

SUMMARY
Histone methyltransferase KMT2D harbors frequent loss-of-function somatic point mutations in several tumor types, including melanoma. Here, we identify KMT2D as a potent tumor suppressor in melanoma through an in vivo epigenome-focused pooled RNAi screen and confirm the finding by using a genetically engineered mouse model (GEMM) based on conditional and melanocyte-specific deletion of KMT2D. KMT2D-deficient tumors show substantial reprogramming of key metabolic pathways, including glycolysis. KMT2D deficiency aberrantly upregulates glycolysis enzymes, intermediate metabolites, and glucose consumption rates. Mechanistically, KMT2D loss causes genome-wide reduction of H3K4me1-marked active enhancer chromatin states. Enhancer loss and subsequent repression of IGFBP5 activates IGF1R-AKT to increase glycolysis in KMT2D-deficient cells. Pharmacological inhibition of glycolysis and insulin growth factor (IGF) signaling reduce proliferation and tumorigenesis preferentially in KMT2D-deficient cells. We conclude that KMT2D loss promotes tumorigenesis by facilitating an increased use of the glycolysis pathway for enhanced biomass needs via enhancer reprogramming, thus presenting an opportunity for therapeutic intervention through glycolysis or IGF pathway inhibitors.
In Brief
Through an in vivo epigenome-focused pooled RNAi screen, Maitituoheti et al. identify KMT2D as a tumor suppressor in melanoma. KMT2D-deficient tumors show substantial reprogramming of key metabolic pathways by reduction of H3K4me1-marked active enhancers, conferring sensitivity to glycolysis and IGFR inhibitors in melanoma with KMT2D-inactivating mutations.
Graphical Abstract
INTRODUCTION
An important theme that has emerged from the cancer genome sequencing studies in the past decade is genetic alterations in epigenetic regulators implicating the epigenome as an important player in cancer progression (Shen and Laird, 2013; Watson et al., 2013). Loss-of-function missense and nonsense point mutations are observed to be highly prevalent across multiple tumor types in two families of chromatin regulators: (1) Histone H3K4 methyltransferase members, including KMT2C and KMT2D (Herz et al., 2012); and (2) SWI/SNF complex members, including SMARCA4, ARID1A, and PBRM1 (Dawson, 2017). Although recent studies have begun to shed light on the roles of these proteins in cancer progression (Dhar et al., 2018; Koutsioumpa et al., 2019; Lin-Shiao et al., 2018; Ortega-Molina et al., 2015; Zhang et al., 2015), we still have limited knowledge of why mutations in these proteins are selected over the course of tumor progression.
We focus our studies on metastatic melanoma that is an aggressive cancer with a 5-year survival of less than 20% (Siegel et al., 2018). In the past decade, the number of people affected by the disease has increased tremendously (Siegel et al., 2018). Although the landscape of available treatment options has expanded for this disease in the form of immune checkpoint blockade agents and targeted agents (such as BRAFi and MEKi) (Luke et al., 2017), durable responses are observed in only a subset of patients, leading several thousand people dying of this disease every year. Hence, other treatment strategies need to be further explored.
In cutaneous melanoma, mutations in epigenetic regulators, including IDH1/2, EZH2, ARID1A/1B, ARID2, and SMARCA4, have been observed at statistically significant frequencies (Hodis et al., 2012; Krauthammer et al., 2012). However, we have a limited understanding of how specific mutant epigenetic proteins impact melanomagenesis. Functional studies have implicated the involvement of other epigenetic factors, such as JARID1B (Roesch et al., 2010), SETDB1 (Ceol et al., 2011), TET2 (Lian et al., 2012), and histone variants (Kapoor et al., 2010; Vardabasso et al., 2015), in melanoma progression. Systematic functional approaches are needed to elucidate how misregulation of epigenetic regulators impact chromatin states and downstream gene expression programs during various stages of tumorigenesis. A detailed mechanistic understanding of melanomagenesis and the role of epigenetic regulators will also inform therapeutic strategies for patients whose tumors bear these mutations. We isolated KMT2D as the top hit in an in vivo RNAi screen focused on identification of epigenetic regulators that play a tumor-suppressive function in melanoma. KMT2D is a histone H3 lysine 4 (H3K4) methyltransferase that primarily performs monomethylation, H3K4me1, which has been shown to be a marker of enhancer elements (Herz et al., 2012; Lai et al., 2017; Lee et al., 2013; Wang et al., 2016). KMT2D not only marks nucleosome with H3K4me1 but also recruits CBP/p300 that in turn acetylate these nucleosomes and hence lead to activation of these enhancers (Lai et al., 2017; Lee et al., 2013; Wang et al., 2016). Several studies have implicated enhancer aberrations as a hallmark of multiple tumor types, including melanoma (Akhtar-Zaidi et al., 2012; Chapuy et al., 2013; Gelato et al., 2018; Herz et al., 2014; Hu et al., 2016; Lin et al., 2016; Lovén et al., 2013; Mansour et al., 2014; Qian et al., 2014; Sur and Taipale, 2016; Verfaillie et al., 2015). However, most of them have focused on aberrant enhancer activation, and little is known about how enhancer inactivation, which may result from loss of KMT2C/KMT2D function, influences tumor progression. We establish that KMT2D-deficient tumors may exhibit reduced enhancer activity that leads to the deregulation of energy metabolism pathways, including glycolysis, thus providing a strategy for targeting KMT2D mutant cancers.
RESULTS
Identification of Eight Potential Tumor Suppressors including KMT2D through an RNAi Screen
We performed an RNAi screen (Figure 1A) to identify tumor-suppressor epigenetic regulators in melanoma. We used a well-characterized system of TERT-immortalized human primary foreskin melanocytes that harbor stably integrated dominant negative p53, CDK4R24C, and BRAFV600E (Garraway et al., 2005; Fiziev et al., 2017) (passage, n < 15). They are referred as HMEL-BRAFV600E. When injected in nude mice, HMEL-BRAFV600E cells form visible tumors only after 22–24 weeks and with low penetrance (~10%–20%) (Figure 1B). In addition, this line is poised to switch to the tumorigenic state upon additional cooperative driver alterations, such as PTEN loss (Fiziev et al., 2017; Figure 1B). Hence, it is a relevant cell-based system for discovering tumor-promoting events, as it provides a minimal yet sensitized tumorigenic background to identify moderate-to-potent tumor suppressors. We had previously used this system for the discovery of pro-tumorigenic epigenomic changes in melanoma (Fiziev et al., 2017).
Figure 1. RNAi Screen Identifies Potential Melanoma Tumor Suppressor Genes.

(A) Schematic of RNAi screen targeting epigenetic regulators to identify tumor suppressors in melanoma. HMEL-BRAFV600E cells were transfected in a pooled fashion with 475 shRNAs targeting 95 epigenetic regulators (five shRNAs/gene) in 19 experimental pools (25 shRNAs targeting five genes per experimental pool). Cells were orthotopically injected intradermally into the flanks of NCR-nude mice. Tumors that arose before the controls (shNT, shLuc, and shGFP) were sequenced to identify the shRNA sequence.
(B) Kaplan-Meier curve showing tumor-free survival of mouse cohorts orthotopically injected with one million HMEL-BRAFv600E cells transfected with pooled shRNAs from primary screen. Nineteen experimental pools (P1–P19), three negative control pools (shLuc, shGFP, and shNT), and one positive control (shPTEN) were injected in 10 mice each, and tumor formation was monitored over 25 weeks. Mantel-Cox test, *p < 0.05, **p < 0.01, and ***p < 0.001; n = 10 per pool. Only pools that show significant acceleration are shown here. For data on non-significant pools, please see Figure S1A.
(C) List of genes identified from their pools, the week of first appearance of tumor in the pool, and the percentage of mice in respective cohort demonstrating accelerated tumorigenesis over control mice (penetrance).
(D–K) Kaplan-Meier curves showing tumor-free survival of mouse cohorts orthotopically injected with HMEL-BRAFV600E cells stably expressing shRNAs against KMT2D (D), KDM1A (E), APOBEC2 (F), HDAC6 (G), KMT2F (H), SETD4 (I), KAT4 (J), and KDM5B (K). Mantel-Cox test, *p < 0.05, **p < 0.01, and ***p < 0.001; n = 10 per arm.
In the current study, we constructed a short hairpin RNA (shRNA) expression vector library that included 475 shRNAs targeting 95 proteins known to regulate epigenetic processes, including chromatin modification and nucleosome remodeling (Table S1). The HMEL-BRAFV600E cells were transfected with 23 pools of shRNAs individually. Hereafter, “pool” refers to stably transfected HMEL-BRAFv600E cells. Of these pools, 19 experimental pools contained 25 shRNAs each (five shRNAs each for five genes selected randomly). The three negative pools contained one negative-control shRNA each (shGFP, shLacZ, and shNT [non-targeting]) and final pool harbored PTEN shRNA (shPTEN) as a positive control (Figure 1B). Briefly, one million cells were orthotopically injected intradermally in nude mice (10 sites) that were monitored for visible tumor formation over the subsequent 25 weeks (Figures 1B and S1A). Mice injected with cells from eight of the 19 pools and the positive control (shPTEN) displayed a significant acceleration of tumor formation compared with the negative controls. The first occurrence of tumor formation was at 5 weeks, whereas tumor formation did not occur until 22 weeks in the negative-control mice and multiple pools (Figures 1B, 1C, and S1A).
We next identified the shRNAs enriched in DNA of tumors harvested from the mice exhibiting significantly accelerated tumor formation by performing Sanger sequencing of the pLKO amplified region containing shRNA (list of genes in Figure 1C). We identified eight unique shRNAs each from eight pools that significantly accelerated tumor formation. To validate the results of the screen, we knocked down each of the eight candidate genes individually by using at least two independently validated shRNAs in HMEL-BRAFV600E and widely used WM115 (BRAFV600E mutant) melanoma cells (Figures S1B–S1I) and tested tumor formation efficiency (Figures 1D–1K and S1J–S1P). All eight genes (KMT2D, KDM1A, APOBEC2, HDAC6, KMT2F, SETD4, KAT4, and KDM5B) were validated as tumor suppressor candidates, as knockdown of these genes in both HMEL-BRAFV600E and WM115 cells resulted in accelerated tumor formation (p < 0.05) (Figures 1D–1K and S1J–S1P). In addition, knockdown of a subset of these genes in HMEL-BRAFV600E cells also promoted invasion in vitro in a Boyden chamber assay (Figure S1Q). KMT2D was the most potent hit, as mice injected with cells with stable KMT2D knockdown developed tumor appearance at the earliest interval and with the highest penetrance compared with negative controls (Figures 1C, 1D, and 2A). Among the rest, KDM5B has been previously implicated in melanoma for which it is believed to control the maintenance of slow cycling melanoma cells and displays complex phenotypes during tumor progression (Roesch et al., 2010).
