

Abstract

Fabrication of high-performance heterostructure devices requires fundamental understanding of the diffusion dynamics of metal species on 2D materials. Here, we investigate the room-temperature diffusion of Ag, Au, Cu, Pd, Pt, and Ru adatoms on graphene using ab initio and classical molecular dynamics simulations. We find that Ag, Au, Cu, and Pd follow Lévy walks, in which adatoms move continuously within ∼1–4 nm2 domains during ∼0.04 ns timeframes, and they occasionally perform ∼2–4 nm flights across multiple surface adsorption sites. This anomalous diffusion pattern is associated with a flat (<50 meV) potential energy landscape (PEL), which renders surface vibrations important for adatom migration. The latter is not the case for Pt and Ru, which encounter a significantly rougher PEL (>100 meV) and, hence, migrate via conventional random walks. Thus, adatom anomalous diffusion is a potentially important aspect for modeling growth of metal films and nanostructures on 2D materials.
Fabrication of multifunctional metal contacts in heterostructure devices based on two-dimensional (2D) materials necessitates growth of vapor-deposited metal films in a controlled fashion on 2D crystal substrates.1,2 A key step toward achieving this control is to fundamentally understand the dynamics of adatom surface diffusion, the atomic-scale process that leads to island nucleation and thereby largely sets the lateral length scale of film morphology.3,4
During epitaxial
metal and semiconductor growth, at temperatures
that are typical for thin-film deposition experiments, adatom diffusion
is described as a thermally activated chain of uncorrelated jumps,
so that adatoms execute a random walk on the substrate surface.5 Within this framework, the diffusivity of an
adatom transitioning between surface sites separated by a mean square
distance l2 at rate ν is calculated
as 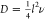 . The temperature-dependent jump rate ν
is typically approximated by the Arrhenius equation, in which the
static (i.e., 0 K) surface migration barrier is determined by ab initio or classical computational methods,6−16 while the jump attempt frequency is estimated via (quasi-)harmonic
transition-state theory approaches.17,18 Alternatively, D can be computed using Einstein’s equation ⟨x2(t)⟩
= 4Dt, where ⟨x2(t)⟩ is the adatom mean square
displacement (MSD) after time t, with ⟨x2(t)⟩
being measured either experimentally or estimated via classical molecular
dynamics simulations.5,19
. The temperature-dependent jump rate ν
is typically approximated by the Arrhenius equation, in which the
static (i.e., 0 K) surface migration barrier is determined by ab initio or classical computational methods,6−16 while the jump attempt frequency is estimated via (quasi-)harmonic
transition-state theory approaches.17,18 Alternatively, D can be computed using Einstein’s equation ⟨x2(t)⟩
= 4Dt, where ⟨x2(t)⟩ is the adatom mean square
displacement (MSD) after time t, with ⟨x2(t)⟩
being measured either experimentally or estimated via classical molecular
dynamics simulations.5,19
At temperatures significantly higher than those used for thin-film synthesis experiments, the above-described notion of random walk can be inadequate20−25 because ⟨x2(t)⟩ may not vary as ∼t. Such nonlinear ⟨x2(t)⟩ versus t dependence is indicative of anomalous diffusion and has been attributed to vibrational entropy modifying the potential energy landscape (PEL) of the substrate surface, thereby allowing for long and correlated adatom jumps.26−31 As 2D materials are intrinsically inert32,33 they often interact weakly with adatoms and their PEL is markedly flat, so that the onset of anomalous diffusion may take place at temperatures that are within experimentally relevant ranges (e.g., room temperature). Moreover, 2D materials, owing to their reduced dimensionality and the smaller density of vibrational modes they host in their lattice, are intuitively expected to dissipate adatom energies less efficiently as compared to bulk crystals, which may further enhance the tendency for anomalous surface diffusion.
In the present work, we explore the propensity of metal adatoms for anomalous diffusion on 2D crystals at room temperature. To this purpose, we study surface migration dynamics of silver (Ag), gold (Au), copper (Cu), palladium (Pd), platinum (Pt), and ruthenium (Ru) adatoms on single-layer graphene (SLG)34 via ab initio and classical (semiempirical) molecular dynamics simulations. The choice of the six metals is motivated by the fact that (i) the reported estimates of their static surface migration barriers on graphene range from 0.004 to 0.188 eV35 and hence represent a multitude of expected dynamic diffusive behaviors and (ii) they are relevant as metal contacts in nanoelectronics, sensing, and catalysis.36−39
We first perform ab initio molecular dynamics (AIMD) simulations of monomer diffusion on SLG for times up to 0.2 ns (see later in the text for simulation details and detailed explanation of the reasons behind the selection of the simulation times for the various adatoms). The trajectories are displayed in Figure 1, which shows that Pt and Ru diffuse within areas of ∼0.04–0.09 nm2. Closer inspection of the trajectories (enlarged sections presented as insets in the Pt and Ru trajectory panels) reveals that the adatoms spend most of the simulation trapped in specific surface sites, and for the case of Pt, random jumps among neighboring adsorption sites are observed. All the above-described features are consistent with a random walk. In contrast, Ag, Au, Cu, and Pd atoms move continuously across the surface without being halted by adsorption sites, diffuse within relatively short-range domains (marked with circles in Figure 1) during time frames of ∼0.04 ns, and occasionally perform long jumps that cover multiple adsorption sites (the distance between adjacent adsorption sites is ∼0.12–0.24 nm). This anomalous behavior corresponds to superdiffusion. Moreover, visual inspection of the trajectories reveals that both the areas of the domains and the jump lengths for Ag and Cu trajectories (the approximate mean values for these quantities are ∼4 nm2 and ∼4 nm, respectively) are significantly larger than those for Au and Pd (∼1 nm2 and ∼2 nm). The distinctly different normal versus superdiffusive behavior exhibited by the various adatoms is further illustrated in Figure S1 in the Supporting Information which plots trajectories of all metals after a 0.02 ns simulation time.
