

Abstract
Amphibians have been disappearing at an unprecedented rate worldwide. Among the proposed contributing factors are infectious diseases. Investigations have focused mainly on ranavirus and chytrids; however, additional agents may be relevant stressors. Two novel batrachoviruses have been discovered (ranid herpesvirus 3 [RaHV-3] and bufonid herpesvirus 1 [BfHV-1]). Their clinical role is still to be clarified; however, both have been associated with obvious skin lesions in their respective hosts. Herein we present 2 consensus PCR protocols that can be used to detect all of the known and, possibly, yet to be discovered batrachoviruses. We targeted a 200 nt long, highly conserved region of the DNA terminase gene. We established a sensitive protocol, which can detect both European batrachoviruses (European batrachovirus PCR protocol; RaHV-3 and BfHV-1) and a panbatrachovirus PCR protocol detecting all known batrachoviruses, including ranid herpesvirus 1 and 2 (RaHV-1, -2). The limit of detection (LOD) for the European batrachovirus protocol was 101 copies of RaHV-3 and 102 copies of BfHV-1 per reaction. The panbatrachovirus protocol could detect all known batrachoviruses with LODs of 103 (RaHV-3, BfHV-1, RaHV-1) to 104 copies (RaHV-2) per reaction. These novel detection tools can be used as a first line of detection when herpesviral infection in amphibians is suspected, followed by additional PCRs with herpesvirus-specific primers in the case of known viral species, or sequencing as in the case of novel batrachoviruses.
Keywords: Alloherpesviridae, amphibian, Batrachovirus, frogs, herpesvirus, PCR, toads, wildlife
Introduction
Herpesviruses are large double-stranded DNA viruses that can infect a wide range of hosts. All of the known herpesviruses are in the order Herpesvirales, which is divided into 3 families. The family Herpesviridae, which includes viral species infecting mammalian, avian, and reptilian hosts; the family Alloherpesviridae, which includes the piscine and amphibian herpesviruses; and the family Malacoherpesviridae, which comprises the invertebrate herpesviruses.2 The family Alloherpesviridae comprises 4 genera: Batrachovirus, Cyprinivirus, Ictalurivirus, and Salmonivirus. All known amphibian herpesviruses are in the genus Batrachovirus.
Until recently, only 2 batrachoviruses were known: ranid herpesvirus 1 (RaHV-1) and ranid herpesvirus 2 (RaHV-2).1 RaHV-1 is the etiologic agent of the Lucké adenocarcinoma of leopard frogs (Lithobates pipiens; https://www.itis.gov/).6,8 RaHV-2, is the only batrachovirus grown in cell culture to date, and was isolated from the urine of a tumor-bearing frog (Lucké adenocarcinoma), although the virus was shown not to have oncogenic potential.3,12
Two new batrachoviruses have been discovered in Switzerland: ranid herpesvirus 3 (RaHV-3) in free-ranging common frogs (Rana temporaria), and bufonid herpesvirus 1 (BfHV-1) in free-ranging common toads (Bufo bufo).9,10 Affected frogs and toads have proliferative dermatitis characterized by epidermal hyperplasia. The discovery of these 2 novel herpesviruses within 2 y suggests that other herpesviral species may be present in free-ranging frogs and toads. Therefore, a broad detection tool that could detect virtually any batrachovirus would be critical for the discovery of the other herpesviruses infecting free-ranging amphibian populations, as well as captive and zoo collections around the world.
Consensus primer PCR protocols have been developed to detect known and unknown human and animal herpesviral sequences, but not for batrachoviruses, to our knowledge.16 The same approach has been followed to develop PCR tests for the detection, among others, of orthopoxviruses, paramyxoviruses, adenoviruses, and a core group of double-stranded DNA viruses.5,11,13,14,17
The basic principle of consensus PCR protocols is the development of a set of degenerate primers designed to target a conserved nucleotide sequence across multiple (viral) species, which, ideally, would allow for the detection of any organism that belongs to a specific taxonomic group, including those yet to be discovered. Accordingly, the aim of our investigation was to develop a consensus PCR protocol that would detect all of the known batrachoviruses (RaHV-1, RaHV-2, RaHV-3, BfHV-1), and ideally any of the yet to be discovered batrachoviruses.
Material and methods
Viruses and viral DNA templates
The sequence of the terminase gene (GenBank accessions RaHV-1: DQ665917.1, RaHV-2: DQ665652.1, RaHV-3: KX832224.1, BfHV-1: MF143550.1) was considered an ideal PCR target given the high level of conservation of this gene across herpesviruses, including Batrachovirus.1,2 Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used to align and compare the selected sequences using standard settings. Regions of the homologous genes characterized by the highest similarities across the 4 viruses were considered for primer design. The candidate primer sequences were obtained from the selected regions from all of the viruses, and in silico assessment of the primers’ features before synthesis was carried out using Oligo Calc (oligonucleotide properties calculator, http://biotools.nubic.northwestern.edu/OligoCalc.html). Genomic DNA (gDNA) was available for RaHV-3 and BfHV-1 from previous studies.9,10 The gDNA of RaHV-1 became available only at the end of our study as a kind gift of Dr. Louise Rollins-Smith (Vanderbilt University); RaHV-2 gDNA was not available throughout our study.
