

Abstract
Von Willebrand disease (VWD) is the most common inherited bleeding disorder and is mainly caused by dominant-negative mutations in the multimeric protein von Willebrand factor (VWF). These mutations may either result in quantitative or qualitative defects in VWF. VWF is an endothelial protein that is secreted to the circulation upon endothelial activation. Once secreted, VWF multimers bind platelets and chaperone coagulation factor VIII in the circulation. Treatment of VWD focuses on increasing VWF plasma levels, but production and secretion of mutant VWF remain uninterrupted. Presence of circulating mutant VWF might, however, still affect normal hemostasis or functionalities of VWF beyond hemostasis. We hypothesized that inhibition of the production of mutant VWF improves the function of VWF overall and ameliorates VWD phenotypes. We previously proposed the use of allele-specific small-interfering RNAs (siRNAs) that target frequent VWF single nucleotide polymorphisms to inhibit mutant VWF . The aim of this study is to prove the functionality of these allele-specific siRNAs in endothelial colony-forming cells (ECFCs). We isolated ECFCs from a VWD type 2A patient with an intracellular multimerization defect, reduced VWF collagen binding, and a defective processing of proVWF to VWF. After transfection of an allele-specific siRNA that specifically inhibited expression of mutant VWF, we showed amelioration of the laboratory phenotype, with normalization of the VWF collagen binding, improvement in VWF multimers, and enhanced VWF processing. Altogether, we prove that allele-specific inhibition of the production of mutant VWF by siRNAs is a promising therapeutic strategy to improve VWD phenotypes.
Keywords: von Willebrand disease, von Willebrand factor, small-interfering RNAs, hemostasis
Introduction
Von Willebrand disease (VWD) is the most common inherited bleeding disorder that clinically affects around 1 in 10,000 people. 1 VWD is mainly characterized by mucocutaneous bleeding, like nose bleeding or menorrhagia. 2 Also, surgical or dental procedures might lead to critical bleeding events. VWD is caused by quantitative or qualitative defects in von Willebrand factor (VWF), a multimeric glycoprotein produced by endothelial cells and megakaryocytes. 3 VWF primarily functions in hemostasis, where ultra-large VWF strings secreted from endothelial cells attract platelets to sites of vascular damage. Furthermore, VWF acts as chaperone for coagulation factor VIII (FVIII) in the circulation, thereby extending the half-life of FVIII. 4 In the past years, also roles of VWF beyond hemostasis have been described. Examples are roles of VWF in inflammation, angiogenesis, and wound healing. 5 6 7
Treatment of VWD is focused on raising VWF plasma levels, most often on demand after a bleeding event, or prior to a planned surgery or dental procedure. 8 Plasma VWF levels can be raised by administration of desmopressin (DDAVP) or VWF-containing concentrates. 9 10 11 12 DDAVP stimulates the release of endogenous VWF from endothelial cells and is the primary choice of treatment in most patients with VWD type 1 and some patients with VWD type 2. In patients with VWD type 2B, DDAVP is contraindicated because released mutant VWF might lead to dangerously deep thrombocytopenia caused by enhanced binding of mutant VWF to platelets. 13 Also, some patients are unresponsive to DDAVP treatment. 10 Patients who do not respond to DDAVP, or for whom DDAVP is contraindicated, are treated with VWF-containing concentrates. 12 Although DDAVP or VWF-containing concentrates are usually sufficient to stop or prevent bleeding, they have only short-term effects and they do not cope with the continuous release of mutant VWF. Also, VWF-containing concentrates cannot prevent thrombocytopenia in VWD type 2B, which is caused by circulating mutant VWF. Furthermore, presence of mutant VWF might affect processes beyond hemostasis in which VWF plays a role. An example is the development of intestinal angiodysplasia resulting in severe intestinal bleeding. This diathesis is more common among VWD patients than the normal population and is suggested to be caused by long-term exposure to mutant VWF that affects angiogenesis. 14 15 VWD patients with recurrent gastrointestinal bleeding are often treated with repeated dosing of VWF-containing concentrates, which is not always effective, and can cause a burden for patients, because they are usually treated several times a week. 16
Since most VWD is caused by dominant-negative mutations in VWF, others and we previously hypothesized that inhibition of production of only mutant VWF might overcome the abovementioned shortcomings of the current treatment modalities. 17 18 Inhibition of mutant VWF only might be accomplished by small-interfering RNAs (siRNAs) that discriminate between two alleles based on one nucleotide mismatch. We recently published the proof of principle of this approach in human embryonic kidney (HEK)293 cells overexpressing VWF alleles. 17 We showed that allele-specific siRNAs that target a heterozygous single-nucleotide polymorphism (SNP) located on the same allele as a dominant-negative VWD type 2A mutation corrects for the VWD phenotype. 17 For this approach, various siRNAs have been selected to target four frequent SNPs in VWF . It was calculated that 74% of the Caucasian population is heterozygous for at least one of these four SNPs and thus might be a candidate for this approach of allele-specific VWF silencing. The ultimate goal is to inject patients with an allele-specific siRNA that targets a heterozygous SNP located on the same allele as the dominant-negative mutation. This allele-specific siRNA should then be encapsulated in an endothelial-targeted vehicle, to target mutant VWF in the endothelium.
HEK293 cells are a good model to prove the principle of allele-specific VWF inhibition and to select for efficient and specific siRNA candidates; however, HEK293 cells do not endogenously produce VWF. We therefore aim in this study to test the approach of allele-specific VWF inhibition by SNP-targeted siRNAs in endothelial colony-forming cells (ECFCs; previously called blood outgrowth endothelial cells or BOECs). ECFCs are cultured endothelial cells that can be isolated from peripheral blood. 19 20 We show that siRNAs are able to selectively inhibit VWF alleles in healthy control ECFCs and that allele-specific siRNAs can improve the VWD phenotype of ECFCs that were isolated from a VWD type 2A patient with the VWF p.Cys1190Tyr mutation.
Methods
Patients and Controls
A VWD type 2A patient with the VWF p.Cys1190Tyr (c.3569G > A) mutation and five healthy control subjects were included in the study. A total of 50 to 70 mL of peripheral blood was drawn for ECFC, plasma, and genomic DNA isolation. The patient's plasma was analyzed for VWF antigen (VWF:Ag), VWF activity, and FVIII activity. VWF:Ag in plasma was determined using the STA LIA VWF:Ag test (Stago, Leiden, the Netherlands) and was analyzed on the Sta-R Max analyzer (Stago) with a commercial STA VWF:Ag calibrator (STA Unicalibrator, Stago) as the reference. VWF activity was determined with the VWF ristocetin-triggered GPIb binding assay (VWF:GPIbR) with the HemosIL AcuStar VWF:RCo reagent (Werfen IL, Breda, the Netherlands). Samples were analyzed on the BIO-FLASH (Werfen) and a commercial calibrator (supplied with the HemosIL AcuStar VWF:RCo) was used as the reference. The FVIII activity was determined using an automated one-stage clotting assay on the STA-R MAX analyzer (Stago) with Sta-immunodef VIII (Stago) and STA-CK Prest 5 (APTT) (Stago) reagents. Commercial normal pool plasma (STA Unicalibrator, Stago) was used as the reference. Plasma characteristics of the patient and controls included in the study are summarized in Table 1 .
