted to identify protein modifications that could favor both the assembly of HR multiprotein complexes and RAD51 loading for DSB repair. These would complete the overall molecular picture and may provide novel predictive biomarkers to monitor therapy response. Moreover, our findings would be strengthened by additional testing of the identified synthetic lethality approach in preclinical cancer models, especially in vivo systems that mimic human tumors, to further corroborate our findings and devise novel therapeutic strategies for clinical settings. Furthermore, in order to enhance and improve future studies, we have planned to develop more effective and safer SMYD3 inhibitors to translate these findings into clinics.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cristiano Simone (cristianosimone73@gmail.com).
Materials Availability
Reagents (peptides, plasmids, and engineered cell lines) generated in this study are available upon request.
Data and Code Availability
In silico data analysis reported in this study were performed as detailed in Transparent Methods section using public repository (i.e., Uniprot, Reactome, cBioPortal), datasets (Colorectal adenocarcinoma, Breast invasive carcinoma, Ovarian serous cystadenocarcinoma, Pancreatic adenocarcinoma of TCGA, PanCancer Atlas), and algorithms (i.e., DISCOVER).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Dr. Francesco Paolo Jori for his helpful discussion during the preparation of the manuscript and editorial assistance, Dr. Vladimir O. Talibov and Prof. Helena Danielson (Department of Chemistry – BMC, Uppsala University, Uppsala, Sweden) for providing the purified SMYD3 protein used in SPR experiments, Valeria Gressani for her technical support, Prof. Ashok Venkitaraman (MRC Cancer Unit, Cambridge, UK) for providing GST-BRCA2 plasmids, Prof. Titia de Lange (The Rockefeller University, New York, USA) for providing GST-ATM plasmids, and Prof. Jeremy Stark (Department of Cancer Genetics and Epigenetics, Beckman Research Institute of the City of Hope, Duarte, California) for providing DR-GFP U2OS cells.
This work was supported by the Fondazione Puglia to P.S., by the Italian Association for Cancer Research (IG grant N. 23794 to C.S., IG grant N.19172 to A.D.R., IG 2018 grant N.21389 to L.O., IG 2018 grant N.21353 to G.C.), by the Italian Ministry of Health “Ricerca Corrente 2018–2020; 2019–2021” to C.S. and “Starting Grant” SG-2019-12371540 to P.S., by the Fondazione Cariplo to G.C., and by the Italian Ministry of Education, University and Research (MIUR) “PRIN - Research Projects of National Relevance“ (PRIN 2017, n. 2017WNKSLRLS4) to C.S.
Author Contributions
C.S. designed the research and wrote the manuscript; P.S., C.F., and V.D. performed the biological and computational assays, analyzed and reviewed the data, and collaborated to write the manuscript; G.B. performed the experiments and reviewed the data and the manuscript for important intellectual content; C.B., S.C., E.F., V.S., V.V., G.F., M.L.S., K.D.M., S.B., U.G., N.P., V.D.M., and E.M. conducted the biological assays; V.G. and M.B. coordinated the experimental work and reviewed the data; G.G. reviewed the manuscript for important intellectual content; A.D.R., G.C., and L.O. collaborated to design the study and write the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101604.
