

Abstract
Lessons Learned
The combination of the antivascular endothelial growth factor receptor 2 monoclonal antibody, ramucirumab, and the type II MET kinase inhibitor, merestinib, is tolerable.
Preliminary efficacy data suggest that the combination may provide clinical benefit to patients with metastatic colorectal cancer (mCRC).
Further development of this combination would likely necessitate the identification of subsets of patients with mCRC where the clinical benefit is of clinical relevance.
Background
This study evaluated safety, preliminary efficacy, and pharmacokinetics of ramucirumab plus merestinib in patients with MCR previously treated with oxaliplatin and/or irinotecan.
Methods
Open‐label phase Ia/b study comprising 3+3 dose‐limiting toxicity (DLT) observation and expansion parts. Treatment was ramucirumab 8 mg/kg on days 1 and 15 and merestinib 80 mg once daily (QD; 28‐day cycle). Primary objective was safety and tolerability. Secondary objectives were pharmacokinetics and preliminary antitumor activity. Exploratory objective was biomarker associations.
Results
Safety findings: DLT (proteinuria) of 7 phase Ia patients (the expansion part started at the initial recommended dose level); 16 patients (70%) with grade ≥3 treatment‐emergent adverse events (TEAEs); 10 patients (43%) with grade ≥3 treatment‐related TEAEs. The most common grade ≥3 treatment‐related TEAEs were fatigue (4 patients [17%]) and increased blood alkaline phosphatase, diarrhea, and hypertension (2 patients each [9%]). One patient discontinued treatment because of cholestatic hepatitis. Geometric mean trough concentrations at cycle 1, day 15, were ramucirumab, 24.8 μg/mL; merestinib, 130 ng/mL. No complete or partial response was seen; 12 patients (52%) achieved stable disease. Median progression‐free survival was 3.3 months (95% confidence interval [CI]: 1.6–4.4). Median overall survival was 8.9 months (95% CI: 3.5–12.7). There were no associations between genetic alterations and efficacy.
Conclusion
Ramucirumab plus merestinib is tolerable and may have clinical benefit in biomarker‐unselected, heavily pretreated patients with mCRC.
Discussion
Therapeutic cotargeting of vascular endothelial growth factor receptor (VEGFR) and c‐MET could provide benefit in the treatment of mCRC.
Ramucirumab, an anti‐VEGFR2 monoclonal antibody, improved overall survival (OS) when added to second‐line FOLFIRI in patients with mCRC. Merestinib is an oral type II MET kinase inhibitor that can also inhibit other receptor tyrosine kinases. Recent findings from the first‐in‐human phase I study indicated that merestinib has a tolerable safety profile and potential anticancer activity as a single agent and in combination with other anticancer agents, including ramucirumab.
The primary objective of this phase Ia/b study was to evaluate the safety and tolerability of ramucirumab plus merestinib in patients with mCRC previously treated with oxaliplatin and/or irinotecan. Secondary objectives were the evaluation of pharmacokinetics and preliminary efficacy. Biomarker associations constituted the exploratory objectives. Treatment comprised ramucirumab 8 mg/kg on day 1 and 15 and merestinib 80 mg QD (28‐day cycle).
A total of 23 patients were enrolled and received one or more doses of study drug. The combination of ramucirumab and merestinib in patients was generally well tolerated (Table 1). One patient discontinued because of a treatment‐related TEAE, and no patients died from TEAEs. One patient experienced a dose‐limiting toxicity (proteinuria). The pattern and incidence of TEAEs were consistent with the expected safety profiles for ramucirumab and merestinib.
Table 1.
Treatment‐related TEAEs a
| TEAE, n (%) | n = 23 | |
|---|---|---|
| Any grade | Grade ≥3 | |
| Any TEAE | 21 (91.3) | 10 (43.5) |
| Fatigue | 7 (30.4) | 4 (17.4) |
| Headache | 7 (30.4) | 0 (0.0) |
| Blood alkaline phosphatase increased | 6 (26.1) | 2 (8.7) |
| Aspartate aminotransferase increased | 6 (26.1) | 1 (4.3) |
| Decreased appetite | 6 (26.1) | 0 (0.0) |
| Alanine aminotransferase increased | 5 (21.7) | 1 (4.3) |
| Proteinuria | 4 (17.4) | 1 (4.3) |
| Hypothyroidism | 4 (17.4) | 0 (0.0) |
| Nausea | 4 (17.4) | 0 (0.0) |
| Edema peripheral | 4 (17.4) | 0 (0.0) |
| Diarrhea | 3 (13.0) | 2 (8.7) |
| Dysphonia | 3 (13.0) | 0 (0.0) |
| Dyspnea | 3 (13.0) | 0 (0.0) |
| Pyrexia | 3 (13.0) | 0 (0.0) |
| Pruritus | 3 (13.0) | 0 (0.0) |
| Hypertension | 2 (8.7) | 2 (8.7) |
Any grade occurring in two or more patients. Presented by preferred term.
After administration of ramucirumab in combination with merestinib, ramucirumab and merestinib concentrations were consistent with corresponding monotherapy, indicating that drug‐drug interaction between these two molecules is unlikely.
Approximately 50% of patients achieved stable disease, suggesting that the combination of ramucirumab plus merestinib may have clinical benefit in this heavily pretreated population. Median progression‐free survival (PFS) was 3.3 months, and median OS was 8.9 months (Fig. 1).
Figure 1.