Figure 1.

Ab initio MD diffusion trajectories for Pt (0.04 ns); Ru (0.02 ns); and Ag, Au, Cu, and Pd (0.2 ns). The circles in the trajectories for Ag, Au, Cu, and Pd mark a short-range diffusion domain (see text for more details).
To estimate the diffusivity D and quantify the
migration dynamics of single adatoms, we compute, for each one of
the anomalous trajectories in Figure 1, the time-averaged adatom mean square displacement40
| 1 |
In eq 1, Δt is the simulation time step, τ
a variable representing
a specific observation time scale, and N the total
number of subtrajectories with a duration τ that can be defined
over the total atomic trajectory (note that two different subtrajectories
defined in this way may partially overlap). For a total simulation
time T and an observation scale τ consisting
of NT and Nτ time steps Δt, respectively, N = NT – Nτ + 1. Thus, each point on a  versus τ curve
corresponds to an
average entailing information on all subtrajectories with a duration
τ. From the latter it follows that the statistical significance
of a given
versus τ curve
corresponds to an
average entailing information on all subtrajectories with a duration
τ. From the latter it follows that the statistical significance
of a given  value decreases with
increasing τ,
because larger τ values yield a smaller number of subtrajectories
available for calculating
value decreases with
increasing τ,
because larger τ values yield a smaller number of subtrajectories
available for calculating  . Moreover, it is worth
pointing out that
the analysis on the data in Figure 1 is based on
. Moreover, it is worth
pointing out that
the analysis on the data in Figure 1 is based on 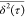 , instead of ⟨x2(t)⟩,
because an accurate
calculation of the latter quantity would require data from multiple
simulated trajectories.
, instead of ⟨x2(t)⟩,
because an accurate
calculation of the latter quantity would require data from multiple
simulated trajectories.
The  versus τ curves (0 < τ ≤
50 ps; a detailed explanation for the selection of τ range is
provided later in the text) for Ag, Au, Cu, and Pd are plotted in
log–log scale in Figure 2. The data reveal that Ag and Cu exhibit larger
versus τ curves (0 < τ ≤
50 ps; a detailed explanation for the selection of τ range is
provided later in the text) for Ag, Au, Cu, and Pd are plotted in
log–log scale in Figure 2. The data reveal that Ag and Cu exhibit larger  values over the entire
τ range, which
is in line with the differences in the jump length magnitudes seen
in Figure 1. Furthermore,
irrespective of the atomic displacement magnitude, the
values over the entire
τ range, which
is in line with the differences in the jump length magnitudes seen
in Figure 1. Furthermore,
irrespective of the atomic displacement magnitude, the  versus τ curves
exhibit a nonlinear
behavior with slopes α larger than one (the α = 1 slope
is represented by the dashed line in Figure 2), which is consistent with the superdiffusive
nature of the atomic trajectories in Figure 1.41 Note that
α ≈ 1 as τ → 50 ps. We attribute this to
the limited statistical significance of
versus τ curves
exhibit a nonlinear
behavior with slopes α larger than one (the α = 1 slope
is represented by the dashed line in Figure 2), which is consistent with the superdiffusive
nature of the atomic trajectories in Figure 1.41 Note that
α ≈ 1 as τ → 50 ps. We attribute this to
the limited statistical significance of 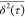 at large τ values, e.g.,
at large τ values, e.g., 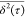 for τ = 50 ps is calculated using
4 subtrajectories (see eq 1).
for τ = 50 ps is calculated using
4 subtrajectories (see eq 1).
Figure 2.

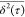 versus τ curves
(plotted in log–log
scale) extracted from the AIMD trajectories presented in Figure 1 for Ag, Au, Cu,
and Pd. All lines exhibit a slope α > 1 which is consistent
with the superdiffusive nature of adatom motion. This is in contrast
to random walk trajectories which exhibit a
versus τ curves
(plotted in log–log
scale) extracted from the AIMD trajectories presented in Figure 1 for Ag, Au, Cu,
and Pd. All lines exhibit a slope α > 1 which is consistent
with the superdiffusive nature of adatom motion. This is in contrast
to random walk trajectories which exhibit a 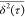 versus τ slope of α = 1 (represented
by the dashed line). A more detailed discussion on the correlation
among α and diffusion nature can be found in the text.
versus τ slope of α = 1 (represented
by the dashed line). A more detailed discussion on the correlation
among α and diffusion nature can be found in the text.
On the basis of the nonlinear behavior established
in Figure 2, we conclude
that
we cannot use the Einstein equation for correlating 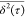 versus τ curves
and D. For this reason, in the remainder of the Letter
we use the continuous
time random walk formalism as an alternative framework to treat anomalous
diffusion.42 This formalism assumes that
versus τ curves
and D. For this reason, in the remainder of the Letter
we use the continuous
time random walk formalism as an alternative framework to treat anomalous
diffusion.42 This formalism assumes that 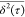 exhibits a power law
dependence on time
exhibits a power law
dependence on time
| 2 |
where the exponent 1 < α < 2 denotes the superdiffusive character of the monomer (α = 2 corresponds to a ballistic trajectory, whereas α = 1 corresponds to a random walk) and Dα is a generalized diffusion coefficient with physical dimensions nm2·ps–α.
