

Abstract
Dysregulated fibroblast growth factor receptor (FGFR) signaling is associated with several cancers, including urothelial carcinoma. Preclinical studies with FGFR inhibitors have shown significant antitumor activity, which has led to clinical evaluation of multiple FGFR inhibitors. Recently, erdafitinib was approved by the U.S. Food and Drug Administration for advanced urothelial carcinoma with FGFR gene alterations as the first molecularly targeted therapy. Additional ongoing clinical trials with other types of FGFR inhibitors have shown encouraging results. This review summarizes the oncogenic signaling of FGFR alterations, completed and ongoing clinical trials of FGFR inhibitors, and resistance patterns.
Implications for Practice
Dysregulated fibroblast growth factor receptor (FGFR) signaling is associated with several cancers, including urothelial carcinoma. Preclinical studies with FGFR inhibitors have shown significant antitumor activity, which has led to clinical evaluation of multiple FGFR inhibitors. Most recently, erdafitinib was approved by the U.S. Food and Drug Administration for advanced urothelial carcinoma with FGFR gene alterations as the first molecularly targeted therapy. Additional ongoing clinical trials with other types of FGFR inhibitors have shown encouraging results. This review summarizes the oncogenic signaling of FGFR alterations, completed and ongoing clinical trials of FGFR inhibitors, and resistance patterns.
Keywords: Urothelial cancer, FGFR alterations, FGFR inhibitors, Erdafitinib, Targeted therapy
Short abstract
Focusing on urothelial malignancies, this review summarizes the oncogenic signaling of FGFR alterations, clinical trials of FGFR inhibitors, and resistance patterns.
Introduction
In the U.S., bladder cancer is the fourth most common malignancy in men, and urothelial histology is the most common subtype [1]. Platinum‐based chemotherapy and checkpoint inhibitors are standard palliative treatment options for metastatic disease. Despite clinical responses with these agents, outcomes are poor. Genomic alterations with constitutive activation of fibroblast growth factor receptors (FGFRs) leading to carcinogenesis are seen in about 20% of patients with advanced urothelial cancer; thus, the exploration of novel targeted therapeutics [2].
FGFRs are a group of transmembrane receptor tyrosine kinases that bind to fibroblast growth factor (FGF) ligands and are involved in cell proliferation, differentiation, and growth, as well as migration and selective apoptosis during embryogenesis and angiogenesis (Fig. 1) [3]. These receptors share similar phylogenetical structures with vascular endothelial growth factor receptors (VEGFRs) and platelet‐derived growth factor receptors [4, 5]. Four distinct transmembrane receptors (FGFR1–4) constitute the FGFR family, and a fifth receptor (FGFRL1) with no intracellular tyrosine kinase activity also binds to FGFs [6]. FGFRL1 is known to promote cell differentiation and has a negative effect on cell proliferation [7].
Figure 1.
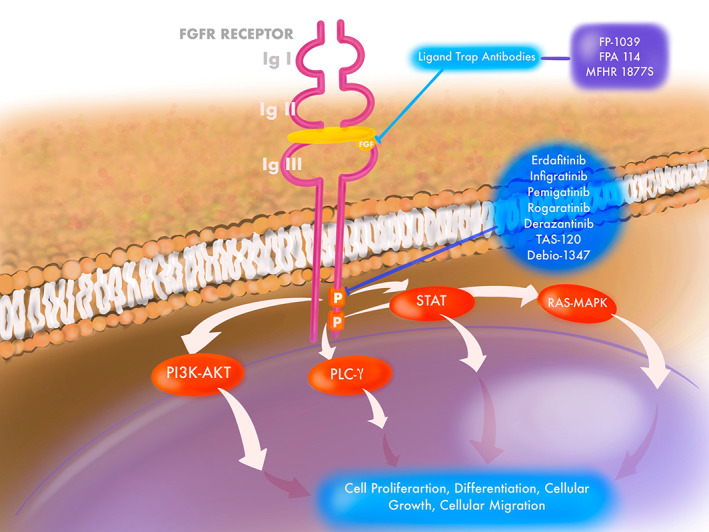
FGFR receptor and signaling with targets of FGFR inhibitors.Abbreviation: FGFR, fibroblast growth factor receptor.
The structure of FGFR is composed of three distinct domains: an extracellular domain, a single‐pass transmembrane domain, and an intracellular tyrosine kinase domain [8]. The extracellular domain contains the FGF‐binding site and has three immunoglobulin (Ig)‐like domains, designated D1–D3 (D2–D3 forms the ligand‐binding domain), and an acid box that connects D1 and D2. A single‐pass transmembrane protein facilitates signal transduction to the intracellular tyrosine kinase domain. Binding of FGF to the FGFR leads to receptor dimerization and autophosphorylation of the tyrosine residues and downstream activation of multiple transduction pathways that include RAS‐dependent mitogen‐activated protein kinase (MAPK) signaling, PI3K/AKT, PLC‐γ, and STAT cascade [8]. Four different genes encode the FGFR family, and because of alternative splicing, a spectrum of isoforms that differ in ligand‐binding specificity have been identified.
Carcinogenic FGF‐FGFR Signaling
Dysregulated FGFR signaling with activation of downstream signal transduction pathways can occur via FGFR gene alterations, increased FGF, leading to autocrine and paracrine signaling, and epithelial‐mesenchymal transition [9]. This dysregulated activation of FGFR leads to oncogenesis and cancer progression [10]. The common genomic alterations leading to FGFR activation include point mutations, gene amplification, gene rearrangements, and fusions [9].
