

 Potent T. brucei methionyl-tRNA synthetase inhibitors built on two novel short and rigid linker systems.
Potent T. brucei methionyl-tRNA synthetase inhibitors built on two novel short and rigid linker systems.
Abstract
Based on crystal structures of Trypanosoma brucei methionyl-tRNA synthetase (TbMetRS) bound to inhibitors, we designed, synthesized, and evaluated two series of novel TbMetRS inhibitors targeting this parasite enzyme. One series has a 1,3-dihydro-imidazol-2-one containing linker, the other has a rigid fused aromatic ring in the linker. For both series of compounds, potent inhibition of parasite growth was achieved with EC50 < 10 nM and most compounds exhibited low general toxicity to mammalian cells with CC50s > 20 000 nM. Selectivity over human mitochondrial methionyl tRNA synthetase was also evaluated, using a cell-based mitochondrial protein synthesis assay, and selectivity in a range of 20–200-fold was achieved. The inhibitors exhibited poor permeability across the blood brain barrier, necessitating future efforts to optimize the compounds for use in late stage human African trypanosomiasis.
Introduction
Human African trypanosomiasis (HAT), also known as sleeping sickness, occurs in Sub-Saharan Africa. HAT is classified by the WHO as a neglected tropical disease.1 It is a vector-borne parasitic disease caused by the protozoan Trypanosoma brucei (T. brucei). The disease progresses in two stages. In the first stage, the parasites multiply in blood and lymph, but have not yet invaded the central nervous system (CNS). Once the parasites cross the blood–brain barrier (BBB) to invade the CNS, the disease enters the second stage. Infection with T. brucei eventually leads to coma and death if not treated.2 Until recently, melarsoprol and eflornithine were the only HAT drugs able to cross the BBB for treatment of late stage HAT. Melarsoprol is derived from arsenic and has many undesirable side effects, while the regimen of eflornithine (combined with nifurtimox) is complex and difficult to administer.3–6 A new orally administered drug that kills parasites by generating reactive nitro intermediates, fexinidazole, also is effective in CNS infection and was recently approved for early and late-stage HAT.7 This is a welcomed advancement, but drugs in new chemical classes with distinct mechanisms of action are still needed to strengthen the armamentarium for HAT treatment and mitigate the risk of drug resistance. To that end, we are working on inhibitors of the methionyl-tRNA synthetase (MetRS), an essential enzyme involved in protein synthesis.
We previously validated MetRS of T. brucei (TbMetRS) as a drug target for new HAT treatment.8,9 RNA interference of TbMetRS demonstrated essentiality, and exposure of T. brucei to MetRS inhibitors resulted in trypanocidal activity both in vitro and in vivo. However, development of new inhibitors for treating the second stage of HAT is challenging. The inhibitors not only need to be potent and selective against the target pathogen, but also have the ability to cross the BBB to access parasites in the CNS. Compounds with high brain permeability represent less than 2% of small molecule drugs.10
Early analogues of compound A (Scheme 1) were reported to inhibit bacterial MetRS.11 We resynthesized compound A and showed potent activity against TbMetRS, and solved its crystal structure in complex with TbMetRS.12 The 3,5-dichlorophenyl moiety of A occupies an enlarged methionine binding pocket, whereas the benzimidazole moiety binds to an auxiliary pocket that opens in the presence of inhibitor. The middle part of A, containing a 1,3-propyldiamine, acts as a linear linker to connect the 3,5-dichlorophenyl and benzimidazole moieties. We have explored variations of this linker either in a linear form or by inclusion of rings.13,14 Note that in those linker variation cases, our compounds still maintained the 1,3,5-substitution pattern on the left-hand aromatic ring. We also noticed in the patent literature that compounds like B developed as antibiotics by the Trius company possess a ring-based linker that connects the left-hand side phenyl ring through a 1,2,4-substitution pattern.15 Therefore we investigated close analogs of B and also obtained an inhibitor-bound crystal structure of TbMetRS.12 Here, we report our exploration of TbMetRS inhibitors with new dihydro-imidazolone and fused ring-based linkers in order to discover new compounds with improved potency and suitability for treating human T. brucei infection.
Scheme 1. New linker design.

Results and discussion
Design and SAR studies of inhibitors with a 5-membered ring linker
We designed our compounds by replacing the pyrimidine ring of compound B with a linear linker of the same length (six atoms), and eventually by shortening the linker and incorporating other ring structures. As a result, we identified compound 1, which contains a linker length of five atoms that is rigidified through a 5-membered 1,3-dihydro-imidazol-2-one ring. Compound 1 was shown to have an IC50 of <40 nM against the TbMetRS enzyme (tested using an ATP depletion assay against 40 nM of enzyme16). When tested against T. brucei parasites, compound 1 was shown to have an EC50 of 350 nM.
In order to understand how compound 1 binds to TbMetRS protein, a crystal structure of compound 1 bound to TbMetRS was solved (Fig. 1 and ESI,† PDB: ; 6MES). Compound 1 occupies the active site of TbMetRS protein in a similar way as compound A. The superposition of TbMetRS·1 and TbMetRS·A (Fig. 1B) revealed that the benzimidazole moiety of compound 1 has shifted toward the center of the linker due to the rigidity of the shorter linker (red circle around C4/C5). Despite this shift the hydrogen bond (2.6 Å) between the carboxylate of Asp287 and the NH on the benzimidazole ring system of compound 1 is maintained (Fig. 1C). The 2,4-dichlorophenyl moiety of compound 1 and 3,5-dichlorophenyl moiety of A overlap almost perfectly in the enlarged methionine binding pocket (Fig. 1B).
Fig. 1. A) TbMetRS·1 structure (PDB: ; 6MES) with difference electron density map (Fo–Fc) calculated by omitting the inhibitor, contoured at 3σ (positive density in grey, negative density in red). B) Superposition of TbMetRS·1 (inhibitor carbon in cyan) and TbMetRS·A (inhibitor carbon in orange). C) Hydrogen bond between the carboxylate of Asp287 and the nitrogen on the benzimidazole ring system of compound 1.

Based on the structure of compound 1 bound to TbMetRS, we designed compound 2 to probe the binding pocket near the linker space by adding an N-ethyl group to the 1,3-dihydro-imidazol-2-one ring of the linker. This resulted in an improvement to an EC50 of 40 nM (Table 1). From here, further SAR studies of the aromatic ring on the right-hand side that occupies the auxiliary pocket were first carried out due to the extra space around C4/C5 positions as shown in Fig. 1B (results in Table 1). Note, that we no longer relied on enzyme inhibition assays (IC50) to rank compounds since most of the compounds presented in the work had IC50s < 40 nM, which is below the concentration of the enzyme used. Instead, parasite growth inhibition (EC50) became the primary assay for rank-ordering compound activity.
Table 1. Structures and associated in vitro testing results for N-ethyl substituted analogs of compound 1.