The task of
extracting relevant values for α and Dα is not straightforward, because each
point at the  versus τ curves
has a different statistical
significance (see eq 1). Thus, following the methodology proposed by Saxton,40 only
versus τ curves
has a different statistical
significance (see eq 1). Thus, following the methodology proposed by Saxton,40 only 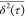 points within an interval
[0: τcut] are considered. The value τcut is a time
cutoff here taken to be equal to 50 ps (see Figure 2), to exclude all points averaged over less
than 4 independent subtrajectories. Moreover, this choice has a physical
justification: τcut is of the same order of magnitude
as the typical time required for an adatom to transition between superdiffusive
domains—note that the trajectories in Figure 1 consist of ∼3–4 domains—and
is thus representative of the adatom migration dynamics at the time
scale of several tens of picoseconds. We also notice in Figure 2 that the slope of log–log
points within an interval
[0: τcut] are considered. The value τcut is a time
cutoff here taken to be equal to 50 ps (see Figure 2), to exclude all points averaged over less
than 4 independent subtrajectories. Moreover, this choice has a physical
justification: τcut is of the same order of magnitude
as the typical time required for an adatom to transition between superdiffusive
domains—note that the trajectories in Figure 1 consist of ∼3–4 domains—and
is thus representative of the adatom migration dynamics at the time
scale of several tens of picoseconds. We also notice in Figure 2 that the slope of log–log 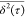 versus τ curves
is not constant.
Hence, in order to extract α we perform a linear fit of the
curves for multiple time intervals [0: τ*] with 0 < τ*≤
50 ps as explained by Kepten et al.43 Then,
the most probable value from the statistical distribution of α
is selected (see Figure S2 in the Supporting Information), from which Dα is calculated
using eq 2.
versus τ curves
is not constant.
Hence, in order to extract α we perform a linear fit of the
curves for multiple time intervals [0: τ*] with 0 < τ*≤
50 ps as explained by Kepten et al.43 Then,
the most probable value from the statistical distribution of α
is selected (see Figure S2 in the Supporting Information), from which Dα is calculated
using eq 2.
Table 1 lists the numerical values of α and Dα for Ag, Au, Cu, and Pd. With regards to the scaling exponents, we see that Ag and Cu have an exponent α = 1.4, whereas for Au α = 1.2, and for Pd α = 1.1. Table 1 also presents the static (0 K) adsorption energies Eads0K (see later in the text for details) of all metals (Pt, Ru, Ag, Cu, Au, and Pd) on the hollow, bridge, and top site of graphene (hollow, above center of a C6 ring; bridge, above midpoint of C=C bond; top, above a C atom). The Eads values are validated against high accuracy static relaxations and are compared to data for both SLG and multilayer graphene from the literature (see Table S1 in the Supporting Information).7,38,44,45 We note that the energy values in Table 1 are shifted with respect to the most stable site for which Eads0K is set equal zero. By comparing α and Eads, we conclude that the propensity of an adatom toward anomalous diffusion can be correlated with the flatness of the surface PEL: (i) for Pt and Ru, the smallest energy difference between adsorption sites ΔEads0K is in the range ∼200 to ∼600 meV, yielding a random walk behavior; (ii) while for Ag, Cu, Au, and Pd, ΔEads is considerably lower (∼1 to ∼50 meV), which offers more and easier transition pathways and promotes superdiffusive behavior, as seen by the α > 1 values. Moreover, the PEL can also be correlated with the degree of anomaly of the different metals that exhibit superdiffusion: the markedly flat PEL for Ag and Cu (ΔEads0K of the order of a few millielectronvolts) induces long atomic jumps, large diffusion domains (∼4 nm and ∼4 nm2, respectively), and α = 1.4; whereas the somewhat rougher PEL of Au and Pd (ΔEads up to 1 order of magnitude larger) results in relatively smaller jump lengths and domain areas for Au and Pd (∼2 nm and ∼1 nm2), and yields α = 1.2 and α = 1.1, respectively. Hence, by computing the static PEL of an adatom on a 2D material, we can qualitatively assess the adatom tendency for exhibiting anomalous diffusion patterns. However, it is important to emphasize that the adatom jump lengths and the specific dynamic diffusion behavior established for the various adatom-on-SLG systems should be considered in relation to the mean separation between islands observed in experiments35
Table 1. Exponents α, Diffusivities (Dα, nm2·ps–α), and Adsorption Energies at 0 and 300 K (Eads0K and Eads, meV) of Metal Adatoms on the Graphene Surfacea.
|
Eads0K |
Eads300K |
|||||||
|---|---|---|---|---|---|---|---|---|
| α | Dα | hollow | bridge | top | hollow | bridge | top | |
| Ag | 1.4(×1.07±1) | 0.016 | 9 | 1 | 0 | 6 | 0 | 3 |
| Au | 1.2(×1.08±1) | 0.007 | 22 | 5 | 0 | ∼33 | 6 | 0 |
| Cu | 1.4(×1.07±1) | 0.024 | 184 | 0 | 8 | ∼171 | 4 | 0 |
| Pd | 1.1(×1.09±1) | 0.008 | 290 | 0 | 53 | ∼660 | 0 | 19 |
| Pt | 1 | 894 | 0 | 215 | ||||
| Ru | 1 | 0 | 641 | 664 | ||||
The adsorption energies refer to the hollow, bridge, and top positions, whereby energies are shifted with respect to the most stable adsorption site for which Eads is set equal zero.
The normal (i.e., random walk) diffusive behavior of Pt and Ru adatoms at 300 K can be explained by the fact that the thermal energy kBT (≈ 25 meV) is signifficantly smaller than the minimum ΔEads0K that separates stable adsorption sites. In contrast, the collective motion of substrate surface atoms may play a decisive role on the dynamics of adatoms interacting weakly with the substrate, whereby the minimum ΔEads may become comparable with the thermal energy kBT. The latter is particularly relevant for strongly anharmonic materials like graphene,46 in which out-of-plane vibrations are relatively larger compared to those on surfaces of bulk crystals. To qualitatively understand the effects induced by graphene vibrations on the PEL, we calculate the adsorption energy of hollow, bridge, and top graphene sites at 300 K (see later in the text for details). Table 1 shows that, for the cases of Ag, Au, and Cu, the numerical accuracy of DFT for adsorption-energy values (a few millielectronvolts) is comparable with the differences between ΔEads0K and ΔEads. The influence of temperature is more pronounced for the case of Pd, where ΔEads300K between bridge and top sites decreases by ∼30 meV compared to ΔEads. This temperature-induced effect may explain the fact that Pd exhibits a diffusion behavior fairly similar to that of Au, despite having a notably larger 0 K PEL corrugation (∼50 vs ∼5 meV). It is important to note that atomic vibrations do not only affect adatom migration dynamics by modifying the effective PEL; they may also alter the adatom preferences for occupying the different adsorption sites because of entropy effects, as explained in the Supporting Information (Section S4 and Table S2).