Table 1. Plasma phenotype of included subjects.
| VWF:Ag (IU/dL) | VWF:GPIbR (IU/dL) | FVIII:C (IU/dL) | VWF:GPIbR/VWF:Ag | |
|---|---|---|---|---|
| ECFC C1 | 105 | 73 | 97 | 0.70 |
| ECFC C2 | 80 | 68 | 98 | 0.85 |
| ECFC C3 | 101 | 87 | 136 | 0.86 |
| ECFC C4 | 108 | 95 | 149 | 0.88 |
| ECFC C5 | 85 | 80 | 144 | 0.94 |
| ECFC 2A | 107 | 27 | 78 | 0.25 |
Abbreviations: VWF, von Willebrand factor; VWF:Ag, VWF antigen; VWF:GPIbR; VWF ristocetin-triggered GPIb binding assay.
Genomic DNA was used to confirm the mutation in the VWD patient and to determine the genotypes in all subjects for four VWF SNPs: rs1800378, rs1063856, rs1063857, and rs1800380. Genotypes were determined by Taqman SNP genotyping assays (Thermo Fisher Scientific, Carlsbad, California, United States).
The study protocol was approved by the institutional ethical review board. Informed consent was obtained from all subjects in accordance with the Declaration of Helsinki.
SNP Phasing
PacBio long-read single-molecule sequencing was used for linkage analysis of the heterozygous SNP and the p.Cys1190Tyr mutation. RNA was isolated from the ECFC cell pellet using the RNeasy Micro Kit (Qiagen, Venlo, the Netherlands). Complementary DNA (cDNA) was synthesized using SuperScript II Reverse Transcriptase (Thermo Fisher Scientific) with primers designed to span a region of 2.8 kb (forward primer: CACCTTCAGTGGGATCTGCC; reverse primer: TTTCAAGACCTCGCTGGTGG). VWF -specific products were amplified using the KAPA HiFi HotStart ReadyMix PCR kit (Roche Diagnostics, Mannheim, Germany). Amplicons were barcoded and SMRTbell adapters were added using the SMRTbell Barcoded Adapter Complete Prep Kit (Pacific Biosciences, Menlo Park, California, United States). Barcoded amplicons were sequenced using a P6-C4 SMRT cell on a Pacific Biosciences RSII sequencer. Error-free circular consensus reads were mapped to the reference, followed by variant calling and resolution of variant phase using WhatsHap suit.
ECFC Isolation and Culture
ECFCs were isolated from peripheral blood that was drawn in Lithium Heparin tubes (ECFC C1, C2, and C4; BD Biosciences, Erembodegem, Belgium) or in Sodium Heparin CPT Mononuclear Cell Preparation tubes (ECFC C3, C5, and 2A; BD Biosciences). Blood was diluted 1:1 in phosphate buffered saline (PBS) and layered over Ficoll-Paque (LUMC Pharmacy, Leiden, the Netherlands), followed by centrifugation. The mononuclear cell fraction was washed twice in PBS supplemented with 10% fetal bovine serum (FBS), after which cells were taken up in Lonza (ECFC C2 and 2A) or PromoCell (ECFCs C1, C3, and C5) endothelial culture medium. Lonza endothelial culture medium consisted of 500 mL EBM-2 medium (Lonza, Breda, the Netherlands) supplemented with the EGM-2 BulletKit (Lonza), 100 mL FBS (GIBCO, Invitrogen, Carlsbad, California, United States), and 7 mL Antibiotic-Antimycotic solution (Sigma-Aldrich #A5955, St. Louis, Missouri, United States). PromoCell endothelial culture medium consisted of 500 mL Endothelial Cell Growth Medium 2 (PromoCell C-22111, Heidelberg, Germany), 50 mL FBS (GIBCO), and 5 mL Antibiotic-Antimycotic solution (Sigma-Aldrich). Mononuclear cells were plated in 48-well plates (Nunc Cell-Culture Treated Multidishes, Nunclon, Roskilde, Denmark or Sarstedt TS plates, Sarstedt, Nümbrecht, Germany) precoated with 50 µg/mL rat tail collagen type I (BD Biosciences). The medium was refreshed every other day until day 21. Passaging and usage of cells are performed as earlier described, 20 but with PromoCell culture medium. For the control ECFCs, a single clone was used for each experiment. A pool of clones was used for the experiments with ECFC 2A.
siRNA Transfections
Custom Silencer Select siRNAs (Life Technologies, Bleiswijk, the Netherlands), a negative control siRNA (siNEG, 4404020, Life Technologies), or an siRNA against VWF (siVWF, s14834, Life Technologies) were transfected in ECFCs using DharmaFECT duo transfection reagent (Dharmacon, Lafayette, Colorado, United States). For transfections, 100,000 to 125,000 cells per well were plated on rat tail collagen (50 µg/mL; BD biosciences) coated wells of a 24-well plate. siRNAs were transfected into ECFCs 24 hours after plating the cells. Before transfection, cells were washed twice with Hanks' Balanced Salt solution (Thermo Fisher Scientific) and once with PromoCell EGM2 culture medium. siRNAs and DharmaFECT duo were separately diluted in Optimem1 (Thermo Fisher Scientific) and mixed together by pipetting. After 20 minutes of incubation, the siRNA–DharmaFECT mixture was complemented with Optimem1 supplemented with 4% FBS. Cells were incubated with 150 µL transfection mixture containing a high concentration of siRNA for 3 hours. After the 3-hour incubation, 250 µL culture medium was added to obtain a final siRNA concentration of 20 nM (unless otherwise stated). The medium was refreshed 24 hours after transfection and every other day thereafter. Six days after transfection, the medium was harvested and cells were lysed with protein lysis buffer containing Optimem1, 0.1% Triton X-100 (Sigma-Aldrich) and a tablet of cOmplete protease inhibitor cocktail with EDTA (Roche Diagnostics, Mannheim, Germany).
VWF Protein Analysis
VWF:Ag levels in conditioned medium and protein lysates were measured by enzyme-linked-immunosorbent serologic assay (ELISA) as described before, 17 with the modification that samples were diluted in PBS containing 0.1% Tween 20 (PBS-tween), or in PBS containing 1% bovine serum albumin (BSA) when the VWF:Ag ELISA was performed simultaneously with the VWF collagen binding (VWF:CB) assay. The amount of unprocessed proVWF in lysates was quantified by ELISA.
For VWF:CB, ELISA plates were coated with 0.3% bovine collagen type I (95%) and III (5%) (StemCell technologies, Cologne, Germany) in PBS. Samples were diluted in dilution buffer (PBS containing 1% BSA) and incubated for 2 hours. To increase the signal of the ELISA, biotin (Sigma-Aldrich) was conjugated to rabbit anti-VWF-IgG (A0082; DAKO, Glostrup, Denmark) to generate rabbit anti-VWF-IgG-Biotin. ELISA plates were incubated with rabbit anti-VWF-IgG-Biotin diluted in dilution buffer for 1 hour. Thereafter, ELISA plates were incubated with Streptavidin-(POLY)horseradish peroxidase (Thermo Fisher Scientific) diluted in dilution buffer for 1 hour. o-Phenylenediamine dihydrochloride (Sigma-Aldrich) was used as a substrate and dissolved in 11 mL substrate buffer (22 mM citric acid, 51 mM phosphate, pH 5.0) with the addition of 11 µL 30% H 2 O 2 . The enzymatic reaction was terminated using 2M H 2 SO 4 . Normal pooled plasma was used as the reference.
