

Abstract
Aging is a major risk factor for numerous human pathologies, including cardiovascular, metabolic, musculoskeletal, and neurodegenerative conditions and various malignancies. While our understanding of aging is far from complete, recent advances suggest that targeting fundamental aging processes can delay, prevent, or alleviate age-related disorders. Cellular senescence is physiologically beneficial in several contexts, but it has causal roles in multiple chronic diseases. New studies have illustrated the promising feasibility and safety to selectively ablate senescent cells from tissues, a therapeutic modality that holds potential for treating multiple chronic pathologies and extending human healthspan. Here, we review molecular links between cellular senescence and age-associated complications and highlight novel therapeutic avenues that may be exploited to target senescent cells in future geriatric medicine.
Cellular Senescence: In the Landscape of an Aging World
Aging (see Glossary) is a course of progressive decline in functional integrity and physiological homeostasis, gradually culminating in tissue dysfunction, organ failure, and, ultimately, organismal death [1]. Advances in healthcare and sanitation have substantially increased human life expectancy, but at the cost of unintentionally prolonged frailty and morbidity. By 2050, with an expected number of 1.6 billion people 65 years of age or older, the frail elderly population will become the major socioeconomic concern worldwide. Insights into key mechanisms that underlie aging are thus critical to control age-related pathologies and to ensure quality of life in old age [2,3].
Recent studies in experimental animals, including yeasts, worms, fruit flies, and mice, suggest the remarkable malleability of aging processes. Biomedical interventions can extend lifespan, ameliorate functional loss, delay progression of age-related diseases, and, in some cases, compress late-life morbidity [4]. Although most animals do not live as long as humans, fundamental aging mechanisms are conserved across diverse evolutionary tracks, providing an opportunity to identify potential targets for maintaining human health in old age or treating serious age-related disorders [3,5]. Regulated by both genes and the environment, aging is highly complex, as partially reflected by the diversity of its hallmarks, including telomere attrition, genomic instability, epigenetic alterations, mitochondrial dysfunction, loss of protein homeostasis (proteostasis), stem cell exhaustion, deregulated nutrient sensing, immune system decline, and cellular senescence [6,7] (Figure 1).
Figure 1. The Hallmarks of Organismal Aging.
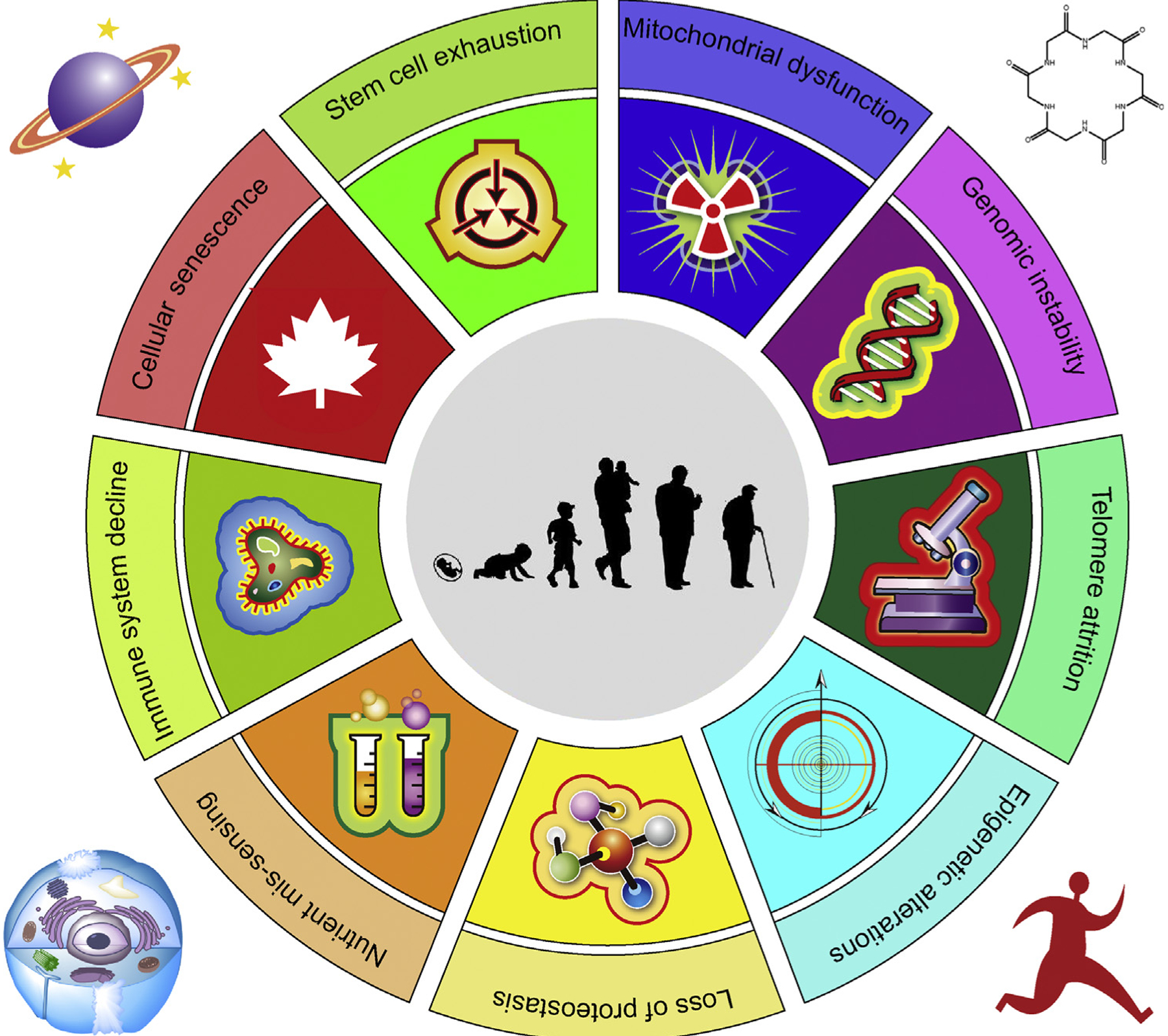
Organismal aging (center) is a complex process contributing to progressive decline of organ functionality and regenerative potential of tissues. The aging clock is governed by interconnected hallmarks of organismal aging. Understanding how the underlying biological mechanisms of aging correlate with and impact longitudinal changes in health trajectories might offer an opportunity to identify resilience mechanisms, their dynamic changes, and their impact on physiological integrity. In geroscience, a research field that is rapidly progressing in the current era of precision medicine, novel antiaging agents developed to delay organismal aging will significantly rely on their safety and effectiveness at the level of cells, basic biological units of an individual (outside the circle).
While some of these age-related hallmarks are generally conserved from yeasts to humans (e.g., deregulated nutrient sensing), others appear to be more specific to vertebrates (e.g., stem cell exhaustion and cellular senescence) [6,8]. It was proposed that aging hallmarks can be divided into three categories: primary, or causes of age-associated damage; counteractive, or responses to the damage; and integrative, or consequences of the responses and culprits of the aging phenotype [9]. Cellular senescence, a cell fate characterized by permanent growth arrest and other phenotypic alterations, including development of a proinflammatory secretome, belongs to the counteractive class. Cellular senescence is indispensable for normal development, tissue homeostasis, wound healing, damage responses, and tumor prevention [10]. However, cellular senescence is also implicated as a major cause of myriad age-related diseases. Accumulating evidence has shown that both prosenescence therapies and antisenescence treatments can be beneficial. In the case of carcinogenesis and during active tissue repair, prosenescence drugs help minimize damage by restraining proliferation and fibrosis, respectively. However, caveats are that cancer cells released from chemotherapy-induced senescence can re-enter cell cycle with a strongly enhanced tumor initiation potential, implying that senescence-associated reprogramming can promote cancer plasticity and aggressiveness [11]. Conversely, antisenescence agents are able to eliminate accumulated senescent cells, restore tissue function, and facilitate organ rejuvenation [12]. Here, we review inherent links between cellular senescence and age-associated pathologies and discuss emerging therapeutic avenues that could accelerate development of novel interventions that target senescent cell subpopulations in the tissue microenvironment.
