

 A novel compound FIPM was developed and labeled with fluorine-18 in vivo visualization of LRRK2 in the brain.
A novel compound FIPM was developed and labeled with fluorine-18 in vivo visualization of LRRK2 in the brain.
Abstract
Mutations that increase leucine-rich repeat kinase 2 (LRRK2) activity in the brain are associated with Parkinson's disease. Here, we synthesized a novel compound 4-(6-fluoro-4-(5-isopropoxy-1H-indazol-3-yl)pyridin-2-yl)morpholine (FIPM) and labeled it with fluorine-18 (18F), to develop a positron emission tomography (PET) tracer for in vivo visualization of LRRK2 in the brain. FIPM showed high in vitro binding affinity for LRRK2 (IC50 = 8.0 nM). [18F]FIPM was prepared in 5% radiochemical yield (n = 5), by inserting 18F into a pyridine ring, followed by removal of the protecting group. After HPLC separation and formulation, [18F]FIPM was acquired with >97% radiochemical purity and 103–300 GBq μmol–1 of molar activity at the end of radiosynthesis. Biodistribution and small-animal PET studies in mice indicated a low in vivo specific binding of [18F]FIPM. While [18F]FIPM presented limited potential as an in vivo PET tracer for LRRK2, we suggested that it can be used as a lead compound for developing new radiotracers with improved in vivo brain properties.
Introduction
Parkinson's disease (PD) is a neurodegenerative disease that is caused by the neuronal cell death in the substantia nigra of the midbrain and affects approximately 2% of the population at 60 years of age.1–3 The symptoms of PD include impaired motor function, tremor, rigidity, impaired balance, and impaired speech. The pathological hallmarks for PD include reduction of dopaminergic neurons and dopamine secretion in the substantia nigra of the brain and the generation of Lewy bodies and intracellular fibrils in the neurons.4 Although efficient therapies are now available, the availability of disease-modifying therapies remains a major challenge for PD.
Mutations that activate leucine-rich repeat kinase 2 (LRRK2) are associated with PD.5 LRRK2 is an enzyme that is encoded by the PARK8 gene in humans and the variants of this gene are associated with increased risk of PD.6–9 Mutations in the LRRK2 gene upregulate the activity of LRRK2 protein and are a main factor for the genetic development of PD. Therefore, LRRK2 is a promising drug target for PD.10–14
Positron emission tomography (PET) imaging using a LRRK2-specific radiotracer can provide a non-invasive modality both for examining the pharmacological roles of LRRK2 in PD and determining target engagement of new LRRK2 inhibitors. The development of a radiotracer for PET imaging of LRRK2 in the brain has gained interest recently. To the best of our knowledge, no useful PET radiotracer has been developed for the clinical visualization of LRRK2 in the human brain yet. Ding et al. have synthesized an LRRK2-specific radiotracer [3H]LRRK-IN-1 (Fig. 1) for in vitro and in vivo evaluations in human brain tissues and rodents.15 Although [3H]LRRK-IN-1 showed low in vivo specific binding to LRRK2, its high density measured in the brain indicated the feasibility of using PET imaging for LRRK2 along with a radiotracer exhibiting higher affinity and specificity. In addition, a patent16 has been reported on the availability of 11C- and 18F-labeled radiotracers specific for LRRK2; however, detailed results of their evaluations have not been reported. Zheng et al. have synthesized [11C]HG-10-102-01 (ref. 17) (Fig. 1), an analog of the patent compounds; however, they did not demonstrate the imaging potentials of the radiotracer. Liang et al. have reported the evaluation results of [11C]GNE-1023 (ref. 18) (Fig. 1). Although this tracer had a high in vitro specific binding for LRRK2, its brain uptake was low. In addition, several radiolabeled analogs19–23 have been developed from [3H]LRRK-IN-1 and PF-06447475 (ref. 24) (Fig. 1), a potent inhibitor for LRRK2 in various pharmacological tests. However, the detailed results of potentials as PET tracers have not been indicated (Scheme 1).
Fig. 1. Chemical structures of reported inhibitors and radiotracers specific for LRRK2.