Quantification of unprocessed proVWF in protein lysates was performed by a sandwich ELISA. ELISA plates were coated with rabbit anti-VWF (Dako) in 100 mM bicarbonate, 500 mM NaCl, at pH 9.0. The ELISA plates were incubated with protein lysates diluted in PBS-tween. Mouse anti-VWFpp conjugated to horseradish peroxidase (CLB-Pro 14.3, Sanquin, Amsterdam, the Netherlands) was used as detection antibody and ELISA plates were incubated with the detection antibody diluted in PBS-tween. o-Phenylenediamine dihydrochloride (Sigma-Aldrich) was used as a substrate and dissolved in substrate buffer with the addition of 11 µL 30% H 2 O 2 . The enzymatic reaction was followed kinetically for 5 minutes. The ratio of unprocessed VWF over the total VWF:Ag levels was used as a measure for the processing of VWF. The average of control samples was set to 1.
VWF multimers were visualized using agarose gel electrophoresis under nonreducing conditions as described before. 17 VWF multimers were visualized using ECL Western Blotting Substrate (Promega, Madison, Wisconsin, United States).
VWF monomers were visualized by western blot under reducing conditions. Protein lysates were reduced using NuPAGE Sample Reducing Agent (Thermo Fisher Scientific) and proteins were separated on a 4 to 12% Bis-Tris gel (Thermo Fisher Scientific). Proteins were transferred to a PVDF membrane (BioRad, Veenendaal, the Netherlands) using a Trans-Blot Turbo Transfer System (BioRad, 1.5A, 15 minutes). Rabbit anti-VWF conjugated to horseradish peroxidase (DAKO, P0226) was used as the detection antibody. The membrane was incubated with the detection antibody diluted in PBS-tween containing 5% nonfat milk (Nutricia, Zoetermeer, the Netherlands). ECL Western Blotting Substrate was used to visualize VWF. Densitometry images were generated and quantified by ImageJ (ImageJ 1.51h, Bethesda, Maryland, United States).
RNA Analysis
RNA isolation, cDNA synthesis, and quantitative polymerized chain reaction (qPCR) analysis with GAPDH as the endogenous reference gene were performed as described before. 20 qPCR was performed to quantify VWF , but also separate VWF alleles. Allele-specific qPCR primers were designed, containing the SNPs on the second last position of the forward primer. Primer sequences and annealing temperatures are shown in Supplementary Table S1 (available in the online version).
Immunofluorescence Staining
ECFCs were fixated using ice-cold methanol and stained as described before. 20 Sheep anti-VWF (Abcam, ab11713), rabbit anti-protein disulphide isomerase (PDI; Stressgen Biotechnologies, San Diego, California, United States), and mouse anti-Golgi matrix protein of 130 kDa (GM130, BD Biosciences) were used as primary antibodies.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, La Jolla, California, United States). Mann–Whitney U -tests were performed to determine significance between specific and aspecific inhibition. One-way ANOVA with Dunnett's multiple comparisons test was used to determine significance between three or four groups. p < 0.05 was considered significant.
Results
Time Course of VWF Inhibition by siVWF in ECFCs
siRNAs are used in this study to inhibit VWF (alleles) in ECFCs. To assess the moment of strongest VWF inhibition on a protein level, but also the duration of VWF inhibition, we transfected ECFCs with an siRNA-targeting total VWF (siVWF) or siNEG and followed the VWF secreted in 24 hours for up to 28 days. Relative VWF inhibition by siVWF was determined by normalization of the VWF:Ag secreted in siVWF-treated cells over the VWF:Ag levels in siNEG-treated cells. The experiment was performed under different conditions, but for all tested conditions the strongest VWF inhibition was observed 6 days after siVWF transfection ( Fig. 1 and Supplementary Fig. S1 [available in the online version]). Furthermore, only after 28 days the VWF levels in siVWF-transfected ECFCs were at the same level as siNEG-transfected ECFCs. Since the strongest effects were observed 6 days after transfection, we decided to consistently measure the effects of the siRNAs on the protein level, 6 days after transfection.
Fig. 1.
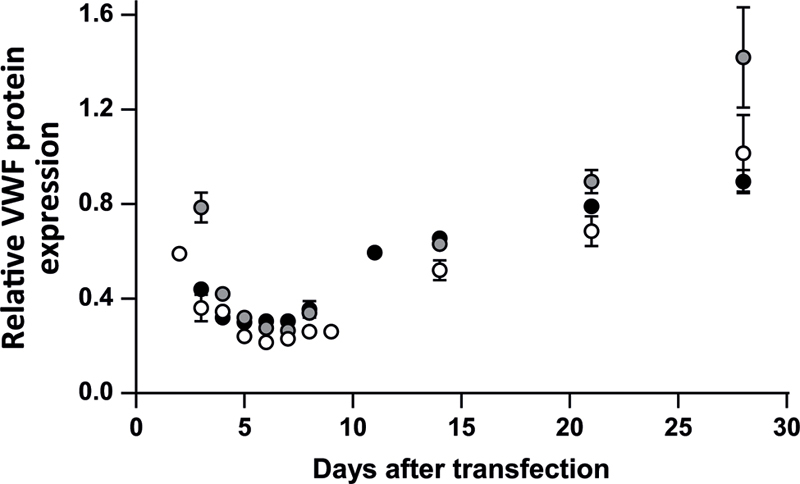
Time course of VWF inhibition by siRNAs in ECFCs. ECFC C3 was plated in a concentration of 125,000 cells per well and cells were transfected with 3 µL DharmaFECT and 10 nM siVWF (inhibits total VWF) or siNEG. 24-hour medium was taken at the time points indicated in the figure. Shown are the VWF:Ag levels measured in cells transfected with siVWF, normalized to the VWF:Ag levels measured in cells transfected with siNEG. The experiments under these conditions were performed three times in duplicate. Separate experiments are shown by different colored circles. Lowest relative VWF protein expression was observed 6 days after transfection. Similar results were observed under slightly different conditions (i.e., cell and DharmaFECT concentration; Supplementary Fig. 1 ). ECFC, endothelial colony-forming cell; nM, nanomolar; siNEG, negative control siRNA; siVWF, siRNA against VWF ; VWF, von Willebrand factor; VWF:Ag, VWF antigen.
Allele-Specific siRNA Inhibition in Healthy Control ECFCs
We previously tested the efficiency and specificity of allele-specific siRNAs that target four common VWF SNPs in VWF-overexpressing HEK293 cells. 17 For the current study, we selected the most effective allele-specific siRNA per SNP target from the previous study. To evaluate the efficiency and specificity of the selected siRNAs on a protein level in ECFCs, siRNAs were transfected in ECFCs that were homozygous for specific SNPs ( Table 2 ). For all siRNAs tested, no or only minor reduction of the production of the untargeted VWF allele was observed as is shown by the relative VWF protein expression in ECFCs that did not harbor the SNP variant corresponding to the transfected siRNA ( Fig. 2A ). On the other hand, a strong VWF knockdown of the targeted allele was observed in ECFCs that contained the SNP variant corresponding to the transfected siRNA ( Fig. 2A ). Knockdown was especially strong and specific for si1451A and si1451G. Results of siRNA transfections with siRNA concentrations of 10 and 50 nM are shown in Supplementary Fig. S2 (available in the online version).