Consensus primers
A 2-step strategy was followed for the design of consensus primers. First, consensus primers that were tailored more closely toward the European batrachoviruses RaHV-3 and BfHV-1 were designed (European batrachovirus PCR protocol). Second, these primers would be modified later, if necessary, to amplify the other 2 known batrachoviruses, RaHV-1 and -2 (panbatrachovirus PCR protocol), which had been discovered and characterized in the United States.6,7,12 In particular, the primer set comprising a forward (FW; EUPB-FW1) and a reverse (RV; EUPB-RV-1) primer (Fig. 1; Table 1) was designed first to amplify the target regions of RaHV-3 and BfHV-1. Variants of these primers were designed later to obtain the highest sensitivity in viral detection for both the European and American batrachoviruses (Fig. 1; Table 1). The expected size of the amplicon was 200 nt for each of the 4 targeted viruses.
Figure 1.

Selected targeted Batrachovirus genome regions and specific primer annealing. The homologous targeted sequence (terminase gene) of the RaHV-1, -2, -3, and BfHV-1 genomes, together with the annealing regions of the selected primers, are shown. The primers used to amplify the positive control amplicons of RaHV-3 and BfHV-1 are underlined; the primers used to amplify the chimeric positive controls of RaHV-1 and -2 are labeled with 2 distinct colors, blue for their genomes and red annealing to the RaHV-3 backbone of the amplicon. Highlighted in gray are the regions corresponding to the annealing sites of the consensus primers included in the developed PCR protocols. Bold indicates the region where the semi-nested reverse primer anneals to the RaHV-3 and BfHV-1 homologous regions. All of the reverse primers are shown with the reverse and complementary sequence (See color version of this figure online).
Table 1.
Consensus primers sets used during the development of the Batrachovirus PCR protocols.
| Primer name | Primer sequence |
|---|---|
| EUPB-FW-1 | 5′-GTRCARGCTCAYAGAAACAAYACATG-3′ |
| EUPB-FW-2 | 5′-GTRCARGCTCAYAGAAACAACACATG-3′ |
| EUPB-FW-3 | 5′-GTRCARGCTCAYMGWAACVMYACRTG-3′ |
| EUPB-FW-4 | 5′-GTRCARGCTCAYMGWAAMVMBACRTG-3′ |
| EUPB-RV-1 | 5′-GGTCCMGARGGYAYAAAATG-3′ |
| EUPB-RV-2 | 5′-GVTCCMGARGGYAHRAAATG-3′ |
| EUPB-RV-3 | 5′-GVWCCMGARGGYAHRAAATG-3′ |
| EUPB-RV-4 | 5′-GVWCCVGARGGYAHRAAATG-3′ |
Bold values indicate the nucleotide substitutions that were carried out on the primers developed during the study to enhance their sensitivity toward the targeted viral sequences.
Positive control synthesis
Following the identification of the candidate regions for each of the selected viral species, specific primers for each of the homologous sequences were designed to obtain specific amplicons to clone into vectors to be used as positive controls. Specifically for RaHV-3 and BfHV-1, a primer set for each virus was designed annealing to a region located 5′ (33/34 nt) and 3′ (43 nt), respectively to the selected target of the degenerate primers (partial sequence of the terminase gene, ORF53-RaHV-3 and ORF39-BfHV-1; Fig. 1). These included, a FW (RaHV-3 Pos. contr. FW) and a RV (RaHV-3 Pos. contr. RV) primer for the amplification of the RaHV-3 positive control, and of a FW (BfHV-1 Pos. contr. FW ) and a RV (BfHV-1 Pos. contr. RV) primer for the amplification of the BfHV-1 positive control, respectively (Fig. 1).
The gDNA of RaHV-1 and -2 were originally not available, and accordingly we designed chimeric amplicons with viral species-specific 5′ and 3′ regions, respectively, spaced by a backbone consisting in the RaHV-3 homologous region. The chimeric primer sets (one each for RaHV-1 and -2) were characterized by either the specific RaHV-1 or RaHV-2 sequences located at the 5′ primer region, and by a perfect match with the homologous RaHV-3 sequence located at the 3′ primer region (chimeric RaHV-1-FW; chimeric RaHV-1-RV; chimeric RaHV-2-FW; chimeric RaHV-2-RV; Fig. 1). The chimeric RaHV-1 and RaHV-2 positive controls were designed to obtain an amplicon of identical size to those expected from the amplification of the RaHV-3 and BfHV-1 targets. Accordingly, although the cloned RaHV-3 and BfHV-1 positive control fragments were longer than those of RaHV-1 and -2, the degenerate primers amplified an amplicon of the same size for all of the targeted viruses (Fig. 1). The positive controls for RaHV-3 and BfHV-1 were obtained using as template their specific gDNA obtained from common frog and common toad infected skin, respectively.9,10 The selected region of the terminase gene of each of the selected viruses was amplified as follows: briefly, 0.5 µL of FW primer (100 µM) and 0.5 µL of RV primer (100 µM) of each primer set was added to a master mix containing 3 µL of 10× reaction buffer, 0.6 µL of a 10 mM dNTPs mix, 0.15 µL of Taq DNA polymerase (0.75 U; Solis BioDyne), 3.75 µL of MgCl2 (25 mM), 1 µL of either RaHV-3 (for RaHV-1, -2, -3) or BfHV-1 DNA suspension (containing 200 ng of template), and 20.5 µL of distilled deionized water, up to a final volume of 30 µL. RaHV-3 template DNA was used to amplify the chimeric amplicon for use as the positive control for RaHV-1 and -2. The targeted positive control amplicons were obtained with a cycling protocol, which included an initial denaturation step of 3 min at 94°C, followed by 40 cycles composed of a 30 s denaturation step at 94°C, a 30 s annealing step at 50°C (for RaHV-3 and BfHV-1) or 55°C (for RaHV-1 and -2), and a 30 s elongation step at 72°C. A final elongation of 10 min at 72°C was carried out to exhaust the polymerase. The PCR product was then resolved in 1.5% agarose gel and examined under ultraviolet light.