Scheme 1. Chemical synthesis of targeted compound FIPM and bromo precursor 1. a) 2-Iodopropane, Cs2CO3, DMF, room temperature (rt), 26 h; b) I2, K2CO3, CH3CN, rt, 4 h; c) trityl chloride, NaH, THF, 0 °C 0.5 h to rt, 25 h; d) (Bpin)2, [(COD)Ir(μ-OMe)]2, dtbpy, THF, 60 °C, 7 h; e) 5, K3PO4·nH2O, Pd(dppf)Cl2·CH2Cl2, DME, reflux, 3.5 h; f) TFA, H2O, CH2Cl2, rt, 6.5 h; g) 5, K2CO3, Pd(PPh3)4, 1,4-dioxane, H2O, 100 °C, 24 h; h) morpholine, Cs2CO3, XantPhos, Pd(OAc)2, dioxane, 100 °C, 4 h.

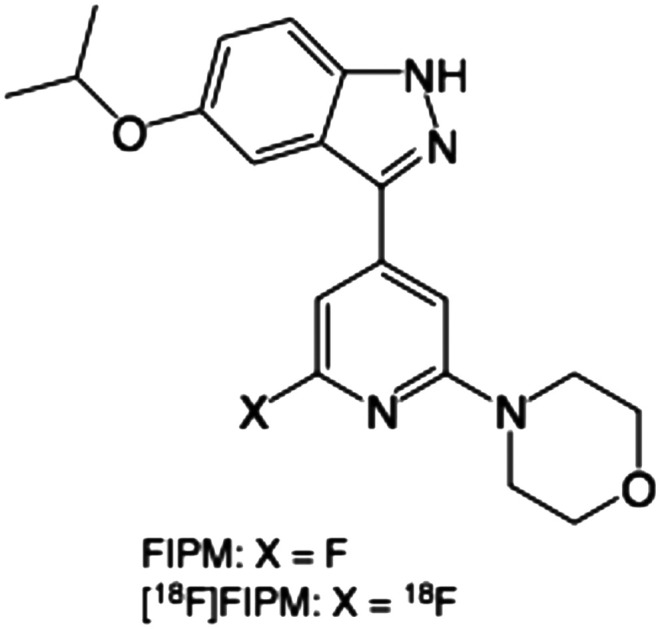
Recently, researchers from Merck have reported 3-(4-pyrimidinyl)indazole analogs as potent inhibitors of LRRK2.25 The optimization for these compounds led to the discovery of MLi-2 (Fig. 1), a highly selective, orally available and brain-penetrant inhibitor of LRRK2.25,26 Due to the favorable pharmacological and physiochemical characteristics of MLi-2, we hypothesized that this scaffold may enable the development of a useful PET tracer to target LRRK2. In this study, we developed a new compound, 4-(6-fluoro-4-(5-isopropoxy-1H-indazol-3-yl)pyridin-2-yl)morpholine (FIPM, Fig. 1), using MLi-2 as a lead compound, with the expectation that FIPM would show high binding affinity for LRRK2 and serve as a promising candidate of PET tracer. We synthesized FIPM and performed an in vitro binding assay for LRRK2. Thereafter, FIPM was labeled with 18F to generate 4-(6-[18F]fluoro-4-(5-isopropoxy-1H-indazol-3-yl)pyridin-2-yl)morpholine ([18F]FIPM, Fig. 1) as a novel radiotracer for LRRK2.
Results and discussion
Chemistry
Two novel compounds FIPM and the bromo precursor 1 for radiolabeling were synthesized following the sequences of reactions shown in Fig. 2.
Fig. 2. Separation (A) and analytic (B) HPLC chromatograms of [18F]FIPM.

The reaction of 5-hydroxyindazole (2) with 2-iodopropane in the presence of Cs2CO3 produced 3, which was reacted with iodine to generate compound 4. The treatment of 4 with trityl chloride and sodium hydride resulted in trityl protection of the N2 position of the indazole to generate 5.25 The coupling of 5 with 6-fluoro-4-(4-(4,4,5,4-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)morpholine (7), which was synthesized by the reaction of fluoropyridine (6) with boron ester, produced 8 at a 27% yield. The removal of the trityl group in 8 with trifluoroacetic acid produced FIPM at a 74% chemical yield. Conversely, the coupling of 6-bromo-4-(4-(4,4,5,4-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)morpholine (9) with 5 in the presence of Pd(dppf)Cl2 produced dibromopyridine (10) at a moderate yield of 38%. Furthermore, 10 was reacted with morphorine to produce bromo precursor 1 at a 22% yield.