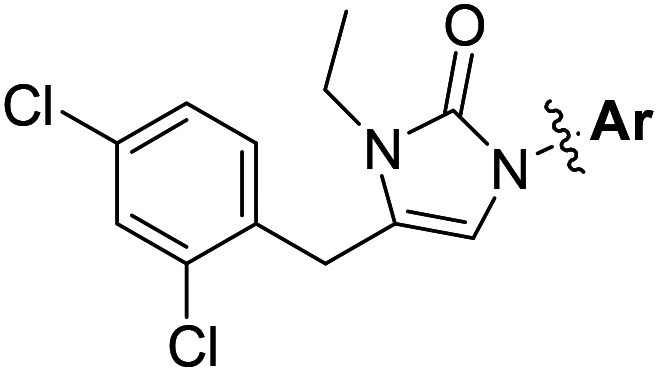
| ||||
| Compound number | Ar | T. brucei a EC50 (nM) | CRL-8155 b CC50 (nM) | HepG2 c CC50 (nM) |
| 2 |
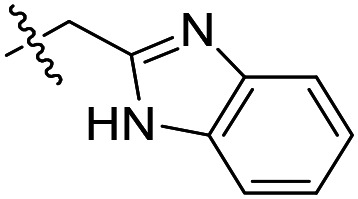
|
40 | >50 000 | >50 000 |
| 3 |
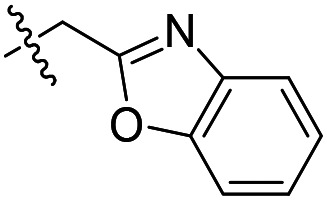
|
>2000 | >50 000 | >50 000 |
| 4 |
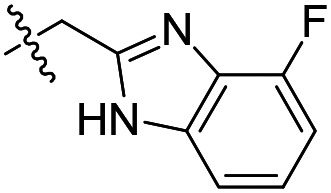
|
31 ± 4 (n = 2) | 45 000 | >50 000 |
| 5 |
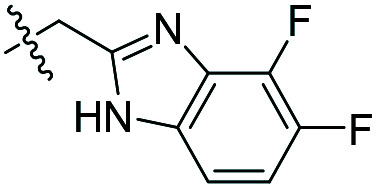
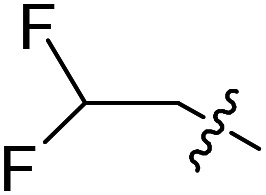
|
26 ± 3 (n = 4) | 41 000 | 40 600 |
| 6 |
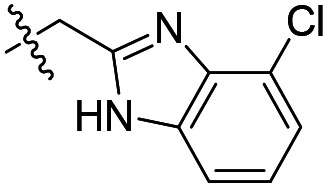
|
210 | >50 000 | >50 000 |
| 7 |
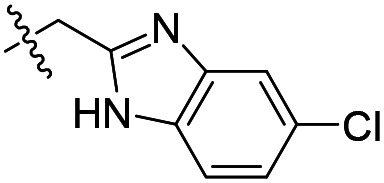
|
69 | 27 100 | 45 300 |
| 8 |
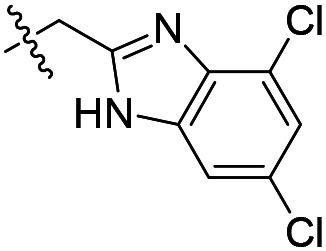
|
1600 | 38 200 | 43 700 |
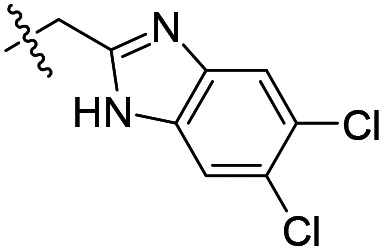
| 9 |
|
660 | 31 600 | 31 300 |
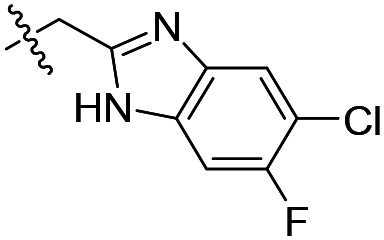
| 10 |
|
220 | 20 900 | 29 800 |
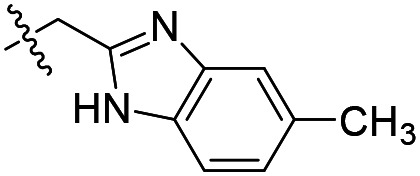
| 11 |
|
110 | >50 000 | >50 000 |
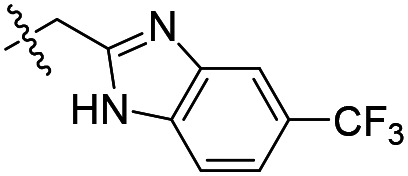
| 12 |
|
>2000 | >50 000 | >50 000 |
| 13 |
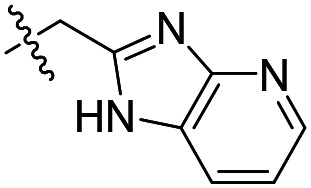
|
17 ± 3 (n = 2) | >50 000 | >50 000 |
| 14 |
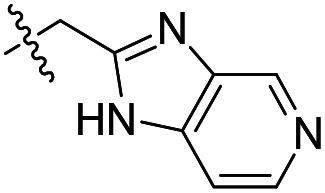
|
390 | >50 000 | >50 000 |
| 15 |
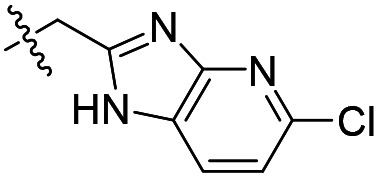
|
1.6 ± 0.2 (n = 5) | >50 000 | >50 000 |
| 16 |
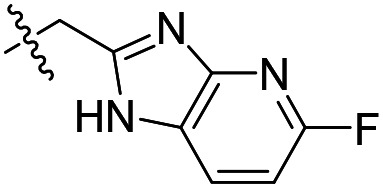
|
3.4 ± 0.1 (n = 3) | >50 000 | >50 000 |
aControl for T brucei EC50 assay average ± SEM: pentamidine (1.7 ± 0.2 nM; n = 14). Compounds with initial EC50 < 30 nM were tested more than once and their EC50s are shown as average ± SEM (number of tests).
bControl for CRL-8155 CC50 assay average ± SEM: quinacrine (7.0 ± 3.7 μM; n = 3).
cControl for HepG2 CC50 assay average ± SEM: quinacrine (7.6 ± 0.9 μM; n = 5).