Figure 2. KMT2D Functions as a Tumor Suppressor in BRAFV600E Mutant Melanomas.

(A and D) Kaplan-Meier curve showing tumor-free survival of mouse cohorts orthotopically injected with WM115 (A) and WM266–4 (D) cells stably expressing shRNAs against KMT2D. Mantel-Cox test, *p < 0.05; n = 10 per arm.
(B) Schematic of KMT2D protein showing missense mutations seen across all melanoma studies deposited in the cBio portal. Green-filled circles indicate missense mutations, black-filled circles indicate truncating mutations, and red circles indicate functional mutations occurring after amino acid residue 4700. Colored boxes within the KMT2D schematic show different protein domains.
(C) Stacked bar chart showing percentage of nevi (n = 18), primary melanoma (n = 62), and metastatic melanoma (n = 22, labeled “Met”) samples with the various intensity (of KMT2D expression) categories (0, 0.5, 1, 2, and 3, as shown in the legend). The p value for the difference in average intensity of KMT2D expression between primary and nevi was <0.05. Similarly, the p value for the difference in average intensity of KMT2D expression between primary and metastatic melanoma was <0.05.
(E) Kaplan-Meier curve of auricular tumor-free survival in KMT2D WT (KMT2D+/+, blue, n = 40), KMT2D heterozygous (KMT2DL/+, green, n = 12), and KMT2D mutant (KMT2DL/L, red, n = 19) mice in an iBIP (Tyr-CreERT2, Rosa26-rtta, TetO-BRAFV600E, PTENL/L, INK/ARFL/L) background that were treated with doxycycline (dox) (2 mg/ml, ad libitum) and 4-OHT (1 μM, topical). The x axis refers to days after 4-OHT application. Mantel-Cox test, *p < 0.05, **p < 0.01, and ***p < 0.001.
(F) Tumor burden of KMT2D WT (KMT2D+/+, blue, n = 40), KMT2D heterozygous (KMT2DL/+, green, n = 12), and KMT2D mutant (KMT2DL/L, red, n = 19) mice in iBIP background at 89 days after 4-OHT application.
(G) Images of Ki-67-stained (standard immunohistochemistry) (40×) melanoma tumors from iBIP;KMT2D+/+ and iBIP;KMT2DL/L mice. Right panel shows percentage of Ki-67-stained cells across five different fields of 100 cells each. Scale bars represent 20 μm.
(H) Table showing the human melanoma lines and iBIP;KMT2D-mouse-model-derived cells that were used in the functional studies through the rest of the figures.
(I) Bar graph showing KMT2D expression levels (n = 3) in WT-m1, Mut-m2, and Mut-m2 + hKMT2D lines (left panel) or in WT-h1, Mut-h1, and Mut-h1 + hKMT2D lines (right panel). The y axis represents fold change of the gene expression compared to 28S and normalized to Mut-m2 or Mut-h1 lines. *** p < 0.001 (unpaired t test comparison between the indicated groups) See Figure S2I for protein levels.
(J) Graph showing tumor volume of nude mice (n = 10 per group) injected with KMT2D mutant murine (Mut-m2) or human (Mut-h1) cells harboring an inducible KMT2D expression vector that leads to KMT2D overexpression upon application of dox. p values represent t test comparison between the indicated groups on the last time point. In this graph, **p < 0.001 and ***p < 0.000001.
In (G) and (I), data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates.
Genetically Engineered Mouse Model (GEMM) Confirms KMT2D Is a Potent Tumor Suppressor in Melanoma
We searched published melanoma genomic studies to identify patients whose tumors harbor genetic aberrations in the potential tumor suppressor genes discovered through the screen. We observed that ~15% of melanoma cases identified harbored missense mutations in KMT2D, whereas ~5%–8% of patients harbored missense mutations in KAT4 (Figures 2B and S2A) (Cancer Genome Atlas Network, 2015b). As KMT2D mutations are prevalent (Cancer Genome Atlas Network, 2015a; da Silva Almeida et al., 2015; Gao et al., 2014; Grasso et al., 2012; Jones et al., 2012; Juhlin et al., 2015; Kandoth et al., 2013; Lawrence et al., 2014; Lin et al., 2014; Lohr et al., 2012; Morin et al., 2011; Parsons et al., 2011; Pasqualucci et al., 2011; Pugh et al., 2012; Rao and Dou, 2015; Sausen et al., 2015; Song et al., 2014) and this gene is increasingly reported to be a potential tumor suppressor across other tumor types (Dhar et al., 2018; Koutsioumpa et al., 2019; Lin-Shiao et al., 2018; Ortega-Molina et al., 2015; Rao and Dou, 2015; Zhang et al., 2015), we next sought to deeply characterize the mechanism of action of KMT2D in melanoma, particularly as the strongest phenotype (fastest tumor growth) in RNAi screen was seen with the KMT2D loss. A subset of the missense mutations in KMT2D were truncating or frameshift insertions/deletions (4.4%) that likely abrogate histone methyltransferase activity (Figure 2B). In addition, 10% of all missense mutations occurred distal to amino acid residue 4700 that were shown to disrupt histone methyltransferase activity in a previous study (Zhang et al., 2015). Together, we categorize these set of mutations—truncating, frameshift, and post4700aa—as “functional” driver mutations for KMT2D. Although we make use of this stringent criteria as a deterministic measure for KMT2D-deficient tumors so that we can delineate its mechanism of action, it is not a reflection of all KMT2D somatic variants that may produce a non-functional KMT2D protein.
First, we checked if, in addition to mutations, KMT2D mRNA and protein levels were also altered in human melanoma. Staining of a tissue microarray (TMA) harboring 100 cases of nevi, primary melanomas, and metastatic melanomas showed a significant progressive loss of KMT2D protein levels in primary and metastatic melanomas (Figures 2C and S2B). A similar trend was also observed in mRNA expression of KMT2D, as identified by the assessment of publicly available melanoma progression transcriptomic datasets (Talantov et al., 2005; Xu et al., 2008; Figure S2C) suggesting KMT2D regulation at both the level of gene expression and somatic mutations.
Functionally, knockdown of KMT2D by two different shRNAs led to increased tumor burden in two clonal variants of HMEL-BRAFV600E, namely, WM115 and WM266–4 cells (Figures 1D, 2A, 2D, and S2D–S2H). In addition, an increase in soft agar colony formation (Figure S2I) and invasion (Figure S2J) was observed in vitro. To further verify the role of KMT2D in melanoma in a specific genetic context, we used a conditional mouse model of KMT2D that harbors Lox sites flanking exons 16–20, thereby leading to deletion of this gene in a tissue-specific manner (Dhar et al., 2018). Melanocyte-specific deletion with Tyr-CreERT2 did not result in the formation of melanomas (data not shown), and thus, these mice were crossed with a previously published doxycycline- and a tamoxifen-inducible mouse model of BRAFV600E melanoma (iBIP = Tyr-CreERT2, Rosa26-rtta, TetO-BRAFV600E, PTENL/L, INK/ARFL/L). Tamoxifen application on the ears of KMT2D mutant iBIP mice resulted in a drastic acceleration of tumorigenesis compared with KMT2D wild-type (WT) iBIP mice (Figures 2E and 2F). Intriguingly, the heterozygous mice also showed significant acceleration of auricular tumor burden (Figures 2E and 2F). Furthermore, increased proliferation and melanocyte origin was confirmed by immunohistochemical analysis (IHC) for Ki-67 and tyrosinase, respectively (Figures 2G and S2K).
Next, we derived cell lines from the tumors of two KMT2D WT (iBIP-KMT2D+/+) and two mutant (Mut, iBIP-KMT2DL/L) models (Figure 2H) and confirmed the genotype of all alleles. The two KMT2D WT iBIP cell lines were labeled as WT-m1 and WT-m2 for WT mouse 1 and 2; and the two KMT2D mutant cell lines were labeled as Mut-m1 and Mut-m2 for mutant mouse 1 and 2 (Figure 2H). These lines were verified for the loss of KMT2D mRNA by qPCR (Figure S2L) and protein by immunofluorescence (Figure S2M). The phenotypes observed in KMT2D mutant lines were dependent on the loss of this gene, as overexpression of full-length KMT2D (Figures 2I and S2M) reduced tumor burden in vivo (Figure 2J) in immunodeficient nude mice. To assess relevance in humans, we performed all of the follow-up experiments in two KMT2D WT human melanoma lines (A375 and RPMI-7951 that are referred to hereafter as WT-h1 and WT-h2) and two KMT2D mutant human melanoma lines (SKMEL-24 and WM278 that are referred to hereafter as Mut-h1 and Mut-h2) (Figures 2H and S2M). Mut-h1 and Mut-h2 harbor truncating mutations at Q2800 and Q2062, respectively (Iorio et al., 2016). Similar to the murine system, overexpression of hKMT2D rescued the tumorigenesis phenotype in the human cell lines (Figures 2I, 2J, and S2M).
Hyperactive Glycolysis in KMT2D Mutant Tumors Is a Targetable Pathway
To determine the molecular phenotype conferred by KMT2D loss, we performed an RNA sequencing (RNA-seq)-based transcriptome profiling experiment in the KMT2D WT and mutant murine melanoma lines. We identified 1,761 genes that were uniquely overexpressed in the KMT2D mutant compared with WT conditions (false discovery rate [FDR] < 0.05, fold change [FC] > 2, n = 3) and 1,443 that were repressed. Genes overexpressed in KMT2D mutant cells were enriched for pathways related to immune response, cell adhesion, and epithelial-to-mesenchymal transition, as well as various metabolic pathways, including the “hexose metabolic pathway” or glycolysis (Figures 3A, 3B, S3A, and S3B; Table S2). Similar pathways, including glycolysis, were also found to be upregulated in KMT2D mutant human melanomas upon analyses of melanoma tumors from a published TCGA study (Figure 3C; Table S2). They included many glycolysis enzymes, including PGAM1, PGK1, ENO1, HK2, GAPDH, TPI1, and LDHA, as well as its upstream regulators (Figure S3C; Table S2). A survey of pan-cancer TCGA data suggested that energy metabolism pathways, including glycolysis, were activated across 6 other tumor types (BLCA [urothelial bladder carcinoma], CESC [cervical squamous cell carcinoma], endocervical adenocarcinoma, HNSC [head and neck squamous cell carcinoma], LUSC [lung squamous cell carcinoma], UCEC [uterine corpus endometrial carcinoma], and STAD [stomach adenocarcinoma]) that harbor functional KMT2D driver mutations (Figures 3D and S3D; Table S2). We observed drastic upregulation of 10 out of 12 glycolysis pathway enzyme genes (GLUT1, HK1, GPI1, PFKA, ALDOC, TPI1, GAPDH, PGK1, PGAM1, and ENO1) by qPCR in KMT2D mutant lines compared with WT lines in both human and murine models (Figures 3E and 3F). Similarly, the rescue of mutant lines with full-length WT KMT2D reduced their expression (Figure 3G). Higher expression of ENO1, PGK1, and PGAM1 was confirmed in KMT2D mutant iBIP melanoma tumors by IHC (Figure 3H). Quantitation of glucose uptake and lactate production confirmed upregulation of glycolysis in the KMT2D mutant lines (Figures 3I and 3J) that was reduced upon KMT2D overexpression (Figure 3K). In addition, mass-spectrometry-based quantitative measurement of glycolysis intermediate metabolites showed higher levels of fructose-1,6-biphosphate, D-glyceraldehyde-3-phosphate, dihydroxy-acetone-phosphate, 1,3-diphosphateglycerate, and pyruvate in two KMT2D mutant murine lines than those of two WT lines (Figure 3L). We also noted a modest increase in some TCA metabolites, amino acids, and sugars, whereas the Pentose Phosphate pathway metabolites either did not change or showed a modest decrease in KMT2D mutant cells compared with WT cells (Figures S3E–S3H). Consistent with this higher glycolysis rate in KMT2D mutants, they grew poorly in low glucose media compared to high glucose media, which likely resulted from rapid exhaustion of glucose in the media (Figures S4A and S4B). A trivial explanation for the increase in glycolysis in KMT2D mutant cells would be the higher proliferative potential of these cells than that of WT cells. However, contradictory to this hypothesis, we observe that KMT2D mutant cells proliferate more slowly than WT cells in vitro (Figures S4A and S4B) despite increased tumorigenesis in vivo. Similarly, re-expression of KMT2D in human and mouse KMT2D mutant cells modestly increased their proliferation (Figure S4C). Together, these data provide the evidence of activation of glycolysis in KMT2D mutant melanomas (Figure 3M) that likely helps meet the increased biomass and energy requirements for increased tumorigenesis.
