

Abstract
Fuchs endothelial corneal dystrophy (FECD) is the most common primary corneal endothelial dystrophy and the leading indication for corneal transplantation worldwide. FECD is characterized by the progressive decline of corneal endothelial cells (CECs) and the formation of extracellular matrix (ECM) excrescences in the Descemet’s membrane (DM), called guttae, that lead to corneal edema and loss of vision. FECD typically manifests in the fifth decades of life and has a greater incidence in women. FECD is a complex and heterogeneous genetic disease where interaction between genetic and environmental factors results in cellular apoptosis and aberrant ECM deposition. In this review, we will discuss a complex interplay of genetic, epigenetic, and exogenous factors in inciting oxidative stress, auto(mito)phagy, unfolded protein response, and mitochondrial dysfunction during CEC degeneration. Specifically, we explore the factors that influence cellular fate to undergo apoptosis, senescence, and endothelial-to-mesenchymal transition. These findings will highlight the importance of abnormal CEC-DM interactions in triggering the vicious cycle of FECD pathogenesis. We will also review clinical characteristics, diagnostic tools, and current medical and surgical management options for FECD patients. These new paradigms in FECD pathogenesis present an opportunity to develop novel therapeutics for the treatment of FECD.
Keywords: Fuchs endothelial corneal dystrophy, corneal endothelium, apoptosis, oxidative stress, mitochondria, guttae
1. Introduction
The corneal endothelium (CE), situated in the posterior layer of the cornea, plays a key role in maintaining the cornea in a state of deturgescence, thus contributing to corneal clarity. The CE is composed of corneal endothelial cells (CECs) that maintain stromal deturgescence by functioning as a selective leaky barrier between, the corneal stroma and aqueous humor (AH), and by actively pumping ions into the AH to allow the osmotic forces to keep the cornea in the state of relative dehydration (Bonanno, 2003; Harris, 1962; Harris and Nordquist, 1955; Maurice, 1957; Mishima, 1982). The CE contains a high density of mitochondria that generate adenosine triphosphate (ATP) for the energy needs of multiple Na+-K+-ATPase and other pumps essential for CE functioning (Bonanno, 2003, 2012; Huang et al., 2003; Miyai, 2018; Zhang et al., 2017a). CECs rest on a specialized basement membrane called Descemet’s membrane (DM), which is secreted by the CECs themselves. The CE is composed of a monolayer of hexagonal CECs derived from neural crest cells (NCCs) that arise from the neuroectoderm (Katikireddy et al., 2016). CECs are post-mitotic cells that are arrested in the G1 phase of the cell cycle, and, while they possess a proliferative capacity, CECs typically do not proliferate in vivo (Joyce, 2003). However, human CEC isolated from both central and peripheral areas of the CE have been shown to proliferate in vitro, where there is a tendency for greater proliferative capacity from peripheral CECs and younger donors (Konomi et al., 2005). It has also been demonstrated that there is an increased endothelial cell density (ECD) in the paracentral and peripheral regions of the human cornea compared to the central cornea (Amann et al., 2003; Schimmelpfennig, 1984). At birth, the human central cornea ECD is approximately 4,000 cells/mm2, which, subsequently, decrease throughout life, with a higher rate of decline during the first 2 years of life (Elbaz et al., 2017). This rapid decrease in ECD begins in utero and continues during the first 2 years of life likely as a result of an increase in corneal diameter with concurrent migration and spreading of CECs during that time period, rather than cell loss (Elbaz et al., 2017). After the first 2 years of life, the corneal diameter stabilizes, and there is a slower decrease in ECD, whereas, by age 5, the ECD is approximately 3,500 cells/mm2 (Elbaz et al., 2017). ECD subsequently decreases throughout adulthood at an average rate of approximately 0.6% per year (Bourne et al., 1997). Progressive endothelial cell loss, which can be exacerbated by either primary or secondary corneal endotheliopathies, leads to loss of barrier function and renders the CE unable to maintain fluid balance, causing fluid accumulation in the cornea, reduction of transparency, and the formation of painful epithelial bullae (Bourne, 2003).
Fuchs endothelial corneal dystrophy (FECD) is the most common primary corneal endothelial dystrophy and the leading indication for corneal transplantation worldwide (Gain et al., 2016). FECD is characterized by the progressive decline of CECs that leads to apoptosis, variation in size (polymegethism) and shape (pleomorphism) in CEC morphology, decreased ECD, and the formation of extracellular matrix (ECM) excrescences called guttae (Jun, 2010; Krachmer et al., 1978; Vedana et al., 2016; Vogt, 1921). The progressive CE damage leads to fluid accumulation and subsequent pathological changes to the corneal stroma and epithelium. Persistent corneal edema causes stromal keratocyte cell death and subepithelial fibrosis, leading to an irregular anterior cornea and vision loss (Hamill et al., 2013). FECD typically manifests in the fifth or sixth decades of life and has a greater incidence in women, at a ratio of 3–4:1 (Afshari et al., 2006; Krachmer et al., 1978; Louttit et al., 2012; Minear et al., 2013). Central corneal guttae have been found in up to 9%–11% of women, while similar findings have been found in only 3.5%–7% of men (Wilson et al., 1988; Zoega et al., 2006). A higher frequency of more advanced FECD has also been reported in women than in men (Hogan et al., 1974). Female sex, in addition to age, is the most significant risk factor for advanced FECD development (Zhang et al., 2013). Furthermore, even though women comprise 75% of patients undergoing corneal transplantation, there is a lack of knowledge as to why women are drastically more affected by FECD than are men (Afshari et al., 2006; Chan et al., 2018). Additional risk factors for FECD other than female sex and age include family history, smoking, and diabetes.
FECD is a complex and heterogeneous genetic disease with variable expressivity and incomplete penetrance. FECD pathogenesis is hypothesized to be an interaction between genetic and environmental factors (Jurkunas, 2018; Schmedt et al., 2012b). Our laboratory, as well as others, has linked an imbalance and dysregulation of the oxidative stress response with the pathogenesis of FECD (Jurkunas et al., 2010). Furthermore, oxidative stress and the subsequent accumulation of mitochondrial and nuclear DNA damage play a critical role in the pathogenesis of FECD and contributes to CEC apoptosis and degeneration (Azizi et al., 2011; Jurkunas, 2018; Jurkunas et al., 2010; Lopez-Otin et al., 2013; Miyai, 2018). FECD pathogenesis also involves abnormal cellular-ECM interactions. While abnormal ECM in the form of guttae, and abnormally expedited cellular death contribute to FECD pathogenesis, it is not clear which comes first, abnormal ECM deposition resulting in increased cellular stress and eventual CEC death, or abnormal CECs with a defective synthetic capacity secreting their own “tombs” composed of guttae (Jurkunas, 2018). Another component of FECD pathogenesis is the activation of an endothelial-to-mesenchymal transition (EMT) state in CECs, where there is a loss of the normal hexagonal CEC mosaic seen in the organized junctional staining of plasma membrane proteins (Katikireddy et al., 2018). Additionally, there is upregulation of EMT- and FECD-related protein markers in FECD patients. Importantly, oxidative stress and DNA damage have been shown to be an inducer of EMT in FECD (Azizi et al., 2011; Halilovic et al., 2016; Jurkunas et al., 2009; Jurkunas et al., 2010; Jurkunas et al., 2008a; Katikireddy et al., 2018).
In this review, we present our current hypothesis and supporting evidence on the vicious cycle of FECD pathogenesis (Figure 1). We believe that cellular stress and abnormal cellular–ECM interactions are the hallmarks of FECD. Our hypothesis is that multiple exogenous factors, particularly oxidative stress, in combination with genetic factors, leads to increased intracellular reactive oxygen species (ROS) production, resulting in mitochondrial dysfunction and alteration in the synthetic capacity of cells, which maintains the vicious cycle of propagating oxidant–antioxidant imbalance in the pathogenesis of FECD (Jurkunas, 2018). Furthermore, this process results in CECs undergoing EMT and cellular senescence, and abnormal ECM deposition of guttae, which further contributes to the pathogenesis of FECD. We also review currently available model systems to study the pathogenesis of FECD, including a novel ultraviolet (UV)-A light induced in vivo non-genetic mouse model of FECD that we have developed. Moreover, we review the clinical characteristics of FECD and the diagnostic tools available for clinical assessment. Lastly, we review the current evidence for the medical and surgical management of FECD, and discuss potential novel therapeutics in development for the treatment of FECD.
Figure 1. The Vicious Cycle of Fuchs Endothelial Corneal Dystrophy (FECD).
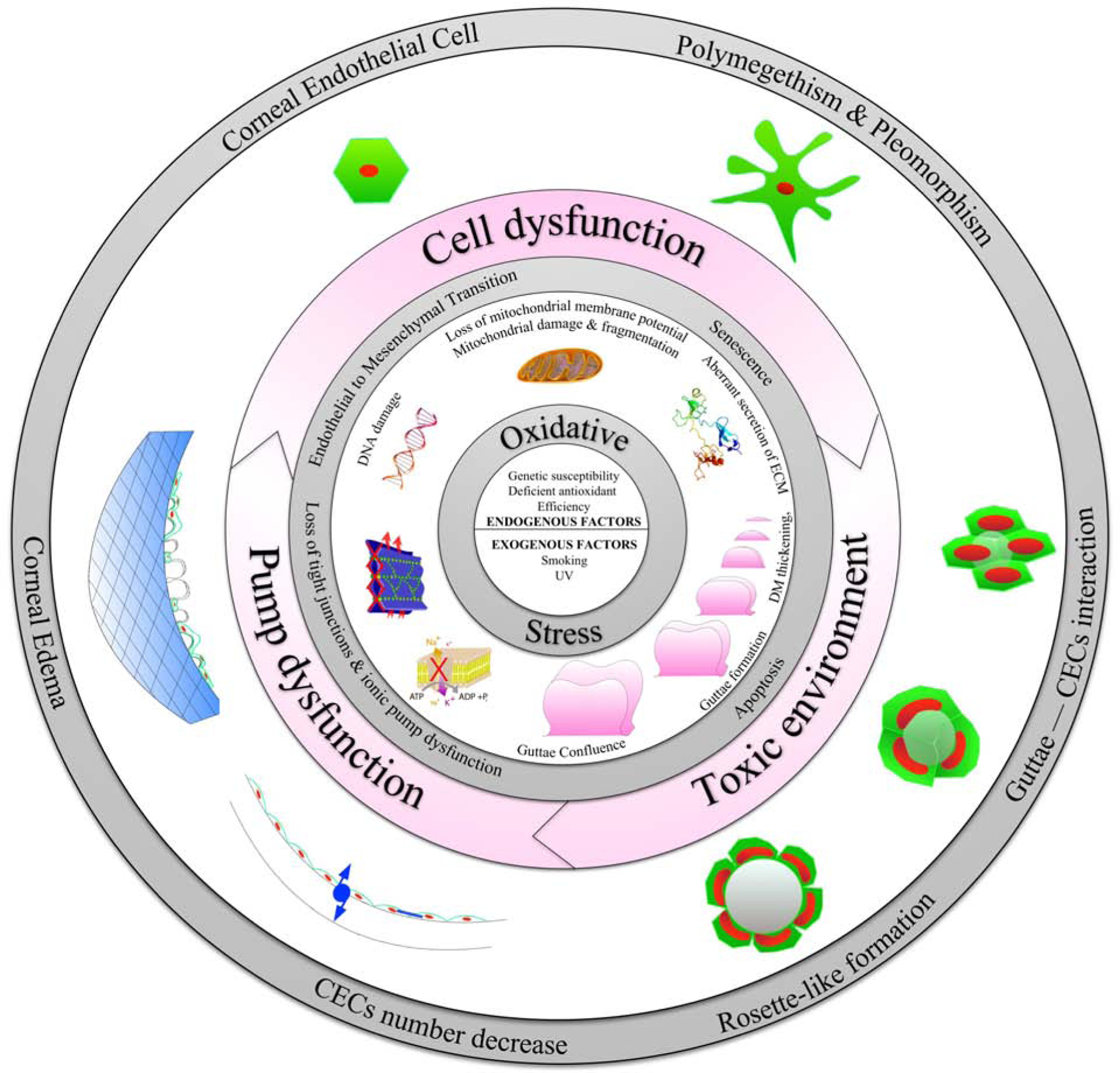
Schematic diagram illustrating the complex interplay between genetic, epigenetic, and exogenous factors in inciting oxidative stress, endothelial to mesenchymal transition, senescence, mitochondrial dysfunction, and apoptosis in corneal endothelial cells (CECs) in FECD. Furthermore, guttae formation contributes to a toxic microenvironment resulting in abnormal CEC-Descemet’s membrane interactions triggering the vicious cycle of FECD pathogenesis.
2. Brief Historic Perspective
In 1910, Ernest Fuchs first described a series of 13 patients with slowly progressive corneal clouding (Fuchs, 1910; Jun, 2010). He observed the appearance of non-inflammatory bilateral diffuse corneal opacities, roughened epithelium with vesicular elevations, and visual fluctuations in these elderly patients. He concluded that the condition was an abnormality of epithelial cells, and he named the condition “dystrophia epithelialis.” Fuchs suggested that the prevalence of “dystrophia epithelialis” would be 13 in 200,000 (Fuchs, 1910; Jun, 2010). Later in 1910, Knapp published a case report of an 82 year old woman with bilateral corneal opacities, for whom one eye decompensated post-cataract surgery (Knapp, 1911). This patient was initially treated with “dionin salve, yellow oxide of mercury salve, subconjunctival injections, hot applications, and bandaging; arsenic, strychnine, and iodide of potash” (Knapp, 1911). Knapp believed that this patient also had “dystrophia epithelialis” and referred to the condition as “dystrophia epithelialis corneae (Fuchs).”
In 1916, with the invention of the slit lamp biomicroscope, Koeppe was able to visualize guttae in the CE of patients with corneal edema (Jun, 2010; Krachmer et al., 1978). In 1920, Kraupa described the CE to have a “crystal like” appearance, and the progressive changes in the CE that lead to corneal edema. In 1921, Vogt described the CE changes as “drop-like endothelial prominences,” which he later called “cornea guttata” (Jun, 2010; Krachmer et al., 1978; Vogt, 1921). The advent of slit lamp biomicroscopy enabled Vogt and Moeschler to first describe “dew-drop-like” elevations on the posterior surface of the cornea, where the singular term “gutta”is the Latin noun for “drop”, and the plural term is “guttae.” The term “guttata” is the adjective form describing the “guttate” cornea (Eghrari and Gottsch, 2010; Friedenwald and Friedenwald, 1925; Vogt, 1921). Vogt also reported the posterior corneal elevations to appear as “powdered bronze” with retroillumination. In 1924, Graves described the dystrophy as bilateral and chronic endothelial degeneration as opposed to previously accepted epithelial degeneration (Graves, 1924). He also noted the central localization of these posterior elevations were becoming more diffuse and peripheral with disease progression and later producing epithelial vacuoles. Similarly, the Friedenwalds, in 1925, described a patient with bilateral CE involvement and unilateral corneal epithelial changes that supported the theory of endothelial pathology preceding epithelial involvement by several years (Friedenwald and Friedenwald, 1925). They proposed that the CE changes and the epithelial dystrophy of Fuchs were not separate entities, but were to be considered part of a single disease. They also noticed that the rate of disease progression was very slow, and that many cases failed to advance beyond the early stages (Friedenwald and Friedenwald, 1925). Subsequent histologic studies confirmed that FECD was a disease of the CE with irregular thickening and excrescences of DM (Goar, 1933).
3. Prevalence and Onset
Central corneal guttae have been shown to be more prevalent with age. As early as 1922, Moeschler reported central corneal guttae in 4.5% of the population over the age of 50 (Krachmer et al., 1978). In 1933, Goar found a 6.62% prevalence of FECD in 800 patients over the age of 20, with an increased prevalence in patients over the ages of 40 and 50 (Goar, 1933). While it was recognized that FECD typically manifests in patients over the age of 50, a few cases have been described as having an earlier onset. In Goar’s 800 patient series, 11 patients were between the age of 30 and 40, where the youngest patient was 32 years old (Goar, 1933). In 1967, Lorenzetti examined 2002 eyes from 1,016 patients and found that 31.5% of patients from the ages of 10–39 and 70.4% of patients from the ages of 40–99 showed some evidence of corneal guttae, the youngest patient with corneal guttae changes being 17 years of age. Approximately 3.9% of patients over the age of 40 have been shown to have confluent guttae (Lorenzetti et al., 1967).
FECD can be divided into early-onset and late-onset forms. Most cases of early-onset FECD present in the third decade; however, an early variant has been described in the first decade (Gottsch et al., 2005a). Late-onset FECD typically manifests in the fifth or sixth decade of life (Borboli and Colby, 2002). The prevalence of the disease seems to vary depending on the ethnicity of the population, ranging from 3.7–11% (Table 1) (Eghrari et al., 2012; Higa et al., 2011; Kitagawa et al., 2002; Lorenzetti et al., 1967; Nagaki et al., 1996; Zoega et al., 2006). In the United States, the prevalence is approximately 3.9–6.62% of the population over the age of 40 (Goar, 1933; Lorenzetti et al., 1967). A higher rate of FECD (21.6%) has been reported on Tangier, an island in the Chesapeake Bay with an isolated population of 500 individuals, with a minimum prevalence of 11% among individuals over the age of 50 when considering all individuals that were not examined (Eghrari et al., 2012). In Iceland, a large population-based study of Caucasians in the Reykjavik Eye Study showed that the prevalence of FECD was 9.2% of the population over the age of 50 (Zoega et al., 2006). In Japan, a lower prevalence rate of 3.3% has been reported (Nagaki et al., 1996). In Monzen-machi, Japan, the prevalence of FECD has been reported to be 3.7% of the population over the age of 50 (Kitagawa et al., 2002). In the southwestern island Kujejima of Japan, a large population based study reported a prevalence rate of 4.1% of the population over the age of 40 (Higa et al., 2011). Based on a Japanese national survey, FECD is an indication for keratoplasty in 1.9% of cases of bullous keratopathy (Shimazaki et al., 2007). In Chinese Singaporeans, the prevalence of FECD has been reported to be 6.6% of the population over the age of 50 (Kitagawa et al., 2002). In the Indian population, about 5.3% of penetrating keratoplasties were from FECD (Dandona et al., 1997).
Table 1. Prevalence of Fuchs Endothelial Corneal Dystrophy by Region.
Prevalence reported as percentage of eyes in respective age and sex category.
| Region | Year | N (eyes) | Age (years) | Prevalence (%) | Ratio F:M | Author | ||
|---|---|---|---|---|---|---|---|---|
| General | Females | Males | ||||||
| North America | ||||||||
| United States (Texas) | 1933 | 800 | >21 | 6.62 | 9.07 | 3.62 | 2.5:1 | (Goar, 1933) |
| United States (Florida) | 1967 | 1348 | >40 | 3.9 | 4 | 3.8 | 1.05:1 | (Lorenzetti et al., 1967) |
| United States (Tangier island) | 2012 | 296 | >30 | 21.6 | 25.3 | 14.1 | 1.79:1 | (Eghrari et al., 2012) |
| Europe | ||||||||
| Iceland (Reykjavik) | 2006 | 1548 | >55 | 9.2 | 11 | 7 | 1.6:1 | (Zoega et al., 2006) |
| Asia | ||||||||
| Singapore (Chinese Singaporean) | 2002 | 920 | >50 | 6.7 | 8.5 | 4.4 | 2:1 | (Kitagawa et al., 2002) |
| Japan | 1996 | 211 | 56–76 | 3.3 | 3.3 | - | - | (Nagaki et al., 1996) |
| Japan (Monzen-machi) | 2002 | 598 | >50 | 3.7 | 5.5 | 1.5 | 3.7:1 | (Kitagawa et al., 2002) |
| Japan (Kujejima Island) | 2011 | 7524 | >40 | 4.1 | 5.8 | 2.4 | 2.4:1 | (Higa et al., 2011) |
Even in Ernest Fuchs’ initial description of FECD, he noted the preponderance of women over men, as 9 out of his 13 patients were women (Fuchs, 1910; Jun, 2010). Goar also found a strong female predominance of FECD, with women being affected 3 times more frequently than were men (Goar, 1933). The prevalence of corneal guttae and FECD has been shown in multiple studies to differ between women and men, with both early- and late-onset FECD having a female predominance. However, early-onset FECD due to the COL8A2 mutation follows a 1:1 ratio; among those older than age 50, there is an expected female to male ratio of 1.2:1 and 1.3:1 due to the greater longevity of women (Gottsch et al., 2005a). Late-onset FECD has been shown to have a female predominance at a ratio of 2.5:1 to 3.5:1 (Afshari et al., 2006; Kitagawa et al., 2002; Zoega et al., 2006). In the Reykjavik Eye Study, a higher prevalence was found in women (11%) than in men (7%) over the age of 50 (Zoega et al., 2006). A follow-up study on the same patient population found that the 7-year cumulative incidence of corneal guttae was estimated to be 15–23% of all eyes studied, with a sex-specific incidence of 8–18% for males and 19–29% for females (Zoega et al., 2013). Similarly, in Japan, a higher prevalence of FECD has been found in women (5.8%) than in men (2.4%) over the age of 40 (Higa et al., 2011). In Chinese Singaporeans, a greater prevalence has also been seen in women (8.5%) as compared to men (4.4%) over the age of 50 (Kitagawa et al., 2002). Other smaller studies have also consistently shown a female predilection for FECD (Cross et al., 1971; Krachmer et al., 1978; Rosenblum et al., 1980). In a multivariate model from the FECD Genetics Multi-Center Study, female sex increased the odds of advanced FECD by 34% (Zhang et al., 2013). Further evidence that supports a female predominance (3.5:1) in FECD is that women represented 77.6% of patients that have undergone a penetrating keratoplasty (PK) for FECD over a 30-year period at Duke University (Afshari et al., 2006). Likewise, a female predominance (1.6:1 to 2:1) has been shown in patients that had undergone endothelial keratoplasty (EK) for primarily FECD at the University of Toronto (Chan et al., 2018; Le et al., 2017).
4. Clinical Presentation and Assessment
FECD is typically a late-onset disease characterized by the slow, progressive degeneration of the CE, leading to corneal edema and vision loss (Figure 2A) (Hamill et al., 2013). In early disease, patients are usually asymptomatic. As FECD progresses, symptoms such as reduced vision and halos appear in the morning, secondary to increased corneal edema after eye closure during the night. In more advanced FECD, symptoms such as decrease in vision, photophobia, epiphora, and pain are present all day as there is increased stromal and epithelial edema with ruptured epithelial bullae, which may lead to a persistent epithelial defect or corneal ulcer. These symptoms can significantly affect the patient’s functioning.
Figure 2. Clinical Features of Fuchs Endothelial Corneal Dystrophy (FECD).
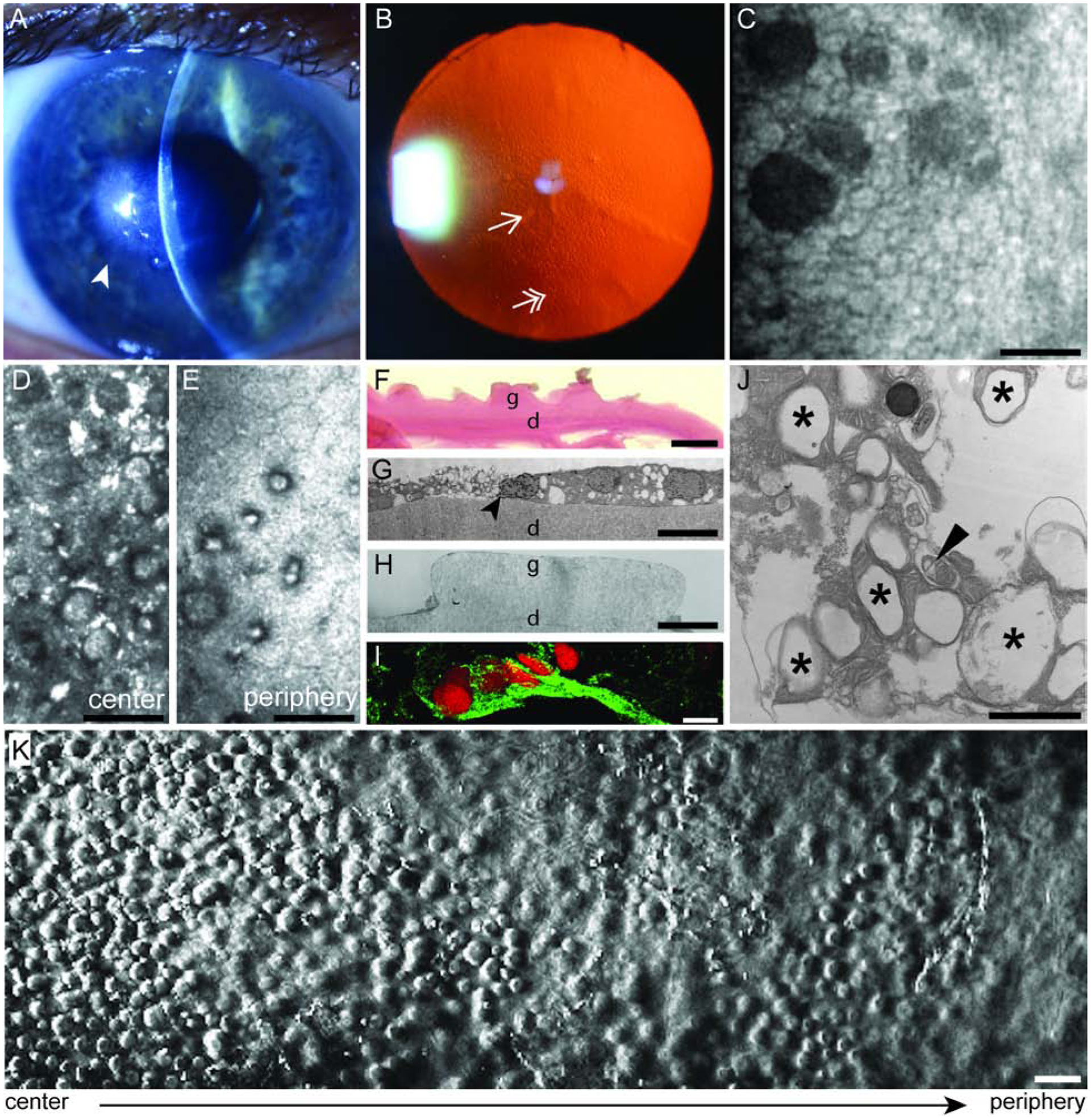
(A) Slit lamp photograph showing central cornea edema (arrowhead) in a patient with FECD. (B) Retroillumination photograph showing confluent central (single arrowhead) and non-confluent peripheral (double arrowhead) guttae. (C) Non-contact specular micrograph showing the corneal endothelium (white) and guttae (dark spaces) in a patient with FECD. Scale bar = 50 μm. (D) In vivo confocal microscopy showing the loss of corneal endothelial cells (CECs) and presence of guttae in the center and (E) periphery of a patient with advanced FECD. Scale bar = 50 μm. (F) Periodic Acid Schiff (PAS) staining of a cornea from a patient with FECD demonstrating excrescences called guttae (g) from Descemet’s membrane (d). Scale bar = 10 μm. (G) Transmission electron microscopy from a patient with FECD showing a CEC undergoing apoptosis (black arrowhead) on Descemet’s membrane (d) and (H) guttae (g). Scale bar = 10 μm. (I) Expression of endothelial mesenchymal marker in a FECD ex vivo specimen. Immunofluorescence distribution, observed by confocal microscopy, of Snail1 protein (green) in the cytoplasm of CECs on a FECD ex vivo specimen. Cells were stained with propidium iodide (red) to localize nuclei. Scale bar = 50 μm. (J) Transmission electron microscopy of FECD ex vivo specimen that demonstrate increased number of autophagic structures in the form of vacuoles (black stars) and autophagosome containing mitochondria (black arrowhead). Scale bar = 1 μm. (K) Phase contrast microscopy of an ex vivo corneal tissue specimen from a patient with FECD that underwent endothelial keratoplasty. A higher density of guttae is observed in the center of the cornea compared to the periphery. Scale bar = 50 μm.
4.1. Clinical Assessment
Examination of the cornea with the slit lamp biomicroscope allows for the direct illumination (diffuse illumination, focal illumination, and specular reflection) and indirect illumination (proximal illumination, sclerotic scatter, and retroillumination) of the different layers of the cornea. Slit-beam illumination with a thin beam produces an optical cross section of the cornea that allows for visualization of the CE and any abnormalities, including epithelial bullae, corneal edema, and guttae. In the cornea, a faint reflection emanates from the posterior corneal surface, and through specular reflection, CEC morphology can be directly assessed through the slit lamp. Retroillumination of the cornea is another important technique that was recognized as early as 1924 to assess and document the distribution and number of guttae (Eghrari et al., 2017a; Gottsch et al., 2006; Graves, 1924). After pupillary dilation, the cornea is assessed with reflected light from the fundus using a small angle between the biomicroscope and the illumination beam, which results in the visualization of individual and confluent guttae from light scattering (Figure 2B) (Gottsch et al., 2006). Retroillumination photography analysis, with either manual or automated counting of guttae, has been shown to be an effective way to document the number and distribution of guttae, and to demonstrate the formation of new guttae and their progression over time (Eghrari et al., 2017a; Gottsch et al., 2006). While a detailed evaluation of the cornea with slit lamp biomicroscopy, including specular reflection and retroillumination, is an essential part of the clinical examination, additional objective diagnostic testing, such as specular microscopy, confocal microscopy, pachymetry, and, most recently, Scheimpflug tomography, are often needed for further evaluation of the CE in FECD.
4.2. Clinical Staging
Guttae are excrescences of abnormal collagen deposited by the CE, and the accumulation of guttae is the first clinical sign of FECD (Figure 2F, 2H) (Arffa, 1991; Laing et al., 1981). As FECD progresses, there are morphological changes in CECs’ hexagonal shape and size, as well as the progressive formation of guttae (Adamis et al., 1993; Laing et al., 1981). Guttae typically originate in the central cornea, and as FECD progresses, they radiate out toward the periphery and are associated with a reduction in ECD, loss of normal CEC morphology, and CEC apoptosis (Figure 2G, 2K) (Kocaba et al., 2018; Zhang and Patel, 2015). FECD usually progresses through well-documented clinical stages, where, in early-stage FECD, there are non-confluent central guttae without any significant corneal opacification or edema (Hamill et al., 2013). In advanced FECD, guttae become confluent, while the DM is thickened, and ECD is significantly decreased (Figure 2F). In addition, visually significant corneal edema appears, leading to subepithelial fibrosis and opacification (Hamill et al., 2013). Four clinical stages of FECD, typically spanning a period of 20–30 years, have been described (Adamis et al., 1993). Stage 1 represents the earliest manifestation of FECD, where the patient is usually asymptomatic. Central corneal guttae, variable amounts of pigment on the posterior cornea, and a gray thickened DM are visible; however, despite these clinical findings, vision remains typically normal, although halos and glare may be present. Stage 2 is defined clinically by a painless decrease in vision and the presence of significant glare, typically worse upon awakening. There is a reduction in ECD, an increase in polymorphism and polymegethism, and guttae become confluent and extend towards the peripheral cornea. The decreased evaporation of tears during sleep results in a reduction in tear osmolarity and can lead to mild corneal stromal edema. Stage 3 is defined by worsening vision and recurrent episodes of pain due to the rupture of epithelial bullae. In stage 4, while there is further reduction of vision, there are fewer episodes of pain, since the chronic severe edema has resulted in the formation of subepithelial scarring, which decreases bullae formation but severely limits vision (Adamis et al., 1993).
Corneal guttae in the rare early-onset FECD due to mutations in COL8A2 are different in morphology than those seen in the more common late-onset FECD. These guttae are small and rounded, and associated with the endothelial center, in contrast to guttae in late-onset FECD, which are larger, sharply peaked, and initially positioned at the edges of the CECs (Gottsch et al., 2005a). Corneal guttae can also be found exclusively in the periphery as a normal finding of the aging population; these are called Hassall-Henle bodies, which do not lead to corneal edema (Hayashi et al., 2002). The clinical appearance of corneal pseudoguttae, which represent transient CEC changes that resemble corneal guttae, can also be seen secondary to inflammation, trauma, toxins, and infections (Nakashima et al., 2007).
FECD disease severity can also be graded subjectively on a 5-step scale (Krachmer grading) through slit lamp biomicroscopy by assessing the confluence and area of guttae, and the presence of corneal stromal or epithelial edema (Table 2) (Krachmer et al., 1978). The Krachmer grading classification scheme was revised from a 5-step scale to a 6-step scale (modified Krachmer grading), where the unaffected were graded 0, intermediate FECD cases were graded 1 to 3, and severe FECD cases were graded 4 to 6 (Table 2) (Louttit et al., 2012). The modified Krachmer grading scale is a simple grading system for the severity of FECD and has been shown to be a reproducible grading system in a multicenter study with histopathological confirmation of FECD (Louttit et al., 2012). Subjective grading of FECD using a modified Krachmer grading scale has also been shown to have moderate inter-observer agreement among corneal specialists (Repp et al., 2013). However, one limitation of this subjective grading scale is that there may be differences in the detection of corneal edema and the area of confluent central guttae, which may lead to inconsistent classification among observers.
Table 2. Krachmer and Modified Krachmer Grading Scales. (Krachmer et al., 1978; Louttit et al., 2012).
Measurements taken at the widest diameter of confluence after rotating the slit beam and measuring the diameter by narrowing the length of the beam.
| Grade | Guttae | Edema |
|---|---|---|
| Krachmer Grading Scale | ||
| 0 | 0 to 12 central guttae | No |
| 1 | >12 central, non-confluent guttae | No |
| 2 | 1 to 2 mm of confluent, central guttae | No |
| 3 | 2 to 5 mm of confluent, central guttae | No |
| 4 | >5 mm of confluent, central guttae | No |
| 5 | >5 mm of confluent, central guttae | Stromal or epithelial edema |
| Modified Krachmer Grading Scale | ||
| 0 | No guttae | No |
| 1 | ≤12 central/paracentral nonconfluent guttae | No |
| 2 | >12 central/paracentral nonconfluent guttae | No |
| 3 | 1 to 2 mm of confluent, central/paracentral guttae | No |
| 4 | 2 to 5 mm of confluent, central/paracentral guttae | No |
| 5 | >5 mm of confluent, central/paracentral guttae | No |
| 6 | >5 mm of confluent, central/paracentral guttae | Stromal and/or epithelial edema |
4.3. Pachymetry
Corneal thickness is another important parameter that clinicians use to monitor FECD and to help with surgical decision making in these patients. It has been previously shown that FECD patients with a preoperative corneal thickness of 640 μm or less (as measured by ultrasonic pachymetry) have a 95% chance that they will not require a corneal transplant (PK) within the first year of cataract surgery (Seitzman et al., 2005). Based on these findings, it was suggested that a change be made to the recommended preoperative pachymetry measurement threshold, from >600 μm to >640 μm, before corneal transplantation be considered (Seitzman et al., 2005). However, these recommendations were made by comparing an initial triple procedure (PK, cataract extraction, and intraocular lens insertion) to cataract surgery alone when determining cost effectiveness and delay in visual rehabilitation in FECD patients. Additional studies are, therefore, needed to evaluate if this threshold is still applicable for EKs. An important consideration when using central corneal thickness (CCT) as an indicator of corneal edema is that, while CCTs >640 μm usually indicate corneal edema, normal corneas without edema may also meet this criterion (Prasad et al., 2011). To address the limitations of the variability in the absolute value of CCT, an objective clinical grading method called the corneal central-to-peripheral thickness ratio (CPTR), which is based on the ratio of the central corneal thickness and the peripheral corneal thickness at 4 mm from the center, has been described (Repp et al., 2013). The CPTR has been shown to be repeatable, highly correlated with the clinical grade of FECD, and having an excellent discrimination between FECD and normal eyes (Repp et al., 2013). Therefore, measurements of CCT should be used as one clinical parameter when assessing FECD patients. Furthermore, there is significant diurnal variation of the cornea in patients with FECD, whereby FECD patients with advanced disease are 31 to 58 μm thicker in the morning, which resolves within 4 hours after eye opening (Fritz et al., 2019). Therefore, time from eye opening to measuring CCT should be considered and may improve the reliability of testing.
4.4. Scheimpflug Tomography
Another objective method of determining subclinical edema in FECD is through Scheimpflug tomography and considering the presence of the following tomographic features: loss of parallel isopachs, displacement of the thinnest point of the cornea, and focal posterior corneal surface depression (Sun et al., 2019). A new classification system for FECD has been proposed, where FECD corneas are classified as having clinically definite edema (based on slit lamp examination), subclinical edema (based on tomographic features without clinically definite edema), or no edema (no slit lamp tomographic features of edema) (Sun et al., 2019). Furthermore, it has been proposed that this classification system, which does not involve CCT, should be considered when evaluating FECD patients prior to cataract surgery or EK.
Scheimpflug imaging can provide an objective measurement of backscatter from both the anterior and posterior corneal surfaces through corneal densitometry. Corneal densitometry has been shown to be higher in FECD eyes compared to controls, even before the onset of clinically visible corneal edema (Alnawaiseh et al., 2016; Chu et al., 2017; Kobashi et al., 2018). An increase in the objective scattering index, followed by anterior corneal densitometry, has the strongest influence of distance visual acuity in eyes with mild FECD suggesting that distance visual acuity is highly associated with light scattering (Kobashi et al., 2018). Furthermore, preoperative corneal backscatter measured through corneal densitometry has been demonstrated to correlate with postsurgical best-spectacle corrected visual acuity after DMEK surgery (Schaub et al., 2019). Therefore, preoperative corneal densitometry may play a role in determining when surgical intervention is indicated in a FECD patient.
4.5. Imaging the Corneal Endothelium
The ability to assess the CE with objective imaging is crucial for the evaluation, monitoring, and guidance of management in FECD. Currently, specular microscopy and in vivo confocal microscopy (IVCM) are the only imaging modalities that allow for high resolution imaging of the CE and guttae (Ong Tone and Jurkunas, 2019). While these imaging modalities provide important diagnostic parameters about corneal ECD and morphology, they are limited in their ability to image central ECD in advanced stages of FECD (Figure 2D).
4.5.1. Specular Microscopy
Specular microscopy is a widely used non-invasive imaging modality that uses specular reflection for the in vivo visualization of the CE (Maurice, 1968; McLaren et al., 2014). Maurice initially developed specular microscopy to study the CE in vivo, and his techniques were further expanded to the use of specular microscope to clinically evaluate and photograph the CE in patients (Bourne and Kaufman, 1976; Laing et al., 1975; Maurice, 1968). In FECD there is variation in size (polymegethism) and shape (pleomorphism) of CECs, decreased ECD, and the formation of guttae (Vedana et al., 2016). Advantages of specular microscopy include its non-contact acquisition technique, rapid image acquisition time, automated focusing technology, and the facility to analyze the CE (McCarey et al., 2008). This non-invasive photographic technique can determine ECD (average number of endothelial cells per mm2), polymegethism (CEC size variations) or coefficient of variation (variation in cell area, calculated by dividing the standard deviation of the cell area by the mean cell area (μm2)), pleomorphism (percentage of hexagonal cells), CCT, and allows the visualization and documentation of guttae. A healthy cornea is expected to have 60% of cells as hexagons (McCarey et al., 2008). These parameters are important in diagnosing and monitoring patients with FECD. They are also important parameters in surgical planning, as patients with an ECD <1,000 cells/mm2, polymegethism or coefficient of variation >0.40, and/or pleomorphism <50% might not tolerate intraocular surgery (Cantor LB, 2017).
In FECD, guttae appear as isolated excrescences of DM with dark hyporeflective round bodies and an occasional central white reflex on specular microscopy (Bigar et al., 1978; Chiou et al., 1999; Laing et al., 1981). This central white specular reflex corresponds to the top of the guttae, where there is a change in the index of refraction between the AH and the surface of the guttae (Bigar et al., 1978). There is also lack of endothelial coverage over the center of the guttae, with some coverage surrounding the periphery of the guttae (Bigar et al., 1978). The specular microscopic appearances of guttae have been correlated to the histological specimens in FECD patients (Bigar et al., 1978). In 1981, using in vivo specular microscopy, Laing described two types of corneal guttae and the progressive morphological changes that occur in FECD, which he classified into five specific stages (Laing et al., 1981). All five stages could be seen in the same cornea clinically free of edema; however, the majority of guttae were typically found at the same progression stage (Laing et al., 1981). While later stages of guttae are often correlated with more advanced cases of FECD, early stages of guttae can be observed in early FECD and late stages of guttae in late FECD (Laing et al., 1981). In stage 1, the earliest form of corneal guttae, a dark structure with a single sharply defined bright spot at its center is seen, and is smaller than an individual CEC. The excrescence is not located adjacent to the CEC boundary, and the overlying CEC, as well as adjacent CECs, appear normal in morphology (Laing et al., 1981). In stage 2, the excrescence is larger and approximately the size of an individual CEC, and the adjacent CECs appear stretched, forming a rosette pattern. The CECs beyond the rosette appear normal (Laing et al., 1981). In stage 3, the excrescence is significantly larger and can be five to ten times the size of a CEC. The excrescence affects many CECs, and adjacent CECs are distinctly abnormal. At this stage, two types of excrescences have been described: a smooth round excrescence with a central well-circumscribed bright spot, and a rough excrescence with a wavy central bright spot. In stage 4, many individual excrescences that have coalesced into a multi-lobed structure containing many bright spots are present. While non-adjacent CECs can retain their normal shape, they tend to be larger in size. In stage 5, there is loss of the organized mosaic pattern of CECs, and there are no recognizable cells or cell boundaries. The typical pattern of light-gray cells surrounded by dark boundaries is reversed, where there now is a dark interior surrounded by a bright boundary, presumably from the deposition of collagenous material (Figure 2C) (Laing et al., 1981). While specular microscopy is an excellent tool in the evaluation of FECD, it has a limited role in advanced FECD, since it is difficult to acquire reliable images in patients with significant corneal edema due to increased light scattering in the stroma from collagen lamellae and keratocytes (Laing et al., 1981; Maurice, 1974).
4.5.2. Confocal Microscopy
In 1990, confocal microscopy was developed into a rapid contact-based imaging modality that allowed for the visualization of all corneal layers in vivo (Cavanagh et al., 1990). IVCM generates a clear image of the CE mosaic that allows for the visualization of CECs and guttae (Hara et al., 2003; Mustonen et al., 1998b). IVCM has several advantages compared to specular microscopy including its ability to image of all layers of the cornea and to analyze the CE through corneal edema (Grupcheva et al., 2001; Hara et al., 2003; Mustonen et al., 1998b; Niederer and McGhee, 2010; Ong Tone et al., 2019a). The optical arrangement of the confocal microscope, where the illumination and detection paths share the same focal plane, is called confocal and avoids the limitations in image quality due to the defocused light typically encountered with conventional light microscopy (Niederer and McGhee, 2010; Wiegand et al., 1995). Therefore, IVCM can produce clearer images of the CE in advanced FECD with significant corneal edema, partly due to the use of a coupling gel to reduce light scattering from the corneal epithelium (Chiou et al., 1999; Hara et al., 2003; Mustonen et al., 1998a; Ong Tone et al., 2019a).
In 1993, Kaufman first described confocal microscopic findings in FECD, where guttae appeared as dark round bodies (20–400 μm) with an occasional central white reflex (5–10 μm) (Chiou et al., 1999; Grupcheva et al., 2001; Kaufman et al., 1993; Mustonen et al., 1998a). Due to its high resolution (lateral optical resolution of 1–2 μm; depth optical resolution 10 μm), IVCM has the ability to image all corneal layers and, thus, can detect pathological changes in FECD in all corneal layers (Mustonen et al., 1998a). In FECD, in addition to the guttae and CEC changes seen in the CE, other prominent changes occur in the posterior layers that can be visualized with IVCM such as a thickened DM, which appears as an abnormal diffuse acellular reflection, and dark bands in the thickened DM (Hernandez-Quintela et al., 1998; Mustonen et al., 1998a). Furthermore, other changes in more anterior corneal layers can be visualized, such as epithelial bullae, cystic lesions, decreased sub-basal corneal nerves, and increased immune dendritic cell density (Aggarwal et al., 2018; Hernandez-Quintela et al., 1998; Mustonen et al., 1998a; Schrems-Hoesl et al., 2013). Increased anterior corneal backscatter as detected by IVCM has also been reported in eyes with moderate and advanced FECD, with similar trends in early FECD before the onset of clinically evident edema (Amin et al., 2014).
In advanced FECD, the central CE layer is largely replaced by confluent guttae, rendering central ECD inaccurate or undetectable (Figure 2D, 2K) (Fujimoto et al., 2014; Syed et al., 2017). Therefore, central ECD does not correlate well with disease grade 3 or higher (modified Krachmer grading scale) (Fujimoto et al., 2014). Since the CE is damaged more severely in the center than in the periphery in FECD, the peripheral zone is particularly important in these advanced FECD cases where central ECD is unreliable (Figure 2D, 2E, 2K). In these advanced cases, IVCM is superior to specular microscopy since it can provide high quality images of the CE mosaic through corneal edema, and is also capable of imaging both the central and peripheral cornea (Ong Tone et al., 2019a; Syed et al., 2017). Additionally, peripheral ECD has been shown to be the best predictor of disease severity and has the highest number of correlations with other clinical markers such as central ECD, logMAR best-corrected visual acuity (BCVA), clinical disease grade, and CCT (Syed et al., 2017).
4.6. Optical Coherence Tomography
Anterior segment optical coherence tomography (AS-OCT) produces high-resolution and 2-dimensional cross-sectional images based on the emission and reflection of light (low-coherence interferometry). Three types of AS-OCT are currently used: time-domain AS-OCT, spectral-domain based AS-OCT and swept-source AS-OCT (Ang et al., 2018; Jancevski and Foster, 2010). High resolution AS-OCT images allows for the identification of the corneal layers and visualizing corneal changes during the progression of FECD, such as a hyper-reflective, thickened, irregular, and multilayered DM (Kaluzny et al., 2009). In advanced cases of FECD, epithelial bullae, thickening of the epithelium, and subepithelial fibrosis can be observed (Kaluzny et al., 2009). Moreover, ultra-high-resolution AS-OCT and recently 3D AS-OCT, which produces cross-sectional and en-face images of the cornea, allow imaging and evaluation of DM characteristics and thickness in vivo and may represent a valuable support in setting of FECD diagnosis and staging (Iovino et al., 2018; Shousha et al., 2010).
AS-OCT also allows for the measurement of corneal thickness, which is helpful in monitoring the severity of disease. AS-OCT is helpful in the postoperative follow-up of endothelial keratoplasty (DMEK and DSAEK) to assess and identify potential causes of early endothelial graft detachment and to assess the graft-host interface (Satue et al., 2016; Tarnawska and Wylegala, 2010; Yeh et al., 2013).
Intraoperative AS-OCT has recently been used to visualize and assess graft orientation in DMEK surgery without using a DM orientation mark. This allows for faster graft positioning with less graft manipulation, and the identification of interface fluid that is clinically undetectable under the surgical microscope, particularly in the presence of severe corneal edema (Ide et al., 2010; Knecht et al., 2010; Saad et al., 2015).