In urothelial carcinoma, FGFR3 alterations have been documented in nearly 60% of low‐grade noninvasive papillary urothelial carcinoma of the bladder [11] and 26.7% of overall urothelial carcinoma [12]. The incidence in upper tract high‐grade urothelial cancer is 35.6% [13]. Of the FGFR3 alterations seen in patients with urothelial cancer, base substitutions are most common (84%) [14]. S249C, the most common mutation in bladder urothelial cancer, affects the extracellular domain, stabilizing receptor dimerization, and eventually leads to downstream signal transduction [11, 15]. Other notable mutations commonly observed are R248C, Y375C, G372C, and N542S [16, 17, 18]. FGFR3‐FGFR3‐TACC3 fusion is a common fusion. This fusion protein is formed by tandem duplication on chromosome 4, resulting in fusion of the FGFR3 gene with transforming acidic coiled‐coil containing protein 3 (TACC3). Other fusion proteins are FGFR3‐TNIP2, FGFR1‐NTM, and FGFR3‐JAKMIP1. FGFR1, FGFR2, and FGFR4 are also altered, to a lesser degree, in urothelial carcinoma, where copy number variation and point mutation are the usual forms of alteration.
The presence of these carcinogenic, activating gene alterations provide a novel therapeutic opportunity for the evaluation of selective FGFR inhibitors in urothelial malignancies (Table 1).
Table 1.
Summary of FGFR inhibitors with results in urothelial cancer
| Agent(s) and dose | Mechanism of action | Study design | Biomarker and cohort | Response rate | Median OS | Median PFS | Most common toxicity (any grade) |
|---|---|---|---|---|---|---|---|
| Infigratinib (BGJ398); 125 mg/d, 3 wk on/1 wk off | Oral, selective FGFR 1–3 tyrosine kinase inhibitor | Phase I, single arm, open‐label, n = 67 | FGFR3 alteration; metastatic urothelial cancer after 1 or 2 lines of therapy | CR + PR: 25.4%; PD: 23.9%; SD: 50% | 7.75 mo | 3.75 mo | Hyperphosphatemia, elevated creatinine, fatigue, constipation, anemia, decreased appetite |
| Erdafitinib; 8 mg daily with option of dose escalation to 9 mg | Oral, selective, tyrosine kinase inhibitor of FGFR1–4 | Phase II, open‐label, single arm, n = 99 | Susceptible FGFR3 mutation or FGFR2/3 fusions; metastatic urothelial cancer with disease progression on one line of prior platinum‐containing chemotherapy, including within 12 mo of neoadjuvant or adjuvant platinum‐containing chemotherapy | CR + PR: 40%; PD: 18%; SD: 39%; unknown: 2% | 13.8 mo | 5.5 mo | Hyperphosphatemia, stomatitis, diarrhea, dry mouth, decreased appetite, dysgeusia |
| Pemigatinib (INCB054828); 13.5 mg/d on 21‐d cycle (2 wk on/1 wk off) | Selective, potent, oral inhibitor of FGFR1–3 | Phase II, open label, n = 100 | FGFR/FGF alterations; metastatic or unresectable urothelial cancer progression after 1 or more lines of prior therapy or are platinum ineligible | ORR: 25% | N/A | N/A | Diarrhea, alopecia, fatigue, constipation, dry mouth |
| Rogaratinib; 800 mg twice daily | Oral, selective inhibitor of FGFR 1–4 kinase activity | Phase I, open‐label, n = 51 | Overexpression of tumor FGFR 1–3 mRNA; advanced urothelial cancer progression after standard of care treatment | CR + PR: 24%; SD: 49%; PD: 27% | N/A | 100 d | Hyperphosphatemia, diarrhea, decreased appetite |
| Rogaratinib 800 mg twice daily vs. chemotherapy (docetaxel, paclitaxel, or vinflunine); Interim analysis | Oral, selective inhibitor of FGFR 1–4 kinase activity | Phase II/III, randomized, open‐label study, n = 175 | FGFR1–3 mRNA overexpression and/or FGFR3‐activating mutations and/or translocations; metastatic urothelial cancer in patients who received prior platinum chemotherapy | Rogaratinib: CR + PR: 19%; SD: 28%; PD: 31%; chemotherapy: CR + PR: 19%; SD: 35%; PD: 24% | Rogaratinib: 8.3 months (95% CI 6.5, NE); chemotherapy: 9.8 months (95% CI 6.8, NE) | Rogaratinib: 2.7 months (95% CI, 1.6–4.6); chemotherapy: 3.2 months (95% CI, 2.7–4.4); 1‐sided p value = .86) | Rogaratinib: diarrhea, hyperphosphatemia, decreased appetite, nausea. Chemotherapy: constipation, fatigue, anemia, decreased appetite, neutropenia |
Abbreviations: CI, confidence interval; CR, complete response; FGFR, fibroblast growth factor receptor; N/A, not applicable; NE, Not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Infigratinib
Infigratinib is an oral, selective, ATP‐competitive FGFR 1–3 tyrosine kinase inhibitor. In a phase I clinical trial, the safety and antitumor activity of infigratinib were evaluated in 132 patients with solid tumors [19]. In the dose‐expansion phase of the study, patients were selected based on FGFR gene alterations, of whom 9.1% had urothelial cancer. Based on a better side effect profile, the 125 mg dose given on a 3‐weeks on/1‐week off schedule was recommended for phase II studies. In the FGFR3‐mutated urothelial cohort, the overall response rate (ORR) was 38%, and 75% achieved disease control [19].