Positive control cloning
The obtained amplicons were gel purified with a conventional kit (NucleoSpin gel and PCR clean-up; Macherey-Nagel) according to the manufacturer’s instructions and then cloned into an expression vector (pGEM-T Easy vector; Promega) according to standard protocols. Briefly, 100 ng of the purified amplicon, resuspended in 2 µL of distilled deionized water, was added to a mix containing 1 µL of pGEM-T Easy vector, 1 µL of T4 DNA ligase (Promega), 1 µL of 10× ligation buffer, and 6 µL of distilled deionized water up to 10 µL, and then incubated at 4°C overnight.
The ligation product was used to transform Stellar competent cells (Takara Bio Europe), according to the manufacturer’s instructions. Briefly, after incubating the ligation mix with the competent cells on ice for 30 min, cells underwent heat shock in a water bath at 42°C for 45 s. The transformed cells were then incubated in a shaker (at 200 rpm) in 500 µL of SOC medium at 37°C for 60 min. Bacteria were finally plated onto lysogeny broth (LB) agar plates containing 40 µL of IPTG (100 mM solution; MilliporeSigma), 80 µL of XGal (20 mg/mL; MilliporeSigma), and 30 µL of ampicillin 100 mg/mL (MilliporeSigma). Plates were incubated at 37°C overnight.
Plasmid purification
Positive colonies were selected and expanded in LB containing 100 mg/mL of ampicillin at 37°C overnight. Minipreps were obtained according to a standard protocol; midiprep preparation was carried out (PureYield plasmid-midiprep system; Promega) according to the manufacturer’s instructions.18 The presence of the amplicon was assessed by excising it from the vector by restriction digestion. Briefly, 5 µL of the DNA suspension, obtained as described above, was admixed with 2 µL of 10× buffer, 1 µL of EcoRI (Promega), 12 µL of distilled deionized water, and incubated at 37°C for 2 h. The digestion was then resolved in 1.5% agarose gel and examined under ultraviolet light. Positive plasmids were submitted to Microsynth (Balgach, Switzerland) for automated Sanger sequencing.
Titration and sensitivity assessment
The cloned target sequences were titrated to determine the sensitivity of the developed PCR platforms when tested against each of them. Following quantification of the DNA (average of 10 consecutive measurements, Nanodrop One spectrophotometer; Thermo Fisher Scientific), the number of plasmid copies/ng of DNA, for each of the viral species considered, was determined on the basis of the weight of the specific plasmid according to the following formula: number of copies = (amount (in ng) × [6.022 × 1023] (molecules/mole))/(length × [1 × 109] (ng/g) × 650 (g/mole of base pair)), using the DNA Copy Number and Dilution Calculator (www.thermofisher.com; ng = nanograms; 6.022 × 1023 = Avogadro’s number (molecules/mole); 650 = weight of a mole of a DNA base pair in a gram).
Following the determination of the weight of each vector containing the specific target sequence of each virus, 10-fold serial dilutions ranging from 105 to 10−2 copies/µL were carried out for RaHV-3 and BfHV-1. For the titration of RaHV-1 and -2, the starting dilutions contained up to 108 copies/µL. The purified target plasmids were used as template for the PCR reactions. The PCR reaction mix was composed of 3 µL of 10× buffer, 0.6 µL of dNTPs (10 mM), 0.5 µL of each FW and RV primer (100 µM), 0.15 µL of Taq DNA polymerase (0.75U; Solis BioDyne), 3.75 µL of MgCl2 (25 mM), 1 µL of DNA suspension, and 20.5 µL of distilled deionized water, up to a total volume of 30 µL (standard conditions). The DNA template dilutions ranged from 108 to 10−2 of plasmid copies/µL, as described above. The targeted amplicons were obtained with the same cycling protocol described above (positive controls), with a standard annealing temperature of 50°C (standard conditions). The PCR products were then resolved in 1.5% agarose gel and examined under ultraviolet light.
Consensus PCR protocol optimization
Magnesium (MgCl2) concentration of the reaction mix, as well as the annealing temperatures of the PCR platforms developed during our investigation, were optimized to improve the sensitivity of the PCR protocol. Different concentrations of MgCl2 (3.125; 3.5; 4; 4.5; 5 mM) and a range of annealing temperatures (45–51°C) were tested. Various amounts of dimethylsulfoxide (DMSO; 0.5, 1, 2, 3, 4, 5 µL), were added to the PCR reaction (maintaining the same final volume) in order to improve its sensitivity.
Semi-nested format for consensus PCR protocol for RaHV-3 and BfHV-1
A semi-nested PCR protocol was also implemented to improve the PCR sensitivity to RaHV-3 and BfHV-1. Following the first amplification run according to the protocol described above, we designed an internal RV primer (SN-RV-1: 5′-ATTTTGAATCTTTGRTCAAACC-3′; 100 µM) annealing in the region between nucleotide 70,811 and 70,790 of RaHV-3, and between nucleotide 50,557 and 50,536 of BfHV-1 of the targeted amplicon (Fig. 1). The amplification protocol was identical in both runs, and to that of the single-run protocol described above. Similarly, 1 µL of each target dilution was used as the DNA template in the first PCR run; 2 µL of each amplification product were used as template in the second PCR run. The final volume of the reaction mix was 30 µL in both runs.
