

Abstract
Background
The increasing molecular characterization of colorectal cancers (CRCs) has spurred the need to look beyond RAS, BRAF, and microsatellite instability (MSI). Genomic alterations, including ERBB2 amplifications and mutations, POLE mutations, MSI, and NTRK1–3 fusions, have emerged as targets for matched therapies. We sought to study a clinically annotated Chinese cohort of CRC subjected to genomic profiling to explore relative target frequencies.
Methods
Tumor and matched whole blood were collected from 609 Chinese patients with CRC. Extracted DNA was analyzed for all classes of genomic alterations across 450 cancer‐related genes, including single‐nucleotide variations (SNVs), short and long insertions and deletions (indels), copy number variations, and gene rearrangements. Next‐generation sequencing–based computational algorithms also determined tumor mutational burden and MSI status.
Results
Alterations in TP53 (76%), APC (72%), and KRAS (46%) were common in Chinese patients with CRC. For the first time, the prevalence of NTRK gene fusion was observed to be around 7% in the MSI‐high CRC cohort. Across the cohort, MSI was found in 9%, ERBB2 amplification in 3%, and POLE pathogenic mutation in 1.5% of patients. Such results mostly parallel frequencies observed in Western patients. However, POLE existed at a higher frequency and was associated with large tumor T‐cell infiltration.
Conclusion
Comparing to the Western counterparts, POLE mutations were increased in our cohort. The prevalence of NTRK gene fusion was around 7% in the MSI‐high CRC cohort. Increased adoption of molecular profiling in Asian patients is essential for the improvement of therapeutic outcomes.
Implications for Practice
The increasing use of genomic profiling assays in colorectal cancer (CRC) has allowed for the identification of a higher number of patient subsets benefiting from matched therapies. With an increase in the number of therapies, assays simultaneously evaluating all candidate biomarkers are critical. The results of this study provide an early support for the feasibility and utility of genomic profiling in Chinese patients with CRC.
Keywords: Colorectal cancer, DNA polymerase ε, Tropomyosin receptor kinase, ERBB2 amplification, Tumor mutational burden
Short abstract
The emergence of precision medicine has identified genomic variants, such as NTRK gene fusion, microsatellite instability (MSI), HER2 amplification, and POLE pathogenic mutation, as potential agonistic biomarkers for immune or targeted therapies. This article examines NTRK, HER2, and POLE in a cohort of Chinese patients with colorectal cancer.
Introduction
Colorectal cancer (CRC) is one of the most common malignancies in China, accounting for 376,000 new cases and 191,000 deaths in 2015 alone [1]. Despite remarkable progress in biological understanding, currently, chemotherapy remains the evidence‐based standard for first‐line therapy in advanced disease. Regimens consisting of 5‐fluorouracil, irinotecan, or oxaliplatin with biologic agents targeting epidermal growth factor receptor (EGFR) or vascular endothelial growth factor remain the global standard. With current therapies, the median overall survival (OS) of patients with metastatic CRC (mCRC) in the past few decades has improved to over 30 months [2]. In spite of significant improvements in mCRC treatment, the most effective treatment for a patient is relatively empiric, mainly based on clinical considerations. Standard biomarkers, including RAS/BRAF and microsatellite status, both inform prognosis and predict benefit, or lack thereof, from biologic agents [3]. The emergence of precision medicine has identified genomic variants such as NTRK gene fusion, microsatellite instability (MSI), HER2 amplification, and POLE pathogenic mutation as potential agonistic biomarkers for immune or targeted therapies.
The recent approval of MSI and mismatch repair deficiency (dMMR) as a biomarker for immunotherapy further reinforces the concept that precision treatment can significantly extend the survival of certain patients. Microsatellites are short tandem repeats of the same base or simple sequence scattering throughout both coding and noncoding regions in the genome. Microsatellite instable tumors are tightly associated with defects in the DNA mismatch repair system, which also causes elevated tumor mutational burden (TMB). A clinical trial of patients with MSI‐high (MSI‐H)/dMMR mCRC treated with nivolumab has shown a median OS of over 33 months. Additionally, a basket trial treating patients with various dMMR cancers with pembrolizumab has demonstrated a median OS of over 36 months [4, 5].
Initially identified in colorectal and papillary thyroid carcinomas in the 1980s, NTRK fusions have recently been attractive biomarkers for targeted therapies research, leading to two drugs approved by the U.S. Food and Drug Administration (FDA), larotrectinib and entrectinib. The NTRK genes (NTRK1, NTRK2, and NTRK3) encode the tropomyosin receptor kinase (TRK) family of proteins including TRKA, TRKB, and TRKC, respectively [6]. NTRK gene fusions are the most common mechanisms of oncogenic TRK activation through intrachromosomal or interchromosomal rearrangements. The chimeric oncoprotein is characterized by the NTRK kinase's constitutive activation through the ligand‐independent dimmer of domains generated by partner genes. For instance, ETV6‐NTRK3 gene fusion has a prevalence of >90% in secretory breast carcinoma [7, 8], mammary analog secretory carcinoma [9], and infantile fibrosarcoma [10]. However, it can be detected in <1% of common cancers such as lung adenocarcinomas, head and neck squamous cell cancer, and colorectal cancer. So far, approximately 80 different 5' NTRK gene fusion partners have been identified in a broad range of human tumor types, many of which have not yet been systematically characterized [11, 12].
DNA polymerase ε (POLE) pathogenic mutation is another gene alteration causing extremely high TMB. The DNA polymerase proofreading is essential for ensuring the fidelity of DNA replication. Mutations in proofreading domains of POLE impair the correction of wrongly paired bases and the fidelity of DNA replication. Cancers with POLE proofreading variants have among the highest mutation burden. The frequency of clinically significant polymerase mutations is around 1% in the Western CRC population [13]. Importantly, previous reports have shown an extraordinary response to immune checkpoint inhibition in POLE‐mutated endometrial adenocarcinoma [14], CRC [15], and glioblastoma [16] with high TMB. Although the frequency of POLE mutations is low, it may become increasingly important as the role of immune checkpoint inhibitors evolves in CRC.