Based on the phase I trial, Pal and coworkers studied infigratinib in 67 patients with metastatic urothelial carcinoma (UC) and prespecified FGFR3 alterations [20]. Most patients had received prior antineoplastic therapy. Overall, 17 patients (25.4%) achieved partial (PR) or complete response (CR), and progressive disease was seen in 16 patients (23.9%). Interestingly, 8 out of 67 patients with upper tract UC (UTUC) achieved 100% disease control (1 CR, 3 PR, and 4 stable disease) [21]. With the caveat of a small cohort, this differential response was attributed to a different FGFR mutation pattern compared with bladder UC. The median progression‐free survival (PFS) and overall survival (OS) were 3.75 and 7.75 months, respectively. About 68.7% of patients developed grade 3/4 adverse events including hyperlipasemia, hypophosphatemia, palmar‐plantar erythrodysesthesia, fatigue, and anemia. Cell‐free DNA testing for FGFR gene alterations on therapy revealed four patients developed FGFR3 gatekeeper resistance mutations, such as V443L, V443M, and L496V. There were no novel recurrent mutations in 22 patients who had disease progression while on treatment. There was an association with decreased FGFR3 allele fraction in patients who were on infigratinib for longer duration and a greater decrease in tumor size in comparison with baseline. A phase III clinical trial is currently evaluating infigratinib in patients with UTUC or bladder UC after surgery in the adjuvant setting (NCT04197986) [21]. Another early phase study is evaluating infigratinib before surgery for patients with UTUC to make the surgical procedure easier and/or less extensive (NCT04228042).
Erdafitinib
Erdafitinib is an oral, potent FGFR 1–4 tyrosine kinase inhibitor. The antitumor activity of erdafitinib was evaluated in a phase II clinical trial in patients with unresectable or metastatic urothelial cancer harboring a prespecified FGFR3 mutation or FGFR2/3 fusion [22]. In the early part of the study, patients were randomized 1:1 to receive an intermittent or continuous dose of erdafitinib. Subsequently, the continuous 8 mg daily dosing was chosen for further evaluation. In the continuous dosing group, 99 patients were started at an 8 mg dose, of which 41 patients had dose escalation to 9 mg on day 14, as there were no significant adverse events and phosphate level was <5.5 mg/dL. Eleven patients were treatment naive, and the rest had received at least one line of therapy. The investigator‐assessed ORR was 40% (95% confidence interval [CI], 31–50). Thirty‐six (49%) of 74 patients with FGFR3 mutations had a response, and 4 patients (16%) of 25 with FGFR 2/3 fusion had a response. Of the 22 patients who received prior immunotherapy, the response rate was 59% with erdafitinib. After a follow up of 2 years, the median PFS was 5.5 months (95% CI, 4.2–6.0), and the median OS was 11.3 months [23]. The most common treatment‐related adverse events (AEs) included hyperphosphatemia (77%), stomatitis (58%), diarrhea (50%), and dry mouth (46%) [22]. Grade 3–4 AEs included hyponatremia (11%), stomatitis (10%), and asthenia (7%). Central serous retinopathy, an adverse effect associated with FGFR inhibitors, was seen in 21% of patients, of which 3% were grade 3. Most of these side effects were reversible with dose interruption or reduction. Biomarkers of primary or secondary resistance to erdafitinib were not reported. Based on this efficacy data, the U.S. FDA granted accelerated approval for erdafitinib in adult patients with metastatic UC with susceptible FGFR3 or FGFR2 genetic alterations [24].
In a phase Ib/II clinical trial (NORSE study), the safety and antitumor activity of erdafitinib in combination with cetrelimab (an IgG4 anti–programmed cell death protein 1 [PD‐1] inhibitor) was evaluated in patients with metastatic urothelial harboring susceptible FGFR2/3 alternations [25]. Patients were enrolled after progression on one or more lines of therapy including platinum‐based chemotherapy. Of the 15 patients enrolled in the study, no dose‐limiting toxicities were noted. Ten patients experienced grade 3 AEs. Three patients had serious unrelated AEs, which lead to death in two patients. The combination of erdafitinib (8 mg with uptitration to 9 mg) with cetrelimab was deemed safe for further evaluation. In the seven patients treated with the recommended phase II dose, ORR was 71%. This combination is further evaluated in a randomized phase II clinical trial (NCT03473743).
A randomized, phase II study is ongoing to further evaluate the efficacy of erdafitinib in comparison with vinflunine, docetaxel, or pembrolizumab in patients with advanced urothelial cancer with FGFR gene aberrations (NCT03390504). The efficacy of erdafitinib in comparison with intravesical chemotherapy is also currently being evaluated in high risk, BCG refractory, nonmuscle invasive bladder cancer with FGFR gene alterations (NCT04172675).