Assay specificity
To determine the specificity of the 2 PCR protocols (European batrachovirus and panbatrachovirus PCR protocols) that we developed, both protocols were tested against a batch of known positive and negative controls from amphibians and irrelevant controls from other animal species. More specifically, we used 5 DNA samples obtained from common frogs confirmed positive for RaHV-3 by a previously developed conventional PCR and 5 confirmed negative samples from common frogs.9 We also tested 10 confirmed positive samples obtained from common toads positive for BfHV-1 by a previously developed conventional PCR and 5 confirmed negative samples.10 Additionally, we tested with both protocols 5 samples from common foxes (Vulpes vulpes) confirmed positive for canine distemper virus (Canine morbillivirus), and 5 samples from blackbirds (Turdus merula) confirmed positive for Usutu virus. Finally, we tested a sample of a tortoise herpesvirus (Testudinid alphaherpesvirus 3) DNA, obtained from infected cell cultures (Terrapene heart cells, ATCC CCL-50). A total of 100 ng of total DNA (or complementary DNA) of each sample was used for this test. The samples were tested at the same conditions described above used for the plasmid controls.
Results
Target sequences
A highly conserved region (153,618–153,817 nt) was identified in the terminase gene of RaHV-1 and in the homologous regions of RaHV-2 (162,581–162,780 nt), RaHV-3 (70,708–70,907), and BfHV-1 (50,454–50,653 nt; Fig. 1). All of the homologous selected regions (amplicon size) spanned 200 nt.
The corresponding regions were amplified with specific primers and cloned into a vector to obtain the species-specific positive controls that would have been used in the PCR reactions (Table 2). The RaHV-3 and BfHV-1 inserts were longer than the RaHV-1 and -2 homologs. This was secondary to the strategy adopted to amplify the target regions of RaHV-3 and BfHV-1 with a few extra buffering nucleotides, both 5′ and 3′ to the target regions. This was considered ideal in order to avoid the occurrence of mutations at the site of annealing of the consensus primers. This strategy could not be adopted when cloning the sequences of RaHV-1 and -2, which were not available as native genomes but were obtained as chimeric positive controls starting from a backbone RaHV-3 sequence carrying 5′ and 3′ of the specific sequences of RaHV-1 and -2, respectively. Although the inserts had different lengths, the amplified region was 200 nt long across all viral species. The identity of each positive control sequence was confirmed by Sanger sequencing.
Table 2.
Main features of the plasmids used as positive controls during the development of the Batrachovirus PCR protocols.
| Unit | RaHV-1 | RaHV-2 | RaHV-3 | BfHV-1 | |
|---|---|---|---|---|---|
| Total plasmid length | nt | 3,200 | 3,200 | 3,324 | 3,324 |
| Insert length | nt | 200 | 200 | 324 | 324 |
| Plasmid MW | ng | 2.654 × 10−9 | 2.654 × 10−9 | 2.757 × 10−9 | 2.757 × 10−9 |
| Plasmid copies | ng | 2.895 × 108 | 2.895 × 108 | 2.787 × 108 | 2.787 × 108 |
Consensus PCR
The first consensus primer set designed (EUPB-FW-1, EUPB-RV-1) spanned 70,708–70,733 nt (FW) and 70,907–70,888 nt (RV) of the RaHV-3 genome, and 50,454–50,479 nt (FW) and 50,653–50,634 nt (RV) of the BfHV-1 genome, respectively (Table 1). The FW primer of the first European batrachovirus primer set (EUPB-FW-1; European batrachovirus PCR protocol) contained 22 of 26 nt identical to the homologous regions of RaHV-3 and BfHV-1. The RV primer (EUPB-RV-1) contained 16 of 20 nt identical to the corresponding RaHV-3 and BfHV-1 specific sequences, respectively (Table 1). This primer set successfully amplified both RaHV-3 and BfHV-1 homologous terminase regions at standard conditions. However, the RaHV-3 amplicon yielded a stronger band than that of BfHV-1, once the PCR products were resolved in agarose, consistent with a likely different sensitivity of the consensus primers toward the 2 distinct targets (RaHV-3 and BfHV-1). More specifically, the minimal number of plasmid copies necessary to obtain a detectable BfHV-1 amplicon band was ~ 10-fold higher than that needed for RaHV-3 amplification, with > 300 copies of BfHV-1 target per reaction needed to obtain a detectable amplification versus ~ 40 copies of RaHV-3 target per reaction (Table 3).
Table 3.
Primer combinations and assay sensitivities assessed during the development of the Batrachovirus PCR protocols.
| Primers set | Viral species | |||
|---|---|---|---|---|
| RaHV-1 | RaHV-2 | RaHV-3 | BfHV-1 | |
| EUPB-FW-1/EUPB-RV-1 | c > 108*§ | c ≥ 106 | c > 101 (40 copies) | c > 102 (300 copies) |
| EUPB-FW-1/EUPB-RV-2 | c ≥ 106 | c ≥ 108* | 105 ≥ c > 101† | 105 ≥ c > 102 |
| EUPB-FW-1/EUPB-RV-3 | c ≥ 106 | c ≥ 108* | 105 ≥ c > 101† | 105 ≥ c > 102 |
| EUPB-FW-2/EUPB-RV-1 | c > 106§ | c ≥ 108 | c ≥ 101‡ | c ≥ 102‡ |
| EUPB-FW-2/EUPB-RV-2 | 106 ≥ c > 104† | c > 108*§ | 105 ≥ c > 101† | 105 ≥ c > 102† |
| EUPB-FW-2/EUPB-RV-3 | 106 ≥ c > 104† | c ≥ 108* | 105 ≥ c > 101† | 105 ≥ c > 102† |
| EUPB-FW-3/EUPB-RV-1 | c > 108*§ | c > 108*§ | 105 ≥ c > 101† | 105 ≥ c > 102† |
| EUPB-FW-3/EUPB-RV-2 | c ≥ 104 | c ≥ 107 | c ≥ 102 | c ≥ 103 |
| EUPB-FW-3/EUPB-RV-3 | c ≥ 105 | c ≥ 106 | c ≥ 104 | c ≥ 105 |
| EUPB-FW-4/EUPB-RV-4 | c ≥ 103‡ | c ≥ 104‡ | c ≥ 103‡ | c ≥ 103‡ |
c = copies.