Figure 3. KMT2D Mutated Tissues Exhibit Aberrant Activation of Glycolysis.

(A) Top five Gene Ontology (GO) terms for upregulated genes (FDR < 0.05, FC > 2) between KMT2D mutant murine cells and KMT2D WT cells by total RNA-seq analysis.
(B) Enrichment plot for HALLMARK glycolysis pathway in all differentially expressed genes (FDR < 0.05, FC > 2) between KMT2D mutant murine cells and KMT2D WT cells by total RNA-seq analysis. Each black bar represents a gene in the pathway.
(C) GSEA plot of the HALLMARK glycolysis pathway in differentially expressed genes between human primary melanomas with low versus high KMT2D expression (n = 10 for each group) from the TCGA-SKCM cohort. Each black bar represents a gene in the pathway.
(D) GSEA of different MSigDB energy metabolism pathways in differentially expressed genes between KMT2D mutant (carrying truncation, frameshift and post4700aa missense) and WT human tumors from six TCGA tumor groups where KMT2D mutant tumors are n > 10.
(E–G) Bar graph showing the relative expression pattern (n = 3 for each sample) of 12 glycolysis enzyme genes (compared to 28S) in KMT2D mutant and WT murine (E) and human (F) cells (details of the system in Figure 2H) as well as in Mut-m1 and Mut-h1 cells with dox-inducible rescue of full-length WT KMT2D expression (G). *p < 0.05, **p < 0.001, ***p < 0.0001. Unpaired t test p values were calculated for each mutant sample versus both WT samples separately, and a higher p value is shown in the figure when both were significant.
(H) Immunohistochemistry images demonstrating expression of ENO1, PGK1, and PGAM1, encoded by three glycolysis genes, in iBIP;KMT2D+/+ and iBIP;KMT2DL/L melanoma tumors. Scale bars represent 20 μm.
(I–K) Graph showing measurement of glucose uptake and lactate production (n = 3 for each sample) in KMT2D mutant and WT murine (I) and human (J) cells (details of the system in Figure 2H) as well as in the KMT2D mutant mouse Mut-m1 and human Mut-h1 cells with dox-inducible rescue of full-length WT KMT2D expression (K).
(L) Bar graph showing relative levels (n = 3 for each sample) of various metabolite intermediates produced during the glycolysis pathway, as measured by selected reaction monitoring tandem mass spectrometry. *p < 0.05, **p < 0.01, ***p < 0.001.
(M) Schematic of the glycolysis pathway showing aberrantly activated glycolysis enzymes (in red) and metabolites (in blue) of KMT2D mutant compared to WT conditions.
In (E)–(G) and (I)–(L), data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates.
Next, we tested whether the aberrantly activated glycolysis pathway contributed to the increased tumorigenic potential of KMT2D mutant melanomas. Inhibition of the glycolysis pathway using three different inhibitors—2-DG (glucose competitor), pomhex (an ENO1 inhibitor; Lin et al., 2018), and lonidamine (Hexokinase inhibitor—selectively reduced the proliferation of KMT2D mutant melanoma cells compared with that of KMT2D WT melanoma cells in both murine as well as human systems (Figures 4A–4D, S4D, and S4E; Table S3). This effect was more pronounced in low glucose conditions than in high glucose media (Figures S4F and S4G). This preferential effect of 2-DG on KMT2D mutant murine and human cell lines was rescued with the expression of WT KMT2D (Figures 4E and 4F). Consistent with the in vitro data, tumors formed by xenotransplantation of KMT2D mutant lines were more sensitive to 2-DG treatment in nude mice (Figures 4G and 4H). Importantly, we did not observe preferential growth inhibition of KMT2D mutant murine cells comparwith WT cells by a OxPhos inhibitor, IACS-10759 (Figure S4H). Together, these data suggest that upregulated glycolysis is an important contributor to enhanced tumorigenesis in KMT2D mutant melanomas and suggest a potential therapeutic strategy in this genetic context.
Figure 4. Inhibition of Glycolysis Preferentially Impacts KMT2D Mutant Cells.

(A–D) Growth curves for KMT2D mutant and WT murine (A and C) and human (B and D) melanoma cells treated with various concentrations of 2-deoxy-D-glucose (A and B) or pomhex (C and D). Relative confluence at 96 h posttreatment are plotted, and IC50 values are shown in the accompanying table.
(E and F) Growth curves for KMT2D mutant mouse Mut-m2 (E) and human Mut-h1 (F) melanoma cells that express inducible KMT2D (with 10 mg/ml doxycycline application). Relative confluence at 96-h posttreatment are plotted, and IC50 values are shown in the accompanying table.
(G and H) Line plot showing average tumor volumes for mice (n = 10 per group) injected with KMT2D mutant and WT murine (G) and human (H) melanoma cells and treated with 2-DG (500 mg/kg) every other day. p values represent t test comparison between the indicated groups on the last time point. In this graph, **p < 0.001 and ***p < 0.000001.
H3K4me1-Marked Enhancer Reprogramming Occurs in KMT2D Mutant Melanoma
We examined total and genome-wide levels of H3K4 marks, as KMT2D is known to harbor histone methyltransferase activity toward multiple H3K4 methylation states and impacts H3K27ac patterns (Dhar et al., 2018; Dorighi et al., 2017; Lee et al., 2007, 2013; Wang et al., 2016). KMT2D mutant murine cells harbored lower levels of total H3K4me1 and H3K27ac marks than those in WT cells (Figures 5A and S5A), and H3K4me1 levels were elevated upon KMT2D re-expression (Figure S5B). Immunohistochemistry staining of a TCGA melanoma tumor TMA suggests that KMT2D expression levels correlate with those of H3K4me1, H3K27ac, and H3K4me3 (Figures 5B and S5C). We also observed a significant loss of H3K4me1 but not H3K27Ac in KMT2D mutant Cancer Cell Line Encyclopedia (CCLE) pan-cancer cell lines (Figures 5C and S5D). Consistently, mass-spectrometry-based quantitation of histone modifications in KMT2D mutant murine cells showed a modest loss of H3K4me1, H3K27ac, and H3K4me3 (Figure S5E). Next, we determined chromatin states in murine melanoma KMT2D mutant and WT tumors using chromatin immunoprecipitation sequencing (ChIP-seq) for the histone modifications H3K4Me1 (enhancers), H3K4Me3 (promoters), H3K27Ac (active), H3K79Me2 (transcription), and H3K27Me3 (polycomb-repressed) (Maunakea et al., 2010), in line with studies from the NIH Roadmap project (Kundaje et al., 2015). Chromatin state calls using a 10-state ChromHMM model representing various epigenomic states, including promoters (states 3 and 4), enhancers (states 2, 5, and 6), polycomb repressed (state 9), transcribed (state 1), and unmarked (states 8 and 10) (Figure 5D). Chromatin state transition between KMT2D mutant and WT cells identified state 6 (active enhancer, H3K4me1 high) to 10 (low) as the most prominent transition that was associated with a loss of H3K4me1- and H3K27ac-based enhancers (Figure 5E). The other two prominent changes (i.e., state 2 [transcribed enhancer] to 1 [transcribed], and state 3 [transcribed 5′ and 3′ promoter] to 2 [transcribed enhancer]) were associated with the loss of H3K4me1 and H3K4me3, respectively (Figure 5E). An examination of average intensities of individual H3K4me1 and H3K27ac marks across the genome showed a significant reduction of H3K4me1 and little/no change in H3K27ac (Figures 5F and 5G). Similarly, we observed changes in H3K4me1-based superenhancer regions but not those called by the H3K27ac signal (Figures S5F and S5G). Importantly, we also noticed a pronounced increase in the average intensities of H3K27me3 peaks in KMT2D mutants compared with WT samples on enhancer loci that lose the H3K4me1 mark that could imply a transcriptional repression of a subset of genes (Figure 5H). H3K27me3 peaks also showed modest genome-wide enrichment (Figure S5H) that could be due to loss of function of H3K27me3-specific demethylase, KDM6A, which is known to be an obligate partner of KMT2D (Lee et al., 2007). On the contrary, we did not notice much change in H3K79me2 and H3K4me3 enrichment between KMT2D WT and mutant tumors (Figures S5I and S5J). We compared the overlapping H3K27ac and H3K4me1 peaks between KMT2D mutant and WT cells to identify 7,555 active enhancer peaks that were lost in mutant cells (Figure 5H). These lost active enhancer peaks were associated with important melanoma regulatory genes in immune pathways, apoptosis signaling pathway, and p53 pathway by glucose deprivation (Figure S5K). These data suggest that KMT2D loss results in significant reprogramming of the enhancer landscape in melanoma.
Figure 5. KMT2D Loss Is Associated with Loss of H3K4me1-Marked Enhancers.

(A) Western blot showing total H3K4me1, H3K27ac, H3K4me3, and H3 in KMT2D mutant (Mut-m1, Mut-m2, Mut-h1, and Mut-h2) and WT (WT-m1, WT-m2, WT-h1, and WT-h2) murine (right) and human (left) melanoma cells.