The Biological Complexity of Cellular Senescence
First described in 1961 by American cell biologists Hayflick and Moorhead, cellular senescence, a term from the Latin word senex which means “old man”, was originally observed in normal human diploid fibroblasts that could no longer divide after a finite number of divisions in culture, a phenomenon later referred to as the ‘Hayflick limit’ [13]. After decades of research, it is clear that cellular senescence indeed represents a state of stable cell cycle arrest arising in response to irreparable damage, intracellular or extracellular insults and can be considered a hallmark of aging [14]. According to the type of stresses and/or stimuli, cells can develop replicative senescence, oncogene-induced senescence (OIS), stress-induced premature senescence (including therapy-induced senescence), or programmed senescence.
Variability of Stimuli and Heterogeneity in Phenotype
Cells can enter senescence after exposure to various types of stimuli, including genotoxic treatment, epigenetic changes, perturbed proteostasis, mitochondrial dysfunction, oncogene activation, inflammation, and/or tissue damage signals. While some of these factors can also result from senescence, improved experimental techniques and new animal models have expanded our knowledge about causes and consequences of cellular senescence. However, universal markers that would allow identification of senescent cells with strong sensitivity and specificity remain largely lacking [15]. Despite the link between senescent cells and organismal aging, senescence and aging should not be considered synonymous. Cells can undergo senescence regardless of organismal age and are subject to induction of senescence by various signals, even those not involving telomere attrition. Being essential to halt the propagation of damaged cells, promote tissue repair, and limit tumor progression, cellular senescence represents an example of evolutionary antagonistic pleiotropy, which has biologically evolved with both beneficial and detrimental effects on the health of organisms [12]. Senescent cells contribute to normal development and maintains tissue homeostasis, but it is also implicated as a major cause of multiple age-related pathologies, even with a limited percentage (b1%) in body tissues [16].
Senescent cells often exhibit a persistent DNA damage response (DDR), engagement of cyclin-dependent kinase inhibitors (CDKIs), and alterations in metabolic activity. Further, senescent cells can develop structural changes, including an enlarged and flattened morphology, altered composition of the plasma membrane, accumulation of lysosomes and mitochondria, and a remarkable nuclear expansion [17] (Figure 2). However, some of these senescence-associated molecular and morphological features can also be found in other cell states and/or conditions, such as upregulation of the tumor suppressor p16INK4a, activation of DDR, and changes in heterochromatin [18,19]. Transcriptomic signatures of senescent cells can be stress- and/or cell type-dependent and the phenotype of senescent cells tends to be highly heterogeneous and dynamic, issues that need to be considered for future development and selection of therapies against senescent cells [15,20].
Figure 2. Hallmarks of Cellular Senescence.
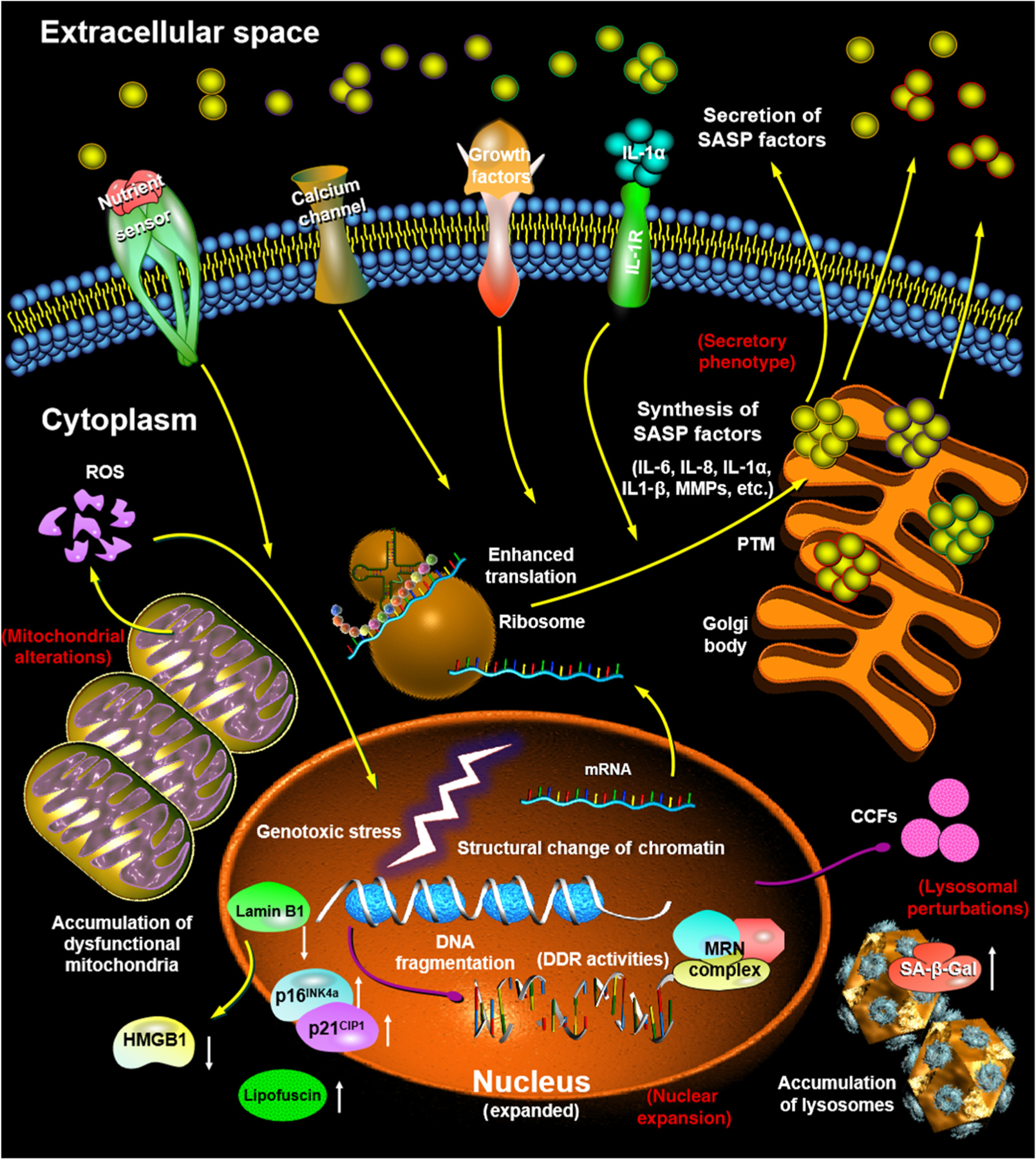
Distinct alterations in molecular pathways of cellular senescence result in morphological changes. Senescent cells are enlarged, flattened, and have an irregular cell shape (not shown). Their nuclear integrity is compromised due to the loss of lamin B1 and exclusion of the high mobility group protein B1 (HMGB1), accompanied by the emergence of cytoplasmic chromatin fragments (CCFs). Senescent cells have increased lysosomal content, as manifested by elevated β-galactosidase (SA-β-gal) activity, with an increased number of large but dysfunctional mitochondria that produce high levels of reactive oxygen species (ROS). There is usually an enhanced level of lipofuscin in the cytoplasm and upregulation of p16INK4a/p21CIP1 in the nucleus. Biosynthesis and extracellular secretion of a large number of proinflammatory and proapoptotic factors [senescence-associated secretory phenotype (SASP)] is robust and constitutes one of the major features of senescent cells. Yellow arrows: signal transduction or molecular traffic; purple arrows: DNA fragment transportation; white arrows: up- or downregulation of expression or activities. Red text: representative typical hallmarks of senescent cells. Different colors of the outlines of SASP factors indicate various types of secreted molecules (e.g., cytokines, chemokines, and growth factors). Abbreviations: DDR, DNA damage response; MMP, matrix metalloproteinase; PTM, post-translational modification.