The amplification of the human epidermal growth factor receptor 2 (HER2) gene, a member of the EGFR family of tyrosine kinase receptors, is an established target in the treatment of patients with breast and gastric cancers. Recently, the HERACLES and MyPathway trials have shown that HER2 gene amplification can serve as an effective biomarker for dual‐targeted therapy with trastuzumab plus either lapatinib or pertuzumab in the treatment of refractory mCRC. Both trials demonstrated that, in treatment‐refractory HER2‐positive metastatic colorectal cancer, targeting HER2 may represent a therapeutic option [17, 18].
To date, the examination of NTRK, HER2, and POLE in Asian patients has not been well represented. In order to overcome this gap in knowledge, we therefore sought to explore NTRK, HER2, and POLE in a cohort of Chinese patients with CRC.
Materials and Methods
Genomic Profiling, Mutation Calling, and MSI/TMB Calculation
Comprehensive genomic profiling was performed by next‐generation sequencing with a panel of 450 cancer‐related genes, as previously reported on samples collected from October 2016 to March 2019 [19]. More than 97% samples were from the 3A hospitals (top tier) across China; 24% of all samples were from the south, 26% from the north, 31% from the west, and 19% from the east. All patients provided written informed consent. To this end, both formalin fixed paraffin embedded (FFPE) tumor and paired peripheral blood samples from Chinese patients with CRC were used. In brief, DNA was extracted from unstained FFPE sections with a tumor content of no less than 20%. Subsequently, DNA was fragmented to ~250 base pairs by sonication. A library was constructed with KAPA Hyper Prep Kit (KAPA Biosystems; Roche, Basel, Switzerland). Then hybridization capture was performed with a custom panel containing about 24,000 individually synthesized 5'‐biotinylated DNA probes. Genomic alterations, including single base substitution, short and long indels, copy number variations, and gene rearrangement and fusions, were assessed. Next‐generation sequencing–based algorithms also determined the TMB and microsatellite status.
Immunohistochemistry
Immunohistochemistry (IHC) staining procedure was performed as previously described [20]. In brief, deparaffinization, rehydration, and target retrieval were performed. These steps were followed by incubation with the monoclonal PD‐L1 (Clone 22C3; Dako, Agilent Technologies, Santa Clara, CA) and CD3 (Kit‐0003, MXB Biotechnologies, Fuzhou, China) antibodies. Slides were then incubated with a ready‐to‐use visualization reagent consisting of horseradish peroxidase molecules coupled with a secondary antibody. The enzymatic conversion of the subsequently added chromogen, followed by the enhancer, resulted in the precipitation of a visible reaction product at the antigen's site. The specimens were then counterstained with hematoxylin and coverslipped. Results were interpreted using a light microscope. For the development of the finalized assay, the sensitivity was optimized with minimum nonspecific staining by adjusting both the primary antibody concentration and reagent incubation times.
T‐cell infiltration of tumors was assessed by semiquantitative estimation of the density of CD3‐positive cells as previously described: (1) no, or sporadic cells; (2) moderate numbers of cells; (3) abundant occurrence of cells; and (4) highly abundant occurrence of cells [21]. PD‐L1 expression in CRC is determined by using combined positive score (CPS), which is the number of PD‐L1 staining cells (tumor cells, lymphocytes, macrophages) divided by the total number of viable tumor cells, multiplied by 100 [22].
Results
Characteristics of Patients with Colorectal Cancer
In the present study, a cohort of 609 patients with CRC comprising 372 men (median age, 61 years) and 237 women (median age, 60 years) had their FFPE tumor samples and paired blood controls undergo NGS comprehensive genome profiling with a 450‐gene panel. The majority (46%) of the patients were stage IV, followed by 25% stage III, 22% stage II, and 6% stage I. Nine percent of the patients (55/609) were MSI‐H determined by either polymerase chain reaction (PCR)/IHC‐ or NGS‐based algorithm or both. Forty‐six percent of the patients carried variants in the KRAS gene, whereas only 8% carried BRAF mutations (Table 1). MSI‐H CRCs were found far less commonly at late stage IV (20%) versus microsatellite stable (MSS) CRCs (49%; Fig. 1A).
Table 1.
Characteristics of patients with colorectal cancer
| Total (n = 609) | n (%) |
|---|---|
| Gender | |
| Male a | 372 (61) |
| Female b | 237 (39) |
| Disease stage | |
| I | 37 (6) |
| II | 135 (22) |
| III | 153 (25) |
| IV | 282 (46) |
| Unknown | 2 (/) |
| MMR/MSI | |
| dMMR/MSI‐H | 55 (9) |
| pMMR/MSS | 554 (91) |
| KRAS | |
| Wild type | 327 (54) |
| Mutant | 282 (46) |
| BRAF | |
| Wild type | 561 (91) |
| Mutant | 48 (8) |
Median age: 61 years.
Median age: 60 years.
Abbreviations: dMMR, mismatch repair deficiency; MMR, mismatch repair; MSI, microsatellite instability; MSI‐H, MSI‐high; MSS, microsatellite stable; pMMR, mismatch repair proficiency.
Figure 1.
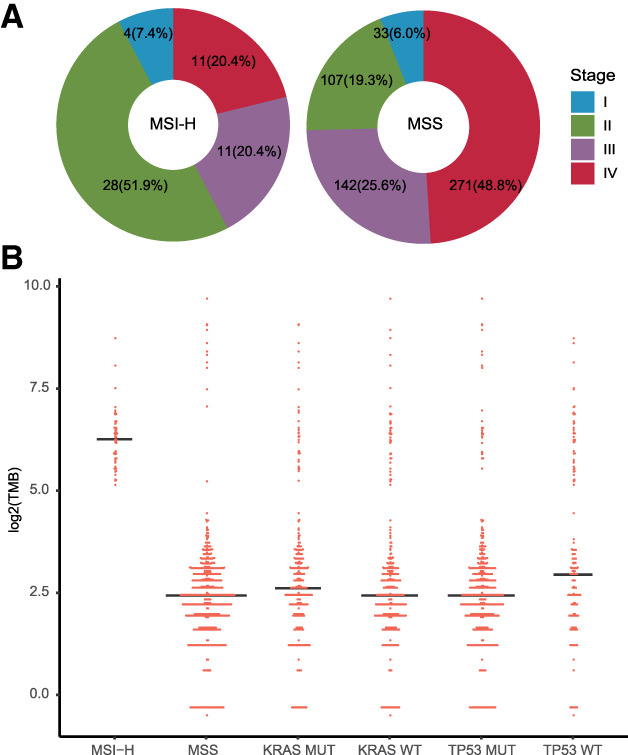
TMB's association with MSI, TP53, and KRAS mutations. (A): MSI‐H colorectal cancers (CRCs) were found more commonly at early stage I or stages II and III (81%) versus the MSS CRC subcohort (51%). (B): Microsatellite statuses and TMB are shown for each tumor (n = 609). The MSI‐H subcohort's median TMB (n = 55) is 59.4 mutations per megabase (mut/Mb), which is much higher than the MSS subcohort (n = 554) at 5.4 mut/Mb, with the exclusion of patients with POLE pathogenic mutation. KRAS mutations previously shown, causing elevated TMB in lung cancer, did not result in higher TMBs in CRC. Wild‐type TP53 was associated with a marginally higher TMB, mainly because of the relatively enriched MSI‐H cases in this cohort. Abbreviations: MSI, microsatellite instability; MSI‐H, MSI‐high; MSS, microsatellite stable; MUT, mutant; TMB, tumor mutational burden; WT, wild type.