Pemigatinib
Pemigatinib is a potent, oral, selective inhibitor of FGFR1–3. Necchi and colleagues presented an interim analysis of phase II clinical trial (FIGHT‐201) in patients with advanced or metastatic urothelial cancer who previously progressed on one or more lines of therapy or are platinum ineligible [26]. Sixty‐four patients with FGFR3 mutation or fusion were assigned to cohort A, and 36 patients with other FGF/FGFR genetic mutations were assigned to cohort B and received pemigatinib. ORR was 25% (95% CI, 14%–40%), including unconfirmed PR in cohort A, and one patient had unconfirmed PR with FGF10 amplification. The common treatment‐related adverse events included diarrhea, alopecia, fatigue, and constipation. Hyperphosphatemia was observed in 68% and 64% in cohort A and B, respectively. The efficacy of pemigatinib in combination with pembrolizumab is compared with the standard of care chemotherapy or immunotherapy in patients with cisplatin‐ineligible urothelial cancer in a current phase II randomized study (FIGHT‐205, NCT04003610).
Rogaratinib
Rogaratinib is an oral, potent, selective FGFR1–4 inhibitor. Preclinical studies showed response to rogaratinib correlated with high FGFR mRNA expression in tumors [27].
Schuler and colleagues conducted a phase I dose‐escalation and dose‐expansion trial in patients with urothelial carcinoma, head and neck squamous cell cancer, non‐small‐cell lung cancer, and other solid tumor types [28, 29]. In the dose‐expansion phase, among all patients with FGFR mRNA‐positive tumors treated with rogaratinib, 74 had urothelial carcinoma. In the entire cohort, 15 patients (15%) had an objective response (OR; 95% CI, 8.6–23.5), and 10 of 15 (67%) patients with FGFR mRNA‐overexpressing tumors without FGFR genetic alteration had an OR. In the urothelial cancer cohort, 20.8% had an OR, with one patient achieving CR. The disease control rate was 68.1%. The most common adverse events were hyperphosphatemia, diarrhea, and decreased appetite. The most common grade 3 or greater AEs were fatigue, increased lipase, dyspnea, anemia, and urinary tract infection. In the urothelial cancer cohort, elevated creatinine of any degree was seen in 64% of patients, including one patient who had transient acute tubular necrosis, which resolved with dose interruption and reduction.
The FORT‐1 study evaluated the efficacy of rogaratinib in comparison with chemotherapy (docetaxel, paclitaxel, or vinflunine) in patients with metastatic urothelial cancer who received prior cisplatin‐based chemotherapy [30]. Patients were selected based on either FGFR1,3 mRNA overexpression and/or FGFR‐3–activating mutations or translocations. Of the 175 patients enrolled in the study, 87 received rogaratinib, and the remaining were randomized to receive chemotherapy. The ORRs were 19.5% and 19.3% (1‐sided p = .56), and mPFS was 2.7 (95% CI, 1.6–4.2) and 2.9 (95% CI, 2.6–4.2) months with rogaratinib and chemotherapy, respectively. Grade 3–4 AEs were seen in 47% and 56% in the rogaratinib and chemotherapy cohorts. On exploratory analysis in patients with FGFR3 mutations or fusion, ORR was 52.4% for rogaratinib, and with chemotherapy it was 26.7%.
FORT‐2 trial is evaluating the safety and antitumor activity of rogaratinib (800 mg and 600 mg doses) along with atezolizumab in cisplatin‐ineligible patients with metastatic urothelial cancer and FGFR1 or 3 mRNA overexpression [31]. In the preliminary analysis, rogaratinib at 600 mg was deemed a maximum tolerated dose based on lower incidence of adverse events. The preliminary efficacy data is promising while we await further results from the dose‐expansion cohort. In BLASST‐3 clinical trial, the safety and efficacy of rogaratinib is evaluated in BCG refractory nonmuscle invasive bladder cancer (NCT04040725).
Derazantinib
Derazantinib is a novel, potent, ATP competitive multikinase inhibitor of FGFR 1–3 and colony stimulating factor 1 receptor (CSF1R) kinase [32]. By reducing CSF1‐stimulated CSF1R phosphorylation in macrophages, derazantinib is speculated to have a regulatory effect on tumor‐associated macrophages [33]. It is hypothesized that the depletion of tumor‐associated macrophages with CSF1R inhibitors would have a synergistic effect with checkpoint inhibitors [34]. FIDES‐02 clinical trial is currently evaluating the safety and antitumor activity of single‐agent derazantinib or in combination with atezolizumab in patients with urothelial cancer and FGFR aberrations [35].
TAS‐120
TAS‐120 is a selective irreversible inhibitor for FGFR 1–4 [36]. Meric‐Bernstam and colleagues presented preliminary results of an ongoing phase I study in patients with solid tumors and FGFR aberrations [37]. The study included a total of 134 patients with primary CNS tumors (n = 24), bladder cancer (n = 21), breast cancer (n = 17), colorectal cancer (n = 15), gastroesophageal cancer (n = 9), and other cancers. In the dose‐escalation phase, a 20 mg per day oral dose of TAS‐120 was considered safe and exhibited clinical activity in various tumors. Hyperphosphatemia was the most common side effect. Partial responses were seen in breast cancer, bladder cancer, primary CNS tumor, and gastroesophageal cancer.