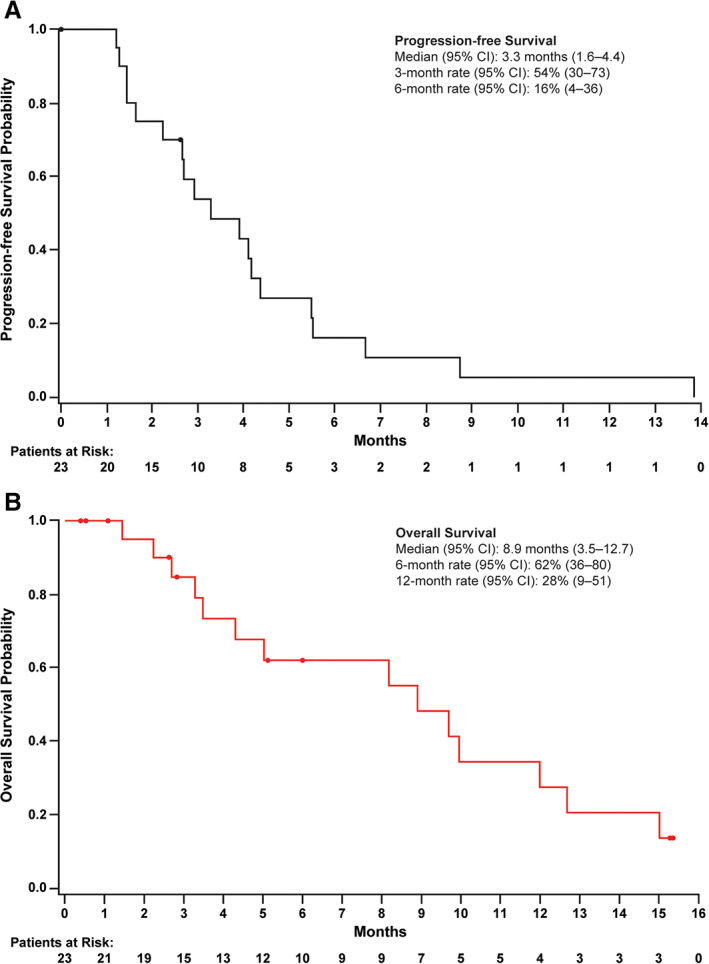
Kaplan‐Meier plots of progression‐free survival and overall survival. Circles indicate censored patients. (A): Progression‐free survival. (B): Overall survival.
There were no associations between genetic alterations and efficacy; however, there was a significant association between VEGF‐A plasma concentrations and OS.
In conclusion, we found that the combination of ramucirumab 8 mg/kg every 2 weeks plus merestinib 80 mg QD is tolerable and may provide clinical benefit in biomarker‐unselected, heavily pretreated patients with mCRC.
Trial Information
| Disease | Colorectal cancer |
| Stage of Disease/Treatment | Metastatic/advanced |
| Prior Therapy | 2 prior regimens |
| Type of Study | Phase I, 3+3 dose‐limiting toxicity (phase Ia) and expansion (phase Ib) |
| Primary Endpoint | Safety and tolerability |
| Secondary Endpoints | Pharmacokinetics, efficacy, biomarker associations (exploratory) |
| Additional Details of Endpoints or Study Design | |
| This was a phase Ia/b, multicenter (three sites across the U.S. and France), nonrandomized, open‐label study. The study comprised a phase Ia DLT observation part (for one treatment cycle) and a phase Ib expansion part. The DLT observation part followed a 3+3 design, in which if one of three patients experienced a DLT, three additional patients were to be enrolled. Patients who completed the observation part without a DLT continued study treatment until a criterion for discontinuation was met. The expansion part started when up to one of the six patients treated developed a DLT. In the expansion period, 15 additional patients were to be enrolled at the recommended dose for the expansion cohort identified in the DLT observation part. Thus, the treatment regimen during the expansion part was the same as during the DLT observation part. | |
| The main inclusion criteria were: age ≥18 years; advanced or mCRC; measurable disease at the time of study enrollment; had received prior second‐line treatment with oxaliplatin and/or irinotecan (no prior immune checkpoint inhibitors had been administered and patients with RAS wild‐type CRC must also have received prior treatment with an epidermal growth factor receptor monoclonal antibody); Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1; and adequate organ function. The main exclusion criteria were significant gastrointestinal bleeding within 3 months and significant venous thromboembolic events or any arterial thromboembolic events within 6 months prior to enrollment; uncontrolled hypertension; treatment with chronic nonsteroidal anti‐inflammatory drug or antiplatelet therapy at the time of enrollment; or other serious uncontrolled medical disorders. | |
| The primary endpoints for safety and tolerability were DLTs and TEAEs. DLTs were defined as grade 4 hematologic toxicity persisting >5 days; grade ≥3 febrile neutropenia; grade 4 thrombocytopenia (unless recovered in 24 hours and in the absence of bleeding) or grade 3 thrombocytopenia complicated with grade ≥2 bleeding; grade 3 nonhematologic toxicity occurring despite maximal supportive medical management; or any other clinically significant toxicity deemed by the investigator and the sponsor's clinical research physician to be dose limiting (such as grade 2 seizures or severe tremors). TEAEs were coded per the Medical Dictionary for Regulatory Activities version 21.1. For phase Ib, the final analysis occurred approximately 1 year after the last patient received his or her first dose of study treatment. Predetermined adverse events of special interest (AESIs) included infusion‐related reactions, hypertension, proteinuria, thromboembolic events, bleeding or hemorrhagic events, gastrointestinal perforation, reversible posterior leukoencephalopathy syndrome, congestive heart failure, fistula formation, surgery and impaired wound healing, and liver failure or liver injury. | |
| Pharmacokinetic endpoints were the minimum concentrations of ramucirumab (serum) and merestinib (plasma), which were analyzed using a validated enzyme‐linked immunosorbent assay and a validated protein precipitation method, respectively. | |
| Preliminary efficacy endpoints included overall response rate (proportion of patients who achieved a complete response [CR] or partial response [PR] as their best overall response) and PFS. OS was also evaluated. Scans for restaging were performed every 6 weeks (±7 days) for the first 6 months after enrollment and every 9 weeks (±7 days) thereafter until radiographic disease progression, death, or study completion, whichever occurred first. | |