In vitro binding affinity and lipophilicity
The in vitro binding assay using FIPM for LRRK2 was performed according to a standard protocol.26 The binding affinity (IC50) was measured using different concentrations of FIPM to inhibit activity of G2019S-mutated LRRK2. FIPM exhibited potent binding affinity (IC50) for LRRK2, which was determined to be 8.0 nM for LRRK2. For comparison, the in vitro binding affinity of PF-06447475, the mostly used LRRK2 inhibitor, was also measured under identical experimental conditions. PF-06447475 showed similar binding affinity (IC50: 10. 2 nM) for LRRK2 as FIPM.
The value of lipophilicity (log D) was measured by the shake-flask method. The log D value of [18F]FIPM following radiolabeling with 18F was 1.16 (n = 1).
Radiosynthesis
Owing to the robust binding affinity of FIPM for LRRK2, we labeled FIPM with 18F to synthesize [18F]FIPM. Prior to the radiosynthesis of [18F]FIPM using an automated synthesis system, we determined the two-step reaction conditions of [18F]fluorination and deprotection (Scheme 2). The [18F]fluorination of 1 with [18F]F– was attempted only using K2CO3/Kryptofix222, which is a typical combination for the routine production of 18F-radiopharmaceuticals.27,28 The reaction mixture that was heated at 150 °C for 20 min produced [18F]8 with low and poorly-reproducible [18F]fluorinating efficiency. Subsequently, tetrabutylammonium mesylate (TBAOMs) was used instead of K2CO3/Kryptofix222, based on our recent experiences for introducing nucleophilic 18F into the benzene ring.29,30 However, unexpectedly, the [18F]fluorination efficiency of 1 with [18F]TBAF was not improved. Conversely, several research groups have used organic bases to increase the [18F]fluorination efficiency of bromopyridine with [18F]F– in the presence of K2CO3/Kryptofix222, which generates the quaternary ammonium cation as a leaving group.31,32 Here, two organic bases: 1,4-diazabicyclo[2,2,2]octane (DABCO) and quinuclidine, were added to the [18F]fluorination reaction mixture of the bromo precursor 1 with [18F]F–, respectively. While the use of quinuclidine did not increase the [18F]fluorination yield of 1, the use of DABCO improved the reaction efficiency and produced a [18F]fluorinated yield of 32%, determined using HPLC based on the total used [18F]F–. Therefore, the [18F]fluorination reaction conditions were fixed as shown in Scheme 2.
Scheme 2. Radiosynthesis of [18F]FIPM. a) [18F]F–/Kryptofix222 and K2CO3, DABCO, DMSO, 150 °C, 20 min; b) 6 mol L–1 HCl, 110 °C, 20 min.

Trifluoroacetic acid and hydrochloric acid were used to remove the trityl group from [18F]8. Both acids achieved complete deprotection of the trityl group in [18F]8 to yield [18F]FIPM. We selected 6 mol L–1 HCl for deprotection of the trityl group to reduce the corrosion risk of the synthesis system.
We performed the radiosynthesis of [18F]FIPM using a home-made automated synthesis system.33 [18F]FIPM was produced as an injectable solution in averaged 5% radiochemical yield (n = 5, based on [18F]F–) after the [18F]fluorination, deprotection, HPLC separation (Fig. 2A), and formulation. Starting from 7.4 GBq [18F]F–, 0.1–0.3 GBq [18F]FIPM was produced within 100 min (n = 5) of averaged synthesis time from the end of bombardment. The identity of [18F]FIPM was confirmed in the final product solution by co-injection with unlabeled FIPM onto the analytical HPLC column. The radiochemical purity of [18F]FIPM was greater than 97% (Fig. 2B) and the molar activity was 103–300 GBq μmol–1 at the end of synthesis. No significant UV peak corresponding to the unreacted bromo precursor 1 was observed on the analytical HPLC chart for the final product (Fig. 2B). Moreover, [18F]FIPM did not undergo radiolysis at room temperature for 90 min after formulation. The analytical results complied with our in-house quality control/assurance specifications of radiopharmaceuticals.