As shown in Table 1, the hydrogen bond donor property of the Ar group is important. A change of the benzimidazole group to an oxazole analog, which abolishes the donor in compound 3, led to loss of inhibitory activity. With compounds 4 to 12 the tolerance of various small hydrophobic substitutions on the benzimidazole ring was explored, partly guided by our previous SAR studies of TbMetRS inhibitors.9,13,14 Our previous analysis of the auxiliary binding pocket indicated that for inhibitors with a linear flexible linker such as compound A only a small F-substitution at C5 on the benzimidazole ring might fit well.13 Here, due to the binding position shift of inhibitor 1 and the resulting slightly larger space around C4/C5 (Fig. 1B), we believe that other halogens or a methyl group are worth exploring. As shown in Table 1, single Cl, F, or up to two F atoms at C4 and/or C5 were well tolerated by the TbMetRS auxiliary binding pocket, but two Cl atoms on C4/C6 or C5/C6 positions were not. For C5 substitution, Me was slightly worse than Cl for EC50 while CF3 led to a significant loss of potency. The reason for the significant loss in potency of the CF3 analog (12) may be due to space limitation of the auxiliary binding pocket. A CH3 or Cl substitution at C5 would put the carbon or chloride atom at ∼3.2 Å away from the carbonyl oxygen of Val471 of TbMetRS. Yet for CF3 substitution at C5, the F to O distance would be a very unfavorable 2.8 Å. In summary, the SAR of small hydrophobic substituents on the benzimidazole ring fits the expectation from the structural data shown in Fig. 1B. Compounds 13 and 14 demonstrated the influence of replacing the benzimidazole ring with imidazo-pyridine, for which the imidazo[4,5-b]pyridine ring (13) showed better results than the imidazo[4,5-c]pyridine analog (14). The largest improvements in potency were obtained with compounds 15 and 16 with either a Cl or a F substitution next to the pyridine N. These two inhibitors had single digit nanomolar potency in T. brucei EC50 assays. All inhibitors were also tested against two mammalian cell lines for general cytotoxicity. As shown in Table 1, all compounds were generally of low toxicity with CC50s at or above 20 μM against either CRL-8155 and HepG2 cells.
Based on the potent compounds 15 and 16, further SAR exploration was carried out by replacing the N-ethyl on the 1,3-dihydro-imidazol-2-one ring (R1) as well as at the C4 position of the left-hand phenyl ring (R3), which occupies the methionine substrate binding site of TbMetRS (Table 2).
Table 2. Structures and associated in vitro results for additional compounds with 1,3-dihydro-imidazol-2-one linker.
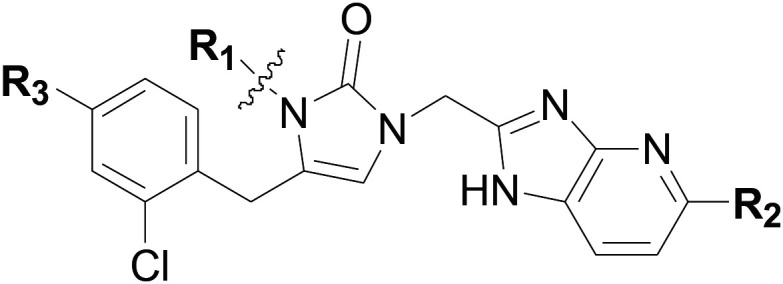
| ||||||
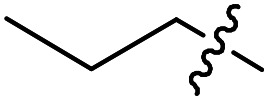
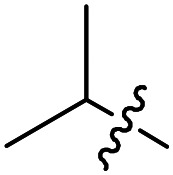
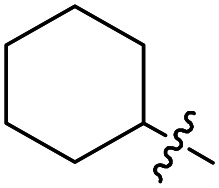
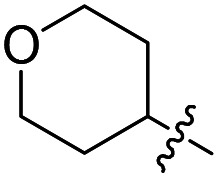
| Compound number | R1 | R2 | R3 | T. brucei a EC50 (nM) | CRL-8155 c CC50 (nM) | HepG2 d CC50 (nM) |
| 17 |
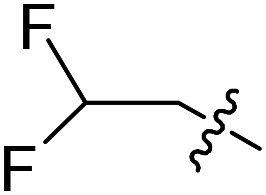
|
Cl | Cl | 3.0 ± 0.5 (n = 2) | >50 000 | >50 000 |
| 18 |
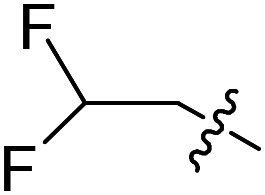
|
F | Cl | 5.9 ± 2.3 (n = 2) | 33 300 | >50 000 |
| 19 |
|
Cl | Cl | 1.8 < 0.9 b | 37 800 | 26 400 |
| 20 |
|
F | Cl | 5.1 ± 0.8 (n = 3) | >50 000 | >50 000 |
| 21 |
|
F | Cl | 2.3 ± 1.3 (n = 2) | >50 000 | >50 000 |
| 22 |
|
F | Cl | 16 ± 5 (n = 3) | 30 400 | 33 200 |
| 23 |
|
F | Cl | 5.8 ± 2.3 (n = 2) | >50 000 | >50 000 |
| 24 |
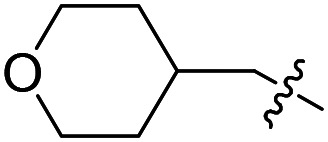
|
F | Cl | 3.0 ± 1.8 (n = 2) | >50 000 | >50 000 |
| 25 |
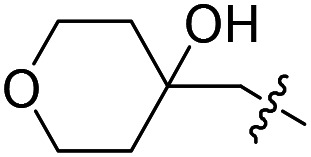
|
F | Cl | 4.6 ± 2.0 (n = 2) | >50 000 | >50 000 |
| 26 |
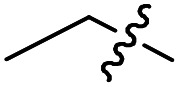
|
Cl | OMe | 1.0 ± 0.2 (n = 9) | >50 000 | >50 000 |
| 27 |
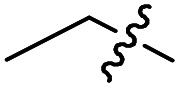
|
F | OMe | 1.8 ± 0.4 (n = 2) | >50 000 | >50 000 |
| 28 |
|
Cl | OMe | 3.6 ± 2.2 (n = 2) | >50 000 | >50 000 |
| 29 |
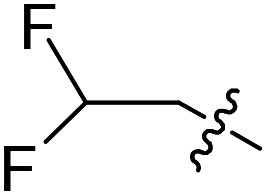
|
F | OMe | 4.6 ± 1.7 (n = 2) | >50 000 | >50 000 |
aControl for T brucei EC50 assay average ± SEM: pentamidine (2.0 ± 0.3 nM; n = 24).
bTwo values, unable to average.
cControl for CRL-8155 CC50 assay average ± SEM: quinacrine (5.1 ± 1.0 μM; n = 7).
dControl for HepG2 CC50 assay average ± SEM: quinacrine (9.1 ± 0.8 μM; n = 7).
Compounds 17 to 25 cover variations to the N-substitutions (R1). It appears that many short chains and substitutions are tolerated, with EC50s against the parasites mostly maintained at single digit nanomolar range, although bulky hydrophobic substituents such as a cyclohexyl (22) led to slight loss of potency. Based on the crystal structure of compound 1 in complex with TbMetRS, these results are not surprising because the linker N-substitution points to a solvent-exposed region. Therefore, it makes sense that a wide range of substitutions are tolerated without loss of potency. Compounds 26 to 29 have a methoxy group as the R3 instead of Cl, which led to increased potency against the parasites compared to the Cl analogs, with the lowest EC50 value reaching 1 nM. General cytotoxicity for all compounds in Table 2 is minor, as most compounds have CC50s above 50 μM against the two mammalian cell lines tested.