The 108 suspensions contained 2–4 × 108 copies per microliter.
Values between 2 different dilutions refer to the lowest and highest dilution, which yielded a positive and negative signal, respectively. Specifically, for these primer combinations, no intermediate dilutions were tested.
Recommended primer combinations along with their sensitivities.
No amplification was obtained in the PCR reaction using the plasmid copy number shown in the table, and no additional dilution was considered.
Consensus PCR optimization (European batrachovirus PCR protocol)
BfHV-1
The first objective of the optimization was to increase the sensitivity toward the BfHV-1 homologous terminase sequence, without, ideally, losing any sensitivity toward the RaHV-3 homologous sequence. A range of annealing temperatures was considered; however, no significant improvements in sensitivity were obtained. A similar outcome was obtained with the different concentrations of MgCl2 (magnesium scanning). Finally, a semi-nested format was developed to attempt to increase the sensitivity of the reaction using a new RV primer (SN-RV-1; Fig. 1). Similarly, this additional strategy did not yield significant improvements in sensitivity and was not pursued further (data not shown).
Secondary to the unsatisfactory results obtained with both the annealing temperature and magnesium scanning, a new FW primer was designed (Table 1: EUPB-FW-2) replacing the original FW primer (Table 1: EUPB-FW-1) in the consensus PCR reaction at standard conditions. The new FW primer contained one additional nucleotide substitution at position 21 (Y to C). The new primer set, including the original RV primer (EUPB-RV-1) and the new FW primer (EUPB-FW-2), at standard PCR conditions (annealing temperature and MgCl2) yielded a marked increase in sensitivity toward BfHV-1, which could be detected for the first time using < 100 copies per reaction as template (Table 3). Remarkably, the same primer set had increased sensitivity toward RaHV-3 as well, with a limit of detection (LOD) that was putatively less than ~ 10 copies per reaction (Table 3; Fig. 2B). Interestingly, a positive signal was obtained with as low as 100 copies of plasmid of RaHV-3 and as low as 101 copies of plasmid of BfHV-1 per reaction (Table 3; Fig. 2A); however, this corresponded to an apparent suboptimal (not proportional) drop in the intensity of the bands on the gel than the expected one. This was interpreted as a possible difference between the 2 consecutive dilutions (101–100 RaHV-3; 102–101 BfHV-1), which might have been less than an actual log. Accordingly, we decided to select conservatively the upper dilutions for each viral species as the LOD of the PCR protocol, although our results suggest that the sensitivity could be slightly higher. Major overestimation of the dilutions was considered unlikely, given that the 2 highest dilutions tested (10−1 and 10−2 copies per reaction) did not yield any positive signal on gel (Fig. 2A, B).
Figure 2.

European batrachovirus PCR protocol. The titrations (105–10−2 copies per reaction) of the consensus PCR carried out using the European batrachovirus PCR protocol (EUPB-FW2/EUPB-RV1) are shown. PCR amplification to A. BfHV-1 and B. RaHV-3. The number of the template copies per reaction are shown over the amplicon lines. The 100-nt ladder (Promega) is shown on the right of each panel.
RaHV-3
Following the application of the semi-nested protocol for the amplification of BfHV-1, the same protocol was also applied to RaHV-3. Similar to the result with BfHV-1, no improvement in the sensitivity was achieved toward this target.
Optimization of the consensus PCR for RaHV-1 and RaHV-2 (panbatrachovirus PCR protocol)
Once a consensus PCR able to amplify both the homologous regions of the RaHV-3 and BfHV-1 terminase gene with a satisfactory sensitivity (< 100 copies of target for both viruses per reaction) was set up, we focused on optimizing the current protocol to amplify the remaining 2 known batrachoviruses, RaHV-1 and -2. Accordingly, the selected primers were annealing to the homologous regions of RaHV-1 and -2, namely 153,618–153,643 nt (FW) and 153,617–153,598 nt (RV) of RaHV-1, and 162,581–162,606 nt (FW) and 162,780–162,761 nt (RV) of RaHV-2, respectively.
Using the first primer set derived from the European batrachovirus PCR protocol (EUPB-FW-1, EUPB-RV-1) at standard conditions, we could detect no less than 106 copies of the chimeric RaHV-2 target per reaction, and no chimeric RaHV-1 target could be detected, even using a larger amount of target plasmid (108 copies) per reaction (Table 3).
RaHV-1
The lack of RaHV-1 amplification prompted us to attempt to first increase the sensitivity of the protocol towards RaHV-1. Initially, temperature optimization was carried out by reducing the annealing temperature to 45°C with the first primer set (EUPB-FW-1/EUPB-RV-1), but no detection of the RaHV-1 target could be obtained (data not shown).
