

Abstract
Background
Patients with multicentric Castleman disease (MCD) who are negative for human immunodeficiency virus and human herpesvirus 8 are considered to have idiopathic MCD (iMCD). The clinical presentation of iMCD varies from mild constitutional symptoms to life‐threatening symptoms or death. The treatment strategy varies from “watchful waiting” to high‐dose chemotherapy. This diverse clinical presentation calls for a classification stratification system that takes into account the severity of the disease.
Subjects, Materials, and Methods
We analyzed the clinical, laboratory, and pathologic abnormalities and treatment outcomes of 176 patients with iMCD (median follow‐up duration 12 years) from the U.S. and China to better understand the characteristics and prognostic factors of this disease. This discovery set of iMCD results was confirmed from the validation set composed of additional 197 patients with iMCD organized from The International Castleman Disease Consortium.
Results
Using these data, we proposed and validated the iMCD international prognostic index (iMCD‐IPI), which includes parameters related to patient characteristics (age > 40 years), histopathologic features (plasma cell variant), and inflammatory consequences of iMCD (hepatomegaly and/or splenomegaly, hemoglobin <80 g/L, and pleural effusion). These five factors stratified patients according to their performance status and extent of organ dysfunction into three broad categories: low risk, intermediate risk, and high risk. The iMCD‐IPI score accurately predicted outcomes in the discovery study cohort, and the results were confirmed on the validation study cohort.
Conclusion
This study represents the largest series of studies on patients with iMCD in the field and proposed a novel risk‐stratification model for iMCD‐IPI that could be used to guide risk‐stratified treatment strategies in patients with iMCD.
Implications for Practice
Patients with idiopathic multicentric Castleman disease (iMCD) can benefit from care based on clinical symptoms and disease severity. This study in 176 patients with iMCD constructed an iMCD‐IPI score based on five clinical factors, including age >40 years, plasmacytic variant subtype, hepatomegaly and/or splenomegaly, hemoglobin <80 g/L, and pleural effusion, and stratified patients into three risk categories: low risk, intermediate risk, and high risk. The predictive value was validated in an independent set of 197 patients with iMCD from The International Castleman Disease Consortium. The proposed novel model is valuable for predicting clinical outcome and selecting optimal therapies using clinical parameters.
Keywords: Castleman disease, Idiopathic multicentric Castleman disease, Prognosis, Treatment, Risk stratification, International prognostic index
Short abstract
This cooperative study analyzed the clinical, laboratory, and pathologic variables of patients with idiopathic multicentric Castleman disease (iMCD), resulting in a proposal for an iMCD international prognostic index that can help physicians agree on a treatment algorithm for iMCD.
Introduction
Castleman disease, first defined by Benjamin Castleman in 1954 [1], is a rare heterogeneous lymphoproliferative disorder of unknown etiopathogenesis commonly presenting with enlarged non‐neoplastic lymphadenopathy. Castleman disease is classified as unicentric or multicentric (MCD) based on the severity of clinical manifestations and the extent of pathologic involvement. Unicentric Castleman disease is typically a slow‐growing solitary mass occurring at a single anatomic site that can be successfully surgically removed [2, 3]. In contrast, MCD involves multiple lymph node stations, and symptoms include recurrent episodes of diffuse lymphadenopathy, systemic symptoms (also called B‐symptoms, including weight loss, fever, and/or fatigue), anemia, edema, hypoalbuminemia, and/or multiple organ system dysfunction [2, 4]. These symptoms can be fatal without proper treatment owing to dysregulated cytokine storming events. Castleman disease can also be subtyped based on characteristic histopathologic features, hyaline‐vascular (HV), plasma cell (PC), or mixed variant that has both HV and PC features [4, 5]. Human immunodeficiency virus (HIV) and human herpesvirus 8 (HHV‐8) are known to cause MCD, but approximately 50%–95% of patients with MCD are negative for HIV and HHV‐8; this is defined as idiopathic MCD (iMCD) [6, 7, 8, 9].
The clinical presentation of iMCD varies from mild constitutional symptoms to life‐threatening cytokine storms, organ failure, or death. Laboratory hallmarks include leukocytosis, anemia, thrombocytosis or thrombocytopenia, elevated erythrocyte sedimentation rate, increased C‐reactive protein (CRP) and fibrinogen, hypergammaglobulinemia, and hypoalbuminemia [9]. iMCD often responds to initial treatment but frequently relapses; thus, the treatment strategy varies from “watchful waiting” to high‐dose chemotherapy, and numerous treatment options have been proposed for patients with iMCD, but no effective regimen has been discovered [2, 10, 11, 12].
This diverse clinical presentation calls for a classification stratification system that takes into account the severity of the disease, allowing physicians to make precise treatment recommendations that are based on distinct iMCD subtypes. However, there is little consensus on classification approaches. The proposed TAFRO subtype, referring to thrombocytopenia (T), anasarca (A), fever (F), reticulum fibrosis of the bone marrow (R), and organomegaly (O), generally was accompanied by normal gamma‐globulin levels but severe clinical symptoms and poor outcomes [13]. The Castleman Disease Collaborative Network sought to stratify patients into two broad categories—severe and nonsevere—based on performance status and extent of organ dysfunction. In that stratification system, patients were considered to have severe iMCD if they met at least two of the following five criteria: Eastern Cooperative Oncology Group performance status ≥2; stage IV renal dysfunction (estimated glomerular filtration rate <30; creatinine >3.0); anasarca, ascites, or pleural/pericardial effusion effects of hypercytokinemia or low albumin; hemoglobin ≤8.0 g/dL; and pulmonary involvement or interstitial pneumonitis dyspnea. Patients who did not meet at least two of these criteria were considered to have nonsevere iMCD [14].
In retrospective case series of iMCD, several characteristics were associated with a poor clinical outcome, including the PC variant subtype, age > 40 years, TAFRO subtype, organ dysfunction, and inflammation levels [9, 15]. These analyses led to the proposal of a few prognostic indices, as described above, but none of these indices have been validated and/or widely adapted in clinical practice. Because iMCD is a complex disease with an annual incidence of only 1,000–1,500 cases in the U.S. and more frequent in Asian countries [16], iMCD presents a management challenge for most physicians, especially without a risk‐stratification model to help evaluate patient's condition and prognosis, and there are no existing recommendations for the available treatment modalities that consider disease severity in treatment decision‐making processes.
In the current study, we analyzed the clinical, laboratory, and pathologic variables of 176 patients with iMCD organized from 12 major academic medical centers in China and the U.S. to better understand the characteristics and prognostic factors of iMCD. This cooperative study culminated in a proposal for an iMCD international prognostic index (iMCD‐IPI). The results were further validated in 197 patients with iMCD organized from six academic medical centers in Japan, France, China, and the U.S. This simple, validated, and accurate system, similar to that proposed for lymphoma, can help physicians agree on a treatment algorithm for iMCD.