Besides AIMD, we employ classical molecular
dynamics (CMD) to model
multiple Ag trajectories of 3 ns and a long one of 30 ns (see later
in the text for details). By using the same methodology as that in
the AIMD simulations to analyze the 30 ns long trajectory (see Figure S3 in the Supporting Information), we
extract a  versus τ exponent
α = 1.3,
which shows that CMD also predicts anomalous diffusion behavior for
Ag. This can be understood in light of the shallow PEL that CMD yields
(see Table S3 in the Supporting Information), which is consistent with AIMD (i.e., millielectronvolt difference
between adsorption sites). Because both simulation approaches yield
a qualitatively consistent physical behavior, we take advantage of
the higher computational efficiency of CMD, to gather statistics and
further our understanding of the type of diffusion that adatoms perform
on SLG.
versus τ exponent
α = 1.3,
which shows that CMD also predicts anomalous diffusion behavior for
Ag. This can be understood in light of the shallow PEL that CMD yields
(see Table S3 in the Supporting Information), which is consistent with AIMD (i.e., millielectronvolt difference
between adsorption sites). Because both simulation approaches yield
a qualitatively consistent physical behavior, we take advantage of
the higher computational efficiency of CMD, to gather statistics and
further our understanding of the type of diffusion that adatoms perform
on SLG.
We analyze the trajectories by computing the ensemble
mean 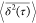 of the time-averaged 3 ns trajectories
and compare it with the ⟨x2(t)⟩ of the same trajectories in order
to confirm or refute the ergodic character47 of the diffusion dynamics. Figure 3a shows that the two averages differ by a small factor
from the simulations start until 0.1 ns, after which they converge.
This discrepancy at short and medium time scales indicates that, because
of the tendency of adatoms to perform long jumps, the expansion rate
of the area that an adatom may visit is greater than its mean diffusion
rate. The adatom is, thus, unable to access all points of the phase
space, which renders the motion nonergodic.
of the time-averaged 3 ns trajectories
and compare it with the ⟨x2(t)⟩ of the same trajectories in order
to confirm or refute the ergodic character47 of the diffusion dynamics. Figure 3a shows that the two averages differ by a small factor
from the simulations start until 0.1 ns, after which they converge.
This discrepancy at short and medium time scales indicates that, because
of the tendency of adatoms to perform long jumps, the expansion rate
of the area that an adatom may visit is greater than its mean diffusion
rate. The adatom is, thus, unable to access all points of the phase
space, which renders the motion nonergodic.
Figure 3.

(a) Ensemble average  and ⟨x2(t)⟩ for the 30 CMD diffusion
trajectories (3 ns long) and (b) jump length distribution for the
30 ns long CMD trajectory of Ag at 300 K.
and ⟨x2(t)⟩ for the 30 CMD diffusion
trajectories (3 ns long) and (b) jump length distribution for the
30 ns long CMD trajectory of Ag at 300 K.
Moreover, we calculate the adatom jump length probability distribution
| 3 |
of the 30 ns run for time sampling intervals ΔtS = 7.5, 10, 12.5, and 15 ps, and fit a regression line for each of the p(l) data sets, which are plotted in Figure 3b. For all the cases, the slope of the regression lines takes values 1.4 < μ < 1.7. We conclude from these results that the adatom motion is an intermediate case between ballistic motion (μ = 1) and random walk (μ = 3).
The nonergodic character and the specific jump length distribution established by the analysis presented in Figure 3 indicate that Ag adatoms follow a fractal movement pattern known as Lévy walk,41,42,48 which is also observed in other phenomena in physical and biological sciences, including light propagation in optical media,49 animal foraging,50 or human travel.51 We expect that Au, Cu, and Pd also perform a Lévy walk, owing to the qualitative resemblance of their AIMD trajectories to that of Ag, as well as because for Ag, Au, Cu, and Pd we find that α > 1. Lévy walks have an effective upper limit for their jump lengths,52 which allows treating the correlated long-range adatom motion between diffusive domains (marked with circles in Figures 1 and S3a) as a random walk53 and hence extracting effective diffusivities from the Einstein equation. These diffusivities, however, cannot be directly correlated with static activation barriers as diffusive domains are not adsorption sites with an associated PEL. Moreover, the relevance of effective diffusivities should be seen in relation to the mean density of surface defects,54−56 because these defects may hinder the atomic motion and inhibit the transition to quasi-random walk behavior.