Accordingly, we adopted all of the primers previously designed for the amplification of the European batrachoviruses (EUPB-FW-1, EUPB-RV-1, EUPB-FW-2) along with new variants (Table 1). In particular, we developed a new FW primer variant (EUPB-FW-3), which was characterized by 5 nt substitutions in comparison with the original EUPB-FW-1 (Table 1). The substitutions were located at positions 13 (A to M), 15 (A to W), 19 (A to V), 20 (A to M), and 24 (A to R). Furthermore, 2 new RV primer variants were designed as well and were named EUPB-RV-2 and EUPB-RV-3. EUPB-RV-2 was characterized by substitutions at positions 2 (G to V), 14 (Y to H), and 15 (A to R). EUPB-RV-3 was characterized by the same substitutions present in EUPB-RV-2, with an additional substitution at position 3 (T to W; Table 1).
All of the primers available (EUPBs) were tested in multiple combinations in standard conditions (annealing temperature: 50°C). The combinations EUPB-FW-1/EUPB-RV-2, EUPB-FW-1/EUPB-RV-3, EUPB-FW-3/EUPB-RV-2, EUPB-FW-3/EUPB-RV-3, EUPB-FW-2/EUPB-RV-2, and EUPB-FW-2/EUPB-RV-3 successfully detected RaHV-1, whereas the combinations EUPB-FW-3/EUPB-RV-1 and EUPB-FW-2/EUPB-RV-1 did not. The combinations EUPB-FW-1/EUPB-RV-2, EUPB-FW-1/EUPB-RV-3, EUPB-FW-2/EUPB-RV-2, and EUPB-FW-2/EUPB-RV-3 produced weak amplification of RaHV-1 and were not pursued further. The combinations EUPB-FW-3/EUPB-RV-2 and EUPB-FW-3/EUPB-RV-3 had the strongest amplification and, accordingly, a full titration (from 108 to 10−1 copies per reaction) was carried out for each of them. Following titration, the primer sets EUPB-FW-3/EUPB-RV-2 and EUPB-FW-3/EUPB-RV-3 were the most sensitive, detecting as few as 104 and 105 copies of target per reaction, respectively (Table 3).
RaHV-2
The LOD of RaHV-2 with the original primer set (EUPB-FW-1/EUPB-RV-1) was 106 copies per reaction. The combinations of primer sets EUPB-FW-1/EUPB-RV-2, EUPB-FW-1/EUPB-RV-3, EUPB-FW-3/EUPB-RV-2, EUPB-FW-3/EUPB-RV-3, EUPB-FW-1/EUPB-RV-3, and EUPB-FW-2/EUPB-RV-3 successfully detected the target plasmid, whereas the combinations of the primer sets EUPB-FW-3/EUPB-RV-1 and EUPB-FW-2/EUPB-RV-2 did not (Table 3). The strongest signals were obtained with the primer sets EUPB-FW-3/EUPB-RV-2 and EUPB-FW-3/EUPB-RV-3, which, following full titration (from 108 to 10−1 copies per reaction), corresponded to as few as 107 and 106 copies per reaction, respectively.
EUPB-FW and RV-4 primer set
Although the primers EUPB-FW-3/EUPB-RV-2 and EUPB-FW-3/EUPB-RV-3 allowed the amplification of both RaHV-1 and -2, the LOD was considered relatively high. Accordingly, a new set of primers was designed, trying to specifically address the limited sensitivity toward RaHV-2. The original EUPB-FW-3 and EUPB-RV-3 were further modified, and the EUPB-FW-4 and RV-4 primers were then derived. The new FW primer EUPB-FW-4 was characterized by 2 nt substitutions at positions 18 (C to M) and 21 (Y to B). The single additional substitution present in the new RV primer EUPB-RV-4 was at position 6 (M to V). This new set of primers allowed the amplification of RaHV-2 for the first time with as few as 104 copies per reaction, 2 logs more sensitive than with the previously best-performing primer set (Fig. 3B; Table 3). The sensitivity could not be further improved by adding DMSO to the PCR reaction.15 On the contrary, DMSO at concentrations > 1 µL per reaction mix showed an inhibitory effect (data not shown).
Figure 3.

Panbatrachovirus PCR protocol. The titrations (105 through 10−1 copies per reaction) of the consensus PCR carried out using the panbatrachovirus PCR protocol (EUPB-FW-4/EUPB-RV-4) are shown. PCR amplification to A. RaHV-1; B. RaHV-2; C. BfHV-1; D. RaHV-3. The number of the template copies per reaction are shown over the amplicon lines. The 100-nt ladder (Promega) is between panels A and B and between panels C and D.
EUPB-FW-4 and RV-4 were tested versus RaHV-1 as well, detecting as few as 103 copies of the target per reaction, and was the most sensitive primer combination tested toward this target (Fig. 3A; Table 3).
Assessing the sensitivity of the primers developed for RaHV-1 and -2 (panbatrachovirus PCR protocol) versus RaHV-3 and BfHV-1
BfHV-1
The most sensitive primer sets that were developed toward RaHV-1 and -2 (EUPB-FW-3/EUPB-RV-2, EUPB-FW-3/EUPB-RV-3, EUPB-FW-4/EUPB-RV-4) were tested also versus BfHV-1. The sensitivity ranged from 103 (EUPB-FW-3/EUPB-RV-2, EUPB-FW-4/EUPB-RV-4) to 105 copies per reaction (EUPB-FW-3/EUPB-RV-3; Fig. 3C; Table 3).
RaHV-3
Similarly, the same primer sets (EUPB-FW-3/EUPB-RV-2, EUPB-FW-3/EUPB-RV-3, EUPB-FW-4/EUPB-RV-4) were tested also versus RaHV-3, and the sensitivities ranged from 102 (EUPB-FW-3/EUPB-RV-2) to 104 (EUPB-FW-3/EUPB-RV-3), with 103 copies per reaction detected by the pairs EUPB-FW-4/EUPB-RV-4 (Fig. 3D; Table 3).