Secretory Phenotype
In most cases, one of the major hallmarks of senescent cells is active secretion of numerous soluble factors, collectively termed the senescence-associated secretory phenotype (SASP) (or senescence-messaging secretome) (Figure 2) [21–23]. Though the SASP was first reported in 2008, several pioneer studies already noticed the secretion activity of senescent cells [24–26]. While not typically detectable in some cases of cellular senescence, such as those induced by merely ectopic expression of p16INK4a or p21CIP1, the SASP can be separable from the growth arrest and represents a temporally regulated dynamic program of senescence that develops through a rapid DDR-associated phase, an early self-amplification phase, and a late ‘mature’ phase, the latter indeed representing the most widely studied SASP signature [27]. Extracellular vesicles, a group of active players in intercellular communication, were recently found to be critical components of senescent cell secretome, an intriguing topic that awaits extended investigation [28].
The SASP is responsible for many pathophysiological effects of senescent cells and such a senescence-associated inflammasome can even reinforce and spread senescence in a paracrine or endocrine manner [16,29]. Further, the SASP is able to recruit immature immunosuppressive myeloid cells to solid tumors and stimulate tumorigenesis by driving angiogenesis and metastasis [30–33]. In solid tumors, the SASP can simultaneously drive cancer resistance and establish an immunodeficient niche by upregulating PD-L1 expression in cancer cells, thereby activating the immune checkpoint and exacerbating pathologies even after therapeutic intervention [34].
The SASP is regulated by a complicated network in senescent cells, including enhancer remodeling and activation of transcription factors such as NF-kB, c/EBPβ, and GATA4 [35,36] and signaling pathways such as those involving cytoplasmic kinases mTOR, p38MAPK, and TAK1 [37–40]. Recent studies have further revealed that upstream signals inducing activation of the SASP can include cytoplasmic chromatin fragments and transposable elements, which engage the cGAS/GAMP-STING axis and trigger a type 1 interferon response, a process potently supported by downregulation of cytoplasmic DNases such as DNase2 and TREX1 [41–45].
Mitochondrial Alterations
Senescent cells display remarkable changes in mitochondrial function, dynamics, and morphology. Upon cellular senescence, mitochondria are less functional, with reduced membrane potential, increased proton leak, enhanced enzyme release, elevated mass, and more tricarboxylic acid (TCA) cycle metabolites [46,47]. Notably, there is an increased number of mitochondria in senescent cells, resulting from the accumulation of old and dysfunctional mitochondria due to deficient mitophagy (mitochondrial clearance) [48,49]. This phenomenon is partially correlated with reduced mitochondrial fission and enhanced fusion, a pattern that likely represents a mechanism to protect mitochondria from mitophagy-mediated damage and circumvent cell apoptosis [17,50]. Interestingly, senescent cells have a transient upregulation of PGC-1α and PGC-1β, two essential regulators of mitochondrial biogenesis that are subsequently downregulated [50,51]. Thus, mitochondrial biogenesis per se does not seem to play a significant role in the increase of mitochondrial content during cellular senescence, while the mitochondrial mass increase caused by mitophagy deficiency may represent a distinctive mechanism [50].
Despite their enhanced abundance, mitochondria in senescent cells usually have a compromised ability to produce ATP [49,52]. Instead, senescent cells can exhibit a partial Warburg shift, tending to generate more reactive oxygen species (ROS), which cause protein and lipid damage, induce telomere shortening, and promote DDR activation [53] (Figure 2). Targeting specific components or activities of mitochondria such as Complex I assembly, the electron transport chain (ETC), mitochondrial fission rates, the process of biogenesis, mitochondrial sirtuin molecules, and/or disruption of the TCA cycle can trigger, or sometimes prevent, senescence [46,54–57]. Further, modulation of the AMP:ATP or ADP:ATP ratio during senescence consolidates cell-cycle arrest by activating AMP-activated protein kinase (AMPK), a cytoplasmic sensor that detects environmental or nutritional stress and maintains energy balance [52,58].
Dysfunctional mitochondria accumulate with aging and are potential drivers of age-related phenotypes, particularly in postmitotic tissues enriched with muscle cells and neurons [59,60]. Although mitophagy in senescent cells appears to suppress the SASP [54], inhibition of the ETC can induce cellular senescence with a distinct SASP that lacks several key proinflammatory factors such as IL-6 and IL-8 [57]. Cells undergoing mitochondrial dysfunction-associated senescence exhibit reduced NAD+/NADH ratios, causing metabolic reprogramming responsible for both growth arrest and absence of an IL-1α-potentiated SASP via AMPK-mediated p53 activation [57].
Senescent cells can release factors to upregulate CD38, a NADase expressed on the surface of endothelial and inflammatory cells and involved in NAD+ consumption, thus allowing depletion of NAD+ in tissues during aging [61]. Further, decreased ratios of NAD+/NADH can alter the activity of poly-ADP ribose polymerase (PARP) and sirtuins, molecules involved in activation of the SASP master regulator NF-kB [52]. However, as occurring in other cellular processes, mitochondrial dysfunction does not appear to be a consistent biomarker of cellular senescence, nor is it clear whether senescent cells are responsible for declining mitochondrial function with aging and/or in age-related disorders [12].
Lysosomal Perturbations
Lysosomal biogenesis is subject to modulation by cellular energetic or degradative activities, while lysosomes functionally interact with mitochondria to maintain mitochondrial homeostasis [62,63]. Senescent cells have lysosomes that are increased in both number and size, with augmented cytoplasmic granularity [64]. Specifically, increased lysosomal number may represent an autonomous attempt of senescent cells to balance the gradual accumulation of dysfunctional lysosomes via production of new lysosomes. This balance is sustained during OIS through the TOR-autophagy spatial-coupling compartment (TASCC), which orchestrates cellular synthesis of SASP factors [65]. As autophagy-associated degradation also decreases during cellular senescence, increased lysosomal content does not necessarily reflect enhanced activity in these cells [63]. The lysosome–mitochondrial axis degrades, causing declined mitochondrial turnover and elevated ROS production, while the latter induces perturbations in organelles, including lysosomes, leading to a vicious feedback loop promoting accumulation of more damage [63].
Senescence-associated β-galactosidase (SA-β-gal) activity is the first reported and most commonly used lysosomal activity-related marker to assess cellular senescence [66,67]. There is an age-dependent increase of SA-β-gal positivity in dermal fibroblasts and epidermal keratinocytes in skin samples of human donors, constituting a line of in situ evidence that senescent cells accumulate with age in vivo [66]. Although SA-β-gal induction during senescence can be partially attributed to increased expression of the lysosomal β-galactosidase protein, SA-β-gal is neither required for nor specific to senescence, nor a determinant of the senescent phenotype [17,68,69].
Resistance to Apoptosis
Senescent cells are inherently resistant to apoptosis. Senescent cells tend to be protected from death induced by intrinsic or extrinsic proapoptotic signals, a feature allowing them to survive for a relatively long time and maintain viability even under stressful conditions [70,71]. In response to multiple types of stimuli, cells become senescent and upregulate antiapoptotic proteins such as Bcl-2, Bcl-xL, and Bcl-w, at least in some types of senescent cells [72,73]. However, once exposed to apoptosis inducers, senescent cells are able to sustain Bcl-2 expression, a capacity associated with chronic activation of the transcription factor cAMP response element-binding protein (CREB) [74]. Simultaneously, expression of the proapoptotic factor Bax is restrained by the repressive histone mark H4K20me3, in line with increased activity of Suv420h2 histone methyltransferase, which targets H4K20 [75].
Recent studies revealed the involvement of other prosurvival networks, where key nodes of these networks include ephrins, PI3K, p21, and plasminogen activated inhibitor-2 [73]. Expression of FOXO4 is enhanced in senescent cells and prevents apoptosis by sequestering p53 in the nucleus, while an artificially designed FOXO4 peptide (FOXO4-DRI) can selectively and potently target senescent cells for p53-dependent apoptosis [76]. The CDKI molecule p21 protects senescent cells against death by dampening JNK and caspase signaling upon persistent DNA damage [77]. HSP90 promotes the survival of senescent cells via stabilization of p-Akt, while the HSP90 inhibitor 17-DMAG can drive senescent cell elimination, extend healthspan and delay the onset of several age-related symptoms. Among these antiapoptotic molecules, Bcl-2 family members are the most studied and considered as promising targets to manipulate senescent cells, although nonsenescent cell types, particularly blood cells, also exhibit upregulation of these antiapoptotic regulators [78].