Design and SAR studies of inhibitors with fused-ring linkers
Further work to obtain inhibitor-bound protein crystals successfully produced a new structure for the TbMetRS·26 complex (PDB: ; 6CML). As shown in Fig. 2A, compound 26 binds in the same way as compound 1, with its N-ethyl group pointing to solvent exposed area, confirming the SAR described above with a large variety of N-substitutions being tolerated for this place. At the same time, inspection of the N-ethyl group as well as the torsional angles of the 2-Cl-4-MeO-phenyl group with respect to the linker 1,3-dihydro-imidazol-2-one ring prompted us to design inhibitors with a fused ring linker, as shown in Fig. 2B. Several fused ring linker compounds were synthesized to test the hypothesis that these modifications would be tolerated, and would give rise to a new scaffold to provide additional opportunities for lead-compound optimization (Table 3).
Fig. 2. A) Superposition of TbMetRS·1 (inhibitor carbon in cyan) and TbMetRS·26 (inhibitor carbon in magenta). B) Design of inhibitors with a fused-ring linker based on 26.

Table 3. Exploration of fused rings as potential linker.
| ||||
| Compound number | Ar | T. brucei a EC50 (nM) | CRL-8155 b CC50 (nM) | HepG2 c CC50 (nM) |
| 30 |
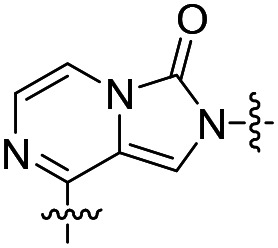
|
31 | >50 000 | >50 000 |
| 31 |
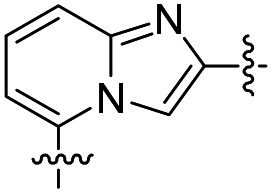
|
24 ± 6 (n = 2) | 13 000 | >50 000 |
| 32 |
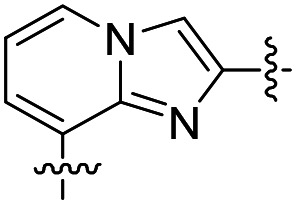
|
>2000 | ND d | ND |
| 33 |
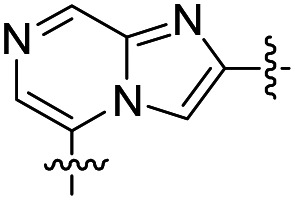
|
32 | >50 000 | >50 000 |
aControl for T brucei EC50 assay average ± SEM: pentamidine (1.8 ± 0.3 nM, n = 7).
bControl for CRL-8155 CC50 assay average ± SEM: quinacrine (4.8 ± 1.0 μM, n = 4).
cControl for HepG2 CC50 assay average ± SEM: quinacrine (10.0 ± 2.0 μM, n = 3).
dND: not determined.
The results were encouraging. Three of the four fused ring linkers compounds have potent EC50s against T. brucei parasites (compounds 30, 31, and 33), validating the design strategy. Compound 32 is the exception, as it lost activity against the parasites. Its linker would have placed an N atom (as compared to a CH in 31) in close proximity to the Asp287 of TbMetRS that makes a hydrogen bond to the imidazopyridine moieties of the inhibitors. We believe that this pointing of two hydrogen bond acceptors to each other may be the cause of loss of inhibitory activity of 32.
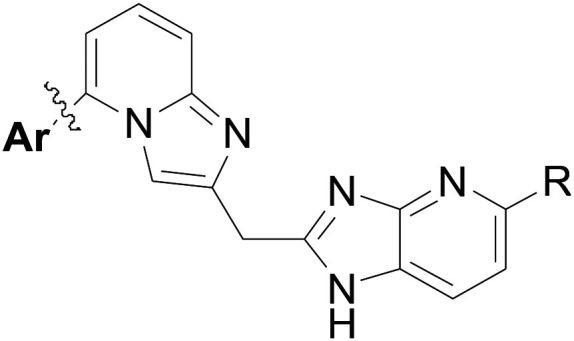
Building on these results, we performed additional SAR studies in an attempt to further improve potencies of inhibitors with fused ring linkers (Table 4). We chose to study the SAR with the imidazo[1,2-a]pyridine moiety of 31 due to better synthetic feasibility.
Table 4. Additional inhibitors with 2,5-substituted imidazo[1,2-a]pyridine linker.
| |||||
| Compound number | Ar | R | T. brucei a EC50 (nM) | CRL-8155 b CC50 (nM) | HepG2 c CC50 (nM) |
| 34 |
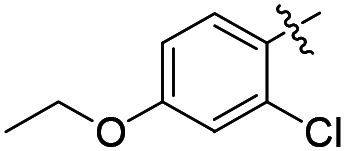
|
Cl | 1900 | ND d | ND |
| 35 |
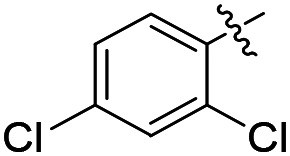
|
Cl | 64 | 16 100 | 43 200 |
| 36 |
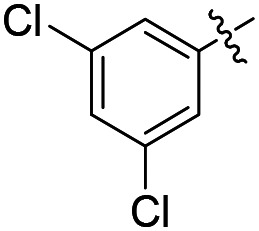
|
Cl | 196 | ND | ND |
| 37 |
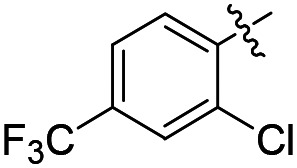
|
Cl | 1780 | ND | ND |
| 38 |
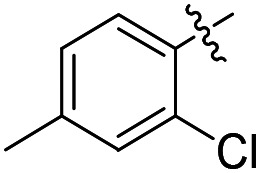
|
Cl | >2000 | ND | ND |
| 39 |
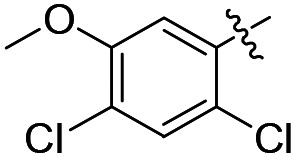
|
Cl | 3.5 ± 0.1 (n = 2) | 9500 | >50 000 |
| 40 |
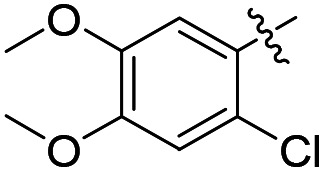
|
Cl | 3.1 ± 0.5 (n = 6) | 19 000 | 28 200 |
| 41 |
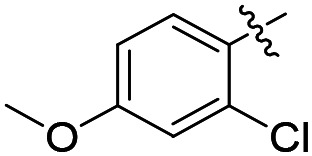
|
F | 59 | 15 700 | >50 000 |
| 42 |
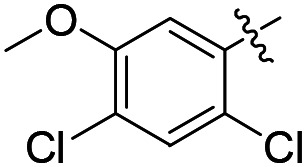
|
F | 8.4 ± 3.1 (n = 2) | 10 400 | >50 000 |
| 43 |
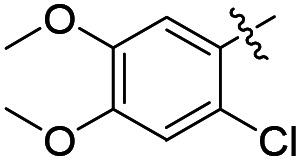
|
F | 7.2 ± 1.7 (n = 4) | >25 000 | >25 000 |
aControl for T brucei EC50 assay average ± SEM: pentamidine (2.4 ± 0.3 nM; n = 10).
bControl for CRL-8155 EC50 assay average ± SEM: quinacrine (6.1 ± 0.6 μM; n = 5).
cControl for HepG2 EC50 assay average ± SEM: quinacrine (10.9 ± 1.1 μM; n = 4).
dND: not determined.