Table 2. ECFCs used per siRNA for testing siRNA efficiency and specificity.
| Protein analysis | RNA analysis | ||
|---|---|---|---|
| siRNA | ECFC used to test siRNA efficiency | ECFC used to test siRNA specificity | ECFC used to test siRNA efficiency and specificity |
| si1451A | C3 (1451AA) | C1 (1451GG) | C5 (1451AG) |
| si1451G | C1 (1451GG) | C3 (1451AA) | |
| si2365A | C3 (2365AA) | C2 (2365GG) | C5 (2365AG) |
| si2365G | C2 (2365GG) | C3 (2365AA) | |
| si2385T | C3 (2385TT) | C2 (2385CC) | C5 (2385TC) |
| si2385C | C2 (2385CC) | C3 (2385TT) | |
| si2880A | C4 (2880AA) | C1 (2880GG) | C5 (2880AG) |
| si2880G | C1 (2880GG) | C4 (2880AA) | |
Abbreviations: ECFC, endothelial colony-forming cell; siRNA, small interfering RNA.
Note: Genotype is indicated between brackets.
Fig. 2.
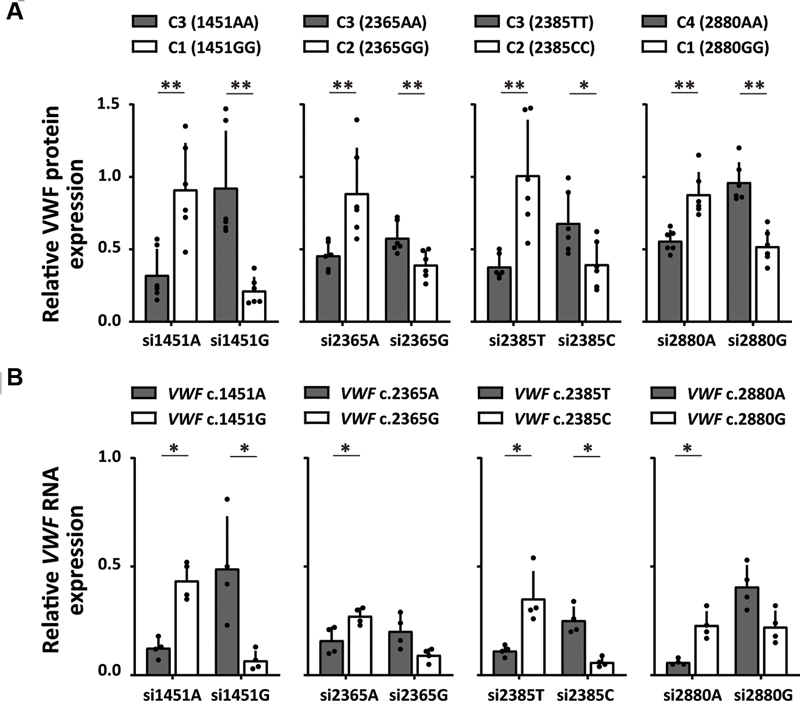
Allele-specific inhibition of VWF in healthy control ECFCs. ( A ) Relative VWF protein expression of ECFCs transfected with allele-specific siRNAs. Shown are the total VWF:Ag levels (conditioned medium + protein lysates) measured in ECFCs transfected with specific siRNAs, normalized to the total VWF:Ag levels measured in the same ECFCs transfected with siNEG. Shown are the mean ± 1 SD of three independent experiments performed in duplicate. Mann–Whitney, * p < 0.05, ** p < 0.01. ( B ) Relative VWF RNA expression of ECFC C5 transfected with allele-specific siRNAs. Shown are the RNA expression levels of VWF alleles of ECFC C5 transfected with specific siRNAs, normalized to the expression level of the same allele measured in ECFC C5 transfected with siNEG. Shown are the mean ± 1 SD of two independent experiments performed in duplicate. Mann–Whitney, * p < 0.05. ECFC, endothelial colony-forming cell; nM, nanomolar; qPCR, quantitative PCR; siNEG, negative control siRNA; SD, standard deviation; si1451G, indicates “small interfering RNA against VWF c.1451G,” all siRNAs are indicated according to this principle; VWF, von Willebrand factor; VWF:Ag, VWF antigen.
The allele-specific siRNAs have been designed with the aim to target heterozygous SNPs located on the same allele as a dominant-negative mutation and thereby improve disease phenotypes. However, the ability of the siRNAs to inhibit VWF alleles in heterozygous ECFCs can only be assessed on the RNA level. We therefore tested the allele-specific siRNAs in an ECFC line that is heterozygous for all four SNPs (ECFC C5; Table 2 ) and performed allele-specific qPCR to determine the relative RNA expression of either of the VWF alleles. On the RNA level, a stronger overall knockdown of VWF was observed compared with what was observed on the protein level, both of the targeted and untargeted alleles ( Fig. 2B ). However, for most siRNAs clear specificity for its targeted allele remained. Only si2365A and si2365G showed minor specificity for its targeted allele. This is in line with results in HEK293 cells. 17
Correction of a VWD Type 2A Multimerization Phenotype by an Allele-Specific siRNA
After proving that allele-specific siRNAs can discriminate between VWF alleles in healthy control ECFCs, we investigated whether allele-specific VWF inhibition could also correct a VWD phenotype in ECFCs. ECFCs from a VWD type 2A patient with the VWF p.Cys1190Tyr mutation and reduced high molecular weight (HMW) multimers were successfully isolated (ECFC 2A; plasma phenotype in Table 1 ). Heterozygous SNPs are essential for allele-specific inhibition of mutant VWF , and genotyping of genomic DNA revealed that the patient is heterozygous for one of the four selected SNPs: VWF c.1451A|G. PacBio long-read sequencing was performed to identify the phasing of this SNP, and showed that VWF c.1451A was located on the same allele as the dominant-negative mutation p.Cys1190Tyr (c.3569A) ( Fig. 3A ). Therefore, ECFCs should be transfected with si1451A to inhibit the mutant allele and correct for the VWD type 2A phenotype. Treatment of ECFC 2A with si1451G should reduce expression of the wild-type allele and is expected to deteriorate the phenotype.
Fig. 3.