Contribution of Cellular Senescence to Age-Related Pathologies
Organismal aging is a complex process that drives progressive impairment of functionality and exhausts regenerative potential of tissues [4]. Senescent cells passively accumulate in organs during aging, contributing to tissue dysfunction and pathological conditions [79]. Senescence is therefore a defining feature of diverse human age-related disorders, whereas its contribution is mediated via several coalescing effects, with the SASP often popping up on top of the list (Box 1).
Box 1. Implications of Cellular Senescence in Physiology and Age-Related Diseases.
Within tissues, senescent cells can play distinctive roles depending on the pathophysiological context. As a primary function, replicative senescence prevents the expansion of precancerous transformed cells, thus providing a major barrier to tumorigenesis. Senescent cells participate in fundamental processes by promoting tissue repair and regeneration as a critical step in normal morphogenesis during embryonic development and promote tissue remodeling in various organ types [14]. Senescent cells are also subject to autonomous elimination in vivo, by recruiting phagocytic immune cells and mobilizing their progenitor cells, activities that engage innate immune surveillance [80,128]. However, physiological clearance of senescent cells is substantially compromised during aging and these cells gradually accumulate, forming a chronic proinflammatory niche in the local microenvironment and underpinning diverse pathological conditions.
Recent studies proved that cell senescence is implicated in a large number of age-related pathologies, including but not limited to cancer, renal dysfunction, diabetes mellitus, nonalcoholic fatty liver disease, cardiovascular disorders, fibrosis, osteoarthritis, obesity, type 2 sarcopenia, and neurodegenerative disorders [9,14,36,88]. Genetic ablation or pharmacological elimination of p16INK4a-positive senescent cells from progeroid and aged animal models alleviates tissue dysfunction, averts physical frailty, and increases organismal healthspan [16,129]. Further, senescent cell clearance effectively restrains the progression of adipose atrophy, atherosclerosis, cancer, cataracts, cardiomyocyte hypertrophy, osteoarthritis, renal glomerulosclerosis, sarcopenia, tumorigenesis, tau-dependent disorder, and cognitive decline [82,83,87,97,130,131]. Despite technical challenges and practical difficulty in pharmacologically clearing senescent cells at tissue and organ levels, research advances have underpinned the development pipelines of senolytics, a collection of pharmacological agents that can selectively and preferentially deplete senescent cells under in vivo conditions [132]. Together, cell senescence is causative of multiple human age-related disorders, while the emerging arsenal of senolytics holds the potential to deliver exciting solutions to optimize therapeutic interventions in future clinics.
As damage accumulates in tissues and organs, the number of senescent cells and their SASP continue to augment. This process can be resolved by physiological processes, particularly clearance of senescent cells by the immune system [80,81]. Paradoxically, however, aging itself is accompanied by a decline in immune function, termed immunosenescence, a degenerative process that limits senescent cell clearance and exacerbates local inflammation. Studies applying genetic systems or pharmacological agents to ablate senescent cells have successfully demonstrated that in vivo depletion of senescent cells reduces inflammation across tissues, even in humans [16,82–85]. Future efforts are expected to explore further the likely causal links among the SASP, chronic inflammation, tissue dysfunction, and organismal aging.
Targeting Senescent Cells to Alleviate Age-Related Pathologies
Advanced age remains the major risk factor for most chronic diseases and functional deficits in humans. The impetus to develop senolytics began in 2004 with an article by the Sharpless group [86]. Specifically, manipulations that increase healthspan and lifespan in mice were found to be effective in delaying accumulation of cells with SA-β-gal elevation and p16INK4a expression [86].
The healthspan of experimental animals can be enhanced by killing senescent cells with a transgenic suicide gene, such as INK-ATTAC, for inducible elimination of p16INK4a-positive senescent cells upon drug administration [87]. Achieving the same goal using small molecules would have a tremendous impact on the quality of life and the burden of age-related chronic pathologies. There is growing interest in searching avenues to target senescent cells therapeutically [88]. Several promising approaches focusing on either clearance of senescent cells or dampening of their proinflammatory activities are taking shape. Considerable efforts are invested in the discovery of chemical compounds or phytochemicals that selectively induce the death of senescent cell by apoptosis [89] (Box 2).
Box 2. Challenges and Opportunities in the Development of Effective Senolytics.
To date, a major challenge for senotherapy is to target the right cells at the right time, thus avoiding side effects on nonsenescent cells. Although in some animal experiments senolytics were applied for a short time, this was sufficient to reduce the burden of senescent cells. To avoid potential adverse effects of senolytics (such as navitoclax), therapeutic protocols can be designed with intermittent rather than long or lasting administration. Clinical trials are initiated to help translate the intervention from bench to the bedside, with the aim to prove the efficacy and safety of senolytics in humans. The first-in-human pilot study (with D + Q) suggested that senolytics can alleviate physical dysfunction in IPF patients, with nonserious events being primarily mild to moderate, though there were respiratory symptoms, skin irritation, and gastrointestinal discomfort [133]. Together, the data support evaluation of such agents in larger randomized controlled trials for senescence-related pathologies.
Targeted elimination of senescent cells appears to be a promising therapeutic approach for decreasing tissue damage, promoting repair, and facilitating regeneration. Repurposing drugs applied to target fundamental aging processes represents a promising strategy for proceeding, since previous clinical experience with these drugs can provide valuable information about dosing, safety, tolerability, drug interactions, and side effects, accelerating regulatory approvals. Examples include new clinical trials underway with such drug candidates as senolytics, as well as mTOR and AMPK inhibitors [123,134]. If successful, current trials for treatments of age-related diseases could eventually be extended to clinical prevention for at-risk elderly people, once a consensus is reached on surrogate endpoints. However, such preventative pharmacological interventions for humans could be ideally initiated later in life, with an aim of reducing the risk of potential side effects of long-term medications.
Bcl-2-Targeting Senolytics
Several intracellular prosurvival pathways (senescent cell antiapoptotic pathways) have been identified and can be used for directed targeting to eliminate senescent cells. This includes, for example, the Bcl-2 family, p53/p21 pathway, PI3K/AKT axis, receptor tyrosine kinases (RTKs), HIF-1α, and HSP90 [72,78,90]. To date, many identified senolytics are against members of the Bcl-2 family. Antiapoptotic proteins of the Bcl-2 family are well investigated as potentially exploitable drug targets [91] and they play multiple roles in cell death modulation via regulation of apoptosis and autophagy [92]. Directly targeting these proteins results in programmed death of senescent cells [72,90] (Figure 3A).
Figure 3. Therapeutic Strategies That Can Be Exploited to Target Senescent Cells.