Debio‐1347
Debio‐1347 is a small, oral molecule that selectively inhibits the ATP binding site of FGFR1–3 [38]. A phase I clinical trial evaluated the safety and antitumor activity of debio‐1347 in 58 patients with solid tumors including breast (21%), biliary tract (14%), bladder (10%), uterine (9%), squamous cell lung cancer (7%), gastric cancer (7%), prostate cancer (3%), and cervical cancer (3%) with known FGFR 1–3 alterations [39]. FGFR1 amplification was most common (40%), followed by FGFR fusion (21%), FGFR2 mutation (12%), and FGFR3 mutation (17%). All‐cause treatment emergent adverse event (TEAEs) included hyperphosphatemia (76%), diarrhea (41%), nausea (40%), fatigue (40%), constipation (38%), decreased appetite (33%), and nail changes (31%). Grade 3 or above TEAEs included hyperphosphatemia (21%), anemia (12%), and dyspnea (5%). Two of the six patients with urothelial cancer had a partial response. Further studies with this compound are being planned in a molecular‐selected cohort.
Vofatamab (B‐701)
Vofatamab is a selective anti‐ FGFR3 receptor monoclonal antibody that is being evaluated in patients with metastatic urothelial cancer in second‐line setting [40]. In the preliminary analysis of 55 patients, vofatamab monotherapy, or in combination with docetaxel, was shown to be well tolerated. The most common side effects were decreased appetite, diarrhea, fever, asthenia, and fatigue. An objective response was seen in seven patients. The final analysis of this study is awaited.
Resistance mechanisms for FGFR inhibitors
Several mechanisms are involved in resistance to FGFR inhibitors and can be classified into three main categories: activation of bypass signaling pathways, alternative or secondary FGFR mutations, and intratumor heterogeneity. As previously mentioned, MAPK, PI3K/AKT, EGFR, PLC‐γ, and STAT are involved in downstream signaling, and upregulation of any of these pathways can bypass the blockade and lead to drug resistance. For example, EGFR upregulation plays an important role in intrinsic and acquired resistance to FGFR inhibitors. In a study by Herra and colleagues on urothelial tumor cells harboring FGFR3 S249C mutation, cells showed upregulation of EGFR signaling and downregulation of FGFR3 after exposure to PD173074 and gefitinib [34]. Additionally, cells with FGFR3‐TACC3 fusions acquire resistance by activating EGFR/HER3‐dependent PI3K‐AKT signaling pathways [41, 42]. Other mechanisms by which tumor cells develop resistance is by acquiring different mutations that can alter drug binding sites (gatekeeper mutations). FGFR1 V561M, FGFR2 V564F/I, FGFR3 V555M, and FGFR4 V550E/L are all documented mutations that arise de novo or become predominant during treatment [41, 43]. Intratumor heterogeneity in which tumors contain different subcolonies and FGFR‐independent clones can play a role in treatment response [44, 45].
Discussion
For select patients with urothelial cancer and FGFR gene aberrations, FGFR inhibitors are a valuable addition to therapeutic options. Currently, erdafitinib is the only drug approved in the U.S., and other agents are under clinical evaluation. It is imperative to note that not all FGFR alterations are associated with response to FGFR inhibitors. Clinical responses have been most notable with FGFR3 alterations versus FGFR1/2 alterations. The genomic and disease characteristics of patients with durable responses should be thoroughly examined, as this can provide valuable insight in the selection of patients. Additionally, the primary and secondary resistance mechanisms of FGFR inhibitors can be explored by suing circulating cell‐free DNA, as it can aid in subsequent treatment strategies [46]. The testing modality to identify FGFR gene aberrations has been variable across the studies. For instance, with erdafitinib, mutations and fusions of FGFR2/3 were analyzed based on RNA sequencing of tumor samples using RT PCR assay, whereas with rogaratinib it was based on overexpression of FGFR mRNA. Currently, it remains unclear which biomarker is ideal for identifying patients with these aberrations.
The common trend of adverse effects due to the class effect of FGFR inhibitors include hyperphosphatemia, ocular disorders such as dry eye, blurry vision, lacrimation, conjunctivitis, and central serous retinopathy. Additionally, gastrointestinal side effects such as diarrhea, stomatitis, dry mouth, and so on and nail changes such as onycholysis, nail dystrophy, and paronychia seem to be commonly associated with FGFR inhibitors. In most studies with FGFR inhibitors, these side effects improved with dose interruption and dose reduction.
Increased renal phosphate reabsorption due to inhibition of renal tubular FGF23 leads to elevated serum phosphate levels with FGFR inhibitors. Early phase studies with erdafitinib showed improved disease outcomes if serum phosphate levels increased to 5.5 mg/dL [47]. Patients are recommended to restrict phosphorus in the diet to 600–800 mg per day, and oral phosphate binders should be initiated if serum phosphate level rises above 7 mg/dL.
Central serous retinopathy (CSR) results from fluid accumulation between the neural retina and retinal pigment epithelium (RPE) [48]. FGFR inhibitors disrupt the normal ocular FGFR–MAPK pathway leading to dysfunction of RPE and resulting in CSR. This disorder presents as visual impairment in both eyes and can be asymptomatic if edema is limited to the fovea. For patients starting erdafitinib, monthly ophthalmological examinations are recommended during the first 4 months and then every 3 months to monitor ocular toxicities. Ocular lubricants are recommended for dry eyes. There should be proper communication between the oncologist and ophthalmology, and erdafitinib should be immediately discontinued for visual acuity worse than 20/40 (grade ≥ 3 ocular toxicity as per CTCAE v4.03) [49]. Similar guidelines are being followed for the other FGFR inhibitors in clinical evaluation.