Subjects, Materials, and Methods
This multicenter retrospective study was designed to develop a risk‐stratification model for patients with iMCD. To establish the discovery study cohort, we consecutively enrolled adult patients (age ≥18 years) with iMCD treated at one of 12 large medical centers in China and the U.S. between January 2005 and February 2017. Treatment was based on the severity of clinical symptoms, varying from watchful waiting to curative‐intent combination chemotherapy regimens. For our analysis, the diagnosis of iMCD was reviewed and verified on the basis of demographics, disease characteristics, laboratory test results, and pathologic features, using international, evidence‐based consensus diagnostic criteria for iMCD [17]. Of these enrolled patients, 35 patients were diagnosed as mixed subtype because they displayed mixed pathologic features for both HV and PC presentations, and the clinical and laboratory features of these cases met the criteria of the consensus diagnostic guidelines [17]. Patients were excluded if the histologic subtype was uncertain or if they were diagnosed with unicentric Castleman disease. We excluded patients with concomitant malignancies, HHV‐8 infection and/or HIV infection, or POEMS (polyneuropathy, organomegaly, endocrinopathy, M‐protein, and skin pigmentation) syndrome, systemic lupus erythematosus (positive for anti–double‐stranded DNA or anti‐extractable nuclear antigen) and patients without laboratory testing and/or clinical evaluation to verify the iMCD diagnosis. The study was approved by the institutional review board and conducted in accordance with the Declaration of Helsinki.
A total of 176 patients had detailed clinical data at diagnosis and treatment regimens available for analysis in the discovery cohort, including three patients who met the diagnose criterion of TAFRO syndrome. Clinical, laboratory, and diagnostic materials from involved lymph nodes, tissues, or organs were evaluated in accordance with generally accepted guidelines to confirm the diagnosis of iMCD [17]. iMCD was defined as the involvement of at least two lymph nodes in at least two separate regions in patients who were HIV negative and HHV‐8 negative. Patient data included demographics, clinical manifestations, laboratory test results, associated autoimmune disorders, treatment, and clinical follow‐up. B‐symptoms (as a clinical manifestation) were defined as fevers, night sweats, or weight loss of ≥10% in the previous 6 months.
Treatments included siltuximab (anti–interleukin [IL]‐6 monoclonal antibodies), rituximab (anti‐CD20 monoclonal antibodies), tocilizumab (anti–IL‐6 receptor monoclonal antibodies), thalidomide, radiotherapy, prednisone, cyclophosphamide (CTX), and chemotherapy. Dosing for single‐agent therapies was as follows: 11 mg/kg siltuximab intravenously every 3 weeks per protocol or every 6 weeks at the investigator's discretion; 375 mg/m2 rituximab intravenously weekly for 4 weeks; 8 mg/kg tocilizumab intravenously every 4 weeks per protocol; 0.5–1 mg/kg prednisone; 0.2–0.4 g/day CTX; 100–150 mg/day thalidomide; or 15–25 Gy dose radiotherapy. Chemotherapy included CHOP (CTX, hydroxyl doxorubicin, hydrochloride, vincristine, and prednisone) or CHOP‐like chemotherapy regimens or CHOP plus rituximab (R‐CHOP). The dose, order, and regimen of drugs administered varied across patients.
Follow‐up information was generated from a review of each visit record until the time of last follow‐up or death. Castleman Disease Collaborative Network response criteria, based on evaluation of biochemical, lymph node, and symptom response, were used to assess treatment response. These criteria were also used by independent radiologists who reviewed our results.
We also evaluated overall survival (OS). OS was defined as the duration from the date of diagnosis to death or last follow‐up. Treatment failure was defined as relapse from complete response, progression from a partial response or during treatment, or death from any cause. Relapse or progression was determined by the Lugano classification. Follow‐up was assessed through February 28, 2018.
Patient characteristics and treatment outcomes were summarized using descriptive statistics. The HV, PC, and mixed variant subtype groups were compared using chi‐square, Fisher's exact, and Mann‐Whitney U tests. We used the Kaplan‐Meier method to perform univariate analyses of possible prognostic factors with OS, and survival curves were compared using the log‐rank test. A multivariable Cox proportional hazards model was used to identify independent prognostic factors for OS. A p value <.05 was considered statistically significant. All statistical analyses were performed using SPSS software (version 21.0; IBM, Armonk, NY).
Results
Demographic Characteristics
Demographic and clinical characteristics of the 176 patients in our cohort are summarized in Table 1. Histopathologic and radiologic findings were used to classify the iMCD as HV subtype (n = 46), PC variant (n = 95), or mixed variant (n = 35). The ethnic origins of the cohort included white (n = 26), African American (n = 5), and Asian (n = 145). There were 89 men and 87 women with a mean age of 46 years (range: 18–74 years) at the time of diagnosis. Patients older than 40 years were much more likely to have PC variant iMCD than HV iMCD (p = .026).
Table 1.