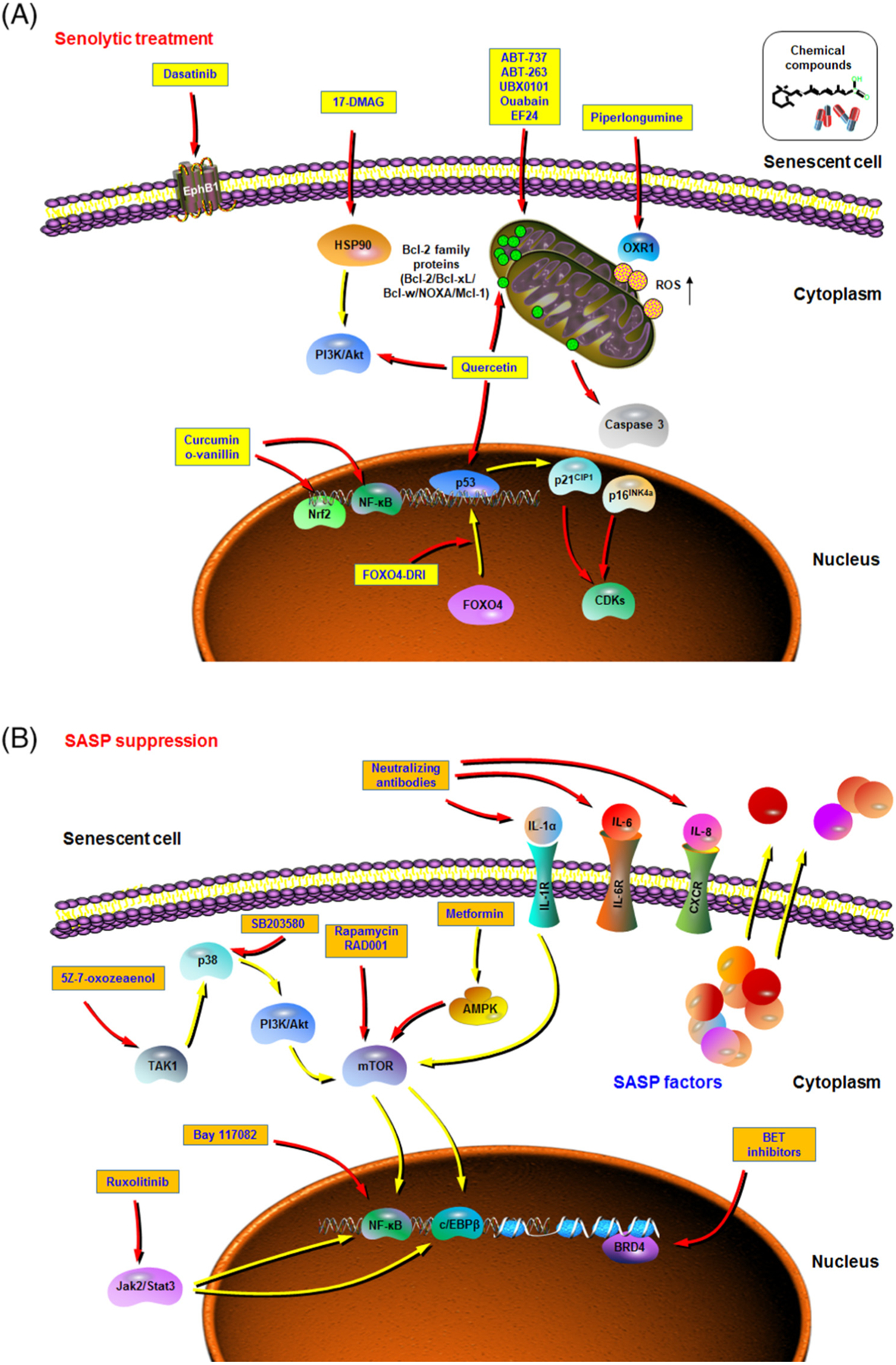
Aging is the leading risk factor for many chronic diseases, including but not limited to cancers, metabolic syndrome, and musculoskeletal, cardiovascular, and neurodegenerative disorders. Recent studies suggest that targeting fundamental aging mechanisms may be a better solution than targeting each chronic pathology individually in order to increase healthspan and delay multimorbidity. Pilot efforts have recently been made to therapeutically target cellular senescence, including selectively eliminating senescent cells by inducing their passive apoptosis (senolytic treatment) (A), and modulating the senescence-associated secretory phenotype (SASP) (SASP suppression, or senostatics) by blocking the SASP development or interfering with the function of its key components in the tissue microenvironment (B). In each figure, yellow arrows represent stimulating or promotive actions, while red arrows are suppressive or counteractive. Agents within yellow or orange boxes are exemplifying senolytics (A) and SASP inhibitors (B), respectively. Abbreviations: AMPK, AMP-activated protein kinase; OXR1, oxidation resistance 1; ROS, reactive oxygen species.
ABT-737 is a Bcl-2 Homology 3 (BH3) domain mimetic that blocks the interaction between antiapoptotic molecules Bcl-2/Bcl-w/Bcl-XL and BH3 domain-containing proapoptotic proteins, thus inducing cell apoptosis [93,94]. ABT-737 efficiently removes senescent cells in lungs of irradiated mice and skin epidermis of transgenic mice [72], but it has limited bioavailability. Navitoclax (ABT-263), an orally available analog of ABT-737 [95], eliminates some types of senescent cells from sublethally irradiated mice and naturally aged mice, including senescent muscle/hematopoietic stem cells, but not senescent fat cell progenitors [90,96]. Navitoclax can also deplete senescent foam cell macrophages from atherosclerotic lesions, hence avoiding disease exacerbation [97]. Further, navitoclax eliminates senescent cardiomyocytes and limits profibrotic protein expression in aged mice, resulting in improved myocardial remodeling and diastolic function as well as overall survival following myocardial infarction [98].
However, certain hematological toxicities, including neutropenia and thrombocytopenia, of Bcl-2 family inhibitors were observed [99], raising safety issues that may impede the development of these inhibitors into future clinical interventions; more specific agents hold the potential to be used as superior candidate senolytics. Intra-articular administration of the Bcl-2-targeting agent UBX0101 selectively removes senescent cells in articular cartilage and synovium after anterior cruciate ligament transection (ACLT), alleviating post-traumatic osteoarthritis, reducing pain while increasing cartilage development in aged mice [83]. The cardiac glycoside (CG) ouabain can work as a novel senolytic agent with broad activity, while senescent cells are subject to ouabain-induced apoptosis, a process mediated partially by induction of the proapoptotic NOXA, a Bcl-2 family member [100]. Simultaneously, another study revealed that senescent cells have a slightly depolarized plasma membrane and higher concentrations of H+, thus being more susceptible to the action of CGs [101]. With several clinical trials ongoing, the efficacy of CGs as promising senolytics in ameliorating age-related chronic conditions, including arthritis and pulmonary fibrosis, awaits validation [102].
Other Senolytics
Recent studies explored alternative routes to effectively target senescent cells while avoiding the cytotoxic side effects of Bcl-2 inhibitors, a strategy substantiated by application of chemicals that inhibit other senescence-associated prosurvival pathways (Figure 3A). For instance, a combination of a pan-tyrosine kinase inhibitor, dasatinib, and a natural flavonoid quercetin, kills senescent cells but not their proliferating or quiescent, differentiated counterparts [73]. Combinatorial treatment with these two compounds (D + Q) selectively eliminates primary preadipocytes and human umbilical vein endothelial cells that are senescent in culture and alleviate senescent cell-related symptoms in several pathological conditions, including idiopathic pulmonary fibrosis (IPF), osteoporosis, Alzheimer’s disease (AD), and obesity-related metabolic disorders such as insulin resistance and diabetes, as well as neuropsychiatric complications [84,103–108]. In the recent clinical trial of senolytics, D + Q improved physical function in patients with IPF, a fatal senescence-associated disorder [85]. Specifically, D + Q lowered senescent cell burden in adipose tissues within 11 days, caused significant reduction of skin epidermal p16INK4a+ and p21CIP1+ cells, and lowered circulating levels of SASP factors including IL-6, IL-1α, and MMP9/12 [85].
Curcumin and o-vanillin clear senescent intervertebral disc cells, minimize the SASP associated with inflammation and back pain, and increase matrix synthesis, effects mediated through the Nrf2 and NF-κB pathways [109].
In a recent study to test the senolytic activity of ten flavonoids, fisetin appeared to be the most potent in reducing the number of senescent cells in murine and human adipose tissues, while administration of fisetin to aged mice restored tissue homeostasis, reduced age-related pathology, and extended median and maximum lifespan [110]. However, fisetin has limited senolytic efficacy for senescent IMR90 cells, human lung fibroblasts, or primary human preadipocytes, suggesting its cell type-specific senolytic value [111]. In contrast, piperlongumine, a major alkaloid from long peppers and some other medicinal plants, preferentially killed senescent human WI38 fibroblasts generated by ionizing radiation, replicative exhaustion, or oncogene RAS expression [112]. Oxidation resistance 1 (OXR1), an important antioxidant protein regulating the expression of many antioxidant enzymes, is upregulated in senescent WI38 cells, while piperlongumine binds OXR1 directly and induces its degradation via the ubiquitin-proteasome system to induce cell death [113]. EF24, a curcumin analog, is a senolytic against multiple types of human senescent cells, including fibroblasts, endothelial cells, and renal epithelial cells by restraining Bcl-xL and Mcl-1 expression in target senescent cells [114]. Berberine (BBR), a natural alkaloid found in Coptis chinensis and with a long medical history in both Ayurvedic and traditional Chinese medicine, can extend replicative lifespan and improve morphology of human fetal lung diploid fibroblast lines, including 2BS and WI38, while oral administration of BBR in mice generated significantly improved healthspan, fur density, and behavioral activity [115].