Supplemental Information
References
- Apostolou P., Papasotiriou I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer. 2017;9:331–335. doi: 10.2147/BCTT.S111394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bottino C., Peserico A., Simone C., Caretti G. SMYD3: an oncogenic driver targeting epigenetic regulation and signaling pathways. Cancers (Basel) 2020;12:E142. doi: 10.3390/cancers12010142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.A., Foreman K., Harriss J., Chhaya D., Zhu L., Edwards M., Shaaban S., Tucker H. C-terminal domain of SMYD3 serves as a unique HSP90-regulated motif in oncogenesis. Oncotarget. 2015;6:4005–4019. doi: 10.18632/oncotarget.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchtel K.M., Vogel Postula K.J., Weiss S., Williams C., Pineda M., Weissman S.M. FDA approval of PARP inhibitors and the impact on genetic counseling and genetic testing practices. J. Genet. Couns. 2018;27:131–139. doi: 10.1007/s10897-017-0130-7. [DOI] [PubMed] [Google Scholar]
- Carreira A., Hilario J., Amitani I., Baskin R.J., Shivji M.K.K., Venkitaraman A.R., Kowalczykowski S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–1043. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee G., Jimenez-Sainz J., Presti T., Nguyen T., Jensen R.B. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016;44:5256–5270. doi: 10.1093/nar/gkw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.J., Tsai C.H., Wang P.Y., Teng S.C. SMYD3 promotes homologous recombination via regulation of H3K4-mediated gene expression. Sci. Rep. 2017;7:3842. doi: 10.1038/s41598-017-03385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock-Rada A.M., Medjkane S., Janski N., Yousfi N., Perichon M., Chaussepied M., Chluba J., Langsley G., Weitzman J.B. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012;72:810–820. doi: 10.1158/0008-5472.CAN-11-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedon P.C., Goldberg I.H. Free-radical mechanisms involved in the formation of sequence- dependent bistranded DNA lesions by the antitumor antibiotics bleomycin, neocarzinostatin, and calicheamicin. Chem. Res. Toxicol. 1992;5:311–332. doi: 10.1021/tx00027a001. [DOI] [PubMed] [Google Scholar]
- Fabini E., Manoni E., Ferroni C., Del Rio A., Bartolini M. Small-molecule inhibitors of lysine methyltransferases SMYD2 and SMYD3: current trends. Future Med. Chem. 2019;11:901–921. doi: 10.4155/fmc-2018-0380. [DOI] [PubMed] [Google Scholar]
- Fabini E., Talibov V.O., Mihalic F., Naldi M., Bartolini M., Bertucci C., Del Rio A., Danielson U.H. Unveiling the biochemistry of the epigenetic regulator SMYD3. Biochemistry. 2019;58:3634–3645. doi: 10.1021/acs.biochem.9b00420. [DOI] [PubMed] [Google Scholar]
- Falck J., Coates J., Jackson S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Fei X., Ma Y., Liu X., Meng Z. Overexpression of SMYD3 is predictive of unfavorable prognosis in hepatocellular carcinoma. Tohoku J. Exp. Med. 2017;243:219–226. doi: 10.1620/tjem.243.219. [DOI] [PubMed] [Google Scholar]
- Fenizia C., Bottino C., Corbetta S., Fittipaldi R., Floris P., Gaudenzi G., Carra S., Cotelli F., Vitale G., Caretti G. SMYD3 promotes the epithelial-mesenchymal transition in breast cancer. Nucleic Acids Res. 2019;47:1278–1293. doi: 10.1093/nar/gky1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A., Stark J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- Gupta M., Iyer R., Fountzilas C. Poly(ADP-Ribose) polymerase inhibitors in pancreatic cancer: a new treatment paradigms and future implications. Cancers (Basel) 2019;11:1980. doi: 10.3390/cancers11121980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto R., Furukawa Y., Masashi M., Iimura Y., Pittella Silva F., Li M., Yagyu R., Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004;6:731–740. doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- Hamamoto R., Pittella Silva F., Tsuge M., Toshihiko Nishidate T., Katagiri T., Nakamura Y., Furukawa Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006;97:113–118. doi: 10.1111/j.1349-7006.2006.