In summary, we study, via AIMD and CMD simulations, the diffusion of Ag, Au, Cu, Pd, Pt, and Ru adatoms on monolayer graphene at 300 K, a temperature that is relevant for thin-film synthesis experiments. We find that, while Pt and Ru exhibit typical features of a random walk, Ag, Au, Cu, and Pd move continuously across the surface unhindered by adsorption sites, and follow a superdiffusive motion pattern known as Lévy walk. This motion is characterized by a wide distribution of jump lengths, often on the order of several nanometers. We also establish that the type of motion undergone by the adatoms is correlated with the PEL they experience on the SLG surface: diffusion resembles a random walk when the energy differences between adsorption sites are of the order of hundreds of millielectronvolts and turns into superdiffusive when these differences become of the order of 10 meV. The overall results of our work highlight that knowledge of static (i.e., 0 K) surface migration barriers that are readily available6−16 and govern the kinetics of random walks between adjacent adatom sites is not sufficient to describe the complex surface diffusion dynamics of adatoms on 2D-materials that exhibit flat PELs. Instead, modeling approaches that take into account the superdiffusive nature of adatom motion are required for guiding knowledge-based synthesis of metal-layers nanostructures with controlled morphology35,57−59 in nanoelectronic, optoelectronic, and catalytic devices based on weakly interacting substrates.36−39
Computational methods
We use AIMD simulations to study atomistic diffusion mechanisms with density functional theory (DFT) accuracy. All AIMD simulations are performed using the Vienna ab initio simulation package (VASP).60 Core electrons are replaced by the projector augmented wave (PAW) pseudopotentials and the generalized gradient approximation of Perdew, Burke, and Ernzerhof (PBE).61 We use an energy cutoff of 400 eV for the plane waves and a 3 × 3 × 1 Γ-centered k-point mesh for the Brillouin zone integration. The monolayer graphene substrate is implemented as a hexagonal supercell containing 72 carbon atoms, between two vacuum layers of 10 Å thickness, and periodic boundary conditions are established in all three directions. We model the van der Waals forces using the DFT-D3 method with Becke–Johnson (BJ) damping,62 which accurately describes the properties of both graphite and diamond.63 All simulations are spin-polarized. Initial test simulations showed that Ru and Pt exhibit diffusion patterns that are encountered in classical homoepitaxial growth systems, while Ag, Au, Cu, and Pd diffuse considerably faster and execute jumps across multiple adsorption sites. Hence, we focus on Ag, Au, Pd, and Cu adatoms, the diffusion of which is simulated for 0.2 ns, while reference simulations are performed for Pt and Ru adatoms for times of 0.02 and 0.04 ns, respectively.
We calculate the PEL at 0 K by computing the ground-state energies Eads of the monomers on the hollow, top, and bridge sites using standard DFT static structure relaxations, and the PEL at room temperature, by averaging Eads over the configurations of the AIMD simulations for which the monomer is found on each of the main adsorption sites.
AIMD runs are complemented by CMD simulations of Ag adatom diffusion on a graphene sheet of 680 atoms. These simulations are performed using LAMMPS.64 The C–C and Ag–C interactions are described using AIREBO65 and Lennard-Jones66 potentials, respectively. The AIREBO potential is well-established in the literature for simulating carbon-based materials67−70 and, together with Lennard-Jones, is used to study the diffusion of noble metals (including Ag) clusters on graphite surfaces.71 More information about CMD validation can be found in the Supporting Information. We run a long simulation of 30 ns and 30 statistically independent simulations of 3 ns, both with a time step of 0.25 fs. We use canonical sampling (Nosé–Hoover thermostat) of the configurational space for all MD simulations. AIMD and CMD outputs are visualized using the Ovito freeware.72
Acknowledgments
V.G. acknowledges Mr. Davide Gambino (Linköping University) for useful discussions regarding spin polarization. V.G., M.Z., and K.S. acknowledge Linköping University (“LiU Career Contract, Dnr-LiU-2015-01510, 2015-2020”), the Swedish research council (contract VR-2015-04630) and the ÅForsk foundation (contract ÅF 19-137) for financial support. V.G., M.Z., D.S., and K.S. acknowledge the Olle Engkvist foundation (contract SOEB 190-312) for financial support. Simulations and data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC) partially funded by the Swedish Research Council through Grant Agreement No. VR-2015-04630.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c02375.
Comparison of 0.02 ns long AIMD trajectories (S1); calculation of scaling exponent α (S2); high-accuracy calculation of static (0 K) adsorption energies (S3); entropy effects on diffusion dynamics (S4); potential energy landscape using classical interatomic potentials and CMD simulation analysis (S5) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Allain A.; Kang J.; Banerjee K.; Kis A. Electrical Contacts to Two-Dimensional Semiconductors. Nat. Mater. 2015, 14, 1195–1205. 10.1038/nmat4452. [DOI] [PubMed] [Google Scholar]