4.7. Artificial Intelligence
During the last decade, corneal imaging has greatly evolved with the development of several techniques allowing measurement of numerous corneal parameters. One of the challenges is how to integrate all this information from complementary multimodal imaging devices. Machine learning with artificial intelligence may allow for earlier identification of patients with FECD, predict which FECD patients are at high risk for progression, and help guide clinical decision making (Ambrosio and Guerra, 2019; Lopes et al., 2019).
Machine learning and artificial intelligence is also being explored as a tool in corneal endothelial keratoplasty, where in combination with intraoperative OCT, residual interface fluid volume can be detected (Lopes et al., 2019). In the postoperative period, graft dislocation can be quantified by using a convolutional neural network (Lopes et al., 2019).
5. Barrier and Pump Dysfunction
The CE plays a key role in preserving the cornea in a state of deturgescence, or relative dehydration, to maintain corneal clarity. The primary functions of the CE are to form a selective passive barrier through simple and facilitated diffusion that allows leakage of solutes and nutrients from the AH to the avascular cornea, and to continuously pump ions to counteract the continuous leakage of fluid into the corneal stroma to maintain the delicate water balance required for adequate corneal hydration (Bonanno, 2003; Harris, 1962; Harris and Nordquist, 1955; Maurice, 1957; Mishima, 1982). The ability of the CE to maintain corneal hydration and transparency occurs through a “pump-leak” mechanism initially described by Maurice (Bonanno, 2003; Maurice, 1972). To achieve this, CECs generate an outward osmotic driving force through primary and secondary active transport mechanisms that generate a net ion flux from the corneal stroma to the anterior chamber side of the CE to counteract an inward imbibition pressure, which is generated by corneal stromal glycosaminoglycans (Barfort and Maurice, 1974; Bonanno, 2003; Maurice, 1972; Mishima, 1982). The CE fluid pump is dependent on the presence of bicarbonate (HCO3−) and chloride (Cl−) (Bonanno, 2003, 2012). The osmotic force to preserve the cornea’s relative state of deturgescence is maintained by the CE pump function that consists of the primary active transport of the Na+-K+ ATPase pump, which actively transports fluid out of the cornea and back into the AH, and numerous secondary membrane ion transporters, including solute carrier family 4 member 11 (SLC4A11), sodium bicarbonate cotransporter-1, Na+: 2HCO3- (pNBCe1), Na+:K+:2Cl cotransporter (NKCC1), Cl-/HCO3- exchanger (AE2), sodium proton exchanger-1; Na+/H+ (NHE1), and the isoforms of monocarboxylate transporters (MCTs) 1, 2, and 4 (Bonanno, 2003, 2012; Geroski and Edelhauser, 1984; Jalimarada et al., 2014; Li et al., 2016). Normal CECs express various levels of these ion transporters including, from most to least abundant, SLC4A11, Na+-K+ ATPase, pNBCe1, NHE1, and MCTs 1, 2, and 4 (Jalimarada et al., 2014).
The CE also maintains a passive cell-to-cell contact barrier through focal tight junction proteins such as zonular occludens 1 (ZO-1) and occludin, which are located at the apical aspect of the lateral membranes. These cell-to-cell contacts are further strengthened through adhesion proteins, such as N-(neuronal) cadherin, E-(epithelial) cadherin, α-catenin, β-catenin, and γ-catenin, which connect to a circumferential band of actin filaments located in the sub-membranous cytoplasmic complex towards the apical aspect of CECs. These cell-to-cell contacts help maintain cell shape and also mediate cellular migration (Joyce, 2003). Neighboring CECs are also functionally coupled through gap junction proteins such as connexin-43. CECs also express numerous proteins that are important in mediating cell-to-substrate adhesions, which include vinculin, talin, β3-, alpha-v-, and beta-5 integrins (Joyce, 2003).
In FECD, there is progressive CEC death ultimately leading to loss of barrier and pump function causing corneal edema and formation of painful epithelial bullae (Bourne, 2003). In FECD patients with an increased CCT but without epithelial edema, it has been shown that there is an increased permeability of CECs as compared to controls, while no significant difference in pump rate has been found (Burns et al., 1981). These results suggest that impairment of barrier function in the CE may precede pump dysfunction in FECD, which results in increased fluid inflow to the cornea without a sufficient compensatory increase in pump function (Burns et al., 1981). In contrast, it has also been shown that there is no difference in the permeability of CECs in FECD patients compared to controls, which suggests that pump dysfunction is largely responsible for corneal edema (Wilson and Bourne, 1988). In early stages of FECD, there is a compensatory increase in Na+-K+ ATPase density on the basolateral membrane of the CE as compared to controls, because the CEC actively transport fluid out of the cornea and back into the AH to maintain corneal clarity (Geroski et al., 1985). However, there is a gradual decline in Na+-K+ ATPase density as FECD progresses in severity (McCartney et al., 1989). Real-time polymerase chain reaction-(RT-PCR) has detected that FECD CECs show significant downregluation of Na+-K+ ATPase as compared to normal samples, along with MCT 1 and 4 downregulation, which has been postulated to play a role in CE fluid secretion. These findings further support the fact that the significant downregulation of ion transporters indicates compromised CE pump function in FECD (Jalimarada et al., 2014). Furthermore, histopathological examination of FECD corneas has revealed aberrant deposition of ECM and CEC bodies surrounding the base of the guttae, as well as extremely thinned but intact CEC membranes overlying the guttae. These findings suggest that it is unlikely that CEC pump function is intact in the areas overlying guttae, since no organelles are seen (Bergmanson et al., 1999).
SLC4A11 belongs to the bicarbonate transport family, and SLC4A11 mutations have been associated with corneal hereditary endothelial dystrophy (CHED) and FECD (Siddiqui et al., 2014; Vithana et al., 2008). Reduced expression and loss-of-function of bicarbonate transporters in CECs have been shown to compromise pump function (Gottsch et al., 2003; Groger et al., 2010). Altered SLC4A11 expression has also been shown to affect pH regulation, bicarbonate and lactic acid transport, and CEC pump function. Furthermore, SLC4A11 knockout mice accumulate sodium chloride in the corneal stroma and develop corneal edema (Groger et al., 2010). Recent evidence has shown that SLC4A11 mutants have altered H+ flux properties, rather than impaired protein trafficking. These findings suggest that altered functional activity of some SLC411 mutants, rather than membrane trafficking, may be responsible for the phenotype seen in patients with corneal endothelial dystrophy with these mutations (Li et al., 2019). CECs derived from SLC4A11 knockout mice demonstrate active ion transport, except for NH3:H+ transporter activity (Zhang et al., 2017b). Furthermore, glutaminolysis (glutamine catabolism) is severely impaired in CECs deficient in NH3:H+ cotransporter SLC4A11. Importantly, glutaminolysis generates a large amount of the ATP through the citric acid cycle to maintain CE function (Zhang et al., 2017a). These findings support that SLC4A11 is part of an important metabolic mechanism that provides CECs with their energy requirements to maintain high levels of transport activity, and suggest that FECD can be a disorder resulting from altered glutamine catabolism, as is seen in SLC4A11-associated degenerative disease.
In summary, these findings suggest that both barrier and pump function of the CE are important in maintaining the cornea in a state of deturgescence and that dysfunction of barrier and pump function can lead to the corneal edema seen in FECD.
6. Genetic Mutations
Although it has been more than one hundred years since Clegg initially reported the hereditary nature of FECD, it was not until the early 1950s that subsequent reports of the inheritability of FECD started emerging (Levitt and Lloyd, 1952; Mortelmans, 1952; Stocker, 1953). Further reports examining multiple families were described in the 1970s, including a report by Cross and colleagues that predicted an autosomal dominant mutation as the possible etiology of FECD (Cross et al., 1971). In 1978, Krachmer and colleagues further supported this when they published their findings from a prospective study showing that FECD demonstrated a strong familial tendency, where 50% of families of the proband were affected. They emphasized that this number was likely higher, since many of the families in which only the proband was affected were inadequately examined (Krachmer et al., 1978). The autosomal dominant mode of inheritance was proposed independently by Magovern and colleagues, and Rosenblum and colleagues (Magovern et al., 1979; Rosenblum et al., 1980). Magovern and colleagues observed complete penetrance but failed to demonstrate 50% segregation, whereas Rosenblum and colleagues reported a high degree of penetrance and variable expressivity in their respective cohorts. The former familial observation included the identification of patients in their sub-teen age group and represents a subset of patients demonstrating an early-onset Fuchs-like phenotype. In 2012, the FECD Genetics Multi-Center Study examined a large collection of FECD-affected individuals and their families in the United States (Louttit et al., 2012). This study was the first to show that the clinical phenotype of guttae and severe disease were highly heritable, with heritability estimates of 30.4% to 39.2%, in the general Caucasian population. The genetic basis of FECD is complex and heterogeneous, with variable expressivity and incomplete penetrance (Cross et al., 1971; Krachmer et al., 1978; Weiss et al., 2015). The strongest genetic association with FECD is a non-coding trinucleotide CTG repeat expansion within the third intron of the transcription factor 4 (TCF4) gene (Wieben et al., 2012). Other less common genetic mutations have also been associated with FECD including COL8A2, DMPK, SLC4A11, ZEB1/TCF8, LOXHD1, AGBL1, KANK4, LAMC1, ATP1B1, RAD51, FEN1, XRCC1, NEIL1, TGFBI, CLU, PITX2, PTPRG, FASLG, and KCNJ13 (Table 3).
Table 3.
Genetic Mutations Associated with Fuchs Endothelial Corneal Dystrophy.
| FECD | Gene | Protein | Mutation | Selected References |
|---|---|---|---|---|
| Early-onset | COL8A2 | α2 subunit of collagen VIII | Q455K L450W |
(Biswas et
al., 2001) (Gottsch et al., 2005a; Liskova et al., 2007) |
| Late-onset | TCF4 (E2–2) | Class I basic helix-loop-helix transcription factor | CTG repeat within third intron | (Mootha et al, 2014; Wieben et al, 2012) (Baratz et al, 2010; Li et al, 2015) (Afshari et al, 2017) |
| DMPK | Myotonic dystrophy protein kinase | CTG repeat within the 3’ UTR | (Mootha et al, 2017; Winkler et al, 2018) | |
| SLC4A11 | NaBC1 co-transporter | E399K G709E T754M c.99–100delTC |
(Vithana et al, 2008) | |
| TCF8/ZEB1 | Zinc-finger transcription factor | N696S N78T Q810P Q840P A905G P649A rs77516068 rsl49166539 IVS2+276 S234S E733K A818V L947Stop |
(Mehta et al,
2008) (Riazuddin et al, 2010) |
|
| L0XHD1 | Highly conserved protein consisting entirely of polycystin/lipoxygenase/alpha-toxin (PLAT) domains | R547C rs450997 |
(Riazuddin et
al., 2012) (Afshari et al., 2017) |
|
| AGBL1 | Cytosolic glutamate decarboxylase | R1028Stop c.2969G>C |
(Riazuddin et al., 2013) | |
| KANK4 | Cytoskeleton regulation by regulating actin polymerization | rs79742895 | (Afshari et al., 2017) | |
| LAMC1 | Extracellular matrix glycoprotein | rs3768617 R490W | (Afshari et al., 2017) (Wieben et al., 2018) | |
| LINC00970/ATP1B1 | β-subunit of the Na+ −K+ATPase | rsl022114 | (Afshari et al., 2017) | |
| DNA Repair enzymes | ||||
| Extracellular Matrix | ||||
| Mitochondrial | ||||
| Other |
6.1. International Classification of Corneal Dystrophies (IC3D): FECD
The most recent IC3D classification categorizes FECD into 2 forms: a rare early-onset FECD (typical age of onset in the third decade of life) and a more common late-onset FECD (typical age of onset in the fifth or sixth decades of life). The genetic locus of early-onset FECD has been mapped to 1p34.3-p32 (FECD1) while the genetic loci of late-onset FECD have been mapped to 13pter-q12.13 (FECD2), 18q21.2-q21.3 (FECD3), 20p13-p12 (FECD4), 5q33.1-q35.2 (FECD5), 10p11.2 (FECD6), 9p24.1-p22.1 (FECD7), and 15q25 (FECD8) (Weiss et al., 2015).
6.2. Early-Onset FECD
6.2.1. COL8A2
The availability of the human genome sequence has propelled the identification of disease-causing variants (Table 3). Linkage analyses have been utilized to delineate the genomic loci that segregate with the disease in familial FECD cases. The COL8A2 gene is the only gene that has been associated with early-onset FECD and is rarely associated with late-onset FECD (Biswas et al., 2001). The COL8A2 gene encodes the 703 amino acid α2 subunit of collagen VIII, a short-chain collagen that is a component of DM secreted by CECs (Biswas et al., 2001). A genome-wide search of a 3-generation family with early-onset FECD identified a locus on chromosome 1p34.3-p32 (Biswas et al., 2001). Four possibly pathogenic variants in COL8A2 (R155Q, R304Q, R434H, and Q455K) were identified by Sanger sequencing following refinement of the linkage interval to chromosome 1p34.3-p32 (Biswas et al., 2001). However, one of these variants, R155Q, was later identified in unaffected individuals in a Japanese cohort, suggesting that this variant is non-pathogenic (Kobayashi et al., 2004). While the Q455K variant is associated with early-onset FECD, the 2 variants R304Q and R434H have been associated with late-onset FECD; however, these could also be benign variants (Biswas et al., 2001; Gottsch et al., 2005a). The use of linkage analysis to identify variants that may be causal was utilized to revisit the previously reported familial case described by Magovern and colleagues (Magovern et al., 1979). The L450W mutation was detected in individuals who were previously identified with an early-onset FECD (Gottsch et al., 2005a; Magovern et al., 1979). The L450W mutation segregated with the disease as a dominant mutant with 100% penetrance, and was present in all affected individuals and none of the unaffected individuals. The L450W mutation was also identified later in second pedigree from a Caucasian British family, providing further evidence of the autosomal dominant inheritance pattern of this mutation (Liskova et al., 2007). The mutations identified thus far have been linked exclusively to early-onset FECD, and none to posterior polymorphous corneal dystrophy patients (Yellore et al., 2005). Additional variants have been detected in various ethnicities, including A35A, G495G, T502M, P575L, and P586P; however, these are considered non-pathogenic or of unknown association (Aldave et al., 2006; Kobayashi et al., 2004; Mok et al., 2009).
6.3. Late-Onset FECD
The identification of possibly causal variants within COL8A2 in unrelated familial cases helped propel the use of linkage to identify multiple loci that segregated with individuals with late-onset FECD. The first late-onset FECD locus, FCD1, was identified in a single large multi-generation family, where the region possibly harboring causal variants was mapped to 13pTel-13q12.13. This single locus spans 26.4 Mb and contains the chromosome 13 nucleolus organizer, the centromere, and 44 protein-encoding genes, whereby sequencing of 10 genes did not yield any causal variants (Sundin et al., 2006b). A second genetic locus, FCD2, spanning 18q21.2-q21.32, was identified in 3 unrelated families with late-onset FECD. Although the critical interval was different, no polymorphic markers were found within or near the FCD1 locus (Sundin et al., 2006a). The progression of FCD2-linked individuals was far lower (5%), than in FCD1 (22%), highlighting loci-dependent phenotypic differences in disease progression. FCD2 also showed a dominant Mendelian trait, with 85% penetrance and a 15% phenocopy rate (Sundin et al., 2006a). A third genetic locus, FCD3, with a defined critical interval between 5q33.1-q35.2, was identified in a single large family with multiple affected individuals and revealed a comparatively milder severity with respect to the age at onset and rate of progression (Riazuddin et al., 2009). A fourth genetic locus, FCD4, mapped to 9p22.1-p24.1 was identified in one family where the affected individuals also harbored the causal TCF8 (Q840P) allele. Out of the 12 affected individuals in this family, the presence of the Q840P allele sufficiently explained the causality in only 7 affected individuals, and further linkage analysis delineated FCD4 in the remaining 5 affected individuals who were negative for the Q840P mutation in TCF8. Furthermore, individuals who harbored both the Q840P mutation as well as FCD4 had a severe FECD phenotype, corresponding to the Krachmer grading of 6 bilaterally, which was significantly higher than that of the individuals with either one of the two alleles (Riazuddin et al., 2010b). The presence of the Q840P allele was sufficient but not necessary for disease pathogenesis in this pedigree.
Most previous FECD genetic studies have involved only a few large multi-generational families. In 2009, a genome-wide linkage analysis with a single nucleotide polymorphism (SNP) panel was performed on 92 FECD individuals from 22 families to identify regions of genetic linkage in FECD and to analyze affected individuals for mutations in the COL8A2 gene. The previously reported mutations in the COL8A2 gene were not found; however, potential linkage regions on chromosome 1, 7, 15, 17, and X were identified (Afshari et al., 2009).
6.4. Causal Variants Linked to FECD
6.4.1. TCF4 (E2–2)
Although a number of loci have been identified and include linkage intervals and causal variants, the contribution of these loci to the total genetic load of FECD accounts only for a very small fraction of FECD cases. Linkage studies are used to delineate loci in familial cases, whereas genome-wide association studies (GWAS) can identify the possible association of SNPs to diseases. Baratz and colleagues took an unbiased approach to probe the causality of FECD, even though a number of loci (FCD1–4) were already associated and linked to FECD. The GWAS analysis was performed on a total of 280 FECD cases divided into a discovery set and an independent replication set based on the most significant observations, and compared with 410 controls (Baratz et al., 2010). The GWAS analysis revealed one region within 18q21.2 with multiple SNPs that showed a significant association with FECD. This region included a locus that involved gene transcription factor 4 (TCF4), and also encompassed the previously identified FCD2 (Sundin et al., 2006a). While many SNPs were found to be associated with FECD, four SNPs (rs17595731, rs613872, rs9954153, and rs2286812) in TCF4 were found to be independently associated with FECD (Baratz et al., 2010). TCF4 encodes a ubiquitously expressed class I basic helix-loop-helix transcription factor called E2–2 that forms either homo- or heterodimers with other basic helix-loop-helix proteins. TCF4, through Ephrussi (E-box) promoter sequences within target genes, are involved in cellular growth and differentiation (Flora et al., 2007; Skerjanc et al., 1996). Individuals that are homozygotes for the disease TCF4 variant are at 30-fold increased odds of having FECD as compared to control subjects. Of note, the TCF4 gene has also been called immunoglobulin factor 2 and SL3–3 enhancer factor 2, and the transcription factor 7-like 2 gene encodes a protein called T-cell factor 4, which has created some confusion in the literature. Subsequent association and linkage studies have further supported the highly significant association between TCF4 (rs613872) and FECD, with a large odds ratio of between 3.6 to 9.5 (Baratz et al., 2010; Igo et al., 2012; Kuot et al., 2012; Kuot et al., 2017; Mootha et al., 2014; Okumura et al., 2019a; Rao et al., 2017; Riazuddin et al., 2011). In 2015, a meta-analysis was performed to investigate the association between the genetic polymorphism of TCF4 (4 SNPs: rs17595731, rs613872, rs9954153, and rs2286812) and the risk of FECD (Li et al., 2015). Thirteen studies were included, and the pooled results showed a significant association between all 4 SNPs and the risk of FECD, with the strongest positive association between the TCF4 rs613872 polymorphism. Furthermore, a recent GWAS on a total of 2,075 FECD cases and 3,342 controls identified the TCF4 SNP rs784257 as the most significant SNP, responsible for 21.9% of the variation in FECD within their discovery sample (Afshari et al., 2017). They also found a strong linkage disequilibrium between rs784257 and the previously reported variant rs613872 in the third intron of TCF4; moreover, both SNPs were in moderate disequilibrium with the expanded form of the CTG18.1 trinucleotide repeat in TCF4 (Afshari et al., 2017). Their sex-stratified analysis also identified that the TCF4 variant rs784257 conferred a higher risk of FECD on men than women (Afshari et al., 2017). In addition to a sex-stratified risk with TCF4 variants, there have also been reported differences of association based on ethnic background. A meta-analysis of 8 publications examining 5 SNPs in TCF4 found that the TCF4 variant rs613872 was strongly associated with FECD in Caucasians but not in Chinese, suggesting that there is ethnic diversity in FECD SNP associations (Lau et al., 2014). Intriguingly, a SNP located with the CTG repeat sequence (10th CTG repeat rs143743309) has been reported; however, the significance of this remains to be determined (Wieben et al., 2014).
6.4.1.a. CTG Repeats
In 1997, Breschel and colleagues in their search for polymorphic CTG repeats as candidate genes for bipolar disorder, first identified the CTG repeat expansion within the third intron of TCF4 located on 18q21.1 (Breschel et al., 1997). Notably, they identified the expanded allele in approximately 3% of their study population. In 2012, the strong association between a non-coding CTG trinucleotide repeat expansion within the third intron of TCF4 and FECD was first reported. It was found that a repeat length >50 was present in 79% of FECD cases as compared to 3% of normal controls; moreover, there was a 79% sensitivity and 96% specificity of >50 repeats identifying FECD in the cohort (Wieben et al., 2012). It was also observed that the expanded repeat was more specific than the previously identified SNP rs613873, which was only 79% specific. These findings were independently replicated, where the CTG 18.1 allele (CTG repeat length ≥40) conferred significant risk for FECD (32.3 odds ratio), and co-segregated with the trait in 52% of families with complete penetrance and 10% with incomplete penetrance (Mootha et al., 2014). Overall, there is strong evidence that CTG repeat expansion in the TCF4 gene is a common causal variant that confers a significant risk of developing FECD. While the CTG repeat expansion polymorphism has been shown to have a stronger association than the SNP rs613872 polymorphism, both together would likely predict better susceptibility to FECD (Kuot et al., 2017).
There appear to be regional differences based on ethnicity in the percentage of FECD patients that have the CTG repeats. In largely Caucasian populations from Europe, the United States, or Australia, CTG repeat expansion prevalence rates between 51% and 79% have been reported in FECD patients (Afshari et al., 2017; Kuot et al., 2017; Mootha et al., 2014; Vasanth et al., 2015; Wieben et al., 2014; Wieben et al., 2012; Zarouchlioti et al., 2018). In contrast, FECD patients from Singapore with an ethnic Chinese background, Indians, Japanese, and African-Americans have lower CTG repeat expansion prevalence rates, between 17.3% and 35% (Nakano et al., 2015; Rao et al., 2017; Xing et al., 2014). Furthermore, there is evidence that these differences are not solely based on region, but that ethnicity plays a large contributory role. In the United States, studies have demonstrated that there is a similar rate of mild FECD among African-Americans as compared to Caucasians; however, African-Americans are less likely to progress to a more severe phenotype requiring keratoplasty (Mahr et al., 2016). Furthermore, African-Americans with FECD are less inclined than Caucasians to demonstrate the CTG repeat expansion; 35% of African-American cases and 62.5% of Caucasian cases have the CTG repeat expansion (Eghrari et al., 2017b). In the Chinese population in Singapore, while there is a lower prevalence rate of CTG repeat expansion, there is still a significant association with FECD, with an estimated odds ratio of 66.5 (Xing et al., 2014).
A correlation between the size of the repeat (and zygosity) and the severity of FECD has also been reported, where a threshold of mono-allelic expansion to 103 CTG repeats has been identified; above this, the allele confers causality in 17.8% of FECD cases (Vasanth et al., 2015). A regression analysis showed a significant correlation between disease severity and age in patients with either a mono-allelic expansion or a bi-allelic expansion of >40 CTG repeats. However, a large inter-generational expansion of the CTG18.1 allele or earlier onset of the disease was not observed in a large multi-generational family, as can be seen in other trinucleotide repeat expansion disorders such as Huntington disease and myotonic dystrophy (Vasanth et al., 2015). Additional studies have also corroborated these findings, where FECD disease severity, as determined by the Krachmer grading scale, was greater in FECD patients with CTG repeat expansion (Soliman et al., 2015). Furthermore, it has also been shown that FECD patients with a CTG repeat expansion >40, as compared to those without, are at greater risk for FECD progression and the development of Threshold Disease at 5 years from the time of diagnosis (defined as CCT >700 μm, ECD <700 cells/mm2, or need for keratoplasty) (Soh et al., 2019). However, Okumura and colleagues found no significant association between the size of the repeat and clinical parameters (age, sex, visual acuity, and CCT) in a large cohort of 398 German patients with FECD (Okumura et al., 2019a).
An important observation is that, in many studies, a small percentage of healthy controls have the CTG repeat expansion without any clinical evidence of FECD. The prevalence rate of the CTG expansion has been reported between 0% to 5% in healthy individuals of Chinese, Japanese, and Indian ethnicity, and 3% to 6% in European and Caucasian ethnicities (Vasanth et al., 2015). An intriguing study would be to follow these asymptomatic individuals with no clinical evidence of FECD to see if they develop FECD.
6.4.1.b. TCF4 Gene Expression and RNA Foci
Several studies have investigated the expression profile of TCF4 in FECD samples and have obtained differing results. Two studies did not find any significant alterations in TCF4 expression levels in FECD endothelial samples (with or without CTG repeat expansion) as compared to controls (Mootha et al., 2015; Oldak et al., 2015). In contrast, another study detected a reduction in TCF4 gene expression in FECD CECs, suggesting that a loss of function of TCF4 may contribute to FECD (Foja et al., 2017). In contrast, a recent study by Okumura and colleagues demonstrated that TCF4 was significantly upregulated in FECD CECs, regardless of repeat expansion status, as compared to controls (Okumura et al., 2019b). Further studies are needed to elucidate how TCF4 expression levels contribute to the pathogenesis of FECD.
The location of the CTG repeat in an intron led to the hypothesis that RNA toxicity played a role in the pathogenesis of FECD, as seen in other repeat expansion neurodegenerative diseases, such as myotonic dystrophy type 1 and 2, fragile X-associated tremor/ataxia syndrome, C9OR72-associated amyotrophic lateral sclerosis, and frontotemporal dementia (Du et al., 2015; Mootha et al., 2015). CTG repeat expansion in the TCF4 gene leads to the sequestration of the mRNA-splicing factor MBNL1 (muscleblind-like 1) and MBNL2 in RNA foci, leading to mis-splicing of essential MBNL1-regulated mRNAs (ADD3, INF2, SORBS1, GNAS, FGFR1, MBNL1, and MBNL2)(Rong et al., 2019; Zarouchlioti et al., 2018). Furthermore, a comparative analysis of the gene expression profiles of FECD patients with or without CTG repeat expansion showed widespread mis-splicing in FECD patients with CTG repeats and identified 39 genes that were significantly different between the 2 cohorts, including MBNL1, NUMA1, PPFIBP1, INF2, SCARB1, SYNE1, ADD3, and MBNL2 (Wieben et al., 2018). Overall, these findings highlighted the importance of RNA toxicity and mis-splicing in FECD. Both sense and anti-sense RNA foci are detectable in FECD CE from patients with a TCF4 repeat expansion. The sense expanded repeat transcripts are the predominant species, and approximately 2 sense foci can be detected in each CEC. An antisense oligonucleotide (ASO) has also been developed to target the mutant-repetitive RNA, which has been shown to decrease foci formation and reversal of mis-splicing in ex-vivo FECD specimens (Hu et al., 2018). Similarly, another ASO complimentary to the CTG repeats has also been shown to reduce RNA foci formation and rescue abnormal MBNL1 nuclear localization and downstream mis-splicing in cell lines derived from FECD patients (Zarouchlioti et al., 2018). Importantly, ASO can be targeted to CECs after intraocular administration of ASO, highlighting the potential for ASO-mediated therapy for FECD.
Another mechanism that has been proposed is the synthesis of repeat-associated non-ATG (RAN) translation products. RAN translation occurs through unconventional protein translation that does not require an initiating ATG (Soragni et al., 2018). RAN translation products are detected in the CE from FECD patients with CTG repeat expansions and have also been shown to be toxic to CECs (Soragni et al., 2018). Another proposed mechanism links CTG repeat expansion, oxidative stress, and DNA damage. It has been hypothesized that the CTG repeat expansion in FECD renders the CECs more susceptible to oxidative damage due to the guanine-rich repeats (Wieben et al., 2012). Environmental stresses, such as cold, heat, hypoxia, and oxidative stress, have been shown to increase mutagenesis, particularly in long trinucleotide repeats, and also to involve DNA re-replication (Chatterjee et al., 2015). While the pathway for stress-induced trinucleotide repeat expansion mutagenesis has been shown to be important in human cells as an effective way to promote adjustments in protein activity and gene expression, it remains to be implicated specifically in FECD (Chatterjee et al., 2015).
6.4.1.c. DMPK
FECD is a repeat expansion disorder with a similar proposed mechanism to that of other neurodegenerative repeat expansion disorders such as myotonic dystrophy type 1 (DM1), where there is a repeat expansion within the 3’-untranslated region of DMPK (dystrophia myotonia protein kinase). An increased prevalence (46%) of FECD in patients with DM1 has been reported, suggesting that DM1 patients are at an increased risk for FECD over the general population (Mootha et al., 2017). A similar phenotype of nuclear RNA foci co-localizing with MBNL1 can also be detected in CECs of FECD patients with DM1. Similar findings were also seen in a prospective study evaluating DM1 patients and their families for FECD (Winkler et al., 2018). An increased prevalence (36%) was also observed in DM1 families, and both diseases co-segregated in their analysis. Furthermore, none of the affected FECD probands studied were positive for repeat expansion in TCF4. While these studies demonstrate that DMPK repeat expansion mutations contribute to the genetic burden of FECD, they are uncommon and likely play a small contributory role to FECD overall.
6.4.2. SLC4A11
SLC4A11 encodes the protein NaBC1, a co-transporter normally located on the cell surface, and has been implicated in various cellular functions, including borate transport, water flux, regulation of cellular pH, and antioxidative stress (Patel and Parker, 2015). Mutations in SLC4A11 were first identified in families with CHED (Vithana et al., 2006). Three missense changes, namely, E399K, G709E, and T754M, and one deletion mutation (c.99–100delTC) were later identified in a screen of FECD patients that contributed to the first set of causal variants for FECD (Vithana et al., 2008). In this study, approximately 5% of FECD seen in patients of Chinese ethnicity from Singapore and Hong Kong was attributed to SLC4A11 mutations. Seven additional heterozygous mutations in SLC4A11 (E167D, R282P, Y526C, V575M, G583D, G742R, and G834S) have been linked to FECD in sporadic and familial cases in patients of primarily northern European descent (Riazuddin et al., 2010a). Based on this study, approximately 2% of FECD in northern Europeans is attributable to SLC4A11. Three additional mutations in SLC4A11 (W240S, V507I, and T434I) in sporadic late-onset and familial early-onset FECD cases have also been identified (Soumittra et al., 2014). Based on their study, approximately 11% of FECD cases in South Asians was attributable to SLC4A11. While multiple mutations in SLC4A11 have been linked to CHED and FECD, one report identified a novel homozygous mutation in SLC4A11 (C386*) in a CHED patient, whose mother harbored the same heterozygous mutation and demonstrated features of late-onset FECD (Kim et al., 2015). This report suggests that the same genetic mutation can result in two different clinical phenotypes (CHED and FECD), and supports the notion that corneal endothelial dystrophies are a phenotypic continuum with genetic overlap.
6.4.3. ZEB1/TCF8
Zinc-finger E-box binding homeobox 1 (ZEB1) (also known as AREB6, TCF8, δEF-1, BZP, and ZFHEP) is a zinc-finger transcription factor that has been implicated in embryonic development, regulation of collagen type 1 expression, and repression of an epithelial phenotype (Krafchak et al., 2005). A homozygous frameshift mutation in ZEB1 on chromosome 10 was first identified in a family diagnosed with posterior polymorphous corneal dystrophy that segregated with the disease along with 4 nonsense and a frameshift mutation in sporadic cases (Krafchak et al., 2005). One heterozygous missense mutation (N696S) was initially found in 1 FECD patient of Chinese ethnicity in Singapore (Mehta et al., 2008). Five additional heterozygous missense variants (N78T, Q810P, Q840P, A905G, and P649A) in ZEB1 were linked to late-onset FECD in sporadic and familial cases seen in patients of northern European descent. Based on their study, approximately 2% of FECD cases were attributable to mutations in ZEB1 (Riazuddin et al., 2010a). Three SNPs (rs77516068, rs149166539, and IVS2+276) and five additional mutations (S234S, E733K, A818V, L947Stop, and Q840P) in ZEB1 were identified in a northern Indian population of FECD. The novel variant IVS2+276 C/T was seen in 14% of their FECD patients (Gupta et al., 2015).
6.4.4. LOXHD1
The advent of next generation sequencing helped investigators couple their linkage analysis with exome or genome sequencing to identify causal loci. Families with FECD-affected individuals who were previously linked to a locus in 18q were followed up with a custom exome capture that spanned all annotated exons within a 36 Mb interval. The sequencing of coding exons within 134 genes in this region revealed a missense change in LOXHD1 (lipoxygenase homology domains 1) (R547C) that was predicted to be pathogenic (Riazuddin et al., 2012). A subsequent GWAS on FECD cases further supported the association between LOXHD1 (top SNP rs450997) and FECD (Afshari et al., 2017). The function of the LOXHD1 protein has not been clearly established, but it is known to encode a highly conserved protein consisting entirely of polycystin/lipoxygenase/alpha-toxin domains and is thought to target proteins to the plasma membrane. Immunohistochemical analysis of human CE from a FECD patient with the R547C LOXHD1 mutation revealed aggregates of LOXHD1 in the CE and throughout the thickened DM. Furthermore, expression of various LOXHD1 mutant alleles in ARPE-19 cells also revealed cytoplasmic aggregates similar to those seen in the FECD cornea specimens, and it is hypothesized that these cytoplasmic aggregates are cytotoxic to CECs (Riazuddin et al., 2012).
6.4.5. AGBL1
The use of next-generation sequencing technology has also been used to study a large family with late-onset FECD, where linkage was unable to conclusively identify the locus responsible for FECD, except for a modest signal in 3p and 15q following a genome-wide scan. The whole exome capture sequencing of two patients and one unaffected family member led to the identification of a c.3082C>T mutation in AGBL1 (ATP/GTP binding protein-like 1) that leads to premature truncation (R1028*). The identification of the premature truncation in AGBL1 led to further probing of the complete cohort and resulted in the identification of a missense mutation, c.2969G>C in AGBL1 (Riazuddin et al., 2013). Based on their data, AGBL1 accounts for 1–2% of the genetic burden of FECD. AGBL1 encodes a cytosolic glutamate decarboxylase that is expressed in CE and can biochemically interact with TCF4. Furthermore, mutations in AGBL1 can lead to abnormal nuclear localization and diminished interaction with TCF4 (Riazuddin et al., 2013). The authors speculate that TCF4 may be an enzymatic target of AGBL1 and that this interaction may provide a potential mechanism for FECD.
6.4.6. KANK4, LAMC1, ATP1B1
In a GWAS on FECD cases and controls of European ancestry, 3 novel loci associated with FECD have been identified: KANK4 (KN motif- and ankyrin repeat domain-containing protein 4), LAMC1 (laminin gamma-1), and LINC009970/ATPB1 (Na+- K+ transporting ATPase, beta-1 polypeptide) (Afshari et al., 2017). The previously identified locus at TCF4 (rs784257) showed the strongest association with FECD and explained 21.9% of the variants in their FECD cohort. Significant associations were also seen within an intronic region in the KANK4 gene (rs79742895), an intergenic region between LINC00970 and the ATP1B1 (ATPase transporting subunit beta 1) gene (rs1022114) and an intronic region within the LAMC1 gene. A sex-stratified analysis identified that the risk-associated major allele G of LAMC1 variant rs3768617 conferred an increased risk of FECD to women (odds ratio (OR)=1.52) and accounted for the 1.74% risk of FECD to women, as compared to the 0.23% risk to men. In contrast, the TCF4 variant rs784257 showed an increased risk of FECD to men (OR=7.56) as compared to women (OR=5.06). A rare variant in the LAMC1 (R490W) has also been identified in a FECD cohort without CTG trinucleotide repeat expansion, thus further supporting the association between LAMC1 and FECD (Wieben et al., 2018). The functional significance of the identification of these 3 novel loci has yet to be completely elucidated. ATP1B1 and LAMC1 are both highly expressed in normal and FECD CE, while KANK4 shows minimal expression. It has been postulated that all 3 genes play a role in regulating fluid transport, especially ATP1B1, which encodes the β-subunit of Na+- K+ ATPase, and that decreased expression of ATP1B1 may lead to corneal edema in FECD. Decreased LAMC1 expression may lead to abnormal basement membrane and ECM composition, contributing to dysfunctional or reduced cell-to-cell contact between CECs. While less is known about the function of KANK4, it is also believed to be involved in the maintenance of tissue integrity and cell-to-cell contact (Afshari et al., 2017).
6.4.7. DNA Repair Enzymes (RAD51, FEN1, XRCC1, NEIL1, LIG3)
Several studies have reported that there are genetic polymorphisms in various DNA repair enzymes that are associated with FECD. The RAD51 gene encodes a central protein involved in homologous recombination and repair of double-strand breaks in humans. In a cohort of FECD patients in Poland, the RAD51 mutants c.−61G>T, c.−98G>C, and C allele were associated with an increased FECD occurrence with an odds ratio of 2.50, 1.85, and 1.90, respectively (Synowiec et al., 2013). Another DNA repair enzyme called Flap endonuclease 1 (FEN1) has also been associated with FECD. FEN1 is an important nuclease that plays a crucial role in long-patch base-excision repair and is also implicated in the resolution of trinucleotide repeat-sequence-derived secondary structures. In a cohort of FECD patients in Poland, there was an association found between the FEN1 SNP rs4246215 g.61564299G>T and FECD (Wojcik et al., 2014). Subsequent studies have also implicated X-ray repair cross complementing group 1 (XRCC1) with FECD (Wojcik et al., 2015). XRCC1 is an important component of base-excision repair and acts as a scaffold protein for other DNA base-excision repair proteins. In a cohort of FECD patients in Poland, the A/A genotopye and A allele of the c.1196A>G polymorphism of the XRCC1 gene were associated with an increased FECD occurrence. A weak association between the polymorphism g.46438521G>C in the Nei Like DNA Glycosylase 1 (NEIL1) gene and FECD was also detected. Other DNA repair enzymes such as PARP-1 and POLG have also been investigated but have not been found to be associated with an increased occurrence of FECD (Wojcik et al., 2015). Additionally, FECD has been linked to the polymorphisms rs1052536 and rs3135967 of the DNA base excision repair gene LIG3, encoding DNA ligase III, which is involved in the repair of oxidative damaged DNA (Synowiec et al., 2015). These polymorphisms in DNA repair enzymes may increase the susceptibility of mitochondria to oxidative stress and predispose affected individuals to the development of FECD.
6.4.8. Extracellular Matrix (TGFBI, CLU)
Overexpression of ECM proteins including CLU (clusterin) and TGFBI (Transforming growth factor, beta-induced) have been associated with FECD (Jurkunas et al., 2009; Jurkunas et al., 2008a; Kuot et al., 2012). In a cohort of FECD patients and controls of Australian Caucasians, CLU SNP rs17466684 and one haplotype of TGFBI were associated with increased OR of FECD (OR=1.85 and OR=2.29, respectively) (Kuot et al., 2012).
6.4.9. Other Genetic Mutations (PITX2, FASLG, PTPRG, KCNJ13)
PITX2 is a homeobox gene that encodes transcription factors that control the expression of other genes that play a crucial role in the development of ocular structures during embryogenesis. There is some limited evidence that PITX2 may also be involved with FECD. DNA linkage analysis in a 5-generation pedigree co-expressing Axenfeld-Rieger anomaly or syndrome and FECD revealed 2 hetero-allelic DNA variants in PITX2 (g.20913G>T and IVS2+8delCinsGTT) that segregated together and were present in all Axenfeld-Rieger anomaly or syndrome patients (Kniestedt et al., 2006). It has been suggested that the g.20913G>T variant could be the cause of FECD in these patients; however, a different co-segregating gene mutation was also possible.
FASLG (FAS ligand) induces a cell death signalling cascade through the FAS (apoptosis stimulating fragment), and this is a crucial pathway in regulating apoptosis in many tissues, including the eye. The regulation of apoptosis has been shown to be important in the pathogenesis of FECD (Li et al., 2001; Liu et al., 2016a). In a cohort of FECD patients from Poland, the G allele of the c.−671A>G polymorphism in FASLG was associated with an increased risk of FECD, while the A allele was associated with a decreased risk of FECD. These studies raise the possibility that other apoptotic regulators may be implicated in the pathogenesis of FECD.
PTPRG (protein tyrosine phosphatase receptor type G) is a protein tyrosine phosphatase that can regulate many different cellular functions, including growth, differentiation, cell motility, cellular adhesion, and ion-channel control (Baratz et al., 2010). A strong association between the polymorphisms rs7640737 and rs10490775 in PTPRG and FECD was found but did not reach genome-wide significance in a GWAS (Baratz et al., 2010). Subsequent studies did not find any evidence for a genetic association between PTPRG and FECD (Kuot et al., 2012; Lau et al., 2014; Wang et al., 2014).
KCNJ13 encodes protein Kir7.1, an inwardly rectifying potassium channel, and a mutation leading to an arginine-to-tryptophan substitution at residue 162 (R162W) has been associated with snowflake vitreoretinal degeneration, increased retinal detachment, retinal degeneration, and FECD (Hejtmancik et al., 2008). While it is plausible that mutations in the KCNJ13 gene can lead to abnormal K+ conductance in CE, this mechanism remains to be investigated.
6.4.10. Mitochondrial DNA (A10398G, Haplogroup 1, TSPOAP1)
Mitochondrial DNA (mtDNA) is susceptible to oxidative stress, and several studies have examined the role of mtDNA and its association with FECD (Miyai, 2018). Mitochondria isolated from both CEC and peripheral blood leukocytes from FECD patients have been found to be more susceptible to mtDNA damage and to have altered mtDNA damage repair and mtDNA copy numbers (Czarny et al., 2014; Halilovic et al., 2016). mtDNA variants A10398G and haplogroup I have been significantly associated with a protective effect for FECD in patients of European descent (Li et al., 2014). In a comparative analysis of gene expression profiles from FECD patients with or without CTG trinucleotide repeat expansion, rare variants in the mitochondrial protein TSPOAP1 (peripheral-type benzodiazepine receptor-associated protein 1) were identified in a FECD patient without CTG trinucleotide repeat expansion and family members (Wieben et al., 2018). In FECD patients with CTG trinucleotide repeat expansion, mis-splicing of TSPOAP1 has been observed, which led to a 280 amino acid deletion of a motif that interacts with TSPO, a mitochondrial outer membrane protein involved with transport of cholesterol into the mitochondria (Wieben et al., 2018). TSPOAP1 has also been shown to bind to TCF4 and is thought to be an important regulator of mitophagy (Wieben et al., 2018). mtDNA missense mutations typically associated with Leber’s hereditary optic neuropathy, mt15257 (G to A) in the cytochrome b sub-unit of complex III and mtDNA 4216 (T to C) in the ND1 subunit of complex 1, have also been reported in a FECD patient with concurrent sensorineural hearing loss, diabetes, cardiac conduction defects, ataxia, and hyperreflexia (Albin, 1998).
6.4.11. Epigenetic Modifications (DNA Methylation and miRNAs)
Epigenetic modifications have been shown to lead to heritable changes and regulation in transcriptional activity of gene expression without altering the DNA sequence itself (Kelly et al., 2010). Epigenetic modifications include DNA methylation, histone acetylation, histone methylation, and microRNA (miRNA) expression. Moreover, these processes can occur in normal tissues and become dysfunctional in diseases (Kelly et al., 2010). DNA methylation occurs almost exclusively on cytosine residues within CpG dinucleotides (phosphodiester bond linking adjacent cytosine and guanosine nucleotides), particularly in CpG-enriched areas of the genome called CpG islands (Otteson, 2011). DNA methylation can increase or silence gene expression depending on its gene location. DNA methylation has been implicated in regulating the development of ocular tissues, and DNA methyltransferases, which mediate DNA methylation, are expressed in the cornea (Bonnin et al., 2014; Otteson, 2011). A genome-scale study of DNA methylation demonstrated that there is a distinct alteration in DNA methylation patterns in FECD as compared to control samples (Khuc et al., 2017). A total of 10,961 differentially methylated probes were found in FECD samples as compare to controls, where the majority (59%) of these probes were hypermethylated, while 41% were hypomethylated. DNA hypermethylation was associated in genes involved in hematopoietic cell differentiation, ROS metabolic processes, and phosphorus metabolic processes, whereas DNA hypomethylation was disproportionately observed in genes involved with fluid and ion transport and cytoskeletal organization in FECD samples (Khuc et al., 2017). No difference in methylation pattern was observed with respect to age or sex. Overall, these findings suggest that alterations in DNA methylation likely contribute to FECD pathogenesis through abnormal regulation of genes involved with endothelial pump function, metabolic processes including ROS, and cytoskeleton structure.
miRNAs are small, endogenous, noncoding 20- to 24-nucleotide-long RNAs that can post-transcriptionally modulate gene expression through complementary targeting of highly conserved sites in 3’ untranslated regions of mRNAs. They can post-transcriptionally regulate gene expression through mRNA destabilization and cleavage or by direct translation repression (Matthaei et al., 2014). A miRNA profile study in FECD and normal CECs demonstrated that there is widespread downregulation of 87 miRNAs in FECD. These miRNAs included DICER1 (which itself is not an miRNA but encodes an endoribonuclease critical to miRNA biogenesis) and three miR-29 family members (miR-29a-3p, miR-29b-2–5p, and miR-29c-5p). Furthermore, this corresponded with increased transcriptional and translational expression of ECM-associated miR-29 targets (collagen I, collagen IV, and laminin) in FECD samples (Matthaei et al., 2014). It has also been shown in an immortalized FECD cell line that overexpression of miR-29b results in the reduction of COL1A1, COL4A1, and LAMC1 mRNA and protein expression (Toyono et al., 2016). These findings suggest that altered miRNA regulation contributes to FECD pathogenesis through increased subendothelial ECM deposition.
Environmental factors such as chemical pollutants, dietary components (folate, vitamin B6, vitamin B12, betaine, methionine, and choline), temperature changes, and other external stressors have been shown to modulate the establishment and maintenance of epigenetic modifications (Feil and Fraga, 2012). In FECD, several environmental factors such as smoking, UV light, and dietary intake have been associated or proposed to affect FECD pathogenesis. Whether these environmental factors lead to epigenetic modifications in FECD pathogenesis remains to be investigated.
7. Effect of Environmental Factors
7.1. Smoking
Smoking is an independent risk factor for ocular diseases and has a dose-dependent effect on ocular tissues (Cheng et al., 2000; Galor and Lee, 2011; Lois et al., 2008; Nita and Grzybowski, 2017; Solberg et al., 1998; Ye et al., 2012; Zhang et al., 2013). It leads to an increase in free radicals and a decrease in antioxidants in the blood, AH, and ocular tissue, suggesting that eyes of smokers are at increased risk from free radical and oxidative damage (Cheng et al., 2000). Several studies have reported an association between smoking and FECD. A population-based cross-sectional study in Reykjavik, Iceland, found that smoking more than 20-pack years is associated with an increased number of corneal guttae as detected by specular microscopy (Zoega et al., 2006). Zhang and colleagues have also demonstrated that smoking is significantly associated with the development of advanced FECD (grades 4–6) in a Caucasian population, increasing the odds of advanced FECD development by 30% (Zhang et al., 2013). However, there are studies that have not found similar associations. A population-based study conducted on a southwestern island in Japan found no significant correlation between corneal guttae and smoking (Higa et al., 2011). Despite some contradicting evidence, it is possible that smoking leads to deleterious effects of ROS production in the CE of the eye, thus, could be considered as an additional risk factor for FECD.