The Genomic Alteration Landscape of Colorectal Cancer
The median TMB of MSI‐H CRC samples at 59.4 (range, 35–264.9) mutations per megabase (mut/Mb) was significantly higher than the MSS subgroup's 5.4 (range, 0.7–37.1) mut/Mb, with the exclusion of pathogenic POLE mutation samples (p < .0001; Fig. 1B) [23].
Genomic profiling of the 609 Chinese patients with CRC revealed a typical CRC genomic alteration landscape with the most frequent mutations observed in TP53 (76%), APC (72%), and KRAS (46%). However, these results had a lower frequency compared with a similar study on FFPE samples in a U.S. cohort [24, 25]. Recurrent somatic mutations in genes including ACVR2A, ACVR1B, TGFBR2, CTNNB1, ARID1A, and mismatch repair–related genes (MSH6, MSH2, and MLH1) were observed more frequently in MSI‐H tumors, whereas APC and TP53 were less frequently alternated than they were in MSS CRC (Fig. 2). Fifteen patients with MSI‐H CRC carried germline pathogenic mutations in mismatch repair–related genes, such as frameshift in MSH2 (Q170Rfs*4), truncations in MLH1 (R226* and E609*), SNVs in MLH1 (R265C and G67R), and splice site mutation in MLH1 (c.1038+1G>T). Although mutated TP53 or KRAS has been reported associated with higher TMB in lung cancer, neither of these mutations had elevated TMB in patients with CRC (Fig. 1B).
Figure 2.
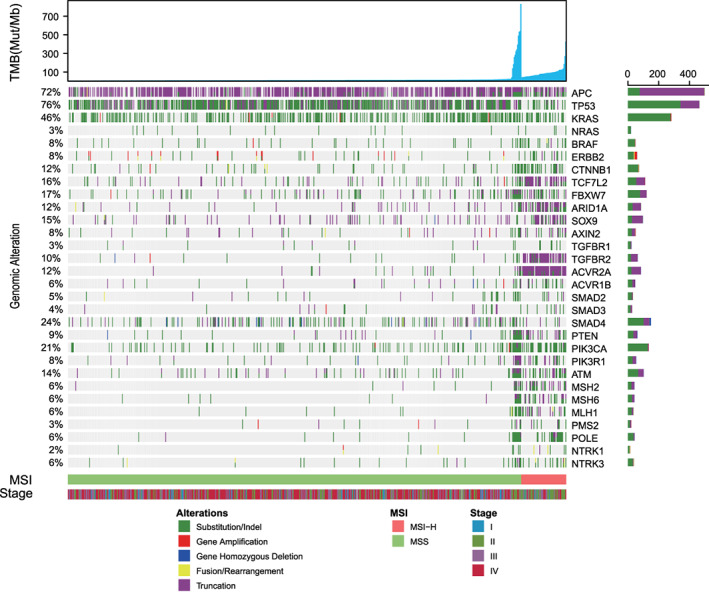
Landscape of colorectal cancer genomic alterations. A total of 609 patients with colorectal cancer were classified into two groups based on their microsatellite status. The remaining pathologic and genomic information was plotted accordingly. In each subcohort, patients were arranged by their TMB values in ascending order, as follows: individuals with extremely high TMB at the MSS section carrying POLE pathogenic mutations; patients in MSI segment displaying overall elevated mutational burden. The bottom half of the figure shows that MSS colorectal cancer's genomic alteration was TP53, APC, and KRAS, whereas the high frequency mutated genes in the MSI counterpart were ACVR2A, ACVR1B, TGFBR2, CTNNB1, ARID1A, and mismatch repair–related genes (i.e., MSH6, MSH2, and MLH1). Abbreviations: MSI, microsatellite instability; MSI‐H, MSI‐high; MSS, microsatellite stable; mut/Mb, mutations per megabase; TMB, tumor mutational burden.
The signal pathway analysis in 609 patients with CRC revealed the presence of dysregulations in well‐defined pathways (i.e., WNT, MAPK, PI3K, TGF‐β, and P53 pathways). In line with prior reports, the latter were related to cell proliferation, genomic stability, and apoptosis [25, 26]. We found that the WNT signaling pathway was altered in 84% of MSS CRCs and 98% of MSI‐H CRCs (p = .002) because of the higher incidence of activating mutations of CTNNB1 and deletions in TCF7L2, SOX9, ARID1A, and FBXW7. Interestingly, APC's mutation in the WNT signaling pathway was frequently found in both MSS (73%) and MSI‐H (64%) CRCs. Such observations suggest that, in both cohorts, APC mutation was the dominant alteration affecting this pathway. It has been shown that, in CRC, the TGF‐β signaling pathway is also extensively deregulated. Genomic alterations in the TGF‐β pathway's genes, including TGFBR1, TGFBR2, ACVR2A, ACVR1B, SMAD2, SMAD3, and SMAD4, were found in 37% of the MSS and 96% of the MSI‐H CRCs (p < .00001). Additionally, genes such as TGFBR2 and ACVR2A, containing microsatellite sites in their coding regions, were mutated at a very high frequency in the MSI‐H group compared with the MSS group (76% vs. 3%, p < .00001 and 93% vs. 4%, p < .00001, respectively). Furthermore, we evaluated the TP53 pathway, which is potentially targetable by a newly developed drug (AZD1775), displaying promising clinical efficacy in head and neck squamous cell carcinoma [27]. Of note, alterations in TP53 and ATM, a kinase that phosphorylates and activates TP53 after DNA damage, was found in 62% of MSI‐H and 84% of MSS CRCs (Fig. 3). Additionally, the KRAS G12C variant was discovered in 16 (2.6%) patients. Importantly, this variant is a potential target for a small molecule such as AMG 510 [28].