Compounds 34–40 all have a chloro substituted imidazo[4,5-b]pyridine moiety on the right hand side of the inhibitor, while various substitutions on the phenyl ring (Ar group) were tried. Compounds with two phenyl substituents (34–38) did not show improvements over the starting compound 31. With three substituents (39 and 40), we obtained potent inhibitors against the parasites with activities close to the best compounds with 1,3-dihydro-imidazol-2-one based linkers, such as 26 as shown in Table 2. Compounds 41–43 are F substituted imidazo[4,5-b]pyridine analogs of compounds 31, 39, and 40, showing comparable potencies as the Cl analogs.
Selectivity over human mitochondrial MetRS and BBB penetration properties
As shown above, through structure-based design, we obtained potent inhibitors of TbMetRS with single digit nanomolar potency against parasite growth and low general toxicity to two mammalian cell lines. However, in vitro measurement of mammalian cell toxicity does not accurately reflect potential mitochondrial toxicity,17 necessitating more direct assessment due to the close homology of the TbMetRS to the human mitochondrial MetRS. Note that human cells have two MetRS enzymes, one in the cytosol (classified as type 2) and one in the mitochondria (type 1).18 We previously tried to differentiate the selectivity using enzyme inhibition assays,9 however, since the inhibitors described in this work often exhibit enzyme IC50s below the concentration of enzyme used, we changed to a cell-based assay in which the EC50 against mitochondrial protein synthesis in mammalian cells are measured using HepG2 cells and an antibody against a mitochondrial protein (COX-1).19 The results are shown in Table 5 for 11 compounds from two different linker systems. For selectivity over the human mitochondrial MetRS, a margin of 20–200-fold was obtained. While only one compound with the 1,3-dihydro-imidazol-2-one linker showed selectivity over 100-fold (17), the fused ring linker compounds have more examples (3 out of 4 listed) with selectivity over 100-fold. The results suggest that inhibitors with a rigid fused-ring linker may have advantages over the single ring linker series for designing future selective inhibitors of TbMetRS. The capacity of compounds to cross the BBB was determined by measuring concentrations in mouse brains 60 min after IP injection to determine the brain/plasma ratio. Unfortunately, all the compounds exhibited poor BBB penetration, with only compounds 15 and 20 showing modest brain/plasma ratios (9.6% and 5.5%, respectively). The rest of the compounds all have brain/plasma ratios of <1% (Table 5).
Table 5. Selectivity over inhibition of human mitochondrial protein synthesis and brain plasma ratio.
| Compound number | Human mitochondrial protein synthesis EC50 (nM) | Selectivity index a | Brain/plasma ratio at 60 min (%) |
| Compounds with 1,3-dihydro-imidazol-2-one linker | |||
| 15 | 42 b | 26 | 9.6 |
| 16 | 290 b | 85 | 0.1 |
| 17 | 430 b | 143 | 0 |
| 18 | 175 b | 30 | 0 |
| 20 | 195 b | 38 | 5.5 |
| 26 | 39 b | 39 | 0 |
| 27 | 75 b | 42 | 0 |
| Compounds with fused ring linker | |||
| 39 | 507 | 145 | 0 |
| 40 | 478 | 154 | 0.5 |
| 42 | 1750 | 203 | 0.6 |
| 43 | 316 | 45 | 0 |
aSelectivity index is defined as EC50 against human mitochondrial protein synthesis divided by EC50 against T. brucei parasite growth.
bAs reported previously.19 Control for mitochondrial protein synthesis inhibition EC50 assay average ± SEM: chloramphenicol (7.9 ± 1.6 μM, n = 9).
Conclusions
Structure-based design allowed us to obtain potent TbMetRS inhibitors with a short 1,3-dihydro-imidazol-2-one based linker and with fused aromatic ring linkers. Many of the inhibitors demonstrated nanomolar potency in parasite growth inhibition assays while exhibiting low general toxicity to mammalian cell lines. For selectivity over human mitochondrial MetRS, cell-based mitochondrial protein synthesis assay indicated that 1–2 orders of magnitude in selectivity can be achieved. However, achieving BBB permeability remains a major challenge for both series of compounds, and further optimization will be necessary to identify potential candidates for treatment of second stage HAT.
Experimental section
Chemical synthesis of inhibitors
Unless otherwise stated, all chemicals were purchased from commercial suppliers and used without further purification. The microwave irradiation was performed in CEM Discover System. Flash chromatography was performed using a CombiFlash system.
Procedure for the synthesis of compound 1 (Scheme 2)
Scheme 2. Synthetic route to compound 1. Reagents and conditions (a) HOBt, EDC, NaBH4. (b) TFA, DCM; (c) tert-butyl 2-(isocyanatomethyl)-1H-benzo[d]imidazole-1-carboxylate, (d) Dess–Martin reagent; (e) formic acid, 50 °C.