There are conflicting reports about the presence of FGFR alterations and response to checkpoint inhibitors (CPI). It is postulated that FGFR3 alterations, which are enriched in luminal type UC, are associated with lower T‐cell infiltration and predict poor response to CPI [50]. This effect was demonstrated in a small cohort of patients treated with erdafitinib, where the responses were poor with CPI in patients harboring FGFR2/3 alterations [22]. However, in another study of patients with urothelial cancer treated with CPI, the FGFR3 mutant or wild type status did not alter response [51]. Clinical trials comparing the efficacy of FGFR inhibitors with CPI or in combination with CPI are ongoing (Table 2). Additionally, FGFR inhibitors are also being evaluated in the neoadjuvant, adjuvant setting of muscle‐invasive bladder cancer (MIBC), UTUC, and BCG refractory non‐MIBC.
Table 2.
Summary of ongoing clinical trials with FGFR inhibitors as monotherapy or in combination with other agents in urothelial cancer
| Study, NCT identifier | Agent(s) | Study design | Estimated sample | FGFR genetic aberrations | Study cohort | Primary endpoint(s) |
|---|---|---|---|---|---|---|
| NCT04045613, FIDES‐02 | Derazantinib monotherapy or in combination with atezolizumab | Phase Ib/II, open label | 303 | Required, FGFR 1–3 mutations per fusion | Metastatic urothelial cancer, cisplatin ineligible | RP2D of derazantinib with atezolizumab; ORR based on RECIST 1.1 |
| NCT03914794 | Pemigatinib | Phase II, open label, window of opportunity study | 43 | Not required | Low‐ or intermediate‐risk NMIBC tumors prior to TURBT | Complete response on pathology |
| NCT02872714, FIGHT‐201 | Pemigatinib | Phase II, open label | 240 | Required | Metastatic urothelial cancer in first‐ or second‐line therapy | ORR in patients with FGFR3 mutations based on RECIST 1.1 |
| NCT04003610, FIGHT‐205 | Pemigatinib + pembrolizumab vs. pemigatinib vs. standard of care (chemotherapy or pembrolizumab) | Phase II, open‐label, randomized, multicenter | 372 | Required, FGFR3 mutation or rearrangement | First‐line, metastatic or unresectable urothelial carcinoma in cisplatin‐ineligible patients | PFS |
| NCT03473743 | Erdafitinib vs. erdafitinib + cetrelimab (anti‐PD‐1 monoclonal antibody) | Phase Ib/II, randomized, open label | 150 | Required | First‐line metastatic urothelial carcinoma, cisplatin ineligible | DLTs, overall response rate by RECIST 1.1 |
| NCT03390504, THOR study | Erdafitinib vs. chemotherapy (docetaxel or vinflunine) vs. pembrolizumab | Phase III, open label, randomized | 631 | Required | Metastatic urothelial carcinoma with disease progression after 1 or 2 prior treatments | OS |
| NCT02052778 | TAS‐120 | Phase I/II, single arm, open label, multicohort | 371 | Required, advanced urothelial carcinoma with FGFR3 fusions or FGFR3 activating mutations | Patients with solid tumor including a cohort of advanced UC | ORR by RECIST 1.1 |
| NCT03834220, FUZE study | Debio 1347 | Phase II, single arm, open label Basket study | 125 | FGFR 1–3 fusion | Solid tumor FGFR alterations | ORR |
| NCT04040725, (BLASST)‐3 | Rogaratinib | Phase II, open label | 33 | Required, FGFR1 or FGFR3 gene overexpression | High‐risk NMIBC unresponsive to BCG | Complete response on TURBT |
| NCT03410693, FORT‐1 | Rogaratinib vs. chemotherapy (docetaxel, paclitaxel or vinflunine) | Phase II/III, randomized, open label | 175 | FGFR1 or 3 gene alterations | Metastatic urothelial carcinoma, received prior platinum‐containing chemotherapy | OS |
| NCT03473756, FORT‐2 | Rogaratinib + atezolizumab vs. placebo + atezolizumab | Phase Ib/II, randomized, blinded | 210 | High FGFR1 or 3 mRNA expression levels | Treatment naïve locally advanced or metastatic urothelial cancer | DLTs, AEs, and PFS |
| NCT04197986, PROOF 302 | Infigratinib vs. placebo | Phase III, randomized, blinded | 218 | FGFR 3 genetic alterations | Adjuvant therapy after definitive surgery for invasive urothelial cancer | DFS |
| NCT04228042 | Infigratinib | Phase I/II | 20 | With and without FGFR3 alterations | Prior to surgery for UTUC | Safety and ORR |
Abbreviations: AE, adverse event; DLT, dose limiting toxicity; FGFR, fibroblast growth factor receptor; NMIBC, non‐muscle invasive bladder cancer; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; RP2D, recommended phase 2 dose; TURBT, transurethral resection of bladder tumor; UC, urothelial cancer; UTUC, upper tract urothelial cancer.
With the availability of a predictive biomarker, physicians now have the potential to use a targeted therapeutic agent with a novel mechanism of action in their armamentarium to treat urothelial cancer.