Figure 3.
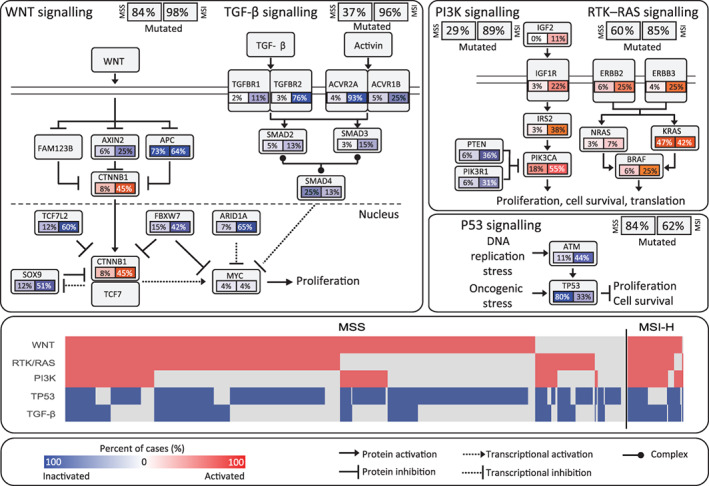
Frequently deregulated signaling pathways in colorectal cancer (CRC). MSI‐H and MSS CRC samples with confirmed genetic variants in related signal pathways such as WNT, TP53, RAS, and TGF‐β were analyzed separately. Alteration frequencies of activated (red) and inactivated (blue) alterations are expressed as a percentage of all cases in each cohort. Samples with alterations in five pathways—WNT, RTK/RAS, PIK3K, TP53, and TGF‐β—are plotted in the middle panel. Abbreviation: MSI, microsatellite instability; MSI‐H, MSI‐high; MSS, microsatellite stable.
High Incidence of NTRK Gene Fusion in Patients with MSI‐H CRC
NTRK fusions occur at low frequencies in most types of cancers. CRC is among the tumor types that have been shown to harbor NTRK fusions with frequency around 1% [12]. Consistently with previous reports, in the present study, six NTRK gene fusion cases were identified of 609 samples. However, we observed that the prevalence of NTRK fusion was around 7% in MSI‐H CRCs (4/55), which was 20‐fold higher than in the MSS CRC (0.36%) subcohort (p = .0008). The four NTRK fusions in the MSI‐H cohort are well‐characterized functional chimeric TPR (e21)–NTRK1 (e11), TPM3 (e7)–NTRK1 (e9), and two cases of ETV6 (e5)–NTRK3 (e15). The two NTRK fusions in the MSS cohort were LMNA (e4)–NTRK1 (e10) and TPM3 (e7)–NTRK1 (e9). The NTRK fusions were more frequent in older patients (median age, 72 years; range, 57–76 years vs. 60 years; range, 17–96 years; p = .03). NTRK fusions have been known as critical drivers for cancer development. With this in mind, we further analyzed the genomic alterations of six CRC cases with NTRK fusions to evaluate the co‐occurrence of driver gene mutation. We observed the inactivation of tumor suppressor genes such as APC, TP53, ACVR2A, and TGFBR2. However, the activating variants of oncogenes (e.g., KRAS and BRAF) were not identified in patients with either MSS or MSI‐H CRC with NTRK fusion (Fig. 4). In addition, except for three MSI‐H CRCs with NTRK fusions that were at stage II, the rest of tumor samples with presumably productive fusions were at either stage III or stage IV (Table 2). The gene rearrangement events were significantly more prevalent in the MSI‐H (6/55, 10.9%) than in the MSS (6/554, 1.1%) cohort (p = .0003).
Figure 4.
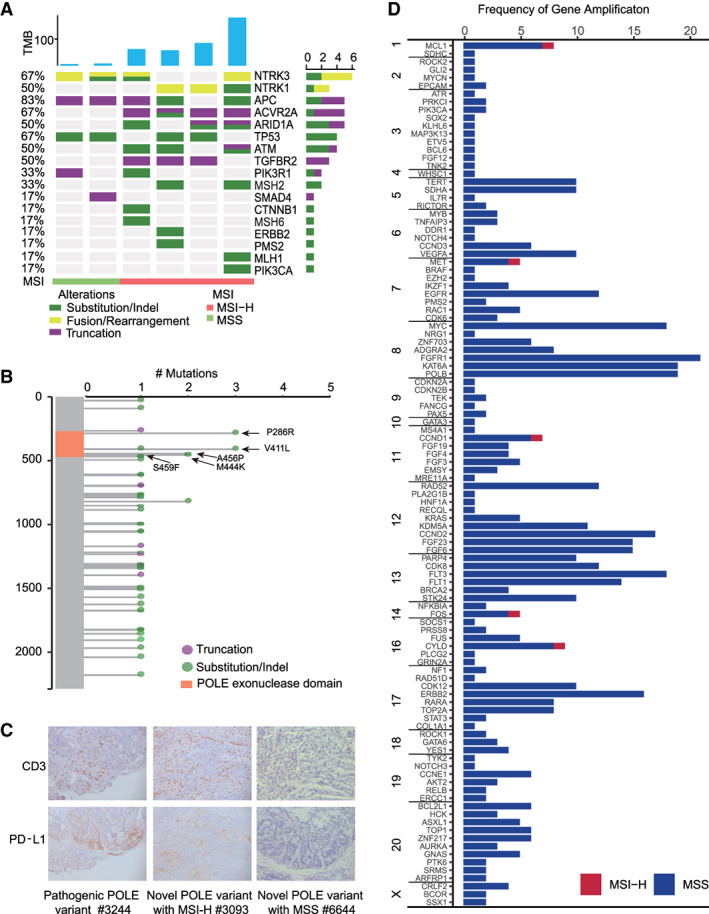
Variants of NTRK and POLE. (A): Genomic profiling of NTRK gene fusion–positive colorectal cancer (CRC). The NTRK‐positive cancer lacks other activating gene variants and is enriched in MSI‐H CRC. (B): The mutations in the DNA polymerase proofreading exonuclease domain encoded by exons 9 to 14 (residues 268–471), causing exceptional high mutational burden, are designated as pathogenic. The remaining mutations in the nonproofreading region appear to be benign. (C): Immunohistochemical analysis of CD3 and PD‐L1 for CRC carrying POLE pathogenic mutation. A highly abundant infiltration of CD3‐positive lymphocytes was observed in all five CRC samples carrying POLE pathogenic mutation. Additionally, a positive staining for PD‐L1 was found in #3634 (TPS, 5%) and #3244 (TPS, 60%). (D): The red and blue bars represent gene amplification events occurring in MSI‐H and MSS CRC, respectively. The event of gene amplification occurred much less frequently in the MSI‐H (5 events in 55 patients) than in the MSS (668 events in 554 patients) cohort. Abbreviations: MSI, microsatellite instability; MSI‐H, MSI‐high; MSS, microsatellite stable; TMB, tumor mutational burden; TPS, tumor proportion score.