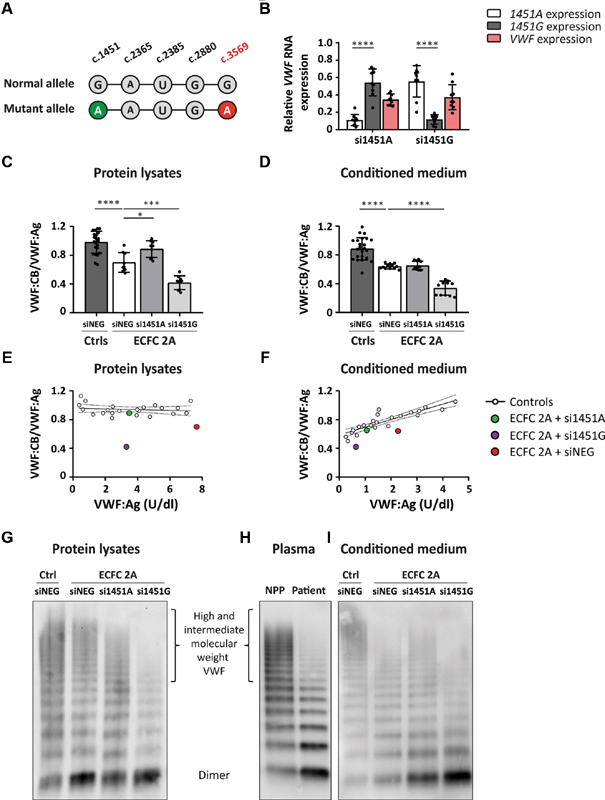
Correction of the VWF multimerization defect in ECFC 2A. ( A ) Phasing of the heterozygous SNP, VWF c.1451A|G, with the dominant-negative mutation VWF p.Cys1190Tyr (c.3569A). PacBio sequencing revealed that VWF c.1451A is located on the same allele as VWF c.3569A. ( B ) Relative VWF RNA expression of ECFC 2A transfected with 20 nM si1451A or si1451G. Shown are the RNA expression levels of VWF alleles c.1451A and c.1451G as well as the total VWF expression. Expression levels were determined in RNA lysates of ECFC 2A transfected with si1451A or si1451G, normalized to the expression level of the same allele measured in ECFCs transfected with siNEG. Shown are the mean ± 1 SD of three independent experiments performed in triplicate. Mann–Whitney (c.1451A vs. c.1451G expression), **** p < 0.0001. VWF:CB/VWF:Ag determined in ( C ) protein lysates and ( D ) conditioned medium of ECFC C1, C2, and C3, transfected with siNEG and ECFC 2A transfected with siNEG, si1451A, and si1451G. Shown are the mean ± 1 SD of three independent experiments performed in triplicate for ECFC 2A and the mean ± 1 SD of three independent experiments performed in duplicate for ECFC C1, C2, and C3. One-way ANOVA, * p < 0.05, *** p < 0.001, **** p < 0.0001. Normal reference line of VWF:CB/VWF:Ag plotted against the VWF:Ag levels measured in ( E ) protein lysates and ( F ) conditioned medium of ECFC C1, C2, and C3 transfected with siNEG, siVWF, and various allele-specific siRNAs. Every white dot represents the average of three independent experiments performed in duplicate of an ECFC line transfected with a specific siRNA. Included in the graphs are the average of VWF:CB/VWF:Ag of three experiments performed in triplicate in which ECFC 2A was transfected with si1451A ( green ), si1451G ( purple ), or siNEG ( red ). VWF multimerization analysis of ( G ) protein lysates, ( H ) plasma, and ( I ) conditioned medium. Protein lysates and conditioned medium were harvested from a healthy control ECFC line transfected with siNEG and ECFC 2A transfected with si1451A, si1451G, and siNEG. ECFC, endothelial colony-forming cell; qPCR, quantitative PCR; siNEG, negative control siRNA; si1451G, indicates “small interfering RNA against VWF c.1451G,” all siRNAs are indicated according to this principle; VWF, von Willebrand factor; VWF:Ag, VWF antigen; VWF:CB, VWF collagen binding.
Treatment of ECFC 2A with either si1451A or si1451G resulted in allele-specific VWF inhibition, as is shown by the allelic VWF expression determined in RNA, 48 hours after transfection ( Fig. 3B ). Inhibition of the mutant allele resulted in a 53 ± 6% reduction of total VWF secreted in the conditioned medium, 6 days after transfection. To assess whether allele-specific inhibition of mutant VWF resulted in phenotypic improvements, we first subjected protein lysates and conditioned medium to the VWF:CB assay, an assay that is able to detect VWF multimerization defects. 21 As expected, we observed both in protein lysates and in conditioned medium of ECFC 2A a lower VWF:CB/VWF:Ag ratio compared to samples derived from healthy control ECFCs ( Fig. 3C, D ). This VWF:CB defect was almost corrected in protein lysates of ECFC 2A transfected with si1451A. Contrarily, inhibition of expression of wild-type VWF by si1451G clearly worsened the VWF:CB phenotype ( Fig. 3C ). Remarkably, in the conditioned medium of ECFC 2A transfected with si1451A, no correction of the VWF:CB defect was observed. Nevertheless, inhibition of the wild-type allele by si1451G did result in significantly lower VWF:CB/VWF:Ag as compared with siNEG-transfected ECFC 2A ( Fig. 3D ).We wondered why inhibition of mutant VWF did not result in correction of VWF:CB/VWF:Ag in the conditioned medium as it did in protein lysates. We hypothesized that downregulation of VWF in general leads to reduced VWF:CB/VWF:Ag in the conditioned medium. To assess this hypothesis, VWF:CB/VWF:Ag was determined in the conditioned medium and protein lysates of ECFC C1, C2, and C3 transfected with allele-specific siRNAs, siNEG, and siVWF. We observed in the conditioned medium, but not in protein lysates, that inhibition of VWF production by (allele-specific) siRNAs resulted in a gradual decrease of VWF:CB/VWF:Ag ( n = 6 for each siRNA in control ECFCs; Fig. 3E, F ). When the VWF:CB/VWF:Ag ratios of ECFC 2A transfected with siNEG, si1451A, or si1451G were plotted against the VWF:CB/VWF:Ag ratios of healthy control ECFCs, we observed that the VWF:CB/VWF:Ag ratio of ECFC 2A transfected with si1451A (inhibition of mutant allele) shifted toward the reference line of the healthy control ECFCs ( n = 9 for ECFC 2A). Whereas inhibition of the normal allele by si1451G resulted in further deterioration of VWF:CB/VWF:Ag (in the conditioned medium as well as in protein lysates, Fig. 3E, F ).
Although multimerization defects can be detected by the VWF:CB ELISA, the assay gives no information on the exact multimerization pattern. To assess the multimerization pattern, plasma, conditioned medium, and protein lysates were subjected to the VWF multimer analysis. In the conditioned medium, a slight decrease of HMW VWF was observed in the patient-derived ECFCs compared with control ECFCs ( Fig. 3I ). This was comparable to the multimerization defect observed in the patient's plasma ( Fig. 3H ). Inhibition of mutant VWF resulted in a slight increase in HMW VWF. Inhibition of production of wild-type VWF clearly worsened the multimerization pattern in the conditioned medium ( Fig. 3I ). No clear decrease of HMW VWF was observed in the protein lysates of ECFC 2A compared with the protein lysates of healthy control ECFCs. However, a clear increase in the intensity of the dimer band was apparent ( Fig. 3G ). Also, a slight change in the running pattern is visible, i.e., VWF from ECFC 2A seems to migrate slightly slower than the VWF from the control ECFC. After inhibition of mutant VWF by si1451A, the intensity of the dimer band clearly decreased; however, this coincided with a small decrease in HMW VWF. Furthermore, VWF migration shifted toward the pattern of the control ECFC. When the wild-type allele was inhibited by si1451G, the multimerization pattern deteriorated with a decrease in HMW VWF and an increase in the intensity of the dimer band.