Author Contributions
Conception/design: Rohan Garje
Manuscript writing: Rohan Garje, Mohammad Obeidat, Josiah An, Yousef Zakharia
Final approval of manuscript: Rohan Garje, Yousef Zakharia
Disclosures
Yousef Zakharia: Amgen, Roche Diagnostics, Novartis, Jansen, Eisai, Exelixis, Castle Bioscience, Array, Bayer, Pfizer, Clovis, EMD Serono (C/A), NewLink Genetics, Pfizer, Exelixis, Eisai (RF‐institutional clinical trial support), Jansen (Other‐data safety monitoring committee). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgments
The authors from University of Iowa are supported by National Institutes of Health grant P30 CA086862.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Knowles MA, Hurst CD. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat Rev Cancer 2015;15:25–41. [DOI] [PubMed] [Google Scholar]
- 3. Itoh N, Ornitz DM. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J Biochem 2010;149:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tiong KH, Mah LY, Leong CO. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis 2013;18:1447–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarabipour S. Parallels and distinctions in FGFR, VEGFR, and EGFR mechanisms of transmembrane signaling. Biochemistry 2017;56:3159–3173. [DOI] [PubMed] [Google Scholar]
- 6. Plotnikov AN, Schlessinger J, Hubbard SR et al. Structural basis for FGF receptor dimerization and activation. Cell 1999;98:641–650. [DOI] [PubMed] [Google Scholar]
- 7. Trueb B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell Mol Life Sci 2011;68:951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dieci MV, Arnedos M, Andre F et al. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov 2013;3:264–279. [DOI] [PubMed] [Google Scholar]
- 9. Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer 2017;17:318–332. [DOI] [PubMed] [Google Scholar]
- 10. Dienstmann R, Rodon J, Prat A et al. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann Oncol 2014;25:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tomlinson DC, Hurst CD, Knowles MA. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene 2007;26:5889–5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. AACR Project GENIE Consortium . AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7:818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sfakianos JP, Cha EK, Iyer G et al. Genomic characterization of upper tract urothelial carcinoma. Eur Urol 2015;68:970–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ross JS, Wang K, Khaira D et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer 2016;122:702–711. [DOI] [PubMed] [Google Scholar]
- 15. Sarabipour S, Hristova K. FGFR3 unliganded dimer stabilization by the juxtamembrane domain. Journal of molecular biology. 2015;427:1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dodurga Y, Tataroglu C, Kesen Z et al. Incidence of fibroblast growth factor receptor 3 gene (FGFR3) A248C, S249C, G372C, and T375C mutations in bladder cancer. Genet Mol Res 2011;10:86–95. [DOI] [PubMed] [Google Scholar]
- 18. Helsten T, Elkin S, Arthur E et al. The FGFR landscape in cancer: Analysis of 4,853 tumors by next‐generation sequencing. Clin Cancer Res 2016;22:259–267. [DOI] [PubMed] [Google Scholar]
- 19. Nogova L, Sequist LV, Perez Garcia JM et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1‐3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase I, dose‐escalation and dose‐expansion study. J Clin Oncol 2017;35:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pal SK, Rosenberg JE, Hoffman‐Censits JH et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1‐3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov 2018;8:812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pal SK, Bajorin D, Dizman N et al. Infigratinib in upper tract urothelial carcinoma versus urothelial carcinoma of the bladder and its association with comprehensive genomic profiling and/or cell‐free DNA results. Cancer 2020;126:2597–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loriot Y, Necchi A, Park SH et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 2019;381:338–348. [DOI] [PubMed] [Google Scholar]
- 23. Siefker‐Radtke AO, Necchi A, Park SH et al. ERDAFITINIB in locally advanced or metastatic urothelial carcinoma (mUC): Long‐term outcomes in BLC2001. J Clin Oncol 2020;38(15 suppl):5015a. [Google Scholar]
- 24. FDA grants accelerated approval to erdafitinib for metastatic urothelial carcinoma. Washington, DC: Food and Drug Adminstration; 2019. [Google Scholar]
- 25. Moreno V, Loriot Y, Valderrama BP et al. Does escalation results from phase Ib/II Norse study of erdafitinib (ERDA) + PD‐1 inhibitor JNJ‐63723283 (Cetrelimab [CET]) in patients (pts) with metastatic or locally advanced urothelial carcinoma (mUC) and selected fibroblast growth factor receptor (FGFR) gene alterations. J Clin Oncol 2020;38(6 suppl):511a. [Google Scholar]
- 26. Necchi A, Pouessel D, Leibowitz‐Amit R et al. Interim results of fight‐201, a phase II, open‐label, multicenter study of INCB054828 in patients (pts) with metastatic or surgically unresectable urothelial carcinoma (UC) harboring fibroblast growth factor (FGF)/FGF receptor (FGFR) genetic alterations (GA). Ann Oncol 2018;29(suppl 8):900P. [Google Scholar]