- Schulman D. S.; Arnold A. J.; Das S. Contact Engineering for 2D Materials and Devices. Chem. Soc. Rev. 2018, 47, 3037–3058. 10.1039/C7CS00828G. [DOI] [PubMed] [Google Scholar]
- Petrov I.; Barna P. B.; Hultman L.; Greene J. E. Microstructural Evolution During Film Growth. J. Vac. Sci. Technol., A 2003, 21, S117–S128. 10.1116/1.1601610. [DOI] [Google Scholar]
- Sarakinos K. A Review on Morphological Evolution of Thin Metal Films on Weakly-Interacting Substrates. Thin Solid Films 2019, 688, 137312. 10.1016/j.tsf.2019.05.031. [DOI] [Google Scholar]
- Antczak G.; Ehrlich G.. Surface Diffusion: Metals, Metal Atoms, and Clusters; Cambridge University Press: Cambridge, U.K., 2010. [Google Scholar]
- Tang Y.; Zhang H.; Shen Z.; Zhao M.; Li Y.; Dai X. The Electronic and Diffusion Properties of Metal Adatoms on Graphene Sheets: a First-Principles Study. RSC Adv. 2017, 7, 33208–33218. 10.1039/C7RA04519K. [DOI] [Google Scholar]
- Amft M.; Lebegue S.; Eriksson O.; Skorodumova N. Adsorption of Cu, Ag, and Au Atoms on Graphene Including van der Waals Interactions. J. Phys.: Condens. Matter 2011, 23, 395001. 10.1088/0953-8984/23/39/395001. [DOI] [PubMed] [Google Scholar]
- Hupalo M.; Liu X.; Wang C.-Z.; Lu W.-C.; Yao Y.-X.; Ho K.-M.; Tringides M. C. Metal Nanostructure Formation on Graphene: Weak versus Strong Bonding. Adv. Mater. 2011, 23, 2082–2087. 10.1002/adma.201100412. [DOI] [PubMed] [Google Scholar]
- Appy D. V.; Lei H.; Wang C. Z.; Tringides M. C.; Liu D.-J.; Evans J. W.; Thiel P. A. Transition Metals on the (0001) Surface of Graphite: Fundamental Aspects of Adsorption, Diffusion, and Morphology. Prog. Surf. Sci. 2014, 89, 219–238. 10.1016/j.progsurf.2014.08.001. [DOI] [Google Scholar]
- Manadé M.; Viñes F.; Illas F. Transition Metal Adatoms on Graphene: a Systematic Density Functional Study. Carbon 2015, 95, 525–534. 10.1016/j.carbon.2015.08.072. [DOI] [PubMed] [Google Scholar]
- Han Y.; Evans J. Adsorption and Diffusion of Ru Adatoms on Ru(0001)-supported Graphene: Large-Scale First-Principles Calculations. J. Chem. Phys. 2015, 143, 164706. 10.1063/1.4934349. [DOI] [PubMed] [Google Scholar]
- Sun X.; Wang Z.; Fu Y. Q. Adsorption and Diffusion of Sodium on Graphene with Grain Boundaries. Carbon 2017, 116, 415–421. 10.1016/j.carbon.2017.01.024. [DOI] [Google Scholar]
- Hasegawa S.; Kunisada Y.; Sakaguchi N. Diffusion of a Single Platinum Atom on Light-Element-Doped Graphene. J. Phys. Chem. C 2017, 121, 17787–17795. 10.1021/acs.jpcc.7b01241. [DOI] [Google Scholar]
- Tang Y.; Chen W.; Li C.; Li W.; Dai X.-Q. Geometric Stability, Electronic Structure, and Intercalation Mechanism of Co Adatom Anchors on Graphene Sheets. J. Phys.: Condens. Matter 2015, 27, 255009. 10.1088/0953-8984/27/25/255009. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wang C. Z.; Hupalo M.; Yao Y. X.; Tringides M. C.; Lu W. C.; Ho K. M. Adsorption and Growth Morphology of Rare-Earth Metals on Graphene Studied by Ab Initio Calculations and Scanning Tunneling Microscopy. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 245408. 10.1103/PhysRevB.82.245408. [DOI] [Google Scholar]
- Suarez-Martinez I.; Felten A.; Pireaux J. J.; Bittencourt C.; Ewels C. P. Transition Metal Deposition on Graphene and Carbon Nanotubes. J. Nanosci. Nanotechnol. 2009, 9, 6171–6175. 10.1166/jnn.2009.1557. [DOI] [PubMed] [Google Scholar]
- Eyring H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. 10.1063/1.1749604. [DOI] [Google Scholar]
- Vineyard G. H. Frequency Factors and Isotope Effects in Solid State Rate Processes. J. Phys. Chem. Solids 1957, 3, 121–127. 10.1016/0022-3697(57)90059-8. [DOI] [Google Scholar]
- Michely T.; Krug J.. Islands, Mounds and Atoms; Springer-Verlag: Berlin, 2004. [Google Scholar]
- Chen L. Y.; Ying S. C. Dynamics of Adatoms on Solid Surfaces. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 49, 13838–13847. 10.1103/PhysRevB.49.13838. [DOI] [PubMed] [Google Scholar]
- Ferrón J.; Miranda R.; de Miguel J. J. Atomic Jumps During Surface Diffusion. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 79, 245407. 10.1103/PhysRevB.79.245407. [DOI] [Google Scholar]
- Antczak G.; Ehrlich G. Long Jumps in Diffusion of Iridium on W(110). Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71, 115422. 10.1103/PhysRevB.71.115422. [DOI] [Google Scholar]
- Senft D. C.; Ehrlich G. Long Jumps in Surface Diffusion: One-Dimensional Migration of Isolated Adatoms. Phys. Rev. Lett. 1995, 74, 294–297. 10.1103/PhysRevLett.74.294. [DOI] [PubMed] [Google Scholar]
- Sangiovanni D.; Edström D.; Hultman L.; Petrov I.; Greene J.; Chirita V. Ti Adatom Diffusion on TiN(001): Ab Initio and Classical Molecular Dynamics Simulations. Surf. Sci. 2014, 627, 34–41. 10.1016/j.susc.2014.04.007. [DOI] [Google Scholar]