7.2. Ultraviolet Light
The cornea is constantly exposed to solar UV light, which is suspected to play an important role in the etiology of various ocular pathologies through increased oxidative stress and DNA damage (Cadet et al., 1983). UV light is a known environmental risk factor that acts as an exogenous oxidizer to generate ROS (Cadet et al., 1983). UV radiation that reaches the surface of the earth consists of approximately 95% UV-A, 320–380 nm light, and, while it is weakly absorbed by native DNA, it can result in the development of UV-A-induced ROS and subsequent oxidative and DNA damage. In contrast to UV-A, ultraviolet B (UV-B, 280–320 nm) is directly absorbed by cellular macromolecules, including DNA. Thus, UV-A acts as a major oxidizing component of the sunlight, whereas UV-B is a major DNA damaging component of sunlight (Cadet et al., 1983; Pourzand and Tyrrell, 1999). Recently, the effect of UV-A has been shown to be an important etiologic factor in FECD pathogenesis, explaining the predominantly central location of cell loss and guttae formation in FECD patients. Futhermore, the studies showed that UVA jumpstarts a cascade of enzymes involved in estrogen metabolism and genotoxicity causing an increased female susceptibility for FECD development (Liu, PNAS 2020; Miayajima, Free Rad Biol 2019).
Human CECs are post-mitotic cells and have a limited capacity to regenerate if damaged through lifelong exposure to UV radiation. The CE is susceptible to UV-A-induced oxidation-related toxicity as shown by accumulation 8-hydroxy-2’-deoxyguanosine (8-OHdG) in nDNA and mDNA, and higher levels of cytosolic S-glutathionylated proteins in the CE (Zinflou and Rochette, 2017). In the central cornea, CECs are directly exposed to UV light and receive higher doses of UV irradiation than do CECs in the peripheral cornea. In FECD, CECs in the central and paracentral zones are more severely damaged compared to the peripheral zones of the cornea (Fujimoto et al., 2014; Syed et al., 2017; Yanbaeva et al., 2007). Furthermore, there is evidence showing that guttae are more likely to increase their distribution pattern horizontally rather than vertically in FECD, likely due to the increased exposure to UV-light through the palpebral fissure (Rosenblum et al., 1980). Additionally, the inferotemporal cornea is often the most severely affected area in FECD patients, likely due to the lack of protection by the eyelid and nose from UV light (Adamis et al., 1993; Cullen et al., 1984; Fujimoto et al., 2014; Pitts et al., 1977).
An epidemiological survey of cataracts comparing Chinese Singaporeans and Japanese (Noto and Amami) found that the prevalence of cataracts in Singapore subjects was significantly higher than that of Japanese subjects in the same age group. This was possibly attributable to the increased ambient UV levels in Singapore, which is one of the highest in the world and 2 times higher than that in Noto, Japan (Sasaki et al., 2001). Similarly, the incidence of corneal guttae and FECD has also been shown to be significantly higher in Singaporeans (6.6%) than in the Japanese (3.3–4.1%) (Higa et al., 2011; Kitagawa et al., 2002). These epidemiological studies provide some evidence that support the association between UV levels and FECD likely due to the proximity to the equator.
In vitro models using cultured CECs can provide further insight into the adverse effects of UV-A irradiation on the CE. UV-A irradiation induces oxidative stress, which subsequently leads to decreased antioxidant gene expression and increased oxidative damage and apoptosis in CECs. One of the key transcription factors that regulates antioxidant defense is nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2-regulated antioxidant defense plays a key role in response to UV-A-induced oxidative stress. Lower fluences of UV-A have also been shown to induce the Nrf2-regulated antioxidant defense by promoting Nrf2 nuclear translocation, upregulating its target genes hemeoxygenase-1 (HO-1), and NAD(P)H:quinone oxidoreductase 1, indicating that even near-environmental levels of UV-A affect normal CECs. Higher fluences of UV-A directly lead to apoptosis by activating p53 and caspase3. These studies suggest that UV-A is involved in the pathogenesis of FECD, but that CECs deficient in antioxidant defense may be particularly susceptible to UV-A irradiation (Liu et al., 2016a).
Similarly, in vivo studies have shed a light on the formative effect of UV-A light on development of FECD. We have shown, for the first time, that UV-A, by causing DNA damage, induces the FECD phenotype in mice. UVA induced fluence–dependent disruption of the monolayer with reduction in cell density and caused formation of guttae-like lesions, where guttae-like bright ‘deposits’ were surrounded by dark areas of lost cells comparable with the morphological alternations seen in human FECD cornea. Interestingly, we detected that female mice exhibited greater CE cell loss, including loss of ZO-1-stained junctional contacts, and corneal edema after UV-A irradiation than did male mice, suggesting a possible sex-dependent effect. This study provides a first line of evidence on the environmental factor, such as UV light, involvement in FECD development, adding to the body of evidence on the etiology and possible prevention of this common corneal condition (Liu et al., 2018).
7.3. Nutrition
Oxidative stress has been associated in the development of various ocular diseases such as age-related macular degeneration (ARMD), cataract, and glaucoma, and has prompted studies investigating dietary supplementation with vitamins and antioxidants to prevent disease progression (Agte and Tarwadi, 2010; Aslam et al., 2014; Evans, 2008; Group et al., 2012; Hammond and Johnson, 2002; Moriarty-Craige et al., 2005; Tarwadi et al., 2008). There is some epidemiological and molecular evidence that environmental factors, such as UV light and smoking, increase oxidative stress in CECs and contribute to the pathogenesis of FECD. Based on these observations, there may be an association between nutrition and FECD development. Thus, future studies identify dietary and lifestyle factors that influence disease development and shed light on prevention and treatment of FECD.
8. Hormones
Even though FECD is more prevalent in women than in men, there is still a lack of knowledge of why there are these sex-driven differences, and sex hormones are likely to play a role in this female predominance. Intriguingly, the greatest sex disparity in FECD prevalence occurs in the 50 to 59-year-old age group in women, which represents the peri- and post-menopausal period, suggesting a potential role for hormonal changes in FECD pathogenesis (Patel et al., 2018). Since sex-steroid receptors such as estrogen receptor beta (ERβ), androgen receptor, and progesterone receptor, are expressed in CECs of both sexes, it is possible that these receptors influence the function of CECs (Hadeyama et al., 2002; Suzuki et al., 2001). ERβ expression in FECD ex-vivo corneal specimens show a subcellular localization pattern, whereas in normal ex-vivo corneal specimens, ERβ has distinct expression patterns within the same cornea, from predominantly nuclear to predominantly cytoplasmic. These differences in expression patterns of ERβ suggest that there are distinct roles for ERβ in normal and FECD CE (Fard et al., 2017). However, the expression profile of ERβ receptors between women and men with FECDs has not been investigated.
Recent studies suggest a possible role of reactive estrogen quinones in FECD pathogenesis (Miyajima et al., 2019). Estrogens, such as estradiol and estrone, are oxidatively metabolized by cytochrome P450 enzymes, mainly CYP1B1, to form catechol estrogens, which are further oxidized to genotoxic estrogen quinones (Hayes et al., 1996). These quinones, in turn, react with DNA to form depurinating DNA adducts and apurinic sites in DNA leading to development of significant mutations and cytotoxicity (Zahid et al., 2006). Previous studies showed that acquired DNA damage causes apoptosis in FECD (Halilovic et al., 2016). As a part of cellular antioxidant defense, NAD(P)H: quinone oxidoreductase 1 (NQO1), which is transcriptionally regulated by Nrf2, detoxifies reactive estrogen metabolites. NQO1 converts estrogen quinones back to catechol estrogens (Gaikwad et al., 2007) which can be neutralized by catechol-O-methyltransferase (COMT) (Zahid et al., 2007). A fine balance in the levels of estrogen metabolizing and detoxifying enzymes is critical for maintaining the estrogen metabolite homeostasis (Singh et al., 2005). We detected marked upregulation of CYP1B1 and decline in Nrf2-NQO1 and COMT in FECD tissues, indicating dysregulation of estrogen metabolite pathways in FECD (Jurkunas et al., 2010), and NQO1 (Miyajima et al., 2020; Miyajima et al., 2019). We have shown experimentally that loss of NQO1 causes catechol estrogen-induced DNA damage and apoptosis in CECs; thus, pointing to the potential role of estrogen metabolism in sex-driven differences seen in FECD (Miyajima et al., 2020; Miyajima et al., 2019).
9. Associated Diseases
9.1. Keratoconus
The association between keratoconus (KC) and FECD has been reported in the literature in various case reports (Darlington et al., 2001; Jurkunas and Azar, 2006; Lipman et al., 1990; Orlin et al., 1990). While the underlying etiology leading to the manifestation of both KC and FECD in the same patient remains unknown, oxidative stress, the formation of ROS, and mitochondrial dysfunction are known to play important roles in the pathogenesis of both diseases (Nita and Grzybowski, 2016; Vallabh et al., 2017; Wojcik et al., 2013a). Polymorphisms in FLAP endonuclease (g.61564299G>T and c.−441G>A) and LIG3 (g.29661G>A and g.29059C>T), which encodes DNA ligase III, have been associated with KC and FECD (Synowiec et al., 2015; Wojcik et al., 2013b). Both of these genes are important components of the base excision repair pathway that mediates the repair of oxidative DNA damage.
It is important to note that corneal edema due to FECD can be obscured by concurrent corneal thinning caused by KC, which may lead to normalized readings of corneal pachymetry and an underestimation of disease severity. This may lead to unexpected postoperative visual outcomes after cataract and refractive surgeries. Therefore, routine preoperative topography and specular microscopy is recommended for patients diagnosed with KC and FECD to minimize unwanted postsurgical outcomes (Gupta et al., 2017; Jurkunas and Azar, 2006).
9.2. Glaucoma
The association between FECD and glaucoma is controversial, with conflicting published reports (Buxton et al., 1967; Imre and Bogi, 1979; Magovern et al., 1979; Roberts et al., 1984). It was initially reported that aqueous outflow was decreased in FECD eyes as compared to normal controls, suggesting that abnormalities in the trabecular meshwork might be involved in the disease process (Buxton et al., 1967). However, subsequent studies did not find that aqueous outflow decreased in FECD (Roberts et al., 1984), and an association between FECD and glaucoma has not been found (Magovern et al., 1979). More recent studies have also resulted in conflicting findings. Patients with severe FECD have been associated with more glaucoma and/or ocular hypertension than have healthy controls, prompting the recommendation of close intraocular pressure monitoring in FECD patients (Nagarsheth et al., 2012). In contrast, another study found no association between FECD and glaucoma (Rice et al., 2014). It has also been suggested that there is no increased risk of primary open-angle glaucoma in patients with FECD, and that increased glaucoma in FECD patients is secondary to mechanical changes due to Schlemm’s canal post-PK or EK, or steroid-induced glaucoma (Ali et al., 2011). Two studies have also reported that FECD patients have a high incidence of angle-closure glaucoma secondary to a high incidence of hypermetropia and a shallow anterior chamber (Loewenstein et al., 1994; Pitts and Jay, 1990). However, the association between angle-closure glaucoma and FECD was not found in another study (Brooks et al., 1994).
From a clinical perspective, it is important to note that, since FECD leads to a change in corneal biomechanical properties, including corneal hysteresis, corneal resistance factor, and CCT it may lead to an underestimation error in intraocular pressure measurement, which, if not recognized, might delay the diagnosis of glaucoma (del Buey et al., 2009).
9.3. Age-Related Macular Degeneration
FECD and ARMD share the same risk factors, including female sex, advanced age, and smoking (Matthaei et al., 2018). In FECD patients undergoing Descemet membrane endothelial keratoplasty (DMEK) compared to consecutive patients without corneal pathology, a general age-dependent presence of macular drusen has been found; however, no correlation between macular drusen and FECD has been observed (Matthaei et al., 2018). These results are consistent with findings from the Reykjavik study, where no increased prevalence of ARMD was found in patients 55 years or older with primary central corneal guttae (Zoega et al., 2006). In contrast, a prospective study found an increased prevalence of ARMD in FECD patients than in controls without any corneal pathology. This study suggests that the presence of corneal guttae in a patient might imply an increased risk for ARMD (Rao et al., 1998). The diverging outcomes of these studies might be related to different sample populations and grading methodologies. Further studies are needed to validate the findings of these preliminary studies. However, FECD and ARMD both show cellular degeneration with deposition of periodic acid-Schiff -positive ECM, in the form of guttae and drusen, respectively. Furthermore, the CE and the retinal pigment epithelium both contain monolayers of post-mitotic polygonal cells of neuroectodermal origin. These findings could suggest cells with a common embryological origin may result in a similar pathological manifestation of disease.
9.4. Cardiovascular Disease
CECs and the cardiovascular system are derived from cranial NCC-derived mesenchymal cells. Based on the embryological commonalities of both tissues, a global survey and correlative meta-analysis was performed investigating the correlation between adult corneal ECD and mortality rates secondary to diseases affecting cardiac NCC-derived cardiovascular structures. A significant negative correlation was found between ECD and mortality due to coronary heart disease, hypertension, and all-cause cardiac disease, suggesting a possible association between FECD and cardiovascular diseases (Scherer, 2018). Moreover, a retrospective study investigated whether there is a clinical association between FECD and atherosclerosis by analyzing the prevalence of cardiovascular diseases in 27 FECD patients and 27 age- and sex-matched controls. An increased cardiovascular disease risk was observed in the FECD patients as compared to controls (44% versus 11%), suggesting that there was a common endothelial factor abnormality. However, the embryological lineage of CECs differs from that of vascular endothelium. Therefore, these associations may be more related to a patient’s susceptibility to environmental factors that lead to premature degeneration in all tissues, including the CE, retina, and cardiovascular tissues (Olsen, 1984).
9.5. Diabetes Mellitus
A lack of association between diabetes and increased risk of advanced FECD has been reported. However, in this study, diabetes increased CCT among FECD cases and controls (Zhang et al., 2013). However, prior studies had already described the association between diabetes and increased CCT (Lee et al., 2006; Luo et al., 2019; Su et al., 2008; Urban et al., 2007). The specific pathophysiology of how diabetes leads to increased CCT remains unknown; however, it has been hypothesized to be due to abnormally functioning CECs and reduced ECD. Consistent with this hypothesis is the fact that FECD patients with diabetes have an increased CEC loss following DMEK surgery, as compared to those without diabetes (Price et al., 2017). Hyperglycemia has been associated with CEC morphological changes (Larsson et al., 1996; Schultz et al., 1984), increased CEC permeability, and decreased CEC pump rates (Lass et al., 1985). Hyperglycemia has also been shown to increase production of mitochondrial superoxide, leading to DNA damage and depletion of the cellular antioxidant nicotinamide adenine dinucleotide (Folli et al., 2011; Rolo and Palmeira, 2006). These findings suggest that additional oxidative stress from diabetes may lead to accelerated CEC death in FECD patients, where CEC homeostasis is already impaired (Azizi et al., 2011; Jurkunas et al., 2010; Tangvarasittichai and Tangvarasittichai, 2018).
10. Models for Studying FECD
10.1. Ex-vivo Specimens, Primary Cultures, Immortalized Cell Lines Derived from FECD Specimens
Study of proteomic and genetic differences on the cellular level was jumpstarted by comparing the ex vivo normal and FECD human specimens. Since FECD cells are nonproliferative in vivo, the stimulation of the division in vitro may induce a set of genes that do not represent the homeostasis of normally post-mitotic cells. Furthermore, isolation of FECD cells from its DM potentially precludes investigation of cell-matrix interactions that are crucial in understanding pathogenesis of FECD (Jurkunas, 2018). Therefore, our initial studies on proteomic analysis of FECD DM and CEC ex vivo revealed changes in ECM composition and antioxidant defence that sparked the formulation of new hypotheses on the cellular stress response and oxidative stress in FECD pathogenesis (Jurkunas et al., 2009; Jurkunas et al., 2010; Jurkunas et al., 2008a; Jurkunas et al., 2008b). Since then, ex vivo specimens have been utilized throughout various experimental designs and used to discern new treatment paradigms by targeting oxidative stress and TCF4 CTG repeat expansions ex vivo (Benischke et al., 2017; Hu et al., 2018; Mootha et al., 2015; Ziaei et al., 2013a). Recently, we have developed a model to study CEC migration and interaction with guttae in ex vivo tissues by using live cell imaging (Kocaba et al., 2018; Ong Tone et al., 2019c). This methodological advance is set to investigate not only static changes captured during specimen processing but the dynamic interaction of cells and ECM during various experimental conditions aimed at unravelling FECD pathogenesis.
Human corneal endothelial cells (HCECs) are terminally differentiated somatic cells derived from NCCs and are arrested in a post-mitotic state with a very low proliferative potential in vivo. Primary HCEC cultures also have a very low proliferative potential, and there is a limited ability to passage them in vitro, as they undergo rapid cellular senescence or EMT, which results in a fibroblast-like transformation and loss of the endothelial phenotype (Bartakova et al., 2018; Peh et al., 2015b; Peh et al., 2017; Roy et al., 2015). While it is possible to use primary HCEC cultures derived from either normal or FECD tissues as a model to study FECD pathogenesis, there are significant drawbacks including the cells’ limited mitotic capacity and loss of characteristics in vitro (Schmedt et al., 2012a; Zaniolo et al., 2012). Furthermore, a phenomenon after cultivation of FECD CECs has been described, whereby there is a loss of the FECD phenotype and FECD CECs become similar to healthy CECs. This may be attributable to either a selection of the healthiest peripheral CECs for culture, and/or the absence of a pathological environment (abnormal DM, guttae, and/or AH), and/or the presence of pro-survival growth factors in the culture medium (Theriault et al., 2018). Regardless, despite the limitations, primary CECs derived from FECD patients undergoing EK provide a valuable model to study FECD pathogenesis, especially if early cell passages are used (Gendron et al., 2016; Haydari et al., 2012; Katikireddy et al., 2016; Zaniolo et al., 2012; Zarouchlioti et al., 2018).
One strategy to establish long-term cultures of CECs has been to transform them using viral oncogenes such as SV40 (simian virus 40) large T antigen, HPV (human papilloma virus) E6/E7, or overexpression of mutant CDK4 (Bednarz et al., 2000; Griffith et al., 1999; Wilson et al., 1993; Yokoi et al., 2012). Another method for immortalization of CECs independent of viral oncogenes has been to overexpress human telomerase reverse transcriptase. We have previously reported the generation of an immortalized highly uniform subpopulation of endothelial cells derived from primary cells harvested from a 21-year-old male donor, HCEnC-21T, that retains a high proliferative activity, corneal endothelial morphology, endothelial markers, and functionality (Schmedt et al., 2012a). Furthermore, we have generated multiple SV40-immortalized FECD cell lines, with and without TCF4 CTG repeat expansion, from specimens derived from patients undergoing EK (Ong Tone et al., 2019b). We observed that TCF4 CTG repeat expansion is retained during immortalization and is consistent with patient blood analyses. We also observed that there is variability in morphological and functional phenotypes among the FECD cell lines, suggesting heterogeneity among FECD patients. Other research groups have also generated SV40 or human telomerase reverse transcriptase-immortalized FECD lines (Benischke et al., 2017; Hu et al., 2018; Okumura et al., 2017a; Okumura et al., 2017c; Okumura et al., 2015b). These immortalized FECD cell lines provide a valuable model system to investigate the pathogenesis of and potential treatments for FECD.
We have also developed a model to study the interplay between CECs and guttae in the pathogenesis of FECD, where immortalized normal human endothelial cells (HCEnC-21T) are seeded on DM with guttae from FECD-affected corneas (Kocaba et al., 2018). This model allowed us to demonstrate that guttae and the ECM play significant roles in the pathogenesis of FECD, as they had the capacity to sicken completely healthy cells. However, guttae did not induce all of the intercellular perturbations known to occur in FECD, suggesting that the guttae microenvironment is only partially contributory to the pathogenesis of FECD. Future studies using immortalized FECD cell lines with different genetic backgrounds could provide further insight into the complex pathogenesis of FECD.
10.2. Animal Models
One of the potential limitations of primary culture or immortalized cell lines derived from FECD specimens is that these tissues are typically collected during EK from more advanced FECD patients and may not entirely reflect the early pathophysiology of FECD. With in vivo animal models, there is an opportunity to study the early pathophysiology of FECD as well as its progression to more advanced stages. In vivo animal models allow for genetic manipulation through knockout and knockin mouse models, as well as the investigation of novel treatments for FECD.
10.2.1. Genetic Mouse Models
The genetic basis of FECD has allowed different research groups to generate various genetically engineered mice as a model to study human endothelial diseases. Jun and colleagues generated homozygous mutant knockin mice with a point mutation causing a glutamine-to-lysine substitution at amino acid position 455 (Q455K) in the COL8A2 gene, which has been shown to cause early-onset FECD in humans (Jun et al., 2012). These homozygous mutant knockin mice have showed a phenotype similar to that of early-onset FECD, including altered CEC morphology, CEC loss, and guttae formation. They also observed that the predominant defect was a dilated ER, suggesting that ER stress and UPR-associated apoptosis play a role in the pathogenesis of FECD (Jun et al., 2012). Meng and colleagues also generated a second homozygous mutant knockin mouse with a point mutation causing a leucine-to-tryptophan substitution at amino acid position 455 (L450W) in the COL8A2 gene. The Col8a2 L450W knockin mouse also displayed features of FECD but exhibited a milder phenotype when compared to that of the Col8a2 Q455K knockin mouse (Meng et al., 2013). Two additional mouse models targeting COL8A2 have been generated. Hopfer and colleagues generated homozygous double knockout mice for COL8A1 and COL8A2, and observed dysgenesis of the anterior segment of the eye with a globoid, keratoglobus-like protrusion of the anterior chamber with thinned corneal stroma and DM (Hopfer et al., 2005). CECs were also enlarged and reduced in number, and had a decreased proliferative activity in vitro. Puk and colleagues generated mutant Aca23 mice with a point mutation causing a glycine-to-aspartate substitution at amino acid position 257 (G257D) in the COL8A2 gene. They also observed dysgenesis of the anterior segment of the eye with a larger anterior chamber, axes, and thinner corneas (Puk et al., 2009). However, while there was a reduction in the size of the corneal epithelium, stroma, and DM, the endothelium was unaltered and no guttae were observed. The phenotypic differences observed in the four COL8A2-mutant mice were likely a result of the different pathological mechanisms seen with the complete reduction of COL8A2, as compared to the expression of abnormally mutated COL8A2, with the L450W and Q455K substitutions representing the closest similarities to FECD in humans. Overall, these findings support a pathogenic mechanism, whereby early-onset FECD is a result of ER stress and UPR-associated apoptosis, rather than a loss of function of COL8A2.
Genetically engineered mice have also been generated to investigate the role of various genes (SLC4A1, ZEB1, and TCF4) that have been associated with late-onset FECD. Lopez and colleagues have generated homozygous mutant SLC4A11 knockout mice that showed a mild corneal phenotype, with normal CE morphology and without any opacification or edema; however, there was an increased absolute and relative height of the corneal basal epithelial cells (Lopez et al., 2009). The lack of a severe phenotype in the cornea could be attributable to compensatory mechanisms in the knockout mouse or to the fact that NaBCl does not play a direct role in maintaining corneal clarity in mice, despite its strong association to FECD and CHED2 in humans. Liu and colleagues have generated homozygous and heterozygous mutant ZEB1 knockout mice that show ectopic expression of epithelial genes in the CE and keratocytes. These mice also show more features of posterior polymorphous corneal dystrophy, rather than that seen in FECD, including abnormal corneal and keratocyte proliferation, corneal thickening, and corneolenticular and iridocorneal adhesions, suggesting that ZEB1 may play a role in the suppression of an epithelial phenotype (Liu et al., 2008). While TCF4 heterozygous mice are viable, TCF4 homozygous knockout mice die within 24 hours after birth, which limits our ability to study the CE (Flora et al., 2007; Zhuang et al., 1996). TCF4 homozygous knockout mice have no gross anatomical defects, including no specific ocular abnormalities, although disrupted hindbrain development was observed (Flora et al., 2007). However, the utility of TCF4 homozygous knockout mice to study FECD may be limited, since TCF4 genetic mutations include either CTG repeat expansion or SNPs, rather than loss of function. Furthermore, haploinsufficiency of the TCF4 gene in humans leads to Pitt-Hopkins syndrome, a neurodevelopmental disorder characterized by severe intellectual disability, seizures, microcephaly, constipation, and a distinctive facial gestalt, which are not features of FECD (Marangi and Zollino, 2015).
10.2.2. UV-A-Induced In Vivo Mouse Model of FECD
CE is exposed to UV light throughout life and, due to post-mitotic arrest, is susceptible to acquired oxidative damage. Our group established a non-genetic UV-A-based mouse model that recapitulates the morphological and molecular changes of FECD and provides the model of the late-onset FECD based on the physiologic outcome of CEC susceptibility to oxidative stress (Liu et al., 2020). The irradiation of mouse corneas with UV-A light (365-nm wavelength) caused morphological changes and loss of CECs, thickening of DM and formation of guttae-like lesions interspersed around the degenerating cells. The reduction in cell density was accompanied by corneal edema. Interestingly, female mice showed a more pronounced cell loss and corneal swelling than male mice, characteristic of greater prevalence of late-onset FECD in females. UV-A irradiation upregulated ROS levels in mouse aqueous humor and caused nDNA and mtDNA damage seen in FECD human specimens ex vivo (Halilovic et al., 2016). The sex-dependent effect of UVA was driven by the activation of estrogen metabolizing enzyme CYP1B1 and formation of reactive estrogen metabolites and estrogen-DNA adducts in female but not male mice (Liu et al., 2020; Liu et al., 2016b; Liu et al., 2018). Because FECD is a genetically heterogeneous disease, the model that utilizes a unifying mechanism of cell loss due to oxidative stress, provides a tool to study potential therapeutic interventions for all forms of FECD, regardless of genotype.
11. Apoptosis, Oxidative Stress, Cell Biology, and Molecular Mechanisms
11.1. Apoptosis
Apoptosis, or programmed cell death, is characterized by cell shrinkage, membrane blebbing, chromatin condensation, and DNA fragmentation (Elmore, 2007; Kerr et al., 1972; Nikoletopoulou et al., 2013). Programmed cell death occurs with minimal damage to the surrounding cells during homeostasis, development, and wound healing (White, 1996). Excessive apoptosis has been associated with several disease processes, including neurodegeneration, autoimmune disorders, ischemic damage, aging, and cancer (Chan et al., 1997; Giordano et al., 1997; Johnson, 1994; Li et al., 1999; Mochizuki et al., 1996; Tomei and Umansky, 1998; Wyllie, 1985). The CE and the posterior third of the cornea are derived from neuroectoderm, suggesting that the pathogenesis of FECD may be similar to other neurodegenerative diseases (Adamis et al., 1993; Adamis et al., 1985; Zhu et al., 2014). Furthermore, CECs are arrested in a post-mitotic state, rendering them susceptible to ROS-induced apoptosis triggered by aging and oxidative stress, as is seen in neurodegenerative diseases (Simonian and Coyle, 1996). Another programmed form of necrotic cell death called necroptosis can occur in vivo, particularly in neurodegenerative diseases, and it has also been shown that there is crosstalk between necroptosis and apoptosis (Nikoletopoulou et al., 2013). However, necroptosis has yet to be implicated in FECD.
There are at least 2 pathways that ultimately induce programmed cell death: (1) the extrinsic and (2) intrinsic pathways (Figure 3). The extrinsic pathway involves cell surface death receptors such as tumor necrosis factor alpha, Fas and tumor necrosis factor alpha-related apoptosis inducing ligand receptors, while the intrinsic pathway involves mitochondrial signalling and cytochrome c release (Nikoletopoulou et al., 2013). Both extrinsic and intrinsic pathways lead to the activation of cysteine aspartyl proteases called caspases that result in mitochondrial membrane permeabilization, chromatin condensation, and DNA fragmentation, which elicits the apoptotic phenotype. The intrinsic mitochondrial pathway of apoptosis is regulated by the Bcl-2 family, which can be divided into 3 groups based on function: (1) anti-apoptotic: Bcl-XL, Bcl-2, Bcl-W, and Mcl-1; (2) proapoptotic: Bax and Bak, and (3) both anti- and pro-apoptotic Bcl-2 members (Nikoletopoulou et al., 2013). Activation of the intrinsic mitochondrial pathway ultimately results in mitochondrial outer membrane permeabilization, which allows for soluble proteins such as cytochrome c to diffuse into the cytosol and activate initiator caspase 9, which subsequently activates caspases 3 and 7 to propagate apoptotic signalling (Nikoletopoulou et al., 2013; Ryter et al., 2007). Excessive apoptosis has been implicated in the pathogenesis of FECD as demonstrated by increased DNA fragmentation and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays in FECD CE (Borderie et al., 2000; Jurkunas et al., 2010; Kocaba et al., 2018; Li et al., 2001; Szentmary et al., 2005, 2008). Transmission electron microscopy (TEM) and Fas, FasL, Bcl-2, and Bax immunohistochemical analysis of FECD ex-vivo specimens have also revealed increased apoptosis as compared to control specimens (Borderie et al., 2000; Li et al., 2001). Mitochondrial fragmentation, cytochrome c release and activation of apoptosis have also been detected in FECD ex-vivo specimens (Halilovic et al., 2016). The major downstream effectors of cellular apoptosis, caspase 9 and caspase 3, have also been demonstrated to be increased in FECD ex-vivo specimens as compared to controls (Engler et al., 2010). Thus, these studies on FECD ex vivo specimens provide strong evidence that excessive apoptosis plays an important role in the pathogenesis of FECD (Figure 3).
Figure 3. The Pathogenesis of Fuchs Endothelial Corneal Dystrophy (FECD).
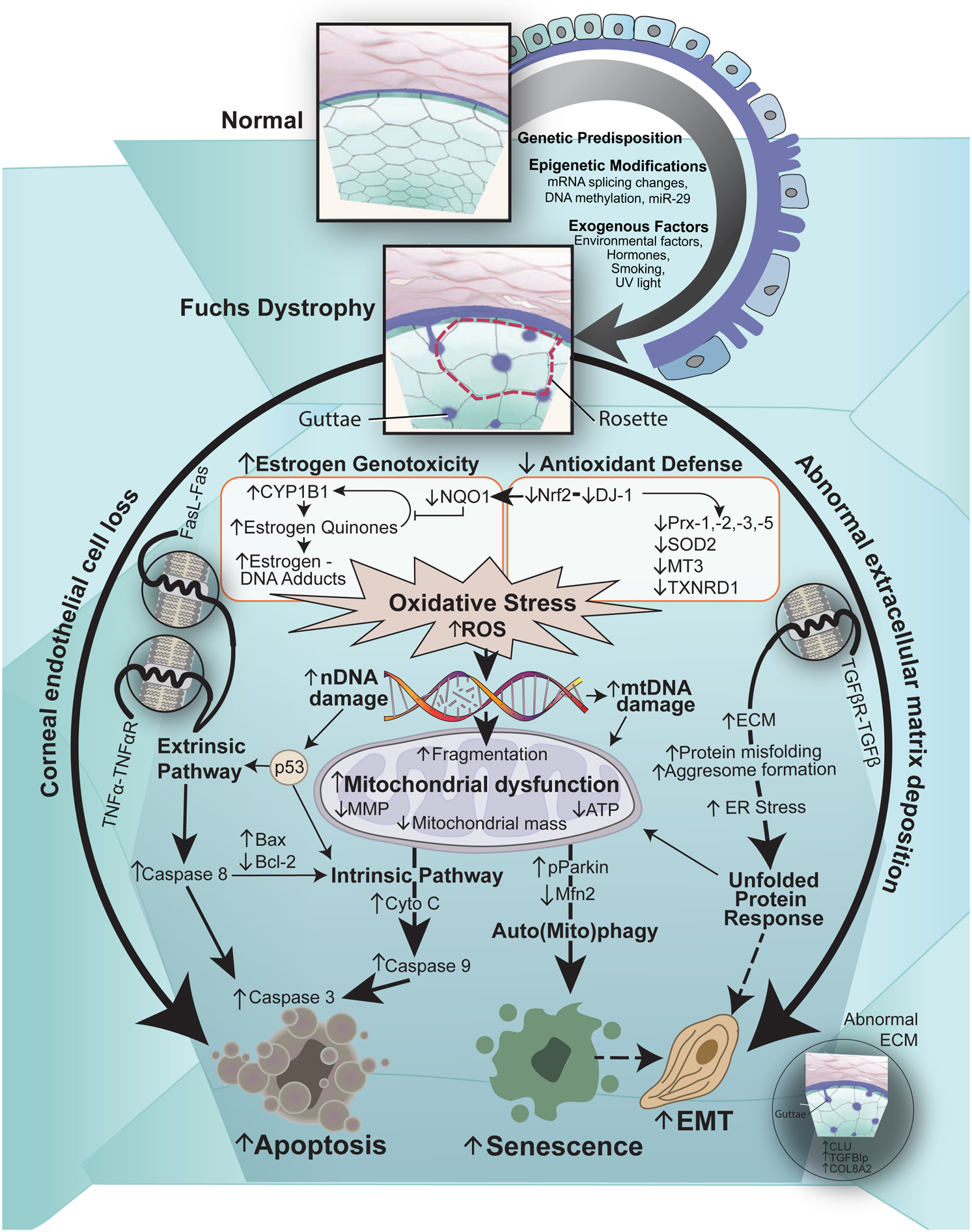
Schematic diagram illustrating our hypothesis on the vicious cycle of FECD pathogenesis. Cellular stress leading to corneal endothelial cell (CEC) loss and abnormal extracellular matrix (ECM) deposition, called guttae, and altered cellular-ECM interactions are the hallmarks of FECD. Genetic predisposition, epigenetic modifications and multiple exogenous factors leads to increased intracellular oxidative stress and reactive oxygen species (ROS) production, resulting in nuclear DNA (nDNA) and mitochondrial DNA (mDNA) damage, and mitochondrial dysfunction and alteration in the synthetic capacity of cells, which maintains the vicious cycle of propagating oxidant–antioxidant imbalance in the pathogenesis of FECD. Furthermore, CECs undergo apoptosis through both the extrinsic and intrinsic pathways, endothelial to mesenchymal transition (EMT) through the unfolded protein response and cellular senescence through auto(mito)phagy. Legend: mRNA=messenger RNA; miR-29= microRNA-29; UV=ultraviolet; TNFα= tumor necrosis factor alpha; TNFαR= tumor necrosis factor alpha receptor; FasL= Fas ligand; TGFβ= transforming growth factor beta; TGFβR= transforming growth factor beta receptor; Nrf= NF-E2 (nuclear factor erythroid 2) related factor; DJ-1= protein deglycase; NQO1= NAD(P)H: quinone oxidoreductase 1; Prx= peroxiredoxins; SOD2= superoxide dismutase 2; MT3= metallothionein-3; TXNRD1= thioredoxin reductase 1; MMP= ; ATP= ;ER= endoplasmic reticulum; pParkin= phosphoParkin; Mfn2= mitofusin 2; Cyto C= cytochrome C; Bax= Bcl-2-associated X protein; Bcl-2= B-cell lymphoma 2; CLU= clusterin; TGFBIp= transforming growth factor beta-induced protein.
The p53 protein is an important tumor suppressor that can be activated as a result of cellular stress from DNA damage, aberrant growth signals, chemotherapeutic drugs, UV light, and protein kinase inhibitors. Activated p53 has been implicated in numerous signalling pathways including cell-cycle progression, angiogenesis, tumor growth suppression, cellular differentiation, maintenance of genetic stability, and apoptosis (Vogelstein et al., 2000). With respect to apoptosis, activation of p53 through phosphorylation induces transcription of components of the death receptors and mitochondrial pathways, such as Fas, Bax, PUMA, and NOXA, to promote programmed cell death (Nikoletopoulou et al., 2013; Vogelstein et al., 2000). Activated phospho-p53 can also directly promote mitochondrial outer membrane permeabilization, where, upon stress, a cytoplasmic pool of p53 translocates to the mitochondrial surface where it interacts with both anti- and pro-apoptotic Bcl-2 family members to inhibit or activate their functions, leading to apoptosis (Nikoletopoulou et al., 2013). In FECD ex vivo CE specimens, there is a twofold increase in p53 expression and an increase in nuclear colocalization of p53 as compared to controls. Furthermore, there is an accumulation of activated phospho-p53 in the cytoplasm of the majority of FECD cells at baseline, before manifestation of the late stages of apoptosis, as detected by TUNEL assay. These findings support the notion that p53 plays a central role in CEC cell death in FECD (Azizi et al., 2011).
Mechanistic studies using CEC derived from normal patients have also shown increased levels of cytochrome c, mitochondrial fragmentation, caspase 9, caspase 3, and apoptosis following exposure to menadione (Halilovic et al., 2016). Similarly, FECD cells are also more susceptible to oxidative-stress-induced apoptosis mediated by tert-butyl hydroperoxide, a potent inducer of oxidative stress, as compared to normal cells. Furthermore, in ex vivo specimens, while normal CECs were found to be resistant to tert-butyl hydroperoxide -mediated apoptosis, FECD CECs displayed a 21% increase in TUNEL-positive cell numbers. Moreover, both p53 protein levels and the percentage of p53-positive CECs were increased in FECD CE. Increased activated phospho-p53 has also been detected in normal CECs after tert-butyl hydroperoxide treatment or in untreated FECD CECs, suggesting that oxidative stress induces apoptosis in FECD. This study was the first report describing p53’s critical role in the cell death seen in FECD, and provided a mechanism for the p53-regulated increase in oxidative-stress-induced apoptosis in FECD (Azizi et al., 2011). Subsequent studies have also shown that FECD CECs are more susceptible to p53-regulated apoptosis from oxidative stress (Bitar et al., 2012; Liu et al., 2014a).
There is strong evidence that CECs undergo excessive apoptosis as a late manifestation in the pathogenesis of FECD. There are multiple apoptotic pathways, including the extrinsic receptor-dependent, intrinsic mitochondrial-dependent, and UPR-dependent pathways that likely converge to mediate CEC apoptosis in FECD (Figure 3). While a caspase-independent pathway that results in apoptosis-like programmed cell death has been described, it has not been reported in FECD (Nikoletopoulou et al., 2013). It is possible that CECs in FECD, in an attempt to counteract the intracellular stresses and pro-apoptotic signalling, produce aberrant ECM that contributes to guttae formation and creates a further toxic environment for the CECs, which further propagates FECD pathogenesis (Figure 1 and Figure 3) (Jurkunas, 2018).
11.2. Oxidative Stress
11.2.1. Oxidant-Antioxidant Imbalance
CE is especially prone to oxidative damage due to life-long exposure to light and high oxygen demand due to exuberant metabolic activity, as well as the non-proliferative nature of the cells. CE cells have the second (after retinal photoreceptors) highest aerobic metabolic rate in the eye, rending them to be highly susceptible to the damaging effects of oxidative stress. Oxidative stress occurs when the balance is disrupted due to depletion of antioxidants or excess accumulation of ROS, or both. In response, cells function to counteract the oxidant effects and restore redox balance by activating or silencing genes encoding defensive enzymes or antioxidants, transcription factors, stress-induced proteins, and apoptotic pathways. There is ample evidence to suggest that oxidative stress plays a key role in the pathogenesis of FECD. Early studies have detected advanced glycation end products (AGEs), non-enzymatically glycated proteins known to be associated with increased cellular oxidative stress in FECD DM and CE (Wang et al., 2007). Previous studies have evaluated the relative amounts of cytotoxic byproducts of ROS in FECD corneas and detected increased amounts of nitrotyrosine, a byproduct of ROS, in FECD CE (Buddi et al., 2002). Even though oxidative stress markers have been observed in corneas from older subjects, the increased levels of 8-OHdG, a marker of oxidative damage to DNA, in FECD CE as compared to age-matched controls, point to a pathophysiological mechanism that bypasses normal aging and causes the distinct oxidative stress-induced phenotype seen in FECD (Arnheim and Cortopassi, 1992; Joyce et al., 1998; Jurkunas et al., 2010; Mecocci et al., 1993).
11.2.2. Antioxidant Defense
Cellular generation of ROS, such as superoxide anion, hydrogen peroxide, hydroxyl radical, and nitric oxide is counteracted with innate antioxidant defense systems. Non-enzymatic defenses include the compounds with intrinsic antioxidant properties such as vitamins, glutathione, and β-carotene. The major enzymatic antioxidant systems in the CE are superoxide dismutases (SODs), catalase, glutathione-s-transferases, and peroxidases, including peroxiredoxins. Initial proteomic studies have revealed downregulation of Prx-2, −3 and −5 in FECD samples as compared to normal controls (Jurkunas et al., 2008b).
Peroxiredoxins (Prxs) are a class of antioxidants with anti-apoptotic properties that function by removing cellular hydrogen peroxide. Six isoforms have been identified, and four (Prx-2, −3, −5, and −6) are expressed in human CECs in various subcellular locations. Prx-2 is a cytosolic protein that blocks hydrogen peroxide-induced apoptosis upstream of Bcl-2 through inhibition of mitochondrial cytochrome c release to the cytosol (Zhang et al., 1997). Prx-3 is a mitochondrion-specific peroxidase that plays a role in antioxidant defense in the mitochondrial respiratory chain. Transcriptome analyses have shown that Prx-3 is downregulated in FECD (Jurkunas et al., 2008b). Reduced levels of Prx-3 have been shown to increase mitochondrial damage via membrane potential collapse, cytochrome c release, and caspase activation. Prx-5 has been localized to mitochondria, peroxisomes, and the cell nucleus and has an anti-apoptotic function that protects against intracellular ROS production via a p-53-dependent pathway (Zhou et al., 2000). Prx-6 plays an important role in regulating intracellular ROS balance and in regenerating oxidized membrane phospholipids.(Wahlig et al., 2018b) The significantly decreased expression of Prx-2, −3 and −5 in FECD suggests that oxidant-antioxidant imbalance contributes to the pathogenesis of FECD, prompting a more general approach to the antioxidant defense in FECD (Figure 3).
Human oxidative stress and antioxidant RT-PCR arrays have been performed comparing FECD and normal control CECs and detected decreased expression of genes related to antioxidant, apoptosis, oxidative stress cell signaling and the oxidative stress response (Jurkunas et al., 2010). The antioxidants genes found to be dysregulated in FECD included Prx-2, Prx-5, thioredoxin reductase 1 (TXNRD1), metallothionein 3 (MT3), and superoxide dismutase 2 (SOD2). The genes involved in the oxidative stress response and signaling included dual specificity phosphatase 1, oxidative-stress responsive 1, and angiopoietin-like 7. The genes involved in apoptosis included BCL2/adenovirus E1B 19 kd interacting protein 3 and glyceraldehyde-3-phosphate dehydrogenase. No compensatory increase in the level of antioxidants such as catalase or glutathione peroxidases was found (Jurkunas et al., 2010). In addition to increased oxidative stress, there was a significant decrease in the protective Nrf-2-mediated antioxidant response, which further contributes to the oxidant-antioxidant imbalance seen in FECD (Jurkunas et al., 2010). Serial analysis of gene expression in the CE of FECD samples have also demonstrated decreased levels of transcripts related to antioxidant, anti-apoptotic, mitochondrial and pump function (Gottsch et al., 2003).
11.2.3. Nrf2 Transcription Factor
The antioxidant response element (ARE) (TGCTGA(G/C)TCAGCA), also known as Maf recognition element, is activated by the Cap’n’Collar family of transcription factors, primarily NF-E2 (nuclear factor erythroid 2) related factors −1, and −2 (Nrf1 and Nrf2) (Moi et al., 1994; Motohashi et al., 2002; Ohtsuji et al., 2008). These transcription factors heterodimerize with small Maf proteins, bind ARE sequences and cause a coordinated upregulation of antioxidant and xenobiotic-metabolizing enzyme genes during oxidative stress (Blank, 2008; Ishii et al., 2000; Shih et al., 2003). Many antioxidant genes contain the ARE in their promoters, such as SODs, Prxs, glutathione transferases, metallothionein proteins, and the Nrf2 gene itself. Activation of Nrf2 transcription has been linked to cytoprotection, and Nrf-mediated ARE activation has been studied in multiple tissues. In the absence of stress, Nrf2 is degraded rapidly by the ubiquitin-proteosome pathway. In an oxidized state, ROS and electrophiles bind to sulfhydryl residues of Keap1, resulting in the release and translocation of Nrf2 into the nucleus, where it induces ARE-dependent gene expression. In the state of oxidative stress, Nrf2 also acts as its own positive regulator by targeting ARE in its own promoter. The cytoplasmic stability of Nrf2 is highly dependent on its stabilizer, the DJ-1 (protein deglycase) protein. A hyperoxidized state can activate the whole army of antioxidant defense systems, components of which become susceptible to the deleterious effects of ROS by inadvertent oxidative modifications.
In FECD, the majority of downregulated antioxidant genes harbor ARE sequence in their promoter regions (Jurkunas, Bitar et al. 2010). By carrying out the analysis of transciption factors that regulate the ARE promoter region, we detected a deficiency in Nrf2-DJ-1-regulated antioxidant defense, pointing to the cause of oxidant-antioxidant imbalance in FECD (Figure 3) (Bitar et al., 2012; Liu et al., 2014a). DJ-1 is a cytoplasmic stabilizer of Nrf2 that enhances nuclear localization of Nrf2, required for Nrf2 to bind to ARE-regulated antioxidant defense. Nrf2 nuclear localization in response to oxidative stress occurred in normal CECs in a DJ-1-dependent mechanism, and this response was abolished with DJ-1 small interference RNA (siRNA)-mediated knockdown (Bitar et al., 2012). We detected loss of DJ-1 and lack of nuclear localization of Nrf2 in FECD cells. Normal CECs were able to upregulate DJ-1 protein synthesis in response to oxidative stimuli, while FECD CECs were not. Oxidative modification of DJ-1 at cysteine 106 resulted in increased DJ-1 degradation in FECD CECs compared to normal CECs. Furthermore, the habitual retention of Nrf2 in the cytoplasm by the Keap1/Cul3 pathway was not altered in cells with DJ-1 knockdown, suggesting that DJ-1 regulates Nrf2 through a Keap1-Cul3 independent pathway (Liu et al., 2014a).
Additionally, DJ-1 is also a negative regulator of p53 transcriptional activity, where it translocates to the nucleus, represses p53-dependent gene transcription, and inhibits apoptosis (Fan et al., 2008; Vasseur et al., 2012). The finding that DJ-1 levels are significantly reduced and phospho-p53 levels are increased in FECD suggests that the decreased DJ-1 expression renders FECD CECs more susceptible to p53-regulated apoptosis from oxidative stress (Bitar et al., 2012; Liu et al., 2014a). Furthermore, siRNA-mediated downregulation of DJ-1 in normal CECs increases caspase 3 and phospho-p53 activation in response to UV-A oxidative stress (Liu et al., 2014a). These findings indicate that DJ-1 expression protects CECs from UV-A-mediated oxidative stress and p53-dependent apoptosis and suggest that DJ-1 deficiency is an important cause of the decreased Nrf2-mediated defense in FECD. In addition to increased oxidative stress, a significant decrease in the protective Nrf-2-mediated antioxidant response has been noted, which further contributes to the oxidant-antioxidant imbalance seen in FECD (Jurkunas et al., 2010).