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloman W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011;18:748–754. doi: 10.1038/nsmb.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon O.H., Kim D., Choi J. Novel function of human ADAM15 disintegrin-like domain and its derivatives in platelet aggregation. Thromb. Res. 2007;119:609–619. doi: 10.1016/j.thromres.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Jerzak K.J., Mancuso T., Eisen A. Ataxia-telangiectasia gene (ATM) mutation heterozygosity in breast cancer: a narrative review. Curr. Oncol. 2018;25:e176–e180. doi: 10.3747/co.25.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanduc D. Protein information content resides in rare peptide segments. Peptides. 2010;31:983–988. doi: 10.1016/j.peptides.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Kataya A., Schei E., Lillo C. MAP kinase phosphatase 1 harbors a novel PTS1 and is targeted to peroxisomes following stress treatments. J. Plant Physiol. 2015;179:12–20. doi: 10.1016/j.jplph.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Kieken F., Jović M., Tonelli M., Naslavsky N., Caplan S., Sorgen P.L. Structural insight into the interaction of proteins containing NPF, DPF, and GPF motifs with the C-terminal EH-domain of EHD1. Protein Sci. 2009;18:2471–2479. doi: 10.1002/pro.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Heo K., Kim J.H., Kim K., Choi J., An W. Requirement of histone methyltransferase SMYD3 for estrogen receptor-mediated transcription. J. Biol. Chem. 2009;284:19867–19877. doi: 10.1074/jbc.M109.021485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.M., Kim K., Schmidt T., Punj V., Tucker H., Rice J.C., Ulmer T.S., An W. Cooperation between SMYD3 and PC4 drives a distinct transcriptional program in cancer cells. Nucleic Acids Res. 2015;43:8868–8883. doi: 10.1093/nar/gkv874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg T.A., Wang L., Zimmermann M.T., Chambwe N., Gao G.F., Cherniack A.D., Fan H., Shen H., Way G.P., Greene C.S. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23:239–254.e6. doi: 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L., Altmannova V., Spirek M., Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunizaki M., Hamamoto R., Fabio Pittella Silva F., Yamaguchi K., Nagayasu T., Shibuya M., Nakamura Y., Furukawa Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007;67:10759–10765. doi: 10.1158/0008-5472.CAN-07-1132. [DOI] [PubMed] [Google Scholar]
- Kusalik A., Trost B., Bickis M., Fasano C., Capone G., Kanduc D. Codon number shapes peptide redundancy in the universal proteome composition. Peptides. 2009;30:1940–1944. doi: 10.1016/j.peptides.2009.06.035. [DOI] [PubMed] [Google Scholar]
- Lee J.H., Paull T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- Lee M., Daniels M.J., Venkitaraman A.R. Phosphorylation of BRCA2 by the polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene. 2004;23:865–872. doi: 10.1038/sj.onc.1207223. [DOI] [PubMed] [Google Scholar]
- Luo X.G., Zhang C.L., Zhao W.W., Liu Z.P., Liu L., Mu A., Guo S., Wang N., Zhou H., Zhang T.C. Histone methyltransferase SMYD3 promotes MRTF-A-mediated transactivation of MYL9 and migration of MCF-7 breast cancer cells. Cancer Lett. 2014;344:129–137. doi: 10.1016/j.canlet.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Maréchal A., Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013;5:a012716. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur P.K., Reynoird N., Khatri P., Jansen P.W.T.C., Wilkinson A.W., Liu S., Barbash O., Van Aller G.S., Huddleston M., Dhanak D. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe N., Lord C.J., Tutt A.N.J., Martin N., Smith G.C.M., Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of poly (ADP-ribose) polymerase: an issue of potency. Cancer Biol. Ther. 2005;4:934–936. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- Nahta R. Molecular mechanisms of trastuzumab-based treatment in HER2-overexpressing breast cancer. ISRN Oncol. 2012;2012:428062. doi: 10.5402/2012/428062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam A.S., Yin Y., von Marschall Z., Fisher L.W. Efficient trafficking of acidic proteins out of the endoplasmic reticulum involves a conserved amino terminal IleProVal (IPV)-like tripeptide motif. Connect. Tissue Res. 2014;55:138–141. doi: 10.3109/03008207.2014.923852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A., Linder M.E. Identification of a novel prenyl and palmitoyl modification at the caax motif of cdc42 that regulates RhoGDI binding. Mol. Cell Biol. 2013;33:1417–1429. doi: 10.1128/MCB.01398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Pascal J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst) 2018;71:177–182. doi: 10.1016/j.dnarep.2018.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penson R.T., Valencia R.V., Cibula D., Colombo N., Leath C.A., Bidziński M., Kim J.W., Nam J.H., Madry R., Hernández C. Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): a randomized phase III trial. J. Clin. Oncol. 2020;38:1164–1174. doi: 10.1200/JCO.19.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peserico A., Germani A., Sanese P., Barbosa A.J., Di Virgilio V., Fittipaldi R., Fabini E., Bertucci C., Varchi G., Pat Moyer M. A SMYD3 small-molecule inhibitor impairing cancer cell growth. J. Cell. Physiol. 2015;230:2447–2460. doi: 10.1002/jcp.24975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli N., Daly M.B., Feldman G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010;12:245–259. doi: 10.1097/GIM.0b013e3181d38f2f. [DOI] [PubMed] [Google Scholar]
- Pierce A.J., Johnson R.D., Thompson L.H., Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proserpio V., Fittipaldi R., Ryall J.G., Sartorelli V., Caretti G. The methyltransferase SMYD3 mediates the recruitment of transcriptional cofactors at the myostatin and c-Met genes and regulates skeletal muscle atrophy. Genes Dev. 2013;27:1299–1312. doi: 10.1101/gad.217240.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss D.J., Schwikowski B. Predicting protein-peptide interactions via a network-based motif sampler. Bioinformatics. 2004;20:i274–i282. doi: 10.1093/bioinformatics/bth922. [DOI] [PubMed] [Google Scholar]
- Rizzolo P., Zelli V., Valentina Silvestri V., Virginia Valentini V., Zanna I., Bianchi S., Masala G., Spinelli A.M., Tibiletti M.G., Russo A. Insight into genetic susceptibility to male breast cancer by multigene panel testing: results from a multicenter study in Italy. Int. J. Cancer. 2019;145:390–400. doi: 10.1002/ijc.32106. [DOI] [PubMed] [Google Scholar]
- Rosen L.S., Jacobs I.A., Burkes R.L. Bevacizumab in colorectal cancer: current role in treatment and the potential of biosimilars. Target Oncol. 2017;12:599–610. doi: 10.1007/s11523-017-0518-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarris M.E., Moulos P., Haroniti A., Giakountis A., Talianidis I. Smyd3 is a transcriptional potentiator of multiple cancer-promoting genes and required for liver and colon cancer development. Cancer Cell. 2016;29:354–366. doi: 10.1016/j.ccell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- Shen J.P., Ideker T. Synthetic lethal networks for precision oncology: promises and pitfalls. J. Mol. Biol. 2018;430:2900–2912. doi: 10.1016/j.jmb.2018.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Sudnitsyna M.V., Mymrikov E.V., Seit-Nebi A.S., Gusev N.B. The role of intrinsically disordered regions in the structure and functioning of small heat shock proteins. Curr. Protein Pept. Sci. 2012;13:76–85. doi: 10.2174/138920312799277875. [DOI] [PubMed] [Google Scholar]