- Sangiovanni D. Copper Adatom, Admolecule Transport, and Island Nucleation on TiN(001) via Ab Initio Molecular Dynamics. Appl. Surf. Sci. 2018, 450, 180–189. 10.1016/j.apsusc.2018.04.191. [DOI] [Google Scholar]
- Kürpick U.; Kara A.; Rahman T. S. Role of Lattice Vibrations in Adatom Diffusion Ulrike. Phys. Rev. Lett. 1997, 78, 1086–1089. 10.1103/PhysRevLett.78.1086. [DOI] [Google Scholar]
- Boisvert G.; Mousseau N.; Lewis L. J. Comment on “Role of Lattice Vibrations in Adatom Diffusion. Phys. Rev. Lett. 1998, 80, 203–203. 10.1103/PhysRevLett.80.203. [DOI] [Google Scholar]
- Kürpick U.; Kara A.; Rahman T. S.; et al. Kürpick et al. Reply. Phys. Rev. Lett. 1998, 80, 204–204. 10.1103/PhysRevLett.80.204. [DOI] [Google Scholar]
- Mei A. B.; Hellman O.; Wireklint N.; Schlepütz C. M.; Sangiovanni D. G.; Alling B.; Rockett A.; Hultman L.; Petrov I.; Greene J. E. Dynamic and Structural Stability of Cubic Vanadium Nitride. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 91, 054101 10.1103/PhysRevB.91.054101. [DOI] [Google Scholar]
- Sangiovanni D. G.; Mei A. B.; Edström D.; Hultman L.; Chirita V.; Petrov I.; Greene J. E. Effects of Surface Vibrations on Interlayer Mass Transport: Ab Initio Molecular Dynamics Investigation of Ti Adatom Descent Pathways and Rates from TiN/TiN(001) Islands. Phys. Rev. B: Condens. Matter Mater. Phys. 2018, 97, 035406 10.1103/PhysRevB.97.035406. [DOI] [Google Scholar]
- Smirnova D.; Starikov S.; Leines G. D.; Liang Y.; Wang N.; Popov M. N.; Abrikosov I. A.; Sangiovanni D. G.; Drautz R.; Mrovec M. Atomistic Description of Self-Diffusion in Molybdenum: A Comparative Theoretical Study of Non-Arrhenius Behavior. Phys. Rev. Mater. 2020, 4, 013605 10.1103/PhysRevMaterials.4.013605. [DOI] [Google Scholar]
- Iacopi F.; Boeckl J. J.; Jagadish C.. 2D Materials; Elsevier, 2016; Chapter 1, pp 1–33. [Google Scholar]
- Noori K.; Cheng N.; Xuan F.; Quek S. Dielectric Screening by 2D Substrates. 2D Mater. 2019, 6, 035036 10.1088/2053-1583/ab1e06. [DOI] [Google Scholar]
- Novoselov K. S.; Geim A. K.; Morozov S. V.; Jiang D.; Zhang Y.; Dubonos S. V.; Grigorieva I. V.; Firsov A. A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. 10.1126/science.1102896. [DOI] [PubMed] [Google Scholar]
- Liu X.; Han Y.; Evans J. W.; Engstfeld A. K.; Behm R. J.; Tringides M. C.; Hupalo M.; Lin H.-Q.; Huang L.; Ho K.-M.; et al. Growth Morphology and Properties of Metals on Graphene. Prog. Surf. Sci. 2015, 90, 397–443. 10.1016/j.progsurf.2015.07.001. [DOI] [Google Scholar]
- Lee J.-H.; Choi H. K.; Yang L.; Chueng S.-T. D.; Choi J.-W.; Lee K.-B. Non-Destructive Real-Time Monitoring of Enhanced Stem Cell Differentiation using a Graphene-Au Hybrid Nanoelectrode Array. Adv. Mater. 2018, 30, 1802762. 10.1002/adma.201802762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuwen L.; Xu F.; Xue B.; Luo Z.; Zhang Q.; Bao B.; Su S.; Weng L.; Huang W.; Wang L. General Synthesis of Noble Metal (Au, Ag, Pd, Pt) Nanocrystal Modified MoS2 Nanosheets and the Enhanced Catalytic Activity of PdMoS2 for Methanol Oxidation. Nanoscale 2014, 6, 5762–5769. 10.1039/C3NR06084E. [DOI] [PubMed] [Google Scholar]
- Tan C.; Huang X.; Zhang H. Synthesis and Applications of Graphene-Based Noble Metal Nanostructures. Mater. Today 2013, 16, 29–36. 10.1016/j.mattod.2013.01.021. [DOI] [Google Scholar]
- Chae S. S.; Jang S.; Lee W.; Jung D. W.; Lee K. H.; Kim J. D.; Jeong D.; Chang H.; Hwang J. Y.; Lee J.-O. Ultrathin Metal Crystals: Growth on Supported Graphene Surfaces and Applications. Small 2018, 14, 1801529. 10.1002/smll.201801529. [DOI] [PubMed] [Google Scholar]
- Saxton M. J. Single-Particle Tracking: The Distribution of Diffusion Coefficients. Biophys. J. 1997, 72, 1744–1753. 10.1016/S0006-3495(97)78820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlesinger M.; Zaslavsky G.; Klafter J. Strange Kinetics. Nature 1993, 363, 31–37. 10.1038/363031a0. [DOI] [Google Scholar]
- Metzler R.; Jeon J.-H.; Cherstvy A. G.; Barkai E. Anomalous Diffusion Models and their Properties: Non-Stationarity, Non-Ergodicity, and Ageing at the Centenary of Single Particle Tracking. Phys. Chem. Chem. Phys. 2014, 16, 24128–24164. 10.1039/C4CP03465A. [DOI] [PubMed] [Google Scholar]
- Kepten E.; Weron A.; Sikora G.; Burnecki K.; Garini Y. Guidelines for the Fitting of Anomalous Diffusion Mean Square Displacement Graphs from Single Particle Tracking Experiments. PLoS One 2015, 10, e0117722. 10.1371/journal.pone.0117722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lii-Rosales A.; Han Y.; Jing D.; Tringides M. C.; Thiel P. A. Search for Encapsulation of Platinum, Silver, and Gold at the Surface of Graphite. Phys. Rev. Research 2020, 2, 033175 10.1103/PhysRevResearch.2.033175. [DOI] [Google Scholar]
- Lii-Rosales A.; Han Y.; Yu K. M.; Jing D.; Anderson N.; Vaknin D.; Tringides M. C.; Evans J. W.; Altman M. S.; Thiel P. A. Reverse-Engineering of Graphene on Metal Surfaces: a Case Study of Embedded Ruthenium. Nanotechnology 2018, 29, 505601. 10.1088/1361-6528/aae1e3. [DOI] [PubMed] [Google Scholar]
- Zakharchenko K.; Katsnelson M.; Fasolino A. Finite Temperature Lattice Properties of Graphene beyond the Quasiharmonic Approximation. Phys. Rev. Lett. 2009, 102, 046808 10.1103/PhysRevLett.102.046808. [DOI] [PubMed] [Google Scholar]