(B) Bar chart showing immunohistochemistry-based staining of melanoma tumors (MDACC TCGA samples) in a tissue microarray for KMT2D, H3K4me1 (left panel), and H3K27ac (right panel). KMT2D intensity was grouped into low (score of 1 or 1.5, n = 10), mid (score of 2, n = 57), and high (score of 3, n = 33) groups, and percent positive cores with similar intensity groups for H3K4me1 or H3K27ac were plotted on y axis.
(C) Boxplot showing relative levels of H3K4me1 in KMT2D mutant (n = 126) or WT (n = 293) cells using the mass spectrometry data from the CCLE database. The bottom and the top rectangles indicate the first quartile (Q1) and third quartile (Q3), respectively. The horizontal lines in the middle signify the median (Q2), and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively.
(D) Emission probabilities of the 10-state ChromHMM model on the basis of ChIP-seq profiles of five histone marks (shown in x axis). Each row represents one chromatin state, and each column corresponds to one chromatin mark. The intensity of the color in each cell reflects the frequency of occurrence of that mark in the corresponding chromatin state on the scale from 0 (white) to 1 (red). States were manually grouped and given candidate annotations.
(E) Heatmap showing the fold enrichment of chromatin state transitions between KMT2D mutant (KMT2DL/L) and WT (KMT2D+/+) samples for the 10-state model defined by the ChromHMM. Color intensities represent the relative fold enrichment. Blue box and arrow point to active enhancer state switch.
(F and G) Heatmaps (left panels) and average intensity curves (right panels) of ChIP-seq reads (RPKM, reads per kilobase of transcript, per million mapped reads) for H3K4me1 (F) and H3K27ac (G) at typical enhancer regions. Enhancers are shown in a 10-kb window centered on the middle of the enhancer in iBIP;KMT2D+/+ and iBIP;KMT2DL/L melanoma tumors.
(H) Venn diagram showing unique or shared H3K4me1 and H3K27Ac co-enriched active enhancers sites in iBIP;KMT2D+/+ and iBIP;KMT2DL/L melanoma tumors.
Upregulated Insulin Growth Factor (IGF) Signaling Regulates Glycolysis in KMT2D Mutants
To understand how enhancer loss may lead to observed metabolic reprogramming, we overlapped changes in gene expression between KMT2D WT and mutant-murine-tumor-derived lines with the changes in active enhancer patterns. Of the 7,555 active enhancer loci that display a loss of intensity in KMT2D mutant tumors compared with WT, 1,165 were located nearby (±200 Kb) genes with decreased expression (Figure 6A). We found a significant association between the loss of expression and loss of H3K4me1 patterns in nearby loci (Figure S6A). These genes were enriched for those involved in various phosphorylation-mediated cell signaling events and are bona fide or putative tumor suppressors (Figures S6B and S6C). Of these, we focused on the IGF signaling pathway that is known to play major roles in regulating metabolic pathways (such as glycolysis) by activation of AKT (Wang et al., 2015; Figures S6B and S6C). Indeed, we observed higher levels of pAKT (S473) and pIGF1R (Y1198) in KMT2D mutant murine and human lines (Figure 6B) as well as KMT2D mutant iBIP tumors (Figure 6C), suggesting aberrant activation of the IGF-AKT-glycolysis pathway. Examination of Reverse Protein Phase Array (RPPA) data from CCLE database (Li et al., 2017) across all cancer types showed that KMT2D mutant cell lines (harboring functional driver mutations) showed higher levels of pS473 and pT308 forms of AKT compared to KMT2D WT (and high expressing) lines (Figure 6D). However, in contrast to observations in pancreatic cancer (Koutsioumpa et al., 2019), we observed reduced levels of the phosphorylated form of mTOR (mammalian target of rapamycin) in KMT2D mutant versus WT cells in the CCLE database RPPA data (Figure S6D). The functional significance of activation of IGF1R signaling was further tested by the treatment of cells with an IGF-1R inhibitor (linsitinib), which reduced the expression of glycolysis genes in KMT2D mutant murine and human cell lines (Figures 6E and 6F). Importantly, treatment of KMT2D mutant murine and human cell lines with linsitinib preferentially reduced the proliferation of KMT2D mutant cell lines both in vitro (Figure 6G) and in vivo (Figures 6H and 6I). This finding was recapitulated in the analysis of all cancer cell lines (from Sanger Cell Line database) for which linsitinib sensitivity data were available. Cells harboring KMT2D functional driver mutations displayed significantly lower IC50 (half maximal inhibitory concentration) values for linsitinib treatment than for cells that harbor high levels of KMT2D (and have WT protein) (Figure 6J). Finally, linsitinib-treated tumors showed a drastic reduction in the expression of glycolysis genes, proliferation marker Ki-67, and pAKT levels (Figure S6E). These data establish activation of the IGF1R-AKT-glycolysis axis in KMT2D-deficient cancer cells.
Figure 6. Aberrant Activation of IGF and AKT Signaling in KMT2D Mutants Confers Sensitivity to IGFR Inhibitor.

(A) Venn diagram showing the overlap between differentially expressed genes and lost active enhancer loci in KMT2D mutant (Mut-m2) melanoma cells compared with KMT2D WT (WT-m1).
(B) Western blot showing the expression of pAKT, AKT, pIGF1R, and IGF1R in KMT2D mutant and WT murine (left panel) and human (right panel) cells. Vinculin is used as a loading control.
(C) Immunohistochemistry images for pAKT and pIGF1R demonstrating their overexpression in iBIP;KMT2D+/+ and iBIP;KMT2DL/L melanoma tumors. Scale bars represent 20 μm.
(D) Boxplot showing RPPA-based protein levels of AKT, pAKT (S473), and pAKT (T308) in CCLE cell lines (all cancer types) that harbor KMT2D functional mutations (n = 15) versus those that harbor WT and high levels of KMT2D (n = 15; RPKM ≥ 10).
(E and F) Bar graph showing relative expression pattern of 12 glycolysis enzyme genes (compared to 28S) (n = 3 for each sample) in KMT2D mutant and WT murine (E) and human (F) cells treated with vehicle or linsitinib (1 μM) for 24 h. t test, *p < 0.05, **p < 0.01. Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates.
(G) Growth curves for KMT2D mutant and WT murine (left) and human (right) melanoma cells treated with different concentrations of linsitinib. Relative confluence at 96-h posttreatment are plotted, and IC50 values are shown in the accompanying table.
(H and I) Line plot showing average tumor volumes for mice (n = 5 per group) injected with KMT2D mutant and WT murine (H) and human (I) melanoma cells and treated with linsitinib (25mg/kg) or vehicle (30% PEG-400) every other day. p values represent t test comparison between the indicated groups on the last time point. In this graph, * p < 0.05, ** p < 0.01, *** p < 0.0001.
(J) Boxplot showing log(IC50) values for linsitinib in the KMT2D mutant (n = 77) and KMT2D WT-high (n = 155) cell lines (all cancer types) from the GDC data (Sanger Cell Line Project).
In the boxplots (D) and (J), the bottom and the top rectangles indicate Q1 and Q3, respectively. The horizontal lines in the middle signify the Q2, and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively.
Loss of a Distal Enhancer of IGFBP5 in KMT2D Mutants Regulates IGF Signaling and Expression of Glycolysis Enzyme Genes
We next searched for putative regulators of the IGF signaling that lose active enhancers and gene expression in KMT2D mutants specifically to identify those that may be responsible for the metabolic reprogramming phenotypes observed in KMT2D-deficient tumors. We focused on IGFBP5 as it is a known negative regulator of IGF1R signaling and acts as a tumor suppressor in melanoma by regulation of AKT and IGF1R signaling (Wang et al., 2015). We found the loss of H3K4me1 signals on proximal and distal enhancers were associated with IGFBP5 in KMT2D mutant tissues (Figure 7A), whereas other IGFBPs did not show a significant change (Figure S7A). Examination of Hi-C-based higher order chromatin interaction data showed that IGFBP5 may be located in a TAD (Tandem Adjacent Domain), thus promoting the interaction between this distal enhancer and the IGFBP5 gene (Figure 7B). Consistently, IGFBP5 expression was also lost in KMT2D mutant murine and human cell lines (Figure 7C), whereas several other IGFBPs showed inconsistent patterns (Figure S7B). Consistently, IGFBP5 expression was significantly reduced in KMT2D mutant human and murine melanoma tumors (Figures 7D, S7C, and S7D). Consistently, we also noted a positive correlation between IGFBP5 expression and KMT2D expression in the metastatic tumors in the TCGA melanoma study (Figure S7E; Cancer Genome Atlas Network, 2015b). Our recent study in lung cancer identified KMT2D-mediated regulation of Per2 expression as the central node in the regulation of glycolysis enzymes (Alam et al., 2020). However, we did not find any difference in Per2 expression in KMT2D WT versus mutant murine or human melanoma tumors (Figures S7C and S7D). We also did not observe any change in TSC1 expression in KMT2D mutant versus WT cell lines (Figure S7C).
Figure 7. Loss of Distal Enhancers of IGFBP5 in KMT2D Mutant Cells Partially Contributes to Its Phenotypes.

(A) IGV snapshot showing RNA-seq, H3K27Ac, and H3K4me1 ChIP-seq signal tracks for genomic locus harboring IGFBP5. Note the loss of blue peaks in the region surrounding the IGFBP5 gene.
(B) Interaction map from mouse embryonic stem cells (ESCs) showing an IGFBP5-containing locus, demonstrating an interaction between the IGFBP5 gene with proximal and distal enhancers. The highlighted blue off-diagonal interaction (box with arrow) points to a downstream enhancer that exhibits a selective loss of H3K4me1 signal in KMT2D mutant samples.
(C) Western blot showing expression of IGFBP5 and vinculin in KMT2D mutant and WT murine (top panel) and human (bottom panel) cells.
(D) Boxplot showing expression of IGFBP5 in the melanoma TCGA samples that harbor functional mutations (nonsense, frameshift or post4700aa) (n = 15) or WT copies for KMT2D (n = 15, RPKM > 10). The bottom and the top rectangles indicate Q1 and Q3, respectively. The horizontal lines in the middle signify Q2, and the vertical lines that extend from the top and the bottom of the plot indicate the maximum and minimum values, respectively. * denotes p <0.05.
(E) western blot showing expression of IGFBP5, pAKT, AKT, pIGF1R, and IGF1R in KMT2D mutant and WT murine control or IGFBP5 overexpressing cells. Vinculin is used as a loading control.
(F and G) Bar graph showing relative expression pattern of 12 glycolysis enzyme genes (compared to 28S) (n = 3 for each sample) in KMT2D mutant and WT murine (F) and human (G) cells overexpressing empty vector or IGFBP5. Standard t test, *p < 0.05, **p < 0.01, ***p < 0.001. Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates.