Baseline demographic and disease characteristics of patients with iMCD (176 cases)
| Demographics and characteristics | Total | Mixed subtype | HV subtype | PC subtype | |||||
|---|---|---|---|---|---|---|---|---|---|
| N | n (%) | N | n (%) | N | n (%) | N | n (%) | p value | |
| Age, years | 176 | 35 | 46 | 95 | |||||
| ≤40 | 51 (28.98) | 10 (28.57) | 19 (41.30) | 22 (23.16) | .0261 | ||||
| >40 | 125 (71.02) | 25 (71.43) | 27 (58.70) | 73 (76.84) | |||||
| Gender | 176 | 46 | 95 | ||||||
| Male | 89 (50.57) | 35 | 20 (57.14) | 21 (45.65) | 48 (50.53) | .5873 | |||
| Female | 87 (49.43) | 15 (42.86) | 25 (54.35) | 47 (49.47) | |||||
| Ethnicity | 176 | 35 | 46 | 95 | |||||
| White | 26 (14.77) | 1 (2.86) | 16 (34.78) | 9 (9.47) | .0005 | ||||
| African American | 5 (2.84) | 0 (0.00) | 0 (0.00) | 5 (5.26) | |||||
| Asian | 145 (82.39) | 34 (97.14) | 30 (65.22) | 81 (85.26) | |||||
| B‐symptom | 176 | 58 (32.95) | 35 | 7 (20.00) | 46 | 8 (17.39) | 95 | 43 (45.26) | .0012 |
| Pleural effusion | 176 | 24 (13.64) | 35 | 5 (14.29) | 46 | 3 (6.52) | 95 | 16 (16.84) | .0924 |
| Hepatomegaly and/or splenomegaly | 176 | 39 (22.16) | 35 | 6 (17.14) | 46 | 7 (15.22) | 95 | 26 (27.37) | .1101 |
| Mass >5 cm | 176 | 25 (14.20) | 35 | 1 (2.86) | 46 | 10 (21.74) | 95 | 14 (14.74) | .2996 |
| Fever | 176 | 36 (20.45) | 35 | 11 (31.43) | 46 | 3 (6.52) | 95 | 22 (23.16) | .0153 |
| Fatigue | 176 | 37 (21.02) | 35 | 11 (31.43) | 46 | 8 (17.39) | 95 | 18 (18.95) | .8232 |
| Pain | 176 | 30 (17.05) | 35 | 12 (34.29) | 46 | 3 (6.52) | 95 | 15 (15.79) | .1221 |
| WBC, ×109/L | 174 | 34 | 46 | 94 | |||||
| ≤4 | 22 (12.64) | 7 (20.59) | 6 (13.04) | 9 (9.57) | .5331 | ||||
| 4–10 | 126 (72.41) | 23 (67.65) | 37 (80.43) | 66 (70.21) | .1976 | ||||
| >10 | 26 (14.94) | 4 (11.76) | 3 (6.52) | 19 (20.21) | .0366 | ||||
| Hemoglobin, g/L | 174 | 34 | 46 | 94 | |||||
| <120 | 92 (52.87) | 19 (55.88) | 14 (30.43) | 60 (63.83) | .0002 | ||||
| <100 | 58 (33.33) | 13 (38.24) | 5 (10.87) | 34 (36.17) | .0017 | ||||
| Platelets, ×109/L | 174 | 34 | 46 | 94 | |||||
| <100 | 19 (10.91) | 6 (17.65) | 0 (0.00) | 13 (13.83) | .0081 | ||||
| 100–300 | 97 (55.75) | 25 (73.53) | 32 (69.57) | 40 (42.55) | .0027 | ||||
| >300 | 58 (33.33) | 3 (8.82) | 14 (30.43) | 41 (43.16) | .1336 | ||||
| Elevated LDH | 150 | 24 (16.00) | 33 | 5 (15.15) | 45 | 6 (13.33) | 72 | 13 (18.06) | .5004 |
| Elevated β2‐MG | 109 | 73 (66.97) | 27 | 18 (66.67) | 29 | 15 (51.72) | 53 | 40 (75.47) | .0287 |
| Elevated ESR | 100 | 62 (62.00) | 28 | 16 (57.14) | 25 | 9 (36.00) | 47 | 37 (78.72) | .0003 |
| Decreased ALB | 93 | 35 (37.63) | 1 | 0 (0.00) | 32 | 3 (9.37) | 60 | 32 (53.33) | .0001 |
| Elevated CRP | 40 | 16 (40.00) | 0 | 0 (0.00) | 19 | 4 (21.05) | 21 | 12 (57.14) | .0200 |
| Elevated AKP | 96 | 26 (27.08) | 29 | 7 (24.14) | 26 | 5 (19.23) | 41 | 14 (34.15) | .1869 |
Bolded p values are statistically significant.
Abbreviations: β2‐MG, β2‐microglobulin, mg/L; AKP, alkaline phosphatase; ALB, albumin, g/dL; CRP, C‐reactive protein, mg/dL; ESR, erythrocyte sedimentation rate, mm/hour; HV, hyaline‐vascular; iMCD, human immunodeficiency virus– and human herpesvirus 8–negative multicentric Castleman disease, idiopathic MCD; LDH, lactate dehydrogenase, IU/L; PC, plasma cell; WBC, white blood cell count.
Clinical Manifestations
Most patients were symptomatic at diagnosis with fever, splenomegaly, edema, effusion, or respiratory symptoms. Patients with the PC variant subtype presented with symptomatic complaints more frequently than patients with the HV subtype (58% compared with 39%, p = .0366). Patients with the PC variant subtype had higher rates of B‐symptoms (45%) than those with the HV subtype (17%, p = .0012). There were no significant differences between the HV and PC variant subtypes in terms of hepatomegaly and/or splenomegaly, pleural effusion, mass >5 cm, fatigue, or pain (Table 1). Patients with mixed subtype showed variable features between the HV and PC subtypes.
Laboratory Findings
The major biological abnormalities among the patients in our cohort were anemia, thrombocytopenia or thrombocytosis, high serum CRP, low serum albumin, and hypergammaglobulinemia. Although all patients in the cohort frequently had symptoms of systemic inflammation, those with the PC variant were more symptomatic than those with the HV subtype and specifically presented with more inflammatory symptoms. Of the 174 patients, whose platelet counts were measured, a platelet count of <100 × 109/L occurred in 13/94 patients with the PC variant subtype (14%) and none of those with the HV subtype (p = .0081). Elevation of β2‐microglobulin, CRP, and erythrocyte sedimentation rate and decreased albumin were significantly more common in patients with the PC variant subtype than in those with the HV variant subtype (p > .05; Table 1). Patients with the PC variant subtype were also much more likely to experience anemia than were patients with the HV subtype (p = .0002).
Treatment Outcomes
Treatments differed considerably among the patients (Table 2). Fifty patients were under watchful waiting, 46 patients received single‐agent therapy with prednisone, CTX, siltuximab, tocilizumab, rituximab, tocilizumab, or thalidomide, and 2 patients received radiotherapy alone. Among these patients, 17 received prednisone; 8 had a response, 7 had no response, and 2 had treatment failure. Seven patients received CTX; two had a response, three had no response, and two had treatment failure. Nine patients were treated with siltuximab; five had a response, two had no response, and two had treatment failure but nonetheless reported improvement in symptoms and quality of life. Five patients received rituximab; three had a response and two had treatment failure. Three patients received tocilizumab; one had a response, one had no response, and one had treatment failure. Five patients received thalidomide; two had a response, two had no response, and one had treatment failure.
Table 2.