- Frenkel D.; Smit B.. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: San Diego, CA, 1996. [Google Scholar]
- Zaburdaev V.; Denisov S.; Klafter J. Lévy Walks. Rev. Mod. Phys. 2015, 87, 483–530. 10.1103/RevModPhys.87.483. [DOI] [Google Scholar]
- Barthelemy P.; Bertolotti J.; Wiersma D. A Lévy Flight for Light. Nature 2008, 453, 495–498. 10.1038/nature06948. [DOI] [PubMed] [Google Scholar]
- Humphries N.; et al. Environmental Context Explains Levy and Brownian Movement Patterns of Marine Predators. Nature 2010, 465, 1066–1069. 10.1038/nature09116. [DOI] [PubMed] [Google Scholar]
- Brockmann D.; Hufnagel L.; Geisel T. The Scaling Laws of Human Travel. Nature 2006, 439, 462–465. 10.1038/nature04292. [DOI] [PubMed] [Google Scholar]
- Maruyama Y.; Murakami J. Truncated Lévy Walk of a Nanocluster Bound Weakly to an Atomically Flat Surface: Crossover from Superdiffusion to Normal Diffusion. Phys. Rev. B: Condens. Matter Mater. Phys. 2003, 67, 085406 10.1103/PhysRevB.67.085406. [DOI] [Google Scholar]
- Sancho J. M.; Lacasta A. M.; Lindenberg K.; Sokolov I. M.; Romero A. H. Diffusion on a Solid Surface: Anomalous is Normal. Phys. Rev. Lett. 2004, 92, 250601. 10.1103/PhysRevLett.92.250601. [DOI] [PubMed] [Google Scholar]
- Cançado L. G.; Jorio A.; Ferreira E. H. M.; Stavale F.; Achete C. A.; Capaz R. B.; Moutinho M. V. O.; Lombardo A.; Kulmala T. S.; Ferrari A. C. Quantifying Defects in Graphene via Raman Spectroscopy at Different Excitation Energies. Nano Lett. 2011, 11, 3190–3196. 10.1021/nl201432g. [DOI] [PubMed] [Google Scholar]
- Lucchese M.; Stavale F.; Ferreira E. M.; Vilani C.; Moutinho M.; Capaz R. B.; Achete C.; Jorio A. Quantifying Ion-Induced Defects and Raman Relaxation Length in Graphene. Carbon 2010, 48, 1592–1597. 10.1016/j.carbon.2009.12.057. [DOI] [Google Scholar]
- Dresselhaus M. S.; Jorio A.; Souza Filho A. G.; Saito R. Defect Characterization in Graphene and Carbon Nanotubes using Raman Spectroscopy. Philos. Trans. R. Soc., A 2010, 368, 5355–5377. 10.1098/rsta.2010.0213. [DOI] [PubMed] [Google Scholar]
- Lü B.; Almyras G. A.; Gervilla V.; Greene J. E.; Sarakinos K. Formation and Morphological Evolution of Self-Similar 3D Nanostructures on Weakly Interacting Substrates. Phys. Rev. Mater. 2018, 2, 063401 10.1103/PhysRevMaterials.2.063401. [DOI] [Google Scholar]
- Gervilla V.; Almyras G. A.; Thunström F.; Greene J. E.; Sarakinos K. Dynamics of 3D-Island Growth on Weakly-Interacting Substrates. Appl. Surf. Sci. 2019, 488, 383–390. 10.1016/j.apsusc.2019.05.208. [DOI] [Google Scholar]
- Gervilla V.; Almyras G. A.; Lü B.; Sarakinos K. Coalescence Dynamics of 3D Islands on Weakly-Interacting Substrates. Sci. Rep. 2020, 10, 2031. 10.1038/s41598-020-58712-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresse J.; Furthmüller J. Efficiency of Ab Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [Google Scholar]
- Perdew J.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Mosyagin I.; Gambino D.; Sangiovanni D.; Abrikosov I.; Caffrey N. Effect of Dispersion Corrections on Ab Initio Predictions of Graphite and Diamond Properties Under Pressure. Phys. Rev. B: Condens. Matter Mater. Phys. 2018, 98, 174103. 10.1103/PhysRevB.98.174103. [DOI] [Google Scholar]
- Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- Stuart S. J.; Tutein A. B.; Harrison J. A. A Reactive Potential for Hydrocarbons with Intermolecular Interactions. J. Chem. Phys. 2000, 112, 6472–6486. 10.1063/1.481208. [DOI] [Google Scholar]
- Neek-Amal M.; Asgari R.; Tabar M. R. R. The Formation of Atomic Nanoclusters on Graphene Sheets. Nanotechnology 2009, 20, 135602. 10.1088/0957-4484/20/13/135602. [DOI] [PubMed] [Google Scholar]
- Varshney V.; Patnaik S. S.; Roy A. K.; Froudakis G.; Farmer B. L. Modeling of Thermal Transport in Pillared-Graphene Architectures. ACS Nano 2010, 4, 1153–1161. 10.1021/nn901341r. [DOI] [PubMed] [Google Scholar]
- Safina L.; Baimova J.; Mulyukov R. Nickel Nanoparticles inside Carbon Nanostructures: Atomistic Simulation. Mech. Adv. Mater. Mod. Process 2019, 5, 2. 10.1186/s40759-019-0042-3. [DOI] [Google Scholar]
- Koukaras E.; Kalosakas G.; Galiotis C.; Papagelis K. Phonon Properties of Graphene Derived from Molecular Dynamics Simulations. Sci. Rep. 2015, 5, 12923. 10.1038/srep12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Reddy C. D.; Jiang J.; Baimova J. A.; Dmitriev S. V.; Nazarov A. A.; Zhou K. Morphology and In-Plane Thermal Conductivity of Hybrid Graphene Sheets. Appl. Phys. Lett. 2012, 101, 211909. 10.1063/1.4767388. [DOI] [Google Scholar]
- Yousefi P.; Abbaspour M.; Sokhanvaran V. A Comparative Study of Graphite and CNT Supported Au-Ag, Au-Pd, Au-Pt and Au-Rh Nanoalloys using MD Simulation. J. Mol. Liq. 2019, 280, 87–96. 10.1016/j.molliq.2019.02.045. [DOI] [Google Scholar]
- Stukowski A. Visualization and Analysis of Atomistic Simulation Data with OVITO the Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2010, 18, 015012 10.1088/0965-0393/18/1/015012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.