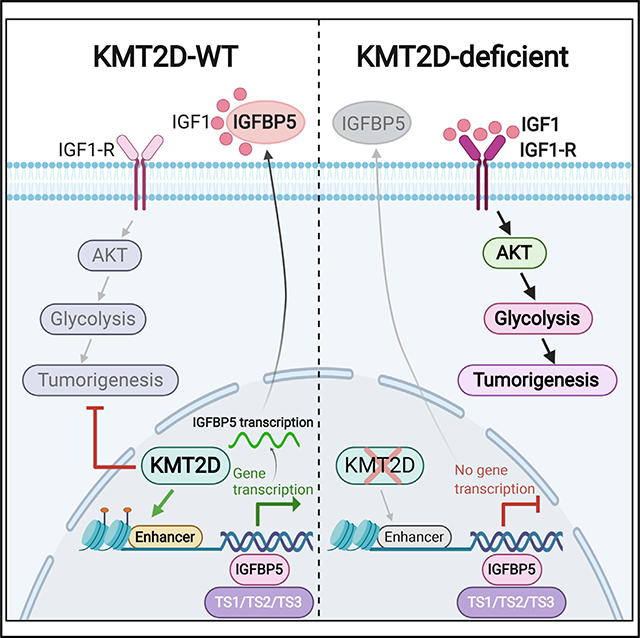
(H) Model of molecular mechanism of KMT2D-loss-mediated promotion of human melanoma. We suggest that KMT2D loss reprograms a subset of enhancers for important regulators of tumor suppressor pathways, including IGFBP5 (and others denoted by TS prefix). Lower levels of IGFBP5 upon KMT2D loss amplifies IGF signaling, thus activating AKT-mediated metabolic reprogramming, including activation of glycolysis.
Knockdown of IGFBP5 in the murine cell line using two shRNAs (30%–50%) led to an increased expression of glycolysis enzymes (Figure S7F). Importantly, epistasis experiments revealed that IGFBP5 overexpression in murine melanoma cells decreased levels of IGF1R and AKT phosphorylation (Figure 7E) as well as glycolysis genes (Figures 7F and 7G) in KMT2D mutant murine and human cells compared with their WT counterparts. Taken together, the data presented in this manuscript establish a model of KMT2D function in cancer for which KMT2D acts as a tumor suppressor by enhancer reprogramming on tumor suppressor genes, such as IGFBP5, that regulate key pathways, such as IGF1R signaling, leading to metabolic rewiring (Figure 7H).
DISCUSSION
Through an unbiased RNAi screen in vivo, we identified and validated eight epigenetic modifiers (KMT2D, KDM1A, APOBEC2, HDAC6, KMT2F, SETD4, KAT4, and KDM5B) whose loss can significantly accelerate tumor growth. Interestingly we identified four enzymes regulating H3K4 methylation, namely, KMT2D (Herz et al., 2012), KDM5B (Seward et al., 2007), KMT2F (Wysocka et al., 2003), and KDM1A (Shi et al., 2004), as hits in this screen, suggesting important roles for H3K4 methylation reprogramming during tumorigenesis. Similarly, KAT4 and HDAC6 suggest important roles for histone acetylation in melanomagenesis. Indeed, our previous study showed drastic deregulation of chromatin states harboring H3K4 and histone acetylation during pre-malignant to malignant transition in melanoma (Fiziev et al., 2017). As the strongest phenotypes were observed for KMT2D, we deeply studied its mechanism of action in melanoma.
Although the somatic loss-of-function mutations in KMT2D are observed across many malignancies (Cancer Genome Atlas Network, 2015a; da Silva Almeida et al., 2015; Gao et al., 2014; Grasso et al., 2012; Jones et al., 2012; Juhlin et al., 2015; Kandoth et al., 2013; Lawrence et al., 2014; Lin et al., 2014; Lohr et al., 2012; Morin et al., 2011; Parsons et al., 2011; Pasqualucci et al., 2011; Pugh et al., 2012; Sausen et al., 2015; Song et al., 2014), it is unclear why these mutations are selected over the course of tumor evolution. Our study suggests that enhancer reprogramming by KMT2D loss may rewire metabolic pathways for increased energy and biomass needs of cancer cells. We observed drastic deregulation of multiple metabolic pathways in KMT2D mutant melanomas in both human and murine systems. Consistently, we observed a preferential dependence of KMT2D mutant cells’ growth on glycolysis compared with WT cells. The glycolysis pathway serves as a central node for various needs of a proliferating cells (Lunt and Vander Heiden, 2011). It is required for a small fraction of energy needs (two ATPs per cycle) and, more importantly, for the production of biomass needed for cell doubling. For example, glucose-6-phosphate provides a gateway to nucleotide biosynthesis and dihydroxyacetone phosphate acts as a starting substrate for the lipid biosynthesis pathway. Increased pyruvate production due to high glycolysis provides a substrate for the OxPhos pathway (to generate 36 ATPs), which is also upregulated in the KMT2D mutant cells, thereby leading to enhanced ATP production. Finally, 3-phosphoglycerate and other OxPhos metabolites provide a substrate for amino acid biosynthesis. Therefore, upregulated glycolysis in KMT2D mutant cells contributes to several different biomass and energy needs to enhance tumorigenesis.
Data shown here along with our recent data in lung cancer (Alam et al., 2020) show the dependence of KMT2D mutant cancers on glycolysis and, critically, will inform future clinical studies testing potent glycolysis-blocking inhibitors in this genetic context. Our data also suggest the potential use of IGF receptor blocking molecules, such as linsitinib, which is being tested in clinical trials (Iams and Lovly, 2015), in the KMT2D mutant patient population. However, further work may be needed to better stratify the functional driver mutations in KMT2D because it is likely to harbor higher mutations due to its excessive length (~42 kb). Therefore, some of the observed somatic mutations may be passenger events, especially in cancers with a high mutation burden, such as melanoma and lung cancers (Lawrence et al., 2013). In addition to mutations, KMT2D expression levels may also need consideration while stratifying patients for such therapies, as many metastatic and primary melanomas show little to no expression of KMT2D.
Although we show an important role for glycolysis, many other metabolic pathways, such as oxidative phosphorylation and fatty acid metabolism, are also highly upregulated in KMT2D mutant cancers. The publicly available CRISPR screening platform Achilles (Tsherniak et al., 2017) suggests a dependency of KMT2D mutant melanomas on specific genes in these other metabolic pathways that need further exploration. Indeed, a recent study suggested enhanced fatty acid metabolism in pancreatic cancer (Koutsioumpa et al., 2019). Although we focused this study on metabolic reprogramming functions of KMT2D, it is likely only one of the many factors contributing to the growth of KMT2D-deficient cells and their selection. Future studies will shed light on other aspects of KMT2D biology, including its role in other pathways, such as immune microenvironment.
KMT2D is a member of the COMPASS (complex of proteins associated with set1) complex that is thought to be critical for depositing H3K4me3 (Hu et al., 2013; Sze and Shilatifard, 2016). Furthermore, some studies, such as one by Dhar et al. (2018) suggest a role of KMT2D in H3K4me3 regulation. However, several other studies suggest KMT2D to be a major regulator of the H3K4me1 mark that marks poised enhancers (Cho et al., 2012; Dorighi et al., 2017; Herz et al., 2012; Hu et al., 2013; Lee et al., 2013; Raman and Rai, 2018; Wang et al., 2016). In a subset of enhancers, H3K4me1 recruits CBP/p300 enzymes in turn activation their target genes (Lai et al., 2017); however, a complete understanding of this mode of active enhancer regulation is still lacking. Our data suggest that KMT2D is a major regulator of H3K4me1 in melanoma. Because it appears that the extent of H3K4me1 loss is more than H3K27ac loss at the global level, it is possible that in a subset of enhancers other histone acetylations (than H3K27ac) may be involved in enhancer activation in KMT2D mutant melanomas. Indeed, evidence for roles of the other histone acetylations in enhancer activation has been previously demonstrated (Pradeepa et al., 2016). Our previous study also showed drastic losses of chromatin states harboring multiple different histone acetylations, including H2BK5ac and H4K5ac, and H3K4me1/2/3 in early stages of tumorigenic transition in melanoma (Fiziev et al., 2017). Nonetheless, the ChIP-seq data for active enhancers, for which we probe the co-occupancy of H3K27ac and H3K4me1, clearly suggest that many active enhancers are lost in KMT2D-deficient cells in the murine system (Figure 5H). Indeed, locus-specific changes in chromatin states are the determinants of expression of a specific gene. Because enhancers are shown to be cell type specific (Kundaje et al., 2015), KMT2D-loss-mediated enhancer misregulation could be responsible for different downstream mechanisms upon KMT2D deficiency in different cancer types (such as Per2 in lung cancer, IGFBP5 in melanoma, or SLC2A3 and TSC1 in pancreatic cancer) (Koutsioumpa et al., 2019).
We noted that KMT2D mutant cells grew slower than WT cells in vitro; however, they proliferated faster in vivo and formed aggressive tumors. There may be several reasons for why KMT2D mutant cells did not grow faster than WT cells in vitro; however, they formed aggressive tumors in vivo. First, due to the rapid consumption of glucose because of their faster metabolism (Figures S4A and S4B), KMT2D mutant cells likely slow once nutrients are exhausted from the media. Indeed, the establishment of KMT2D mutant cells from GEMM (iBIP;KMT2DL/L) tumors required a repeated change of DMEM media with high glucose every 3–4 h (Figures S4A and S4B). Second, other factors in the tumor microenvironment (which are lacking in the in vitro culture conditions) may play important roles in supporting the growth of KMT2D mutant cells. Indeed, pathway analysis of the differential gene expression data suggests that idea as well. We noted immune pathways to be enriched in KMT2D-deficient tumors. Other events, such as hypoxia, that occur more prominently under in vivo conditions, may also play important roles in promoting the growth of KMT2D mutant cells. Indeed, HIF1α expression was modestly higher (1.5- to 2-fold) in KMT2D mutant cells compared with WT cells. Third, it is possible that KMT2D loss promotes the fitness of slow cycling cells in vitro and that these fitter cells rapidly proliferate once they encounter an optimal environment (i.e., tumor microenvironment) in vivo. Indeed, the evidence for a role for KMT2D in clonal fitness in the hepatic disease was recently shown (Zhu et al., 2019).
Overall, our study provides evidence for the dependency of the KMT2D mutant melanomas on glycolysis and the IGF pathway by enhancer reprogramming. These results suggest a potential therapeutic strategy in the patients with melanoma harboring mutations in this epigenetic regulator.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information and reagents should be directed to and will be fulfilled by the Lead Contact, Kunal Rai (krai@mdanderson.org).