Targeting the SASP
While the adverse effects of senescent cells can be counteracted by senolytics, specific control of their detrimental components may represent an alternative strategy. Senescent cells manifest a robust, profound, and stable proinflammatory secretory activity with the expression spectrum largely conserved between different cell types, cell states, and stress types, although there are variations in the SASP signature, a form of heterogeneity driven by certain unknown mechanism(s) [20]. The SASP comprises a large array of soluble factors, expression of which is regulated by several complicated signaling networks, ultimately converging on transcription machineries such as NF-κB and c/EBPβ [36].
As the IL-1α/IL-1R axis can promote the SASP development through signaling upstream of NF-κB, application of neutralizing antibodies against this axis is sufficient to control NF-κB transcriptional activity and dampen the SASP [85]. Several mTOR inhibitors, including rapamycin and its analogs like RAD001, can abrogate the SASP by suppressing protein translation of the membrane-bound IL-1α and diminish the protumorigenic capacity of senescent cells, an effect demonstrated by preclinical trials [39,40]. Metformin, an LKB1-AMPK pathway agonist and FDA-approved chemical for type 2 diabetes, prevents NF-κB from translocating to nucleus and restricts its transcriptional activity [116]. In mice, therapeutic inhibition of the SASP with metformin can reduce destructive retinal neovascularization, a condition of pathological angiogenesis in retinopathy [117].
Since Jak2/Stat3 signaling mediates the c/EBPβ transcriptional activity, treatment with a Jak inhibitor ruxolitinib (FDA approved) results in higher bone mass and strength, better bone microarchitecture, improved cardiovascular function, enhanced insulin sensitivity, and reduced frailty compared with vehicle-treated mice, suggesting a novel treatment strategy not only for osteoporosis, but also for multiple age-related comorbidities [118]. In senescent cells, TAK1 activates p38 and PI3K/Akt/mTOR pathways to support a persistent SASP signaling via engagement in a dual-feedforward mechanism, while pharmacologically targeting TAK1 minimizes cancer resistance and promotes tumor regression in vivo [40]. Thus, multiple lines of evidence support that targeting the SASP itself without radically removing senescent cells from the microenvironment can reduce the deleterious effects of senescent cells, while preserving them in their original niches to sustain tissue structure and physiological integrity (Figure 3B).
Concluding Remarks
Improved health conditions of all ages and a continued increase in human life expectancy are central goals of contemporary biomedical research and should be celebrated as achievements of modern civilization. However, our healthspan has not increased as much as lifespan [119]. The global extension of 5.0 years in total life expectancy between 2000 and 2015 has been accompanied by an increase in healthspan expectancy of only 4.6 years [3]. People now live much longer than in history, particularly in ages not shaped by natural selection; therefore, advanced adult age is the major risk factor for many chronic conditions [120]. Thus, minimizing the length and severity of late-life morbidity should be a major aim for the near future, which can be referred to as ‘compression of morbidity’ [3]. Genetic, epigenetic, environmental, and pharmacological interventions hold the potential for extending healthspan, ameliorating age-related pathologies, and, in some cases, compressing late-life morbidity, for example as achieved via dietary restriction [121,122].
Cellular senescence is associated with resistance to cell death, intracellular production, and extracellular secretion of myriad bioactive molecules, the SASP. Cellular senescence is physiologically beneficial during embryonic development, tissue remodeling, and wound healing, although in these contexts senescent cells are eventually removed by immune cells [123,124]. However, in the course of aging, senescent cells persist and their presence causes tissue damage and drives the etiology of various human disorders. A recent study investigating premature aging phenotypes in mice with deficiency of excision repair cross complementing-group 1 (ERCC1) suggests that these phenotypes and shortened healthspan or lifespan are partially caused by stem cell depletion via apoptosis promoted by senescent cells [125]. Selective elimination of senescent cells, or disruption of the SASP, can restore tissue homeostasis and increase healthspan and lifespan in mice [3,9,83,126,127]. However, as the SASP may also play physiologically beneficial roles [124], caution needs to be exercised when targeting the SASP. Although continued work is warranted to address several open but key issues in related fields (see Outstanding Questions), early results suggest these agents may be effective in humans, with clinical trials ongoing for treatment of AD (Phase I/II, NCT04063124), IPF (Phase I, NCT02874989), diabetic kidney disease (Phase II, NCT02848131), and hematopoietic stem cell transplant survivors at increased risk of premature aging (NCT02652052) (each involving a D + Q regimen).
Outstanding Questions.
Are there cell type-specific and/or universal triggers and responding pathways that drive different secretomes and how do they correlate with distinct age-related disorders?
What types of cells, once senescent and displaying a typical SASP, are the most influential in affecting the functional integrity of tissues and organs?
What is the biochemical mechanism that allows senescent cells to affect health conditions efficiently, as aging-related impairment in physical function occurs even if senescent cells count for less than 1% throughout the body? Is such an effect mediated mainly by the SASP, or any specific metabolite of these cells, or both?
Where is the energy supporting biosynthesis of numerous SASP factors mainly from, when senescent cells exhibit a significantly reduced ATP production level in mitochondria? Is this special energy source or machinery targetable to manipulate the activity or fate of senescent cells?
Do senescent cells escape from routine immune surveillance via expression of certain molecules that resemble those engaged in immune checkpoint, as in the case of cancer patients (e.g., PD-L1/L2, B7-H3/H4, VISTA)? Such mechanisms could synergize with or even function completely independently of the immunosuppression mechanisms we have already known.
How strong is the impact of current senolytics on the vitality of proliferating or quiescent cells in the tissue microenvironment? Is it possible to identify specific cytoplasmic, membrane, intracellular, or extracellular markers of senescent cells to guide development of the next generation of senolytics with increased selectivity and safety, but with reduced off-target effects in vivo?
Can a set of medical criteria be established for assessing the outcome of the first-wave senolytics currently in human clinical trials, guiding ongoing studies to screen, or designing new senolytics with established pipelines (e.g., animal models and drug platforms) and directing their future translation for clinical purposes?
Highlights.
Aging is the largest risk factor for most pathologies, ranging from cancer to neurodegenerative disorders.
Senescent cells accumulate in organs during aging, promote tissue dysfunction, and cause pathological manifestations, with senescence as a defining feature of myriad aging-related diseases.
Senescent cells display hallmark features, including the senescence-associated secretory phenotype (SASP), a major driver of pathologies, and alterations to the structure and function of organelles.
Targeted elimination of senescent cells has emerged as a promising therapeutic solution to ameliorate tissue damage and promote repair and regeneration.
In the scope of clinical medicine, advances that identify key biochemical pathways, specifically those differentiating senescent cells from their proliferating counterparts, would positively affect pathological and aging processes.
Acknowledgments
The authors apologize to colleagues whose work in aging, cellular senescence, and chronic diseases could not be cited due to space limitations. We are grateful to members of the Sun Laboratory for inspiring discussions and insightful comments about the manuscript. This work is supported in part by grants from the National Key Research and Development Program of China (2016YFC1302400), the National Natural Science Foundation of China (NSFC) (81472709, 31671425, 31871380), the Key Lab of Tissue Microenvironment and Tumor of Chinese Academy of Sciences, the National 1000 Young Talents Research Program of China, the University and Locality Collaborative Development Program of Yantai (2019XDRHXMRC08), and the U.S. Department of Defense (DoD) Prostate Cancer Research Program (PCRP) (Idea Development Award PC111703) to Y.S.; CRUK (A12011), Breast Cancer Now (2012MayPR070; 2012NovPhD016), the Medical Research Council of the United Kingdom (MR/N012097/1), the Cancer Research UK Imperial Centre, Imperial ECMC and NIHR Imperial BRC to E.W-F.L.; and US National Institutes of Health grants R37-AG013925 and P01 AG062413, the Connor Fund, Robert J. and Theresa W. Ryan, and the Noaber Foundation to J.L.K.