Efficacy of drug treatment for iMCD
| Treatment | Cases, n | First‐line | Second‐line | Response, n (%) | No response, n (%) | Treatment failure, n (%) |
|---|---|---|---|---|---|---|
| Watch and wait | 50 | 50 | 0 | 32 (64.00) | 8 (16.00) | 10 (20.00) |
| Prednisone | 17 | 17 | 0 | 8 (47.06) | 7 (41.18) | 2 (11.76) |
| CTX | 7 | 5 | 2 | 2 (28.57) | 3 (42.86) | 2 (28.57) |
| CHOP/CHOP‐like | 55 | 45 | 10 | 23 (41.82) | 15 (27.27) | 17 (30.91) |
| R‐CHOP | 22 | 18 | 4 | 14 (63.64) | 3 (13.64) | 5 (22.73) |
| Siltuximab | 9 | 5 | 4 | 5 (55.56) | 2 (22.22) | 2 (22.22) |
| R‐only | 5 | 4 | 1 | 3 (60.00) | 0 (0.00) | 2 (40.00) |
| Tocilizumab | 3 | 1 | 2 | 1 (33.33) | 1 (33.33) | 1 (33.33) |
| Thalidomide | 5 | 2 | 3 | 2 (40.00) | 2 (40.00) | 1 (20.00) |
| Radiotherapy | 2 | 1 | 1 | 1 (50.00) | 0 (0.00) | 1 (50.00) |
| Others | 6 | 0 | 6 | 4 (66.67) | 1 (16.67) | 1 (16.67) |
Response = complete remission and partial remission; No response = stable disease; Failure = progressive disease and death.
Abbreviations: CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CTX, cyclophosphamide; iMCD, human immunodeficiency virus– and human herpesvirus 8–negative multicentric Castleman disease, idiopathic MCD; R, rituximab.
Seventy‐seven patients underwent systemic chemotherapy, of whom 55 received CHOP or CHOP‐like chemotherapy and 22 received R‐CHOP. Among the 22 patients who received R‐CHOP, 14 had a response, 3 had no response, and 5 had treatment failure. Among the 55 patients who received CHOP or CHOP‐like chemotherapy, 23 had a response, 15 had no response, and 17 had treatment failure.
Survival Analysis
iMCD comprises heterogeneous subgroups, and the prognosis can vary both among and between these subgroups. Because prognostic factors for iMCD are not well established, we sought to predict iMCD prognosis by using clinical parameters. Detailed survival data were available for 148 patients in the discovery cohort, including 85 patients with PC subtype, 42 patients with HV subtype, and 21 patients with mixed subtype. In the univariate analysis for OS, significant predictive factors were age >40 years, the presence of hepatosplenomegaly and/or splenomegaly, hemoglobin <80 g/L, the presence of pleural effusion, and the PC variant subtype (Fig. 1). Other factors analyzed, including sex, extranodal involvement, constitutional symptoms, proteinuria, hepatitis B virus infection, clinical complications, and corticosteroid use, were not significant. In the multivariable analysis, all of these factors remained significant predictors of OS except for the PC variant subtype (Table 3).
Figure 1.
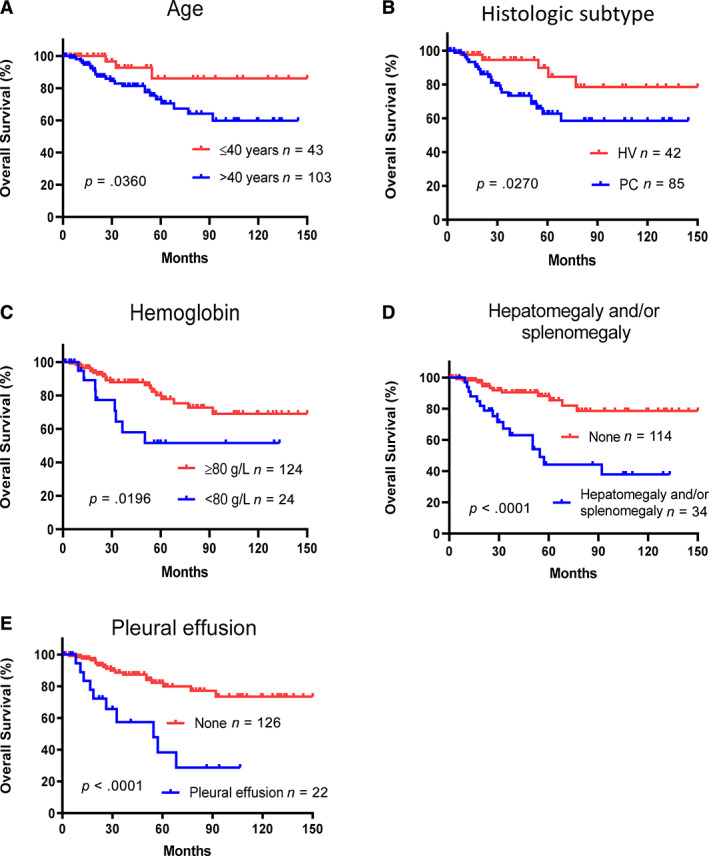
Prognostic significance of clinical features in 148 patients with idiopathic multicentric Castleman disease. Age >40 years (A), plasmacytic variant subtype (B), hemoglobin <80 g/L (C), hepatosplenomegaly and/or splenomegaly (D), and pleural effusion (E) were associated with poor overall survival in the univariate analysis.Abbreviations: HV, hyaline‐vascular subtype; PC, plasma cell subtype.
Table 3.
Multivariate analysis of prognostic factors for overall survival in iMCD (148 cases)
| Factor | OS in patients with iMCD | ||
|---|---|---|---|
| HR | 95% CI | p value | |
| Age >40 years | 2.755 | 0.999–7.596 | .050 |
| Histologic type (PC variant) | 0.964 | 0.602–1.542 | .877 |
| Hepatomegaly and/or splenomegaly | 3.272 | 1.437–7.450 | .005 |
| Hemoglobin <80 g/L | 2.366 | 1.004–5.577 | .049 |
| Pleural effusion | 2.840 | 1.154–6.986 | .023 |
Bolded p values are statistically significant.
Abbreviations: CI, confidence interval; HR, hazard ratio; iMCD, human immunodeficiency virus– and human herpesvirus 8–negative multicentric Castleman disease, idiopathic MCD; OS, overall survival; PC, plasma cells.
Development of iMCD‐IPI
The data in this study provided sufficient prognostic data to develop a risk‐stratification model for iMCD. We used the five factors identified in our univariate and multivariable analysis as independent predictors of OS (age >40 years, PC variant, hepatomegaly and/or splenomegaly, hemoglobin <80 g/L, and pleural effusion) and as clinicopathologic parameters for an iMCD risk‐stratification model. The presence of each factor added one point to a possible five‐point score, which was then categorized into one of three risk levels: a score of 0 or 1 indicated low risk, 2 or 3 indicated intermediate risk, and 4 or 5 indicated high risk. In the discovery cohort, there were 56 patients with low risk, 81 with intermediate risk, and 11 with high risk, and these risk stratifications reflected OS in patients who had survival data available (Fig. 2). An iMCD‐IPI score of 4 or 5 (high risk) predicted poor survival, and the median OS of low‐risk patients was significantly higher than that of high‐risk patients (135.27 months compared with 50.15 months; Fig. 2). The percentage of 5‐year overall survival in the low‐risk, intermediate‐risk, and high‐risk groups were 97.37%, 72.18%, and 20.0%, respectively.