| Exploratory endpoints included PFS and OS by biomarker status. Plasma collected at baseline was evaluated for circulating cell‐free tumor DNA using the Archer® Reveal ctDNA™ 28‐gene next generation sequencing assay (ArcherDX, Inc., Boulder, CO), which evaluates 28 genes including HRAS (exons 2, 3), KRAS (exons 2, 3, 4), NRAS (exons 2, 3), and TP53 (all exons). Samples were classified as HRAS, KRAS, NRAS, or TP53 mutation positive if the analysis identified genetic variants that are confirmed somatic/germline mutations in publications annotated in COSMIC [1] or predicted somatic/germline mutations via the FATHMM algorithm [2]; samples were classified as negative if analysis identified no variants or only variants of unknown significance. VEGF‐A, VEGF‐D, and soluble VEGFR3 (sVEGFR3) concentrations were evaluated as described previously [3] in plasma samples collected at baseline. | |
| Safety analyses included all patients who received one or more doses of ramucirumab and/or merestinib. Efficacy analyses included all patients who received any quantity of study treatment (intent‐to‐treat analysis). Pharmacokinetic analyses included all patients who received one or more doses of ramucirumab and merestinib and had baseline and one or more postbaseline evaluable pharmacokinetic samples. PFS and OS were estimated by Kaplan‐Meier methodology. Cox regression analysis was performed to evaluate the association between response outcomes and biomarkers (treated as binary outcomes; mutation positive vs. negative for genetic markers and high vs. low for VEGF‐A, VEGF‐D, and sVEGFR3 [dichotomized at the median concentration for each marker]). Multiplicity adjustment was applied, adjusted for all biomarkers. The data cutoff date was November 5, 2018. Statistical analyses were performed using SAS software (version 9.4; SAS Institute, Cary, NC). | |
| Investigator's Analysis | Drug tolerable, hints of efficacy |
Drug Information
| Drug 1 | |
| Generic/Working Name | Merestinib |
| Trade Name | NA |
| Company Name | Eli Lilly and Company |
| Drug Type | Multikinase inhibitor |
| Drug Class | Small molecule |
| Dose | 80 mg |
| Route | oral (po) |
| Schedule of Administration | Continuous QD |
| Drug 2 | |
| Generic/Working Name | Ramucirumab |
| Trade Name | Cyramza® |
| Company Name | Eli Lilly and Company |
| Drug Type | VEGFR2 inhibitor |
| Drug Class | Antibody |
| Dose | 8 mg/kg |
| Route | IV |
| Schedule of Administration | Day 1 and day 15 over a 28‐day cycle |
Patient Characteristics
| Number of Patients, Male | 13 |
| Number of Patients, Female | 10 |
| Stage | All metastatic/advanced |
| Age | Median (range): 59 (18–79) years |
| Number of Prior Systemic Therapies | Median (range): 4 (1–10) |
| Performance Status: ECOG |
0 — 4 1 — 19 2 — 3 — Unknown — |
Detailed Patient Characteristics (n = 23)
| Male sex, n (%) | 13 (56.5) |
| Age, median (range), yr | 59 (18–79) |
| Age group, n (%) | |
| <65 yr | 14 (60.9) |
| ≥65 yr | 9 (39.1) |
| Race, n (%) | |
| Black | 5 (21.7) |
| White | 18 (78.3) |
| ECOG Performance Status, n (%) | |
| 0 | 4 (17.4) |
| 1 | 19 (82.6) |
| Primary tumor site, n | |
| Colon | 14 (60.9) |
| Rectum | 8 (34.8) |
| Unknown | 1 (4.3) |
| Primary tumor side, n (%) | |
| Left | 17 (73.9) |
| Right | 5 (21.7) |
| Unknown | 1 (4.3) |
| Prior surgery, n (%) | 22 (95.7) |
| Prior radiotherapy, n (%) | 8 (34.8) |
| Prior systemic therapy, n (%) | 23 (100) |
| Neoadjuvant | 6 (26.1) |
| Adjuvant | 15 (65.2) |
| Locally advanced setting | 1 (4.3) |
| Metastatic setting | 23 (100) |
| 1 regimen | 1 (4.3) |
| 2 regimens | 3 (13.0) |
| ≥3 regimens | 19 (82.6) |
| Cancer Types or Histologic Subtypes | Colon, 14; rectum, 8; unknown, 1 |
| Outcome Notes | |
| A total of 23 patients were enrolled (7 patients in phase Ia, 16 patients in phase Ib) and received at least one dose of study drug. Of these patients, 16 (70%) discontinued study treatment because of progressive disease (PD). Other causes for discontinuation of study treatment were physician decision (3 patients [13%]; 1 due to clinical decline, 1 due to cancer worsening, and 1 due to nonradiologic progression), withdrawal by patient (2 patients [9%]), TEAE (1 patient [4%], grade 4 cholestatic hepatitis), and death due to study disease (1 patient [4%]). The median (range) OS follow‐up time was 15.3 months (0.4–15.3). All patients had received prior systemic therapy in the metastatic setting, with a median (range) of 4 (1–10) prior regimens. The most common (received by ≥7 patients) prior systemic anticancer therapies in the metastatic setting were irinotecan (22 patients [96%]), 5 FU/capecitabine (22 patients [96%]), bevacizumab (18 patients [78%]), oxaliplatin (17 patients [74%]), cetuximab/panitumumab (13 patients [57%]), regorafenib (8 patients [35%]), and TAS 102 (7 patients [30%]). Exposure and dose modifications are summarized in Table 2. One patient had a ramucirumab dose reduction due to a TEAE of weight decreased. One patient each had a merestinib dose reduction due to TEAEs of blood bilirubin increased and fatigue, respectively. No patients underwent a postdiscontinuation surgical procedure. One patient (4.3%) received radiotherapy and four patients (17.4%) received a postdiscontinuation systemic therapy with investigational antineoplastic agents. | |
Table 2.