Final assessment
The best overall amplification of all of the known batrachoviruses could be achieved with the EUPB-FW-4/EUPB-RV-4 primer set (panbatrachovirus PCR protocol), with a sensitivity of 103 target copies per reaction for all viral species, with the exception of RaHV-2, which could be detected with 104 target copies per reaction (Fig. 3; Table 3).
The ideal protocol for the European batrachoviruses (European batrachovirus PCR protocol) was determined to include the EUPB-FW-2/EUPB-RV-1 primer set (101 copies of RaHV-3 and 102 copies of BfHV-1, per reaction; Fig. 2; Table 3).
Assay specificity
All unrelated samples and confirmed negative amphibian samples were confirmed negative with the selected protocols (data not shown). The results of PCR amplification of the confirmed positive RaHV-3 and BfHV-1 samples obtained with the protocols developed with our research investigation are shown in Figure 4. Prior to testing with the European and panbatrachovirus PCR protocols, all of these samples were re-tested with the original PCR protocol specifically developed for each of the selected targets (RaHV-3 and BfHV-1).9,10 Specifically, for the RaHV-3 samples, 4 of 5 were re-confirmed positive by the original specific PCR protocol, whereas 1 sample had only a very faint and poorly detectable band (Fig. 4A), suggesting possible degradation while stored. Interestingly, the European batrachovirus PCR protocol was able to confirm all 5 samples as positive, whereas the panbatrachovirus PCR protocol could not amplify the same sample that the original specific protocol could not confirm as well. The 10 confirmed positive samples from common toads all tested positive by the original specific PCR protocol, whereas 8 of 10 samples were confirmed with the European batrachovirus PCR protocol (80%) and 7 of 10 (70%) with the panbatrachovirus PCR protocol (Fig. 4). One of the samples that tested negative with the European batrachovirus PCR protocol and 2 of the samples that tested negative with the panbatrachovirus PCR protocols were only weakly amplified by the original specific PCR protocol.
Figure 4.
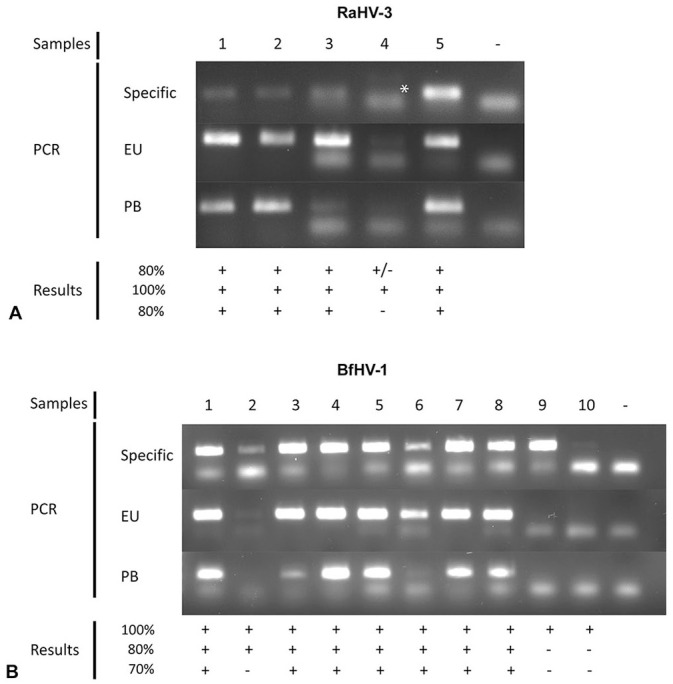
Assay specificity test. Confirmed positive A. RaHV-3 and B. BfHV-1 samples were tested using either their respective original PCR protocol (Specific) or the European batrachovirus protocol (EU) or the panbatrachovirus PCR protocol (PB). Sample numbers, along with a negative control (−), are listed at the top of the figure; the results (+ or −) are shown at the bottom of the figure. An asterisk highlights an inconclusive amplification of a RaHV-3 sample by the original specific PCR protocol.9
Discussion
Our strategy of developing consensus PCR protocols was successful, and we were able to establish 2 protocols, one with high sensitivity to the European isolates and another with a broader spectrum that was able to amplify all known batrachoviruses.
Our European batrachovirus PCR protocol detected < 102 copies of BfHV-1 and as few as 101 of RaHV-3 per reaction. The higher sensitivity toward RaHV-3 was evident from the very first trials and could not be improved by changing the magnesium concentration, the annealing temperature, or by adding DMSO. An increase in sensitivity was achieved by resolving as many nt mismatches as possible in the sequence of the primers including also a “trial and error” strategy. However, RaHV-3 always showed a lower LOD than BfHV-1. The reason for this different sensitivity is not clear; however, the homologous sequence of the terminase gene of BfHV-1 targeted by the FW consensus primer has a GC content of 46%, whereas the corresponding homologous sequence of RaHV-3 has a GC content of 38%. The lower GC content of the RaHV-3 homologous sequence than of BfHV-1 might promote less stringent annealing conditions, favoring the amplification of this target.4
The panbatrachovirus PCR protocol successfully detected all known batrachoviruses. This protocol always showed a relatively lower sensitivity to RaHV-2, which could be significantly improved only with the development of the EUPB-FW-4/EUPB-RV-4 primer set and the consequent resolution of 2 mismatches in the FW primer, both within the 3′ half of the primer and accordingly, highly critical for binding.