Improved VWF Processing after Allele-Specific Inhibition of Mutant VWF in ECFC 2A
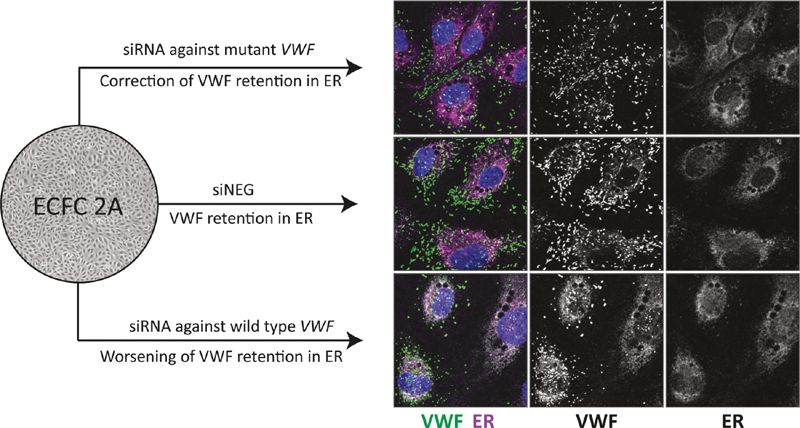
The multimerization pattern of protein lysates of ECFC 2A showed an increase in the intensity of the dimer band. This suggests that the processing of VWF is affected. Dimerization of VWF takes place in the endoplasmic reticulum (ER), after which VWF multimers are formed in the Golgi. 22 To test whether VWF is retained in the ER, we performed a co-staining of VWF and ER marker PDI. Indeed, a clear overlap of VWF and PDI staining is observed in ECFC 2A transfected with siNEG, indicating ER retention ( Fig. 4A ; ECFC 2A + siNEG, second column). This was not observed in a control ECFC transfected with siNEG ( Fig. 4A ; control + siNEG, first column). No colocalization of VWF with the Golgi was observed, indicating pure ER retention for this mutation ( Supplementary Fig. 3 , available in the online version). When the mutant allele was inhibited by si1451A, retention of VWF in the ER was clearly decreased (best observed in the VWF grayscale image) and many cells showed no ER retention at all ( Fig. 4A ; ECFC 2A + si1451A, third column). Inhibition of wild-type VWF by si1451G resulted in a rather severe cellular phenotype, with increased ER retention ( Fig. 4A ; ECFC 2A + si1451G, fourth column).
Fig. 4.
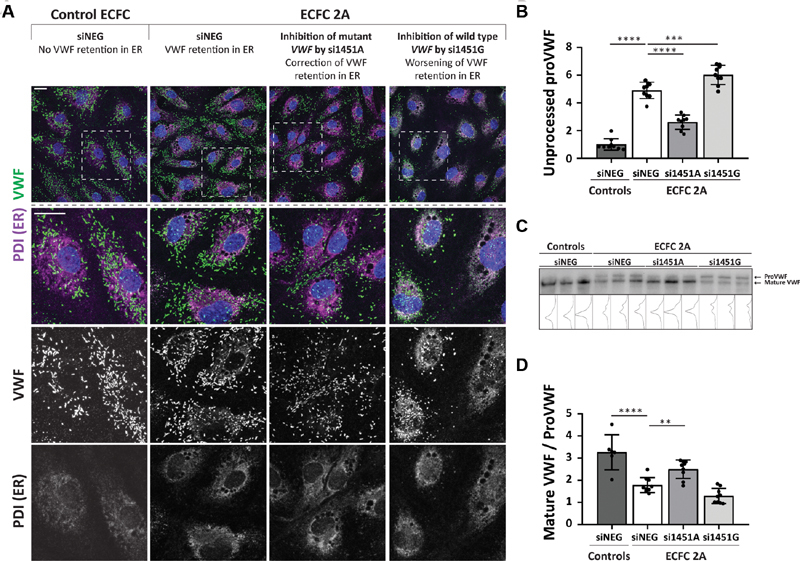
Correction of the VWF processing defect in ECFC 2A. ( A ) Confocal images of a healthy control ECFC transfected with siNEG and ECFC 2A transfected with siNEG, si1451A, and si1451G. ECFCs were stained for VWF, PDI (ER), and nuclei. The upper row shows an overview image with VWF ( green ), PDI ( magenta ), and nuclei ( blue ). The second row shows a zoom-in of the upper image. Colocalization between VWF and PDI is shown in gray . The third and fourth rows show grayscale images of VWF and PDI staining, respectively. Scale bar represents 20 µm. Images were taken with the Leica TCS SP8 X WLL converted confocal microscope equipped with a 63x/1.40 NA Plan Apo oil immersion objective. ( B ) Quantification of unprocessed proVWF measured by ELISA in protein lysates of control ECFCs transfected with siNEG or ECFC 2A transfected with siNEG, si1451A, or si1451G. Shown are the mean ± 1 SD of three independent experiments performed in triplicate for ECFC 2A and the mean ± 1 SD of nine randomly selected protein samples of ECFC C1, C2, and C3. The average of the control ECFCs is set to one. One-way ANOVA, *** p < 0.001, **** p < 0.0001. ( C ) Western blot of protein lysates of control ECFCs transfected with siNEG or ECFC 2A transfected with siNEG, si1451A, or si1451G. Protein lysates were run under reduced conditions on a 4–12% Bis-Tris gel. Shown are protein lysates of ECFC C1, C2, and C3 transfected with siNEG and the three samples of a triplicate experiment for ECFC 2A transfected with siNEG, si1451A, and si1451G. ( D ) Quantification of the western blot shown in panel (C). Shown is the mean ± 1 SD of quantified western blots performed on protein lysates of three independent experiments performed in triplicate for ECFC 2A and the mean ± 1 SD of six protein lysate samples of ECFC C1, C2, and C3. One-way ANOVA, ** p < 0.01, **** p < 0.0001. ECFC, endothelial colony-forming cell; ER, endoplasmic reticulum; PDI, protein disulphide isomerase; si1451G, indicates “small interfering RNA against VWF c.1451G,” all siRNAs are indicated according to this principle; siNEG, negative control siRNA; VWF, von Willebrand factor; VWF:Ag, VWF antigen.
VWF is produced as proVWF that is dimerized in the ER. After dimerization of proVWF in the ER, VWF is translocated to the Golgi where the propeptide is cleaved from VWF by furin. 23 Increased ER retention of VWF results in an increase in unprocessed proVWF in the cells. This can be quantified by an ELISA that measures the amount of propeptide that is still bound to VWF. Protein lysates of ECFC 2A showed a 4.9-fold increase in unprocessed VWF compared with protein lysates of control ECFCs. This was 1.9-fold decreased after inhibition of production of the mutant allele by si1451A ( Fig. 4B ). Compared with control ECFCs, sixfold more unprocessed VWF was present in ECFC 2A in which the wild-type allele was inhibited by si1451G.
A western blot in which the samples are run under reducing conditions can be used as an alternative method to identify defects in the processing of VWF. In this western blot, an increase in the intensity of the unprocessed proVWF band was detected in protein lysates of ECFC 2A compared with control ECFCs ( Fig. 4C ). The intensity of the unprocessed proVWF band decreased after inhibition of the mutant allele by si1451A. An increase in the intensity of the unprocessed proVWF band was observed when the wild-type allele was inhibited by si1451G. Quantification of the western blot confirms the defective processing of VWF and improvement of the phenotype after transfection of si1451A that inhibited expression of the mutant allele ( Fig. 4D ).
Discussion
Allele-specific siRNAs can be used to selectively inhibit mutant alleles to improve disease phenotypes. Previously, we have shown that allele-specific siRNAs that distinguish two VWF alleles based on one nucleotide mismatch can specifically inhibit VWF alleles in HEK293 cells, and that these siRNAs can improve a VWD phenotype. 17 Here, we aimed to extend the proof of concept of allele-specific VWF inhibition to an ex vivo setting by the use of patient-derived and healthy control ECFCs. In this study, we prove that allele-specific siRNAs can inhibit single VWF alleles based on one nucleotide mismatch in healthy control ECFCs. This was shown both on protein and RNA levels. Also, inhibition of the mutant allele in ECFCs isolated from a VWD type 2A patient resulted in clear improvements in the patient's cellular phenotype ( Fig. 5 ).
Fig. 5.
Visual Summary: Allele-specific inhibition of mutant von Willebrand factor (VWF) by small-interfering RNAs (siRNAs) in von Willebrand disease type 2A endothelial colony-forming cells results in correction of VWF retention in the endoplasmic reticulum (ER).