Table 2.
Gene fusion events that were predicted to generate functional chimeric proteins
| ID | Gene pair | MSI | TMB | Stage | KRAS/BRAF | Novel |
|---|---|---|---|---|---|---|
| 4080 | FFP‐SEPT14 | MSS | 10.1 | III | N/A | Reported |
| 5172 | ERBB2‐GRB7 | MSS | 8 | III | N/A | Reported |
| 6444 | LMNA‐NTRK1 | MSS | 6.2 | IV | N/A | Reported |
| 4089 | MAP3K9‐ROS1 | MSS | 6.1 | IV | N/A | Novel |
| 1959 | STARD3‐ERBB2 | MSS | 3 | III | N/A | Novel |
| 8050 | TPM3‐NTRK1 | MSS | 8.5 | III | N/A | Reported |
| 2105 | CUL1‐BRAF | MSI‐H | 82.8 | III | BRAF | Reported |
| 3943 | ETV6‐NTRK3 | MSI‐H | 59.4 | III | N/A | Reported |
| 8115 | ETV6‐NTRK3 | MSI‐H | 180.3 | II | N/A | Reported |
| 9942 | FGFR2‐PIBF1 | MSI‐H | 73.5 | III | N/A | Novel |
| 7363 | TPM3‐NTRK1 | MSI‐H | 82.8 | II | N/A | Reported |
| 3950 | TPR‐NTRK1 | MSI‐H | 54.9 | II | N/A | Reported |
Twelve gene fusion events in this cohort that were predicted to generate functional chimeric proteins are listed in this table. Three of them, including MAP3K9‐ROS1, STARD3‐ERBB2, and FGFR2‐PIBF1, had not been reported previously.
Abbreviations: MSI, microsatellite instability; MSI‐H, MSI‐high; MSS, microsatellite stable; N/A, not altered; TMB, tumor mutational burden.
High Incidence of POLE Mutation in Chinese Patients with CRC
We identified a high incidence (6%, 39/609) of Chinese patients with CRC harboring POLE somatic mutations either confirmed by the COSMIC and MKSCC database or novel (Fig. 2) [29]. Despite the high frequency of somatic mutations, we did not find any germline pathogenic POLE variants. Specifically, we observed only one benign splice site mutation (c.2865‐4delT) in our cohort. The recurrent variants within the DNA polymerase proofreading exonuclease domain encoded by exons 9 to 14 (residues 268–471) are known to cause an exceptionally high mutational burden in CRC [30]. As a consequence, they were designated as pathogenic mutations. In the present cohort, the frequency of POLE pathogenic mutations, including ten cases of V411L, P286R, M444K, A456P, and a dual mutation S459F/Y468C, was 1.8% (11/609), which is higher than 1% in the European counterpart (Fig. 4B) [13]. The genome of patients carrying recurrent POLE pathogenic mutations was hypermutated with extremely high TMB compared with the whole cohort (median 361 vs. 5 mut/Mb). Of note, all of them were MSS (Table 3). Interestingly, four POLE pathogenically mutated CRCs carried MSH2/MSH6/PSM2‐truncated variants, however, with a stable microsatellite. We therefore assumed that these truncated variants were monoallelic, and the combination of POLE pathogenic mutation and the dMMR might be detrimental to the cancer cells. Moreover, 17 novel somatic variants of uncertain significance (VUS) of POLE were found in 13 patients (13/609, 2%), and all of these variants were located at the regions out of the DNA polymerase proofreading exonuclease domain (Table 3). Of note, of these individuals, 11 were associated with MSI‐H status. The general association of these novel VUS mutations with MSI‐H status might indicate the collateral damage caused by the deficiency of a mismatch repair system. And the high TMB scores of these individuals, except for one with novel somatic POLE variants, resembled the feature of MSI‐H rather than the POLE pathogenic mutation [31]. The sample (#9104) with TMB at 421 mut/Mb carried three mutations at exon 31 (T1322A), exon 43 (E1968G), and exon 23 (P886S), which may cause the defect in proofreading function by altering the tertiary structure of POLE. We subsequently performed anti–PD‐L1 and CD3 IHC staining on 12 available CRC tissue samples including seven pathogenic POLE mutations, three VUS POLE mutations with MSI‐H status, and two VUS POLE mutations with MSS status. We found the presence of abundant/high abundant CD3‐positive lymphocytes infiltration in six of seven CRC tumor samples with pathogenic POLE mutations and all three MSI‐H CRC samples with POLE mutations but not in MSS CRC samples with POLE mutations. Additionally, elevated PD‐L1 expression was observed in six of seven CRC tumor samples with pathogenic POLE mutations and in three cases was particularly high (ID: 3634, CPS, 25; ID: 4371, CPS, 30; and ID: 3244, CPS, 80; Fig. 4C). In additional, the three MSI‐H CRC samples with POLE mutations also displayed elevated PD‐L1 expression, but not the MSS CRC samples with POLE mutations (Table 3).
Table 3.