11.3. Mitochondria
Mitochondria are pivotal to CEC bioenergetic function and survival. Over the past two decades, numerous studies have demonstrated the association between mitochondrial dysfunction and FECD.
11.3.1. Mitochondrial and Nuclear DNA Damage
Mitochondrial dysfunction represents a hallmark in the process of aging resulting from oxidative stress, which plays a critical role in the pathogenesis of FECD (Figure 3) (Jurkunas, 2018; Jurkunas et al., 2010; Lopez-Otin et al., 2013). Due to the direct exposure of mitochondria to the ROS-generating electron transport chain (during oxidative phosphorylation) and lack of histone protection, mtDNA is specifically prone to oxidative stress and DNA damage (Johns, 1995; Jurkunas et al., 2010). Mitochondria are the core energy source of the endothelial ion pumps and are essential for endothelial cellular function (Zhang et al., 2017a). Cumulative mitochondrial damage and dysfunction is a causative factor of degenerating CE in FECD and mechanistically similar to loss of post-mitotic cells in other organ systems, providing evidence of the neuro(endo)-degenerative aspect of the condition (Borboli and Colby, 2002; Calvani et al., 2013; Zhu et al., 2014).
Initial investigation on the role of mitochondria in the degenerative process of FECD revealed accumulation of 8-ODdG, a marker of oxidized DNA lesions, primarily in mtDNA of CECs clustered around the rosettes. This finding indicated that mtDNA is more susceptible to oxidative stress and mitochondrial damage in FECD (Jurkunas et al., 2010). Importantly, the colocalization of oxidative DNA damage, mostly to mitochondria, was found to be specific to FECD, as similar findings were not detected in the CE from pseudophakic bullous keratopathy patients (Jurkunas et al., 2010). Further studies utilized quantitative polymerase chain reaction assays to specifically quantify mtDNA and nDNA damage. They detected that in FECD ex vivo specimens, mtDNA harbored an 81% increase in DNA lesion frequency and nDNA harbored a 99% increase in lesion frequency compared with normal corneal specimens (Halilovic et al., 2016). Furthermore, loss of mitochondrial density and loss of mitochondrial membrane potential along with activation of caspase-driven apoptosis was detected in FECD.
Subsequent studies utilized modeling of intracellular oxidative stress by exposing CECs to menadione, a quinone that generates intracellular ROS and induces rosette formations, which are characteristic morphological changes of FECD ex-vivo. Both FECD specimens and cell lines exhibited extensive mtDNA and nuclear DNA (nDNA) damage at baseline, while menadione treatment resulted in an increase in mitochondrial superoxide levels and led to mtDNA and nDNA damage in normal CECs, which was rescued with N-acetyl-cysteine (NAC) pre-treatment. Moreover, menadione also disrupted the inner mitochondrial membrane potential and led to mitochondrial dysfunction in normal CECs, while FECD CECs displayed mitochondrial dysfunction at baseline. Mitochondrial fragmentation, cytochrome c release, and activation of apoptosis via caspase 9 and 3 were detected in FECD specimens and normal CECs treated with menadione. The greater accumulation of acquired mtDNA and nDNA damage found in FECD, as compared to normally aging, CECs provided the first evidence that lifelong accumulation of DNA damage has detrimental effects on mitochondrial bioenergetics, leading to mitochondrial fragmentation, along with activation of intrinsic apoptosis via caspase cleavage and cytochrome c release (Halilovic et al., 2016). TEM of FECD CE revealed unusual abundance of degenerated and swollen mitochondria with lack of normal cristae (Benischke et al., 2017) and large amounts of abnormal prominent rough ER (Zhang et al., 2006). The degenerating cells harboured abundant vacuoles indicative of autophagic structures and autophagosomes that contained degraded mitochondria, representing mitophagy process (Figure 2J) (Benischke et al., 2017). These data suggested greater accumulation of acquired mtDNA and nDNA damage in FECD than observed in normal aging of the CE and provide the first evidence that lifelong accumulation of DNA damage leads to disruption of mitochondrial bioenergetics, mitochondrial fragmentation, and induction of CEC apoptosis (Figure 3).
Mitochondrial dysfunction and the accumulation of mtDNA and nDNA damage raise the possibility that mtDNA and nDNA might be more susceptible to mutagenesis in FECD CECs (Albin, 1998; Czarny et al., 2014; Jurkunas et al., 2010). It has been reported that there is a higher ratio of the common mtDNA 4977 bp deletion, a higher copy number, and increased mtDNA damage in FECD CECs as compared to normal controls (Czarny et al., 2014). However, another study found no significant differences in mtDNA 4977 bp deletion levels between FECD and normal controls (Gendron et al., 2016). Increased mtDNA levels and telomere shortening were found in FECD specimens but not in cultured FECD CECs. The increased mtDNA levels in FECD specimens was attributed to increased mitochondrial mass by the remaining CECs to maintain ATP production during increased Na+-K+ ATPase activity, in an effort to maintain corneal clarity. Intriguingly, cultured FECD CECs and normal CECs did not differ in mtDNA levels, telomere length, antioxidant-oxidant gene expression, or susceptibility to oxidative stress-induced apoptosis. These findings suggest that a normal phenotype of CEC could be restored from cultured FECD CECs through the selection of less affected FECD CECs (Gendron et al., 2016).
mtDNA damage has been shown to play an important role in cellular aging, whereby aging senescent cells accumulate oxidative mtDNA damage that subsequently results in altered gene expression of components of the respiratory chain (Gredilla, 2010). Mitochondrial dysfunction of the respiratory chain results in increased production of ROS, propagating further oxidative damage (Nita and Grzybowski, 2016). In FECD patients, there is a reduction in cytochrome oxidase activity, a complex IV enzyme of the mitochondrial electron transport chain, which has been shown to be clinically correlate with central corneal edema (Tuberville et al., 1986). Reduced levels of the electron transport chain subunits, complexes I and V, have also been reported and have been shown to result in sustained mitochondrial depolarization and mitochondrial dysfunction (Benischke et al., 2017).
Epidemiological studies have linked different mtDNA variants with the susceptibility of FECD, where the 10398G allele and Haplogroup I appear to confer significant protective effects for FECD (Li et al., 2014). Rare variants in the mitochondrial protein TSPOAP1 have also been reported in FECD (Wieben et al., 2018). Patients with the mtDNA A3243G point mutation display CEC polymegethism and mild guttae. Based on these findings it has been suggested that CEC polymegethism may be a biomarker of mitochondrial disease specifically in patients with the mtDNA A3243G mutation, which typically presents as a spectrum of syndromes ranging from maternally inherited diabetes and deafness to mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (Bakhoum et al., 2018). Several studies have reported a link between mitochondrial diseases, such as Kearns-Sayre syndrome and Pearson syndrome, and endothelial dysfunction similar to FECD (Boonstra et al., 2002; Chang et al., 1994; Kasbekar et al., 2013; Kim et al., 2016; Ohkoshi et al., 1989).
11.3.2. Mitochondrial Quality Control: Mito(Auto)phagy
Autophagy is a complex self-cannibalization mechanism that occurs in response to ER stress and oxidative stress and involves the engulfment of cytoplasmic material and intracellular organelles within autophagosomes, which fuse with lysosomes to form autolysosomes. Autophagy is primarily considered to have a cytoprotective function, especially in post-mitotic differentiated cells such as neurons and cardiomyocytes; however, it can also promote cell death (Nikoletopoulou et al., 2013). In FECD specimens, there is an upregulation of the autophagy marker DNA-damage regulated autophagy marker 1 (Dram1) as compared to control specimens. These findings are similar to those observed in a mouse model of early-onset FECD, where increased autophagy has been observed (Meng et al., 2013). Furthermore, lithium treatment has been found to increase CEC survival and autophagy, suggesting that autophagy may contribute to increased CEC survival (Kim et al., 2013). Both autophagy and apoptosis are important pathways that interact through a complex cross-talk that can be activated by similar stimuli (Nikoletopoulou et al., 2013). This complex interplay between autophagy and apoptosis can ultimately influence a cell’s fate to undergo cell death or to survive.
Our finding of mitochondrial fragmentation in FECD suggested that abnormal mitochondrial dynamics play a role in its pathogenesis, prompting us to further investigate the mitochondrial quality control system in CE (Halilovic et al., 2016; Jurkunas, 2018). The mitochondrial quality control system is a dynamic cellular protective mechanism triggered by mitochondrial damage. Mitochondria are dynamic organelles that exist in mobile networks and are continually undergoing fusion and fission (Miyai, 2018). One key process of mitochondrial quality control is mitophagy, a mitochondria-specific autophagy pathway that selectively eliminates damaged mitochondria. Fission or fragmentation precedes and supports the process of mitophagy by dividing and concentrating depolarized mitochondria, while also downregulating fusion mediators to prevent reintegration and to support mitophagy (Chang et al., 2016; Youle and van der Bliek, 2012). Thus, a critical function of fission-fusion dynamics is the spatial separation of dysfunctional mitochondria from the mitochondrial network, their engulfment in autophagosomes, and subsequent degradation after fusion with lysosomes (Figge et al., 2012). Loss of mitochondrial membrane potential activates the PTEN-induced putative kinase 1 (PINK)-Parkin-dependent pathway, whereby PINK1 accumulation on the outer mitochondrial membrane depolarizes mitochondria and signals mitochondrial dysfunction to Parkin, a ubiquitin ligase. PINK1 activates Parkin by phosphorylation, which, in turn, attaches ubiquitin to the outer mitochondrial membrane proteins such as mitofusin (Mfn2). Subsequently, Mfn2 degradation leads to mitochondrial fragmentation and complete removal of the damaged mitochondria by the autolysosome (Koyano et al., 2014; Narendra et al., 2010; Okatsu et al., 2013).
FECD cells are characterized by the loss of mitochondrial membrane potential, which is one of the main inducers of mitophagy (Halilovic et al., 2016). Our previous study provided the first evidence that the process of mitophagy plays a role in the pathogenesis of FECD (Benischke et al., 2017). An ultrastructural analysis of FECD CE specimens demonstrated an extensive number of autophagosomes containing mitochondria, providing evidence of disproportionate engulfment and degradation of mitochondria by mitophagy. Loss of mitochondrial mass was detected by MitoTracker green, and a decrease in mitochondrial density was detected by TEM in FECD CE, with and without TCF4 gene trinucleotide repeat expansions, as compared with normal specimens. In addition, autophagosome marker LC3 and lysosomal protein lysosome-associated membrane protein 1 were found to be upregulated, while decreased levels of Mfn2 in FECD specimens were found. Remarkably, the decline of the fusion protein Mfn2 suggested that dysfunctional FECD mitochondria with lower mitochondrial membrane potential downregulate fusion mediators to prevent the formation of functional networks and upregulate mitochondrial fission and the mitophagy pathway, leading to a lower density of mitochondria in FECD CE (Benischke et al., 2017). In FECD specimens, there is an accumulation of PINK1 and phospho-Parkin, along with a loss of total Parkin and total Drp1. Furthermore, intracellular oxidative stress induces Parkin-mediated mitochondrial fragmentation, whereby endogenous PINK1 and Drp1 are sequestered and degraded by mitophagy during degenerative loss of CECs in FECD (Miyai et al., 2019).
While DNA damage and excessive mitophagy activation have been detected in FECD, the mechanism of aberrant mitochondrial dynamics in CEC loss has not been completely elucidated and requires further investigation. Currently, it is still unknown if the extensive mitophagy observed in FECD rescues cells reacting to oxidative stress or if it even leads to more harm due to a substantial loss of a critical mass of mitochondria and activation of the apoptosis also seen in FECD (Jurkunas, 2018). Therefore, future research is needed to further investigate the mitochondrial quality control processes in FECD, particularly the fission-dominant morphology and low density of mitochondria through mitophagy upregulation.
11.4. Endoplasmic Reticulum Unfolded Protein Response
Apoptosis can also be initiated by the unfolded protein response (UPR), secondary to endoplasmic reticulum (ER) stress. Protein folding is important for overall cellular functioning, and accumulation of misfolded proteins can lead to ER stress and contribute to cellular toxicity. UPR-mediated apoptosis occurs through caspase 4, caspase 3, and caspase 9 (Szegezdi et al., 2006). In FECD, TEM has shown markedly increased dilated rough ER and increased expression of immunohistochemical markers of the UPR, such as α subunit of eukaryotic initiation factor 2, HSPA5 (GRP78 or BiP), C/EBP homologous protein (CHOP), in ex vivo specimens. Furthermore, increased levels of caspase 9 and caspase 3, markers of cellular apoptosis, are also present (Engler et al., 2010). These observations in FECD ex vivo specimens suggest that ER stress leads to UPR-mediated CEC apoptosis in the pathogenesis of FECD. Subsequent studies using two knockin mouse models of early-onset FECD, Col8A2(L450W/L450W) and Col8A2(Q455K/Q455K), showed FECD phenotypes and increased UPR by enlarged rough ER and upregulation of UPR markers HSPA5 (GRP78 or BiP) and CHOP (Jun et al., 2012; Meng et al., 2013). Furthermore, in these Col8A2 knockin mouse models of early-onset FECD, CECs undergo early UPR-mediated apoptosis via caspases 12 and 9 (Jun et al., 2012; Morishima et al., 2002). In a comparative analysis of FECD and normal specimens, quantitative-PCR and a UPR-specific PCR array testing 84 UPR pathway genes demonstrated altered expression of 43 (51%) UPR-related genes, where 13 of the altered genes showed significant changes (Jalimarada et al., 2014).
The involvement of the UPR in both early-onset (animal model) and late-onset forms of FECD suggests that multiple dysfunctional pathways may converge onto the UPR as a final pathway to mediate CEC apoptosis (Engler et al., 2010). Indeed, activation of transforming growth factor-beta (TGFβ) signaling in FECD has been shown to lead to the chronic overload of ECM proteins to the ER, which ultimately results in the formation of unfolded proteins and apoptotic signaling through the UPR (Okumura et al., 2017a). Furthermore, the TGFβ signalling pathway inhibitor (TGFβR1 inhibitor SB431542) suppresses aggresome accumulation, UPR, and apoptosis in FECD and has been identified as a potential therapeutic for FECD (Okumura et al., 2017a). Activation of TGFβ signalling induced mitochondrial membrane potential depolarization, which was reversed with the TGFβ inhibitor SB431542 (Okumura et al., 2017a). These findings provided evidence that TGFβ induced ER stress and simultaneous mitochondrial dysfunction, result in the activation of the apoptotic pathway in FECD (Figure 3) (Okumura et al., 2017a). Furthermore, CHOP may be acting as a “communicator” between the ER and mitochondria during apoptosis. Activation of CHOP by the UPR induces ER stress, which is, in turn, transmitted to mitochondria associated with alterations in both mitochondrial morphology and function, and initiates the intrinsic apoptosis pathway (Okumura et al., 2017c).
Furthermore, two markers of ER stress/UPR activation, CHOP and Grp78, have been shown to be significantly downregulated at the mRNA and protein levels after NAC treatment of Col8a2(L450W/L450W) knockin mice compared to wild-type mice, suggesting that the increased survival effect of NAC observed in this FECD model is associated with reduced ER stress/UPR activation (Kim et al., 2014). This finding presents in vivo evidence of a novel potential medical treatment for FECD acting via its effects on oxidative and ER stress (Kim et al., 2014).
11.5. Extracellular matrix
11.5.1. Endothelial-to-Mesenchymal Transition
Epithelial-to-mesenchymal transition or endothelial-to-mesenchymal transition (EMT) is a complex process that occurs during embryonic development, cell migration or invasion, wound healing, cancer progression, tissue fibrosis, and scar formation in many organ systems in response to a pathological stress (Campbell and Casanova, 2016). During EMT, there is a loss of intercellular contacts and cellular polarity, and the acquisition of a fibroblastic phenotype. EMT results in activated mesenchymal-like cells that upregulate EMT markers, such as vimentin, Snail1, collagen 1A1, laminin, fibronectin, and N-cadherin, and also leads to the production of excessive collagen-rich ECM. There is increasing genetic evidence implicating abnormal ECM regulation and EMT in the pathogenesis of both the early-form (COL8A2) and late-form of FECD (TCF4, ZEB1, LAMC1) (Afshari et al., 2017; Baratz et al., 2010; Biswas et al., 2001; Mehta et al., 2008; Riazuddin et al., 2010b). In FECD, it has been shown that the pathobiology involves abnormal cell-matrix interactions and expedited cellular aging leading to cell death and senescence. However, whether the abnormal cell-matrix deposition in the form of guttae precedes cellular aging, or whether diseased CECs secrete abnormal cell-matrix that leads to more cell death, still remains to be completely elucidated. We have previously demonstrated that there is activation of EMT in ex-vivo FECD specimens, and that there is a loss of organized junctional staining of plasma membrane-bound N-cadherin and of the normal hexagonal CEC mosaic (Katikireddy et al., 2018). Furthermore, in both FECD ex-vivo specimens and in our established cellular in vitro model, we have observed upregulation of EMT-and FECD-related markers such as Snail1 (Figure 2I), N-cadherin, ZEB1, fibronectin, and TGFβI.
TGFβ is an important regulator of EMT and induces a pro-fibrotic effect by increasing ECM synthesis, inhibiting collagenolysis, and promoting proliferation of fibroblasts during wound healing. TGFβ isoforms TGFβ1 and TGFβ2, as well as TGFβ receptors TGFβR1 and TGFβR2, have been shown to be elevated in the CE of FECD patients (Okumura et al., 2017a). Activation of TGFβ signaling in FECD leads to the chronic overload of ECM proteins to the ER, which ultimately results in the formation of unfolded proteins and apoptotic signalling through the UPR (Okumura et al., 2017a). Furthermore, TGFβ1 has been shown to induce ROS, cell death, and EMT during rosette formation in vitro, resulting in upregulation of EMT- and FECD-associated markers (Katikireddy et al 2017). TGFβ activation results in increased expression of fibronectin, collagen type 1, ZEB1, and Snail1 in FECD, as well as increasing aggresome formation (Okumura et al., 2017a; Okumura et al., 2015b). The TGFβ signalling pathway inhibitor, TGFβR1 inhibitor SB431542, suppresses aggresome accumulation, UPR, and apoptosis in FECD and has been identified as a potential therapeutic for FECD.
The TGFBI gene encodes the TGFβ-induced protein (TGFBIp), an ECM protein with an N-terminus secretory signal sequence that interacts with collagens, fibronectin, and integrins to mediate cell adhesions (Runager et al., 2008). Mutations in the TGFBI gene have been implicated in several corneal stromal dystrophies such as Lattice dystrophy type I, Reis-Bückler dystrophy, Theil-Behnke corneal dystrophy, granular corneal dystrophy type II (Avellino corneal dystrophy), and granular corneal dystrophy type I (Runager et al., 2008; Yamamoto et al., 2000). In FECD, TGFBI mRNA expression and age-dependent proteolytic processing of TGFBIp into 57-kDa and 39-kDa fragments are increased as compared to that in normal controls; moreover, TGFBIp also forms aggregates in the lower portions of guttae (Figure 3) (Jurkunas et al., 2009).
Clusterin (CLU) is a ubiquitous heterodimeric, di-sulfide-linked glycoprotein that is found at tissue-fluid interfaces and is involved in the maintenance of normal cell-ECM interactions (Tung et al., 1992). There are various forms of CLU that undergo posttranslational modifications, including nuclear CLU (nCLU), presecretory CLU (pre-sCLU), and secreted CLU (sCLU), and are targeted to different subcellular compartments. Pre-sCLU is an unglycosylated form that is targeted to the ER and Golgi, where it is glycosylated and cleaved to form α-and β-subunits. Mature sCLU is a glycosylated heterodimer also composed of α- and β-subunits linked by disulfide bonds. nCLU does not undergo extensive α- and β-cleavage or glycosylation and is thought to localize to the nucleus to be involved with DNA repair (Caccamo et al., 2003; Koch-Brandt and Morgans, 1996; Reddy et al., 1996; Yang et al., 2000). In FECD, CLU mRNA expression and both the pre-sCLU and nCLU forms are increased as compared to that in normal controls, whereas the sCLU form is not (Jurkunas et al., 2008a). While there is clearly a dysregulation in CLU mRNA expression and expression of its various posttranslational forms, the exact mechanism by which it affects CECs is not completely understood. CLU expression can be anti-apoptotic or pro-apoptotic, depending on the form expressed. In response to stressors, such as ionizing radiation or increased TGFβ activation, nCLU can translocate to the nucleus and promote apoptosis in stressed cells (Caccamo et al., 2003; Koch-Brandt and Morgans, 1996; Reddy et al., 1996; Yang et al., 2000). In contrast, sCLU has been shown to be cytoprotective and have anti-apoptotic properties. It is possible that in FECD, CLU can either promote or inhibit apoptosis of CECs depending on the isoform expressed (Jurkunas et al., 2008a). CLU also localizes at the cell membranes of CECs, forming a rosette-like pattern around guttae in FECD ex vivo specimens, which could represent CLU’s role in cell clustering under stress conditions (Jurkunas et al., 2008a). CLU has also been co-localized with TGFBIp in guttae, where there is increased TGFBIp expression in the center of the guttae and increased CLU expression closer to the apical side of the endothelium (Jurkunas et al., 2009). Based on these studies, is it possible that CECs in FECD are clumping through increased CLU expression and are adhering to their substrate through TGFBIp, which is progressively disrupted as guttae form, thereby leaving the CECs susceptible to apoptosis (Figure 3) (Jurkunas et al., 2009).
TCF4, also known as E2–2, through E-box promoter sequences within target genes are implicated in diverse cellular processes including proliferation, differentiation, cell migration, and EMT (Sobrado et al., 2009). While there is strong genetic evidence supporting the association between TCF4 SNPs and CTG repeat expansion in FECD, there is no clear mechanistic link between TCF4 and EMT. TCF4 also plays an important role in TGFβ signalling and has been shown to upregulate ZEB1. ZEB1, also known as TCF8, is a zinc-finger transcription factor that binds to the E2 box DNA sequence and is involved in EMT through E-cadherin repression, TGFβ activation, and fibroblast growth factor (FGF) signalling (Krafchak et al., 2005; Miyazono, 2009). In FECD, EMT-inducing genes, including ZEB1 and Snail1, are highly expressed and are involved in the excessive production of ECM proteins such as type 1 collagen and fibronectin via TGFβ-dependent signalling (Okumura et al., 2015b). TCF4 can regulate ZEB1 expression, and both genes have been implicated in FECD. Currently, the RNA toxicity hypothesis in FECD supports a model whereby CTG repeat expansion in the TCF4 gene leads to the sequestration of the mRNA-splicing factor MBNL1 in RNA foci, leading to mis-splicing of essential MBNL1 regulated mRNAs (Du et al., 2015). Intriguingly, MBNL1 plays an important role in the activation of EMT in TGFβ-dependent endocardial cell invasion (LeMasters et al., 2012). While MBNL1 sequestration has been shown in FECD to lead to mis-splicing of downstream targets such as ADD3, INF2, SORBS1, GNAS, FGFR1, MBNL1, and MBNL2, its specific role in EMT in FECD remains to be investigated. One possibility may be that CTG repeat expansions in TCF4 lead to MBNL1 sequestration and subsequent mis-splicing of MBNL1 and other targets leading to EMT and a mesenchymal-like phenotype.
Snail proteins (Snail1, Snail2, and Snail3) are zinc-finger proteins that are downstream targets in different signalling pathways that mediate EMT (Barrallo-Gimeno and Nieto, 2005). We have previously shown that Snail 1 is highly expressed in in FECD CECs, and its activation leads to upregulation of its downstream targets fibronectin, N-cadherin, and ZEB1. These findings are consistent with another study that showed upregulation of ZEB1 and Snail1, but not Snail2 or Snail3, in FECD (Katikireddy et al., 2018; Okumura et al., 2015b).
Oxidative stress and DNA damage have been shown to be a major inducer of EMT in various pathological conditions including FECD (Azizi et al., 2011; Halilovic et al., 2016; Jurkunas et al., 2009; Jurkunas et al., 2010; Jurkunas et al., 2008a; Katikireddy et al., 2018). We have demonstrated that increased oxidative stress via application of menadione, a potent generator of ROS, leads to rosette formation and upregulation of EMT- and FECD-related markers, similar to what is observed in FECD specimens (Katikireddy et al., 2018). These findings support a mechanism by which intracellular oxidative stress, generated through intrinsic or extrinsic factors such as UV light, leads CECs to undergo EMT and upregulate pro-fibrotic proteins that ultimately lead to ECM deposition, guttae formation, cytoskeletal rearrangements, loss of cell-cell junctions, and, subsequently, further cell death (Jurkunas, 2018). Interestingly, clustered regularly interspaced short palindromic repeats (CRISPR)/CAS9 knockout of NQO1, a metabolizing enzyme of menadione, was found to potentiate EMT, highlighting the role of oxidative stress in EMT induction (Katikireddy et al., 2018).
11.5.2. Epithelial Metaplasia
Epithelial metaplasia of the CE in FECD has been suggested to represent a non-specific response of distressed CEC as seen with posterior polymorphous dystrophy, CHED, and iridocorneal endothelial syndrome (Hidayat and Cockerham, 2006). Scanning electron microscopy from FECD specimens has shown the presence of fibroblast-like cells on DM, with some flat CECs appearing to transform into a fibroblastic-phenotype (Waring et al., 1978). Cytokeratins, which are intermediate filaments that are typically present in almost all cells of epithelial origin, have also been shown to be expressed in CECs from FECD corneas (Hidayat and Cockerham, 2006). Specifically, CECs from FECD corneas show strong expression using antibodies against pancytokeratin and CK 7, both which are not normally present in CECs (Hidayat and Cockerham, 2006). Vimentin, which is also an intermediate filament expressed in tissues of mesenchymal origin, is also expressed in FECD CECs, but is also seen in normal CECs as well (Hidayat and Cockerham, 2006). Overall, FECD CECs show features of epithelial, fibroblastic, and mesenchymal-like transformation, and this may represent a dedifferentiation to a more primitive cell type as seen in other tissues (Hidayat and Cockerham, 2006).
11.5. Senescence/Decreased Proliferation
Cellular senescence is a biological process whereby cells lose their proliferative capacity through permanent exit from the cell cycle. Despite senescence’s playing an important role in normal development, maintaining tissue homeostasis, and limiting tumor progression, it is also associated with a wide range of age-related diseases. This cellular response prevents the proliferation of aged cells with shortened telomeres, as well as cells containing genomic instability from ROS-mediated DNA damage (often termed stress-induced premature senescence), ultimately halting the replication of an impaired genome. It is also thought that mitochondrial dysfunction may contribute to senescence. In addition to growth arrest, senescent cells undergo morphological changes, becoming enlarged and flattened, and often produce a pro-fibrotic/pro-inflammatory secretome that has deleterious effects on tissue microenvironments (termed senescence-associated secretory phenotype) (Coppe et al., 2010). Senescent cells are also characterized by an increase in senescence-associated beta-galactosidase (SA-β-GAL) activity and overexpression of cyclin-dependent kinase inhibitors p21 and p16.
CECs have minimal proliferative capacity in vivo, explaining why their cell density decreases with age, as they are thought to be arrested in the G1 phase of the cell cycle (Joyce et al., 1996a; Joyce et al., 1996b). Progression through each stage of the cell cycle is controlled by cyclin-dependent kinases (CDKs), which must bind specific cyclins to be activated; for example, cyclin D is required for progression from the G1-S phase. Cell cycle arrest occurs through the activity of CDK inhibitors such as p21 and p16, through inactivation of the cyclin-CDK complex. Specifically, for G1-phase arrest, p16 cyclin/CDK-induced hyperphosphorylation of the retinoblastoma protein Rb is required. Both p16 and p21 are expressed in normal CECs (Enomoto et al., 2006; Song et al., 2008). The mechanisms by which these cells are maintained in a non-replicative state include lack of growth factors (Tripathi et al., 1991), the presence of TGFβ in AH (Chen et al., 1999), and active suppression through contact inhibition within the monolayer (Joyce et al., 1998; Senoo et al., 2000). Breaking the cell-cell contacts and providing a cell with growth media stimulates proliferation ex vivo (Senoo et al., 2000), indicating that CECs are in a state of quiescence in vivo, as opposed to being senescent. Studies using both an ex vivo wound model (Senoo and Joyce, 2000) and a primary culture of human CECs (Zhu and Joyce, 2004) have shown age-related differences in relative growth capacity in favor of younger cells. Furthermore, regional differences also exist in older cells, with peripheral cells maintaining a higher proliferative capacity (Mimura and Joyce, 2006), likely due to age-related increases in nuclear oxidative DNA damage (Joyce and Harris, 2010; Joyce et al., 2009), and potentially contributing to the pathogenesis of corneal dystrophies.
It is widely thought that premature senescence is part of the cellular response in FECD pathogenesis, although there is currently a lack of conclusive evidence. Matthaei and colleagues developed a transgenic Col8a2 knockin mouse model of early-onset FECD to examine the endothelial expression profile of specific cellular stress response-related proteins (Matthaei et al., 2012). CECs from mutant animals showed evidence of senescence, including significant upregulation of cdkn1a (p21) expression, increased expression of nuclear p21 and its transcription factor p53, elevated SA-β-GAL staining, and senescence-like morphology with the presence of large multi-nucleated cells. These data correlate with increased p21 and p53 levels in human FECD CE as compared to that in normal controls (Azizi et al., 2011; Matthaei et al., 2012). Similarly, p16 has been shown to be upregulated in FECD (Cui et al., 2018; Matthaei et al., 2014). Senescence is evident when culturing primary endothelial cells obtained from FECD tissue, where the cells stain for SA-β-GAL and rarely progress through the initial passages. This process may explain why viable cells become hypertrophic in the diseased endothelium, and this impaired cellular function may subsequently contribute to other detrimental processes including EMT. Preliminary data from our laboratory have shown that that UV-A irradiation induces both senescence and EMT in human CECs in vitro. Furthermore, UV-A light-induced DNA damage results in proliferating cells becoming arrested in the G2/M phase of the cell cycle; moreover, genes associated with EMT (ACTA2 and Snail2) and senescence (CDKN1a) are expressed significantly higher in these G2/M cells than in cells in G0/G1 phases (White et al., 2019). Additionally, simulating this process by arresting cells in G2/M with the CDK1 inhibitor RO-3306 induces EMT, indicating that senescence may precede and lead to fibrosis (Figure 3).
Despite this evidence of senescence in FECD, there is still a lack of understanding of its molecular mechanisms or its involvement in the disease pathogenesis. One of the problems encountered when studying cellular senescence is the difficulty in visualizing these cells in vitro, and more so in vivo. SA-β-GAL is the staining method most widely associated with senescent cells, although this can sometimes be unreliable, since slight deviations in pH level during the staining procedure can influence the results. Other markers used include Ki67 and minichromosome maintenance-2. Given that these proteins are expressed solely in proliferating cells, their lack of activity does not distinguish between quiescence and senescence. Interestingly, our lab has recently shown that a portion of mouse CECs express Ki67 following UV-A irradiation in vivo, indicating activation of the cell cycle in these post-mitotic cells (White et al., 2019). It is an intriguing possibility that the damaged quiescent cells activate the cell cycle before deciding to undergo apoptosis (Liu et al., 2016a) or surviving in a senescence-like state following sub-cytotoxic stress in order to maintain an intact monolayer. These dysfunctional cells may have detrimental effects by directly undergoing EMT or releasing pro-fibrotic mediators that induce EMT in surrounding cells, ultimately leading to ECM and guttae formation (Figure 3).
11.6. Corneal Endothelial Cell-Descemet’s Membrane Interactions
11.6.1. Descemet’s Membrane
Descemet’s membrane (DM), named after French physician Jean Descemet in 1770 (Descemet et al., 1770), corresponds to the basement membrane secreted by CECs (Kabosova et al., 2007) and is located between the posterior stroma and the hexagonal monolayer of CECs. DM forms a compact layer composed of ECM proteins, including fibronectin, laminin, collagen types IV and VIII, and proteoglycans, including heparin sulfate, dermatan sulfate, and keratin sulphate (Joyce, 2003; LeBleu et al., 2007). Its composition and ultrastructure vary throughout life (Levy et al., 1996), thus forming distinct stratified layers over time (Murphy et al., 1984). On its stromal side, a normal DM is composed of fibronectin and collagen type IV (chains α1 and α2) and VIII, whereas the endothelial side is composed of entactin, laminin, perlecan, and collagen type IV (chains α3 and α6) (Levy et al., 1996; Ljubimov et al., 1995).
The anterior part of DM is formed during fetal development (approximately 12 weeks gestation until birth), acquiring a complex lamellar structure. By 16 weeks gestation, thin, short filaments composed from polypeptide chains of collagen VIII α1 and α2 subunits deposit perpendicularly along the DM (Ljubimov et al., 1995; Murphy et al., 1984; Sawada et al., 1990; Stephan et al., 2004) forming a striated pattern defined as the anterior banded layer (ABL). At 32 weeks gestation, the ABL is regularly organized with collagen VIII α1 and α2 subunits (Gottsch et al., 2005b). This characteristic deposition process progresses from one layer at 12 weeks gestation, to ten layers at the end of the second trimester, to 30–40 layers at birth (Murphy et al., 1984). At birth, DM is about 3 μm thick; moreover, collagen VIII secretion decreases when the cells progress from a proliferative to a non-proliferative state, while collagen IV secretion persists (Hopfer et al., 2005; Kabosova et al., 2007). The secreted ECM then loses its striated pattern and becomes a non-striated posterior layer, termed the posterior non-banded layer (PNBL), defined as a postnatal layer (Levy et al., 1996). The thickness of the ABL remains stable with aging (Waring, 1982), while PNBL thickness gradually increases by 0.1 μm per year in a non-lamellar manner and gradually thickens throughout adult life, reaching approximately 10 μm by age 80 (Hayashi et al., 2002; Johnson et al., 1982; Joyce, 2003; Murphy et al., 1984; Waring et al., 1982; Xia et al., 2016b).
Topographical DM analysis using scanning electron microscopy, TEM and atomic force microscopy reveals a fine meshwork, with a rich felt-like surface of intertwined fibers containing pores (average size, 38 nm) (Abrams et al., 2000; Di Mundo et al., 2017; Hayashi et al., 2002; Last et al., 2009; Ojeda et al., 2001). The DM is elastic, solid, and resistant to the proteolytic action of metalloproteases. Its porous structure allows for water permeability and contributes to corneal stromal dehydration.
11.6.2. DM in FECD: from DM Thickening to Guttae Formation
In FECD, with respect to DM, the ABL remains unchanged, while the PNBL becomes less electron dense and has a fine-grained structure that lacks periodicity; posterior to this layer is another fine-grained banded layer that undergoes marked thickening and ECM deposition, leading to the formation of guttae (Gottsch et al., 2005b). In addition to a thickened and multi-layered DM, the presence of ECM excrescences, termed guttae, appear first in the central cornea and progressively spread throughout the corneal periphery. In early-onset FECD (Col8a2(L450W/L450W)), thickening of the ABL predominates, and ultrastructure images support its thickening as a combination of an internal collagenase layer (ICL) and a posterior striated layer (also known as the posterior banded layer (PBL) or posterior collagenous layer located at the more posterior DM. The ICL represents a layer of dense, widely spaced collagen, and PBL exhibits a horizontal striated pattern that grows to 12 μm in thickness (Gottsch et al., 2005b). However, in late-onset FECD, the ABL remains unchanged, while there is a gradual transition from the PNBL to the PBL, interspersed with banded patterns named “wide-spaced collagens,” similar to those in the ABL (Bourne et al., 1982; Levy et al., 1996; Waring, 1982). In late-onset FECD, guttae are larger than those seen in early-onset FECD, and the ICL is not present in the DM. Oxytalan fibers, components of the ECM that are not present in normal corneas, have been observed in FECD, keratoconus, and several other corneal dystrophies. Oxytalan can be detected in the subepithelial stroma but is most abundant under the CE. Furthermore, oxytalan fibers surround guttae but are not detected within either the guttae bodies or in the DM (Alexander et al., 1981). The precise implication of these findings remains unknown.
Fibronectin deposition is one of the first steps in the pathogenesis of late-onset FECD, preceding deregulation of laminin and type IV collagen (Goyer et al., 2018). These findings are supported by a recent study using an early-onset mouse model of FECD expressing a mutant COL8A2 protein (Col8a2(L450W/L450W) and Col8a2(Q455K/Q455K)), where alterations in DM tissue compliance preceded phenotypic changes in CEC counts and morphology (Leonard et al., 2019). These observations led to the notion that both early- and late-onset FECD have similar initial pathways that lead to FECD pathogenesis. The accumulation of glycation end products (Jurkunas et al., 2008b; Wang et al., 2007), and the dysregulation of several proteins have been identified in DM from FECD patients (Table 4). Furthermore, proteomics analysis shows a high degree of variability among FECD specimens, which might reflect the large spectrum of genetic susceptibilities in FECD (Goyer et al., 2018).
Table 4:
Gene Expression and Proteomics in Fuchs Endothelial Corneal Dystrophy
| Up-regulation | Down-regulation | References |
|---|---|---|
| COL8A2 | (Poulsen
et al., 2014) (Gottsch et al., 2005b) |
|
| COL8A1 COL4 Laminin |
(Gottsch et al., 2005b) | |
| Fibronectin | (Gottsch
et al., 2005b) (Matthaei et al., 2014) |
|
| TCF4 TCF8 SLC4A11 Apoliprotein D Keratocan Matrilin-3 Decorin EMILIN-1 Complement C3 |
COL2A2 COL3A1 COL4A4 Fibrillin-1 |
(Poulsen et al., 2014) |
| TGFBI Clusterin |
(Poulsen
et al., 2014) (Jurkunas et al., 2009) (Weller et al., 2014) |
|
| Agrin | (Poulsen
et al., 2014) (Weller et al., 2014) |
|
| COL3 COL7 COL 15 COL 16 Fibulin-2 Versican |
(Weller et al., 2014) | |
| Prx-2 Prx-3 Prx-5 |
(Jurkunas et al., 2008) | |
| N0X4 CDKN2A ARHGAP18 ETS1 AGBL1 C0L1A1 COL3A1 |
CDKN2A-inhibitorIDl | (Matthaei et al., 2014) |
| LOXHD1 | (Riazuddin et al, 2012) (Matthaei et al., 2014) |
Guttae formation represents one of the hallmarks of FECD. Laing and colleagues have previously defined five stages of guttae formation based on guttae size, cell abnormalities, guttae coalescence, and contour (Laing et al., 1981). Son and colleagues have suggested that guttae might be formed via a cytoplasmic accumulation of proteins within the ER, inducing a subsequent UPR. They proposed two potential mechanisms of guttae formation: (1) the first model involves collections of intracellular material growing inside a CEC and then fusing with the basal cell membrane to create a “pore” through which proteinaceous material is externalized towards the DM, forming the gutta stem, and (2) in the second model, CECs directly secret the proteinaceous material onto DM, and the gutta are formed outside the CECs (Son et al., 2014). Interestingly, Xia and colleagues, using TEM analysis of DMs from FECD patients, identified three types of FECD-DMs: (1) type I DM harbors the three typical layers, ABL, PNBL, and PBL, containing abnormally deposited collagen, guttae, occasional 10–20 nm fibrils, and amorphous substances (from stromal side to endothelial side: ABL, PNBL, PBL, and guttae); (2) type II DM contains an additional posterior fibrillar layer (PFL), which is present over the PBL and the guttae, showing a loose matrix of collagen with a fibril diameter of 20–40 nm; (from stromal side to endothelial side: ABL, PNBL, PBL, guttae, and PFL), and (3) type III DM contains two layers of guttae, where one layer is buried under the PFL, as in type II, while the other layer protrudes above the PFL as in type I (from stromal side to endothelial side: ABL, PNBL, PBL, guttae, PFL, guttae). They also identified the short fibrillar type VIII collagen as one of the major components of the abnormally secreted posterior collagenous layer of DM (Xia et al., 2016a).
The molecular basis leading to the formation of guttae and subsequent CEC apoptosis has not been completely elucidated. We previously demonstrated that there was a dysregulation of protein synthesis and/or secretion in FECD CECs that ultimately lead to abnormal ECM deposition, guttae formation, and cellular apoptosis (Jurkunas et al., 2008a). A subsequent study demonstrated that the cell adhesion protein TGFBIp, an ECM pro-adhesive protein that can interact with collagens, fibronectin, and integrins, was overexpressed and colocalized to guttae with CLU in FECD specimens (Jurkunas et al., 2009). However, TGFBIp formed aggregates at the base of the guttae next to DM, while CLU localized on the top of the TGFBIp-stained areas, closer to the level of the CEC nuclear plane. While these studies do not provide direct functional roles for TGFBIp and CLU, their localization to guttae and abnormal ECM suggest that they may play roles in CEC apoptosis and the pathogenesis of FECD (Figure 3).
11.6.3. Dynamic Reciprocity between DM and CECs
It has been well described that the intrinsic biophysical characteristics of ECM affect cells and play a vital role in cellular behavior (Ali et al., 2016; Hynes, 2009). The ECM is present in all tissues, and, in addition to providing mechanical support, is a major component of the cellular microenvironment that ultimately influences cell differentiation, migration, activation, behavior, and death (Lu et al., 2011). Changes in ECM composition due to the aberrant production of ECM proteins affect the physiological activity of cells and may lead to functional impairment. For example, aberrant production of ECM has been associated with the kidney vascular endothelial dysfunction seen in diabetes (Grutzmacher et al., 2013), as well as neuronal degeneration (Fielder et al., 2017). Several studies support the notion that, in FECD, the DM ECM homeostasis is compromised, which ultimately leads to the formation of guttae and the disorganization of ECM collagen fibrils (Poulsen et al., 2014). Since DM is one of the major sources of biophysical stimuli for CECs, a better understanding of guttae formation and the interplay among guttae, ECM, and CECs is needed, and will likely provide further insight into the pathogenesis of FECD.
In our previous work, we hypothesized that guttae size might influence the interaction between CECs and DM. To further investigate the relationship between CECs and their interactions with the abnormal ECM in FECD, cells from a human CEC line, HCEnC-21T, were seeded on decellularized normal and FECD DM. Our results suggested that guttae size highly influences normal CEC behavior. Indeed, using time-lapse microscopy of HCEnC-21T cells seeded on FECD-DM, as compared to those in normal-DM, the migratory process revealed a delay in cellular attachment of HCEnC-21T cells to FECD-DM (Kocaba et al., 2018). These results were consistent with a study by Rizwan and colleagues, who demonstrated, using a synthetic guttae FECD model, that guttae dimensions, density, and spacing alter the behavior of normal CECs, thus negatively influencing cell monolayer formation (Rizwan et al., 2016). In addition, we were able to identify three growth patterns with respect to guttae diameter: (1) in the first pattern, small guttae (<15 μm) were covered by a normal monolayer of CECs; (2) in the second pattern, medium-sized guttae (15–30 μm) were covered by an elongated cytoplasm, with nuclei surrounding the guttae, and (3) in the third pattern, large guttae (>30 μm) were not covered by CECs and formed a rosette-like pattern (Kocaba et al., 2018). We also observed that medium and large-sized guttae induced TUNEL-positive apoptosis in a rosette pattern, similar to that seen in FECD ex vivo specimens (Azizi et al., 2011; Kocaba et al., 2018). Laser capture microscopy and dissection of CECs adjacent to large guttae revealed an upregulation of EMT-markers, such as N-cadherin, Snail1, and alpha smooth muscle actin, as well as involvement of the oxidant-antioxidant pathway through nicotinamide adenine dinuleotide phosphate oxidase 4. Therefore, these findings support a mechanism by which guttae induce an oxidative stress response, senescence, cellular apoptosis, and an EMT-phenotype in a size-dependent manner. However, an important observation was that guttae did not induce all the changes typically seen in FECD ex vivo specimens, such as mitochondrial dysfunction, abnormal UPR, and DNA damage, suggesting that, while guttae play an important role in FECD pathogenesis, it is not the sole abnormality. Hence, we propose a mechanism whereby aberrant ECM deposition is an additional extrinsic factor, contributing to the vicious cycle of FECD pathogenesis, and is secondary to underlying intrinsic factors that predispose CECs to additional intrinsic and extrinsic factors (Figure 1 and Figure 3) (Jurkunas, 2018; Kocaba, 2018; Kocaba et al., 2018). Based on this mechanism, an underlying primary abnormality in the cell make it susceptible to additional intrinsic or extrinsic stressors, and results in the secretion of abnormal ECM and the formation of guttae, which create a toxic environment for the cell that eventually leads to cell death (Figure 1 and Figure 3).
The term dynamic reciprocity has been used to describe the ongoing, bidirectional interactions that occur between cells and their microenvironment, particularly the ECM (Bissell and Aggeler, 1987; Bissell et al., 1982). CECs require a normal and intact DM to function normally, and, in turn, healthy CECs are crucial for normal ECM production. The dynamic reciprocity between CECs and the ECM in FECD has not been extensively investigated, and a greater understanding of this process will likely yield insight into the pathogenesis of FECD. Furthermore, since the deposits on DM do not remodel over time, cross-sectional examinations might serve as a record of endothelial development and pathology (Murphy et al., 1984; Waring, 1982), similar to strata studies in geology, where DM likely represents “memories” of CEC behavior over time (Bissell and Aggeler, 1987; Bissell et al., 1982). Future studies could separately analyze DM, CECs, and CEC-DM interactions, to further elucidate the dynamic reciprocity in the CE and the alterations that occur in FECDs. Additionally, proteomic analysis is needed to determine the exact composition of the toxic microenvironment formed by guttae of various sizes.
12. Treatment
Management of FECD depends on the stage of the disease and the patient’s symptoms. In early FECD, patients are usually asymptomatic, and observation without any medical or surgical intervention is indicated. As FECD progresses, symptoms such as reduced vision, halos, and glare occur and can be managed conservatively with topical hypertonic saline drops or ointment. In more advanced FECD, surgical intervention, typically EK, is indicated.