SNPs in VWF with a high minor allele frequency have been used in this study as a target to inhibit VWF alleles. This approach was chosen since with only a small number of SNPs, a large percentage of the patient population can be reached. From this study it became evident that especially si1451A and si1451G have high potency as allele-specific siRNAs, and both siRNAs proved successful to, respectively, correct or deteriorate a VWD type 2A phenotype ex vivo. Whether the other siRNAs have the same ability to correct for VWD phenotypes remains to be unanswered. It is however unlikely that si2365A or si2365G have similar effects as si1451A or si1451G. Fortunately, exclusion of VWF c.2365A|G does not reduce the fraction of the targeted patient population, since VWF c.2365A|G is in almost complete linkage disequilibrium with VWF c.2385C|T. 24 Furthermore, the efficacy of the allele-specific siRNAs in this study may be improved by alternating the chemical modification of the siRNAs. 25 Also, more SNP targets could be tested to increase the percentage of the targeted patient population.
The ability of the allele-specific siRNAs to correct a VWD phenotype was tested in ECFCs isolated from a VWD type 2A patient with the VWF p.Cys1190Tyr mutation. This mutation is characterized by a clearly defined laboratory phenotype with reduced HMW multimers and reduced VWF:CB. 26 Inhibition of mutant VWF by si1451A resulted in clear improvements in VWF processing and the VWF:CB/VWF:Ag in protein lysates. Remarkably, no increase of VWF:CB/VWF:Ag was observed in the conditioned medium of ECFC 2A in which expression of the mutant allele was inhibited ( Fig. 3D ). It appeared, however, that also a reduced VWF:CB/VWF:Ag was observed in healthy control ECFCs transfected with siRNAs that reduced the overall expression of VWF ( Fig. 3F ). This decrease in VWF:CB/VWF:Ag might be a result of the formation of shorter Weibel–Palade bodies after inhibition of VWF, as was observed by Ferraro et al. 27 When the VWF:CB/VWF:Ag of ECFC 2A transfected with si1451A was plotted against the normal reference line, VWF:CB/VWF:Ag was almost normalized. Also, the multimerization pattern of VWF in the conditioned medium showed an increase in HMW VWF in ECFCs transfected with si1451A compared with the siNEG-transfected cells. The reason for discrepancy between the VWF:CB assay and the VWF multimer analysis remains uncertain. It is possible that the VWF:CB assay is sensitive for altered ratios between HMW and low-molecular-weight VWF. Indeed, the decrease in VWF:CB after inhibition is undesirable. Further in vivo studies should reveal whether reduced VWF:CB after downregulation of VWF is also apparent in vivo and what the potential consequences of this effect are.
ECFCs are used in this study as an ex vivo cell model for VWD. ECFCs are the only source of proliferative endothelial cells that can be isolated from patients by a peripheral blood venepuncture. 28 The use of ECFCs has the advantage that the cells are proliferative, have generally a high VWF production, and are easy to transfect. 29 However, also clear variations between various ECFC cell lines emerge. 20 Most importantly, clear variations in VWF expression exist between ECFC lines and it was therefore suggested to be cautious when comparing different ECFC lines. 20 When allele-specific siRNAs are tested in (patient-derived) ECFCs, the effects are determined in ECFCs transfected with either an allele-specific siRNA or a negative control siRNA. Any effects of the allele-specific siRNA compared with the negative control siRNA are an effect of the siRNA and not due to potential variability of the ECFC, as the cells are used as their own control. Therefore, we are confident to draw conclusions on the effects of allele-specific siRNAs on ECFCs from a single patient. To assess the efficiency and specificity of the allele-specific siRNAs on a protein level, we had no choice but to compare different healthy control ECFC lines ( Fig. 2A ). Although the ECFC lines in this study were carefully selected based on their proliferative state and VWF production, differences between ECFC lines could not be avoided. This was also reflected in variations in the efficiency of siVWF to inhibit VWF production in ECFCs. For example, we reproducibly observed a stronger relative VWF inhibition by siVWF in ECFC C1 compared with ECFC C2 (data not shown). No firm conclusions can therefore be drawn on the efficiency and specificity of the allele-specific siRNAs on a protein level that have been tested in the different healthy control ECFC lines ( Fig. 2A ).
The use of siRNAs as a therapeutic application requires persistence of the siRNA activity over a longer period of time. Clinical trials with siRNAs that target liver-expressed genes showed persistent siRNA-mediated downregulation of several genes for more than a month. 30 31 32 In ECFCs, downregulation of VWF was observed for up to 28 days. Furthermore, siRNA-mediated inhibition of Icam2 and Tie2 in mouse endothelium by siRNAs that were complexed in polymeric nanoparticles or a cationic lipoplex delivery system, respectively, resulted in persistent, and around 80%, Icam2 and Tei2 inhibition for more than 21 days after a single dose. 33 34 These results of long-term gene inhibition are promising for further developments of siRNA therapeutics for endothelial genes, like VWF .
To conclude, we show that allele-specific siRNAs are effective in the inhibition of single VWF alleles in ECFCs. Inhibition of expression of the mutant allele in ECFCs isolated from a VWD type 2A patient with the VWF p.Cys1190Tyr mutation resulted in clear improvements of the cellular phenotype. Further studies in VWD mouse models are needed to translate the positive ex vivo results to an in vivo model, and to show whether the allele-specific siRNAs are able to correct a bleeding phenotype.
Acknowledgments
The authors would like to thank Petra Noordijk and Lejla Mahic (LUMC, Department of Internal Medicine, Clinical Epidemiology, the Netherlands) for performing DNA isolations, and Suzan de Boer (LUMC, Department of Internal Medicine, Thrombosis and Hemostasis, the Netherlands) for ECFC isolations. We also would like to thank Emile de Meijer (LUMC, Human Genetics, the Netherlands) for PacBio sequencing sample preparation.
Funding Statement
Funding This study was financially supported by a research grant from the Landsteiner Foundation for Blood Transfusion Research (grant 1504).
Conflict of Interest J.B. received research funding from CSL Behring and is an employee of Sobi. F.A. received research funding from CSL Behring and a travel grant from Sobi. F.W.G.L received research funding from CSL Behring, Takeda/Shire, and uniQure. He is member of a DSMB for Roche. He is consultant for Takeda, Biomarin, and uniQure of which fees go to the institution. J.E. received research funding from CSL Behring. J.E. reports grants from Landsteiner Foundation for Blood Transfusion Research, during the conduct of the study.other authors declare no conflicts of interest.
Authors’ Contributions
A.d.J. designed the study, performed the experiments and data analyses, and wrote the manuscript. R.J.D. performed the experiments. J.B., F.A., and F.W.G.L. were involved in the control and patient inclusions. S.Y.A. contributed to the analysis of PacBio sequencing data. B.J.M.v.V. contributed to discussions and reviewing of the manuscript. J.E. designed the study, interpreted the data, and contributed to writing of the manuscript. All authors revised and approved the final version of the manuscript.
What is known about this topic?
Continued circulation of mutant VWF negatively affects hemostasis in VWD patients.
Most VWDs are caused by dominant-negative mutations.
siRNAs can discriminate between two alleles with only one nucleotide mismatch and can therefore inhibit mutant alleles.
What does this paper add?
Inhibition of VWF in ECFCs by siRNAs results in reduced VWF levels up to 21 days in cultured endothelial cells.
siRNAs allow allele-specific inhibition of VWF alleles in ECFCs.
siRNA-mediated inhibition of mutant VWF in patient-derived ECFCs results in clear improvements in the cellular phenotype.