Figure 2.
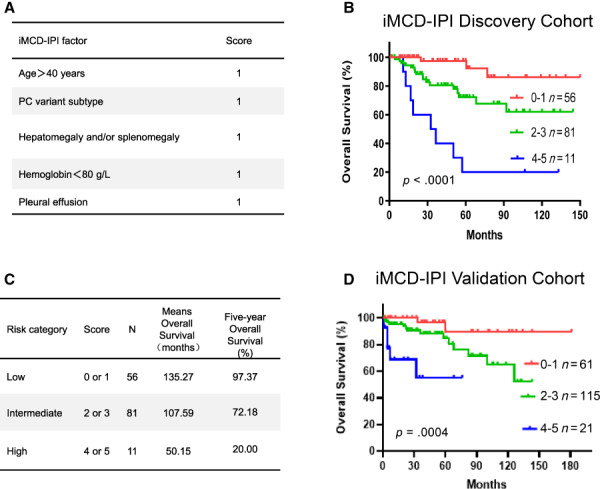
The iMCD‐IPI model for risk stratification in patients with iMCD. (A): iMCD‐IPI scoring system. (B): Overall survival curves for the 148 patients with survival data available, according to risk group as defined by the iMCD‐IPI. (C): Median survival of patients in each iMCD‐IPI risk category. (D): Overall survival curves for 197 patients from the validation cohort to test and confirm the iMCD‐IPI predictive model.Abbreviations: iMCD‐IPI, idiopathic multicentric Castleman disease international prognostic index; PC, plasma cell variant.
iMCD‐IPI Scores and Symptoms
When patients in our cohort were stratified according to the iMCD‐IPI model, those with high‐risk and intermediate‐risk scores presented with symptomatic complaints, especially inflammatory symptoms, more frequently than those with low‐risk scores. High‐risk patients had higher rates of B‐symptoms (82%), hepatomegaly and/or splenomegaly (91%), and pleural effusion and/or ascites (73%) than low‐risk patients (p < .0001; Table 4). Among the 174 patients whose platelet counts were measured, a platelet count of <100 × 109/L occurred in 14/91 intermediate‐risk patients (15%) and 2/72 low‐risk patients (3%) compared with 3/11 high‐risk patients (27%; p = .0075). Elevation of CRP and erythrocyte sedimentation rate and decreased albumin were significantly more common in high‐risk and intermediate‐risk patients than in low‐risk patients (p < .05). High‐risk and intermediate‐risk patients were also much more likely to experience anemia than were low‐risk patients (p < .05).
Table 4.
Using the iMCD‐IPI model to revalue the clinicopathologic factors for patients with iMCD
| Total number | Low risk | Intermediate risk | High risk | p value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Factor | N | n (%) | N | n (%) | N | n (%) | N | n (%) | |
| Age | 176 | 73 | 92 | 11 | |||||
| ≤40 | 51 (28.98) | 34 (46.58) | 16 (17.39) | 1 (9.09) | <.0001 | ||||
| >40 | 125 (71.02) | 39 (53.42) | 76 (82.61) | 10 (90.91) | |||||
| Gender | 176 | 73 | 92 | 11 | |||||
| Male | 89 (50.57) | 36 (49.32) | 46 (50.00) | 7 (63.64) | .6672 | ||||
| Female | 87 (49.43) | 37 (50.68) | 46 (50.00) | 4 (36.36) | |||||
| Ethnicity | 176 | 73 | 92 | 11 | |||||
| White | 26 (14.77) | 17 (23.29) | 9 (9.78) | 0 (0.00) | .0190 | ||||
| African American | 5 (2.84) | 0 (0.00) | 5 (5.43) | 0 (0.00) | .0954 | ||||
| Asian | 145 (82.39) | 56 (76.71) | 78 (84.78) | 11 (100.00) | .1144 | ||||
| Pathologic subtype | 176 | 73 | 92 | 11 | |||||
| HV | 46 (26.14) | 39 (53.42) | 7 (7.61) | 0 (0.00) | <.0001 | ||||
| PC | 95 (53.98) | 9 (12.33) | 75 (81.52) | 11 (100.00) | <.0001 | ||||
| Mixed | 35 (19.89) | 25 (34.25) | 10 (10.87) | 0 (0.00) | .0002 | ||||
| B‐symptom | 176 | 58 (32.95) | 73 | 15 (20.55) | 92 | 34 (36.96) | 11 | 9 (81.82) | <.0001 |
| Pleural effusion | 176 | 24 (13.64) | 73 | 0 (0.00) | 92 | 16 (17.39) | 11 | 8 (72.73) | <.0001 |
| Hepatomegaly and/or splenomegaly | 176 | 39 (22.16) | 73 | 2 (2.74) | 92 | 27 (29.35) | 11 | 10 (90.91) | <.0001 |
| Mass >5 cm | 176 | 25 (14.20) | 73 | 12 (16.44) | 92 | 13 (14.13) | 11 | 0 (0.00) | .3464 |
| WBC, ×109/L | 174 | 72 | 91 | 11 | |||||
| ≤4 | 22 (12.64) | 8 (11.11) | 12 (13.19) | 2 (18.18) | .7855 | ||||
| 4–10 | 126 (72.41) | 57 (79.17) | 62 (68.13) | 7 (63.64) | .2342 | ||||
| >10 | 26 (14.94) | 7 (9.72) | 17 (18.68) | 2 (18.18) | .2677 | ||||
| Hemoglobin, g/dL | 174 | 72 | 91 | 11 | |||||
| <12 | 93 (53.45) | 27 (37.50) | 57 (62.64) | 9 (81.82) | .0009 | ||||
| <10 | 58 (33.33) | 13 (18.06) | 37 (40.66) | 8 (72.73) | .0002 | ||||
| <8 | 25 (14.37) | 0 (0.00) | 19 (20.88) | 6 (54.55) | <.0001 | ||||
| Platelets, ×109/L | 174 | 72 | 91 | 11 | |||||
| <100 | 19 (10.92) | 2 (2.78) | 14 (15.38) | 3 (27.27) | .0075 | ||||
| 100–300 | 97 (55.75) | 49 (68.06) | 43 (47.25) | 5 (45.45) | .0229 | ||||
| >300 | 58 (33.33) | 21 (29.17) | 34 (37.36) | 3 (27.27) | .4943 | ||||
| Elevated LDH | 150 | 24 (16.00) | 68 | 8 (11.76) | 79 | 16 (20.25) | 3 | 0 (0.00) | .2805 |
| Elevated β2‐MG | 109 | 73 (66.97) | 44 | 25 (56.82) | 62 | 45 (72.58) | 3 | 3 (100.00) | .1101 |
| Elevated ESR | 100 | 62 (62.00) | 41 | 18 (43.90) | 55 | 40 (72.73) | 4 | 4 (100.00) | .0044 |
| Decreased ALB | 91 | 33 (36.26) | 37 | 6 (16.22) | 45 | 20 (44.44) | 9 | 7 (77.78) | .0007 |
| Elevated CRP | 40 | 16 (40.00) | 19 | 3 (15.79) | 20 | 13 (65.00) | 1 | 0 (0.00) | .0052 |
| Elevated AKP | 96 | 26 (27.08) | 42 | 7 (16.67) | 52 | 18 (34.62) | 2 | 1 (50.00) | .1146 |
Bolded p values are statistically significant.