12.1. Medical Treatment
12.1.1. Dehydration Treatment
In FECD, as the cornea decompensates and imbibes water, the cornea swells and becomes edematous. Patients with FECD can report worse vision in the morning due to overnight lid closure, which improves throughout the day as evaporation decreases corneal edema. Anecdotal evidence supports the use of a blow dryer directed towards the anterior surface of the eye to help facilitate evaporation and to expedite the decrease in the corneal edema that occurs throughout the day (Costagliola et al., 2013). Hyperosmotic sodium chloride (NaCl) drops or ointments (2% or 5%) can be topically administered to help decrease corneal edema. There are currently many formulations of hyperosmotic NaCl available for treatment of corneal edema including, but not limited to, Muro 128 (5% NaCl), Ocusalin (5% NaCl), Omnisorb (5 mg/mL NaCl with 0.1 mg/mL benzalkonium chloride), and ODM5 (5% NaCl and 0.15% sodium hyaluronate) (Dutescu et al., 2015). Hypertonic saline has been used to treat corneal edema secondary to FECD as well as other conditions, such as bullous keratopathy, herpes simplex keratitis, and corneal hydrops (Marisi and Aquavella, 1975). There have been both animal and human studies conducted that support the use of hypertonic saline to reduce corneal edema (Insler et al., 1987). In an ex vivo model of corneal edema, Omnisorb and Ocusalin hypertonic saline drops have been shown to significantly reduce corneal edema by approximately 260–280 μm (Dutescu et al., 2015). Improvements in corneal edema and BCVA have also been demonstrated in patients with the use of hypertonic saline (Marisi and Aquavella, 1975; Rouland, 2015). A few studies have investigated the role of corneal collagen cross-linking in FECD to increase the rigidity of the cornea and decrease corneal edema; however, they have not found that collagen cross-linking is an effective long-term treatment for corneal edema in FECD (Cordeiro Barbosa et al., 2010; Ucakhan and Saglik, 2014).
12.1.2. Steroids
The role of topical steroids in reducing corneal edema in FECD has not been extensively investigated. Dexamethasone has been shown to increase Na+-K+ ATPase activity and pump function in cultured mouse and bovine CECs by increasing the expression of its Na+-K+ ATPase alpha1-subunit (Chen et al., 2006; Hatou et al., 2009). However, there is limited evidence that topical steroids are beneficial for the treatment of stromal edema in patients with FECD (Wilson et al., 1988). There is one case report demonstrating that subconjunctival administration of dexamethasone in a FECD patient post-cataract surgery can lead to a decrease in CCT and corneal edema (Chikamoto et al., 2008).
12.1.3. Bandage Contact Lenses
Bandage contact lenses can be used in more advanced FECD for symptomatic relief from painful epithelial bullae and epithelial defects, and can also help with re-epithelialization of the corneal surface by providing a protective layer between the cornea and the eyelids (Bergmanson et al., 1999). An important consideration when selecting a bandage contact lens is the oxygen transmissibility (Dk/t), which should be high to allow adequate oxygenation of the cornea (Blackmore, 2010). Topical prophylaxis with an antibiotic should also be considered given the higher risk of infection with prolonged bandage contact lens use. The safety and efficacy of a hyper osmotic contact lens (Hyper-CL Lens) in subjects with corneal edema (ClinicalTrials.gov Identifier: NCT02660151) is currently under investigation and may be beneficial for FECD patients with corneal edema.
12.2. Surgical Treatment
12.2.1. Endothelial Keratoplasty and Penetrating Keratoplasty
There are many factors to consider when deciding which surgical intervention is warranted in a patient with FECD, including stage of disease, a patient’s symptoms, a patient’s lifestyle and expectations, a surgeon’s experience, cost, as well as access to surgical instruments and donor tissue (Figure 4). Until the introduction of partial thickness endothelial keratoplasty (EK), surgical intervention was indicated only in FECD patients with advanced disease, and these patients would undergo a full-thickness penetrating keratoplasty (PK). PK is a surgical procedure, whereby a full-thickness central circular section of the cornea, typically between 7.0 mm and 8.5 mm in diameter, is removed from the host and replaced with a corresponding full-thickness donor cornea. The surgical outcomes of PK in FECD patients have been acceptable, where the majority of patients showed significant improvement in visual acuity after PK (Price et al., 1991). However, PK has also been associated with higher intraoperative risks, such as a choroidal hemorrhage, a higher risk of post-operative graft rejection, and a longer period of visual recovery requiring suture removal and management of post-operative astigmatism with a scleral or rigid gas permeable contact lens (Tan et al., 2012). Improvements in surgical techniques over the past few decades, particularly EK, have resulted in earlier surgical intervention in FECD patients. FECD is the most common indication for keratoplasty worldwide (39%) (Gain et al., 2016). However, there are regional differences in these percentages (Bigan et al., 2018; Flockerzi et al., 2018; Gain et al., 2016). In addition to the global increase in the number of keratoplasties being performed, there is a trend in many countries towards increased numbers of EKs and decreased numbers of PKs (Almeida et al., 2018; Bigan et al., 2018; Chan et al., 2018; Flockerzi et al., 2018; Frigo et al., 2015; Gogia et al., 2015; Mohamed et al., 2014; Rezaei Kanavi et al., 2016; Tan et al., 2009; Ting et al., 2012). For example, in the United States, there were almost 13,000 more cases of EK than PK in 2018. Furthermore, more recently in certain countries, such as the United States, Canada, and Germany, there has been a shift towards DMEK as compared to Descemet stripping automated endothelial keratoplasty (DSAEK) (Chan et al., 2018; Flockerzi et al., 2018).
Figure 4. Current, Frontier and Developing Therapies for Fuchs Endothelial Corneal Dystrophy (FECD).
Legend: PK= penetrating keratoplasty; DSAEK= Descemet stripping automated endothelial keratoplasty; DMEK= Descemet membrane endothelial keratoplasty; DWEK= Descemetorhexis without endothelial keratoplasty; DSO= Descemet stripping only; UT-DSAEK= ultrathin-DSAEK; NT-DSAEK= nanothin-DSAEK; ROCKi= Rho kinase inhibitor; TGFβ= transforming growth factor beta; EK= endothelial keratoplasty; DMT= Descemet membrane transfer; RNAi= RNA interference; miRNA= micro RNA; CRISPR= clustered regularly interspaced short palindromic repeats.
Despite the widespread adoption of EK, there are still many countries around the world where PK is the preferred surgical procedure, even for endothelial disease (Dong et al., 2016; Hong et al., 2015; Rezaei Kanavi et al., 2016). This has largely been attributed to the lack of surgical training in EK, reduced surgical volumes, and a shortage of donor tissue (Dong et al., 2016; Hong et al., 2015). While studies have found that EK (DSAEK) induces less astigmatism, has better wound stability, and results in faster visual rehabilitation as compared to PK, some studies have found no differences in graft rejection rate and BCVA improvement (Price et al., 2013; van Rooij et al., 2018). Another study showed that, while DSAEK in FECD achieved better BCVA and lower refractive error than did PK, DSAEK also had a higher risk of graft failure (Greenrod et al., 2014). A Cochrane review including three randomized controlled trials comparing EK and PK for FECD did not show any significant differences in BCVA at two years. However, EK did result in a lower incidence of higher order aberrations and increased endothelial cell loss than did PK (Nanavaty et al., 2014).
An ideal EK should produce a refractive neutral outcome and not induce any change in corneal astigmatism or corneal power, should result in a healthy donor endothelium that resolves all edema with a tectonically stable globe that is safe from injury and infection, and an optically pure cornea (Terry, 2003). While DSAEK and DMEK are currently the most common methods of performing EK, previous surgical techniques aimed at endothelial replacements have been described including posterior lamellar keratoplasty and deep lamellar endothelial keratoplasty (Melles et al., 1998; Tan et al., 2012; Terry, 2003; Tillett, 1956). However, posterior lamellar keratoplasty and deep lamellar endothelial keratoplasty still require a large scleral wound and a deep scleral-corneal lamellar pocket (Terry, 2003). With the introduction and popularity of DSAEK and DMEK, both posterior lamellar keratoplasty and deep lamellar endothelial keratoplasty have been largely replaced. Both DSAEK and DMEK are widely performed, with the choice of surgical procedure depending on multiple factors, including the patient’s anatomy, surgical comorbidities, previous ocular history, surgeon’s preference, and access to donor tissue. The goal of EK in FECD is to surgically replace abnormal DM, guttae, and diseased CECs with healthy CECs and DM free of guttae, such that there is a reversal of corneal edema and visual function restoration. In both procedures, a descemetorhexis is first performed to strip the DM and the CE (Melles et al., 1998). In DSAEK, the donor posterior stroma with DM and CECs is transplanted into the recipient stromal bed, and the graft is kept in place with air or 20% sulphur hexafluoride, and, in some cases, sutures are used to further support the positioning of the graft. In DMEK, only the donor DM and CE is transplanted into the recipient stromal bed, and the graft is kept in place with air or 20% sulphur hexafluoride. While DMEK allows for better visual acuity outcomes, superior patient satisfaction, and possibly less graft rejection, it also has a steep learning curve, is more technically challenging due to tissue handling and graft unscrolling, and has a higher rebubbling rate as compared to DSAEK (Marques et al., 2019; Pavlovic et al., 2017; Singh et al., 2017; Stuart et al., 2018; Zhu et al., 2018b). Furthermore, in a cost-effectiveness analysis conducted in a surgical center in the United States, performance of DMEK instead of DSAEK provided greater utility, with the gain of an extra 0.4 quality-adjusted life-years over a 15-year period (Gibbons et al., 2019).
Although previous studies have compared DMEK to DSAEK, the DSAEK groups typically included thicker grafts, which likely contributed to the worse visual outcomes (Neff et al., 2011). Better visual outcomes have been reported with ultrathin DSAEK (<130 μm) compared to conventional DSAEK; however, DMEK still provides superior visual acuity outcomes compared to ultrathin DSAEK (Busin et al., 2013; Chamberlain et al., 2019; Dickman et al., 2016; Duggan et al., 2019). In an attempt to generate thinner precut DSAEK tissues, nanothin DSAEK (≤50 μm) has also been developed and has been shown to have comparable visual outcomes and complication rates as compared to DMEK (Kurji et al., 2018). These results support the notion that thinner donor tissue is an important factor in determining visual outcomes in EK.
12.2.1.1. Tissue-Sparing EK
In the United States and other developed countries, cornea donor supply currently meets the demand; however, there is a global donor shortage that limits corneal transplantations to approximately 1 out of 70 potential patients (Gain et al., 2016). Different surgical methods have been developed in an effort to overcome the global donor shortage including hemi-DMEK and quarter-DMEK (Figure 4).
Hemi-DMEK is a modification of the DMEK technique that uses a full diameter untrephinated, approximately 12 mm, semi-circular graft (Gerber-Hollbach et al., 2016; Lam et al., 2014; Lam et al., 2015; Lie et al., 2016). Since one donor graft is bisected, one donor’s tissues can be used for two patients. In this technique, an 8.0 to 9.0 mm descemetorhexis is completed and the semi-circular donor graft is inserted and positioned in the donor eye. The semi-circular graft has an equally large surface area and can be unfolded and positioned in a fashion similar to that in a standard DMEK graft (Lam et al., 2014; Lam et al., 2015). In a case series of 3 eyes with FECD that underwent hemi-DMEK, all corneas cleared within 6 months of surgery, and at 12 months, visual outcomes were similar to those seen with standard DMEK (Lam et al., 2014; Lam et al., 2015). The denuded stromal areas adjacent to the hemi-DMEK graft were initially edematous and devoid of cells but cleared by 6 months, at which peripheral ECD was observed, suggesting that endothelial cell migration leads to the re-distribution of CECs over both DM and denuded stroma.
Quarter-DMEK is a modification of the hemi-DMEK technique, whereby 4 grafts are obtained from one donor cornea (Baydoun et al., 2018; Muller et al., 2017; Zygoura et al., 2018). In quarter-DMEK, an untrephinated, full-size donor DM is divided into 4 equally sized grafts, which allows for transplantation into 4 recipients. A 7.0 to 8.0 mm descemetorhexis is performed, and the graft is unrolled and positioned over the denuded stromal bed. Quarter-DMEK results in good visual outcomes, similar to those seen with conventional standard DMEK at 6 months; moreover, this technique may potentially quadruple the availability of endothelial grafts (Zygoura et al., 2018). However, there is a steep decline in ECD within the first postoperative month following quarter-DMEK as compared to that with conventional DMEK. This is hypothesized to occur due to incomplete/ongoing cellular migration within the donor graft. Furthermore, there may be differences in cellular migration patterns, as there was a tendency of corneas to clear adjacent to the cut edges, while the “limbal” rounded edge lagged behind (Zygoura et al., 2018). An in vitro study investigating central and peripheral CEC migration patterns from quarter-DMEK grafts has shown that CECs migrate over the central radial cut edges; however, there is a decrease in migration activity towards the far periphery. No migration of CECs was observed along the outer peripheral edge, possibly due to differences in the ECM (Miron et al., 2018).
Another technique that has been developed to reduce donor shortage is split cornea transplantation, whereby a single donor cornea is used for 2 recipients by combining deep anterior lamellar keratoplasty and DMEK surgeries on the same day, or within 72 hours (Heindl et al., 2011a; Heindl et al., 2011b).
12.2.2. Descemet Membrane Endothelial Transfer (DMET), Descemet Membrane Transfer (DMT), and Descemetorhexis Without Endothelial Keratoplasty (DWEK)/Descemet Stripping Only (DSO)
Previous studies have reported corneal clearance in eyes that developed spontaneous graft detachment within the first few weeks of DMEK or graft removal after DSAEK, likely as a result of CEC migration from the donor graft onto the recipient stroma (Balachandran et al., 2009; Ziaei et al., 2013b). However, spontaneous corneal clearing over a 6-month period was reported in an eye that underwent DMEK for FECD and had a “free-floating” DM roll within hours after surgery, long before CEC migration could have been completed. Hence, this case suggested that solely the presence of donor CECs on a carrier (in this case DM) within the anterior chamber is sufficient to repopulate the CE of the recipient posterior stroma (Dirisamer et al., 2012a; Dirisamer et al., 2012b). This technique is termed DMET and involves the insertion a “free-floating” DM graft, where the upper edge of the graft is fixated within the limbal incision to create contact between the donor and recipient tissues (Birbal et al., 2018). It is hypothesized that the mechanism of action of DMET is to stimulate a host endothelial migratory response rather than to supply the receipt cornea with additional CECs (Birbal et al., 2018). Ultimately, DMET likely has a limited role in the treatment of FECD when compared to DMEK due to a prolonged period of 3 to 6 months for corneal clearing, a lower ECD, and a high rate of corneal decompensation requiring retransplantation (Birbal et al., 2018).
Another technique termed, DMT, has been described, where a decellularized DM is transplanted following descemetorhexis (Bhogal et al., 2017; Soh and Mehta, 2018). DMT replaces the damaged DM with guttae in a FECD patient with a decellularized healthy DM, and allows the remaining peripheral CECs to migrate and repopulate the central CE. In a rabbit model, DMT promoted CEC wound healing, migration, proliferation, and restoration of corneal clarity (Bhogal et al., 2017). A case report of the first-in-human trial of DMT for the treatment of FECD reported successful implantation of an acellular DM after a central 5.0-mm descemetorhexis was performed in a 56-year old Chinese female with FECD. At postoperative month 6, the graft remained attached, her central ECD was 889 cells/mm2, her BCVA was 6/7.5, and she had been taken off all topical medications (Soh and Mehta, 2018). While the initial result of DMT is encouraging, larger cases series or a clinical trial is needed to determine its efficacy and potential benefits when compared to other methods of EK. However, DMT could address the shortage of donor tissues for transplantation, since a larger donor corneal tissue pool would become available, because DMT does not require donor CECs; multiple grafts could, therefore, be prepared from a single donor, since smaller, 5.0-mm grafts are used (Soh and Mehta, 2018). Furthermore, postoperative management of DMT likely would not require chronic topical immunosuppression with steroids. However, further clinical studies with longer follow-ups are needed to confirm this observation.
In 2012, the first definitive case of spontaneous corneal clearing with CEC repopulation after Descemet stripping without donor endothelial cell implantation was performed on a 34-year old female patient with posterior polymorphous corneal dystrophy and FECD-like endothelial changes (Shah et al., 2012). In this case, the corneal cleared within 6 months and attained good uncorrected visual acuity. The proposed mechanism for the central endothelial clearing was that CEC migration occurred from the periphery. Another case of long-term corneal clearing after spontaneous resolution of corneal edema in an iatrogenic endothelial trauma patient with FECD was reported and further supports the possibility of surgically treating FECD with descemetorhexis only (Koenig, 2013). Subsequent studies have further explored this technique termed Descemetorhexis without endothelial keratoplasty (DWEK), also referred to as Descemet stripping only (DSO), in patients with FECD and has shown promising, yet sometimes unpredictable results (Arbelaez et al., 2014; Bleyen et al., 2013; Borkar et al., 2016; Davies et al., 2018; Davies and Pineda, 2019; Garcerant et al., 2019; Ham et al., 2011; Huang et al., 2018; Iovieno et al., 2017; Kaufman et al., 2018; Koenig, 2015; Moloney et al., 2017). In certain cases, persistent corneal edema after DWEK remained, requiring further surgical intervention with EK (Arbelaez et al., 2014; Bleyen et al., 2013; Borkar et al., 2016; Ham et al., 2011; Iovieno et al., 2017; Koenig, 2015). However, in other cases DWEK resulted in complete clearance of corneal edema and improvement of visual acuity (Borkar et al., 2016; Davies et al., 2018; Huang et al., 2018; Iovieno et al., 2017; Moloney et al., 2017).
Given the discrepancies of the previous studies, it is important to identify any preoperative, intraoperative, and postoperative factors that can predict corneal clearance after DWEK. We previously performed a retrospective chart review of 17 eyes from 13 patients with FECD that underwent central 4.0-mm DWEK to study preoperative factors that contribute to corneal clearance after DWEK. Corneal clearance occurred in 14 out of 17 eyes, with a mean time to resolution of corneal edema of approximately 3 months. There was no difference in baseline age, ECD, or pachymetry between eyes that cleared and those that did not. All eyes that failed to clear had DWEK performed using an initial 360-degree scoring technique followed by stripping, while no eyes failed to clear that had undergone stripping by scoring 2 clock hours, followed by a complete Descemetorhexis manoeuvre. Furthermore, 3 out of the 4 eyes that underwent the 360-degree scoring technique failed to clear (Davies et al., 2018). This finding highlights the importance of surgical technique in the success of DWEK, and that manual stripping can result in small DM detachments with corresponding areas of edema and/or can result in stromal scarring that inhibits the central migration of CECs; however, in our cases, there was a steady increase in central ECD over time in all eyes. The importance of the recipient stromal surface has also been reported, where areas of stromal surface irregularity seemed to impede CEC migration and resulted in localized persistent edema after DWEK (Arbelaez et al., 2014).
Descemetorhexis size is likely another important surgical factor that contributes to the success of corneal clearance after DWEK. Initial cases of persistent corneal edema after DWEK for FECD created larger Descemetorhexis of 6.0 to 9.0 mm in size (Arbelaez et al., 2014; Ham et al., 2011; Koenig, 2015). Certain cases of persistent corneal edema after DWEK for FECD did not report the Descemetorhexis size, and it can be presumed that a larger Descemetorhexis was created, as it typically is in DMEK (Bleyen et al., 2013). In other studies, where DWEK resulted in complete clearance of corneal edema and improvement of visual acuity in the majority of FECD patients, a smaller 4.0-mm Descemetorhexis was performed (Borkar et al., 2016; Davies et al., 2018; Huang et al., 2018; Iovieno et al., 2017; Moloney et al., 2017). An increased Descemetorhexis size of 2.0 mm would more than double the surface area required by the remaining CECs to repopulate (Borkar et al., 2016). It is possible that a Descemetorhexis size greater than 4.0 mm in diameter is too large to allow the remaining CECs to migrate, repopulate the entire surface area of the CE, or adequately clear the cornea in the majority of FECD patients.
Patient factors likely contribute to the success of corneal clearance after DWEK, as even in studies where a small 4.0-mm Descemetorhexis was performed, cases of persistent corneal edema were observed (Borkar et al., 2016; Davies et al., 2018; Huang et al., 2018; Iovieno et al., 2017). In our study, both eyes of 4 patients underwent DWEK, and the eye pairs demonstrated similar clearing times, suggesting that other environmental factors such as growth factors in the anterior chamber or genetic factors may also influence the rate of CEC migration and corneal clearance. Furthermore, certain eyes showed rapid corneal clearance and CEC repopulation within a month of surgery, while in other cases corneal clearance took up to 6 to 8 months, suggesting that patient factors contribute to the response to DWEK (Borkar et al., 2016; Davies et al., 2018; Huang et al., 2018; Iovieno et al., 2017; Moloney et al., 2017). Patient selection may have also impacted surgical outcomes, where, in one study, patients were excluded if they had a peripheral ECD <1000 cells/mm2 and central edema (Moloney et al., 2017). Results of preoperative pachymetry have been weakly correlated with a reduced response rate and final visual outcomes after DWEK (Borkar et al., 2016; Huang et al., 2018). Additional patient factors, such as age, sex, smoking history, and genetic factors, and how these predict CEC migration and response to DWEK have not been thoroughly investigated. However, using an ex vivo human corneal culture model, the presence of an intact DM, young donor age, and supplementation with the Rho kinase inhibitor Y-27632 have been found to promote CEC migration (Soh et al., 2016).
Corneal clearance after DWEK is likely based on the migration of normal-appearing CECs from the periphery towards the center of the cornea, where the Descemetorhexis is performed. In early FECD, the central CE is typically affected, while the peripheral cornea is normal appearing. A similar pattern is observed with increasing rosettes, composed of degenerating CECs around guttae, in the central cornea as compared to the periphery (Jurkunas, 2018). Based on these observations, the question arises of why peripheral CECs do not migrate spontaneously towards the central cornea to replace degenerating CECs in FECD without surgical interventions such as Descemetorhexis. It is possible that the peripheral NCC-derived CEC progenitors are kept inactive through contact inhibition (Joyce, 2003; Katikireddy et al., 2016). Following Descemetorhexis contact inhibition is lifted, stimulating the peripheral cornea to replace the central CE through proliferation and migration of NCC-derived progenitors and mature CECs. The cytokine TGFβ may also contribute to maintaining the CE monolayer in a contact-inhibited state and suppressing the spontaneous proliferation and migration of peripheral CECs. TGFβ is detected in the AH and has been shown to be a mediator of G1-phase arrest of CECs in vivo (Joyce, 2003). Furthermore, TGF-β and TGF-β receptors are normally expressed in CECs and are upregulated in FECD (Joyce, 2003; Okumura et al., 2017a). Therefore, it is possible that in FECD, degenerating CECs located in the central cornea are secreting TGFβ into the AH to inhibit the G1/S transition phase through paracrine signalling. Following Descemetorhexis and removal of the central degenerating CECs, TGFβ signalling is reduced, and the peripheral CECs can progress through the G1/S transition phase.
Another important consideration in understanding why peripheral CECs do not migrate spontaneously towards the central cornea is the size of guttae and DM in FECD. DM has been shown to act as a depot for growth factors including FGF (Joyce, 2003). Thus, it is possible that DM sequesters and concentrates inhibitory factors that suppress central cell migration. Furthermore, we have previously shown that the diameter of guttae is an important determinant of CEC behavior, including cell migration and monolayer formation. While small guttae did not impede cell growth and monolayer formation, large guttae inhibited monolayer formation where the apices of the guttae were not covered by CECs (Kocaba et al., 2018). Therefore, the higher density and increased size of guttae in the central cornea may be actively inhibiting CEC cellular migration and monolayer formation through physical and chemical mechanisms. With Descemetorhexis and the removal of DM and the large central guttae, the remaining healthier CECs could be released from these inhibitory mechanisms and be allowed to migrate and reform a monolayer.
In this FECD model, peripheral cornea NCC-derived CEC progenitors are kept inactive in G1-phase arrest through contact inhibition, increased TGFβ inhibitory signalling, and physical and chemical signalling from guttae and DM. Following Descemetorhexis, these inhibitory signals are released, and CECs can progress through the G1/S transition phase as well as migrate centrally to replace and reform a functional CE.
While there are limitations to DWEK, including delayed visual recovery and possibility of a subsequent surgery, the benefit of eliminating the risk of rejection, reduced surgical complications, and adverse events, as well as independence from donor tissues suggest that there is a role for DWEK in selected FECD patients. Future studies are required to help identify patients that would be ideal DWEK candidates.
12.3. Regenerative Treatment
12.3.1. Drugs/Novel Molecules
12.3.1.1. ROCK Inhibitors
In 2009, in an effort to develop a clinically applicable and efficient method of culturing human CECs free of animal-origin pathogens, Okumura and colleagues identified a specific Rho-associated coiled-coil-containing protein kinase (ROCK) inhibitor Y-27632 as a new tool for culturing CECs (Okumura et al., 2009). ROCKs (ROCK1 and ROCK2) are serine/threonine kinases that act as downstream effector proteins of the small Rho GTPase, and are involved in cytoskeleton organization, cell migration, cell-cell adhesion, proliferation, cell cycle control, and apoptosis (Riento and Ridley, 2003). ROCK1 and ROCK2 inhibition with Y-27632 promotes cell adhesion, inhibits apoptosis and increases proliferation of monkey and human CECs (Okumura et al., 2016a; Okumura et al., 2014b; Okumura et al., 2009; Pipparelli et al., 2013). ROCK inhibitors have also been shown to promote corneal endothelial wound healing in an in vitro scratch assay and an in vivo transcorneal freezing rabbit model, likely through increased cellular migration and proliferation (Okumura et al., 2011). Based on these studies, ROCK inhibitors have been further investigated as a topical eye drop for the treatment of corneal endothelial diseases. The topical administration of the ROCK inhibitor Y-27632 promoted CEC wound healing in rabbit and primate models, and also improved central corneal edema in pseudophakic bullous keratopathy and FECD patients (Okumura et al., 2015a; Okumura et al., 2013b). Topical ROCK inhibitor Y-27632 following corneal endothelial denudation with transcorneal freezing has also been shown to improve corneal edema and visual acuity in a FECD patient, where 6 months after treatment BCVA improved from 20/63 to 20/16 and CCT improved from 703 μm to 568 μm (Koizumi et al., 2013). Subsequent studies have also identified Y-39983 as a more potent ROCK inhibitor (ROCK1 and ROCK2) than Y-27632, and that both compounds promote CEC proliferation and facilitate CE wound healing through upregulation of cyclin D and downregulation of p27Kip1 via phosphatidylinositol 3-kinase signalling to mediate G1/S progression (Okumura et al., 2014b). Another ROCK inhibitor (ROCK1 and ROCK2), H-1152, has also been shown to be more potent than Y-27632 for promoting CEC migration and proliferation; topical administration of H-1152 has been shown to reduce corneal edema in a rabbit model (Meekins et al., 2016).
The Rho kinase (ROCK1 and ROCK2) inhibitor ripasudil hydrochloride hydrate (Glanatec ophthalmic solution 0.4%, Kowa Co Ltd., Nagoya, Japan) was approved in Japan in 2014 for the treatment of glaucoma and ocular hypertension. Topical administration of ripasudil in healthy subjects leads to transient morphological changes of CECs such as indistinct cell borders and pseudo-guttae, likely due to the protrusion formation along intracellular bodies caused by the reduction of actomyosin contractility in CECs (Nakagawa et al., 2015; Okumura et al., 2015c). Ripasudil has also been shown to promote CEC wound healing similarly to that seen in previous studies with Y-27632 (Okumura et al., 2016b). Ripasudil and Y-27632 have also been used as salvage agents in cases of DWEK with a 4.0-mm Descemetorhexis, where the cornea failed to clear (Moloney et al., 2017). It has been postulated that the differences in ability to salvage between ripasudil and Y-27632 is due to the differing inhibitions of ROCK1 and ROCK2 (Moloney et al., 2017). In another retrospective study comparing DMEK to DWEK, 3 out of 12 patients that underwent DWEK elected to use ripasudil 6 times a day for 2 to 3 weeks. All 3 DWEK patients showed subjective improvement in vision and corneal clearance after the initiation of ripasudil (Huang et al., 2018). Future studies, including an ongoing phase IIa prospective blinded placebo-controlled randomized controlled trial (ClinicalTrials.gov Identifier: NCT03575130) will provide more information on the safety and efficacy of ripasudil 0.4% after DWEK in patients with moderate to advanced FECD.
In addition to the development of ROCK inhibitors as a topical eye drop for the treatment of corneal endothelial diseases, ROCK inhibitors have been shown to convert CECs into a phenotype that is capable of regenerating in vivo endothelial tissue and are now being utilized as compounds to optimize CEC-based therapies (Koizumi et al., 2012; Koizumi et al., 2014; Okumura et al., 2014a, 2017b; Okumura et al., 2012; Peh et al., 2019).
12.4. Tissue Engineering
12.4.1. Corneal Endothelial Cell-Based Therapies (Tissue-Engineered Constructs and Cell Suspension Injection)
Tissue engineering has allowed for the development of two strategies that may eventually replace conventional corneal transplantation using donor corneas: tissue-engineered endothelial keratoplasty (TE-EK), composed of a cultured CEC sheet on a scaffold and direct injection of cultured CECs in the form of a cell suspension (Okumura et al., 2017b; Peh et al., 2019). These cell-based therapies will become increasingly important, as there is already a global corneal donor shortage (Gain et al., 2016). This will potentially allow CECs derived from a single donor cornea to treat multiple patients requiring EK; moreover, it has been estimated that 1 pair of donor corneas could generate 28 tissue-engineered constructs (Tan et al., 2014). However, both of these strategies require the ability to cultivate and expand primary CECs in an in vitro setting, which is challenging, given that primary human CECs have a very low proliferative potential and a limited ability to be passaged when they undergo rapid cellular senescence and EMT, and acquire fibroblastic-like cell morphology (Bartakova et al., 2018; Peh et al., 2015b; Peh et al., 2017; Roy et al., 2015). Various isolation and culture methods have been developed to optimize human cultured CECs; most of these have focused on the composition of the basal culture medium supplemented with different compounds, including L-ascorbic acid 2-phosphate to protect against oxidative damage, activation of p120-catenin-Kaiso signalling to alter contact inhibition, SB431542 and bone morphogenetic protein-7 to inhibit TGFβ signalling, and Y-27632 to inhibit ROCK signalling (Kimoto et al., 2012; Okumura et al., 2013a; Peh et al., 2015a; Peh et al., 2011; Peh et al., 2015b; Peh et al., 2019; Shima et al., 2011; Zhu et al., 2012). The propagation of human CECs via a dual media approach to prevent potential EMT has been described using M5-Endo medium for the attachment phase and M4-F99 medium for the proliferative phase (Peh et al., 2015b). This dual media approach can be further enhanced with the addition of Y-27632, where increased cell-substrate adhesion, cellular proliferation, cell survival, and cryopreservation are observed (Peh et al., 2015a). Overall, the inclusion of Y-27632 using a dual media approach can increase overall human CEC yield by 1.96- to 3.36-fold (Peh et al., 2015a). In addition to having high yields for cultured human CECs, it is important to generate a reproducibly homogenous population of human CECs that are functional and devoid of cell-state transition properties (Hamuro et al., 2016; Toda et al., 2017). Flow cytometry can be used to identify the percentage of a specified cultured effector cell population (known as the E-ratio) sharing the same phenotype as mature human CECs by selecting for the surface markers clusters-of-differentiation (CD)44−CD166+CD133-CD105−CD24−CD26−. The continuous presence of Y-27632 and the absence of SB431542 in the basal culture medium (modified Opti-MEM I reduced serum media referred to as C medium) has also been demonstrated to generate high E-ratios with no karyotype abnormalities. Other important factors for maintaining a high E-ratio include seeding cell density during culture passages and donor age younger than 29 years old (Toda et al., 2017).
An important consideration when developing the technology of cultured human CECs as a therapeutic for humans is that culture conditions should be xeno-free to reduce the risk of contamination and transfer of pathogens. Human CECs should be isolated and propagated under good manufacturing practices conditions for cell-based therapies in humans, using good manufacturing practices-grade instead of research-grade reagents and techniques (Okumura et al., 2016c; Peh et al., 2017; Peh et al., 2019). These detailed culture protocols have allowed for the isolation and expansion of primary human CECs for both TE-EK and cell suspension injection therapy, which has already demonstrated promising results in patients with bullous keratopathy and FECD (Kinoshita et al., 2018).
For TE-EK, cultured human CECs are grown and seeded onto a scaffold carrier, stabilized in vitro, and then transplanted into the recipient eye, similar to the procedures employed in DSAEK. Various scaffold carriers have been investigated, including amniotic membranes, collagen type 1 sheets, silk fibroin, laser dissected 100-μm thick human corneal stromal lenticules with intact DM, and the temperature-responsive polymer poly(N-isopropylacrylamide), a fully synthetic hydrogel based on polyethylene glycol and polycaprolactone that are cross-linked with sebacic acid (Ishino et al., 2004; Koizumi et al., 2012; Koizumi et al., 2007; Liu et al., 2014b; Mimura et al., 2004; Ozcelik et al., 2014; Peh et al., 2019; Proulx and Brunette, 2012; Sumide et al., 2006; Williams et al., 2018). Using a rabbit corneal transplantation model, where DM and CE are removed, human CEC sheets grown on amniotic membrane or collagen sheets have been shown to be functional in vivo, with a cell density and morphology similar to that of normal CE (Ishino et al., 2004; Mimura et al., 2004; Sumide et al., 2006). Normal morphology, as shown by ZO-1 and Na+-K+ ATPase immune-staining patterns, and in vivo functionality of cultured CECs grown on collagen carriers to reduce corneal edema have also been demonstrated in a primate corneal transplantation model, where the ability of CECs to proliferate in severely limited (Koizumi et al., 2012; Koizumi et al., 2007). Similar results have also been shown with other carriers such as donor posterior corneal stromal lenticules (Koizumi et al., 2012; Peh et al., 2017; Peh et al., 2019). While these findings using TE-EK in animal models are promising, no clinical trials in humans have been completed. However, a first-in-man clinical trial has been approved in Singapore to use TE-EK for the treatment of FECD as well as all forms of pseudophakic or aphakic bullous keratopathy (Clinical Trial Certificate: CTC1800013) (Peh et al., 2019).
Another cell-based approach to replacing CE is to deliver cultured human CECs as a cell suspension directly to the anterior chamber through an intracameral injection following the removal of the native CECs by scrapping the DM. Initial animal studies investigating intracameral administration of cultured CECs have produced conflicting results (Bostan et al., 2016; Mimura et al., 2003). In a feline model of corneal transplantation, while injection of CECs resulted in superior outcomes as compared to controls, it was still inferior to that of an intact endothelium, indicating incomplete functionality of the transplanted CE. The authors also found that injection of Y-27632 with CECs following scraping formed the healthiest endothelium. Furthermore, labelling of injected CECs demonstrated only sparse integration with the newly regenerated CE, suggesting that the repopulation of the denuded areas was most likely composed of peripheral unscraped areas rather than injected CECs (Bostan et al., 2016). Additional studies have used both rabbit and primate models of corneal transplantation, and have found that intracameral administration of CECs in combination with Y-27632 is capable of forming a monolayer with normal expression of ZO-1 and Na+-K+ ATPase, and yields a functional CE in vivo (Okumura et al., 2012; Okumura et al., 2016c; Peh et al., 2019). Overall, these studies demonstrate that cell-based therapy with human CECs delivered as a cell suspension in the anterior chamber can yield a functional CE in vivo and is well tolerated. Furthermore, while various strategies have been used to target cultured human CECs to DM, including the use of a magnetic field and iron-endocytosed CECs or superparamagnetic microspheres incorporated into CECs; the inclusion of a ROCK inhibitor to enhance adhesion to the ECM and cell engraftment is crucial to future cell-based therapies for FECD (Mimura et al., 2003; Okumura et al., 2016c; Patel et al., 2009; Peh et al., 2019). A landmark clinical trial by Kinoshita and colleagues investigated whether injection of cultured human CECs supplemented with Y-27632 into the anterior chamber could increase CEC density after removal of abnormal ECM and/or degenerated CECs in the 8-mm diameter central cornea of 11 patients with bullous keratopathy (7 of which were secondary to FECD with moderate to severe diffuse corneal edema) (Kinoshita et al., 2018). After injection of CECs into the anterior chamber, the patients were placed in the prone position for 3 hours. At 24 weeks after cell injection, CEC density greater than 500 cells/mm2 was observed in all 11 patients, CCT less than 630 μm was attained in 10 out of the 11 patients, and improvement in BCVA was seen in 9 out of the 11 patients. The procedure was also safe, as no major adverse events such as anterior uveitis, intraocular infection, or immune rejection, were observed. One patient underwent trabeculotomy for steroid-induced glaucoma 8 months after cell injection. Longer term follow up showed that cell-based therapy with human CECs delivered as a cell suspension and supplemented with Y-27632 produced a well-tolerated functional CE without an immune response during the 2-year follow-up period (Kinoshita et al., 2018). While the corneas of the 7 FECD patients have remained clear for more than 3 years after treatment, the procedure of mechanically scraping DM with a silicone tip to remove abnormal ECM does not remove guttae, and it has been reported that these guttae have persisted for up to at least 2 years in follow-up (Wahlig et al., 2018a). Longer term follow-up in these FECD patients is required, as we have previously demonstrated the important role of guttae in CEC growth and survival, and that guttae have the capacity to sicken completely healthy CECs (Kocaba et al., 2018). Future clinical findings in these 7 FECD patients will provide further clinical insight into the interplay between healthy CECs and guttae.
12.5. Future Therapies
We now have a greater understanding of the pathogenesis of FECD disease, which has allowed for the development of potential novel therapeutics for its treatment. Many of these therapeutics in development are aimed at targeting the most common genetic abnormality in FECD, the CTG repeat expansion within an intronic region of TCF4. Additional therapeutic interventions in development are targeting the oxidative and ER stress responses in CECs, and well as the toxic ECM environment.
12.5.1. Antioxidant Therapies
Oxidative stress and ER stress lead to increased apoptosis of CECs and play a role in the pathogenesis of FECD (Jurkunas et al., 2010). Therefore, targeting the oxidative stress and ER stress pathways is a potential therapeutic treatment for FECD. NAC is a thiol-containing antioxidant and free radical scavenger, and is also a reduced glutathione precursor. NAC has also been shown to increase the survival of cultured CECs exposed to ER and oxidative stress. Initial studies showed that DNA damage, mitochondrial membrane potential and ATP levels can be rescued by NAC in the menadione-induced FECD model in vitro (Halilovic et al., 2016). Additionally, NAC was shown to abolish oxidative stress-induced rosette formation by abrogating EMT in FECD (Katikireddy et al., 2018). Furthermore, systemic administration of NAC also increases CEC survival through an increased antioxidant effect and decreased ER stress in a mouse model of early-onset of FECD (Kim et al., 2014). More recently, NAC was shown to be cytoprotective in UV-A-induced model of late-onset FECD. The systemic administration of NAC in mice diminished CEC loss, corneal edema and macromolecular changes such as DNA damage and apoptosis seen in FECD (Liu et al., 2018). While targeting the oxidative stress and ER stress pathways with NAC has proven successful in in vitro and in vivo animal models, no clinical studies exist investigating its therapeutic role in FECD patients.
In an effort to identify drugs that protect CECs against oxidative stress and the UPR, 640 compounds from a Food and Drug Administration-approved library were screened and characterized. Two compounds, oxotremorine, a muscarinic receptor agonist, and mefenamic acid, a non-steroidal anti-inflammatory drug (NSAID), have been shown to display protective effects in cultured bovine CECs (Kim et al., 2017). Furthermore, additional NSAIDs (celecoxib, diclofenac, fenoprofen, ketoprofen, niflumic acid, piroxicam, tolfenamic acid, and aceclofenac) in the drug library have shown a partial effect against oxidative stress and the UPR (Kim et al., 2017). While some of these NSAIDs are already commercially available in a topical ophthalmic formulation, no clinical studies have been performed to investigate their potential role in the treatment of FECD.
12.5.2. Nrf2 Agonists
The evidence of lost Nrf2-mediated antioxidant defense in various forms of FECD (Guha et al., 2017; Jurkunas et al., 2010) presents a rationale for investigating whether Nrf2 agonists confer cytoprotection in FECD. Sulforaphane (SFN) is a naturally occurring glucosinole found in green cruciferous vegetables and activates Nrf2 by post-translational modification of Nrf2 inhibitor, Keap1. Additionally, many other compounds, including D3T, which also interferes with Nrf2 degradation, can enhance Nrf2 levels. Both compounds showed cytoprotection against oxidative stress induced cell death in FECD cells in vitro. Importantly, direct application of SFN to the FECD post-keratoplasty specimens ex vivo enhanced nuclear localization of Nrf2 and decreased p53-mediated apoptosis, indicating that Nrf2 function can be ‘rescued’ even in end-stage FECD. Additionally, SFN unregulated the Nrf2-depenent antioxidant defense and decreased ROS production; thus, providing a novel therapeutic target for FECD (Ziaei et al., 2013a).
12.5.3. TGFβ Inhibition
TGFβ is an important regulator in EMT and has been implicated in FECD. Expression levels of TGFβ and TGFβ receptors are elevated in FECD, and TGFβ signalling induces a chronic overload of ECM proteins to the ER that leads to the formation of unfolded proteins and the UPR, ultimately leading to apoptosis (Okumura et al., 2017a). Furthermore, TGFβ inhibition supresses aggresome accumulation, the UPR, and apoptosis activation. Therefore, TGFβ inhibition is a potential therapeutic target in FECD. Various therapeutic strategies targeting TGFβ inhibition have already been developed for other diseases, and clinical trials using anti-TGFβ antibodies and TGFβ receptor inhibitors have been conducted (Okumura et al., 2017a). However, whether TGFβ inhibition represents a viable therapeutic strategy in FECD patients remains to be elucidated.
12.5.4. RNA Targeting
12.5.4.1. Anti-Sense Oligonucleotides (ASOs)
ASOs have emerged as a potential therapeutic for intervention in FECD (Du et al., 2015; Hu et al., 2018; Zarouchlioti et al., 2018). ASOs are short, synthetic, single-stranded oligodeoxynucleotides that have been used to either target mRNA, and, subsequently, knockdown protein expression, alter pre-mRNA splicing by sterically blocking splicing factors, block mRNA translation by preventing ribosome recruitment, or bind non-coding RNAs and toxic RNAs associated with disease pathogenesis (Rinaldi and Wood, 2018). In FECD, the location of the CTG repeat in an intronic region has led to the discovery that RNA toxicity plays a role in the pathogenesis of FECD, as is seen in other repeat expansion neurodegenerative diseases such as Huntington’s disease and myotonic dystrophy (Du et al., 2015; Rinaldi and Wood, 2018). ASOs targeting the CTG repeat expansion in TCF4 have been developed and have shown decreased RNA foci formation, rescue of abnormal MBNL1 nuclear localization, and reversal of mis-splicing in cell lines derived from FECD patients and in ex-vivo FECD specimens (Hu et al., 2018; Zarouchlioti et al., 2018). Importantly, ASOs have been targeted to CECs after intravitreal administration in a mouse model (Zarouchlioti et al., 2018). However, there are still many challenges with ASO therapy, including optimizing the delivery method, dosage, dosing time interval, and time of therapeutic intervention to the human CECs in FECD patients (Zarouchlioti et al., 2018). These findings highlight the potential for ASO-mediated therapy for FECD patients with the CTG repeat expansion in TCF4, where patients in the early stage of the disease may benefit the most from therapeutic intervention.
12.5.4.2. RNA Interference (RNAi)
RNA interference (RNAi) includes siRNA, short hairpin RNA, or miRNA that bind to mature, spliced cytosolic mRNAs and target them for removal by argonaute and RNA degradation (Wild and Tabrizi, 2017). These short RNAs are important regulators of gene expression and can be abnormal in many diseases. In FECD, levels of miR-29, a regulator of ECM production, has been shown to be decreased in the CE. Overexpression of miR-29b in ex vivo human CECs have resulted in a decrease in ECM production (Toyono et al., 2016). Therefore, one potential strategy would be to overexpress miR-29b in CECs in vivo to alter ECM production. However, these studies are preliminary and require further investigation.
12.5.5. Genome Editing
12.5.5.1. CRISPR/Cas9
Conventional gene therapy involves the introduction of a functional copy of the gene to treat loss-of-function mutations, usually using a viral delivery system (Moore et al., 2018). While the genetics of FECD is complex and heterogeneous, many of the causal genetic mutations have been identified including the CTG repeat expansion in TCF4, which is the most common. Given that the proposed mechanism of action of the CTG repeat expansion in TCF4 is through a gain-of-function, via RNA toxicity and RAN translation rather than a loss-of-function of TCF4, conventional gene therapy will likely not rescue the phenotype. Instead, genome editing using programmable nucleases that can manipulate the genome in a sequence-specific manner may allow for targeting of the CTG repeat expansion in TCF4 (Zhu et al., 2018a). CRISPR and CRISPR-associated nuclease Cas9 has quickly become the genome editing tool of choice, primarily due to the ease of targeting a specific sequence using simple molecular cloning techniques (Moore et al., 2018). To target specific sequences in mammalian cells using the CRISPR/Cas9 system, the Cas9 nuclease is directed by a single guide RNA containing a 20 nucleotide spacer sequence specific for the target sequence, which is upstream of a protospacer adjacent motif sequence also recognized by Cas9. The CRISPR/Cas9 system creates a double-strand break by the Cas9 endonuclease and will initiate downstream repair pathways either through non-homologous end joining or homology directed repair (Moore et al., 2018). CRISPR/Cas9-mediated genome editing in corneal epithelial cells and stromal keratocytes through intrastromal injection has been reported in mice as well as in cultured human corneal epithelial cells (Courtney et al., 2016; Kitazawa et al., 2017; Moore et al., 2018; Yang et al., 2018). While no studies have shown CRISPR/Cas9-mediated gene editing of the CTG repeats in TCF4 in CECs, CRISPR/Cas9-mediated gene editing has been shown to efficiently and permanently eliminate polyglutamine expansion-mediated neuronal toxicity in a mouse model of Huntington’s disease, a neurodegenerative repeat expansion disorder (Wild and Tabrizi, 2017; Yang et al., 2017). Genome editing in FECD CECs should provide many therapeutic advantages, including accessibility and immunoprivilege (Zhu et al., 2018a). However, challenges exist, including the optimization of the delivery of CRISPR/Cas9 to specific CECs and minimizing or eliminating non-specific off-target events. Viral vectors, particularly adeno-associated viruses, have been extensively studied for corneal transduction and have also been developed into human clinical trials for the treatment of ocular diseases such as Leber’s congenital amaurosis. There are potential challenges for adeno-associated virus–mediated delivery of CRISPR/Cas9, including limitation in transgene size, persistent transgene expression, and specificity of expression (Moore et al., 2018). Other non-viral delivery systems have been reported, including physical delivery methods and chemical delivery methods such as the use of lipid nanoparticles to encapsulate the CRISPR components (Moore et al., 2018). Despite the challenges that exist, genome editing with CRISPR/Cas9 targeting the CTG repeat expansion in TCF4 or other known causative genetic mutations has enormous therapeutic potential in the treatment of FECD and possible prevention, if targeting is performed before any clinical manifestations.
13. Conclusion and Future Directions
In this review, we provided an overview of the clinical characteristics of FECD, as well as the available diagnostic tools for clinical assessment. In addition, we presented our hypothesis on the vicious cycle of FECD pathogenesis, whereby cellular stress and abnormal cellular–ECM interactions are the hallmarks of disease pathogenesis (Figure 1 and Figure 3). In this model, multiple exogenous factors, particularly ultraviolet light and oxidative stress, in combination with genetic factors, such as TCF4 CTG repeat expansion, leads to increased intracellular ROS production, resulting in mitochondrial dysfunction and alteration in the synthetic capacity of cells. Furthermore, CECs undergo senescence, where cells, although post-mitotic, become metabolically active, and lead to a senescence-associated secretory phenotype and secretion of pro-fibrotic mediators that induce EMT during guttae formation. The guttae, the scars of the CECs, in turn create a toxic microenvironment and further propagate the vicious cycle of oxidant–antioxidant imbalance in FECD pathogenesis (Figure 1 and Figure 3).