Exposure and dose modifications
| Parameter | Ramucirumab, n = 23 | Merestinib, n = 23 |
|---|---|---|
| Exposure, median (range) | ||
| Duration of therapy, wk | 10.0 (2.0–60.7) | 12.0 (4.0–60.7) |
| Number of cycles | 3.0 (1.0–15.0) | 3.0 (1.0–15.0) |
| Relative dose intensity, % | 99.4 (73.6–103.1) | 84.8 (50.0–100.0) |
| Dose modifications | ||
| Dose reduction, n (%) | 1 (4.3) | 2 (8.7) |
| Dose delay, n (%) | 12 (52.2) | NA |
| Dose omission, n (%) | 4 (17.4) | 14 (60.9) |
| Dose omission length, a % | 9.4 | 10.3 |
Percentage of dose holding time with respect to total time on treatment.
Abbreviation: NA, not applicable.
Primary Assessment Method
| Title | Safety and tolerability |
| Number of Patients Screened | 29 |
| Number of Patients Enrolled | 23 |
| Number of Patients Evaluable for Toxicity | 23 |
| Number of Patients Evaluated for Efficacy | 23 |
| Evaluation Method | RECIST 1.1 |
Secondary Assessment Method
| Title | Efficacy |
| Number of Patients Screened | 29 |
| Number of Patients Enrolled | 23 |
| Number of Patients Evaluable for Toxicity | 23 |
| Number of Patients Evaluated for Efficacy | 23 |
| Evaluation Method | RECIST 1.1 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 0 (0%) |
| Response Assessment SD | n = 12 (52%) |
| Response Assessment PD | n = 6 (26%) |
| Response Assessment Nonevaluable | n = 5 (22%) |
| (Median) Duration Assessments PFS | 3.3 months, 95% CI: 1.6–4.4 |
| (Median) Duration Assessments OS | 8.9 months, 95% CI: 3.5–12.7 |
| Outcome Notes | |
| Five patients (22%) were nonevaluable for response, because of either lack of postbaseline tumor assessment (4 patients) or discontinuation due to nonradiographic progression. The overall disease control (DC) rate was 52% (95% CI: 31–73). The median duration of DC was 5.5 months (95% CI: 2.9–6.7). Figure 2 summarizes the best percentage change from baseline in tumor size for each patient. Five patients experienced tumor shrinkage and no patient experienced tumor shrinkage ≥30%. | |
Figure 2.
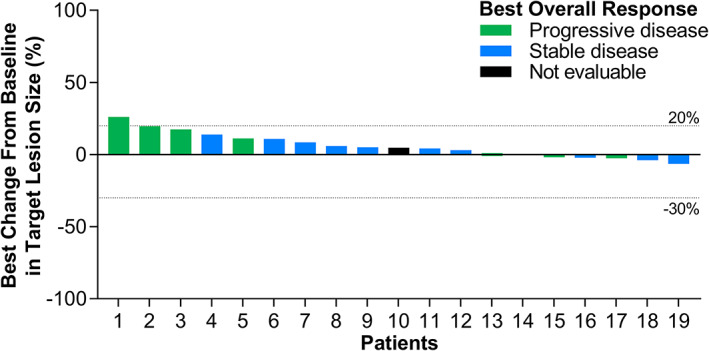
Waterfall plot of best percentage change from baseline in tumor size.
Serious Adverse Events
| Name | Grade | Attribution |
|---|---|---|
| Abdominal pain | 3 | Unrelated |
| Blood bilirubin increased | 3 | Unrelated |
| Hemorrhoidal hemorrhage | 2 | Unrelated |
| Cholestatic hepatitis | 3 | Definite |
| Pulmonary embolism | 3 | Unrelated |
| Small intestinal obstruction | 3 | Unrelated |
| Small intestinal obstruction | 3 | Unrelated |
| Small intestinal obstruction | 2 | Unrelated |
| Upper gastrointestinal hemorrhage | 3 | Unrelated |
| Adverse Events Notes | |
| TEAEs and grade ≥3 TEAEs were reported by 23 (100%) and 16 (70%) patients, respectively (Table 3). No patients died because of a TEAE. A total of six patients (26%) experienced hypothyroidism; all events were grade 1 or 2 (3 patients each). These patients were treated with levothyroxine, and none discontinued study treatment or had a dose reduction due to hypothyroidism. A review of available thyroid stimulating hormone and thyroxine laboratory data from patients did not indicate any pattern associated with study drug treatment. Treatment‐related TEAEs (per investigator assessment) and grade ≥3 treatment‐related TEAEs were reported by 21 (91%) and 10 (43%) patients, respectively (Table 1). The most commonly reported treatment‐related TEAEs were fatigue; headache; increased blood alkaline phosphatase, aspartate aminotransferase, and alanine aminotransferase; and decreased appetite. Fatigue was the most commonly reported grade ≥3 treatment‐related TEAE (4 patients [17%]). The only serious adverse event considered related to the study treatment was grade 3 cholestatic hepatitis in cycle 3. The event occurred 7 days after the last dose of ramucirumab and on the same day as the last scheduled dose of merestinib, which was omitted. This patient discontinued study treatment after the event worsened to grade 4. No other patients discontinued study treatment due to a TEAE. A total of 20 patients (87%) experienced AESIs for ramucirumab, of whom 10 (43%) had grade ≥3 AESIs (Table 4). AESIs experienced by ≥3 patients included liver injury or failure events (liver function abnormalities and other hepatic events), bleeding or hemorrhagic events, proteinuria, hypertension, and thromboembolic events. Fourteen patients (61%) experienced liver function abnormalities, of whom five experienced grade 3 or 4 events. Of these patients, 11 had liver metastases at baseline, and 2 developed liver metastases at progression. The majority of liver events were reversible laboratory abnormalities of mild or moderate severity. Five patients (22%) experienced a bleeding or hemorrhagic event. There were no events of gastrointestinal perforation, reversible posterior leukoencephalopathy syndrome, fistula formation, or surgery and impaired wound healing. |
Table 3.