To a solution of 2-Boc-amino-3-(2,4-dichlorophenyl)propanoic acid (24.5 mg, 0.074 mmol) in DCM (8 mL) was added HOBt (12.3 mg, 0.08 mmol) and EDC (15.4 mg, 0.08 mmol). The mixture was stirred for 30 min and then concentrated in vacuo. The residue was dissolved in THF (8 mL) and NaBH4 (5.6 mg, 0.147 mmol) was added at 0 °C. Then to the mixture was added 0.2 ml of water. The resulting mixture was stirred at 0 °C for 30 min and quenched with MeOH (2 mL). After most solvents were removed, EtOAc (25 mL) was added. The organic phase was washed sequentially with 10% citric acid, saturated NaHCO3, brine and dried over Na2SO4. After solvent was removed, the residue was purified via silica gel chromatography, eluted with MeOH/DCM to give 2-Boc-amino-3-(2,4-dichlorophenyl)propan-1-ol.20 2-Boc-amino-3-(2,4-dichlorophenyl)propan-1-ol (9.6 mg, 0.03 mmol) was dissolved in 5 ml of DCM. To the solution, 1 ml of TFA was added dropwise. The solution was stirred for 40 min at room temperature. The reaction mixture was completely evaporated under vacuum. The residue was dissolved in anhydrous DCM containing DIPEA (0.08 mmol). The solution was cooled to 0 °C and tert-butyl 2-(isocyanatomethyl)-1H-benzo[d]imidazole-1-carboxylate (0.03 mmol) in DCM was added slowly. The reaction was performed at room temperature for 1 h. The solution was diluted with DCM and washed with water, brine, and dried over Na2SO4. After solvent was removed, the residue was purified via silica gel chromatography, eluted with MeOH/DCM to give 1-((1-Boc-1H-benzo[d]imidazol-2-yl)methyl)-3-(-3-(2,4-dichlorophenyl)-1-hydroxypropan-2-yl)urea (yield: 50%). Next, the resulting 1-((1-Boc-1H-benzo[d]imidazol-2-yl)methyl)-3-(-3-(2,4-dichlorophenyl)-1-hydroxypropan-2-yl)urea was dissolved in 5 ml of DCM. To the solution, 1 ml of TFA was added dropwise. The solution was stirred for 40 min at room temperature. The reaction mixture was completely evaporated under vacuum to give 1-((1H-benzo[d]imidazol-2-yl)methyl)-3-((S)-3-(2,4-dichlorophenyl)-1-hydroxypropan-2-yl)urea residue, which was dissolved in THF and Dess–Martin reagent (0.015 mmol) was added. The mixture was stirred at room temperature overnight. The solvent was then removed under vacuum. The residue was dissolved in formic acid and heated at 50 °C overnight to give 1 after purification via silica gel chromatography (yield: 75%).
General procedure for the synthesis of compounds 2–29 (Scheme 3)
Scheme 3. Synthetic route to compound 2. Reagents and conditions (a) triphosgene, Na2CO3, DCM, ethylamine; (b) TFA; (c) ethyl bromoacetate, K2CO3; (d) LiOH, ethanol/water; (e) EDC, benzene-2,3-diamine, pyridine; (f) HOAc, microwave irradiation.

To an ice-cooled suspension of 1-amino-3-(2,4-dichlorophenyl)propan-2-one (0.1 mmol) in anhydrous methylene chloride (10 mL) and saturated NaHCO3 (10 mL), triphosgene (5.9 μl, 0.035 mmol) was added with stirring. After the mixture was stirred at 0 °C for 15 min, the organic layer was separated and ethylamine (0.11 mmol) was added. The mixture was stirred at 0 °C for 30 min and room temperature for 1 h. Then 1 ml of TFA was added and the mixture was stirred at room temperature for 50 min. After most solvent was removed, the mixture was diluted with EtOAc and washed successively with saturated NaHCO3 and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (DCM/MeOH) (yield: 73%). The purified intermediate was dissolved in 10 ml anhydrous acetonitrile, treated with 16.3 μl (0.147 mmol) of ethyl bromoacetate and potassium carbonate (20.3 mg, 0.147 mmol). The mixture was refluxed overnight. After the reaction solvent was evaporated, the residue was dissolved in EtOAc and washed successively with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (dichloromethane/methanol, yield: 82%) and then was dissolved in 1 ml of ethanol and 3 ml of water containing LiOH (0.15 mml). The mixture was stirred for 50 min at room temperature. The solution was acidified with 0.2 N HCl and the solvent completely removed in vacuo. The residue was dissolved in 3 ml of pyridine, then bezene-2,3-diamine (0.20 mmol) and EDC hydrochloride (0.20 mmol) were added. The mixture was stirred at room temperature overnight. The solvent was removed on a rotary evaporator. The residue was dissolved in EtOAc (30 ml) and washed with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was dissolved in 2 ml of acetic acid and the solution was microwave irradiated at 125 °C for 40 min. After the solvent was removed on a rotary evaporator, the product (2) was purified by flash column chromatography (yield: 75%).
Procedure for the synthesis of compound 30 (Scheme 4)
Scheme 4. Synthetic route to compound 30. Reagents and conditions (a) K2CO3, Pd(PPh3)4, DME, H2O, microwave irradiation; (b) K2CO3,tert-butyl 2-bromoacetate; (c) triphosgene; (d), TFA, DCM; (e) HATU, 6-chlororopyridine-2,3-diamine, DIPEA; (f) HOAc, microwave irradiation.

(3-Chloropyrazin-2-yl)methanamine HCl salt (0.087 mmol), 2-chloro-4-methoxyphenylboronic acid (0.175 mmol), potassium carbonate (0.35 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.0043 mmol) in water (0.5 mL) and DME (1.5 mL) were microwave irradiated at 110 °C for 30 min. After most organic solvent was removed in vacuo, the residue was extracted with EtOAc and washed successively with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (MeOH/DCM, yield: 35%) and dissolved in 5 ml of DMF. To the solution potassium carbonate (0.128 mmol) and tert-butyl 2-bromoacetate (0.030 mmol) were added. The mixture was microwave irradiated at 80 °C for 30 min. After organic solvent was removed in vacuo, the residue was purified by flash chromatography on silica gel (yield: 53%) and dissolved in 5 ml of anhydrous DCM. To the solution were added DIPEA (0.021 mmol) and triphosgen (0.008 mmol) at 0 °C the mixture was stirred at 0 °C for 40 min then room temperature for 1 hour.
The solvent was removed on a rotary evaporator. The residue was purified by flash column chromatography (yield: 87%) and treated with 1 ml of DCM and 1 ml of TFA. After the mixture was stirred at room temperature for 2 hours, solvents were completely removed in vacuo. The residue was dissolved in DMF (2 ml). The solution was added DIPEA (10 μl), HATU (0.011 mmol) and 6-chlororopyridine-2,3-diamine (0.014 mmol). The mixture was stirred at room temperature for 3 hours. The solvent was removed on a rotary evaporator. The residue was dissolved in EtOAc (20 ml) and washed with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was dissolved in 1 ml of acetic acid and the solution was microwave irradiated at 125 °C for 40 min. The product was purified by flash column chromatography (yield: 72%).
General procedure for the synthesis of compounds 31–43 (Scheme 5)
Scheme 5. Synthetic route to compound 31. Reagents and conditions (a) ethyl 4-chloroacetoacetate, ethanol, refluxing; (b) 2-chloro-4-methoxyphenylboronic acid, K2CO3, Pd(PPh3)4, DME, H2O, microwave irradiation; (c) LiOH, ethanol/water; (d) EDC, 6-chloropyridine-2,3-diamine, pyridine; (e) HOAc, microwave irradiation.

A mixture of ethyl 4-chloroacetoacetate (0.2 mmol) and 6-bromoylpyridin-2-amine (0.2 mmol) in EtOH (15 mL) was refluxed overnight. After the solution was cooled down, the white precipitate was collected and washed with cold EtOH. Ethyl 2-(5-bromoH-imidazo[1,2-a]pyridin-2-yl)acetate HCl salt was obtained and directly used.