Glossary
- Aging
a course of progressive decline in physical, and often mental, capacities, often considered as adaptation to wear and tear
- Cellular senescence
a cell state characterized by a stable and usually irreversible cell cycle arrest in response to different types of stress and/or damage, including cell replicative exhaustion, oxidative stress, oncogene activation, and therapeutic agents
- Cytoplasmic chromatin fragments
fragments that pinch off from intact nuclei of primary cells during senescence, can activate the innate immunity cytosolic DNA-sensing pathway, and lead to short-term or chronic inflammation
- DNA damage response (DDR)
a cell response to agents that cause single and/or double strand breaks of DNA, with activation of a network of intracellular pathways that sense, signal, and repair DNA lesions in the nucleus
- Healthspan
a period of good health in an individual’s lifetime
- Lifespan
the longest period over which the life of any organism or species may extend
- Metabolic reprogramming
changes in metabolic pathways that alter the bio-energetic profile and metabolism of the cell, including anabolism and catabolism
- Mitochondrial dysfunction
a condition in which the regulation of mitochondrial homeostasis, production of mitochondrial metabolites, membrane potential, and ROS generation is compromised. Mitochondrial dysfunction can decrease the NAD+/NADH ratio and induce a senescent phenotype overlapping yet differing from that caused by other senescence inducers
- Oncogene-induced senescence (OIS)
a subtype of cellular senescence induced by aberrant activation of a specific oncogene such as HRASG12V, which causes cell hyperproliferation followed by subsequent proliferation arrest
- Programmed senescence
a subtype of senescence playing proregenerative or morphogenetic roles in physiological events such as embryogenesis, wound healing, and limb regeneration. It can promote cell turnover, tissue remodeling, and, paradoxically, growth
- Proteostasis
a failure of protein homeostasis caused by accumulation of misfolded proteins within the cell, which eventually compromise cellular function
- Replicative senescence
a subtype of senescence induced by telomeric attrition after multiple cell divisions, with a decrease in cell proliferative potential. It may serve as one of the major safeguards to maintain cellular integrity necessitated by the extended longevity of humans
- Secretome
the set of protein factors secreted by cells into the extracellular space. Once used for senescent cells, it is also referred to as the SASP
- Senescence-associated β-galactosidase (SA-β-Gal)
enhanced lysosomal enzymatic activity usually observed in senescent cells, a feature that allows increased hydrolysis of β-galactoside into monosaccharides. It is frequently used as a biomarker for detection of cellular senescence
- Senescence-associated secretory phenotype (SASP)
a hallmark feature of senescent cells, characterized by robust production and secretion of soluble molecules such as growth factors, cytokines, chemokines, and extracellular matrix metalloproteases
- Senolytics
a group of chemical compounds or phytochemicals that are applied to selectively eliminate senescent cells via induction of programmed cell death
- Stress-induced premature senescence
a subtype of senescence that appears after exposure of cells to a chemical or physical stimulation that induces oxidative stress, DNA damage, and/or mitochondrial injury
- Therapy-induced senescence
a subtype of cellular senescence triggered by therapeutic modalities such as chemotherapy and radiotherapy
Footnotes
Disclaimer Statement
J.L.K. and T.T. have a financial interest related to this research. Patents on senolytic drugs are held by the Mayo Clinic. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies.
References
- 1.Singh PP et al. (2019) The genetics of aging: a vertebrate perspective. Cell 177, 200–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barzilai N et al. (2018) Aging as a biological target for prevention and therapy. JAMA 320, 1321–1322 [DOI] [PubMed] [Google Scholar]
- 3.Partridge L et al. (2018) Facing up to the global challenges of ageing. Nature 561, 45–56 [DOI] [PubMed] [Google Scholar]
- 4.Kirkland JL (2016) Translating the science of aging into therapeutic interventions. Cold Spring Harb. Perspect. Med 6, a025908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkland JL (2013) Translating advances from the basic biology of aging into clinical application. Exp. Gerontol 48, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy BK et al. (2014) Geroscience: linking aging to chronic disease. Cell 159, 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Otin C et al. (2013) The hallmarks of aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodell MA and Rando TA (2015) Stem cells and healthy aging. Science 350, 1199–1204 [DOI] [PubMed] [Google Scholar]
- 9.McHugh D and Gil J (2018) Senescence and aging: causes, consequences, and therapeutic avenues. J. Cell Biol 217, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ovadya Y and Krizhanovsky V (2018) Strategies targeting cellular senescence. J. Clin. Invest 128, 1247–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milanovic M et al. (2018) Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 [DOI] [PubMed] [Google Scholar]
- 12.Gorgoulis V et al. (2019) Cellular senescence: defining a path forward. Cell 179, 813–827 [DOI] [PubMed] [Google Scholar]
- 13.Hayflick L and Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp. Cell Res 25, 585–621 [DOI] [PubMed] [Google Scholar]
- 14.Paez-Ribes M et al. (2019) Targeting senescent cells in translational medicine. EMBO Mol. Med 11, e10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiley CD et al. (2017) Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 16, 1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu M et al. (2018) Senolytics improve physical function and increase lifespan in old age. Nat. Med 24, 1246–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Segura A et al. (2018) Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 [DOI] [PubMed] [Google Scholar]
- 18.Sharpless NE and Sherr CJ (2015) Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 [DOI] [PubMed] [Google Scholar]
- 19.Rodier F and Campisi J (2011) Four faces of cellular senescence. J. Cell Biol 192, 547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernandez-Segura A et al. (2017) Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol 27, 2652–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coppe JP et al. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuilman T et al. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 [DOI] [PubMed] [Google Scholar]
- 23.Acosta JC et al. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 [DOI] [PubMed] [Google Scholar]
- 24.West MD et al. (1989) Replicative senescence of human skin fibroblasts correlates with a loss of regulation and over-expression of collagenase activity. Exp. Cell Res 184, 138–147 [DOI] [PubMed] [Google Scholar]
- 25.Maier JA et al. (1990) Extension of the life-span of human endothelial cells by an interleukin-1 alpha antisense oligomer. Science 249, 1570–1574 [DOI] [PubMed] [Google Scholar]
- 26.Tahara H et al. (1995) Increase in expression levels of interferon-inducible genes in senescent human diploid fibroblasts and in SV40-transformed human fibroblasts with extended lifespan. Oncogene 11, 1125–1132 [PubMed] [Google Scholar]
- 27.Malaquin N et al. (2016) Keeping the senescence secretome under control: molecular reins on the senescence-associated secretory phenotype. Exp. Gerontol 82, 39–49 [DOI] [PubMed] [Google Scholar]
- 28.Takasugi M et al. (2017) Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun 8, 15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acosta JC et al. (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol 15, 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Mitri D et al. (2014) Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 515, 134–137 [DOI] [PubMed] [Google Scholar]
- 31.Eggert T et al. (2016) Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30, 533–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen F et al. (2018) Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat. Commun 9, 4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim YH et al. (2017) Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun 8, 15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Q et al. (2019) Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 18, e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito Y et al. (2017) Spatial and temporal control of senescence. Trends Cell Biol. 27, 820–832 [DOI] [PubMed] [Google Scholar]
- 36.Sun Y et al. (2018) Cellular senescence: the sought or the unwanted? Trends Mol. Med 24, 871–885 [DOI] [PubMed] [Google Scholar]
- 37.Freund A et al. (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herranz N et al. (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol 17, 1205–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laberge RM et al. (2015) MTOR regulates the protumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol 17, 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang BY et al. (2018) The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat. Commun 9, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang H et al. (2017) cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U. S. A 114, E4612–E4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dou ZX et al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gluck S et al. (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol 19, 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi A et al. (2018) Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun 9, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Cecco M et al. (2019) L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaplon J et al. (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112 [DOI] [PubMed] [Google Scholar]
- 47.Passos JF et al. (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol 6, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tai H et al. (2017) Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 13, 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korolchuk VI et al. (2017) Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine 21, 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalle Pezze P et al. (2014) Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput. Biol 10, e1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cunningham JT et al. (2007) mTOR controls mitochondrial oxidative function through a YY1-PGC-1 alpha transcriptional complex. Nature 450, 736–740 [DOI] [PubMed] [Google Scholar]
- 52.Birch J and Passos JF (2017) Targeting the SASP to combat ageing: mitochondria as possible intracellular allies? Bioessays 39, 1600235. [DOI] [PubMed] [Google Scholar]