Study treatment‐emergent adverse events a
| TEAE, n (%) | n = 23 | |
|---|---|---|
| Any grade | Grade ≥3 | |
| Any TEAE | 23 (100.0) | 16 (69.6) |
| Blood alkaline phosphatase increased | 11 (47.8) | 5 (21.7) |
| Fatigue | 10 (43.5) | 5 (21.7) |
| Aspartate aminotransferase increased | 10 (43.5) | 3 (13.0) |
| Headache | 9 (39.1) | 0 |
| Decreased appetite | 9 (39.1) | 0 |
| Alanine aminotransferase increased | 7 (30.4) | 2 (8.7) |
| Abdominal pain b | 6 (26.1) | 1 (4.3) |
| Edema peripheral | 6 (26.1) | 0 |
| Constipation | 6 (26.1) | 0 |
| Hypothyroidism | 6 (26.1) | 0 |
| Hyperbilirubinemia | 5 (21.7) | 2 (8.7) |
| Nausea | 5 (21.7) | 0 |
| Rash c | 5 (21.7) | 0 |
| Proteinuria | 4 (17.4) | 1 (4.3) |
| Dyspnea | 4 (17.4) | 0 |
| Diarrhea | 3 (13.0) | 2 (8.7) |
| Small intestinal obstruction | 3 (13.0) | 2 (8.7) |
| Gamma‐glutamyl transferase increased | 3 (13.0) | 2 (8.7) |
| Hypertension | 3 (13.0) | 2 (8.7) |
| Blood creatine phosphokinase increased | 3 (13.0) | 1 (4.3) |
| Dysphonia | 3 (13.0) | 0 (0.0) |
| Pruritus | 3 (13.0) | 0 (0.0) |
| Pyrexia | 3 (13.0) | 0 (0.0) |
| Somnolence | 3 (13.0) | 0 (0.0) |
| Vomiting | 3 (13.0) | 0 (0.0) |
| Supraventricular tachycardia | 2 (8.7) | 2 (8.7) |
Any grade occurring in two or more patients. Presented by preferred term.
Consolidated term including abdominal pain (4 patients), abdominal pain upper (1 patient), and hepatic pain (1 patient).
Consolidated term including maculopapular rash (3 patients), dermatitis (1 patient), and follicular rash (1 patient).
Abbreviation: TEAE, treatment‐emergent adverse event.
Table 4.
Adverse events of special interest
| AESI a, n (%) | n = 23 | |
|---|---|---|
| Any grade | Grade ≥3 | |
| Any AESI | 20 (87.0) | 10 (43.5) |
| Alanine aminotransferase increased | 9 (39.1) | 2 (8.7) |
| Blood bilirubin increased | 5 (21.7) | 2 (8.7) |
| Proteinuria | 5 (21.7) | 1 (4.3) |
| Gamma‐glutamyl transferase increased | 4 (17.4) | 1 (4.3) |
| Hypertension | 3 (13.0) | 2 (8.7) |
| Blood creatinine increased | 2 (8.7) | 0 |
| Hematuria | 2 (8.7) | 0 |
| Hypoalbuminemia | 2 (8.7) | 0 |
| Pulmonary embolism | 2 (8.7) | 2 (8.7) |
| Ascites | 1 (4.3) | 1 (4.3) |
| Bilirubin conjugated increased | 1 (4.3) | 1 (4.3) |
| Cholestatic hepatitis | 1 (4.3) | 1 (4.3) |
| Ejection fraction decreased | 1 (4.3) | 1 (4.3) |
| Glomerular filtration rate decreased | 1 (4.3) | 0 |
| Hemorrhoidal hemorrhage | 1 (4.3) | 0 |
| Hepatic pain | 1 (4.3) | 0 |
| Jaundice | 1 (4.3) | 0 |
| Upper gastrointestinal hemorrhage | 1 (4.3) | 1 (4.3) |
| Vaginal hemorrhage | 1 (4.3) | 0 |
| Venous thrombosis | 1 (4.3) | 1 (4.3) |
Presented by preferred term.
Abbreviation: AESI, adverse event of special interest.