Supplementary Material
References
- 1.Leebeek F W, Eikenboom J C. Von Willebrand's disease. N Engl J Med. 2016;375(21):2067–2080. doi: 10.1056/NEJMra1601561. [DOI] [PubMed] [Google Scholar]
- 2.Tosetto A, Rodeghiero F, Castaman G. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD) J Thromb Haemost. 2006;4(04):766–773. doi: 10.1111/j.1538-7836.2006.01847.x. [DOI] [PubMed] [Google Scholar]
- 3.Springer T A. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124(09):1412–1425. doi: 10.1182/blood-2014-05-378638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss H J, Sussman I I, Hoyer L W. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand's disease. J Clin Invest. 1977;60(02):390–404. doi: 10.1172/JCI108788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawecki C, Lenting P J, Denis C V. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15(07):1285–1294. doi: 10.1111/jth.13696. [DOI] [PubMed] [Google Scholar]
- 6.Starke R D, Ferraro F, Paschalaki K E. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117(03):1071–1080. doi: 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishihara J, Ishihara A, Starke R D. The heparin binding domain of von Willebrand factor binds to growth factors and promotes angiogenesis in wound healing. Blood. 2019;133(24):2559–2569. doi: 10.1182/blood.2019000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leebeek F WG, Atiq F. How I manage severe von Willebrand disease. Br J Haematol. 2019;187(04):418–430. doi: 10.1111/bjh.16186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannucci P M, Ruggeri Z M, Pareti F I, Capitanio A.1-Deamino-8-d-arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrands' diseases Lancet 19771(8017):869–872. [DOI] [PubMed] [Google Scholar]
- 10.Castaman G, Lethagen S, Federici A B. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European study MCMDM-1VWD. Blood. 2008;111(07):3531–3539. doi: 10.1182/blood-2007-08-109231. [DOI] [PubMed] [Google Scholar]
- 11.Franchini M, Mannucci P M. Von Willebrand factor (Vonvendi®): the first recombinant product licensed for the treatment of von Willebrand disease. Expert Rev Hematol. 2016;9(09):825–830. doi: 10.1080/17474086.2016.1214070. [DOI] [PubMed] [Google Scholar]
- 12.Peyvandi F, Kouides P, Turecek P L, Dow E, Berntorp E. Evolution of replacement therapy for von Willebrand disease: from plasma fraction to recombinant von Willebrand factor. Blood Rev. 2019;38:100572. doi: 10.1016/j.blre.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Kruse-Jarres R, Johnsen J M. How I treat type 2B von Willebrand disease. Blood. 2018;131(12):1292–1300. doi: 10.1182/blood-2017-06-742692. [DOI] [PubMed] [Google Scholar]
- 14.Selvam S, James P. Angiodysplasia in von Willebrand disease: understanding the clinical and basic science. Semin Thromb Hemost. 2017;43(06):572–580. doi: 10.1055/s-0037-1599145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchini M, Mannucci P M. Gastrointestinal angiodysplasia and bleeding in von Willebrand disease. Thromb Haemost. 2014;112(03):427–431. doi: 10.1160/TH13-11-0952. [DOI] [PubMed] [Google Scholar]
- 16.WiN study group . de Wee E M, Sanders Y V, Mauser-Bunschoten E P. Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease. Thromb Haemost. 2012;108(04):683–692. doi: 10.1160/TH12-04-0244. [DOI] [PubMed] [Google Scholar]
- 17.de Jong A, Dirven R J, Oud J A, Tio D, van Vlijmen B JM, Eikenboom J. Correction of a dominant-negative von Willebrand factor multimerization defect by small interfering RNA-mediated allele-specific inhibition of mutant von Willebrand factor. J Thromb Haemost. 2018;16(07):1357–1368. doi: 10.1111/jth.14140. [DOI] [PubMed] [Google Scholar]
- 18.Casari C, Pinotti M, Lancellotti S. The dominant-negative von Willebrand factor gene deletion p.P1127_C1948delinsR: molecular mechanism and modulation. Blood. 2010;116(24):5371–5376. doi: 10.1182/blood-2010-02-268920. [DOI] [PubMed] [Google Scholar]
- 19.Ingram D A, Mead L E, Tanaka H. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104(09):2752–2760. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- 20.de Jong A, Weijers E, Dirven R, de Boer S, Streur J, Eikenboom J. Variability of von Willebrand factor-related parameters in endothelial colony forming cells. J Thromb Haemost. 2019;17(09):1544–1554. doi: 10.1111/jth.14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmerman Program Investigators . Flood V H, Gill J C, Friedman K D. Collagen binding provides a sensitive screen for variant von Willebrand disease. Clin Chem. 2013;59(04):684–691. doi: 10.1373/clinchem.2012.199000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lippok S, Kolšek K, Löf A. von Willebrand factor is dimerized by protein disulfide isomerase. Blood. 2016;127(09):1183–1191. doi: 10.1182/blood-2015-04-641902. [DOI] [PubMed] [Google Scholar]
- 23.van de Ven W J, Voorberg J, Fontijn R. Furin is a subtilisin-like proprotein processing enzyme in higher eukaryotes. Mol Biol Rep. 1990;14(04):265–275. doi: 10.1007/BF00429896. [DOI] [PubMed] [Google Scholar]
- 24.1000 Genomes Project Consortium Auton A, Brooks L D, Durbin R M.A global reference for human genetic variation Nature 2015526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malek-Adamian E, Fakhoury J, Arnold A E, Martínez-Montero S, Shoichet M S, Damha M J. Effect of sugar 2′,4′-modifications on gene silencing activity of siRNA duplexes. Nucleic Acid Ther. 2019;29(04):187–194. doi: 10.1089/nat.2019.0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneppenheim R, Michiels J J, Obser T. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood. 2010;115(23):4894–4901. doi: 10.1182/blood-2009-07-226324. [DOI] [PubMed] [Google Scholar]
- 27.Ferraro F, Kriston-Vizi J, Metcalf D J. A two-tier Golgi-based control of organelle size underpins the functional plasticity of endothelial cells. Dev Cell. 2014;29(03):292–304. doi: 10.1016/j.devcel.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin-Ramirez J, Hofman M, van den Biggelaar M, Hebbel R P, Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat Protoc. 2012;7(09):1709–1715. doi: 10.1038/nprot.2012.093. [DOI] [PubMed] [Google Scholar]
- 29.Medina R J, Barber C L, Sabatier F. Endothelial progenitors: a consensus statement on nomenclature. Stem Cells Transl Med. 2017;6(05):1316–1320. doi: 10.1002/sctm.16-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasi K J, Rangarajan S, Georgiev P. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377(09):819–828. doi: 10.1056/NEJMoa1616569. [DOI] [PubMed] [Google Scholar]
- 31.Ray K K, Landmesser U, Leiter L A. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med. 2017;376(15):1430–1440. doi: 10.1056/NEJMoa1615758. [DOI] [PubMed] [Google Scholar]
- 32.Adams D, Gonzalez-Duarte A, O'Riordan W D. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(01):11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 33.Dahlman J E, Barnes C, Khan O. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol. 2014;9(08):648–655. doi: 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fehring V, Schaeper U, Ahrens K. Delivery of therapeutic siRNA to the lung endothelium via novel Lipoplex formulation DACC. Mol Ther. 2014;22(04):811–820. doi: 10.1038/mt.2013.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.