POLE genomic alterations in patients with colorectal cancer
| ID | Age | Sex | Stage | MSS MSI‐H | TMB | DNA change | AA change | Exon | CD3 | PD‐L1CPS |
|---|---|---|---|---|---|---|---|---|---|---|
| 4371 | 30 | M | I | MSS | 336 | 857C>G | P286R | 9 | 4 | 30 |
| 8186 | 42 | F | II | MSS | 825 | 857C>G | P286R | 9 | / | / |
| 5892 | 23 | M | II | MSS | 525 | 857C>G | P286R | 9 | / | / |
| 0698 | 32 | F | IV | MSS | 387 | 1231G>T | V411L | 13 | 4 | 2 |
| 2231 | 47 | M | IV | MSS | 176 | 1231G>T | V411L | 13 | 2 | 1 |
| 3021 | 68 | M | II | MSS | 132 | 1231G>T | V411L | 13 | 3 | 4 |
| 9759 | 39 | M | III | MSS | 533 | 1331T>A | M444K | 14 | / | / |
| 8381 | 43 | M | II | MSS | 279 | 1331T>A | M444K | 14 | 3 | 5 |
| 3634 | 55 | M | II | MSS | 317 | 1366G>C | A456P | 14 | 3 | 25 |
| 2525 | 36 | M | II | MSS | 254 | 1366G>C | A456P | 14 | / | / |
| 3244 | 52 | M | IV | MSS | 484 | 1376C>T | S459F | 14 | 4 | 80 |
| 1403A>G | Y468C | |||||||||
| 6644 | 62 | M | II | MSS | 10 | 89C>T | p.S30L | 2 | 1 | 0 |
| 1388 | 39 | M | IV | MSS | 19 | 3652G>C | p.V1218L | 30 | 1 | 0 |
| 7414 | 55 | M | II | MSI‐H | 61 | 5021C>T | p.A1674V | 38 | / | / |
| 0366 | 63 | M | IV | MSI‐H | 78 | 2084T>A | p.F695Y | 19 | / | / |
| 5468G>A | p.R1823H | 40 | ||||||||
| 4123 | 45 | F | II | MSI‐H | 85 | 1225A>G | p.R409G | 12 | / | / |
| 9942 | 71 | F | III | MSI‐H | 74 | 2324G>T | p.W775L | 21 | / | / |
| 2460 | 33 | M | II | MSI‐H | 89 | 6119C>T | p.A2040V | 44 | / | / |
| 2786 | 42 | M | II | MSI‐H | 92 | 4005G>T | p.Q1335H | 31 | / | / |
| 4193_4194del | P.Y1398* | 33 | ||||||||
| 3093 | 47 | M | III | MSI‐H | 116 | 4027G>A | p.G1343S | 32 | 4 | 30 |
| 1886 | 75 | F | IV | MSI‐H | 116 | 4493C>T | p.A1498V | 35 | / | / |
| 7281 | 55 | F | II | MSI‐H | 100 | 5464T>A | p.Y1822N | 40 | 4 | 8 |
| 0737 | 43 | F | II | MSI‐H | 124 | 1823C>T | p.A608V | 17 | / | / |
| 9104 | 50 | M | IV | MSI‐H | 421 | 3964A>G | T1322A | 31 | 3 | 1 |
| 5903A>G | p.E1968G | 43 | ||||||||
| 2656C>T | p.P886S | 23 |
Patients with colorectal cancer harboring pathogenic (n = 11, with gray background) and novel (n = 13, with blank background) POLE somatic mutations were listed below. Patients with colorectal cancer (CRC) with pathogenic POLE mutations are younger than the entire CRC cohort (median 42 vs. 61 years), and the majority of them are male (82%, 9/11). The novel POLE variants were mainly associated with MSI‐H status, which indicated collateral damage caused by the deficiency of mismatch repair system. The samples with pathogenic POLE mutations and novel POLE somatic mutations with MSI‐H were associated with upregulated expression of PD‐L1 and elevated density of tumor‐infiltrating lymphocytes.
Abbreviations: AA, amino acids; CPS, combined positive score; F, female; M, male; MSI‐H, MSI‐high; MSS, microsatellite stable; TMB, tumor mutational burden.
ERBB2/HER2 Amplification in Chinese Patients with CRC
About 3% (16/609) of the patients with CRC in this cohort harbored ERBB2/HER2 amplifications, 14 of which were KRAS wild type. Therefore, the 16 patients with MSS CRC and ERBB2 amplification may be candidates for HER2‐targeted therapies (e.g., trastuzumab + lapatinib/pertuzumab/tucatinib) [32]. We also found that all (n = 10) of the CDK12 gene amplifications, located close to ERBB2 at 17q12, were concurrent with ERBB2 amplification. Moreover, all 16 cases of ERBB2 amplification occurred exclusively in MSS CRC (n = 554; Fig. 4D). Specifically, across our cohort, only 5 of 673 gene amplification events took place in MSI‐H CRC (0.1 gene amplification per patient). Such a finding is significantly lower than in the MSS subcohort (1.2 gene amplification per patient; Fig. 4D). This observation likely reflects the types of mutations prone to accumulate in dMMR, with genomic rearrangements being less common through this mechanism. Additionally, 43 confirmed somatic ERBB2 point mutations were detected in 35 patients with CRC (35/609, 6%), including 23 with MSS and 12 with MSI‐H tumors.
Discussion
Here we show the genomic landscape of a Chinese cohort, mostly comprising patients with advanced CRC. We believe that this is the first report highlighting the high frequency of NTRK gene fusion in patients with MSI‐H CRC, as well as the higher incidence of pathogenic POLE mutations in Chinese patients. Importantly, this study has immediate therapeutic implications for these patient subsets. Current CRC biomarkers, including RAS/RAF, can spare patients toxicities from therapies (i.e., EGFR inhibitors) that are not effective in this genomically defined group. Newer biomarkers, including NTRK, HER2, and POLE, are becoming increasingly important. Non‐small cell lung cancer (NSCLC) remains the paradigm for precision medicine, and U.S. guidelines (from the National Comprehensive Cancer Center) now recommend broad panel‐based testing to assess the multiple therapeutic targets in NSCLC. In CRC, there is an increasing need to simultaneously assess multiple candidate biomarkers and spare potentially repeated invasive procedures. Additionally, expanding the portion of patients with CRC who may benefit from immunotherapy approaches will require broad genomic profiling. Our data set adds to the current literature and makes several hypotheses generated by observations in Chinese patients.