Dose‐Limiting Toxicities
| Dose level | Number enrolled | Number evaluable for toxicity | Number with a DLT | Dose‐limiting toxicity information |
|---|---|---|---|---|
| Ramucirumab 8 mg/kg on days 1 and 15 and merestinib 80 mg QD (28‐day cycle) | 7 | 7 | 1 | One male patient, aged 47 years, had a DLT of grade 3 proteinuria. The patient received ramucirumab on cycle 1, days 1 and 15 and merestinib from day 1 to 28 as per the recommended dosing schedule for phase Ia. On day 15, the patient experienced grade 2 treatment‐related proteinuria. By day 27, the event worsened to grade 3 proteinuria. On day 29, ramucirumab was permanently discontinued and merestinib dosing was halted for the following 2 weeks. On day 43, merestinib dosing resumed at the same dose level. Grade 3 proteinuria persisted until day 71, after which the severity of the TEAE decreased to grade 1 until merestinib discontinuation on day 85 due to PD. |
Pharmacokinetics/Pharmacodynamics
| Day 15 of cycle 1 (n = 17) | Geometric mean trough concentration (% coefficient of variation) |
|---|---|
| Ramucirumab | 24.8 μg/mL (53%) |
| Merestinib | 130 ng/mL (88%) |
Assessment, Analysis, and Discussion
| Completion | Study completed |
| Investigator's Assessment | Drug tolerable, hints of efficacy |
Despite major advances in systemic chemotherapy, metastatic colorectal cancer (mCRC), which composes approximately 20% of all colorectal cancer (CRC) at presentation, is associated with a high rate of mortality (the 5‐year survival rate in the U.S. is 14%) [4]. Further optimization of treatment strategies is needed to improve patient survival.
Therapeutic cotargeting of vascular endothelial growth factor receptor (VEGFR) and c‐MET may provide benefit in the treatment of mCRC. Evidence to support such an approach includes the demonstration of cross talk between hepatocyte growth factor/c‐MET signaling and VEGFR in endothelial cells mediating angiogenesis [5], which may play a role in CRC progression. In addition, c‐MET has been implicated in mediating resistance to antiangiogenic therapy [6]. Specifically, an increase in tumor hypoxia (due to an antiangiogenic‐associated decrease in tumor vascularization) has been shown to increase c‐MET expression and, consequently, metastatic potential [7]. Finally, overexpression of the c‐MET protein is common in CRC, occurring in approximately 60% of cases [8], and is associated with advanced tumor stages and poor clinical outcomes [8, 9].
Ramucirumab is approved as a second‐line treatment for mCRC. Merestinib is an oral MET kinase inhibitor that can also inhibit oncokinases, including AXL, RON, MERTK, TYRO3, ROS1, FLT3, MKNK1/2, DDR1/2, and NTRK 1/2/3 [10, 11]. Multiple merestinib targets are potentially actionable in the treatment of CRC. Preclinical studies have shown that merestinib retains inhibitory activity against MET‐activating mutations and that merestinib displays in vivo antitumor effects [10]. More recently, findings from the first‐in‐human phase I study of merestinib indicate that merestinib has a tolerable safety profile and potential anticancer activity at doses of 120 mg once daily (QD) as a single agent and 80 mg QD in combination with other anticancer agents, including ramucirumab [12].
This phase Ia/b study is the first to evaluate the combination of ramucirumab plus merestinib in patients with previously treated mCRC. We found that this combination was generally well tolerated; no patients died because of treatment‐emergent adverse events (TEAEs), and only one patient discontinued because of a treatment‐related TEAE. Furthermore, only one patient experienced a dose‐limiting toxicity, with the recommended dose for the expansion cohort being the initial recommended dose of ramucirumab 8 mg/kg on day 1 and day 15 with merestinib 80 mg QD. The most commonly reported treatment‐related TEAEs (fatigue, headache, increased blood alkaline phosphatase, increased aspartate aminotransferase, decreased appetite, and increased alanine aminotransferase) have been reported in previous studies with either ramucirumab [13] or merestinib [12], and the pattern and incidence of adverse events of special interest (AESIs) observed were consistent with the predetermined AESIs of ramucirumab. Several low‐grade hypothyroidism events were observed, and approximately 40% of patients experienced grade 3 treatment‐related TEAEs. Overall, the pattern and incidence of TEAEs observed are consistent with the expected individual safety profiles for ramucirumab [13, 14, 15] and merestinib [12].
Following administration of ramucirumab 8 mg/kg every 2 weeks in combination with merestinib 80 mg QD, ramucirumab and merestinib plasma concentrations were consistent with corresponding monotherapy [16], indicating that drug‐drug interaction between these 2 molecules is unlikely.
Overall, results from the efficacy analysis, whereby approximately 50% of patients achieved stable disease, suggest that the combination of ramucirumab plus merestinib may have clinical benefit in this heavily pretreated mCRC population. Of note, median progression‐free survival was 3.3 months and median overall survival (OS) was 8.9 months. These values compare well with data in the literature from similar settings [17, 18]; however, our efficacy evaluations are limited by the small number of patients and the lack of a control group, which make it difficult to draw definitive conclusions regarding the magnitude of effect and whether the effect is due to the combination of treatments or just one of the components.
Biomarker findings for each patient are summarized in Figure 3. There were no significant associations between genetic alterations detected at baseline from ctDNA in RAS (KRAS and NRAS; there were no alterations detected for HRAS) and/or TP53, baseline plasma VEGF‐D concentrations, or plasma sVEGFR3 concentrations and efficacy (Table 5). VEGF‐A plasma levels below the median were significantly associated with longer OS (Table 5; hazard ratio, 17.0; p = .006). This finding of longer OS in patients with lower plasma VEGF‐A concentrations (Figure 4) is consistent with a previous report that VEGF‐A is a negative prognostic factor in patients with CRC [19]. The biomarker analyses are limited by the small sample size and the lack of tissue sample analysis.
Figure 3.