Abbreviations: β2‐MG, β2‐microglobulin, mg/L; AKP, alkaline phosphatase; ALB, albumin, g/dL; CRP: C‐reactive protein, mg/dL; ESR, erythrocyte sedimentation rate, mm/hour; HV, hyaline‐vascular; iMCD, human immunodeficiency virus– and human herpesvirus 8–negative multicentric Castleman disease, idiopathic MCD; IPI, international prognostic index; LDH, lactate dehydrogenase, IU/L; PC, plasma cell variant; WBC, white blood cell count.
Each of the five iMCD‐IPI parameters was carefully evaluated according to iMCD‐IPI risk group, including age >40 years, PC variant, hepatomegaly and/or splenomegaly, hemoglobin <80 g/L, and pleural effusion. The proposed model using these parameters could stratify iMCD patients with into low risk, intermediated risk, and high risk (p < .05; Fig. 3). Additional 197 patients with iMCD were organized from The International Castleman Disease Consortium (France, Japan, China, and the U.S.). The group consisted of one third white and two thirds Asian patients, and the mean age of this group was 46 years (range: 18–80 years) at the time of diagnosis. Among these patients, the mean age of patients ≤40 years was 29 years (range: 18–40 years), and that for patients >40 years was 56 years (range: 41–80 years). This consortium was analyzed using the five iMCD‐IPI scores and parameters identical in the discovery set. The risk factors and survival were independently validated and confirmed for the iMCD‐IPI model established from the discovery set (Fig. 2D).
Figure 3.
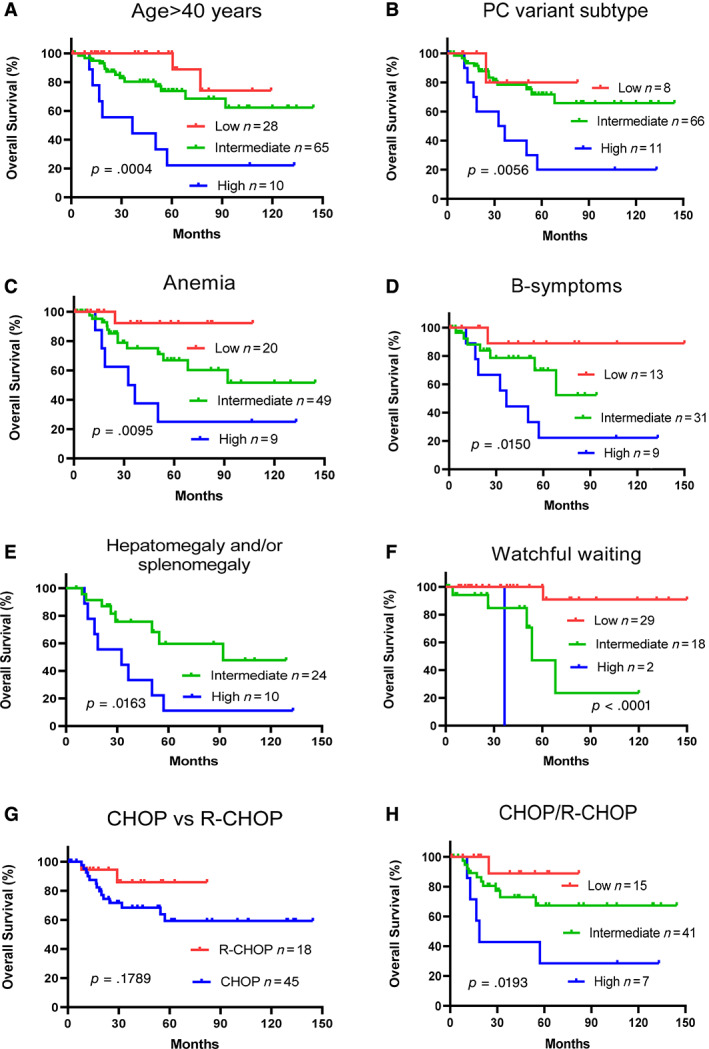
Idiopathic multicentric Castleman disease international prognostic index (iMCD‐IPI) model prognosis prediction in patients with idiopathic (human immunodeficiency virus– and human herpesvirus‐8–negative) multicentric Castleman disease (iMCD) by specific factor or symptom and treatment. iMCD‐IPI risk stratification was accurate for various factors and symptoms: age >40 years (A), PC variant (B), anemia (C), B‐symptoms (D), and splenomegaly (E). (F): Overall survival in patients who received watchful waiting. (G): Overall survival did not differ between patients treated with CHOP and those treated with R‐CHOP. (H): Overall survival in patients who received CHOP or R‐CHOP.Abbreviations: CHOP, cyclophosphamide, hydroxyl doxorubicin, hydrochloride, vincristine, and prednisone; PC, plasmacytic variant; R, rituximab.
iMCD‐IPI Scores and Treatment Options
Treatments for iMCD in our cohort were chosen on the basis of clinical performance and physician experience, in which we examined the treatments chosen for the patients in the cohort according to iMCD‐IPI risk groups. Generally, low‐risk patients most often received prednisone; intermediate‐risk patients received CTX, siltuximab, or rituximab; and intermediate‐risk or high‐risk patients received systemic chemotherapy (CHOP/R‐CHOP).
Among the 50 patients who received watchful waiting, 56% were low risk, 41% were intermediate risk, and 4% were high‐risk patients who died within 32 months. More than 95% of low‐risk patients survived for at least 150 months, whereas only 22% of intermediate‐risk patients survived for 90 months (Fig. 3). Of the 17 patients who received prednisone, 4 were low risk, 2 were intermediate risk, and none were high risk. There were 63 patients who received systemic chemotherapy (CHOP/R‐CHOP) with follow‐up information were more likely to be intermediate risk or high risk: 15 were low risk, 41 were intermediate risk, and 7 were high risk. OS rates at 60 months for patients who received systemic chemotherapy were about 90% for low‐risk patients, 68% for intermediate‐risk patients, and 30% for high‐risk patients (Fig. 3).