Whole body biodistribution studies in mice
To evaluate pharmacokinetics of [18F]FIPM in LRRK2-rich organs (lung, liver, and brain), the biodistribution study using mice was performed at several experimental time points (5, 15, 30, 60 and 120 min) after intravenous injection of [18F]FIPM. The uptake value of radioactivity is expressed as the percentage of the injected dose per gram of wet tissue (% ID per g) (Table 1). At 5 min after the injection, high uptakes (>5% ID per g) were observed in the liver and small intestine, and moderate uptakes (>1% ID per g) were seen in the heart, lung, pancreas, spleen, adrenals, large intestine, muscle, bone. After that, the radioactive accumulation in the blood, heart, pancreas, spleen, kidney and muscle rapidly decreased. The high level of radioactivity in the small intestine and liver was retained up to 60 min after the injection. The pharmacokinetic results suggest that hepatobiliary and intestinal reuptake pathway mainly contributed to the whole body distribution of radioactivity. In the mouse brains, the most interesting organ, the radioactivity uptake of [18F]FIPM was 1.22% ID per g at 5 min. This result indicates that a certain amount of [18F]FIPM could rapidly pass through the blood–brain-barrier (BBB) and enter into the brain, despite low lipophilicity. However, its brain uptake was not as high as we expected. The ratio of radioactivity between the brain and blood did not increase with time after the injection, indicating the in vivo specific binding in the brain was limited.
Table 1. Biodistribution (% ID per g) of [18F]FIPM in mice (means ± SD, n = 3 in each group).
| Organs | Time after injection (min) |
|||||||||
| 5 |
15 |
30 |
60 |
120 |
||||||
| Blood | 0.94 | ±0.04 | 0.52 | ±0.01 | 0.29 | ±0.04 | 0.18 | ±0.02 | 0.12 | ±0.02 |
| Heart | 2.12 | ±0.28 | 0.79 | ±0.01 | 0.42 | ±0.05 | 0.18 | ±0.01 | 0.08 | ±0.01 |
| Lung | 3.48 | ±0.54 | 2.17 | ±0.17 | 1.73 | ±0.44 | 1.42 | ±0.43 | 1.01 | ±0.07 |
| Liver | 14.66 | ±1.07 | 17.39 | ±1.74 | 19.08 | ±1.58 | 16.44 | ±2.17 | 10.55 | ±1.51 |
| Pancreas | 4.01 | ±0.34 | 1.37 | ±0.07 | 0.59 | ±0.08 | 0.33 | ±0.10 | 0.10 | ±0.01 |
| Spleen | 1.45 | ±0.06 | 0.63 | ±0.05 | 0.32 | ±0.00 | 0.22 | ±0.09 | 0.08 | ±0.01 |
| Kidney | 4.57 | ±0.99 | 1.73 | ±0.26 | 1.15 | ±0.21 | 0.68 | ±0.20 | 0.30 | ±0.07 |
| Adrenals | 3.93 | ±0.66 | 2.30 | ±0.16 | 1.81 | ±0.27 | 1.02 | ±0.14 | 0.34 | ±0.10 |
| Stomach | 0.67 | ±0.30 | 0.42 | ±0.27 | 0.67 | ±0.18 | 0.40 | ±0.19 | 1.21 | ±0.95 |
| Small intestine | 8.09 | ±0.59 | 10.56 | ±5.90 | 5.15 | ±1.72 | 5.80 | ±0.84 | 4.61 | ±1.73 |
| Large intestine | 1.03 | ±0.27 | 0.83 | ±0.40 | 0.37 | ±0.09 | 0.35 | ±0.09 | 0.27 | ±0.02 |
| Testes | 0.49 | ±0.04 | 0.40 | ±0.03 | 0.32 | ±0.02 | 0.19 | ±0.02 | 0.09 | ±0.01 |
| Muscle | 1.52 | ±0.22 | 0.69 | ±0.03 | 0.32 | ±0.05 | 0.15 | ±0.05 | 0.06 | ±0.02 |
| Brain | 1.22 | ±0.21 | 0.88 | ±0.36 | 0.26 | ±0.03 | 0.09 | ±0.01 | 0.04 | ±0.01 |
| Bone | 2.22 | ±0.19 | 2.51 | ±0.56 | 3.06 | ±0.29 | 3.43 | ±0.37 | 3.08 | ±0.43 |
PET studies in mice
To determine in vivo specific binding, dynamic PET scans were performed using [18F]FIPM in mice treated with or without unlabeled FIPM (1 mg kg–1). Fig. 3A and C show representative PET images summed between 0 and 60 min in whole body and time-activity curves (TACs) for representative organs after the injection in control mice. In the brain, a rapid entrance of radioactivity was observed peaked at 1.5 min with a standardized uptake value (SUV) of 0.82, which suggested that [18F]FIPM could enter into the brain through the BBB. However, the radioactivity in the brain rapidly decreased afterwards. Among TACs of the investigated organs and tissues, the uptake in the liver was the highest. No significant radioactivity was observed in the bone, which suggested that few defluorination of [18F]FIPM occurred in vivo.