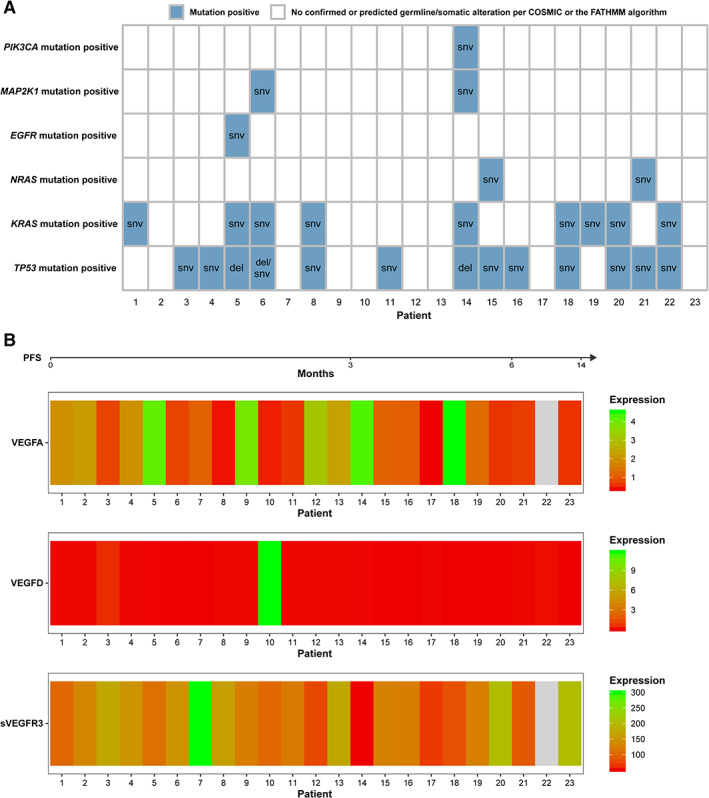
Summary of biomarker findings for each patient. (A): genetic mutations and (B) VEGF expression. For (A), the text in cells shows the specific genetic variant type. For (B), missing expression value is denoted in gray (all units are ng/mL). In both parts of the figure, patients are ordered by ascending PFS. PFS measurement unit is months. All 23 patients had ctDNA and VEGF‐D data available; 22 of 23 patients had VEGF‐A and sVEGFR3 data available.
Abbreviations: del, deletion; PFS, progression‐free survival; snv, single nucleotide variant; sVEGFR3, soluble vascular endothelial growth factor receptor 3; VEGF, vascular endothelial growth factor.
Table 5.
Summary of associations between response outcomes and biomarkers
| Marker | Median concentration (pg/mL) | Hazard ratio a (95% CI) | p‐value | Adjusted p‐value |
|---|---|---|---|---|
| Progression‐free survival | ||||
| TP53 mutation | NA | 1.29 (0.48–3.45) | .613 | 1 |
| RAS mutation | NA | .78 (0.3–2.04) | .619 | 1 |
| RAS and TP53 mutation b | NA | .88 (0.34–2.25) | .786 | 1 |
| VEGF‐A | 1.118 | 2.08 (0.73–5.92) | .166 | .996 |
| VEGF‐D | .043 | .97 (0.38–2.5) | .951 | 1 |
| sVEGFR3 | 126.075 | .92 (0.35–2.43) | .865 | 1 |
| Overall survival | ||||
| TP53 mutation | NA | .75 (0.26–2.18) | .598 | 1 |
| RAS mutation | NA | 1.26 (0.43–3.71) | .671 | 1 |
| RAS and TP53 mutation b | NA | 1.11 (0.38–3.26) | .850 | 1 |
| VEGF‐A | 1.118 | 16.95 (2.04–141.22) | .001 | .006 |
| VEGF‐D | .043 | 1.11 (0.39–3.2) | .843 | 1 |
| sVEGFR3 | 126.075 | 1.22 (0.4–3.7) | .728 | 1 |
The reference group for mutation status is the “mutation negative” group and the reference group for the VEGF biomarkers is the “low concentration” group.
Comparison of patients with concurrent RAS and TP53 mutations versus patients with only RAS mutations, only TP53 mutations, or no mutations.
Abbreviations: CI, confidence interval; NA, not applicable; RAS, KRAS or NRAS; sVEGFR3, soluble vascular endothelial growth factor receptor 3; VEGF, vascular endothelial growth factor.
Figure 4.
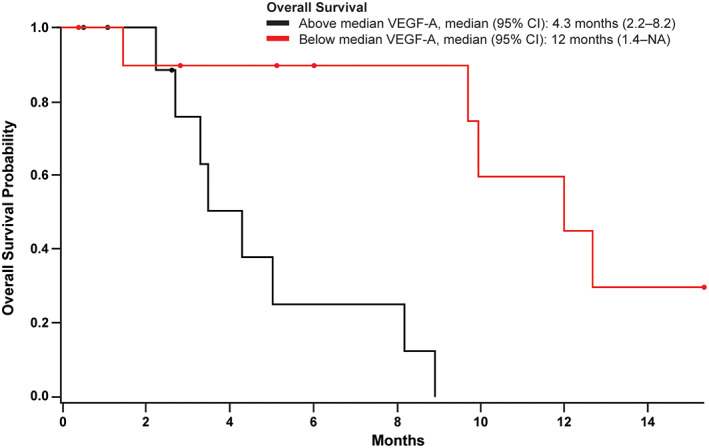
Kaplan‐Meier plot of overall survival by VEGF‐A concentration (above or below the median concentration). Circles indicate censored patients.
Abbreviations: CI, confidence interval; NA, not applicable; VEGF, vascular endothelial growth factor.
We found that the combination of ramucirumab 8 mg/kg every 2 weeks plus merestinib 80 mg QD is tolerable and may have clinical benefit in biomarker‐unselected, heavily pretreated patients with mCRC. However, considering the current development landscape, which is highly competitive for resources, these findings may not be sufficiently promising to warrant further development of the combination in unselected patients.