A mixture of ethyl 2-(5-bromoH-imidazo[1,2-a]pyridin-2-yl)acetate HCl salt (0.097 mmol) 2-chloro-4-methoxyphenylboronic acid (0.147 mmol), potassium carbonate (0.12 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.0015 mmol) in water (0.5 mL) and DME (1.5 mL) was microwave irradiated at 100 °C for 20 min. After most organic solvent was removed in vacuo, the residue was extracted with EtOAc and washed successively with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexane/EtOAc, yield: 57%). To the purified intermediate was added 0.5 ml of ethanol and 1.5 ml of water containing LiOH (0.21 mmol). The mixture was stirred for 50 min at room temperature. The solution was acidified with 0.2 N hydrochloric acid and the solvent completely removed in vacuo. The residue was dissolved in 3 ml of pyridine, then 6-chlororopyridine-2,3-diamine (0.063 mmol) and EDC hydrochloride (0.11 mmol) were added. The mixture was stirred at room temperature overnight. The solvent was removed on a rotary evaporator. The residue was dissolved in EtOAc (30 ml) and washed with water and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was dissolved in 1 ml of acetic acid and the solution was microwave irradiated at 125 °C for 40 min. The product was purified by flash column chromatography (yield: 65%).
Protein expression and purification
Expression and purification of TbMetRS was achieved as reported earlier.12 Briefly, site directed mutagenesis of surface residues 452KKE454 to ARA was performed on the truncated TbMetRS (237–773) in the AVA0421 vector. TbMetRS was expressed in Escherichia coli and purified by Ni-NTA affinity chromatography followed by N-terminal His6-tag overnight cleavage at 4 °C with 3C protease. Upon a second Ni-NTA step, the cleaved TbMetRS was purified from the His6-tagged 3C protease. Purified TbMetRS was obtained upon size-exclusion chromatography (SEC) on a Superdex 75 column (Amersham Pharmacia Biotech) with SEC buffer (25 mM HEPES at pH 7.0, 500 mM NaCl, 2 mM DTT, 5% glycerol, 0.025% NaN3) in the presence of 10 mM l-methionine.
Protein crystallization and crystal soaking
Truncated TbMetRS surface mutant was crystallized and soaked in compounds 1 or 26 as reported previously.12 Protein crystals were obtained by sitting-drop vapor-diffusion equilibrated against a reservoir containing 2.0 M (NH4)2SO4, 0.2 M NaCl and 0.1 M sodium cacodylate pH 6.2–6.8 after 1–2 days at room temperature. Crystallization drops consisted of 1 μL protein at 10 mg mL–1 with 10 mM l-methionine (necessary for crystallization), 1 mM tris(2-carboxyethyl)phosphine and 1 μL of reservoir solution. TbMetRS·Met crystals were soaked for 60 seconds as described before12 in cryo-solution (30% glycerol in SEC buffer) containing 2 mM of the inhibitor in order to obtain TbMetRS·inhibitor complexes. Inhibitor soaked TbMetRS crystals were immediately flash frozen in liquid nitrogen until data collection.
Data collection and structure determination
Crystal diffraction data were collected under cryogenic conditions either at Stanford Synchrotron Radiation Lightsource (SSRL) beamline 12-2 at a wavelength of 1 Å (for compound 26) or in home source facility using a MicroMax-007 HF rotating anode (Rigaku) equipped with VariMax HF (Osmic) monochromator and a Saturn 994 (Rigaku) CCD detector at a wavelength of 1.54 Å (for compound 1). Data processing was done with HKL2000.21 Previously reported structures of TbMetRS14,22 were used as models for phase determination by molecular replacement with Phaser.23 Iterated building, rebuilding and refinement of the structure were performed with Coot24 and REFMAC5.25 The Grade web server26 was used in the generation of the refinement restraints for compounds 1 and 26. The structure validation server MolProbity27 was employed for validation throughout the process of structure determination. The final crystallographic refinement statistics are given in Table S1.† Coordinates and structure factors for TbMetRS in complex with compounds 1 or 26 were deposited in the Protein Data Bank under PDB IDs ; 6MES and ; 6CML, respectively.
Mammalian cell growth inhibition assay
Compounds were screened in a 96-well format against two human cell lines, the lymphocytic cell line CRL-8155 and the hepatic cell line HepG2 as previously described.9 Cell lines were obtained from the ATCC (; www.atcc.org). CRL-8155 cells were grown in RPMI media supplemented with 10% heat inactivated fetal bovine serum (FBS) (R&D Systems, ; www.rndsystems.com), 2 mM l-glutamine, 0.4 mM sodium pyruvate and 100 U mL–1 penicillin/100 μg mL–1 streptomycin at 37 °C with 5% CO2. HepG2 cells were grown in DMEM/F12 media supplemented with 10% heat inactivated FBS, 0.4 mM sodium pyruvate and 100 U mL–1 penicillin/100 μg mL–1 streptomycin at 37 °C with 5% CO2. CRL-8155 or HepG2 cells (30 000 cells/well and 25 000 cells/well respectively) were plated in quadruplicate in 96-well plates and test compounds were added in quadruplicate in serial 3-fold dilutions starting at 50 μM (or 25 μM if dictated by solubility limits). At 48 hours, plates were developed using AlamarBlue (ThermoFisher Scientific) and percent inhibition was calculated by subtracting background and comparing fluorescence to wells without test compound. Background controls contained cells in the presence of 50 μM quinacrine (MP Biomedicals, LLC). EC50; www.collaborativedrug.com). For results listed in Tables 1–4, compounds were screened in a single experiment in quadruplicate.
T. brucei growth inhibition assay
Compounds were tested for anti-trypanosomal activity against T.b. brucei cells (BSF 427 parasites) in HMI-9 media containing 10% heat inactivated FBS 100 U mL–1 penicillin/100 μg mL–1 streptomycin at 37 °C with 5% CO2, as previously described.8 Test compounds were assayed in a 96-well format in triplicate with serial 3-fold dilutions of compound or a pentamidine control against an initial inoculum of 10 000 cells/well and quantified at 48 hours with Alamar Blue (Invitrogen, Waltham, MA). EC50; www.collaborativedrug.com). For results listed in Tables 1–4, compounds were screened in a single experiment with triplicates. Only compounds that showed EC50 below 30 nM were retested one or more times, and the results are shown as averages ± SEM of all independent runs.
Mitochondrial protein synthesis inhibition assay
The methods used for this assay are replicated from a previous publication.19 Briefly, collagen coated 96-well plates (Invitrogen, Waltham, MA) were seeded with 6000 HepG2 cells/well and grown in DMEM/F12 media supplemented with 10% heat inactivated FBS, 0.4 mM sodium pyruvate and 100 U mL–1 penicillin/100 μg mL–1 streptomycin at 37 °C and 5% CO2 for 24 hours prior to adding compounds. At 24 hours, the media was removed and fresh media containing test compound was added to the plates. Compounds were added in serial 3-fold dilutions in duplicate, with a starting concentration of 25 μM. The assay was allowed to incubate with test compounds or a chloramphenicol control for 6 days in the above media conditions, with the compound/media being refreshed every 48 hours. On day 6, cells were fixed with 4% paraformaldehyde and mitochondrial protein synthesis activity was quantified using the MitoBiogenesis™ In-Cell ELISA Kit (Abcam, Cambridge, UK) according to manufacturer's instructions. All EC50; www.collaborativedrug.com).