Materials availability
Any plasmid and cell line generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availabilty
The accession number for the RNA-Seq and ChIP-Seq data reported in this paper is GEO: GSE116921. All codes are available at https://gitlab.com/railab.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
All melanoma cell lines (HMEL-BRAFV600E, A375, RPMI-7951, WM115, SKMEL-24, WM278, 5770, 300, 3417 and 3418) that were used in this study are described in the Key Resources Table, and they were cultured within 10 to 15 passages. Cell culture reagents and other chemicals were purchased from Thermo Fisher Scientific (GIBCO & Hyclone), Sigma-Aldrich, and Fisher Bioreagents.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K4me1 | Abcam | Cat# ab8895; RRID: AB_306847 |
| H3K27Ac | Abcam | Cat# ab4729; RRID: AB_2118291 |
| H3K4me3 | Abcam | Cat# ab8580; RRID: AB_306649 |
| H3K27me3 | Abcam | Cat# ab6002; RRID: AB_305237 |
| H3K79me2 | Abcam | Cat# ab3594; RRID: AB_303937 |
| H3 | Abcam | Cat# ab1791; RRID: AB_302613 |
| KMT2D | Sigma | Cat# HPA035977, RRID: AB_10670673 |
| IGFBP5 | Proteintech | Cat# 55205-1-AP; RRID: AB_2736835 |
| Ki-67 | Cell Signaling Technology | Cat# 9027; RRID: AB_2636984 |
| ENO1 | Proteintech | Cat# 11204-1-AP; RRID: AB_2099064 |
| PGK1 | Proteintech | Cat# 17811-1-AP; RRID: AB_2161218 |
| PGAM1 | Proteintech | Cat# 16126-1-AP; RRID: AB_2160786 |
| AKT | Cell Signaling Technology | Cat# 4691; RRID: AB_915783 |
| pAKT | Cell Signaling Technology | Cat# 9271; RRID: AB_329825 |
| IGF1R | Cell signaling Technology | Cat# 9750; RRID: AB_10950969 |
| pIGF1R | Abcam | Cat# ab39398; RRID: AB_731544 |
| Vinculin | Sigma-Aldrich | Cat# V4139, RRID: AB_262053 |
| Tyrosinase | Abcam | Cat# ab738; RRID: AB_305899 |
| Rabbit-on-Rodent HRP-Polymer | Biocare Medical | Cat# RMR622L |
| Mouse-on-Mouse HRP-Polymer | Biocare Medical | Cat# MM510L |
| Alexa 488-conjugated anti-mouse IgG | Life technologies | Cat# A-11029; RRID: AB_138404 |
| Alexa 488-conjugated anti-rabbit IgG | Life technologies | Cat# A-11037; RRID: AB_2534095 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-Deoxy-D-glucose (2-DG) | Sigma | Cat# D6134; CAS: 154-17-6 |
| Linsitinib | Selleck Chemicals | Cat# HY-10191; CAS: 867160-71-2 |
| Lonidamine | Sigma | Cat# L4900; CAS: 50264-69-2 |
| POMHEX | Gift from Dr. Florian Muller (Lin et al., 2018) | N/A |
| IACS-010759 | Gift from Dr. Joseph R. Marszalek | N/A |
| Deposited Data | ||
| ChIP-seq and RNA-seq for iBIP;KMT2D+/+ and iBIP;KMT2DL/L melanoma tumors | This paper | GEO: GSE 116921 |
| Experimental Models: Cell Lines | ||
| Human: HMEL-BRAFV600E | Gift from Dr. David Fisher’s laboratory | N/A |
| Human: A375 | ATCC | CRL-1619 |
| Human: RPMI-7951 | ATCC | HTB-66 |
| Human: WM115 | ATCC | CRL-1675 |
| Human: WM266-4 | ATCC | CRL-1676 |
| Human: SKMEL-24 | ATCC | HTB-71 |
| Human: WM278 | ATCC | CRL-2809 |
| Mouse: 5770 | This paper | N/A |
| Mouse: 300 | This paper | N/A |
| Mouse: 3417 | This paper | N/A |
| Mouse: 3418 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Kmt2dfl/fl (also called Mll4fl/fl) | Dhar et al., 2018 | N/A |
| inducible BRAF INK/ARF PTEN (iBIP) mice | Kwong et al., 2015 | N/A |
| Xenograft Experiments (Swiss Nude) | Experimental Radiation Oncology at MDACC | Nude |
| Xenograft Experiments (NCr Nude) | Taconic Biosciences | NCRNU-F |
| Oligonucleotides | ||
| Primers for quantitative RT-PCR (human and mouse): See Table S4 | N/A | N/A |
| Recombinant DNA | ||
| pInducer20-KMT2D | This study | N/A |
| shKmt2d-01 (mouse) | Sigma-Aldrich | Cat# TRCN0000239234 |
| shKmt2d-03 (mouse) | Sigma-Aldrich | Cat# TRCN0000239233 |
| Software and Algorithms | ||
| cBio cancer genomics portal | Gao et al., 2013 | http://www.cbioportal.org/ |
| Pyflow-ChIPseq | Terranova et al., 2018 | https://github.com/crazyhottommy/pyflow-ChIPseq |
| Bowtie1 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| samtools | Li et al., 2009 | http://www.htslib.org/ |
| Deeptools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| MACS14 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| ROSE | Lovén et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
| chromHMM | Ernst and Kellis, 2012 | http://compbio.mit.edu/ChromHMM/ |
| EnrichedHeatmap | Gu et al., 2018 | https://bioconductor.org/packages/release/bioc/html/EnrichedHeatmap.html |
| ComplexHeatmap | Gu et al., 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts | Liao et al., 2014 | http://subread.sourceforge.net/ |
| ChIPseeker | Yu et al., 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| TCGAbiolinks | Colaprico et al., 2016 | https://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html |
| GSEA | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/ |
| Snakemake | Köster and Rahmann, 2012 | https://snakemake.readthedocs.io/en/stable/ |
| Pyflow-RNaseq | This paper | https://github.com/crazyhottommy/pyflow-RNaseq |
| IGV | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| ggplot2 | Wickham, 2006 | https://ggplot2.tidyverse.org/ |
| MetaboAnalyst 2.0 | Xia et al., 2012 | https://www.metaboanalyst.ca |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | |
Mouse strains and genetically engineered mouse models (GEMM)
All animal studies were performed according to University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee (IACUC) approved protocols.
GEMM model
The KMT2Dfl/fl mice were previously described (Dhar et al., 2018). KMT2Dfl/fl mutant mice were crossed with iBIP mice (Kwong et al., 2015) (iBIP = Tyr-CreERT2, Rosa26-rtta, TetO-BRAFV600E, PTENL/L, INK/ARFL/L; mixed genetic background of FVB and B6) to generate iBIP;KMT2DL/+ genotype containing mice. The iBIP;KMT2DL/+ male and female mice were mated to generate iBIP;KMT2D+/+, iBIP;KMT2DL/+, and iBIP;KMT2DL/L genotype containing cohorts. Genotypes of these mice were confirmed by a standard PCR-based protocol. The primers used for the genotyping are listed in Table S4. In this model, upon weaning and cohort generation (4–6 weeks old), mice were fed with doxycycline (2mg/ml in 40mg/ml of water, ad libitum) and 4-OHT (1 μM) was applied on the ears at weaning age to generate auricular tumors. Both female and male mice were used in approximately similar numbers. The tumor progression and survival of mouse groups were compared. Tumors were harvested by excision of the lesion and digested for generation of WT-m1, WT-m2, Mut-m1 and Mut-m2 cell lines. For chromatin immunoprecipitation (ChIP) and RNA analyses, the distinct tumors were dissected, washed with ice-cold phosphate-buffered saline solution (PBS) and snap frozen.
Xenograft experiments
For the xenograft experiments (as described below) in the RNAi screen and subsequent validations, we used NCr Nude mice from Taconic. For all inhibitor treatment experiments, we used the Swiss Nude mice. Four to six weeks old females were used to avoid fighting wounds.
Study approval
The care and use of all mice were approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Texas MD Anderson Cancer Center.
METHOD DETAILS
Cell culture, stable cell generation and inhibitor treatment
HMEL-BRAFV600E, A375, RPMI-7951, WM115 and WM266–4 cells were maintained in standard tissue-culture conditions in DMEM media (high glucose) with 10% FBS. SKMEL-24 (Mut-h1) and WM278 (Mut-h2) cells were maintained in the recommended media, except all assays were performed in the same media as for A735 (WT-h1) and RPMI-7951 (WT-h2). Mouse tumor cell lines 300 (WT-m1), 5770 (WT-m2), 3417 (mut-m1) and 3418 (Mut-m2) were isolated from melanoma tumors by digestion in RPMI media (Sigma) using collagenase (2mg/ml) (GIBCO) and dispase (4mg/ml) (GIBCO). Single cell suspension was generated using MACS homogenizer (Milteny Biotec) following manufacturer’s mTumor protocol. Cells were plated in DMEM with high glucose (Sigma/GIBCO) and Glutamax (Sigma/GIBCO) and replenished every 4 h. Once cultures were stable, cells were maintained in DMEM with high glucose and Glutamax. Stable lines expressing shRNAs and ORFs were established by standard lentiviral mediated transduction. All cells were routinely tested for mycoplasma by mycoAlert kit (Lonza) or by PCR.
Inhibitor treatment experiments
For inhibitor treatment experiments, cells were seeded in 96-well plates at a density of 500 cells per well and treated 24-hours later with specific inhibitors. Linsitinib (SelleckChem), 2-Deoxy-D-Glucose (2-DG, Sigma) and Lonidamine (Sigma) were dissolved in DMSO and diluted in cell culture media for treatment. Cell density was measured by imaging every 3–4 h using an IncuCyte live cell analysis system (Essen Bioscience). Once treated cells reached confluence (90%–100%), they were stained with crystal violet and destained in 10% acetic acid. Absorbance was read at 590nM and background-corrected readings for each condition were normalized to vehicle treatments which were plotted to obtain the growth curves. IC50 values were calculated using GraphPad Prism.
RNAi screen
For the screen, 475 shRNAs against 95 chromatin modifiers (Table S1) in pLKO.1 vector were obtained from the Broad Institute/Harvard Medical School core facility. Lentiviruses were produced in HEK293T cells in a 6-well format using pDelta8.2 and pMD2.G (addgene). Viruses for 25 shRNAs (five shRNAs for each of five genes selected at random) were pooled together and the mixture was used to transfect one million cells at MOI of 1. Cells were grown for 3 days and subsequently grafted intradermally into the flanks of NCR-NUDE mice at a density of 1million per flank. All mice experiments were performed per Institutional Animal Care and Use Committee (IACUC) guidelines. Mice were monitored weekly for tumor growth and sacrificed when tumor size reached 2 cm in one dimension. At sacrifice, tumors were harvested and genomic DNA prepared. The segment of pLKO.1 containing shRNA sequence was amplified by PCR and then sequenced to identify the integrated shRNA. Genes corresponding to these shRNAs were noted as hits from the primary screen. We did not observe two different shRNAs for any gen hit. Only one shRNA hit per gene was identified. For validation experiments, stable HMEL-BRAFV600E cells were generated using individual shRNAs, with multiple shRNAs targeting each gene from primary hits. Cell lines generated from the two best shRNAs demonstrating the best knockdown of the corresponding gene (and with at least 50% knockdown) were then injected intradermally in the flanks of NCR-NUDE mice (one million cells/injection). Mice were monitored for tumor formation and growth.