Based on these data, we found that observation and waiting was chosen for most of the patients with low risk in risk stratification, whereas systemic chemotherapy was chosen for those patients with intermediate and high risk, but the outcome of the patients with high risk after chemotherapy was not ideal.
Discussion
The aim of the current study was to establish an evidence‐based risk‐stratification model of OS in patients with iMCD. The current study represents the largest and most comprehensive review of iMCD patient data with the intent to develop a risk score–based clinical model and provide therapy recommendations more practically for iMCD to improve outcomes.
Up to now, clinical, laboratory, and treatment data for patients with iMCD have been dispersed among case reports, small series, and a single randomized controlled trial. However, there are few truly effective therapies for iMCD, and its prognosis has remained mostly unchanged over the past 30 years [4, 18]. Recently, several new treatment modalities, including chemotherapy, rituximab, siltuximab, tocilizumab, anakinra (IL‐1RA agonist), thalidomide, and lenalidomide, have been used and, in some cases, shown efficacy [12, 15, 19, 20, 21, 22]. However, these treatments have substantial toxicities and are costly. A prognostic index would be very helpful for identifying the patients most likely to benefit from these therapies. Using clinical, laboratory, and pathologic data, as well as a review of treatment outcomes with a median follow‐up duration of 12 years, we have performed the most comprehensive evaluation of iMCD to date in China and North America. In particular, our study provides a valuable patient risk‐stratification model based on comprehensive information, and this model should advance our understanding of iMCD and its treatment options.
We observed heterogeneity within patients with iMCD in the cohort. The progression of iMCD is known to vary from indolent to aggressive and immediately life threatening, as reported previously [14, 15, 18]. This heterogeneity makes diagnosis and management a challenge for most practitioners, who rarely encounter iMCD during their careers. The pathogenesis of iMCD is also poorly understood at this time.
Patients with iMCD in our cohort were treated with a variety of regimens. Patients treated with prednisone often showed an initial response, but they also often experienced disease recurrence within 1 to 2 years, which is consistent with data reported previously [15]. CTX as monotherapy led to a 29% response rate. Response rates to CTX have not been previously reported. Rituximab had about a 60% response rate in our cohort, which is higher than that reported previously in the literature for patients with iMCD in the U.S.; only 20% were shown to achieve a complete response [15]. Patients who were treated with rituximab or rituximab‐based therapy had a higher response rate than those treated with CHOP or a CHOP‐like regimen (64% compared with 42%); this percentage is much lower than that reported in the literature for patients with HHV‐8–associated MCD (84%) [23]. Siltuximab and tocilizumab were used in 22 patients and 3 patients, respectively. Of note, the 56% response rate in our series is much higher than the 34% response rate for siltuximab that was observed in the only randomized controlled trial of iMCD [24]. The difference in response may be related to improved patient selection or longer follow‐up time to achieve a response. Thalidomide was only used in five patients, and two patients had response. Recently, immunomodulatory therapy including thalidomide and lenalidomide were reported to be efficacious treatments for iMCD [12], which suggests that these agents may be another potential treatment strategy for iMCD.
The iMCD‐IPI score in our study cohort has important clinical implications. First, the iMCD‐IPI model can further segment iMCD with unfavorable prognostic factors and/or B‐symptoms. These parameters, including age > 40 years, PC variant, hepatomegaly and/or splenomegaly, anemia, and pleural effusion can function as independent factors to enhance better disease severity stratification. Second, a high‐risk iMCD‐IPI score indicates an extremely poor prognosis, which could be considered severe disease status. Third, patients scored as intermediate risk had a better prognosis, more treatment options, and more time to try agents than those scored as high risk. Given the low rates of aggressive progress among low‐risk patients, the feasibility of a watchful waiting strategy is obviously reasonable in this group. Because patients with low‐risk iMCD can later progress to intermediate or high risk, this model can also be used to assess patients when iMCD progresses to determine the appropriate treatment approach at that point. Because OS was significantly different among low‐, intermediate‐, and high‐risk patients, and the factors used to generate iMCD‐IPI scores are simple to collect or routinely checked in hospitals around the world, the iMCD‐IPI model represents a practical and feasible clinical revenue to determine the biological aggressiveness of iMCD. We believe this proposed predictive model can be used to guide risk‐based treatment strategies. To the best of our knowledge, our study is the first one in which clinical factors of iMCD were used to estimate the risk‐stratified score in the field.
Our study had several limitations. First, it was a study of a relatively rare disease, and thus, the availability of data was somewhat limited. Second, only 11 patients in the discovery cohort and 21 patients in the validation cohort were classified into the high‐risk group. In particular, this made it difficult to evaluate the potential treatment options in this group because of the diversity of treatments administered; thus, this should be further assessed in the near future in which more high‐risk patients and more TAFRO patients are enrolled. In addition, we could not compare the monotherapy efficacy such as siltuximab, rituximab, CTX, and prednisone among the three groups. A large multicenter study has been designed prospectively and is being analyzed now in collaboration with EUSA Pharmaceuticals to refine the scores and risk factors used in our model, and these risk factors could potentially be highly valuable when combined with additional risk factors (especially cytokines and chemokines) and treatment options. Although our findings suggest that iMCD‐IPI may help identify those at high risk of relapse or disease progression, a substantial number of low‐risk or intermediate‐risk patients in our study had treatment failure, indicating that iMCD‐IPI may have a low negative predictive value in such a subset of patients. Accurate identification of low‐risk or intermediate‐risk patients at risk of treatment failure remains challenging. A forthcoming large multicenter study has been successfully coordinated by the investigators in the iMCD Consortium Program with next‐generation sequencing and gene expression profiling that might shed new light on the molecular mechanisms of this intriguing disease in the context of risk factors and disease subtypes.
Conclusion
We developed a novel risk‐stratification system for iMCD‐IPI to help guide the development of a standard‐of‐care treatment algorithm. It was found that age > 40 years, PC variant, hepatomegaly and/or splenomegaly, hemoglobin <80 g/L, and pleural effusion are significant risk factors for reduced OS in iMCD. A clinical predictive model was proposed to use these parameters to help predict outcomes and could be used to support optimal therapeutic selection.