Despite the strong epidemiological evidence that FECD affects women more often than men, there remains a lack of understanding of the mechanisms that lead to this female predominant disease. Recent studies suggest an important role for estrogen genotoxicity in mediating sex-associated differences in FECD. Future studies using a non-genetic UV-A-based mouse model, that recapitulates the morphological and molecular changes of FECD, may provide further insight into the pathogenesis and sex-dependent differences in FECD.
We now have a greater understanding of the various pathways that contribute to FECD pathogenesis. While much progress has been made in the medical and surgical management of FECD, particularly with endothelial keratoplasty, there are many novel therapeutics in development for the treatment, and possible prevention, of FECD. We postulate, that development of biologics targeting specific molecular mechanisms involved in cellular degeneration holds promise in FECD therapy in the future.
Article Highlights.
FECD pathogenesis is an interaction between genetic and environmental factors.
FECD has a greater incidence in women; however, there is a lack of knowledge as to why.
There is dysregulation of the oxidative stress response, and mitochondrial and nuclear DNA damage.
There are abnormal cell-extracellular matrix interactions and endothelial-to-mesenchymal transition activation.
A non-genetic ultraviolet A mouse model may provide further insight into FECD pathogenesis.
There has been much advancement in the medical and surgical management of FECD.
Acknowledgments
Funding: This work was supported by a NEI/NIH (R01 EY020581); Shire Research Scholarship; European Society of Cataract & Refractive Surgery Peter Barry Fellowship grant; a Fulbright Scholarship; a Harvard Sachs Grant; a Harvard Vera Bellus Bequest Grant; a French Society of Ophthalmology (Société Française d’Ophtalmologie) Grant; a Hospices Civils de Lyon Grant, and funding from The Kosciuszko Foundation.
We would like to thank Peter Mallen for his help with the illustration.
Declaration of interest:
U.V. Jurkunas has received grants from the NEI/NIH (R01 EY020581), Santen Pharmaceuticals Co, and the Eversight Foundation; has received travel support from Santen Pharmaceuticals Co; and has received consulting fees from Chiesi and a grant from Intelli . S. Ong Tone has received a Shire Research Scholarship. V. Kocaba has received a Fulbright Scholarship, a Harvard Sachs Grant, a Harvard Vera Bellus Bequest Grant, a French Society of Ophthalmology (Société Française d’Ophtalmologie) Grant, and a Hospices Civils de Lyon Grant. M. Böhm has received a European Society of Cataract & Refractive Surgery Peter Barry Fellowship grant. A. Wylegala has received funding from The Kosciuszko Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author’s Statement
We declare that this manuscript is original, has not been published and is not currently considered for publication elsewhere including preprint servers. All previously mentioned authors are independent of any commercial funding or sponsor and have had full access to all data in the study. All authors have no conflicts of interest to declare. All the authors have read the manuscript and have approved this submission. All authors take full responsibility for the integrity of the data and accuracy of the data analysis. I have full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis as well as the decision to submit for publication.
References
- Abrams GA, Schaus SS, Goodman SL, Nealey PF, Murphy CJ, 2000. Nanoscale topography of the corneal epithelial basement membrane and Descemet’s membrane of the human. Cornea 19, 57–64. [DOI] [PubMed] [Google Scholar]
- Adamis AP, Filatov V, Tripathi BJ, Tripathi RC, 1993. Fuchs’ endothelial dystrophy of the cornea. Surv Ophthalmol 38, 149–168. [DOI] [PubMed] [Google Scholar]
- Adamis AP, Molnar ML, Tripathi BJ, Emmerson MS, Stefansson K, Tripathi RC, 1985. Neuronal-specific enolase in human corneal endothelium and posterior keratocytes. Exp Eye Res 41, 665–668. [DOI] [PubMed] [Google Scholar]
- Afshari NA, Igo RP Jr., Morris NJ, Stambolian D, Sharma S, Pulagam VL, Dunn S, Stamler JF, Truitt BJ, Rimmler J, Kuot A, Croasdale CR, Qin X, Burdon KP, Riazuddin SA, Mills R, Klebe S, Minear MA, Zhao J, Balajonda E, Rosenwasser GO, Baratz KH, Mootha VV, Patel SV, Gregory SG, Bailey-Wilson JE, Price MO, Price FW Jr., Craig JE, Fingert JH, Gottsch JD, Aldave AJ, Klintworth GK, Lass JH, Li YJ, Iyengar SK, 2017. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat Commun 8, 14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshari NA, Li YJ, Pericak-Vance MA, Gregory S, Klintworth GK, 2009. Genome-wide linkage scan in fuchs endothelial corneal dystrophy. Investigative ophthalmology & visual science 50, 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshari NA, Pittard AB, Siddiqui A, Klintworth GK, 2006. Clinical study of Fuchs corneal endothelial dystrophy leading to penetrating keratoplasty: a 30-year experience. Archives of ophthalmology (Chicago, Ill. : 1960) 124, 777–780. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Cavalcanti BM, Regali L, Cruzat A, Trinidad M, Williams C, Jurkunas UV, Hamrah P, 2018. In Vivo Confocal Microscopy Shows Alterations in Nerve Density and Dendritiform Cell Density in Fuchs’ Endothelial Corneal Dystrophy. Am J Ophthalmol 196, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agte V, Tarwadi K, 2010. The importance of nutrition in the prevention of ocular disease with special reference to cataract. Ophthalmic Res 44, 166–172. [DOI] [PubMed] [Google Scholar]
- Albin RL, 1998. Fuch’s corneal dystrophy in a patient with mitochondrial DNA mutations. J Med Genet 35, 258–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldave AJ, Rayner SA, Salem AK, Yoo GL, Kim BT, Saeedian M, Sonmez B, Yellore VS, 2006. No pathogenic mutations identified in the COL8A1 and COL8A2 genes in familial Fuchs corneal dystrophy. Investigative ophthalmology & visual science 47, 3787–3790. [DOI] [PubMed] [Google Scholar]
- Alexander RA, Grierson I, Garner A, 1981. Oxytalan fibers in Fuch’s endothelial dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960) 99, 1622–1627. [DOI] [PubMed] [Google Scholar]
- Ali M, Raghunathan V, Li JY, Murphy CJ, Thomasy SM, 2016. Biomechanical relationships between the corneal endothelium and Descemet’s membrane. Exp Eye Res 152, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali ZK, Whitson JT, Mootha VV, Witherspoon SR, Joseph JA, Joseph AM, Hynan LS, Cavanagh HD, 2011. Glaucoma in patients with corneal endothelial dystrophy. Eye & contact lens 37, 332–336. [DOI] [PubMed] [Google Scholar]
- Almeida HG, Hida RY, Kara-Junior N, 2018. Trends in corneal transplantation from 2001 to 2016 in Brazil. Arq Bras Oftalmol 81, 529–538. [DOI] [PubMed] [Google Scholar]
- Alnawaiseh M, Zumhagen L, Wirths G, Eveslage M, Eter N, Rosentreter A, 2016. Corneal Densitometry, Central Corneal Thickness, and Corneal Central-to-Peripheral Thickness Ratio in Patients With Fuchs Endothelial Dystrophy. Cornea 35, 358–362. [DOI] [PubMed] [Google Scholar]
- Amann J, Holley GP, Lee SB, Edelhauser HF, 2003. Increased endothelial cell density in the paracentral and peripheral regions of the human cornea. Am J Ophthalmol 135, 584–590. [DOI] [PubMed] [Google Scholar]
- Ambrosio R Jr., Guerra FP, 2019. Advanced Corneal Imaging for Fuchs Endothelial Corneal Dystrophy. Ophthalmology 126, 205–206. [DOI] [PubMed] [Google Scholar]
- Amin SR, Baratz KH, McLaren JW, Patel SV, 2014. Corneal abnormalities early in the course of Fuchs’ endothelial dystrophy. Ophthalmology 121, 2325–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang M, Baskaran M, Werkmeister RM, Chua J, Schmidl D, Aranha Dos Santos V, Garhöfer G, Mehta JS, Schmetterer L, 2018. Anterior segment optical coherence tomography. Progress in Retinal and Eye Research 66, 132–156. [DOI] [PubMed] [Google Scholar]
- Arbelaez JG, Price MO, Price FW Jr., 2014. Long-term follow-up and complications of stripping descemet membrane without placement of graft in eyes with Fuchs endothelial dystrophy. Cornea 33, 1295–1299. [DOI] [PubMed] [Google Scholar]
- Arffa RC, 1991. Clinical applications of corneal topographic analysis. Semin Ophthalmol 6, 122–132. [DOI] [PubMed] [Google Scholar]
- Arnheim N, Cortopassi G, 1992. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutation research 275, 157–167. [DOI] [PubMed] [Google Scholar]
- Aslam T, Delcourt C, Holz F, Garcia-Layana A, Leys A, Silva RM, Souied E, 2014. European survey on the opinion and use of micronutrition in age-related macular degeneration: 10 years on from the Age-Related Eye Disease Study. Clin Ophthalmol 8, 2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi B, Ziaei A, Fuchsluger T, Schmedt T, Chen Y, Jurkunas UV, 2011. p53-regulated increase in oxidative-stress--induced apoptosis in Fuchs endothelial corneal dystrophy: a native tissue model. Investigative ophthalmology & visual science 52, 9291–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum MF, Wu WP, White EC, Sengillo JD, Sanfilippo C, Morcos MM, Freund KB, Perry HD, Sarraf D, Tsang SH, 2018. Mitochondrial A3243G mutation results in corneal endothelial polymegathism. Graefes Arch Clin Exp Ophthalmol 256, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran C, Ham L, Verschoor CA, Ong TS, van der Wees J, Melles GR, 2009. Spontaneous corneal clearance despite graft detachment in descemet membrane endothelial keratoplasty. Am J Ophthalmol 148, 227–234 e221. [DOI] [PubMed] [Google Scholar]
- Baratz KH, Tosakulwong N, Ryu E, Brown WL, Branham K, Chen W, Tran KD, Schmid-Kubista KE, Heckenlively JR, Swaroop A, Abecasis G, Bailey KR, Edwards AO, 2010. E2–2 protein and Fuchs’s corneal dystrophy. The New England journal of medicine 363, 1016–1024. [DOI] [PubMed] [Google Scholar]
- Barfort P, Maurice D, 1974. Electrical potential and fluid transport across the corneal endothelium. Exp Eye Res 19, 11–19. [DOI] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, Nieto MA, 2005. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132, 3151–3161. [DOI] [PubMed] [Google Scholar]
- Bartakova A, Kuzmenko O, Alvarez-Delfin K, Kunzevitzky NJ, Goldberg JL, 2018. A Cell Culture Approach to Optimized Human Corneal Endothelial Cell Function. Investigative ophthalmology & visual science 59, 1617–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydoun L, Zygoura V, Hsien S, Birbal RS, Spinozzi D, Lie JT, Ham L, Oellerich S, Melles GRJ, 2018. Clinical feasibility of using multiple grafts from a single donor for Quarter-DMEK. Acta Ophthalmol 96, e656–e658. [DOI] [PubMed] [Google Scholar]
- Bednarz J, Teifel M, Friedl P, Engelmann K, 2000. Immortalization of human corneal endothelial cells using electroporation protocol optimized for human corneal endothelial and human retinal pigment epithelial cells. Acta Ophthalmol Scand 78, 130–136. [DOI] [PubMed] [Google Scholar]
- Benischke AS, Vasanth S, Miyai T, Katikireddy KR, White T, Chen Y, Halilovic A, Price M, Price F Jr., Liton PB, Jurkunas UV, 2017. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci Rep 7, 6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmanson JP, Sheldon TM, Goosey JD, 1999. Fuchs’ endothelial dystrophy: a fresh look at an aging disease. Ophthalmic Physiol Opt 19, 210–222. [DOI] [PubMed] [Google Scholar]
- Bhogal M, Lwin CN, Seah XY, Peh G, Mehta JS, 2017. Allogeneic Descemet’s Membrane Transplantation Enhances Corneal Endothelial Monolayer Formation and Restores Functional Integrity Following Descemet’s Stripping. Investigative ophthalmology & visual science 58, 4249–4260. [DOI] [PubMed] [Google Scholar]
- Bigan G, Puyraveau M, Saleh M, Gain P, Martinache I, Delbosc B, Gauthier AS, 2018. Corneal transplantation trends in France from 2004 to 2015: A 12-year review. Eur J Ophthalmol 28, 535–540. [DOI] [PubMed] [Google Scholar]
- Bigar F, Schimmelpfennig B, Hurzeler R, 1978. Cornea guttata in donor material. Archives of ophthalmology (Chicago, Ill. : 1960) 96, 653–655. [DOI] [PubMed] [Google Scholar]
- Birbal RS, Parker J, Dirisamer M, Janicijevic A, Baydoun L, Dapena I, Melles GRJ, 2018. Descemet Membrane Endothelial Transfer: Ultimate Outcome. Cornea 37, 141–144. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Aggeler J, 1987. Dynamic reciprocity: how do extracellular matrix and hormones direct gene expression? Prog Clin Biol Res 249, 251–262. [PubMed] [Google Scholar]
- Bissell MJ, Hall HG, Parry G, 1982. How does the extracellular matrix direct gene expression? J Theor Biol 99, 31–68. [DOI] [PubMed] [Google Scholar]
- Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P, Sutphin JE, Noble B, Batterbury M, Kielty C, Hackett A, Bonshek R, Ridgway A, McLeod D, Sheffield VC, Stone EM, Schorderet DF, Black GC, 2001. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet 10, 2415–2423. [DOI] [PubMed] [Google Scholar]
- Bitar MS, Liu C, Ziaei A, Chen Y, Schmedt T, Jurkunas UV, 2012. Decline in DJ-1 and decreased nuclear translocation of Nrf2 in Fuchs endothelial corneal dystrophy. Investigative ophthalmology & visual science 53, 5806–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore SJ, 2010. The use of contact lenses in the treatment of persistent epithelial defects. Cont Lens Anterior Eye 33, 239–244. [DOI] [PubMed] [Google Scholar]
- Blank V, 2008. Small Maf proteins in mammalian gene control: mere dimerization partners or dynamic transcriptional regulators? J Mol Biol 376, 913–925. [DOI] [PubMed] [Google Scholar]
- Bleyen I, Saelens IE, van Dooren BT, van Rij G, 2013. Spontaneous corneal clearing after Descemet’s stripping. Ophthalmology 120, 215. [DOI] [PubMed] [Google Scholar]
- Bonanno JA, 2003. Identity and regulation of ion transport mechanisms in the corneal endothelium. Prog Retin Eye Res 22, 69–94. [DOI] [PubMed] [Google Scholar]
- Bonanno JA, 2012. Molecular mechanisms underlying the corneal endothelial pump. Exp Eye Res 95, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin N, Belville C, Chiambaretta F, Sapin V, Blanchon L, 2014. DNA methyl transferases are differentially expressed in the human anterior eye segment. Acta Ophthalmol 92, e366–371. [DOI] [PubMed] [Google Scholar]
- Boonstra F, Claerhout I, Hol F, Smit G, van Collenburg J, Meire F, 2002. Corneal decompensation in a boy with Kearns-Sayre syndrome. Ophthalmic Genet 23, 247–251. [DOI] [PubMed] [Google Scholar]
- Borboli S, Colby K, 2002. Mechanisms of disease: Fuchs’ endothelial dystrophy. Ophthalmol Clin North Am 15, 17–25. [DOI] [PubMed] [Google Scholar]
- Borderie VM, Baudrimont M, Vallee A, Ereau TL, Gray F, Laroche L, 2000. Corneal endothelial cell apoptosis in patients with Fuchs’ dystrophy. Investigative ophthalmology & visual science 41, 2501–2505. [PubMed] [Google Scholar]
- Borkar DS, Veldman P, Colby KA, 2016. Treatment of Fuchs Endothelial Dystrophy by Descemet Stripping Without Endothelial Keratoplasty. Cornea 35, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Bostan C, Theriault M, Forget KJ, Doyon C, Cameron JD, Proulx S, Brunette I, 2016. In Vivo Functionality of a Corneal Endothelium Transplanted by Cell-Injection Therapy in a Feline Model. Investigative ophthalmology & visual science 57, 1620–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne WM, 2003. Biology of the corneal endothelium in health and disease. Eye (Lond) 17, 912–918. [DOI] [PubMed] [Google Scholar]
- Bourne WM, Johnson DH, Campbell RJ, 1982. The ultrastructure of Descemet’s membrane. III. Fuchs’ dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960) 100, 1952–1955. [DOI] [PubMed] [Google Scholar]
- Bourne WM, Kaufman HE, 1976. Specular microscopy of human corneal endothelium in vivo. Am J Ophthalmol 81, 319–323. [DOI] [PubMed] [Google Scholar]
- Bourne WM, Nelson LR, Hodge DO, 1997. Central corneal endothelial cell changes over a ten-year period. Investigative ophthalmology & visual science 38, 779–782. [PubMed] [Google Scholar]
- Breschel TS, McInnis MG, Margolis RL, Sirugo G, Corneliussen B, Simpson SG, McMahon FJ, MacKinnon DF, Xu JF, Pleasant N, Huo Y, Ashworth RG, Grundstrom C, Grundstrom T, Kidd KK, DePaulo JR, Ross CA, 1997. A novel, heritable, expanding CTG repeat in an intron of the SEF2–1 gene on chromosome 18q21.1. Hum Mol Genet 6, 1855–1863. [DOI] [PubMed] [Google Scholar]
- Brooks AM, Grant G, Gillies WE, 1994. The significance of anterior chamber depth in Fuchs’ corneal dystrophy and cornea guttata. Cornea 13, 131–135. [DOI] [PubMed] [Google Scholar]
- Buddi R, Lin B, Atilano SR, Zorapapel NC, Kenney MC, Brown DJ, 2002. Evidence of oxidative stress in human corneal diseases. J Histochem Cytochem 50, 341–351. [DOI] [PubMed] [Google Scholar]
- Burns RR, Bourne WM, Brubaker RF, 1981. Endothelial function in patients with cornea guttata. Investigative ophthalmology & visual science 20, 77–85. [PubMed] [Google Scholar]
- Busin M, Madi S, Santorum P, Scorcia V, Beltz J, 2013. Ultrathin descemet’s stripping automated endothelial keratoplasty with the microkeratome double-pass technique: two-year outcomes. Ophthalmology 120, 1186–1194. [DOI] [PubMed] [Google Scholar]
- Buxton JN, Preston RW, Riechers R, Guilbault N, 1967. Tonography in cornea guttata. A preliminary report. Archives of ophthalmology (Chicago, Ill. : 1960) 77, 602–603. [DOI] [PubMed] [Google Scholar]
- Caccamo AE, Scaltriti M, Caporali A, D’Arca D, Scorcioni F, Candiano G, Mangiola M, Bettuzzi S, 2003. Nuclear translocation of a clusterin isoform is associated with induction of anoikis in SV40-immortalized human prostate epithelial cells. Ann N Y Acad Sci 1010, 514–519. [DOI] [PubMed] [Google Scholar]
- Cadet J, Gentner NE, Rozga B, Paterson MC, 1983. Rapid quantitation of ultraviolet-induced thymine-containing dimers in human cell DNA by reversed-phase high-performance liquid chromatography. Journal of chromatography 280, 99–108. [DOI] [PubMed] [Google Scholar]
- Calvani R, Joseph AM, Adhihetty PJ, Miccheli A, Bossola M, Leeuwenburgh C, Bernabei R, Marzetti E, 2013. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biological chemistry 394, 393–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, Casanova J, 2016. A common framework for EMT and collective cell migration. Development 143, 4291–4300. [DOI] [PubMed] [Google Scholar]
- Cantor LB, R. C, Cioffi GA, 2017. External Disease and Cornea. American Academy of Ophthalmology, San Francisco, CA. [Google Scholar]
- Cavanagh HD, Jester JV, Essepian J, Shields W, Lemp MA, 1990. Confocal microscopy of the living eye. CLAO J 16, 65–73. [PubMed] [Google Scholar]
- Chamberlain W, Lin CC, Austin A, Schubach N, Clover J, McLeod SD, Porco TC, Lietman TM, Rose-Nussbaumer J, 2019. Descemet Endothelial Thickness Comparison Trial: A Randomized Trial Comparing Ultrathin Descemet Stripping Automated Endothelial Keratoplasty with Descemet Membrane Endothelial Keratoplasty. Ophthalmology 126, 19–26. [DOI] [PubMed] [Google Scholar]
- Chan CC, Matteson DM, Li Q, Whitcup SM, Nussenblatt RB, 1997. Apoptosis in patients with posterior uveitis. Archives of ophthalmology (Chicago, Ill. : 1960) 115, 1559–1567. [DOI] [PubMed] [Google Scholar]
- Chan SWS, Yucel Y, Gupta N, 2018. New trends in corneal transplants at the University of Toronto. Can J Ophthalmol 53, 580–587. [DOI] [PubMed] [Google Scholar]
- Chang R, Zhou R, Qi X, Wang J, Wu F, Yang W, Zhang W, Sun T, Li Y, Yu J, 2016. Protective effects of aloin on oxygen and glucose deprivation-induced injury in PC12 cells. Brain Res Bull 121, 75–83. [DOI] [PubMed] [Google Scholar]
- Chang TS, Johns DR, Stark WJ, Drachman DB, Green WR, 1994. Corneal decompensation in mitochondrial ophthalmoplegia plus (Kearns-Sayre) syndrome. A clinicopathologic case report. Cornea 13, 269–273. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, Lin Y, Santillan BA, Yotnda P, Wilson JH, 2015. Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc Natl Acad Sci U S A 112, 3764–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Harris DL, Joyce NC, 1999. TGF-beta2 in aqueous humor suppresses S-phase entry in cultured corneal endothelial cells. Investigative ophthalmology & visual science 40, 2513–2519. [PubMed] [Google Scholar]
- Chen WL, Lin CT, Yao CC, Huang YH, Chou YB, Yin HS, Hu FR, 2006. In-vitro effects of dexamethasone on cellular proliferation, apoptosis, and Na+-K+-ATPase activity of bovine corneal endothelial cells. Ocul Immunol Inflamm 14, 215–223. [DOI] [PubMed] [Google Scholar]
- Cheng AC, Pang CP, Leung AT, Chua JK, Fan DS, Lam DS, 2000. The association between cigarette smoking and ocular diseases. Hong Kong Med J 6, 195–202. [PubMed] [Google Scholar]
- Chikamoto N, Takahashi N, Wakuta M, Fujitsu Y, Nishida T, 2008. Recovery of corneal thickness promoted by glucocorticoid administration after phacoemulsification in eyes affected by Fuchs’ dystrophy. Jpn J Ophthalmol 52, 336–339. [DOI] [PubMed] [Google Scholar]
- Chiou AG, Kaufman SC, Beuerman RW, Ohta T, Soliman H, Kaufman HE, 1999. Confocal microscopy in cornea guttata and Fuchs’ endothelial dystrophy. Br J Ophthalmol 83, 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, Hsiao CH, Chen PY, Ma DH, Chang CJ, Tan HY, 2017. Corneal Backscatters as an Objective Index for Assessing Fuchs’ Endothelial Corneal Dystrophy: A Pilot Study. J Ophthalmol 2017, 8747013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J, 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro Barbosa MM, Barbosa JB Jr., Hirai FE, Hofling-Lima AL, 2010. Effect of cross-linking on corneal thickness in patients with corneal edema. Cornea 29, 613–617. [DOI] [PubMed] [Google Scholar]
- Costagliola C, Romano V, Forbice E, Angi M, Pascotto A, Boccia T, Semeraro F, 2013. Corneal oedema and its medical treatment. Clin Exp Optom 96, 529–535. [DOI] [PubMed] [Google Scholar]
- Courtney DG, Moore JE, Atkinson SD, Maurizi E, Allen EH, Pedrioli DM, McLean WH, Nesbit MA, Moore CB, 2016. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther 23, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross HE, Maumenee AE, Cantolino SJ, 1971. Inheritance of Fuchs’ endothelial dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960) 85, 268–272. [DOI] [PubMed] [Google Scholar]
- Cui Z, Zeng Q, Guo Y, Liu S, Wang P, Xie M, Chen J, 2018. Pathological molecular mechanism of symptomatic late-onset Fuchs endothelial corneal dystrophy by bioinformatic analysis. PLoS One 13, e0197750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen AP, Chou BR, Hall MG, Jany SE, 1984. Ultraviolet-B damages corneal endothelium. American journal of optometry and physiological optics 61, 473–478. [DOI] [PubMed] [Google Scholar]
- Czarny P, Seda A, Wielgorski M, Binczyk E, Markiewicz B, Kasprzak E, Jimenez-Garcia MP, Grabska-Liberek I, Pawlowska E, Blasiak J, Szaflik J, Szaflik JP, 2014. Mutagenesis of mitochondrial DNA in Fuchs endothelial corneal dystrophy. Mutat Res 760, 42–47. [DOI] [PubMed] [Google Scholar]
- Dandona L, Ragu K, Janarthanan M, Naduvilath TJ, Shenoy R, Rao GN, 1997. Indications for penetrating keratoplasty in India. Indian J Ophthalmol 45, 163–168. [PubMed] [Google Scholar]
- Darlington JK, Mannis MJ, Segal WA, 2001. Anterior keratoconus associated with unilateral cornea guttata. Cornea 20, 881–884. [DOI] [PubMed] [Google Scholar]
- Davies E, Jurkunas U, Pineda R 2nd, 2018. Predictive Factors for Corneal Clearance After Descemetorhexis Without Endothelial Keratoplasty. Cornea 37, 137–140. [DOI] [PubMed] [Google Scholar]
- Davies E, Pineda R 2nd, 2019. Corneal Tomography Changes and Refractive Outcomes After Descemet Stripping Without Endothelial Keratoplasty. Cornea 38, 817–819. [DOI] [PubMed] [Google Scholar]
- del Buey MA, Cristobal JA, Ascaso FJ, Lavilla L, Lanchares E, 2009. Biomechanical properties of the cornea in Fuchs’ corneal dystrophy. Investigative ophthalmology & visual science 50, 3199–3202. [DOI] [PubMed] [Google Scholar]
- Descemet J, Demours P, Vincent P, 1770. RÈponse de M. Descemet,… ‡ la lettre de M. Demours,… insÈrÈe dans le “Journal de mÈdecine” du mois de novembre dernier. [Google Scholar]
- Di Mundo R, Recchia G, Parekh M, Ruzza A, Ferrari S, Carbone G, 2017. Sensing inhomogeneous mechanical properties of human corneal Descemet’s membrane with AFM nano-indentation. J Mech Behav Biomed Mater 74, 21–27. [DOI] [PubMed] [Google Scholar]
- Dickman MM, Kruit PJ, Remeijer L, van Rooij J, Van der Lelij A, Wijdh RH, van den Biggelaar FJ, Berendschot TT, Nuijts RM, 2016. A Randomized Multicenter Clinical Trial of Ultrathin Descemet Stripping Automated Endothelial Keratoplasty (DSAEK) versus DSAEK. Ophthalmology 123, 2276–2284. [DOI] [PubMed] [Google Scholar]
- Dirisamer M, Ham L, Dapena I, van Dijk K, Melles GR, 2012a. Descemet membrane endothelial transfer: “free-floating” donor Descemet implantation as a potential alternative to “keratoplasty”. Cornea 31, 194–197. [DOI] [PubMed] [Google Scholar]
- Dirisamer M, Yeh RY, van Dijk K, Ham L, Dapena I, Melles GR, 2012b. Recipient endothelium may relate to corneal clearance in descemet membrane endothelial transfer. Am J Ophthalmol 154, 290–296 e291. [DOI] [PubMed] [Google Scholar]
- Dong PN, Han TN, Aldave AJ, Chau HT, 2016. Indications for and techniques of keratoplasty at Vietnam National Institute of Ophthalmology. Int J Ophthalmol 9, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Aleff RA, Soragni E, Kalari K, Nie J, Tang X, Davila J, Kocher JP, Patel SV, Gottesfeld JM, Baratz KH, Wieben ED, 2015. RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J Biol Chem 290, 5979–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan MJ, Rose-Nussbaumer J, Lin CC, Austin A, Labadzinzki PC, Chamberlain WD, 2019. Corneal Higher-Order Aberrations in Descemet Membrane Endothelial Keratoplasty versus Ultrathin DSAEK in the Descemet Endothelial Thickness Comparison Trial: A Randomized Clinical Trial. Ophthalmology 126, 946–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutescu RM, Panfil C, Schrage N, 2015. Osmolarity of prevalent eye drops, side effects, and therapeutic approaches. Cornea 34, 560–566. [DOI] [PubMed] [Google Scholar]
- Eghrari AO, Gottsch JD, 2010. Fuchs’ corneal dystrophy. Expert Rev Ophthalmol 5, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghrari AO, McGlumphy EJ, Iliff BW, Wang J, Emmert D, Riazuddin SA, Katsanis N, Gottsch JD, 2012. Prevalence and severity of fuchs corneal dystrophy in Tangier Island. Am J Ophthalmol 153, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghrari AO, Mumtaz AA, Garrett B, Rezaei M, Akhavan MS, Riazuddin SA, Gottsch JD, 2017a. Automated Retroillumination Photography Analysis for Objective Assessment of Fuchs Corneal Dystrophy. Cornea 36, 44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghrari AO, Vahedi S, Afshari NA, Riazuddin SA, Gottsch JD, 2017b. CTG18.1 Expansion in TCF4 Among African Americans With Fuchs’ Corneal Dystrophy. Investigative ophthalmology & visual science 58, 6046–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz U, Mireskandari K, Tehrani N, Shen C, Khan MS, Williams S, Ali A, 2017. Corneal Endothelial Cell Density in Children: Normative Data From Birth to 5 Years Old. Am J Ophthalmol 173, 134–138. [DOI] [PubMed] [Google Scholar]
- Elmore S, 2007. Apoptosis: a review of programmed cell death. Toxicologic pathology 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kelliher C, Spitze AR, Speck CL, Eberhart CG, Jun AS, 2010. Unfolded protein response in fuchs endothelial corneal dystrophy: a unifying pathogenic pathway? Am J Ophthalmol 149, 194–202 e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto K, Mimura T, Harris DL, Joyce NC, 2006. Age differences in cyclin-dependent kinase inhibitor expression and rb hyperphosphorylation in human corneal endothelial cells. Investigative ophthalmology & visual science 47, 4330–4340. [DOI] [PubMed] [Google Scholar]
- Evans J, 2008. Antioxidant supplements to prevent or slow down the progression of AMD: a systematic review and meta-analysis. Eye (Lond) 22, 751–760. [DOI] [PubMed] [Google Scholar]
- Fan J, Ren H, Fei E, Jia N, Ying Z, Jiang P, Wu M, Wang G, 2008. Sumoylation is critical for DJ-1 to repress p53 transcriptional activity. FEBS Lett 582, 1151–1156. [DOI] [PubMed] [Google Scholar]
- Fard AM, Morales MJ, Patel SP, 2017. Estrogen receptor beta in human corneal endothelium. Investigative ophthalmology & visual science 58, 1448. [Google Scholar]
- Feil R, Fraga MF, 2012. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 13, 97–109. [DOI] [PubMed] [Google Scholar]
- Fielder E, von Zglinicki T, Jurk D, 2017. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J Alzheimers Dis. [DOI] [PubMed] [Google Scholar]
- Figge MT, Reichert AS, Meyer-Hermann M, Osiewacz HD, 2012. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS computational biology 8, e1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flockerzi E, Maier P, Bohringer D, Reinshagen H, Kruse F, Cursiefen C, Reinhard T, Geerling G, Torun N, Seitz B, all German Keratoplasty Registry, C., 2018. Trends in Corneal Transplantation from 2001 to 2016 in Germany: A Report of the DOG-Section Cornea and its Keratoplasty Registry. Am J Ophthalmol 188, 91–98. [DOI] [PubMed] [Google Scholar]
- Flora A, Garcia JJ, Thaller C, Zoghbi HY, 2007. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A 104, 15382–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foja S, Luther M, Hoffmann K, Rupprecht A, Gruenauer-Kloevekorn C, 2017. CTG18.1 repeat expansion may reduce TCF4 gene expression in corneal endothelial cells of German patients with Fuchs’ dystrophy. Graefes Arch Clin Exp Ophthalmol 255, 1621–1631. [DOI] [PubMed] [Google Scholar]
- Folli F, Corradi D, Fanti P, Davalli A, Paez A, Giaccari A, Perego C, Muscogiuri G, 2011. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Current diabetes reviews 7, 313–324. [DOI] [PubMed] [Google Scholar]
- Friedenwald H, Friedenwald JS, 1925. Epithelial Dystrophy of the Cornea. Br J Ophthalmol 9, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigo AC, Fasolo A, Capuzzo C, Fornea M, Bellucci R, Busin M, Marchini G, Pedrotti E, Ponzin D, Group, C.S., 2015. Corneal transplantation activity over 7 years: changing trends for indications, patient demographics and surgical techniques from the Corneal Transplant Epidemiological Study (CORTES). Transplant Proc 47, 528–535. [DOI] [PubMed] [Google Scholar]
- Fritz M, Grewing V, Maier P, Lapp T, Bohringer D, Reinhard T, Wacker K, 2019. Diurnal Variation in Corneal Edema in Fuchs Endothelial Corneal Dystrophy. Am J Ophthalmol 207, 351–355. [DOI] [PubMed] [Google Scholar]
- Fuchs E, 1910. Dystrophia epithelialis cornea. Graefes Arch Clin Exp Ophthalmol, 478–508. [Google Scholar]
- Fujimoto H, Maeda N, Soma T, Oie Y, Koh S, Tsujikawa M, Nishida K, 2014. Quantitative regional differences in corneal endothelial abnormalities in the central and peripheral zones in Fuchs’ endothelial corneal dystrophy. Investigative ophthalmology & visual science 55, 5090–5098. [DOI] [PubMed] [Google Scholar]
- Gaikwad NW, Rogan EG, Cavalieri EL, 2007. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med 43, 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gain P, Jullienne R, He Z, Aldossary M, Acquart S, Cognasse F, Thuret G, 2016. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol 134, 167–173. [DOI] [PubMed] [Google Scholar]
- Galor A, Lee DJ, 2011. Effects of smoking on ocular health. Curr Opin Ophthalmol 22, 477–482. [DOI] [PubMed] [Google Scholar]
- Garcerant D, Hirnschall N, Toalster N, Zhu M, Wen L, Moloney G, 2019. Descemet’s stripping without endothelial keratoplasty. Curr Opin Ophthalmol 30, 275–285. [DOI] [PubMed] [Google Scholar]
- Gendron SP, Theriault M, Proulx S, Brunette I, Rochette PJ, 2016. Restoration of Mitochondrial Integrity, Telomere Length, and Sensitivity to Oxidation by In Vitro Culture of Fuchs’ Endothelial Corneal Dystrophy Cells . Investigative ophthalmology & visual science 57, 5926–5934. [DOI] [PubMed] [Google Scholar]
- Gerber-Hollbach N, Parker J, Baydoun L, Liarakos VS, Ham L, Dapena I, Melles GR, 2016. Preliminary outcome of hemi-Descemet membrane endothelial keratoplasty for Fuchs endothelial dystrophy. Br J Ophthalmol 100, 1564–1568. [DOI] [PubMed] [Google Scholar]
- Geroski DH, Edelhauser HF, 1984. Quantitation of Na/K ATPase pump sites in the rabbit corneal endothelium. Investigative ophthalmology & visual science 25, 1056–1060. [PubMed] [Google Scholar]
- Geroski DH, Matsuda M, Yee RW, Edelhauser HF, 1985. Pump function of the human corneal endothelium. Effects of age and cornea guttata. Ophthalmology 92, 759–763. [DOI] [PubMed] [Google Scholar]
- Gibbons A, Leung EH, Yoo SH, 2019. Cost-Effectiveness Analysis of Descemet’s Membrane Endothelial Keratoplasty Versus Descemet’s Stripping Endothelial Keratoplasty in the United States. Ophthalmology 126, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano C, Stassi G, De Maria R, Todaro M, Richiusa P, Papoff G, Ruberti G, Bagnasco M, Testi R, Galluzzo A, 1997. Potential involvement of Fas and its ligand in the pathogenesis of Hashimoto’s thyroiditis. Science 275, 960–963. [DOI] [PubMed] [Google Scholar]
- Goar EL, 1933. Dystrophy of the Corneal Endothelium (Cornea Guttata), with Report of a Histologic Examination. Trans Am Ophthalmol Soc 31, 48–59. [PMC free article] [PubMed] [Google Scholar]
- Gogia V, Gupta S, Agarwal T, Pandey V, Tandon R, 2015. Changing pattern of utilization of human donor cornea in India. Indian J Ophthalmol 63, 654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottsch JD, Bowers AL, Margulies EH, Seitzman GD, Kim SW, Saha S, Jun AS, Stark WJ, Liu SH, 2003. Serial analysis of gene expression in the corneal endothelium of Fuchs’ dystrophy. Investigative ophthalmology & visual science 44, 594–599. [DOI] [PubMed] [Google Scholar]
- Gottsch JD, Sundin OH, Liu SH, Jun AS, Broman KW, Stark WJ, Vito EC, Narang AK, Thompson JM, Magovern M, 2005a. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of fuchs corneal dystrophy. Investigative ophthalmology & visual science 46, 1934–1939. [DOI] [PubMed] [Google Scholar]
- Gottsch JD, Sundin OH, Rencs EV, Emmert DG, Stark WJ, Cheng CJ, Schmidt GW, 2006. Analysis and documentation of progression of Fuchs corneal dystrophy with retroillumination photography. Cornea 25, 485–489. [DOI] [PubMed] [Google Scholar]
- Gottsch JD, Zhang C, Sundin OH, Bell WR, Stark WJ, Green WR, 2005b. Fuchs corneal dystrophy: aberrant collagen distribution in an L450W mutant of the COL8A2 gene. Investigative ophthalmology & visual science 46, 4504–4511. [DOI] [PubMed] [Google Scholar]
- Goyer B, Theriault M, Gendron SP, Brunette I, Rochette PJ, Proulx S, 2018. Extracellular Matrix and Integrin Expression Profiles in Fuchs Endothelial Corneal Dystrophy Cells and Tissue Model. Tissue Eng Part A 24, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves B, 1924. Report to the Lang Clinical Research Committee, Royal London Ophthalmic Hospital: A Bilateral Chronic Affection of the Endothelial Face of the Cornea of Elderly Persons with an Account of the Technical and Clinical Principles of Its Slit-Lamp Observation. Br J Ophthalmol 8, 502–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredilla R, 2010. DNA damage and base excision repair in mitochondria and their role in aging. Journal of aging research 2011, 257093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenrod EB, Jones MN, Kaye S, Larkin DF, National Health Service B, Transplant Ocular Tissue Advisory, G., Contributing O, 2014. Center and surgeon effect on outcomes of endothelial keratoplasty versus penetrating keratoplasty in the United Kingdom. Am J Ophthalmol 158, 957–966. [DOI] [PubMed] [Google Scholar]
- Griffith M, Osborne R, Munger R, Xiong X, Doillon CJ, Laycock NL, Hakim M, Song Y, Watsky MA, 1999. Functional human corneal equivalents constructed from cell lines. Science 286, 2169–2172. [DOI] [PubMed] [Google Scholar]
- Groger N, Frohlich H, Maier H, Olbrich A, Kostin S, Braun T, Boettger T, 2010. SLC4A11 prevents osmotic imbalance leading to corneal endothelial dystrophy, deafness, and polyuria. J Biol Chem 285, 14467–14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group AR, Chew EY, Clemons T, SanGiovanni JP, Danis R, Domalpally A, McBee W, Sperduto R, Ferris FL, 2012. The Age-Related Eye Disease Study 2 (AREDS2): study design and baseline characteristics (AREDS2 report number 1). Ophthalmology 119, 2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupcheva CN, Craig JP, Sherwin T, McGhee CN, 2001. Differential diagnosis of corneal oedema assisted by in vivo confocal microscopy. Clin Exp Ophthalmol 29, 133–137. [DOI] [PubMed] [Google Scholar]
- Grutzmacher C, Park S, Zhao Y, Morrison ME, Sheibani N, Sorenson CM, 2013. Aberrant production of extracellular matrix proteins and dysfunction in kidney endothelial cells with a short duration of diabetes. Am J Physiol Renal Physiol 304, F19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha S, Chaurasia S, Ramachandran C, Roy S, 2017. SLC4A11 depletion impairs NRF2 mediated antioxidant signaling and increases reactive oxygen species in human corneal endothelial cells during oxidative stress. Sci Rep 7, 4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Kinderyte R, Jacobs DS, Jurkunas UV, 2017. Elimination of Anterior Corneal Steepening With Descemet Membrane Endothelial Keratoplasty in a Patient With Fuchs Dystrophy and Keratoconus: Implications for IOL Calculation. Cornea 36, 1260–1262. [DOI] [PubMed] [Google Scholar]
- Gupta R, Kumawat BL, Paliwal P, Tandon R, Sharma N, Sen S, Kashyap S, Nag TC, Vajpayee RB, Sharma A, 2015. Association of ZEB1 and TCF4 rs613872 changes with late onset Fuchs endothelial corneal dystrophy in patients from northern India. Mol Vis 21, 1252–1260. [PMC free article] [PubMed] [Google Scholar]
- Hadeyama T, Nakayasu K, Ha NT, Nakamura S, 2002. [Expression of estrogen receptors alpha and beta, androgen receptors and progesterone receptors in human cornea]. Nippon Ganka Gakkai zasshi 106, 557–564. [PubMed] [Google Scholar]
- Halilovic A, Schmedt T, Benischke AS, Hamill C, Chen Y, Santos JH, Jurkunas UV, 2016. Menadione-Induced DNA Damage Leads to Mitochondrial Dysfunction and Fragmentation During Rosette Formation in Fuchs Endothelial Corneal Dystrophy. Antioxidants & redox signaling 24, 1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham L, Dapena I, Moutsouris K, Melles GR, 2011. Persistent corneal edema after descemetorhexis without corneal graft implantation in a case of fuchs endothelial dystrophy. Cornea 30, 248–249. [DOI] [PubMed] [Google Scholar]
- Hamill CE, Schmedt T, Jurkunas U, 2013. Fuchs endothelial cornea dystrophy: a review of the genetics behind disease development. Semin Ophthalmol 28, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond BR Jr., Johnson MA, 2002. The age-related eye disease study (AREDS). Nutrition reviews 60, 283–288. [DOI] [PubMed] [Google Scholar]
- Hamuro J, Toda M, Asada K, Hiraga A, Schlotzer-Schrehardt U, Montoya M, Sotozono C, Ueno M, Kinoshita S, 2016. Cell Homogeneity Indispensable for Regenerative Medicine by Cultured Human Corneal Endothelial Cells. Investigative ophthalmology & visual science 57, 4749–4761. [DOI] [PubMed] [Google Scholar]
- Hara M, Morishige N, Chikama T, Nishida T, 2003. Comparison of confocal biomicroscopy and noncontact specular microscopy for evaluation of the corneal endothelium. Cornea 22, 512–515. [DOI] [PubMed] [Google Scholar]
- Harris JE, 1962. Symposium on the cornea. Introduction: Factors influencing corneal hydration. Invest Ophthalmol 1, 151–157. [PubMed] [Google Scholar]
- Harris JE, Nordquist LT, 1955. The hydration of the cornea. I. The transport of water from the cornea. Am J Ophthalmol 40, 100–110. [DOI] [PubMed] [Google Scholar]
- Hatou S, Yamada M, Mochizuki H, Shiraishi A, Joko T, Nishida T, 2009. The effects of dexamethasone on the Na,K-ATPase activity and pump function of corneal endothelial cells. Curr Eye Res 34, 347–354. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Osawa T, Tohyama K, 2002. Comparative observations on corneas, with special reference to Bowman’s layer and Descemet’s membrane in mammals and amphibians. J Morphol 254, 247–258. [DOI] [PubMed] [Google Scholar]
- Haydari MN, Perron MC, Laprise S, Roy O, Cameron JD, Proulx S, Brunette I, 2012. A short-term in vivo experimental model for Fuchs endothelial corneal dystrophy. Investigative ophthalmology & visual science 53, 6343–6354. [DOI] [PubMed] [Google Scholar]
- Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR, 1996. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci U S A 93, 9776–9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindl LM, Riss S, Bachmann BO, Laaser K, Kruse FE, Cursiefen C, 2011a. Split cornea transplantation for 2 recipients: a new strategy to reduce corneal tissue cost and shortage. Ophthalmology 118, 294–301. [DOI] [PubMed] [Google Scholar]
- Heindl LM, Riss S, Laaser K, Bachmann BO, Kruse FE, Cursiefen C, 2011b. Split cornea transplantation for 2 recipients - review of the first 100 consecutive patients. Am J Ophthalmol 152, 523–532 e522. [DOI] [PubMed] [Google Scholar]
- Hejtmancik JF, Jiao X, Li A, Sergeev YV, Ding X, Sharma AK, Chan CC, Medina I, Edwards AO, 2008. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am J Hum Genet 82, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Quintela E, Mayer F, Dighiero P, Briat B, Savoldelli M, Legeais JM, Renard G, 1998. Confocal microscopy of cystic disorders of the corneal epithelium. Ophthalmology 105, 631–636. [DOI] [PubMed] [Google Scholar]
- Hidayat AA, Cockerham GC, 2006. Epithelial metaplasia of the corneal endothelium in Fuchs endothelial dystrophy. Cornea 25, 956–959. [DOI] [PubMed] [Google Scholar]
- Higa A, Sakai H, Sawaguchi S, Iwase A, Tomidokoro A, Amano S, Araie M, 2011. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: the Kumejima study. Archives of ophthalmology (Chicago, Ill. : 1960) 129, 332–336. [DOI] [PubMed] [Google Scholar]
- Hogan MJ, Wood I, Fine M, 1974. Fuchs’ endothelial dystrophy of the cornea. 29th Sanford Gifford Memorial lecture. Am J Ophthalmol 78, 363–383. [DOI] [PubMed] [Google Scholar]
- Hong J, Shi W, Liu Z, Pineda R, Cui X, Sun X, Xu J, 2015. Limitations of Keratoplasty in China: A Survey Analysis. PLoS One 10, e0132268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfer U, Fukai N, Hopfer H, Wolf G, Joyce N, Li E, Olsen BR, 2005. Targeted disruption of Col8a1 and Col8a2 genes in mice leads to anterior segment abnormalities in the eye. FASEB J 19, 1232–1244. [DOI] [PubMed] [Google Scholar]
- Hu J, Rong Z, Gong X, Zhou Z, Sharma VK, Xing C, Watts JK, Corey DR, Mootha VV, 2018. Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs’ dystrophy. Hum Mol Genet 27, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Blanco G, Mercer RW, Fleming T, Pepose JS, 2003. Human corneal endothelial cell expression of Na+,K+-adenosine triphosphatase isoforms. Archives of ophthalmology (Chicago, Ill. : 1960) 121, 840–845. [DOI] [PubMed] [Google Scholar]
- Huang MJ, Kane S, Dhaliwal DK, 2018. Descemetorhexis Without Endothelial Keratoplasty Versus DMEK for Treatment of Fuchs Endothelial Corneal Dystrophy. Cornea 37, 1479–1483. [DOI] [PubMed] [Google Scholar]
- Hynes RO, 2009. The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T, Wang J, Tao A, Leng T, Kymionis GD, O’Brien TP, Yoo SH, 2010. Intraoperative use of three-dimensional spectral-domain optical coherence tomography. Ophthalmic Surg Lasers Imaging 41, 250–254. [DOI] [PubMed] [Google Scholar]
- Igo RP Jr., Kopplin LJ, Joseph P, Truitt B, Fondran J, Bardenstein D, Aldave AJ, Croasdale CR, Price MO, Rosenwasser M, Lass JH, Iyengar SK, Group, F.G.M.-c.S., 2012. Differing roles for TCF4 and COL8A2 in central corneal thickness and fuchs endothelial corneal dystrophy. PLoS One 7, e46742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imre G, Bogi J, 1979. [Reduced production of aqueous humor in Fuch’s corneal dystrophy]. Klin Monbl Augenheilkd 174, 471–474. [PubMed] [Google Scholar]
- Insler MS, Benefield DW, Ross EV, 1987. Topical hyperosmolar solutions in the reduction of corneal edema. CLAO J 13, 149–151. [PubMed] [Google Scholar]
- Iovieno A, Neri A, Soldani AM, Adani C, Fontana L, 2017. Descemetorhexis Without Graft Placement for the Treatment of Fuchs Endothelial Dystrophy: Preliminary Results and Review of the Literature. Cornea 36, 637–641. [DOI] [PubMed] [Google Scholar]
- Iovino C, Fossarello M, Giannaccare G, Pellegrini M, Braghiroli M, Demarinis G, Napoli PE, 2018. Corneal endothelium features in Fuchs’ Endothelial Corneal Dystrophy: A preliminary 3D anterior segment optical coherence tomography study. PLoS One 13, e0207891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M, 2000. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 275, 16023–16029. [DOI] [PubMed] [Google Scholar]
- Ishino Y, Sano Y, Nakamura T, Connon CJ, Rigby H, Fullwood NJ, Kinoshita S, 2004. Amniotic membrane as a carrier for cultivated human corneal endothelial cell transplantation. Investigative ophthalmology & visual science 45, 800–806. [DOI] [PubMed] [Google Scholar]
- Jalimarada SS, Ogando DG, Bonanno JA, 2014. Loss of ion transporters and increased unfolded protein response in Fuchs’ dystrophy. Mol Vis 20, 1668–1679. [PMC free article] [PubMed] [Google Scholar]
- Jancevski M, Foster CS, 2010. Anterior segment optical coherence tomography. Semin Ophthalmol 25, 317–323. [DOI] [PubMed] [Google Scholar]
- Johns DR, 1995. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. The New England journal of medicine 333, 638–644. [DOI] [PubMed] [Google Scholar]
- Johnson DH, Bourne WM, Campbell RJ, 1982. The ultrastructure of Descemet’s membrane. I. Changes with age in normal corneas. Archives of ophthalmology (Chicago, Ill. : 1960) 100, 1942–1947. [DOI] [PubMed] [Google Scholar]
- Johnson EM Jr., 1994. Possible role of neuronal apoptosis in Alzheimer’s disease. Neurobiology of aging 15 Suppl 2, S187–189. [DOI] [PubMed] [Google Scholar]
- Joyce NC, 2003. Proliferative capacity of the corneal endothelium. Prog Retin Eye Res 22, 359–389. [DOI] [PubMed] [Google Scholar]
- Joyce NC, Harris DL, 2010. Decreasing expression of the G1-phase inhibitors, p21Cip1 and p16INK4a, promotes division of corneal endothelial cells from older donors. Mol Vis 16, 897–906. [PMC free article] [PubMed] [Google Scholar]
- Joyce NC, Harris DL, Zieske JD, 1998. Mitotic inhibition of corneal endothelium in neonatal rats. Investigative ophthalmology & visual science 39, 2572–2583. [PubMed] [Google Scholar]
- Joyce NC, Meklir B, Joyce SJ, Zieske JD, 1996a. Cell cycle protein expression and proliferative status in human corneal cells. Investigative ophthalmology & visual science 37, 645–655. [PubMed] [Google Scholar]
- Joyce NC, Navon SE, Roy S, Zieske JD, 1996b. Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Investigative ophthalmology & visual science 37, 1566–1575. [PubMed] [Google Scholar]
- Joyce NC, Zhu CC, Harris DL, 2009. Relationship among oxidative stress, DNA damage, and proliferative capacity in human corneal endothelium. Investigative ophthalmology & visual science 50, 2116–2122. [DOI] [PubMed] [Google Scholar]
- Jun AS, 2010. One hundred years of Fuchs’ dystrophy. Ophthalmology 117, 859–860 e814. [DOI] [PubMed] [Google Scholar]
- Jun AS, Meng H, Ramanan N, Matthaei M, Chakravarti S, Bonshek R, Black GC, Grebe R, Kimos M, 2012. An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Hum Mol Genet 21, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas U, Azar DT, 2006. Potential complications of ocular surgery in patients with coexistent keratoconus and Fuchs’ endothelial dystrophy. Ophthalmology 113, 2187–2197. [DOI] [PubMed] [Google Scholar]
- Jurkunas UV, 2018. Fuchs Endothelial Corneal Dystrophy Through the Prism of Oxidative Stress. Cornea 37 Suppl 1, S50–S54. [DOI] [PubMed] [Google Scholar]
- Jurkunas UV, Bitar M, Rawe I, 2009. Colocalization of increased transforming growth factor-beta-induced protein (TGFBIp) and Clusterin in Fuchs endothelial corneal dystrophy. Investigative ophthalmology & visual science 50, 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas UV, Bitar MS, Funaki T, Azizi B, 2010. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am J Pathol 177, 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas UV, Bitar MS, Rawe I, Harris DL, Colby K, Joyce NC, 2008a. Increased clusterin expression in Fuchs’ endothelial dystrophy. Investigative ophthalmology & visual science 49, 2946–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas UV, Rawe I, Bitar MS, Zhu C, Harris DL, Colby K, Joyce NC, 2008b. Decreased expression of peroxiredoxins in Fuchs’ endothelial dystrophy. Investigative ophthalmology & visual science 49, 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabosova A, Azar DT, Bannikov GA, Campbell KP, Durbeej M, Ghohestani RF, Jones JC, Kenney MC, Koch M, Ninomiya Y, Patton BL, Paulsson M, Sado Y, Sage EH, Sasaki T, Sorokin LM, Steiner-Champliaud MF, Sun TT, Sundarraj N, Timpl R, Virtanen I, Ljubimov AV, 2007. Compositional differences between infant and adult human corneal basement membranes. Investigative ophthalmology & visual science 48, 4989–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluzny BJ, Szkulmowska A, Szkulmowski M, Bajraszewski T, Kowalczyk A, Wojtkowski M, 2009. Fuchs’ endothelial dystrophy in 830-nm spectral domain optical coherence tomography. Ophthalmic Surg Lasers Imaging 40, 198–200. [DOI] [PubMed] [Google Scholar]
- Kasbekar SA, Gonzalez-Martin JA, Shafiq AE, Chandna A, Willoughby CE, 2013. Corneal endothelial dysfunction in Pearson syndrome. Ophthalmic Genet 34, 55–57. [DOI] [PubMed] [Google Scholar]
- Katikireddy KR, Schmedt T, Price MO, Price FW, Jurkunas UV, 2016. Existence of Neural Crest-Derived Progenitor Cells in Normal and Fuchs Endothelial Dystrophy Corneal Endothelium. Am J Pathol 186, 2736–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katikireddy KR, White TL, Miyajima T, Vasanth S, Raoof D, Chen Y, Price MO, Price FW, Jurkunas UV, 2018. NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy. Free Radic Biol Med 116, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman AR, Nose RM, Pineda R 2nd, 2018. Descemetorhexis Without Endothelial Keratoplasty (DWEK): Proposal for Nomenclature Standardization. Cornea 37, e20–e21. [DOI] [PubMed] [Google Scholar]
- Kaufman SC, Beuerman RW, Kaufman HE, 1993. Diagnosis of advanced Fuchs’ endothelial dystrophy with the confocal microscope. Am J Ophthalmol 116, 652–653. [DOI] [PubMed] [Google Scholar]
- Kelly TK, De Carvalho DD, Jones PA, 2010. Epigenetic modifications as therapeutic targets. Nat Biotechnol 28, 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR, 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer 26, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuc E, Bainer R, Wolf M, Clay SM, Weisenberger DJ, Kemmer J, Weaver VM, Hwang DG, Chan MF, 2017. Comprehensive characterization of DNA methylation changes in Fuchs endothelial corneal dystrophy. PLoS One 12, e0175112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EC, Meng H, Jun AS, 2013. Lithium treatment increases endothelial cell survival and autophagy in a mouse model of Fuchs endothelial corneal dystrophy. Br J Ophthalmol 97, 1068–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EC, Meng H, Jun AS, 2014. N-Acetylcysteine increases corneal endothelial cell survival in a mouse model of Fuchs endothelial corneal dystrophy. Exp Eye Res 127, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EC, Toyono T, Berlinicke CA, Zack DJ, Jurkunas U, Usui T, Jun AS, 2017. Screening and Characterization of Drugs That Protect Corneal Endothelial Cells Against Unfolded Protein Response and Oxidative Stress. Investigative ophthalmology & visual science 58, 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Medsinge A, Chauhan B, Wiest C, Scanga H, Monaghan R, Moore WH, Nischal KK, 2016. Coenzyme Q10 in the Treatment of Corneal Edema in Kearns-Sayre: Is There an Application in Fuchs Endothelial Corneal Dystrophy? Cornea 35, 1250–1254. [DOI] [PubMed] [Google Scholar]
- Kim JH, Ko JM, Tchah H, 2015. Fuchs Endothelial Corneal Dystrophy in a Heterozygous Carrier of Congenital Hereditary Endothelial Dystrophy Type 2 with a Novel Mutation in SLC4A11. Ophthalmic Genet 36, 284–286. [DOI] [PubMed] [Google Scholar]
- Kimoto M, Shima N, Yamaguchi M, Amano S, Yamagami S, 2012. Role of hepatocyte growth factor in promoting the growth of human corneal endothelial cells stimulated by L-ascorbic acid 2-phosphate. Investigative ophthalmology & visual science 53, 7583–7589. [DOI] [PubMed] [Google Scholar]
- Kinoshita S, Koizumi N, Ueno M, Okumura N, Imai K, Tanaka H, Yamamoto Y, Nakamura T, Inatomi T, Bush J, Toda M, Hagiya M, Yokota I, Teramukai S, Sotozono C, Hamuro J, 2018. Injection of Cultured Cells with a ROCK Inhibitor for Bullous Keratopathy. The New England journal of medicine 378, 995–1003. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Kojima M, Sasaki H, Shui YB, Chew SJ, Cheng HM, Ono M, Morikawa Y, Sasaki K, 2002. Prevalence of primary cornea guttata and morphology of corneal endothelium in aging Japanese and Singaporean subjects. Ophthalmic Res 34, 135–138. [DOI] [PubMed] [Google Scholar]
- Kitazawa K, Hikichi T, Nakamura T, Sotozono C, Kinoshita S, Masui S, 2017. PAX6 regulates human corneal epithelium cell identity. Exp Eye Res 154, 30–38. [DOI] [PubMed] [Google Scholar]
- Knapp A, 1911. Dystrophia Epithelialis Corneae (Fuchs), with the Report of a Case. Trans Am Ophthalmol Soc 12, 745–747. [PMC free article] [PubMed] [Google Scholar]
- Knecht PB, Kaufmann C, Menke MN, Watson SL, Bosch MM, 2010. Use of intraoperative fourier-domain anterior segment optical coherence tomography during descemet stripping endothelial keratoplasty. Am J Ophthalmol 150, 360–365 e362. [DOI] [PubMed] [Google Scholar]
- Kniestedt C, Taralczak M, Thiel MA, Stuermer J, Baumer A, Gloor BP, 2006. A novel PITX2 mutation and a polymorphism in a 5-generation family with Axenfeld-Rieger anomaly and coexisting Fuchs’ endothelial dystrophy. Ophthalmology 113, 1791 e1791–1798. [DOI] [PubMed] [Google Scholar]
- Kobashi H, Kamiya K, Shimizu K, 2018. Factors Influencing Visual Acuity in Fuchs’ Endothelial Corneal Dystrophy. Optom Vis Sci 95, 21–26. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Fujiki K, Murakami A, Kato T, Chen LZ, Onoe H, Nakayasu K, Sakurai M, Takahashi M, Sugiyama K, Kanai A, 2004. Analysis of COL8A2 gene mutation in Japanese patients with Fuchs’ endothelial dystrophy and posterior polymorphous dystrophy. Jpn J Ophthalmol 48, 195–198. [DOI] [PubMed] [Google Scholar]
- Kocaba V, 2018. Tissue engineering pour la reconstruction cornéenne.