MSI formerly served as a biomarker for FOLFOX‐based adjuvant treatment, as observed in the ACCENT database containing clinical information from around 8,000 patients with colon cancer. Specifically, MSI‐H patients at stage II had a more favorable prognosis and could routinely be spared cytotoxic therapy, whereas patients at stage III were advised to receive adjuvant chemotherapy irrespective of MSI status [33]. Recently, MSI has been approved by the FDA as a pancancer biomarker for the PD‐1 checkpoint inhibitor pembrolizumab, partly because of the extraordinary response of patients with MSI‐H CRC to the therapy [4, 5]. Compared with a small number of mononucleotide MSI markers used by traditional PCR assays, NGS‐based MSI detection aims to scan massive amounts of microsatellite loci more comprehensively [34, 35]. Of note, our NGS‐based MSI determination algorithm, evaluating a large number of instability‐prone microsatellite loci in the CRC genome, yields 100% concordance with either the classic PCR MSI or IHC mismatch repair assays or both. Additionally, the tight association between MSI‐H status and TMB value reinforces the patient selection function of comprehensive genomic profiling for immunotherapy.
Around 1% of patients in this cohort carried NTRK gene fusions, which more frequently occurred in elderly patients (p = .03). NTRK gene fusion occurs at a high frequency in the MSI‐H CRC cohort (7%). This finding is consistent with the previous studies reporting that ALK, RET, ROS1, and NTRK gene fusions are rare and associated with older age and MSI‐H status [36, 37, 38]. The constitutive signal from the NTRK fusion protein seems critical for cancer development because this subset lacks common activating mutations in KRAS or BRAF. Furthermore, it has a lower frequency of CTNNB1‐ and PIK3CA‐activating mutations. Therefore, it may provide this already treatable cohort an additional opportunity for highly effective therapy. Patients with MSI‐H CRC resistant to anti–PD‐1 immunotherapy may benefit from the treatment targeting NTRK fusion by larotrectinib and entrectinib.
Accumulating evidence from clinical trials has suggested that ERBB2 amplification might be a potential biomarker for CRC treatment. The HERACLES trial observed a remarkable 30% objective response rate (ORR) to dual‐targeted therapy (i.e., trastuzumab and lapatinib) in heavily pretreated metastatic CRC with ERBB2 amplification but KRAS codon 12/13 wild type. The HERACLES trial result is also in line with data from the MyPathway basket trial, which observed 38% ORR in patients with refractory disease with the overexpression of HER2 regardless of KRAS status. Together, the preclinical data from both trials supported inhibition of HER2 with trastuzumab plus either lapatinib or pertuzumab as an effective treatment option. These results represent a breakthrough in CRC treatment, even though they apply only to about 3% of Chinese patients with MSS CRC. Although IHC and fluorescence in situ hybridization constitute the current standard for HER2 amplification's assessment, the hybrid‐based comprehensive genomic profiling is a surrogate for the evaluation of this genomic alteration, with a 98.4% overall concordance [39]. An advantage of genomic profiling is the ability to assess the copy number quantitatively, and subset analyses suggest that higher copy number may be associated with improved outcomes [17].
A key observation in our series is the finding that pathogenic POLE mutations exist in 1.8% of Chinese patients with CRC, which is higher than in the Western population. Our finding is preliminary and potentially confounded by the stage distribution. Similar to MSI, patients with POLE‐mutant nonmetastatic disease have lower recurrence rates, as well as metastatic frequency, owing to enhanced immune surveillance [13]. Risk factors for POLE‐mutant CRC are unknown. Future studies should focus on exploring the relationship between a traditional Chinese diet and how potential germline differences increase the risk of POLE mutation. Although the frequency of POLE pathogenic mutation is relatively low, the extraordinary response of patients with CRC and POLE pathogenic mutation to immune checkpoint inhibition makes it an attractive biomarker. CRC tumors with POLE mutations are constantly associated with extremely high TMB. However, a large proportion of these patients are MSS, and standard IHC or PCR alone would oversee them. Hence, the direct sequencing of POLE exons containing either functional domains or the indirect evaluation of TMB value of CRC tissues or both is the ideal way to discover mutations in this gene [31]. The significant mutual exclusion of POLE pathogenic mutation and dMMR indicates that the concurrent alteration of these two DNA proofreading and repair mechanisms might be detrimental to the cancer cell. Our data suggest that testing for POLE mutations in CRC holds the promise of identifying a subgroup of patients who are excellent candidates for immune checkpoint inhibitor therapy. The extensive CD3 infiltration seen in our POLE‐mutant cases is consistent with the immunogenicity of these tumors [40, 41].
Conclusion
In summary, we confirm the feasibility and potential of identifying predictive and prognostic biomarkers in a cohort of Chinese patients with CRC by comprehensive genomic profiling. The observation that POLE mutations may exist at higher frequency requires further validation. If confirmed, this finding would expand the number of patients in China who may benefit from immune‐based approaches.
Author Contributions
Conception/design: Weifeng Wang, Ming Xiao
Provision of study material or patients: Yun Guo, Xian‐ling Guo, Shuang Wang, Xinyu Chen
Collection and/or assembly of data: Yun Guo, Xian‐ling Guo, Shuang Wang, Xinyu Chen
Data analysis and interpretation: Jiaochun Shi, Jian Wang, Kai Wang, Weifeng Wang
Manuscript writing: Samuel J. Klempner, Weifeng Wang, Ming Xiao
Final approval of manuscript: Yun Guo, Xian‐ling Guo, Shuang Wang, Xinyu Chen, Jiaochun Shi, Jian Wang, Kai Wang, Samuel J. Klempner, Weifeng Wang, Min Xiao
Disclosures
Jiaochun Shi: OrigiMed (E); Jian Wang: OrigiMed (E); Samuel J. Klempner: Foundation Medicine, Inc., Eli Lilly & Co., Merck, Bristol‐Myers Squibb, Pieris (C/A), Turning Point Therapeutics (OI); Weifeng Wang: OrigiMed (E). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Contributor Information
Weifeng Wang, Email: wangwf@origimed.com.
Min Xiao, Email: min.xiao@shulan.com.