Author Contributions
Conception/design: Li Yu, Ken H. Young
Provision of study material or patients: Li Yu, Menghan Shi, Qingqing Cai, Paolo Strati, Fredrick Hagemeister, Qiongli Zhai, Ling Li, Xiaosheng Fang, Jianyong Li, Ruifang Sun, Shanxiang Zhang, Hanjin Yang, Zhaoming Wang, Wenbin Qin, Noriko Iwaki, Yasuharu Sato, Lu Zhang, Jian Li, Eric Oksenhendler, Zijun Y. Xu‐Monette, Ken H. Young
Collection and/or assembly of data: Li Yu, Ken H. Young
Data analysis and interpretation: Li Yu, Ken H. Young
Manuscript writing: Li Yu, Ken H. Young
Final approval of manuscript: Li Yu, Menghan Shi, Qingqing Cai, Paolo Strati, Fredrick Hagemeister, Qiongli Zhai, Ling Li, Xiaosheng Fang, Jianyong Li, Ruifang Sun, Shanxiang Zhang, Hanjin Yang, Zhaoming Wang, Wenbin Qin, Noriko Iwaki, Yasuharu Sato, Lu Zhang, Jian Li, Eric Oksenhendler, Zijun Y. Xu‐Monette, Ken H. Young
Disclosures
The authors indicated no financial relationships.
Acknowledgments
K.H.Y was supported by the Hagemeister Lymphoma Foundation, the Gundersen Lutheran Medical Foundation. K.H.Y was also supported by Roche Molecular System, Gilead Sciences, Seattle Genetics, Dai Sanyo, Adaptive Biotechnology, Incyte Pharmaceutical, and HTG Molecular Diagnostics. L.Y. is supported by National Natural Science Foundation of China grants 81460030 and 81770221.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Case Records of the Massachusetts General Hospital . Weekly Clinicopathological Exercises. Richard C. Cabot, Founder; Benjamin Castleman, Editor; Virginia W. Towne, Assistant Editor. N Engl J Med 1954;251:396–400. [DOI] [PubMed]
- 2. Dispenzieri A, Armitage JO, Loe MJ et al. The clinical spectrum of Castleman's disease. Am J Hematol 2012;87:997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman's disease: A systematic review of 404 published cases. Ann Surg 2012;255:677–684. [DOI] [PubMed] [Google Scholar]
- 4. Soumerai JD, Sohani AR, Abramson JS. Diagnosis and management of Castleman disease. Cancer Control 2014;21:266–278. [DOI] [PubMed] [Google Scholar]
- 5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV‐8‐negative, idiopathic multicentric Castleman disease: Novel insights into biology, pathogenesis, and therapy. Blood 2014;123:2924–2933. [DOI] [PubMed] [Google Scholar]
- 6. Polizzotto MN, Uldrick TS, Wang V et al. Human and viral interleukin‐6 and other cytokines in Kaposi sarcoma herpesvirus‐associated multicentric Castleman disease. Blood 2013;122:4189–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nicoli P, Familiari U, Bosa M et al. HHV8‐positive, HIV‐negative multicentric Castleman's disease: Early and sustained complete remission with rituximab therapy without reactivation of Kaposi sarcoma. Int J Hematol 2009;90:392–396. [DOI] [PubMed] [Google Scholar]
- 8. Suda T, Katano H, Delsol G et al. HHV‐8 infection status of AIDS‐unrelated and AIDS‐associated multicentric Castleman's disease. Pathol Int 2001;51:671–679. [DOI] [PubMed] [Google Scholar]
- 9. Talat N, Schulte KM. Castleman's disease: Systematic analysis of 416 patients from the literature. The Oncologist 2011;16:1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fajgenbaum DC, Kurzrock R. Siltuximab: A targeted therapy for idiopathic multicentric Castleman disease. Immunotherapy 2016;8:17–26. [DOI] [PubMed] [Google Scholar]
- 11. Dong Y, Zhang L, Nong L et al. Effectiveness of rituximab‐containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol 2018;97:1641–1647. [DOI] [PubMed] [Google Scholar]
- 12. Zhang L, Zhao AL, Duan MH et al. Phase 2 study using oral thalidomide‐cyclophosphamide‐prednisone for idiopathic Multicentric Castleman disease. Blood 2019;133:1720–1728. [DOI] [PubMed] [Google Scholar]
- 13. Iwaki N, Fajgenbaum DC, Nabel CS et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV‐8‐negative multicentric Castleman disease. Am J Hemtol 2016;91:220–226. [DOI] [PubMed] [Google Scholar]
- 14. van Rhee F, Voorhees P, Dispenzieri A et al. International, evidence‐based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018;132:2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu L, Tu M, Cortes J et al. Clinical and pathological characteristics of HIV‐ and HHV‐8‐negative Castleman disease. Blood 2017;129:1658–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simpson D. Epidemiology of Castleman disease. Hematol Oncol Clin North Am 2018;32:1–10. [DOI] [PubMed] [Google Scholar]
- 17. Fajgenbaum DC, Uldrick TS, Bagg A et al. International, evidence‐based consensus diagnostic criteria for HHV‐8‐negative/idiopathic multicentric Castleman disease. Blood 2017;129:1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oksenhendler E, Boutboul D, Fajgenbaum D et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol 2018;180:206–216. [DOI] [PubMed] [Google Scholar]
- 19. Galeotti C, Tran TA, Franchi‐Abella S et al. IL‐1RA agonist (anakinra) in the treatment of multifocal Castleman disease: Case report. J Pediatr Hematol Oncol 2008;30:920–924. [DOI] [PubMed] [Google Scholar]
- 20. Soudet S, Fajgenbaum D, Delattre C et al. Schnitzler syndrome co‐occurring with idiopathic multicentric Castleman disease that responds to anti‐IL‐1 therapy: A case report and clue to pathophysiology. Curr Res Transl Med 2018;66:83–86. [DOI] [PubMed] [Google Scholar]
- 21. van Rhee F, Casper C, Voorhees PM et al. A phase 2, open‐label, multicenter study of the long‐term safety of siltuximab (an anti‐interleukin‐6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget 2015;6:30408–30419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morra DE, Pierson SK, Shilling D et al. Predictors of response to anti‐IL6 monoclonal antibody therapy (siltuximab) in idiopathic multicentric Castleman disease: Secondary analyses of phase II clinical trial data. Br J Haematol 2019;184:232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uldrick TS, Polizzotto MN, Aleman K et al. Rituximab plus liposomal doxorubicin in HIV‐infected patients with KSHV‐associated multicentric Castleman disease. Blood 2014;124:3544–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Rhee F, Wong RS, Munshi N et al. Siltuximab for multicentric Castleman's disease: A randomised, double‐blind, placebo‐controlled trial. Lancet Oncol 2014;15:966–974. [DOI] [PubMed] [Google Scholar]