Mouse experiments
All mice were kept in specific pathogen free vivarium at the MD Anderson Cancer Center mouse facilities. Mice were fed commercial rodent diet (PicoLab Rodent diet 5053 from Labdiet) and water ad libitum.
Xenograft experiments
Mice were injected with five million cells in one flank each and monitored every other day for tumor growth. When tumor size reached 0.5 cm in one mouse in each treatment arm, all mice were injected with 2-DG (500mg/kg dissolved in PBS) or Linsitinib (25mg/kg dissolved in 30% PEG-400) via intraperitoneal route. Tumor volume was measured every other day.
Genetically engineered mouse model
KMT2D conditional mutant mice were obtained from Dr. MinGyu Lee (Dhar et al., 2018). These mice are engineered with Lox sites flanking exons 16 and 20, resulting in loss of KMT2D protein as previously described (Dhar et al., 2018). KMT2D mutant mice were crossed with iBIP mice (Kwong et al., 2015) (iBIP = Tyr-CreERT2, Rosa26-rtta, TetO-BRAFV600E, PTENL/L, INK/ARFL/L; mixed genetic background of FVB and B6) to generate iBIP;KMT2DL/+ genotype containing mice (as assessed by genotyping for all alleles). The iBIP;KMT2DL/+ male and female mice were mated to generate iBIP;KMT2D+/+, iBIP;KMT2DL/+, and iBIP;KMT2DL/L genotype containing cohorts. These mice were treated with doxycycline systemically (2mg/ml in 40mg/ml of water) by feeding (ad libitum) and 4-OHT (1 μM) was applied on the ears to generate auricular tumors. Mice were observed twice a week for tumor formation and upon tumor appearance, tumor growth was measured every other day. Tumors were harvested by excision of the lesion and digested for generation of WT-m1, WT-m2, Mut-m1 and Mut-m2 cell lines.
RNA-Seq analysis of murine tumor cells
Strand specific libraries were constructed using the ScriptSeq Kit (Epicenter/Illumina). RNaseq data were processed by pyflow-RNaseq (Tang, 2017b), a snakemake based RNaseq pipeline. Raw reads were mapped by STAR (Dobin et al., 2013), RPKM normalized bigwigs were generated by deeptools (Ramírez et al., 2016), and gene counts were obtained by featureCount (Liao et al., 2014). Differential expression analysis was carried out using DESeq2 (Love et al., 2014). Gene set enrichment analysis (GSEA) was done using the GSEA tool (Subramanian et al., 2005) from Broad Institute. The pre-rank mode was used. The signed fold change *–log10(pvalue) metric was used to pre-rank the genes.
ChIP-Seq
Chromatin immunoprecipitation was performed as described earlier (Terranova et al., 2018) with optimized shearing conditions and minor modifications for melanocytes. The antibodies used were: H3K4me1 (Abcam ab8895), H3K27ac (Abcam ab4729), H3K4me3 (Abcam ab8580), H3K79me2 (Abcam ab3594), H3K27me3 (Abcam ab6002). ChIP-seq data were quality controlled and processed by pyflow-ChIPseq (Tang, 2017a), a snakemake (Köster and Rahmann, 2012) based ChIPseq pipeline. Briefly, raw reads were mapped by bowtie1(Langmead et al., 2009) to hg19. Duplicated reads were removed and only uniquely mapped reads were retained. RPKM normalized bigwigs were generated by deep tools (Ramírez et al., 2016) and tracks were visualized with IGV (Robinson et al., 2011). Peaks were called using macs1.4 (Zhang et al., 2008) with a p value of 1e-9. Chromatin state was called using ChromHMM (Ernst and Kellis, 2012) and the emission profile was plotted by ComplexHeatmap (Gu et al., 2016). Heatmaps were generated using R package EnrichedHeatmap (Gu et al., 2018). ChIP-seq peaks were annotated with the nearest genes using ChIPseeker (Yu et al., 2015). Super-enhancers were identified using ROSE (Lovén et al., 2013) based on H3K27ac ChIP-seq data.
Chromatin state analysis
ChromHMM (Ernst and Kellis, 2012) was used with default parameters to derive genome-wide chromatin state maps for all cell types. Input data were binarized using ChromHMM’s BinarizeBed method (Ernst and Kellis, 2012) with a p value cutoff of 1e−4. Chromatin state models were learnt jointly on all data for all five histone marks (H3K4me1, H3K4me3, H3K27ac, H3K79me2 and H3K27me3) from WT-m1 and Mut-m2 tumor cells and a model with 10 states was chosen for detailed analysis. Chromatin state segmentations of WT-m1 and Mut-m212 were produced subsequently by applying this model to the original binarized, quantile normalized or downsampled chromatin data from these cell types.
TCGA RNA-Seq data analysis
TCGA melanoma (SKCM) RNaseq raw counts were downloaded using TCGAbiolinks (Colaprico et al., 2016). The mutation MAF files were downloaded with TCGAbiolinks as well. Mutation status of KMT2D was inferred from the MAF files. Ten SKCM primary tumor samples with wild-type copies of KMT2D and expressing high levels and 10 SKCM primary tumor samples with mutant KMT2D (see supplemental data for samples included in the analysis) were compared using DESeq2, the signed fold change *–log10(pvalue) metric was used to pre-rank the gene list and for GSEA pre-rank analysis. Boxplots were generated using R package ggplot2 (Wickham, 2006).
For the pan cancer analysis, TCGA tumor samples were grouped based on KMT2D gene expression and mutation status: KMT2D mutation free group are samples with high KMT2D expression (among the top quantile) and no somatic mutation; KMT2D mutated group are samples with low KMT2D expression (falling into the bottom quantile) and have either nonsense mutations or missense mutations with the amino acid 4,700. Six tumor types (BLCA, urothelial bladder carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; HNSC, head and neck squamous cell carcinoma; LUSC, lung squamous cell carcinoma; UCEC, uterine corpus endometrial carcinoma; STAD, stomach adenocarcinoma) had adequate sample size (≥10) to be included for differential gene expression analysis and pathway enrichment analysis. TCGA normalized RNaseq read count were processed by Wilcoxon test to identify differentially expressed genes (DEGs) across KMT2D mutation status groups. A cut-off of gene expression fold change of ≥ 2 or ≤ −0.5 and a FDR q-value of < 0.05 were applied to select the most differentially expressed genes. A ranked list of genes was generated based on the product of Wilcoxon test FDR q-values and log2 fold change for all coding genes and processed by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) against the curated gene sets from Molecular Signature Database (MSigDB) (Liberzon et al., 2015) to identify significantly enriched signaling pathways.
Immunohistochemistry
Tumors were fixed in formalin for 24 h, paraffin embedded, sectioned, and stained according to standard procedures. Briefly, endogenous peroxidases were inactivated by 3% hydrogen 673 peroxide. Non-specific signals were blocked using 3% BSA, 10% goat serum in 0.1% Triton X-100. After antigen retrieval in citrate buffer, slides were stained using respective antibodies overnight at 4°C, [ ENO1 (Proteintech, #11204–1-AP), PGK1 (Proteintech #17811–1-AP), PGAM1 (Proteintech, #16126–1-AP), Tyrosinase (Abcam, ab738), pAKT (Cell Signaling, #9271), pIGF1R (Abcam, ab39398)]. After overnight incubation, the slides were washed and incubated with secondary antibody (HRP-polymers, Biocare Medical) for 30 min at room temperature. The slides were washed three times and stained with DAB substrate (ThermoFisher Scientific). The slides were then counterstained with hematoxylin and mounted with mounting medium.
Inducible ectopic expression of KMT2D
Full-length KMT2D was cloned from KMT2D overexpression vector (gift from Dr. Laura Pasqualucci at Columbia University) into the pInducer20 doxycycline inducible lentiviral vector (Addgene 44012). Lentivirus was produced using standard virus production methods by co-transfecting target and packaging plasmids (psPAX2 – Addgene12260 and pMD2.G- Addgene 12259) into HEK293T cells. Cell lines were then transduced with 0.45uM filtered and ultracentrifuge concentrated viral particles with Polybrene (8 μg/ml). After 16 h of transduction, media was changed into fresh regular growth media, and 48 h later selection started using G418 (0.2–0.6μg/ml). After selection was complete in 120 h, cells were termed stably transduced. KMT2D expression was induced for 72 h with doxycycline 2 μg/ml.
Whole Cell Extracts, Acid Extraction and Western Blotting
Cells were harvested in media, washed with PBS, and pelleted. Cell pellets were dissolved in RIPA buffer (25mM Tris PH8, 150mM NaCl, 01% SDS, 0.5% Sodium Deoxycholate, 1% Triton X-100, Protease and phosphatase inhibitor cocktail; bought from Boston bioproducts) and incubated for 30’ on ice before brief sonication followed by centrifugation to remove debris. Supernatant was collected, protein measured using Bradford assay and equal amounts were loaded on the 4%–12% SDS-PAGE gel (Invitrogen). Proteins were transferred to a Nitrocellulose or PVDF membrane which was then blocked in Odyssey Blocking buffer (LiCOR) and incubated with primary antibody overnight in the same buffer. Blots were then washed and probed with HRP-labeled secondary antibodies (Pierce) and developed using a X-ray film (Phenix). For histone marks, we incubated cell pellets in Triton Extraction buffer (PBS containing 0.5% Triton X-100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (w/v) NaN3) for nuclei isolation. Nuclei were subjected to histone extraction by overnight incubation in 0.2N HCl (with protease and phosphatase inhibitors) followed by centrifugation. Rest of the western blotting was done as explained above. Antibodies used were (also listed in Key Resources Table): IGFBP5 (Proteintech, #55205–1-AP), pAKT (Cell Signaling, 9271) Total AKT (CST, #4691), Total IGF-1R (Cell signaling, #9750). Histone antibodies are same as used for ChIP experiments.