- Sun Y., McCorvie T.J., Yates L.A., Zhang X. Structural basis of homologous recombination. Cell. Mol. Life Sci. 2020;77:3–18. doi: 10.1007/s00018-019-03365-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H., Wang R.C., Takai K.K., Yang H., de Lange T. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131:1248–1259. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- Tazzite A., Jouhadi H., Benider A., Nadifi S. BRCA mutational status is a promising predictive biomarker for platinum-based chemotherapy in triple-negative breast. Curr. Drug Targets. 2020 doi: 10.2174/1389450121666200203162541. [DOI] [PubMed] [Google Scholar]
- Telli M.L., Ford J.M. PARP inhibitors in breast cancer. Clin. Adv. Hematol. Oncol. 2010;8:629–635. [PubMed] [Google Scholar]
- Thomenius M.J., Totman J., Harvey D., Mitchell L.H., Riera T.V., Cosmopoulos K., Grassian A.R., Klaus C., Foley M., Admirand E.A. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS One. 2018;13:e0197372. doi: 10.1371/journal.pone.0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge M., Hamamoto R., Pittella Silva F., Ohnishi Y., Chayama K., Kamatani N., Furukawa Y., Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5ʹ flanking region of SMYD3 is a risk factor for human cancers. Nat. Genet. 2005;37:1104. doi: 10.1038/ng1638. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2014;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang Q.T., Liu Y.P., Dong Q.Q., Hu H.J., Miao Z., Li S., Liu Y., Zhou H., Zhang T.C. ATM signaling pathway is implicated in the SMYD3-mediated proliferation and migration of gastric cancer cells. J. Gastric Cancer. 2017;17:295–305. doi: 10.5230/jgc.2017.17.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Weaver D.T. The ups and downs of DNA repair biomarkers for PARP inhibitor therapies. Am. J. Cancer Res. 2011;1:301–327. [PMC free article] [PubMed] [Google Scholar]
- Weinstein I.B., Joe A.K. Mechanisms of disease: oncogene addiction-a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006;3:448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- Wong S.S., Østergaard S., Hall G., Li C., Williams P.M., Stennicke H., Emsley J. A novel DFP tripeptide motif interacts with the coagulation factor XI apple 2 domain. Blood. 2016;127:2915–2923. doi: 10.1182/blood-2015-10-676122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka Y., Suzuki T., Matsuo Y., Tsurita G., Watanabe T., Dohmae N., Nakamura Y., Hamamoto R. Protein lysine methyltransferase SMYD3 is involved in tumorigenesis through regulation of HER2 homodimerization. Cancer Med. 2017;6:1665–1672. doi: 10.1002/cam4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka Y., Suzuki T., Matsuo Y., Nakakido M., Tsurita G., Simone C., Watanabe T., Dohmae N., Nakamura Y., Hamamoto R. SMYD3-mediated lysine methylation in the PH domain is critical for activation of AKT1. Oncotarget. 2016;7:75023–75037. doi: 10.18632/oncotarget.11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Bailis J.M., Johnson S.A., Dilworth S.M., Hunter T. Rapid activation of ATM on DNA flanking double-strand breaks. Nat. Cell Biol. 2007;9:1311–1318. doi: 10.1038/ncb1651. [DOI] [PubMed] [Google Scholar]
- Zhu C.L., Huang Q. Overexpression of the SMYD3 promotes proliferation, migration, and invasion of pancreatic cancer. Dig. Dis. Sci. 2020;65:489–499. doi: 10.1007/s10620-019-05797-y. [DOI] [PubMed] [Google Scholar]
- Zhu H., Wei M., Xu J., Hua J., Liang C., Meng Q., Zhang Y., Liu J., Zhang B., Yu X. PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Mol. Cancer. 2020;19:49. doi: 10.1186/s12943-020-01167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J.N., Wang S.Z., Yang J.S., Luo X.G., Xie J.H., Xi T. Knockdown of SMYD3 by RNA interference down-regulates c-Met expression and inhibits cells migration and invasion induced by HGF. Cancer Lett. 2009;280:78–85. doi: 10.1016/j.canlet.2009.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In silico data analysis reported in this study were performed as detailed in Transparent Methods section using public repository (i.e., Uniprot, Reactome, cBioPortal), datasets (Colorectal adenocarcinoma, Breast invasive carcinoma, Ovarian serous cystadenocarcinoma, Pancreatic adenocarcinoma of TCGA, PanCancer Atlas), and algorithms (i.e., DISCOVER).