Disclosures
Mansoor Saleh: Aileron Therapeutics, Bristol‐Myers Squibb, Eli Lilly and Company, Genentech, Gilead Sciences, Incyte, Merck, Novartis, Sanofi, TRACON Pharma (RF, H); Philippe A. Cassier: AbbVie, Amgen, AstraZeneca, Bayer, Blueprint Medicines, Bristol‐Myers Squibb, Celgene, Eli Lilly and Company, GlaxoSmithKline, Innate Pharma, Janssen, Loxo, Merck Serono, Merck Sharp & Dohme, Novartis, Plexxikon, Roche/Genentech, Taiho Pharmaceutical, Toray Industries, Transgene (RF, H, travel/accommodation expenses); Van K. Morris: Array BioPharma, Bristol‐Myers Squibb, EMD Serono (C/A, RF), Shubham Pant: Arqule, Bristol‐Myers Squibb, Eli Lilly and Company, Five Prime Therapeutics, GlaxoSmithKline, Mirati Therapeutics, Novartis, Onco Response, Red Hill Biopharma, Rgenix, Sanofi‐Aventis, Sanofi US Services Inc., Xencor (RF, H); Ling Gao: Eli Lilly and Company (E, OI); Amanda Long: Eli Lilly and Company (E, OI); Huzhang Mao: Eli Lilly and Company (E, OI); Samuel McNeely: Eli Lilly and Company (E, OI); Erin K. Wagner: Eli Lilly and Company (C/A), BioStat Soutions Inc. (E); Roberto M. Carlesi: Eli Lilly and Company (former E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Tables and Figures
Acknowledgments
These findings were presented in part at the ESMO World Congress on Gastrointestinal Cancer, Barcelona, Spain, 3–6 July 2019. Medical writing assistance was provided by Luke Carey, PhD, CMPP, of ProScribe – Envision Pharma Group, and was funded by Eli Lilly and Company. ProScribe's services complied with international guidelines for Good Publication Practice (GPP3). The authors would like to thank all study participants and Sylwia Szymczak (Eli Lilly and Company) for support with data collection.
Data sharing statement: Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the U.S. and E.U. and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Footnotes
- ClinicalTrials.gov Identifier: NCT02745769
- Sponsor: Eli Lilly and Company
- Principal Investigator: Mansoor Saleh
- IRB Approved: Yes
References
- 1.Catalogue of Somatic Mutations in Cancer (COSMIC). Mutation COSV53361901. Available at https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=94488854. Accessed January 16, 2020.
- 2.Functional Analysis through Hidden Markov Models (fathmm). Available at http://fathmm.biocompute.org.uk/. Accessed January 16, 2020.
- 3. Fuchs CS, Shitara K, Di Bartolomeo M et al. Ramucirumab with cisplatin and fluoropyrimidine as first‐line therapy in patients with metastatic gastric or junctional adenocarcinoma (RAINFALL): A double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol 2019;20:420–435. [DOI] [PubMed] [Google Scholar]
- 4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 5. Sulpice E, Ding S, Muscatelli‐Groux B et al. Cross‐talk between the VEGF‐A and HGF signalling pathways in endothelial cells. Biol Cell 2009;101:525–539. [DOI] [PubMed] [Google Scholar]
- 6. Safaie Qamsari E, Safaei Ghaderi S, Zarei B et al. The c‐Met receptor: Implication for targeted therapies in colorectal cancer. Tumour Biol 2017;39:1010428317699118. [DOI] [PubMed] [Google Scholar]
- 7. Pennacchietti S, Michieli P, Galluzzo M et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003;3:347–361. [DOI] [PubMed] [Google Scholar]
- 8. Liu Y, Yu XF, Zou J et al. Prognostic value of c‐Met in colorectal cancer: A meta‐analysis. World J Gastroenterol 2015;21:3706–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Oliveira AT, Matos D, Logullo AF et al. MET is highly expressed in advanced stages of colorectal cancer and indicates worse prognosis and mortality. Anticancer Res 2009;29:4807–4811. [PubMed] [Google Scholar]
- 10. Yan SB, Peek VL, Ajamie R et al. LY2801653 is an orally bioavailable multi‐kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti‐tumor activities in mouse xenograft models. Invest New Drugs 2013;31:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Konicek BW, Capen AR, Credille KM et al. Merestinib (LY2801653) inhibits neurotrophic receptor kinase (NTRK) and suppresses growth of NTRK fusion bearing tumors. Oncotarget 2018;9:13796–13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He AR, Cohen RB, Denlinger CS et al. First‐in‐human phase I study of merestinib, an oral multikinase inhibitor, in patients with advanced cancer. The Oncologist 2019;24:e930–e942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fuchs CS, Tomasek J, Yong CJ et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro‐oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo‐controlled, phase 3 trial. Lancet 2014;383:31–39. [DOI] [PubMed] [Google Scholar]
- 14. Arnold D, Fuchs CS, Tabernero J et al. Meta‐analysis of individual patient safety data from six randomized, placebo‐controlled trials with the antiangiogenic VEGFR2‐binding monoclonal antibody ramucirumab. Ann Oncol 2017;28:2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noguerido A, Mulet‐Margalef N, Matos I et al. The safety of ramucirumab for the treatment of colorectal cancer. Expert Opin Drug Saf 2018;17:945–951. [DOI] [PubMed] [Google Scholar]
- 16. Ueda S, Satoh T, Gotoh M et al. A phase Ib study of safety and pharmacokinetics of ramucirumab in combination with paclitaxel in patients with advanced gastric adenocarcinomas. The Oncologist 2015;20:493–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grothey A, Van Cutsem E, Sobrero A et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013;381:303–312. [DOI] [PubMed] [Google Scholar]
- 18. Mayer RJ, Van Cutsem E, Falcone A et al. Randomized trial of TAS‐102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–1919. [DOI] [PubMed] [Google Scholar]
- 19. Hegde PS, Jubb AM, Chen D et al. Predictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumab. Clin Cancer Res 2013;19:929–937. [DOI] [PubMed] [Google Scholar]