References
- 1. Chen W, Zheng R, Baade PD et al. Cancer statistics in China, 2015. CA Cancer J Clin 2016;66:115–132. [DOI] [PubMed] [Google Scholar]
- 2. Venook AP, Niedzwiecki D, Lenz HJ et al. Effect of first‐line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild‐type advanced or metastatic colorectal cancer: A randomized clinical trial. JAMA 2017;317:2392–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schirripa M, Lenz HJ. Biomarker in colorectal cancer. Cancer J 2016;22:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Overman MJ, McDermott R, Leach JL et al. Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): An open‐label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 2015;5:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lae M, Freneaux P, Sastre‐Garau X et al. Secretory breast carcinomas with ETV6‐NTRK3 fusion gene belong to the basal‐like carcinoma spectrum. Mod Pathol 2009;22:291–298. [DOI] [PubMed] [Google Scholar]
- 8. Tognon C, Knezevich SR, Huntsman D et al. Expression of the ETV6‐NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002;2:367–376. [DOI] [PubMed] [Google Scholar]
- 9. Skalova A, Vanecek T, Sima R et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6‐NTRK3 fusion gene: A hitherto undescribed salivary gland tumor entity. Am J Surg Pathol 2010;34:599–608. [DOI] [PubMed] [Google Scholar]
- 10. Bourgeois JM, Knezevich SR, Mathers JA et al. Molecular detection of the ETV6‐NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol 2000;24:937–946. [DOI] [PubMed] [Google Scholar]
- 11. Hsiao SJ, Zehir A, Sireci AN et al. Detection of tumor NTRK gene fusions to identify patients who may benefit from tyrosine kinase (TRK) inhibitor therapy. J Mol Diagn 2019;21:553–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cocco E, Scaltriti M, Drilon A. NTRK fusion‐positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018;15:731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Domingo E, Freeman‐Mills L, Rayner E et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol 2016;1:207–216. [DOI] [PubMed] [Google Scholar]
- 14. Mehnert JM, Panda A, Zhong H et al. Immune activation and response to pembrolizumab in POLE‐mutant endometrial cancer. J Clin Invest 2016;126:2334–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong J, Wang C, Lee PP et al. Response to PD‐1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Canc Netw 2017;15:142–147. [DOI] [PubMed] [Google Scholar]
- 16. Johanns TM, Miller CA, Dorward IG et al. Immunogenomics of hypermutated glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 2016;6:1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sartore‐Bianchi A, Trusolino L, Martino C et al. Dual‐targeted therapy with trastuzumab and lapatinib in treatment‐refractory, KRAS codon 12/13 wild‐type, HER2‐positive metastatic colorectal cancer (HERACLES): A proof‐of‐concept, multicentre, open‐label, phase 2 trial. Lancet Oncol 2016;17:738–746. [DOI] [PubMed] [Google Scholar]
- 18. Meric‐Bernstam F, Hurwitz H, Raghav KPS et al. Pertuzumab plus trastuzumab for HER2‐amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open‐label, phase 2a, multiple basket study. Lancet Oncol 2019;20:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao J, Chen L, Li H et al. An accurate and comprehensive clinical sequencing assay for cancer targeted and immunotherapies. The Oncologist 2019;24:e1294–e1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roach C, Zhang N, Corigliano E et al. Development of a companion diagnostic PD‐L1 immunohistochemistry assay for pembrolizumab therapy in non‐small‐cell lung cancer. Appl Immunohistochem Mol Morphol 2016;24:392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dahlin AM, Henriksson ML, Van Guelpen B et al. Colorectal cancer prognosis depends on T‐cell infiltration and molecular characteristics of the tumor. Mod Pathol 2011;24:671–682. [DOI] [PubMed] [Google Scholar]
- 22. Kulangara K, Zhang N, Corigliano E et al. Clinical utility of the combined positive score for programmed death ligand‐1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med 2019;143:330–337. [DOI] [PubMed] [Google Scholar]
- 23. Cicek MS, Lindor NM, Gallinger S et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population‐ and clinic‐based colorectal tumors results from the Colon Cancer Family Registry. J Mol Diagn 2011;13:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ross JS, Wang K, Khaira D et al. Comprehensive genomic profiling of clinically advanced colorectal carcinoma to reveal frequent opportunities for targeted therapies. J Clin Oncol 2015;33:3553–3553. [Google Scholar]
- 25. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dienstmann R, Vermeulen L, Guinney J et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer 2017;17:268. [DOI] [PubMed] [Google Scholar]
- 27. Mendez E, Rodriguez CP, Kao MC et al. A phase I clinical trial of AZD1775 in combination with neoadjuvant weekly docetaxel and cisplatin before definitive therapy in head and neck squamous cell carcinoma. Clin Cancer Res 2018;24:2740–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lanman BA, Allen JR, Allen JG et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J Med Chem 2020;63:52–65. [DOI] [PubMed] [Google Scholar]
- 29. Schrock AB, Fabrizio D, He Y et al. Analysis of POLE mutation and tumor mutational burden (TMB) across 80,853 tumors: Implications for immune checkpoint inhibitors (ICPIs). Ann Oncol 2017;28(suppl 5):V415. [Google Scholar]
- 30. Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev 2014;24:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stadler ZK, Battaglin F, Middha S et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next‐generation sequencing panels. J Clin Oncol 2016;34:2141–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strickler JH, Zemla T, Ou FS et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): Initial results from the MOUNTAINEER trial. Ann Oncol 2019;30(suppl 5):V200. [Google Scholar]
- 33. Sinicrope FA, Mahoney MR, Smyrk TC et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX‐based adjuvant chemotherapy. J Clin Oncol 2013;31:3664–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brahmer JR, Drake CG, Wollner I et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pietrantonio F, Di Nicolantonio F, Schrock AB et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol 2018;29:1394–1401. [DOI] [PubMed] [Google Scholar]
- 37. Yakirevich E, Resnick MB, Mangray S et al. Oncogenic ALK fusion in rare and aggressive subtype of colorectal adenocarcinoma as a potential therapeutic target. Clin Cancer Res 2016;22:3831–3840. [DOI] [PubMed] [Google Scholar]
- 38. Pietrantonio F, Di Nicolantonio F, Schrock AB et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst 2017;109:djx089. [DOI] [PubMed] [Google Scholar]
- 39. Ross DS, Zehir A, Cheng DT et al. Next‐generation assessment of human epidermal growth factor receptor 2 (ERBB2) amplification status: Clinical validation in the context of a hybrid capture‐based, comprehensive solid tumor genomic profiling assay. J Mol Diagn 2017;19:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Gool IC, Eggink FA, Freeman‐Mills L et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res 2015;21:3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Howitt BE, Shukla SA, Sholl LM et al. Association of polymerase e‐mutated and microsatellite‐instable endometrial cancers with neoantigen load, number of tumor‐infiltrating lymphocytes, and expression of PD‐1 and PD‐L1. JAMA Oncol 2015;1:1319–1323. [DOI] [PubMed] [Google Scholar]