- 27. Grünewald S, Politz O, Bender S et al. Rogaratinib: A potent and selective pan‐FGFR inhibitor with broad antitumor activity in FGFR‐overexpressing preclinical cancer models. Int J Cancer 2019;145:1346–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schuler M, Cho BC, Sayehli CM et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: A phase 1 dose‐escalation and dose‐expansion study. Lancet Oncol 2019;20:1454–1466. [DOI] [PubMed] [Google Scholar]
- 29. Kempf E, Penel N, Tournigand C et al. Phase I experience with rogaratinib in patients (pts) with urothelial carcinoma (UC) selected based on FGFR mRNA overexpression. J Clin Oncol 2020;38(6 suppl):527a. [Google Scholar]
- 30. Quinn DI, Petrylak DP, Bellmunt J et al. FORT‐1: Phase II/III study of rogaratinib versus chemotherapy (CT) in patients (pts) with locally advanced or metastatic urothelial carcinoma (UC) selected based on FGFR1/3 mRNA expression. J Clin Oncol 2020;38(6 suppl):489a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosenberg JE, Gajate P, Morales‐Barrera R et al. Safety and preliminary efficacy of rogaratinib in combination with atezolizumab in a phase Ib/II study (FORT‐2) of first‐line treatment in cisplatin‐ineligible patients (pts) with locally advanced or metastatic urothelial cancer (UC) and FGFR mRNA overexpression. J Clin Oncol 2020;38(15 suppl):5014a. [Google Scholar]
- 32. Hall TG, Yu Y, Eathiraj S et al. Preclinical activity of ARQ 087, a novel inhibitor targeting FGFR dysregulation. PLoS One. 2016;11:e0162594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaudhry A, Sternberg CN, De Santis M et al. FIDES‐02, a phase Ib/II study of derazantinib (DZB) as monotherapy and combination therapy with atezolizumab (A) in patients with surgically unresectable or metastaticurothelial cancer (UC) and FGFR genetic aberrations. J Clin Oncol 2020;38(6 suppl):TPS590‐TPS. [Google Scholar]
- 34. Zhu Y, Knolhoff BL, Meyer MA et al. CSF1/CSF1R blockade reprograms tumor‐infiltrating macrophages and improves response to T‐cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quigley D, Alumkal JJ, Wyatt AW et al. Analysis of circulating cell‐free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov 2017;7:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ochiiwa H, Fujita H, Itoh K et al. TAS‐120, a highly potent and selective irreversible FGFR inhibitor, is effective in tumors harboring various FGFR gene abnormalities. Mol Cancer Ther 2014;12:A270a. [Google Scholar]
- 37. Meric‐Bernstam F, Goyal L, Tran B et al. TAS‐120 in patients with advanced solid tumors bearing FGF/FGFR aberrations: A Phase I study. Cancer Res 2019;79(suppl):CT238a. [Google Scholar]
- 38. Nakanishi Y, Akiyama N, Tsukaguchi T et al. The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther 2014;13:2547–2558. [DOI] [PubMed] [Google Scholar]
- 39. Voss MH, Hierro C, Heist RS et al. A Phase I, Open‐label, multicenter, dose‐escalation study of the oral selective FGFR inhibitor debio 1347 in patients with advanced solid tumors harboring FGFR gene alterations. Clin Cancer Res 2019;25:2699–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Necchi A, Castellano DE, Mellado B et al. Fierce‐21: Phase II study of vofatmab (B‐701), a selective inhibitor of FGFR3, as salvage therapy in metastatic urothelial carcinoma (mUC). J Clin Oncol 2019;37(7 suppl):409a. [Google Scholar]
- 41. Herrera‐Abreu MT, Pearson A, Campbell J et al. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3‐mutant cancer. Cancer Discov 2013;3:1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang L, Šuštić T, Leite de Oliveira R et al. A functional genetic screen identifies the phosphoinositide 3‐kinase pathway as a determinant of resistance to fibroblast growth factor receptor inhibitors in FGFR mutant urothelial cell carcinoma. Eur Urol 2017;71:858–862. [DOI] [PubMed] [Google Scholar]
- 43. Chell V, Balmanno K, Little AS et al. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 2013;32:3059–3070. [DOI] [PubMed] [Google Scholar]
- 44. Kim HP, Cho GA, Han SW et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene 2014;33:5434–5441. [DOI] [PubMed] [Google Scholar]
- 45. Vandekerkhove G, Todenhöfer T, Annala M et al. Circulating tumor DNA reveals clinically actionable somatic genome of metastatic bladder cancer. Clin Cancer Res 2017;23:6487–6497. [DOI] [PubMed] [Google Scholar]
- 46. Moss TJ, Rodon Ahnert J, Oakley HD et al. Baseline cfDNA characteristics and evolution of cfDNA profile during treatment with selective FGFR inhibitor TAS‐120. J Clin Oncol 2019;37(15 suppl):3056a. [Google Scholar]
- 47. Tabernero J, Bahleda R, Dienstmann R et al. Phase I dose‐escalation study of JNJ‐42756493, an oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 2015;33:3401–3408. [DOI] [PubMed] [Google Scholar]
- 48. van der Noll R, Leijen S, Neuteboom GHG et al. Effect of inhibition of the FGFR–MAPK signaling pathway on the development of ocular toxicities. Cancer Treat Rev 2013;39:664–672. [DOI] [PubMed] [Google Scholar]
- 49. Erdafitinib . Package insert; 2019. Food and Drug Administration. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212018s000lbl.pdf. Accessed September 10, 2020.
- 50. Sweis RF, Spranger S, Bao R et al. Molecular drivers of the non–T‐cell‐Inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol Res 2016;4:563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang L, Gong Y, Saci A et al. Fibroblast growth factor receptor 3 alterations and response to PD‐1/PD‐L1 blockade in patients with metastatic urothelial cancer. Eur Urol 2019;76:599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]