- Kocaba V, Katikireddy KR, Gipson I, Price MO, Price FW, Jurkunas UV, 2018. Association of the Gutta-Induced Microenvironment With Corneal Endothelial Cell Behavior and Demise in Fuchs Endothelial Corneal Dystrophy. JAMA Ophthalmol 136, 886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch-Brandt C, Morgans C, 1996. Clusterin: a role in cell survival in the face of apoptosis? Progress in molecular and subcellular biology 16, 130–149. [DOI] [PubMed] [Google Scholar]
- Koenig SB, 2013. Long-term corneal clarity after spontaneous repair of an iatrogenic descemetorhexis in a patient with Fuchs dystrophy. Cornea 32, 886–888. [DOI] [PubMed] [Google Scholar]
- Koenig SB, 2015. Planned Descemetorhexis Without Endothelial Keratoplasty in Eyes With Fuchs Corneal Endothelial Dystrophy. Cornea 34, 1149–1151. [DOI] [PubMed] [Google Scholar]
- Koizumi N, Okumura N, Kinoshita S, 2012. Development of new therapeutic modalities for corneal endothelial disease focused on the proliferation of corneal endothelial cells using animal models. Exp Eye Res 95, 60–67. [DOI] [PubMed] [Google Scholar]
- Koizumi N, Okumura N, Ueno M, Kinoshita S, 2014. New therapeutic modality for corneal endothelial disease using Rho-associated kinase inhibitor eye drops. Cornea 33 Suppl 11, S25–31. [DOI] [PubMed] [Google Scholar]
- Koizumi N, Okumura N, Ueno M, Nakagawa H, Hamuro J, Kinoshita S, 2013. Rho-associated kinase inhibitor eye drop treatment as a possible medical treatment for Fuchs corneal dystrophy. Cornea 32, 1167–1170. [DOI] [PubMed] [Google Scholar]
- Koizumi N, Sakamoto Y, Okumura N, Okahara N, Tsuchiya H, Torii R, Cooper LJ, Ban Y, Tanioka H, Kinoshita S, 2007. Cultivated corneal endothelial cell sheet transplantation in a primate model. Investigative ophthalmology & visual science 48, 4519–4526. [DOI] [PubMed] [Google Scholar]
- Konomi K, Zhu C, Harris D, Joyce NC, 2005. Comparison of the proliferative capacity of human corneal endothelial cells from the central and peripheral areas. Investigative ophthalmology & visual science 46, 4086–4091. [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N, 2014. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. [DOI] [PubMed] [Google Scholar]
- Krachmer JH, Purcell JJ Jr., Young CW, Bucher KD, 1978. Corneal endothelial dystrophy. A study of 64 families. Archives of ophthalmology (Chicago, Ill. : 1960) 96, 2036–2039. [DOI] [PubMed] [Google Scholar]
- Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA, Mian S, Nairus T, Elner V, Schteingart MT, Downs CA, Kijek TG, Johnson JM, Trager EH, Rozsa FW, Mandal MN, Epstein MP, Vollrath D, Ayyagari R, Boehnke M, Richards JE, 2005. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet 77, 694–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuot A, Hewitt AW, Griggs K, Klebe S, Mills R, Jhanji V, Craig JE, Sharma S, Burdon KP, 2012. Association of TCF4 and CLU polymorphisms with Fuchs’ endothelial dystrophy and implication of CLU and TGFBI proteins in the disease process. Eur J Hum Genet 20, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuot A, Hewitt AW, Snibson GR, Souzeau E, Mills R, Craig JE, Burdon KP, Sharma S, 2017. TGC repeat expansion in the TCF4 gene increases the risk of Fuchs’ endothelial corneal dystrophy in Australian cases. PLoS One 12, e0183719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurji KH, Cheung AY, Eslani M, Rolfes EJ, Chachare DY, Auteri NJ, Nordlund ML, Holland EJ, 2018. Comparison of Visual Acuity Outcomes Between Nanothin Descemet Stripping Automated Endothelial Keratoplasty and Descemet Membrane Endothelial Keratoplasty. Cornea 37, 1226–1231. [DOI] [PubMed] [Google Scholar]
- Laing RA, Leibowitz HM, Oak SS, Chang R, Berrospi AR, Theodore J, 1981. Endothelial mosaic in Fuchs’ dystrophy. A qualitative evaluation with the specular microscope. Archives of ophthalmology (Chicago, Ill. : 1960) 99, 80–83. [DOI] [PubMed] [Google Scholar]
- Laing RA, Sandstrom MM, Leibowitz HM, 1975. In vivo photomicrography of the corneal endothelium. Archives of ophthalmology (Chicago, Ill. : 1960) 93, 143–145. [DOI] [PubMed] [Google Scholar]
- Lam FC, Baydoun L, Dirisamer M, Lie J, Dapena I, Melles GR, 2014. Hemi-Descemet membrane endothelial keratoplasty transplantation: a potential method for increasing the pool of endothelial graft tissue. JAMA Ophthalmol 132, 1469–1473. [DOI] [PubMed] [Google Scholar]
- Lam FC, Baydoun L, Satue M, Dirisamer M, Ham L, Melles GR, 2015. One year outcome of hemi-Descemet membrane endothelial keratoplasty. Graefes Arch Clin Exp Ophthalmol 253, 1955–1958. [DOI] [PubMed] [Google Scholar]
- Larsson LI, Bourne WM, Pach JM, Brubaker RF, 1996. Structure and function of the corneal endothelium in diabetes mellitus type I and type II. Archives of ophthalmology (Chicago, Ill. : 1960) 114, 9–14. [DOI] [PubMed] [Google Scholar]
- Lass JH, Spurney RV, Dutt RM, Andersson H, Kochar H, Rodman HM, Stern RC, Doershuk CF, 1985. A morphologic and fluorophotometric analysis of the corneal endothelium in type I diabetes mellitus and cystic fibrosis. Am J Ophthalmol 100, 783–788. [DOI] [PubMed] [Google Scholar]
- Last JA, Liliensiek SJ, Nealey PF, Murphy CJ, 2009. Determining the mechanical properties of human corneal basement membranes with atomic force microscopy. J Struct Biol 167, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LC, Ma L, Young AL, Rong SS, Jhanji V, Brelen ME, Pang CP, Chen LJ, 2014. Association of common variants in TCF4 and PTPRG with Fuchs’ corneal dystrophy: a systematic review and meta-analysis. PLoS One 9, e109142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le R, Yucel N, Khattak S, Yucel YH, Prud’homme GJ, Gupta N, 2017. Current indications and surgical approaches to corneal transplants at the University of Toronto: A clinical-pathological study. Can J Ophthalmol 52, 74–79. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, Macdonald B, Kalluri R, 2007. Structure and function of basement membranes. Exp Biol Med (Maywood) 232, 1121–1129. [DOI] [PubMed] [Google Scholar]
- Lee JS, Oum BS, Choi HY, Lee JE, Cho BM, 2006. Differences in corneal thickness and corneal endothelium related to duration in diabetes. Eye (Lond) 20, 315–318. [DOI] [PubMed] [Google Scholar]
- LeMasters KE, Blech-Hermoni Y, Stillwagon SJ, Vajda NA, Ladd AN, 2012. Loss of muscleblind-like 1 promotes invasive mesenchyme formation in endocardial cushions by stimulating autocrine TGFbeta3. BMC Dev Biol 12, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard BC, Jalilian I, Raghunathan VK, Wang W, Jun AS, Murphy CJ, Thomasy SM, 2019. Biomechanical changes to Descemet’s membrane precede endothelial cell loss in an early-onset murine model of Fuchs endothelial corneal dystrophy. Exp Eye Res 180, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt JM, Lloyd RI, 1952. Congenital and familial endothelial defects. Am J Ophthalmol 35, 342–349. [DOI] [PubMed] [Google Scholar]
- Levy SG, Moss J, Sawada H, Dopping-Hepenstal PJ, McCartney AC, 1996. The composition of wide-spaced collagen in normal and diseased Descemet’s membrane. Curr Eye Res 15, 45–52. [DOI] [PubMed] [Google Scholar]
- Li D, Peng X, Sun H, 2015. Association of TCF4 polymorphisms and Fuchs’ endothelial dystrophy: a meta-analysis. BMC Ophthalmol 15, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sun B, Matteson DM, O’Brien TP, Chan CC, 1999. Cytokines and apoptotic molecules in experimental melanin-protein induced uveitis (EMIU) and experimental autoimmune uveoretinitis (EAU). Autoimmunity 30, 171–182. [DOI] [PubMed] [Google Scholar]
- Li QJ, Ashraf MF, Shen DF, Green WR, Stark WJ, Chan CC, O’Brien TP, 2001. The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Archives of ophthalmology (Chicago, Ill. : 1960) 119, 1597–1604. [DOI] [PubMed] [Google Scholar]
- Li S, Hundal KS, Chen X, Choi M, Ogando DG, Obukhov AG, Bonanno JA, 2019. R125H, W240S, C386R, and V507I SLC4A11 mutations associated with corneal endothelial dystrophy affect the transporter function but not trafficking in PS120 cells. Exp Eye Res 180, 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kim E, Bonanno JA, 2016. Fluid transport by the cornea endothelium is dependent on buffering lactic acid efflux. Am J Physiol Cell Physiol 311, C116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YJ, Minear MA, Qin X, Rimmler J, Hauser MA, Allingham RR, Igo RP, Lass JH, Iyengar SK, Klintworth GK, Afshari NA, Gregory SG, Consortium FG, 2014. Mitochondrial polymorphism A10398G and Haplogroup I are associated with Fuchs’ endothelial corneal dystrophy. Investigative ophthalmology & visual science 55, 4577–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie JT, Lam FC, Groeneveld-van Beek EA, van der Wees J, Melles GR, 2016. Graft preparation for hemi-Descemet membrane endothelial keratoplasty (hemi-DMEK). Br J Ophthalmol 100, 420–424. [DOI] [PubMed] [Google Scholar]
- Lipman RM, Rubenstein JB, Torczynski E, 1990. Keratoconus and Fuchs’ corneal endothelial dystrophy in a patient and her family. Archives of ophthalmology (Chicago, Ill. : 1960) 108, 993–994. [DOI] [PubMed] [Google Scholar]
- Liskova P, Prescott Q, Bhattacharya SS, Tuft SJ, 2007. British family with early-onset Fuchs’ endothelial corneal dystrophy associated with p.L450W mutation in the COL8A2 gene. Br J Ophthalmol 91, 1717–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen Y, Kochevar IE, Jurkunas UV, 2014a. Decreased DJ-1 leads to impaired Nrf2-regulated antioxidant defense and increased UV-A-induced apoptosis in corneal endothelial cells. Investigative ophthalmology & visual science 55, 5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Miyajima T, Melangath G, Miyai T, Vasanth S, Deshpande N, Kumar V, Ong Tone S, Gupta R, Zhu S, Vojnovic D, Chen Y, Rogan EG, Mondal B, Zahid M, Jurkunas UV, 2020. Ultraviolet A light induces DNA damage and estrogen-DNA adducts in Fuchs endothelial corneal dystrophy causing females to be more affected. Proc Natl Acad Sci U S A 117, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Vojnovic D, Kochevar IE, Jurkunas UV, 2016a. UV-A Irradiation Activates Nrf2-Regulated Antioxidant Defense and Induces p53/Caspase3-Dependent Apoptosis in Corneal Endothelial Cells. Investigative ophthalmology & visual science 57, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Miyai T, Vojnovic D, Kochevar IE, Jurkunas UV, 2016b. UV-A Irradiation Induced Mouse Model of Fuchs Endothelial Corneal Dystrophy. Investigative ophthalmology & visual science 57. [Google Scholar]
- Liu CL, Miyajima T, Vasanth S, Miyai T, Farinelli B, Jurkunas UV, 2018. N-Acetylcysteine Alleviates Progression of Fuchs Endothelial Corneal Dystrophy in a UVA Irradiation-Induced Mouse Model. Investigative ophthalmology & visual science 59, 1356–1356. [Google Scholar]
- Liu Y, Peng X, Tan J, Darling DS, Kaplan HJ, Dean DC, 2008. Zeb1 mutant mice as a model of posterior corneal dystrophy. Investigative ophthalmology & visual science 49, 1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Teo EP, Adnan KB, Yam GH, Peh GS, Tan DT, Mehta JS, 2014b. Endothelial approach ultrathin corneal grafts prepared by femtosecond laser for descemet stripping endothelial keratoplasty. Investigative ophthalmology & visual science 55, 8393–8401. [DOI] [PubMed] [Google Scholar]
- Ljubimov AV, Burgeson RE, Butkowski RJ, Michael AF, Sun TT, Kenney MC, 1995. Human corneal basement membrane heterogeneity: topographical differences in the expression of type IV collagen and laminin isoforms. Lab Invest 72, 461–473. [PubMed] [Google Scholar]
- Loewenstein A, Hourvitz D, Goldstein M, Ashkenazi I, Avni I, Lazar M, 1994. Association of fuchs’ corneal endothelial dystrophy with angle-closure glaucoma. J Glaucoma 3, 201–205. [PubMed] [Google Scholar]
- Lois N, Abdelkader E, Reglitz K, Garden C, Ayres JG, 2008. Environmental tobacco smoke exposure and eye disease. Br J Ophthalmol 92, 1304–1310. [DOI] [PubMed] [Google Scholar]
- Lopes BT, Eliasy A, Ambrosio R, 2019. Artificial Intelligence in Corneal Diagnosis: Where Are we? Current Ophthalmology Reports 7, 204–211. [Google Scholar]
- Lopez IA, Rosenblatt MI, Kim C, Galbraith GC, Jones SM, Kao L, Newman D, Liu W, Yeh S, Pushkin A, Abuladze N, Kurtz I, 2009. Slc4a11 gene disruption in mice: cellular targets of sensorineuronal abnormalities. J Biol Chem 284, 26882–26896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE, 1967. Central cornea guttata. Incidence in the general population. Am J Ophthalmol 64, 1155–1158. [PubMed] [Google Scholar]
- Louttit MD, Kopplin LJ, Igo RP Jr., Fondran JR, Tagliaferri A, Bardenstein D, Aldave AJ, Croasdale CR, Price MO, Rosenwasser GO, Lass JH, Iyengar SK, Group FGM-CS, 2012. A multicenter study to map genes for Fuchs endothelial corneal dystrophy: baseline characteristics and heritability. Cornea 31, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Takai K, Weaver VM, Werb Z, 2011. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XY, Dai W, Chee ML, Tao Y, Chua J, Tan NYQ, Tham YC, Aung T, Wong TY, Cheng CY, 2019. Association of Diabetes With Central Corneal Thickness Among a Multiethnic Asian Population. JAMA network open 2, e186647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magovern M, Beauchamp GR, McTigue JW, Fine BS, Baumiller RC, 1979. Inheritance of Fuchs’ combined dystrophy. Ophthalmology 86, 1897–1923. [DOI] [PubMed] [Google Scholar]
- Mahr MA, Baratz KH, Hodge DO, Erie JC, 2016. Racial/Ethnic Differences in Rates of Penetrating or Endothelial Keratoplasty for Fuchs Endothelial Corneal Dystrophy Among US Medicare Beneficiaries. JAMA Ophthalmol 134, 1178–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi G, Zollino M, 2015. Pitt-Hopkins Syndrome and Differential Diagnosis: A Molecular and Clinical Challenge. J Pediatr Genet 4, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marisi A, Aquavella JV, 1975. Hypertonic saline solution in corneal edema. Ann Ophthalmol 7, 229–233. [PubMed] [Google Scholar]
- Marques RE, Guerra PS, Sousa DC, Goncalves AI, Quintas AM, Rodrigues W, 2019. DMEK versus DSAEK for Fuchs’ endothelial dystrophy: A meta-analysis. Eur J Ophthalmol 29, 15–22. [DOI] [PubMed] [Google Scholar]
- Matthaei M, Elsner E, Caramoy A, Adler W, Siebelmann S, Schaub F, Skevas C, Liakopoulos S, Bachmann B, Cursiefen C, Heindl LM, 2018. Fuchs endothelial corneal dystrophy and macular drusen: evidence for coincidence? Eye (Lond) 32, 840–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaei M, Hu J, Kallay L, Eberhart CG, Cursiefen C, Qian J, Lackner EM, Jun AS, 2014. Endothelial cell microRNA expression in human late-onset Fuchs’ dystrophy. Investigative ophthalmology & visual science 55, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaei M, Meng H, Meeker AK, Eberhart CG, Jun AS, 2012. Endothelial Cdkn1a (p21) overexpression and accelerated senescence in a mouse model of Fuchs endothelial corneal dystrophy. Investigative ophthalmology & visual science 53, 6718–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DM, 1957. The structure and transparency of the cornea. J Physiol 136, 263–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DM, 1968. Cellular membrane activity in the corneal endothelium of the intact eye. Experientia 24, 1094–1095. [DOI] [PubMed] [Google Scholar]
- Maurice DM, 1972. The location of the fluid pump in the cornea. J Physiol 221, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DM, 1974. A scanning slit optical microscope. Invest Ophthalmol 13, 1033–1037. [PubMed] [Google Scholar]
- McCarey BE, Edelhauser HF, Lynn MJ, 2008. Review of corneal endothelial specular microscopy for FDA clinical trials of refractive procedures, surgical devices, and new intraocular drugs and solutions. Cornea 27, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney MD, Wood TO, McLaughlin BJ, 1989. Moderate Fuchs’ endothelial dystrophy ATPase pump site density. Investigative ophthalmology & visual science 30, 1560–1564. [PubMed] [Google Scholar]
- McLaren JW, Bachman LA, Kane KM, Patel SV, 2014. Objective assessment of the corneal endothelium in Fuchs’ endothelial dystrophy. Invest Ophthalmol Vis Sci 55, 1184–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF, 1993. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Annals of neurology 34, 609–616. [DOI] [PubMed] [Google Scholar]
- Meekins LC, Rosado-Adames N, Maddala R, Zhao JJ, Rao PV, Afshari NA, 2016. Corneal Endothelial Cell Migration and Proliferation Enhanced by Rho Kinase (ROCK) Inhibitors in In Vitro and In Vivo Models. Investigative ophthalmology & visual science 57, 6731–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta JS, Vithana EN, Tan DT, Yong VH, Yam GH, Law RW, Chong WG, Pang CP, Aung T, 2008. Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Investigative ophthalmology & visual science 49, 184–188. [DOI] [PubMed] [Google Scholar]
- Melles GR, Eggink FA, Lander F, Pels E, Rietveld FJ, Beekhuis WH, Binder PS, 1998. A surgical technique for posterior lamellar keratoplasty. Cornea 17, 618–626. [DOI] [PubMed] [Google Scholar]
- Meng H, Matthaei M, Ramanan N, Grebe R, Chakravarti S, Speck CL, Kimos M, Vij N, Eberhart CG, Jun AS, 2013. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Investigative ophthalmology & visual science 54, 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura T, Joyce NC, 2006. Replication competence and senescence in central and peripheral human corneal endothelium. Investigative ophthalmology & visual science 47, 1387–1396. [DOI] [PubMed] [Google Scholar]
- Mimura T, Shimomura N, Usui T, Noda Y, Kaji Y, Yamgami S, Amano S, Miyata K, Araie M, 2003. Magnetic attraction of iron-endocytosed corneal endothelial cells to Descemet’s membrane. Exp Eye Res 76, 745–751. [DOI] [PubMed] [Google Scholar]
- Mimura T, Yamagami S, Yokoo S, Usui T, Tanaka K, Hattori S, Irie S, Miyata K, Araie M, Amano S, 2004. Cultured human corneal endothelial cell transplantation with a collagen sheet in a rabbit model. Investigative ophthalmology & visual science 45, 2992–2997. [DOI] [PubMed] [Google Scholar]
- Minear MA, Li YJ, Rimmler J, Balajonda E, Watson S, Allingham RR, Hauser MA, Klintworth GK, Afshari NA, Gregory SG, 2013. Genetic screen of African Americans with Fuchs endothelial corneal dystrophy. Mol Vis 19, 2508–2516. [PMC free article] [PubMed] [Google Scholar]
- Miron A, Spinozzi D, Bruinsma M, Lie JT, Birbal RS, Baydoun L, Oellerich S, Melles GRJ, 2018. Asymmetrical endothelial cell migration from in vitro Quarter-Descemet membrane endothelial keratoplasty grafts. Acta Ophthalmol 96, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima S, 1982. Clinical investigations on the corneal endothelium-XXXVIII Edward Jackson Memorial Lecture. Am J Ophthalmol 93, 1–29. [DOI] [PubMed] [Google Scholar]
- Miyai T, 2018. Fuchs Endothelial Corneal Dystrophy and Mitochondria. Cornea 37 Suppl 1, S74–S77. [DOI] [PubMed] [Google Scholar]
- Miyai T, Vasanth S, Melangath G, Deshpande N, Kumar V, Benischke AS, Chen Y, Price MO, Price FW Jr., Jurkunas UV, 2019. Activation of PINK1-Parkin-Mediated Mitophagy Degrades Mitochondrial Quality Control Proteins in Fuchs Endothelial Corneal Dystrophy. Am J Pathol 189, 2061–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima T, Melangath G, Zhu S, Deshpande N, Vasanth S, Mondal B, Kumar V, Chen Y, Price MO, Price FW Jr., Rogan EG, Zahid M, Jurkunas UV, 2020. Loss of NQO1 generates genotoxic estrogen-DNA adducts in Fuchs Endothelial Corneal Dystrophy. Free Radic Biol Med 147, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima T, Vasanth S, Melangath G, Deshpande N, Chen YM, Zhu S, Zahid M, Rogan E, Price M, Price FW, Jurkunas UV, 2019. NQO1 downregulation generates genotoxic DNA adducts in in vitro FECD model. Investigative ophthalmology & visual science 60, 2177–2177. [Google Scholar]
- Miyazono K, 2009. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci 85, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki H, Goto K, Mori H, Mizuno Y, 1996. Histochemical detection of apoptosis in Parkinson’s disease. Journal of the neurological sciences 137, 120–123. [DOI] [PubMed] [Google Scholar]
- Mohamed A, Chaurasia S, Murthy SI, Ramappa M, Vaddavalli PK, Taneja M, Garg P, Chinta S, Basu S, Rathi VM, Sangwan VS, 2014. Endothelial Keratoplasty: A Review of Indications at a Tertiary Eye Care Centre in South India. Asia Pac J Ophthalmol (Phila) 3, 207–210. [DOI] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW, 1994. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 91, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok JW, Kim HS, Joo CK, 2009. Q455V mutation in COL8A2 is associated with Fuchs’ corneal dystrophy in Korean patients. Eye (Lond) 23, 895–903. [DOI] [PubMed] [Google Scholar]
- Moloney G, Petsoglou C, Ball M, Kerdraon Y, Hollhumer R, Spiteri N, Beheregaray S, Hampson J, D’Souza M, Devasahayam RN, 2017. Descemetorhexis Without Grafting for Fuchs Endothelial Dystrophy-Supplementation With Topical Ripasudil. Cornea 36, 642–648. [DOI] [PubMed] [Google Scholar]
- Moore CBT, Christie KA, Marshall J, Nesbit MA, 2018. Personalised genome editing - The future for corneal dystrophies. Prog Retin Eye Res 65, 147–165. [DOI] [PubMed] [Google Scholar]
- Mootha VV, Gong X, Ku HC, Xing C, 2014. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs’ endothelial corneal dystrophy. Investigative ophthalmology & visual science 55, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VV, Hansen B, Rong Z, Mammen PP, Zhou Z, Xing C, Gong X, 2017. Fuchs’ Endothelial Corneal Dystrophy and RNA Foci in Patients With Myotonic Dystrophy. Investigative ophthalmology & visual science 58, 4579–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VV, Hussain I, Cunnusamy K, Graham E, Gong X, Neelam S, Xing C, Kittler R, Petroll WM, 2015. TCF4 Triplet Repeat Expansion and Nuclear RNA Foci in Fuchs’ Endothelial Corneal Dystrophy. Investigative ophthalmology & visual science 56, 2003–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty-Craige SE, Adkison J, Lynn M, Gensler G, Bressler S, Jones DP, Sternberg P Jr., 2005. Antioxidant supplements prevent oxidation of cysteine/cystine redox in patients with age-related macular degeneration. Am J Ophthalmol 140, 1020–1026. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y, 2002. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277, 34287–34294. [DOI] [PubMed] [Google Scholar]
- Mortelmans L, 1952. [Familial form of Fuchs’ corneal dystrophy]. Ophthalmologica 123, 88–99. [DOI] [PubMed] [Google Scholar]
- Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M, 2002. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 294, 1–12. [DOI] [PubMed] [Google Scholar]
- Muller TM, Lavy I, Baydoun L, Lie JT, Dapena I, Melles GR, 2017. Case Report of Quarter-Descemet Membrane Endothelial Keratoplasty for Fuchs Endothelial Dystrophy. Cornea 36, 104–107. [DOI] [PubMed] [Google Scholar]
- Murphy C, Alvarado J, Juster R, 1984. Prenatal and postnatal growth of the human Descemet’s membrane. Investigative ophthalmology & visual science 25, 1402–1415. [PubMed] [Google Scholar]
- Mustonen RK, McDonald MB, Srivannaboon S, Tan AL, Doubrava MW, Kim CK, 1998a. In vivo confocal microscopy of Fuchs’ endothelial dystrophy. Cornea 17, 493–503. [DOI] [PubMed] [Google Scholar]
- Mustonen RK, McDonald MB, Srivannaboon S, Tan AL, Doubrava MW, Kim CK, 1998b. Normal human corneal cell populations evaluated by in vivo scanning slit confocal microscopy. Cornea 17, 485–492. [DOI] [PubMed] [Google Scholar]
- Nagaki Y, Hayasaka S, Kitagawa K, Yamamoto S, 1996. Primary cornea guttata in Japanese patients with cataract: specular microscopic observations. Jpn J Ophthalmol 40, 520–525. [PubMed] [Google Scholar]
- Nagarsheth M, Singh A, Schmotzer B, Babineau DC, Sugar J, Lee WB, Iyengar SK, Lass JH, Fuchs’ Genetics Multi-Center Study, G., 2012. Relationship Between Fuchs Endothelial Corneal Dystrophy Severity and Glaucoma and/or Ocular Hypertension. Arch Ophthalmol 130, 1384–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Koizumi N, Okumura N, Suganami H, Kinoshita S, 2015. Morphological Changes of Human Corneal Endothelial Cells after Rho-Associated Kinase Inhibitor Eye Drop (Ripasudil) Administration: A Prospective Open-Label Clinical Study. PLoS One 10, e0136802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M, Okumura N, Nakagawa H, Koizumi N, Ikeda Y, Ueno M, Yoshii K, Adachi H, Aleff RA, Butz ML, Highsmith WE, Tashiro K, Wieben ED, Kinoshita S, Baratz KH, 2015. Trinucleotide Repeat Expansion in the TCF4 Gene in Fuchs’ Endothelial Corneal Dystrophy in Japanese. Investigative ophthalmology & visual science 56, 4865–4869. [DOI] [PubMed] [Google Scholar]
- Nakashima Y, Yoshitomi F, Oshika T, 2007. Clinical evaluation of cornea pseudoguttata. Br J Ophthalmol 91, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanavaty MA, Wang X, Shortt AJ, 2014. Endothelial keratoplasty versus penetrating keratoplasty for Fuchs endothelial dystrophy. Cochrane Database Syst Rev, CD008420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ, 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS biology 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff KD, Biber JM, Holland EJ, 2011. Comparison of central corneal graft thickness to visual acuity outcomes in endothelial keratoplasty. Cornea 30, 388–391. [DOI] [PubMed] [Google Scholar]
- Niederer RL, McGhee CN, 2010. Clinical in vivo confocal microscopy of the human cornea in health and disease. Prog Retin Eye Res 29, 30–58. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N, 2013. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833, 3448–3459. [DOI] [PubMed] [Google Scholar]
- Nita M, Grzybowski A, 2016. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxidative medicine and cellular longevity 2016, 3164734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita M, Grzybowski A, 2017. Smoking and Eye Pathologies. A Systemic Review. Part I. Anterior Eye Segment Pathologies. Curr Pharm Des 23, 629–638. [DOI] [PubMed] [Google Scholar]
- Ohkoshi K, Ishida N, Yamaguchi T, Kanki K, 1989. Corneal endothelium in a case of mitochondrial encephalomyopathy (Kearns-Sayre syndrome). Cornea 8, 210–214. [PubMed] [Google Scholar]
- Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M, 2008. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem 283, 33554–33562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda JL, Ventosa JA, Piedra S, 2001. The three-dimensional microanatomy of the rabbit and human cornea. A chemical and mechanical microdissection-SEM approach. J Anat 199, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N, 2013. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288, 36372–36384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Fujii K, Kagami T, Makiko N, Kitahara M, Kinoshita S, Koizumi N, 2016a. Activation of the Rho/Rho Kinase Signaling Pathway Is Involved in Cell Death of Corneal Endothelium. Investigative ophthalmology & visual science 57, 6843–6851. [DOI] [PubMed] [Google Scholar]
- Okumura N, Hashimoto K, Kitahara M, Okuda H, Ueda E, Watanabe K, Nakahara M, Sato T, Kinoshita S, Tourtas T, Schlotzer-Schrehardt U, Kruse F, Koizumi N, 2017a. Activation of TGF-beta signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dystrophy. Sci Rep 7, 6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Hayashi R, Nakano M, Tashiro K, Yoshii K, Aleff R, Butz M, Highsmith EW, Wieben ED, Fautsch MP, Baratz KH, Komori Y, Ueda E, Nakahara M, Weller J, Tourtas T, Schlotzer-Schrehardt U, Kruse F, Koizumi N, 2019a. Association of rs613872 and Trinucleotide Repeat Expansion in the TCF4 Gene of German Patients With Fuchs Endothelial Corneal Dystrophy. Cornea 38, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Hayashi R, Nakano M, Yoshii K, Tashiro K, Sato T, Blake DJ, Aleff R, Butz M, Highsmith EW, Wieben ED, Fautsch MP, Baratz KH, Komori Y, Nakahara M, Tourtas T, Schlotzer-Schrehardt U, Kruse F, Koizumi N, 2019b. Effect of Trinucleotide Repeat Expansion on the Expression of TCF4 mRNA in Fuchs’ Endothelial Corneal Dystrophy. Investigative ophthalmology & visual science 60, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Inoue R, Okazaki Y, Nakano S, Nakagawa H, Kinoshita S, Koizumi N, 2015a. Effect of the Rho Kinase Inhibitor Y-27632 on Corneal Endothelial Wound Healing. Investigative ophthalmology & visual science 56, 6067–6074. [DOI] [PubMed] [Google Scholar]
- Okumura N, Kay EP, Nakahara M, Hamuro J, Kinoshita S, Koizumi N, 2013a. Inhibition of TGF-β signaling enables human corneal endothelial cell expansion in vitro for use in regenerative medicine. PLoS ONE 8, e58000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Kinoshita S, Koizumi N, 2014a. Cell-based approach for treatment of corneal endothelial dysfunction. Cornea 33 Suppl 11, S37–41. [DOI] [PubMed] [Google Scholar]
- Okumura N, Kinoshita S, Koizumi N, 2017b. Application of Rho Kinase Inhibitors for the Treatment of Corneal Endothelial Diseases. J Ophthalmol 2017, 2646904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Kitahara M, Okuda H, Hashimoto K, Ueda E, Nakahara M, Kinoshita S, Young RD, Quantock AJ, Tourtas T, Schlotzer-Schrehardt U, Kruse F, Koizumi N, 2017c. Sustained Activation of the Unfolded Protein Response Induces Cell Death in Fuchs’ Endothelial Corneal Dystrophy. Investigative ophthalmology & visual science 58, 3697–3707. [DOI] [PubMed] [Google Scholar]
- Okumura N, Koizumi N, Kay EP, Ueno M, Sakamoto Y, Nakamura S, Hamuro J, Kinoshita S, 2013b. The ROCK inhibitor eye drop accelerates corneal endothelium wound healing. Investigative ophthalmology & visual science 54, 2493–2502. [DOI] [PubMed] [Google Scholar]
- Okumura N, Koizumi N, Ueno M, Sakamoto Y, Takahashi H, Hirata K, Torii R, Hamuro J, Kinoshita S, 2011. Enhancement of corneal endothelium wound healing by Rho-associated kinase (ROCK) inhibitor eye drops. Br J Ophthalmol 95, 1006–1009. [DOI] [PubMed] [Google Scholar]
- Okumura N, Koizumi N, Ueno M, Sakamoto Y, Takahashi H, Tsuchiya H, Hamuro J, Kinoshita S, 2012. ROCK inhibitor converts corneal endothelial cells into a phenotype capable of regenerating in vivo endothelial tissue. Am J Pathol 181, 268–277. [DOI] [PubMed] [Google Scholar]
- Okumura N, Minamiyama R, Ho LT, Kay EP, Kawasaki S, Tourtas T, Schlotzer-Schrehardt U, Kruse FE, Young RD, Quantock AJ, Kinoshita S, Koizumi N, 2015b. Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab Invest 95, 1291–1304. [DOI] [PubMed] [Google Scholar]
- Okumura N, Nakano S, Kay EP, Numata R, Ota A, Sowa Y, Sakai T, Ueno M, Kinoshita S, Koizumi N, 2014b. Involvement of cyclin D and p27 in cell proliferation mediated by ROCK inhibitors Y-27632 and Y-39983 during corneal endothelium wound healing. Investigative ophthalmology & visual science 55, 318–329. [DOI] [PubMed] [Google Scholar]
- Okumura N, Okazaki Y, Inoue R, Kakutani K, Nakano S, Kinoshita S, Koizumi N, 2016b. Effect of the Rho-Associated Kinase Inhibitor Eye Drop (Ripasudil) on Corneal Endothelial Wound Healing. Investigative ophthalmology & visual science 57, 1284–1292. [DOI] [PubMed] [Google Scholar]
- Okumura N, Okazaki Y, Inoue R, Nakano S, Fullwood NJ, Kinoshita S, Koizumi N, 2015c. Rho-Associated Kinase Inhibitor Eye Drop (Ripasudil) Transiently Alters the Morphology of Corneal Endothelial Cells. Investigative ophthalmology & visual science 56, 7560–7567. [DOI] [PubMed] [Google Scholar]
- Okumura N, Sakamoto Y, Fujii K, Kitano J, Nakano S, Tsujimoto Y, Nakamura S, Ueno M, Hagiya M, Hamuro J, Matsuyama A, Suzuki S, Shiina T, Kinoshita S, Koizumi N, 2016c. Rho kinase inhibitor enables cell-based therapy for corneal endothelial dysfunction. Sci Rep 6, 26113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Ueno M, Koizumi N, Sakamoto Y, Hirata K, Hamuro J, Kinoshita S, 2009. Enhancement on primate corneal endothelial cell survival in vitro by a ROCK inhibitor. Investigative ophthalmology & visual science 50, 3680–3687. [DOI] [PubMed] [Google Scholar]
- Oldak M, Ruszkowska E, Udziela M, Ozieblo D, Binczyk E, Sciezynska A, Ploski R, Szaflik JP, 2015. Fuchs Endothelial Corneal Dystrophy: Strong Association with rs613872 Not Paralleled by Changes in Corneal Endothelial TCF4 mRNA Level. Biomed Res Int 2015, 640234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen T, 1984. Is there an association between Fuchs’ endothelial dystrophy and cardiovascular disease? Graefes Arch Clin Exp Ophthalmol 221, 239–240. [DOI] [PubMed] [Google Scholar]
- Ong Tone S, Bruha MJ, Bohm M, Prescott C, Jurkunas U, 2019a. Regional variability in corneal endothelial cell density between guttae and non-guttae areas in Fuchs endothelial corneal dystrophy. Can J Ophthalmol 54, 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong Tone S, Jurkunas U, 2019. Imaging the Corneal Endothelium in Fuchs Corneal Endothelial Dystrophy. Semin Ophthalmol 34, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong Tone S, Melangath G, Deshpande N, Jurkunas UV, 2019b. Immortalization of Fuchs Endothelial Corneal Dystrophy (FECD) Corneal Endothelial Cells (CEnC) Retains TCF4 Repeat Expansion. Investigative ophthalmology & visual science 60, 2174–2174. [Google Scholar]
- Ong Tone S, Wylȩgała A, Bohm M, Jurkunas U, 2019c. Increased Corneal Endothelial Cell Migration in Fuchs Endothelial Corneal Dystrophy in a DSO Model, American Academy of Ophthalmology, San Francisco, USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlin SE, Raber IM, Eagle RC Jr., Scheie HG, 1990. Keratoconus associated with corneal endothelial dystrophy. Cornea 9, 299–304. [PubMed] [Google Scholar]
- Otteson DC, 2011. Eyes on DNA methylation: current evidence for DNA methylation in ocular development and disease. J Ocul Biol Dis Infor 4, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcelik B, Brown KD, Blencowe A, Ladewig K, Stevens GW, Scheerlinck JP, Abberton K, Daniell M, Qiao GG, 2014. Biodegradable and biocompatible poly(ethylene glycol)-based hydrogel films for the regeneration of corneal endothelium. Adv Healthc Mater 3, 1496–1507. [DOI] [PubMed] [Google Scholar]
- Patel SP, Gianfagna PA, Ruster TE, Millen AE, 2018. Prevalence of Fuchs’ endothelial corneal dystrophy by age and sex. Investigative ophthalmology & visual science 59, 3774. [Google Scholar]
- Patel SP, Parker MD, 2015. SLC4A11 and the Pathophysiology of Congenital Hereditary Endothelial Dystrophy. Biomed Res Int 2015, 475392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SV, Bachman LA, Hann CR, Bahler CK, Fautsch MP, 2009. Human corneal endothelial cell transplantation in a human ex vivo model. Investigative ophthalmology & visual science 50, 2123–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic I, Shajari M, Herrmann E, Schmack I, Lencova A, Kohnen T, 2017. Meta-Analysis of Postoperative Outcome Parameters Comparing Descemet Membrane Endothelial Keratoplasty Versus Descemet Stripping Automated Endothelial Keratoplasty. Cornea 36, 1445–1451. [DOI] [PubMed] [Google Scholar]
- Peh GS, Adnan K, George BL, Ang HP, Seah XY, Tan DT, Mehta JS, 2015a. The effects of Rho-associated kinase inhibitor Y-27632 on primary human corneal endothelial cells propagated using a dual media approach. Sci Rep 5, 9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh GS, Beuerman RW, Colman A, Tan DT, Mehta JS, 2011. Human corneal endothelial cell expansion for corneal endothelium transplantation: an overview. Transplantation 91, 811–819. [DOI] [PubMed] [Google Scholar]
- Peh GS, Chng Z, Ang HP, Cheng TY, Adnan K, Seah XY, George BL, Toh KP, Tan DT, Yam GH, Colman A, Mehta JS, 2015b. Propagation of human corneal endothelial cells: a novel dual media approach. Cell Transplant 24, 287–304. [DOI] [PubMed] [Google Scholar]
- Peh GSL, Ang HP, Lwin CN, Adnan K, George BL, Seah XY, Lin SJ, Bhogal M, Liu YC, Tan DT, Mehta JS, 2017. Regulatory Compliant Tissue-Engineered Human Corneal Endothelial Grafts Restore Corneal Function of Rabbits with Bullous Keratopathy. Sci Rep 7, 14149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh GSL, Ong HS, Adnan K, Ang HP, Lwin CN, Seah XY, Lin SJ, Mehta JS, 2019. Functional Evaluation of Two Corneal Endothelial Cell-Based Therapies: Tissue-Engineered Construct and Cell Injection. Sci Rep 9, 6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipparelli A, Arsenijevic Y, Thuret G, Gain P, Nicolas M, Majo F, 2013. ROCK inhibitor enhances adhesion and wound healing of human corneal endothelial cells. PLoS One 8, e62095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts DG, Cullen AP, Hacker PD, 1977. Ocular effects of ultraviolet radiation from 295 to 365 nm. Investigative ophthalmology & visual science 16, 932–939. [PubMed] [Google Scholar]
- Pitts JF, Jay JL, 1990. The association of Fuchs’s corneal endothelial dystrophy with axial hypermetropia, shallow anterior chamber, and angle closure glaucoma. Br J Ophthalmol 74, 601–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen ET, Dyrlund TF, Runager K, Scavenius C, Krogager TP, Hojrup P, Thogersen IB, Sanggaard KW, Vorum H, Hjortdal J, Enghild JJ, 2014. Proteomics of Fuchs’ endothelial corneal dystrophy support that the extracellular matrix of Descemet’s membrane is disordered. J Proteome Res 13, 4659–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourzand C, Tyrrell RM, 1999. Apoptosis, the role of oxidative stress and the example of solar UV radiation. Photochemistry and photobiology 70, 380–390. [PubMed] [Google Scholar]