BBB penetration assay in mice
All murine experiments were approved by the University of Washington Institutional Animal Care and Use Committee, IACUC approval code 4248-01 (animal welfare approval number A3464-01). Brain and plasma concentrations of test compounds were determined as previously described.28 Briefly, compounds were administered to Swiss Webster mice in triplicate at 5 mg kg–1via IP injection in a 0.4 mL volume in a vehicle containing 7% Tween80, 5% DMSO, 3% EtOH in saline. At 1 hour post injection, blood was collected and plasma was isolated via centrifugation. At the same time, brains were harvested and frozen for later analysis. Brains were homogenized in water and test compounds were extracted from the brain and plasma samples for analysis via liquid chromatography/tandem mass spectrometry. Calculations for brain concentrations accounted for the 3% volume/weight blood in the brain.29
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI097177 (to EF and FSB) and R01AI084004 (to WGJH). XBA had support from a Fulbright Fellowship. We thank Stewart Turley for providing support for the X-ray data collection. Structure determination benefitted from remote access to resources at the Stanford Synchrotron Radiation Lightsource supported by the U.S. Department of Energy Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515 and by the National Institutes of Health (P41GM103393). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00057d
References
- Neglected tropical diseases, https://www.who.int/neglected_diseases/diseases/en/, (accessed Jan 2020).
- Kennedy P. G. Lancet Neurol. 2013;12:186–194. doi: 10.1016/S1474-4422(12)70296-X. [DOI] [PubMed] [Google Scholar]
- Brun R., Blum J., Chappuis F., Burri C. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- Croft S. L., Barrett M. P., Urbina J. A. Trends Parasitol. 2005;21:508–512. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Wilkinson S. R., Kelly J. M. Expert Rev. Mol. Med. 2009;11:e31. doi: 10.1017/S1462399409001252. [DOI] [PubMed] [Google Scholar]
- Rodgers J. J. Neuroimmunol. 2009;211:16–22. doi: 10.1016/j.jneuroim.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Pelfrene E., Harvey Allchurch M., Ntamabyaliro N., Nambasa V., Ventura F. V., Nagercoil N., Cavaleri M. PLoS Neglected Trop. Dis. 2019;13:e0007381. doi: 10.1371/journal.pntd.0007381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S., Gillespie J. R., Kelley A. M., Napuli A. J., Zhang Z., Kovzun K. V., Pefley R. M., Lam J., Zucker F. H., Van Voorhis W. C., Merritt E. A., Hol W. G., Verlinde C. L., Fan E., Buckner F. S. Antimicrob. Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S., Gillespie J. R., Ranade R. M., Koh C. Y., Kim J. E., Laydbak J. U., Zucker F. H., Hol W. G., Verlinde C. L., Buckner F. S., Fan E. J. Med. Chem. 2012;55:6342–6351. doi: 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvest R. L., Armstrong S. A., Berge J. M., Brown P., Elder J. S., Brown M. J., Copley R. C., Forrest A. K., Hamprecht D. W., O'Hanlon P. J., Mitchell D. J., Rittenhouse S., Witty D. R. Bioorg. Med. Chem. Lett. 2004;14:3937–3941. doi: 10.1016/j.bmcl.2004.05.070. [DOI] [PubMed] [Google Scholar]
- Koh C. Y., Kim J. E., Shibata S., Ranade R. M., Yu M., Liu J., Gillespie J. R., Buckner F. S., Verlinde C. L., Fan E., Hol W. G. Structure. 2012;20:1681–1691. doi: 10.1016/j.str.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Koh C. Y., Ranade R. M., Shibata S., Gillespie J. R., Hulverson M. A., Huang W., Nguyen J., Pendem N., Gelb M. H., Verlinde C. L., Hol W. G., Buckner F. S., Fan E. ACS Infect. Dis. 2016;2:399–404. doi: 10.1021/acsinfecdis.6b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Zhang Z., Barros-Alvarez X., Koh C. Y., Ranade R. M., Gillespie J. R., Creason S. A., Shibata S., Verlinde C., Hol W. G. J., Buckner F. S., Fan E. Eur. J. Med. Chem. 2016;124:1081–1092. doi: 10.1016/j.ejmech.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Finn J. M. and Hilgers M. T., PCT Int. Appl., WO2008098143A2, 2008.
- Pedro-Rosa L., Buckner F. S., Ranade R. M., Eberhart C., Madoux F., Gillespie J. R., Koh C. Y., Brown S., Lohse J., Verlinde C. L., Fan E., Bannister T., Scampavia L., Hol W. G., Spicer T., Hodder P. J. Biomol.J. Biomol. ScreeningScreening. 2015;20:122–130. doi: 10.1177/1087057114548832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin L. D., Hynes J., Dykens J. A., Jamieson J. D., Will Y. Toxicol. Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Gentry D. R., Ingraham K. A., Stanhope M. J., Rittenhouse S., Jarvest R. L., O'Hanlon P. J., Brown J. R., Holmes D. J. Antimicrob. Agents Chemother. 2003;47:1784–1789. doi: 10.1128/AAC.47.6.1784-1789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F. S., Ranade R. M., Gillespie J. R., Shibata S., Hulverson M. A., Zhang Z., Huang W., Choi R., Verlinde C., Hol W. G. J., Ochida A., Akao Y., Choy R. K. M., Van Voorhis W. C., Arnold S. L. M., Jumani R. S., Huston C. D., Fan E. Antimicrob. Agents Chemother. 2019;63:e02061. [Google Scholar]
- Morales-Serna J. A., García-Ríos E., Bernal J., Paleo E., Gaviño R., Cárdenas J. Synthesis. 2011;1375–1382:1375. [Google Scholar]
- Otwinowski Z. and Minor W., in Macromolecular Crystallography, Part A, ed. C. Carter and R. Sweet, Academic Press, 1997, pp. 307–326. [Google Scholar]
- Koh C. Y., Kim J. E., Wetzel A. B., de van der Schueren W. J., Shibata S., Ranade R. M., Liu J., Zhang Z., Gillespie J. R., Buckner F. S., Verlinde C. L., Fan E., Hol W. G. PLoS Neglected Trop. Dis. 2014;8:e2775. doi: 10.1371/journal.pntd.0002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G., Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N., Vagin A. A., Dodson E. J. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Grade Web Server, version 1.001, http://grade.globalphasing.org, (accessed Jan 2020).
- Chen V. B., Arendall, 3rd W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchynskyy A., Gillespie J. R., Herbst Z. M., Ranade R. M., Buckner F. S., Gelb M. H. ACS Med. Chem. Lett. 2017;8:886–891. doi: 10.1021/acsmedchemlett.7b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe H. W., Bodsch W., Hossmann K. A. Acta Neuropathol. 1984;64:37–42. doi: 10.1007/BF00695604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.