The gDNA of neither RaHV-1 nor RaHV-2 was available at the beginning of our experiment and accordingly, a chimeric positive control consisting of a DNA sequence composed of the homologous RaHV-3 backbone with the 5′ and 3′ regions containing the specific viral (RaHV-1 or RaHV-2) nt sequences, was obtained for each of them. Later, the gDNA of RaHV-1 became available. The panbatrachovirus PCR protocol amplified the specific native RaHV-1 genomic target consistently, with the chimeric target being a suitable surrogate of the actual RaHV-1 gDNA and presumptively of RaHV-2.
The European batrachovirus PCR protocol confirmed its high sensitivity and specificity both on RaHV-3 and BfHV-1 from infected or uninfected common frogs and toads, and did not show any nonspecific amplification in the negative and unrelated samples (data not shown). Interestingly, the European batrachovirus PCR protocol was able to amplify an originally RaHV-3 confirmed positive sample that could not be conclusively reamplified by the specific protocol. We speculate that the sample might have been partially degraded, and the degenerate primers might have overcome some inefficiency in the annealing of the specific primers at the target sequence. The panbatrachovirus PCR protocol was as specific as the European batrachovirus protocol, but less sensitive, consistent with observations in ideal testing conditions (purified target plasmid). We are confident that the newly developed PCR protocols will be instrumental in the detection of any of the known batrachovirus in free-ranging and captive amphibians. Furthermore, the broad spectrum of these investigative tools will help to detect yet to be discovered batrachoviruses, contributing to better understanding of herpesvirus-associated amphibian disease ecology and conservation.
Acknowledgments
We thank Dr. Louise Rollins-Smith, Vanderbilt University, for the kind gift of RaHV-1 gDNA. We thank all of the personnel of the Centre for Fish and Wildlife Health (FIWI) of the University of Bern and in particular Drs. Helmut Segner, Marie Pierre Ryser-Degiorgis, and Irene Adrian-Kalchhauser for their support.
Footnotes
Declaration of conflicting interests: The authors declare that they have no conflict of interest concerning the data and information contained in this article.
Funding: This work was supported by grants R212-1468 and R414-1866 from the Swiss Federal Office for the Environment (FOEN) and by the Vontobel Foundation, Zurich.
ORCID iD: Francesco C. Origgi  https://orcid.org/0000-0002-2874-6563
https://orcid.org/0000-0002-2874-6563
References
- 1. Davison AJ, et al. Genome sequences of two frog herpesviruses. J Gen Virol 2006;87:3509–3514. [DOI] [PubMed] [Google Scholar]
- 2. Davison AJ, et al. The order Herpesvirales. Arch Virol 2009; 154:171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Granoff A. Amphibian herpesviruses. In: Roizman B, ed. The Herpesviruses. Vol. 1 Springer, 1983:367–384. [Google Scholar]
- 4. Guido N, et al. Improved PCR amplification of broad spectrum GC DNA templates. PLoS One 2016;11:e0156478–e0156478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hanson LA, et al. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virol J 2006;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lucké B. A neoplastic disease of the kidney of the frog, Rana pipiens. Am J Cancer 1934;20:352. [Google Scholar]
- 7. Lucké B. Carcinoma in the leopard frog: its probable causation by a virus. J Exp Med 1938;68:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McKinnell RG, Carlson DL. Lucké renal adenocarcinoma, an anuran neoplasm: studies at the interface of pathology, virology, and differentiation competence. J Cell Physiol 1997;173: 115–118. [DOI] [PubMed] [Google Scholar]
- 9. Origgi FC, et al. Ranid herpesvirus 3 and proliferative dermatitis in free-ranging wild common frogs (Rana temporaria). Vet Pathol 2017;54:686–694. [DOI] [PubMed] [Google Scholar]
- 10. Origgi FC, et al. Bufonid herpesvirus 1 (BfHV1) associated dermatitis and mortality in free ranging common toads (Bufo bufo) in Switzerland. Sci Rep 2018;8:14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parkin DB, et al. Genotype differentiation of Agamid Adenovirus 1 in bearded dragons (Pogona vitticeps) in the USA by hexon gene sequence. Infect Genet Evol 2009;9:501–506. [DOI] [PubMed] [Google Scholar]
- 12. Rafferty KA., Jr The cultivation of inclusion-associated viruses from Lucké tumor frogs. Ann NY Acad Sci 1965;126:3–21. [DOI] [PubMed] [Google Scholar]
- 13. Ropp SL, et al. PCR strategy for identification and differentiation of smallpox and other orthopoxviruses. J Clin Microbiol 1995;33:2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schatzberg SJ, et al. Broadly reactive pan-paramyxovirus reverse transcription polymerase chain reaction and sequence analysis for the detection of Canine distemper virus in a case of canine meningoencephalitis of unknown etiology. J Vet Diagn Invest 2009;21:844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strien J, et al. Enhancement of PCR amplification of moderate GC-containing and highly GC-rich DNA sequences. Mol Biotechnol 2013;54:1048–1054. [DOI] [PubMed] [Google Scholar]
- 16. VanDevanter DR, et al. Detection and analysis of diverse herpesviral species by consensus primer PCR. J Clin Microbiol 1996;34:1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wellehan JFX, et al. Detection and analysis of six lizard adenoviruses by consensus primer PCR provides further evidence of a reptilian origin for the atadenoviruses. J Virol 2004;78: 13366–13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou C, et al. Mini-prep in ten minutes. Biotechniques 1990;8: 172–173. [PubMed] [Google Scholar]