Metabolomics via selected reaction monitoring tandem mass spectrometry
One 15 cm2 plate of cells (~10–15 million) per sample was extracted with 80% methanol (−80°C) for 15 min. Dried metabolite pellets were resuspended in 20 μL of LC/MS-grade water, and 5 μL aliquots were injected for targeted LC/MS/MS on a 5500 QTRAP hybrid triple-quadrupole mass spectrometer coupled to a Prominence ultrafast liquid chromatography (UFLC) system from 287 selected reaction monitoring (SRM) transitions with positive/negative polarity switching. Samples were separated on a 4.6 mm i.d. × 100 mm Amide XBridge hydrophilic interaction liquid chromatography (HILIC) column at 360 μL/min starting from 85% buffer B (100% ACN) and moving to 0% B over 16 min. Buffer A was 20 mM NH4OH/20 mM CH3COONH4 (pH = 9.0) in 95:5 water/ACN. Q3 peak areas were integrated by use of MultiQuant 2.1 software (AB/SCIEX). MetaboAnalyst 2.0 (Xia et al., 2012; https://www.metaboanalyst.ca) was used to normalize data. All metabolite samples were prepared as biological triplicates.
RT-qPCR
RNA was isolated using RNeasy kit (qiagen) or Trizol (Thermo Fisher) reagent using manufacturer’s protocol. cDNA was prepared using SuperScript III first strand synthesis kit (Thermo Fisher) using 2micrograms of RNA and manufacturer’s protocol. Quantitative PCR was performed using QuantiTect Sybr Green PCR kit in Stratagene’s Mx3000p system. Primers used are listed in Table S4.
Tissue microarray
The tissue microarray containing 100 samples (62 cases of primary melanoma, 22 cases of metastatic melanoma, and 18 nevi) was obtained from US Biomax. The staining for KMT2D antibody (Sigma, Prestige) was performed at the immunohistochemistry core at MD Anderson Cancer Center. Two pathologists read the TMA and consensus scores were assigned to each sample. For the histone modification study, we built and stained a TCGA melanoma tumor TMA of 65 samples. TMA was stained with KMT2D (Sigma), H3K4me1 (Abcam ab8895), H3K27ac (Abcam ab4729), H3K4me3 (Abcam ab8580), antibodies. Each sample was represented in two cores and intensity data were averaged between the two cores.
Mass spectrometry analysis of histone modifications (Mod-Spec)
Histones were acid extracted, derivatized via propionylation, digested with trypsin, newly formed N-termini were propionylated as previously described (Garcia et al., 2007) and then measured three separate times using the Thermo Scientific TSQ Quantum Ultra mass spectrometer coupled with an UltiMate 3000 Dionex nano-liquid chromatography system. The data were quantified using Skyline (MacLean et al., 2010) and represents the percent of each modification within the total pool of that amino acid residue.
QUANTIFICATION AND STATISTICAL ANALYSIS
The two-tailed Student’s t test was used to determine the statistical significance of two groups of data using GraphPad Prism. Data are presented as means ± standard error of the mean (SEM; error bars) of at least three independent experiments or three biological replicates. p values of less than 0.05 were considered statistically significant.
Supplementary Material
Highlights.
KMT2D is a tumor suppressor in melanoma
KMT2D rewires metabolic pathways through enhancer reprogramming
KMT2D loss impairs IGFBP5 enhancers and thereby deprives repression to glycolytic genes
KMT2D mutant melanomas are preferentially sensitive to glycolysis and IGFR inhibition
ACKNOWLEDGMENTS
We thank Marcus Coyle, Curtis Gumbs, and SMF core at MDACC for sequencing support. We thank Yanping Cao, Jill Garvey, Ibarra Ivonne, and Kun Zhao for mouse colony maintenance. The work described in this article was supported by grants from the National Institutes of Health (CA160578 and CA222214 to K.R.; CA157919, CA207109, and CA207098 to M.G.L.; and CA016672 to SMF Core), American Cancer Society (RSG-15–145-01-CDD to F.M.), Center for Cancer Epigenetics at MDACC (K.R.), and MD Anderson Cancer Center (start-up funds to K.R.). The following fellowship support is acknowledged: K.R. (Charles A. King postdoctoral fellowship) and H.A. (Odyssey Fellowship at MD Anderson Cancer Center). The histology work were performed at the Histopathology Core Lab at the UT MD Anderson Cancer Center supported by the NIH National Cancer Institute (P30CA016672).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108293.
REFERENCES
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. (2012). Epigenomic enhancer profiling defines a signature of colon cancer. Science 336, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, Ambati CR, Amin SB, Gu B, Chen TY, et al. (2020). KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37, 599–617.e597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015a). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015b). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferré F, et al. (2011). The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YW, Hong S, and Ge K (2012). Affinity purification of MLL3/MLL4 histone H3K4 methyltransferase complex. Methods Mol. Biol 809, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, Vermeer MH, Rabadan R, Ferrando A, and Palomero T (2015). The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat. Genet 47, 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA (2017). The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, et al. (2018). MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell 70, 825–841.e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD, Garcia BA, Adelman K, and Wysocka J (2017). Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 66, 568–576.e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiziev P, Akdemir KC, Miller JP, Keung EZ, Samant NS, Sharma S, Natale CA, Terranova CJ, Maitituoheti M, Amin SB, et al. (2017). Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression. Cell Rep. 19, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 6 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, et al. (2014). Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet 46, 1097–1102. [DOI] [PubMed] [Google Scholar]
- Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, and Hunt DF (2007). Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc 2, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. [DOI] [PubMed] [Google Scholar]
- Gelato KA, Schöckel L, Klingbeil O, Rückert T, Lesche R, Toedling J, Kalfon E, Héroult M, Lejeune P, Mönning U, et al. (2018). Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 37, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. (2012). The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M, and Ishaque N (2018). EnrichedHeatmap: an R/ Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genomics 19, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, and Shilatifard A (2012). Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 26, 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Hu D, and Shilatifard A (2014). Enhancer malfunction in cancer. Mol. Cell 53, 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Gao X, Morgan MA, Herz HM, Smith ER, and Shilatifard A (2013). The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol 33, 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Zhang Z, Kashiwagi M, Yoshida T, Joshi I, Jena N, Somasundaram R, Emmanuel AO, Sigvardsson M, Fitamant J, et al. (2016). Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev. 30, 1971–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iams WT, and Lovly CM (2015). Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res 21, 4270–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stütz AM, et al. (2012). Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhlin CC, Stenman A, Haglund F, Clark VE, Brown TC, Baranoski J, Bilguvar K, Goh G, Welander J, Svahn F, et al. (2015). Whole-exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Genes Chromosomes Cancer 54, 542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al. (2010). The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster J, and Rahmann S (2012). Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Koutsioumpa M, Hatziapostolou M, Polytarchou C, Tolosa EJ, Almada LL, Mahurkar-Joshi S, Williams J, Tirado-Rodriguez AB, Huerta-Yepez S, Karavias D, et al. (2019). Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut 68, 1271–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet 44, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, et al. (2015). Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J. Clin. Invest 125, 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B, Lee JE, Jang Y, Wang L, Peng W, and Ge K (2017). MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 45, 6388–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, and Shiekhattar R (2007). Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450. [DOI] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, and Ge K (2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2, e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AK, et al. (2017). Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, et al. (2012). Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Satani N, Hammoudi N, Ackroyd JJ, Khadka S, Yan VC, Georgiou DK, Sun Y, Zielinski R, Tran T, et al. (2018). Eradication of ENO1-deleted Glioblastoma through Collateral Lethality. bioRxiv. 10.1101/331538. [DOI] [Google Scholar]
- Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, et al. (2014). Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 46, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al. (2016). Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Shiao E, Lan Y, Coradin M, Anderson A, Donahue G, Simpson CL, Sen P, Saffie R, Busino L, Garcia BA, et al. (2018). KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev. 32, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. (2012). Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 109, 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke JJ, Flaherty KT, Ribas A, and Long GV (2017). Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol 14, 463–482. [DOI] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. (2014). Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, and Zhao K (2010). Epigenome mapping in normal and disease States. Circ. Res 107, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. (2011). Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med 21, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et al. (2011). Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet 43, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GC, Schneider R, and Bickmore WA (2016). Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet 48, 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al. (2012). Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G, Tang Z, Mathé E, Benner C, et al. (2014). B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman AT, and Rai K (2018). Loss of Histone Acetylation and H3K4 methylation promotes melanocytic malignant transformation. Mol. Cell Oncol 5, e1359229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RC, and Dou Y (2015). Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 15, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, and Herlyn M (2010). A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, et al. (2015). Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun 6, 7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward DJ, Cubberley G, Kim S, Schonewald M, Zhang L, Tripet B, and Bentley DL (2007). Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat. Struct. Mol. Biol 14, 240–242. [DOI] [PubMed] [Google Scholar]
- Shen H, and Laird PW (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA Cancer J. Clin 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, et al. (2014). Identification of genomic alterations in oesophageal squamous cell cancer. Nature 509, 91–95. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur I, and Taipale J (2016). The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493. [DOI] [PubMed] [Google Scholar]
- Sze CC, and Shilatifard A (2016). MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med 6, a026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, and Wang Y (2005). Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 11, 7234–7242. [DOI] [PubMed] [Google Scholar]
- Tang M (2017a). pyflow-ChIPseq: a snakemake based ChIP-seq pipeline (Zenodo; ). [Google Scholar]
- Tang M (2017b). pyflow-RNAseq: a snakemake based RNAseq pipeline (Zenodo; ). [Google Scholar]
- Terranova C, Tang M, Orouji E, Maitituoheti M, Raman A, Amin S, Liu Z, and Rai K (2018). An Integrated Platform for Genome-wide Mapping of Chromatin States Using High-throughput ChIP-sequencing in Tumor Tissues. J. Vis. Exp, 56972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardabasso C, Gaspar-Maia A, Hasson D, Pünzeler S, Valle-Garcia D, Straub T, Keilhauer EC, Strub T, Dong J, Panda T, et al. (2015). Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Mol. Cell 59, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. (2015). Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun 6, 6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ding N, Li Y, Cheng H, Wang D, Yang Q, Deng Y, Yang Y, Li Y, Ruan X, et al. (2015). Insulin-like growth factor binding protein 5 (IGFBP5) functions as a tumor suppressor in human melanoma cells. Oncotarget 6, 20636–20649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, and Ge K (2016). Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. USA 113, 11871–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Takahashi K, Futreal PA, and Chin L (2013). Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet 14, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2006). An implementation of the grammar of graphics in R: ggplot. Paper presented at the 2006 use R conference: Book of Abstracts. [Google Scholar]
- Wysocka J, Myers MP, Laherty CD, Eisenman RN, and Herr W (2003). Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 17, 896–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Mandal R, Sinelnikov IV, Broadhurst D, and Wishart DS (2012). MetaboAnalyst 2.0–a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 40, W127–W133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet JP, Wagner SN, Ramaswamy S, Mesirov JP, and Hynes RO (2008). Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol. Cancer Res 6, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. (2015). Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med 21, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Lu T, Jia Y, Luo X, Gopal P, Li L, Odewole M, Renteria V, Singal AG, Jang Y, et al. (2019). Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell 177, 608–621.e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.