- 53.Passos JF et al. (2007) Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Correia-Melo C et al. (2016) Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang P et al. (2013) Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493, 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miwa S et al. (2014) Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun 5, 3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiley CD et al. (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu SL et al. (2019) AMPK alpha 2 knockout enhances tumour inflammation through exacerbated liver injury and energy deprivation-associated AMPK alpha 1 activation. J. Cell. Mol. Med 23, 1687–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Safdar A et al. (2010) Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One 5, e10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wallace DC (2010) Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen 51, 440–450 [DOI] [PubMed] [Google Scholar]
- 61.Chini C et al. (2019) The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochem. Biophys. Res. Commun 513, 486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Settembre C and Ballabio A (2014) Lysosomal adaptation: how the lysosome responds to external cues. Cold Spring Harb. Perspect. Biol 6, a016907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park JT et al. (2018) Adjustment of the lysosomalmitochondrial axis for control of cellular senescence. Ageing Res. Rev 47, 176–182 [DOI] [PubMed] [Google Scholar]
- 64.Robbins E et al. (1970) Morphologic changes accompanying senescence of cultured human diploid cells. J. Exp. Med 131, 1211–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salama R et al. (2014) Cellular senescence and its effector programs. Genes Dev. 28, 99–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dimri GP et al. (1995) A biomarker that identifies senescent human-cells in culture and in aging skin in-vivo. Proc. Natl. Acad. Sci. U. S. A 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee BY et al. (2006) Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5, 187–195 [DOI] [PubMed] [Google Scholar]
- 68.Yegorov YE et al. (1998) Endogenous beta-galactosidase activity in continuously nonproliferating cells. Exp. Cell Res 243, 207–211 [DOI] [PubMed] [Google Scholar]
- 69.Severino J et al. (2000) Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res 257, 162–171 [DOI] [PubMed] [Google Scholar]
- 70.Sasaki M et al. (2001) Senescent cells are resistant to death despite low Bcl-2 level. Mech. Ageing Dev 122, 1695–1706 [DOI] [PubMed] [Google Scholar]
- 71.Sagiv A et al. (2013) Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 32, 1971–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yosef R et al. (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun 7, 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu Y et al. (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryu SJ et al. (2007) Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 14, 1020–1028 [DOI] [PubMed] [Google Scholar]
- 75.Sanders YY et al. (2013) Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 1, 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baar MP et al. (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yosef R et al. (2017) p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 36, 2280–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuhrmann-Stroissnigg H et al. (2017) Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun 8, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tchkonia T et al. (2013) Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest 123, 966–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kang TW et al. (2011) Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 [DOI] [PubMed] [Google Scholar]
- 81.Prata L et al. (2019) Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin. Immunol 40, 101275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baker DJ et al. (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeon OH et al. (2017) Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med 23, 775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palmer AK et al. (2019) Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hickson LJ et al. (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krishnamurthy J et al. (2004) Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest 114, 1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baker DJ et al. (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fane M and Weeraratna AT (2019) How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kirkland JL and Tchkonia T (2015) Clinical strategies and animal models for developing senolytic agents. Exp. Gerontol 68, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang J et al. (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22, 78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Czabotar PE et al. (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol 15, 49–63 [DOI] [PubMed] [Google Scholar]
- 92.Levine B et al. (2008) Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Avsar Abdik E et al. (2019) ABT-737 and erufosine combination against castration-resistant prostate cancer: a promising but cell-type specific response associated with the modulation of anti-apoptotic signaling. Anti-Cancer Drugs 30, 383–393 [DOI] [PubMed] [Google Scholar]
- 94.Garandeau D et al. (2019) Targeting the sphingosine 1-phosphate axis exerts potent antitumor activity in BRAFi-resistant melanomas. Mol. Cancer Ther 18, 289–300 [DOI] [PubMed] [Google Scholar]
- 95.Lever JR and Fergason-Cantrell EA (2019) Allosteric modulation of sigma receptors by BH3 mimetics ABT-737, ABT-263 (navitoclax) and ABT-199 (venetoclax). Pharmacol. Res 142, 87–100 [DOI] [PubMed] [Google Scholar]
- 96.Zhu Y et al. (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Childs BG et al. (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walaszczyk A et al. (2019) Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 18, e12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cang S et al. (2015) ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guerrero A et al. (2019) Cardiac glycosides are broad-spectrum senolytics. Nat. Metab 1, 1074–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Triana-Martinez F et al. (2019) Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun 10, 4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martin N et al. (2020) Cardiac glycosides as senolytic compounds. Trends Mol. Med 26, 1–3 [DOI] [PubMed] [Google Scholar]
- 103.Schafer MJ et al. (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun 8, 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Farr JN et al. (2017) Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23, 1072–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lehmann M et al. (2017) Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J 50, 1602367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ogrodnik M et al. (2019) Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 29, 1061–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Musi N et al. (2018) Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang P et al. (2019) Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci 22, 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cherif H et al. (2019) Curcumin and o-vanillin exhibit evidence of senolytic activity in human IVD cells in vitro. J. Clin. Med 8, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yousefzadeh MJ et al. (2018) Fisetin is a senotherapeutic that extends health and lifespan. Ebiomedicine 36, 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhu Y et al. (2017) New agents that target senescent cells: the flavone, fisetin, and the BCL-X-L inhibitors, A1331852 and A1155463. Aging-Us 9, 955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang YY et al. (2016) Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging-Us 8, 2915–2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang X et al. (2018) Oxidation resistance 1 is a novel senolytic target. Aging Cell 17, e12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li W et al. (2019) The curcumin analog EF24 is a novel senolytic agent. Aging-Us 11, 771–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dang Y et al. (2019) Berberine ameliorates cellular senescence and extends the lifespan of mice via regulating p16 and cyclin protein expression. Aging Cell 19, e13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sena P et al. (2018) Metformin induces apoptosis and alters cellular responses to oxidative stress in Ht29 colon cancer cells: preliminary findings. Int. J. Mol. Sci 19, E1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Oubaha M et al. (2016) Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med 8, 362ra144. [DOI] [PubMed] [Google Scholar]
- 118.Farr JN et al. (2017) Corrigendum: Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23, 1384. [DOI] [PubMed] [Google Scholar]
- 119.Crimmins EM (2015) Lifespan and healthspan: past, present, and promise. Gerontologist 55, 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Niccoli T and Partridge L (2012) Ageing as a risk factor for disease. Curr. Biol 22, R741–R752 [DOI] [PubMed] [Google Scholar]
- 121.Solon-Biet SM et al. (2019) Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat. Metab 1, 532–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hahn O et al. (2019) A nutritional memory effect counteracts benefits of dietary restriction in old mice. Nat. Metab 1, 1059–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Munoz-Espin D et al. (2013) Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 [DOI] [PubMed] [Google Scholar]
- 124.Demaria M et al. (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim DE et al. (2019) Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin. Aging Cell 19, e13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Childs BG et al. (2017) Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov 16, 718–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Childs BG et al. (2015) Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med 21, 1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xue W et al. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ovadya Y et al. (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun 9, 5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Demaria M et al. (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bussian TJ et al. (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Campisi J et al. (2019) From discoveries in ageing research to therapeutics for healthy ageing. Nature 571, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Justice JN et al. (2019) Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mannick JB et al. (2014) mTOR inhibition improves immune function in the elderly. Sci. Transl. Med 6, 268ra179. [DOI] [PubMed] [Google Scholar]