- Prasad A, Fry K, Hersh PS, 2011. Relationship of age and refraction to central corneal thickness. Cornea 30, 553–555. [DOI] [PubMed] [Google Scholar]
- Price FW Jr., Whitson WE, Marks RG, 1991. Progression of visual acuity after penetrating keratoplasty. Ophthalmology 98, 1177–1185. [DOI] [PubMed] [Google Scholar]
- Price MO, Gorovoy M, Price FW Jr., Benetz BA, Menegay HJ, Lass JH, 2013. Descemet’s stripping automated endothelial keratoplasty: three-year graft and endothelial cell survival compared with penetrating keratoplasty. Ophthalmology 120, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MO, Lisek M, Feng MT, Price FW Jr., 2017. Effect of Donor and Recipient Diabetes Status on Descemet Membrane Endothelial Keratoplasty Adherence and Survival. Cornea 36, 1184–1188. [DOI] [PubMed] [Google Scholar]
- Proulx S, Brunette I, 2012. Methods being developed for preparation, delivery and transplantation of a tissue-engineered corneal endothelium. Exp Eye Res 95, 68–75. [DOI] [PubMed] [Google Scholar]
- Puk O, Dalke C, Calzada-Wack J, Ahmad N, Klaften M, Wagner S, de Angelis MH, Graw J, 2009. Reduced corneal thickness and enlarged anterior chamber in a novel ColVIIIa2G257D mutant mouse. Investigative ophthalmology & visual science 50, 5653–5661. [DOI] [PubMed] [Google Scholar]
- Rao BS, Tharigopala A, Rachapalli SR, Rajagopal R, Soumittra N, 2017. Association of polymorphisms in the intron of TCF4 gene to late-onset Fuchs endothelial corneal dystrophy: An Indian cohort study. Indian J Ophthalmol 65, 931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao GP, Kaye SB, Agius-Fernandez A, 1998. Central corneal endothelial guttae and age-related macular degeneration: is there an association? Indian J Ophthalmol 46, 145–147. [PubMed] [Google Scholar]
- Reddy KB, Jin G, Karode MC, Harmony JA, Howe PH, 1996. Transforming growth factor beta (TGF beta)-induced nuclear localization of apolipoprotein J/clusterin in epithelial cells. Biochemistry 35, 6157–6163. [DOI] [PubMed] [Google Scholar]
- Repp DJ, Hodge DO, Baratz KH, McLaren JW, Patel SV, 2013. Fuchs’ endothelial corneal dystrophy: subjective grading versus objective grading based on the central-to-peripheral thickness ratio. Ophthalmology 120, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei Kanavi M, Javadi MA, Motevasseli T, Chamani T, Rezaei Kanavi M, Kheiri B, Safi S, 2016. Trends in Indications and Techniques of Corneal Transplantation in Iran from 2006 to 2013; an 8-year Review. J Ophthalmic Vis Res 11, 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin SA, Eghrari AO, Al-Saif A, Davey L, Meadows DN, Katsanis N, Gottsch JD, 2009. Linkage of a mild late-onset phenotype of Fuchs corneal dystrophy to a novel locus at 5q33.1-q35.2. Investigative ophthalmology & visual science 50, 5667–5671. [DOI] [PubMed] [Google Scholar]
- Riazuddin SA, McGlumphy EJ, Yeo WS, Wang J, Katsanis N, Gottsch JD, 2011. Replication of the TCF4 intronic variant in late-onset Fuchs corneal dystrophy and evidence of independence from the FCD2 locus. Investigative ophthalmology & visual science 52, 2825–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin SA, Parker DS, McGlumphy EJ, Oh EC, Iliff BW, Schmedt T, Jurkunas U, Schleif R, Katsanis N, Gottsch JD, 2012. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet 90, 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin SA, Vasanth S, Katsanis N, Gottsch JD, 2013. Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am J Hum Genet 93, 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin SA, Vithana EN, Seet LF, Liu Y, Al-Saif A, Koh LW, Heng YM, Aung T, Meadows DN, Eghrari AO, Gottsch JD, Katsanis N, 2010a. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum Mutat 31, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin SA, Zaghloul NA, Al-Saif A, Davey L, Diplas BH, Meadows DN, Eghrari AO, Minear MA, Li YJ, Klintworth GK, Afshari N, Gregory SG, Gottsch JD, Katsanis N, 2010b. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet 86, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GD, Wright K, Silverstein SM, 2014. A retrospective study of the association between Fuchs’ endothelial dystrophy and glaucoma. Clin Ophthalmol 8, 2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento K, Ridley AJ, 2003. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4, 446–456. [DOI] [PubMed] [Google Scholar]
- Rinaldi C, Wood MJA, 2018. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 14, 9–21. [DOI] [PubMed] [Google Scholar]
- Rizwan M, Peh GS, Adnan K, Naso SL, Mendez AR, Mehta JS, Yim EK, 2016. In Vitro Topographical Model of Fuchs Dystrophy for Evaluation of Corneal Endothelial Cell Monolayer Formation. Adv Healthc Mater 5, 2896–2910. [DOI] [PubMed] [Google Scholar]
- Roberts CW, Steinert RF, Thomas JV, Boruchoff SA, 1984. Endothelial guttata and facility of aqueous outflow. Cornea 3, 5–9. [PubMed] [Google Scholar]
- Rolo AP, Palmeira CM, 2006. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicology and applied pharmacology 212, 167–178. [DOI] [PubMed] [Google Scholar]
- Rong Z, Hu J, Corey DR, Mootha VV, 2019. Quantitative Studies of Muscleblind Proteins and Their Interaction With TCF4 RNA Foci Support Involvement in the Mechanism of Fuchs’ Dystrophy. Investigative ophthalmology & visual science 60, 3980–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum P, Stark WJ, Maumenee IH, Hirst LW, Maumenee AE, 1980. Hereditary Fuchs’ Dystrophy. Am J Ophthalmol 90, 455–462. [DOI] [PubMed] [Google Scholar]
- Rouland JF, 2015. [Clinical pilot study to evaluate the efficacy of a preservative-free hypertonic ophthalmic solution for patients with symptomatic corneal edema]. J Fr Ophtalmol 38, 800–808. [DOI] [PubMed] [Google Scholar]
- Roy O, Leclerc VB, Bourget JM, Theriault M, Proulx S, 2015. Understanding the process of corneal endothelial morphological change in vitro. Investigative ophthalmology & visual science 56, 1228–1237. [DOI] [PubMed] [Google Scholar]
- Runager K, Enghild JJ, Klintworth GK, 2008. Focus on molecules: Transforming growth factor beta induced protein (TGFBIp). Exp Eye Res 87, 298–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM, 2007. Mechanisms of cell death in oxidative stress. Antioxidants & redox signaling 9, 49–89. [DOI] [PubMed] [Google Scholar]
- Saad A, Guilbert E, Grise-Dulac A, Sabatier P, Gatinel D, 2015. Intraoperative OCT-Assisted DMEK: 14 Consecutive Cases. Cornea 34, 802–807. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Shui YB, Kojima M, Chew SJ, Ono M, Katoh N, Cheng HM, Takahashi N, Sasaki K, 2001. Characteristics of cataracts in the Chinese Singaporean. J Epidemiol 11, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satue M, Idoipe M, Sanchez-Perez A, Liarakos VS, Mateo A, Garcia-Martin E, Polo V, Gavin A, Brito C, 2016. Evaluation of Early Graft Detachment After Descemet Membrane Endothelial Keratoplasty Using New Swept-Source Optical Coherence Tomography. Cornea 35, 1279–1284. [DOI] [PubMed] [Google Scholar]
- Sawada H, Konomi H, Hirosawa K, 1990. Characterization of the collagen in the hexagonal lattice of Descemet’s membrane: its relation to type VIII collagen. J Cell Biol 110, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub F, Gerber F, Adler W, Enders P, Schrittenlocher S, Heindl LM, Cursiefen C, Bachmann BO, 2019. Corneal Densitometry as a Predictive Diagnostic Tool for Visual Acuity Results After Descemet Membrane Endothelial Keratoplasty. Am J Ophthalmol 198, 124–129. [DOI] [PubMed] [Google Scholar]
- Scherer WJ, 2018. Corneal endothelial cell density and cardiovascular mortality : A Global Survey and Correlative Meta-Analysis. Clinical Anatomy 31, 927–936. [DOI] [PubMed] [Google Scholar]
- Schimmelpfennig BH, 1984. Direct and indirect determination of nonuniform cell density distribution in human corneal endothelium. Investigative ophthalmology & visual science 25, 223–229. [PubMed] [Google Scholar]
- Schmedt T, Chen Y, Nguyen TT, Li S, Bonanno JA, Jurkunas UV, 2012a. Telomerase immortalization of human corneal endothelial cells yields functional hexagonal monolayers. PLoS One 7, e51427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmedt T, Silva MM, Ziaei A, Jurkunas U, 2012b. Molecular bases of corneal endothelial dystrophies. Experimental eye research 95, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrems-Hoesl LM, Schrems WA, Cruzat A, Shahatit BM, Bayhan HA, Jurkunas UV, Hamrah P, 2013. Cellular and subbasal nerve alterations in early stage Fuchs’ endothelial corneal dystrophy: an in vivo confocal microscopy study. Eye (Lond) 27, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RO, Matsuda M, Yee RW, Edelhauser HF, Schultz KJ, 1984. Corneal endothelial changes in type I and type II diabetes mellitus. Am J Ophthalmol 98, 401–410. [DOI] [PubMed] [Google Scholar]
- Seitzman GD, Gottsch JD, Stark WJ, 2005. Cataract surgery in patients with Fuchs’ corneal dystrophy: expanding recommendations for cataract surgery without simultaneous keratoplasty. Ophthalmology 112, 441–446. [DOI] [PubMed] [Google Scholar]
- Senoo T, Joyce NC, 2000. Cell cycle kinetics in corneal endothelium from old and young donors. Investigative ophthalmology & visual science 41, 660–667. [PubMed] [Google Scholar]
- Senoo T, Obara Y, Joyce NC, 2000. EDTA: a promoter of proliferation in human corneal endothelium. Investigative ophthalmology & visual science 41, 2930–2935. [PubMed] [Google Scholar]
- Shah RD, Randleman JB, Grossniklaus HE, 2012. Spontaneous corneal clearing after Descemet’s stripping without endothelial replacement. Ophthalmology 119, 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH, 2003. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23, 3394–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima N, Kimoto M, Yamaguchi M, Yamagami S, 2011. Increased proliferation and replicative lifespan of isolated human corneal endothelial cells with L-ascorbic acid 2-phosphate. Investigative ophthalmology & visual science 52, 8711–8717. [DOI] [PubMed] [Google Scholar]
- Shimazaki J, Amano S, Uno T, Maeda N, Yokoi N, Japan Bullous Keratopathy Study, G., 2007. National survey on bullous keratopathy in Japan. Cornea 26, 274–278. [DOI] [PubMed] [Google Scholar]
- Shousha MA, Perez VL, Wang J, Ide T, Jiao S, Chen Q, Chang V, Buchser N, Dubovy SR, Feuer W, Yoo SH, 2010. Use of ultra-high-resolution optical coherence tomography to detect in vivo characteristics of Descemet’s membrane in Fuchs’ dystrophy. Ophthalmology 117, 1220–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui S, Zenteno JC, Rice A, Chacon-Camacho O, Naylor SG, Rivera-de la Parra D, Spokes DM, James N, Toomes C, Inglehearn CF, Ali M, 2014. Congenital hereditary endothelial dystrophy caused by SLC4A11 mutations progresses to Harboyan syndrome. Cornea 33, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonian NA, Coyle JT, 1996. Oxidative stress in neurodegenerative diseases. Annual review of pharmacology and toxicology 36, 83–106. [DOI] [PubMed] [Google Scholar]
- Singh A, Zarei-Ghanavati M, Avadhanam V, Liu C, 2017. Systematic Review and Meta-Analysis of Clinical Outcomes of Descemet Membrane Endothelial Keratoplasty Versus Descemet Stripping Endothelial Keratoplasty/Descemet Stripping Automated Endothelial Keratoplasty. Cornea 36, 1437–1443. [DOI] [PubMed] [Google Scholar]
- Singh S, Chakravarti D, Edney JA, Hollins RR, Johnson PJ, West WW, Higginbotham SM, Cavalieri EL, Rogan EG, 2005. Relative imbalances in the expression of estrogen-metabolizing enzymes in the breast tissue of women with breast carcinoma. Oncol Rep 14, 1091–1096. [PubMed] [Google Scholar]
- Skerjanc IS, Truong J, Filion P, McBurney MW, 1996. A splice variant of the ITF-2 transcript encodes a transcription factor that inhibits MyoD activity. J Biol Chem 271, 3555–3561. [DOI] [PubMed] [Google Scholar]
- Sobrado VR, Moreno-Bueno G, Cubillo E, Holt LJ, Nieto MA, Portillo F, Cano A, 2009. The class I bHLH factors E2–2A and E2–2B regulate EMT. J Cell Sci 122, 1014–1024. [DOI] [PubMed] [Google Scholar]
- Soh YQ, Mehta JS, 2018. Regenerative Therapy for Fuchs Endothelial Corneal Dystrophy. Cornea 37, 523–527. [DOI] [PubMed] [Google Scholar]
- Soh YQ, Peh G, George BL, Seah XY, Primalani NK, Adnan K, Mehta JS, 2016. Predicative Factors for Corneal Endothelial Cell Migration. Investigative ophthalmology & visual science 57, 338–348. [DOI] [PubMed] [Google Scholar]
- Soh YQ, Peh Swee Lim G, Htoon HM, Gong X, Mootha VV, Vithana EN, Kocaba V, Mehta JS, 2019. Trinucleotide repeat expansion length as a predictor of the clinical progression of Fuchs’ Endothelial Corneal Dystrophy. PloS one 14, e0210996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg Y, Rosner M, Belkin M, 1998. The association between cigarette smoking and ocular diseases. Surv Ophthalmol 42, 535–547. [DOI] [PubMed] [Google Scholar]
- Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV, 2015. Correlation of Severity of Fuchs Endothelial Corneal Dystrophy With Triplet Repeat Expansion in TCF4. JAMA Ophthalmol 133, 1386–1391. [DOI] [PubMed] [Google Scholar]
- Son HS, Villarreal G Jr., Meng H, Eberhart CG, Jun AS, 2014. On the origin of ‘guttae’. Br J Ophthalmol 98, 1308–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Wang Y, Xie L, Zang X, Yin H, 2008. Expression of senescence-related genes in human corneal endothelial cells. Mol Vis 14, 161–170. [PMC free article] [PubMed] [Google Scholar]
- Soragni E, Petrosyan L, Rinkoski TA, Wieben ED, Baratz KH, Fautsch MP, Gottesfeld JM, 2018. Repeat-Associated Non-ATG (RAN) Translation in Fuchs’ Endothelial Corneal Dystrophy. Investigative ophthalmology & visual science 59, 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumittra N, Loganathan SK, Madhavan D, Ramprasad VL, Arokiasamy T, Sumathi S, Karthiyayini T, Rachapalli SR, Kumaramanickavel G, Casey JR, Rajagopal R, 2014. Biosynthetic and functional defects in newly identified SLC4A11 mutants and absence of COL8A2 mutations in Fuchs endothelial corneal dystrophy. J Hum Genet 59, 444–453. [DOI] [PubMed] [Google Scholar]
- Stephan S, Sherratt MJ, Hodson N, Shuttleworth CA, Kielty CM, 2004. Expression and supramolecular assembly of recombinant alpha1(viii) and alpha2(viii) collagen homotrimers. J Biol Chem 279, 21469–21477. [DOI] [PubMed] [Google Scholar]
- Stephanongtone, 2018. Statistical Report - Eye Bank Association of America Eye Bank Association of America [Google Scholar]
- Stocker FW, 1953. The endothelium of the cornea and its clinical implications. Trans Am Ophthalmol Soc 51, 669–786. [PMC free article] [PubMed] [Google Scholar]
- Stuart AJ, Romano V, Virgili G, Shortt AJ, 2018. Descemet’s membrane endothelial keratoplasty (DMEK) versus Descemet’s stripping automated endothelial keratoplasty (DSAEK) for corneal endothelial failure. Cochrane Database Syst Rev 6, CD012097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su DH, Wong TY, Wong WL, Saw SM, Tan DT, Shen SY, Loon SC, Foster PJ, Aung T, Singapore Malay Eye Study, G., 2008. Diabetes, hyperglycemia, and central corneal thickness: the Singapore Malay Eye Study. Ophthalmology 115, 964–968 e961. [DOI] [PubMed] [Google Scholar]
- Sumide T, Nishida K, Yamato M, Ide T, Hayashida Y, Watanabe K, Yang J, Kohno C, Kikuchi A, Maeda N, Watanabe H, Okano T, Tano Y, 2006. Functional human corneal endothelial cell sheets harvested from temperature-responsive culture surfaces. FASEB J 20, 392–394. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wacker K, Baratz KH, Patel SV, 2019. Determining Subclinical Edema in Fuchs Endothelial Corneal Dystrophy: Revised Classification using Scheimpflug Tomography for Preoperative Assessment. Ophthalmology 126, 195–204. [DOI] [PubMed] [Google Scholar]
- Sundin OH, Broman KW, Chang HH, Vito EC, Stark WJ, Gottsch JD, 2006a. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Investigative ophthalmology & visual science 47, 3919–3926. [DOI] [PubMed] [Google Scholar]
- Sundin OH, Jun AS, Broman KW, Liu SH, Sheehan SE, Vito EC, Stark WJ, Gottsch JD, 2006b. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Investigative ophthalmology & visual science 47, 140–145. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kinoshita Y, Tachibana M, Matsushima Y, Kobayashi Y, Adachi W, Sotozono C, Kinoshita S, 2001. Expression of sex steroid hormone receptors in human cornea. Current eye research 22, 28–33. [DOI] [PubMed] [Google Scholar]
- Syed ZA, Tran JA, Jurkunas UV, 2017. Peripheral Endothelial Cell Count Is a Predictor of Disease Severity in Advanced Fuchs Endothelial Corneal Dystrophy. Cornea 36, 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synowiec E, Wojcik KA, Izdebska J, Binczyk E, Blasiak J, Szaflik J, Szaflik JP, 2013. Polymorphisms of the homologous recombination gene RAD51 in keratoconus and Fuchs endothelial corneal dystrophy. Dis Markers 35, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synowiec E, Wojcik KA, Izdebska J, Binczyk E, Szaflik J, Blasiak J, Szaflik JP, 2015. Polymorphism of the LIG3 gene in keratoconus and Fuchs endothelial corneal dystrophy. Cellular and molecular biology (Noisy-le-Grand, France) 61, 56–63. [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A, 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports 7, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentmary N, Szende B, Suveges I, 2005. Epithelial cell, keratocyte, and endothelial cell apoptosis in Fuchs’ dystrophy and in pseudophakic bullous keratopathy. Eur J Ophthalmol 15, 17–22. [DOI] [PubMed] [Google Scholar]
- Szentmary N, Szende B, Suveges I, 2008. P53, CD95, cathepsin and survivin pathways in Fuchs’ dystrophy and pseudophakic bullous keratopathy corneas. Histol Histopathol 23, 911–916. [DOI] [PubMed] [Google Scholar]
- Tan DT, Anshu A, Mehta JS, 2009. Paradigm shifts in corneal transplantation. Ann Acad Med Singapore 38, 332–338. [PubMed] [Google Scholar]
- Tan DT, Dart JK, Holland EJ, Kinoshita S, 2012. Corneal transplantation. Lancet 379, 1749–1761. [DOI] [PubMed] [Google Scholar]
- Tan TE, Peh GS, George BL, Cajucom-Uy HY, Dong D, Finkelstein EA, Mehta JS, 2014. A cost-minimization analysis of tissue-engineered constructs for corneal endothelial transplantation. PLoS One 9, e100563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangvarasittichai O, Tangvarasittichai S, 2018. Oxidative Stress, Ocular Disease and Diabetes Retinopathy. Curr Pharm Des 24, 4726–4741. [DOI] [PubMed] [Google Scholar]
- Tarnawska D, Wylegala E, 2010. Monitoring cornea and graft morphometric dynamics after descemet stripping and endothelial keratoplasty with anterior segment optical coherence tomography. Cornea 29, 272–277. [DOI] [PubMed] [Google Scholar]
- Tarwadi KV, Chiplonkar SA, Agte V, 2008. Dietary and nutritional biomarkers of lens degeneration, oxidative stress and micronutrient inadequacies in Indian cataract patients. Clinical nutrition (Edinburgh, Scotland) 27, 464–472. [DOI] [PubMed] [Google Scholar]
- Terry MA, 2003. Deep lamellar endothelial keratoplasty (DLEK): pursuing the ideal goals of endothelial replacement. Eye (Lond) 17, 982–988. [DOI] [PubMed] [Google Scholar]
- Theriault M, Gendron SP, Brunette I, Rochette PJ, Proulx S, 2018. Function-Related Protein Expression in Fuchs Endothelial Corneal Dystrophy Cells and Tissue Models. Am J Pathol 188, 1703–1712. [DOI] [PubMed] [Google Scholar]
- Tillett CW, 1956. Posterior lamellar keratoplasty. Am J Ophthalmol 41, 530–533. [DOI] [PubMed] [Google Scholar]
- Ting DS, Sau CY, Srinivasan S, Ramaesh K, Mantry S, Roberts F, 2012. Changing trends in keratoplasty in the West of Scotland: a 10-year review. Br J Ophthalmol 96, 405–408. [DOI] [PubMed] [Google Scholar]
- Toda M, Ueno M, Hiraga A, Asada K, Montoya M, Sotozono C, Kinoshita S, Hamuro J, 2017. Production of Homogeneous Cultured Human Corneal Endothelial Cells Indispensable for Innovative Cell Therapy. Investigative ophthalmology & visual science 58, 2011–2020. [DOI] [PubMed] [Google Scholar]
- Tomei LD, Umansky SR, 1998. Aging and apoptosis control. Neurologic clinics 16, 735–745. [DOI] [PubMed] [Google Scholar]
- Toyono T, Usui T, Villarreal G Jr., Kallay L, Matthaei M, Vianna LM, Zhu AY, Kuroda M, Amano S, Jun AS, 2016. MicroRNA-29b Overexpression Decreases Extracellular Matrix mRNA and Protein Production in Human Corneal Endothelial Cells. Cornea 35, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RC, Borisuth NS, Tripathi BJ, Fang VS, 1991. Analysis of human aqueous humor for epidermal growth factor. Exp Eye Res 53, 407–409. [DOI] [PubMed] [Google Scholar]
- Tuberville AW, Wood TO, McLaughlin BJ, 1986. Cytochrome oxidase activity of Fuchs’ endothelial dystrophy. Curr Eye Res 5, 939–947. [DOI] [PubMed] [Google Scholar]
- Tung PS, Burdzy K, Wong K, Fritz IB, 1992. Competition between cell-substratum interactions and cell-cell interactions. Journal of cellular physiology 152, 410–421. [DOI] [PubMed] [Google Scholar]
- Ucakhan OO, Saglik A, 2014. Outcome of Two Corneal Collagen Crosslinking Methods in Bullous Keratopathy due to Fuchs’ Endothelial Dystrophy. Case Rep Med 2014, 463905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban B, Peczynska J, Glowinska-Olszewska B, Urban M, Bakunowicz-Lazarczyk A, Kretowska M, 2007. [Evaluation of central corneal thickness in children and adolescents with type I diabetes mellitus]. Klinika oczna 109, 418–420. [PubMed] [Google Scholar]
- Vallabh NA, Romano V, Willoughby CE, 2017. Mitochondrial dysfunction and oxidative stress in corneal disease. Mitochondrion 36, 103–113. [DOI] [PubMed] [Google Scholar]
- van Rooij J, Lucas EH, Geerards AJ, Remeijer L, Wubbels R, 2018. Corneal transplantation for Fuchs endothelial dystrophy: A comparison of three surgical techniques concerning 10 year graft survival and visual function. PLoS One 13, e0203993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanth S, Eghrari AO, Gapsis BC, Wang J, Haller NF, Stark WJ, Katsanis N, Riazuddin SA, Gottsch JD, 2015. Expansion of CTG18.1 Trinucleotide Repeat in TCF4 Is a Potent Driver of Fuchs’ Corneal Dystrophy. Investigative ophthalmology & visual science 56, 4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur S, Afzal S, Tomasini R, Guillaumond F, Tardivel-Lacombe J, Mak TW, Iovanna JL, 2012. Consequences of DJ-1 upregulation following p53 loss and cell transformation. Oncogene 31, 664–670. [DOI] [PubMed] [Google Scholar]
- Vedana G, Villarreal G Jr., Jun AS, 2016. Fuchs endothelial corneal dystrophy: current perspectives. Clin Ophthalmol 10, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, Anand S, Khine KO, Venkataraman D, Yong VH, Salto-Tellez M, Venkatraman A, Guo K, Hemadevi B, Srinivasan M, Prajna V, Khine M, Casey JR, Inglehearn CF, Aung T, 2006. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet 38, 755–757. [DOI] [PubMed] [Google Scholar]
- Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, Venkatraman A, Yam GH, Nagasamy S, Law RW, Rajagopal R, Pang CP, Kumaramanickevel G, Casey JR, Aung T, 2008. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet 17, 656–666. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ, 2000. Surfing the p53 network. Nature 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Vogt A, 1921. Weitere Ergebnisse der Spaltlampenmikroskopie des vordern Bulbusabschnittes. Archives of ophthalmology (Chicago, Ill. : 1960), 63–113. [Google Scholar]
- Wahlig S, Kocaba V, Mehta JS, 2018a. Cultured Cells and ROCK Inhibitor for Bullous Keratopathy. The New England journal of medicine 379, 1184. [DOI] [PubMed] [Google Scholar]
- Wahlig S, Lovatt M, Mehta JS, 2018b. Functional role of peroxiredoxin 6 in the eye. Free Radic Biol Med 126, 210–220. [DOI] [PubMed] [Google Scholar]
- Wang KJ, Jhanji V, Chen J, Law RW, Leung AT, Zhang M, Wang N, Pang CP, Yam GH, 2014. Association of transcription factor 4 (TCF4) and protein tyrosine phosphatase, receptor type G (PTPRG) with corneal dystrophies in southern Chinese. Ophthalmic Genet 35, 138–141. [DOI] [PubMed] [Google Scholar]
- Wang Z, Handa JT, Green WR, Stark WJ, Weinberg RS, Jun AS, 2007. Advanced glycation end products and receptors in Fuchs’ dystrophy corneas undergoing Descemet’s stripping with endothelial keratoplasty. Ophthalmology 114, 1453–1460. [DOI] [PubMed] [Google Scholar]
- Waring GO 3rd, 1982. Posterior collagenous layer of the cornea. Ultrastructural classification of abnormal collagenous tissue posterior to Descemet’s membrane in 30 cases. Archives of ophthalmology (Chicago, Ill. : 1960) 100, 122–134. [DOI] [PubMed] [Google Scholar]
- Waring GO 3rd, Bourne WM, Edelhauser HF, Kenyon KR, 1982. The corneal endothelium. Normal and pathologic structure and function. Ophthalmology 89, 531–590. [PubMed] [Google Scholar]
- Waring GO 3rd, Rodrigues MM, Laibson PR, 1978. Corneal dystrophies. II. Endothelial dystrophies. Surv Ophthalmol 23, 147–168. [DOI] [PubMed] [Google Scholar]
- Weiss JS, Moller HU, Aldave AJ, Seitz B, Bredrup C, Kivela T, Munier FL, Rapuano CJ, Nischal KK, Kim EK, Sutphin J, Busin M, Labbe A, Kenyon KR, Kinoshita S, Lisch W, 2015. IC3D classification of corneal dystrophies--edition 2. Cornea 34, 117–159. [DOI] [PubMed] [Google Scholar]
- White E, 1996. Life, death, and the pursuit of apoptosis. Genes & development 10, 1–15. [DOI] [PubMed] [Google Scholar]
- White T, Deshpande N, Jurkunas UV, 2019. UV-A light induces G2/M phase arrest and subsequent endothelial-mesenchymal transition in Fuchs Endothelial Corneal Dystrophy. Investigative ophthalmology & visual science 60, 2179–2179. [Google Scholar]
- Wieben ED, Aleff RA, Eckloff BW, Atkinson EJ, Baheti S, Middha S, Brown WL, Patel SV, Kocher JP, Baratz KH, 2014. Comprehensive assessment of genetic variants within TCF4 in Fuchs’ endothelial corneal dystrophy. Investigative ophthalmology & visual science 55, 6101–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieben ED, Aleff RA, Tang XJ, Kalari KR, Maguire LJ, Patel SV, Baratz KH, Fautsch MP, 2018. Gene expression in the corneal endothelium of Fuchs endothelial corneal dystrophy patients with and without expansion of a trinucleotide repeat in TCF4. Plos One 13, e0200005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH, 2012. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS One 7, e49083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand W, Thaer AA, Kroll P, Geyer OC, Garcia AJ, 1995. Optical sectioning of the cornea with a new confocal in vivo slit-scanning videomicroscope. Ophthalmology 102, 568–575. [DOI] [PubMed] [Google Scholar]
- Wild EJ, Tabrizi SJ, 2017. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol 16, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R, Lace R, Kennedy S, Doherty K, Levis H, 2018. Biomaterials for Regenerative Medicine Approaches for the Anterior Segment of the Eye. Adv Healthc Mater 7, e1701328. [DOI] [PubMed] [Google Scholar]
- Wilson SE, Bourne WM, 1988. Fuchs’ dystrophy. Cornea 7, 2–18. [PubMed] [Google Scholar]
- Wilson SE, Bourne WM, Brubaker RF, 1988. Effect of dexamethasone on corneal endothelial function in Fuchs’ dystrophy. Investigative ophthalmology & visual science 29, 357–361. [PubMed] [Google Scholar]
- Wilson SE, Lloyd SA, He YG, McCash CS, 1993. Extended life of human corneal endothelial cells transfected with the SV40 large T antigen. Investigative ophthalmology & visual science 34, 2112–2123. [PubMed] [Google Scholar]
- Winkler NS, Milone M, Martinez-Thompson JM, Raja H, Aleff RA, Patel SV, Fautsch MP, Wieben ED, Baratz KH, 2018. Fuchs’ Endothelial Corneal Dystrophy in Patients With Myotonic Dystrophy, Type 1. Investigative ophthalmology & visual science 59, 3053–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik KA, Kaminska A, Blasiak J, Szaflik J, Szaflik JP, 2013a. Oxidative stress in the pathogenesis of keratoconus and Fuchs endothelial corneal dystrophy. Int J Mol Sci 14, 19294–19308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik KA, Synowiec E, Jimenez-Garcia MP, Kaminska A, Polakowski P, Blasiak J, Szaflik J, Szaflik JP, 2013b. Polymorphism of the transferrin gene in eye diseases: keratoconus and Fuchs endothelial corneal dystrophy. Biomed Res Int 2013, 247438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik KA, Synowiec E, Polakowski P, Blasiak J, Szaflik J, Szaflik JP, 2015. Variation in DNA Base Excision Repair Genes in Fuchs Endothelial Corneal Dystrophy. Med Sci Monit 21, 2809–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik KA, Synowiec E, Polakowski P, Glowacki S, Izdebska J, Lloyd S, Galea D, Blasiak J, Szaflik J, Szaflik JP, 2014. Polymorphism of the flap endonuclease 1 gene in keratoconus and Fuchs endothelial corneal dystrophy. Int J Mol Sci 15, 14786–14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie AH, 1985. The biology of cell death in tumours. Anticancer research 5, 131–136. [PubMed] [Google Scholar]
- Xia D, Zhang S, Nielsen E, Ivarsen AR, Liang C, Li Q, Thomsen K, Hjortdal JO, Dong M, 2016a. The Ultrastructures and Mechanical Properties of the Descement’s Membrane in Fuchs Endothelial Corneal Dystrophy. Sci Rep 6, 23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Zhang S, Nielsen E, Ivarsen AR, Liang C, Li Q, Thomsen K, Hjortdal JO, Dong M, 2016b. The Ultrastructures and Mechanical Properties of the Descement’s Membrane in Fuchs Endothelial Corneal Dystrophy. Sci Rep 6, 23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing C, Gong X, Hussain I, Khor CC, Tan DT, Aung T, Mehta JS, Vithana EN, Mootha VV, 2014. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs’ corneal dystrophy in Chinese implies common causal variant. Investigative ophthalmology & visual science 55, 7073–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Okada M, Tsujikawa M, Morimura H, Maeda N, Watanabe H, Inoue Y, Shimomura Y, Kinoshita S, Tano Y, 2000. The spectrum of beta ig-h3 gene mutations in Japanese patients with corneal dystrophy. Cornea 19, S21–23. [DOI] [PubMed] [Google Scholar]
- Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF, 2007. Systemic effects of smoking. Chest 131, 1557–1566. [DOI] [PubMed] [Google Scholar]
- Yang CR, Leskov K, Hosley-Eberlein K, Criswell T, Pink JJ, Kinsella TJ, Boothman DA, 2000. Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc Natl Acad Sci U S A 97, 5907–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Park JW, Zheng D, Xu RH, 2018. Universal Corneal Epithelial-Like Cells Derived from Human Embryonic Stem Cells for Cellularization of a Corneal Scaffold. Transl Vis Sci Technol 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, Sun X, Qin Z, Jin P, Li S, Li XJ, 2017. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest 127, 2719–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, He J, Wang C, Wu H, Shi X, Zhang H, Xie J, Lee SY, 2012. Smoking and risk of age-related cataract: a meta-analysis. Investigative ophthalmology & visual science 53, 3885–3895. [DOI] [PubMed] [Google Scholar]
- Yeh RY, Quilendrino R, Musa FU, Liarakos VS, Dapena I, Melles GR, 2013. Predictive value of optical coherence tomography in graft attachment after Descemet’s membrane endothelial keratoplasty. Ophthalmology 120, 240–245. [DOI] [PubMed] [Google Scholar]
- Yellore VS, Rayner SA, Emmert-Buck L, Tabin GC, Raber I, Hannush SB, Stulting RD, Sampat K, Momi R, Principe AH, Aldave AJ, 2005. No pathogenic mutations identified in the COL8A2 gene or four positional candidate genes in patients with posterior polymorphous corneal dystrophy. Investigative ophthalmology & visual science 46, 1599–1603. [DOI] [PubMed] [Google Scholar]
- Yokoi T, Seko Y, Yokoi T, Makino H, Hatou S, Yamada M, Kiyono T, Umezawa A, Nishina H, Azuma N, 2012. Establishment of functioning human corneal endothelial cell line with high growth potential. PLoS One 7, e29677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid M, Kohli E, Saeed M, Rogan E, Cavalieri E, 2006. The greater reactivity of estradiol-3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: implications for tumor-initiating activity. Chem Res Toxicol 19, 164–172. [DOI] [PubMed] [Google Scholar]
- Zahid M, Saeed M, Lu F, Gaikwad N, Rogan E, Cavalieri E, 2007. Inhibition of catechol-O-methyltransferase increases estrogen-DNA adduct formation. Free Radic Biol Med 43, 1534–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaniolo K, Bostan C, Rochette Drouin O, Deschambeault A, Perron MC, Brunette I, Proulx S, 2012. Culture of human corneal endothelial cells isolated from corneas with Fuchs endothelial corneal dystrophy. Exp Eye Res 94, 22–31. [DOI] [PubMed] [Google Scholar]
- Zarouchlioti C, Sanchez-Pintado B, Hafford Tear NJ, Klein P, Liskova P, Dulla K, Semo M, Vugler AA, Muthusamy K, Dudakova L, Levis HJ, Skalicka P, Hysi P, Cheetham ME, Tuft SJ, Adamson P, Hardcastle AJ, Davidson AE, 2018. Antisense Therapy for a Common Corneal Dystrophy Ameliorates TCF4 Repeat Expansion-Mediated Toxicity. Am J Hum Genet 102, 528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bell WR, Sundin OH, De La Cruz Z, Stark WJ, Green WR, Gottsch JD, 2006. Immunohistochemistry and electron microscopy of early-onset fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans Am Ophthalmol Soc 104, 85–97. [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Patel DV, 2015. The pathophysiology of Fuchs’ endothelial dystrophy--a review of molecular and cellular insights. Exp Eye Res 130, 97–105. [DOI] [PubMed] [Google Scholar]
- Zhang P, Liu B, Kang SW, Seo MS, Rhee SG, Obeid LM, 1997. Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of Bcl-2. J Biol Chem 272, 30615–30618. [DOI] [PubMed] [Google Scholar]
- Zhang W, Li H, Ogando DG, Li S, Feng M, Price FW Jr., Tennessen JM, Bonanno JA, 2017a. Glutaminolysis is Essential for Energy Production and Ion Transport in Human Corneal Endothelium. EBioMedicine 16, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ogando DG, Kim ET, Choi MJ, Li H, Tenessen JM, Bonanno JA, 2017b. Conditionally Immortal Slc4a11−/− Mouse Corneal Endothelial Cell Line Recapitulates Disrupted Glutaminolysis Seen in Slc4a11−/− Mouse Model. Investigative ophthalmology & visual science 58, 3723–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Igo RP Jr., Fondran J, Mootha VV, Oliva M, Hammersmith K, Sugar A, Lass JH, Iyengar SK, Fuchs’ Genetics Multi-Center Study, G., 2013. Association of smoking and other risk factors with Fuchs’ endothelial corneal dystrophy severity and corneal thickness. Investigative ophthalmology & visual science 54, 5829–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kok KH, Chun AC, Wong CM, Wu HW, Lin MC, Fung PC, Kung H, Jin DY, 2000. Mouse peroxiredoxin V is a thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochemical and biophysical research communications 268, 921–927. [DOI] [PubMed] [Google Scholar]
- Zhu AY, Eberhart CG, Jun AS, 2014. Fuchs endothelial corneal dystrophy: a neurodegenerative disorder? JAMA Ophthalmol 132, 377–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu AY, Jaskula-Ranga V, Jun AS, 2018a. Gene Editing as a Potential Therapeutic Solution for Fuchs Endothelial Corneal Dystrophy: The Future Is Clearer. JAMA Ophthalmol 136, 969–970. [DOI] [PubMed] [Google Scholar]
- Zhu C, Joyce NC, 2004. Proliferative response of corneal endothelial cells from young and older donors. Investigative ophthalmology & visual science 45, 1743–1751. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zha Y, Cai J, Zhang Y, 2018b. Descemet stripping automated endothelial keratoplasty versus descemet membrane endothelial keratoplasty: a meta-analysis. Int Ophthalmol 38, 897–905. [DOI] [PubMed] [Google Scholar]
- Zhu YT, Chen HC, Chen SY, Tseng SC, 2012. Nuclear p120 catenin unlocks mitotic block of contact-inhibited human corneal endothelial monolayers without disrupting adherent junctions. J Cell Sci 125, 3636–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Cheng P, Weintraub H, 1996. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2–2, and HEB. Mol Cell Biol 16, 2898–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziaei A, Schmedt T, Chen Y, Jurkunas UV, 2013a. Sulforaphane decreases endothelial cell apoptosis in fuchs endothelial corneal dystrophy: a novel treatment. Investigative ophthalmology & visual science 54, 6724–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziaei M, Barsam A, Mearza AA, 2013b. Spontaneous corneal clearance despite graft removal in Descemet stripping endothelial keratoplasty in Fuchs endothelial dystrophy. Cornea 32, e164–166. [DOI] [PubMed] [Google Scholar]
- Zinflou C, Rochette PJ, 2017. Ultraviolet A-induced oxidation in cornea: Characterization of the early oxidation-related events. Free Radic Biol Med 108, 118–128. [DOI] [PubMed] [Google Scholar]
- Zoega GM, Arnarsson A, Sasaki H, Soderberg PG, Jonasson F, 2013. The 7-year cumulative incidence of cornea guttata and morphological changes in the corneal endothelium in the Reykjavik Eye Study. Acta Ophthalmol 91, 212–218. [DOI] [PubMed] [Google Scholar]
- Zoega GM, Fujisawa A, Sasaki H, Kubota A, Sasaki K, Kitagawa K, Jonasson F, 2006. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology 113, 565–569. [DOI] [PubMed] [Google Scholar]
- Zygoura V, Baydoun L, Ham L, Bourgonje VJA, van Dijk K, Lie JT, Dapena I, Oellerich S, Melles GRJ, 2018. Quarter-Descemet membrane endothelial keratoplasty (Quarter-DMEK) for Fuchs endothelial corneal dystrophy: 6 months clinical outcome. Br J Ophthalmol 102, 1425–1430. [DOI] [PubMed] [Google Scholar]