Fig. 3. Small-animal PET imaging of [18F]FIPM in mice: (A) representative 0–60 min summed PET images for control mouse; (B) for self-blocking mouse (treatment with 1 mg kg–1 FIPM). Time-activity curves (n = 2, means ± range) for [18F]FIPM of mouse in the heart, lung, liver, kidney, brain, muscle, and bone: (C) for control; (D) for self-blocking; (E) area under the radioactivity curves from 30 to 60 min (AUC30–60 min) in the heart, lung, liver, and brain of control and self-blocking mice.

Fig. 3B and D show representative summed-PET images and TACs for several organs in self-blocking mice (pretreatment with unlabeled FIPM). Although there were no significant differences compared to the control, area under the radioactivity curve (AUC) in the liver (47.0 ± 10.9 SUV × min in the control) decreased with roughly 25% by self-blocking (36.8 ± 9.8 SUV × min in the self-blocking) (Fig. 3E). Because it was known that LRRK2 is highly expressed in the liver,34 we suggested that the reduction of uptake may reflect the presence of some in vivo specific binding of FIPM for LRRK2 in the liver.
Conclusions
We developed FIPM as a novel inhibitor of LRRK2. This compound showed high binding affinity for LRRK2, similar to that exhibited by the commonly used PF-06447475. Although [18F]FIPM is not considered ideal for PET tracer development, we performed radiochemistry and preliminary in vivo experiments that may provide insight into the structure–activity relationship study using FIPM as a lead compound. Searching for new candidates with improved in vivo properties in terms of brain penetration, higher binding affinity, and mutant selectivity will be essential in order to move these tracers for in vivo applications.
Experimental part
Materials and methods
Melting points were measured using a micro melting point apparatus (MP-500P, Yanaco, Tokyo, Japan) and were uncorrected. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded using a JEOL-AL-300 spectrometer (JEOL, Tokyo), with tetramethylsilane (TMS) as an internal standard. All chemical shifts (δ) are reported as ppm downfield relative to TMS signal. Signals are quoted as s (singlet), d (doublet), t (triplet), br (broad), or m (multiplet). High-resolution fast atom bombardment mass spectra (HRMS) were acquired using a JEOL NMS-SX102 102A spectrometer. Silica gel column chromatography was performed using Wakosil C-200 (FUJIFILM Wako Pure Chemical Industries, Osaka, Japan). All chemical reagents and solvents were purchased from commercial sources (Sigma-Aldrich, St. Louis, MO; FUJIFILM Wako Pure Chemical Industries, Osaka, Japan; Tokyo Chemical Industries, Tokyo) and used as supplied. For radio-HPLC separation and analysis, effluent radioactivity was monitored using a NaI (Tl) scintillation detector system. Unless otherwise stated, radioactivity was measured with an IGC-3R Curiemeter (Aloka, Tokyo).
Chemical synthesis
4-(6-Fluoro-4-(5-isopropoxy-1-trityl-1H-indazol-3-yl)pyridin-2-yl)morpholine (8)
A mixture of 4-(6-fluoropyridin-2-yl)morpholine25 (6; 114 mg, 0.62 mmol), bis(pinacolato)diboron (316 mg, 1.25 mmol), 2,6-di-tert-butylpyridine (dtbpy; 5 mg, 0.018 mmol) and (1,5-cyclooctadiene)(methoxy)iridium(i) dimer ([(COD)Ir(μ-OMe)]2; 19 mg, 0.029 mmol) in THF (2 mL) was heated at 60 °C for 7 h under N2. After cooling the reaction mixture was concentrated under vacuum to produce 4-(6-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)morpholine (7) for the subsequent reaction without further purification.
A mixture of 3-iodo-5-isopropoxy-1-trityl-1H-indazole25 (5; 246 mg, 0.45 mmol), 7 (164 mg, 0.53 mmol), K3PO4·nH2O (278 mg, 1.31 mmol) and Pd(dppf)Cl2·CH2Cl2 (33 mg, 0.04 mmol) in dimethoxyethane (5 mL) was heated at reflux for 3.5 h under N2. After cooling the reaction mixture was filtered through a pad of celite, and the filter cake was washed with ethyl acetate. The combined filtrate was concentrated under vacuum to generate a crude product, which was purified by silica gel chromatography to generate 8 (74 mg, 27%) as a white solid. Melting point (mp): 228–230 °C. 1H NMR (300 MHz, CDCl3) δ 7.29–7.20 (m, 16H), 6.94 (s, 1H), 6.72 (s, 1H), 6.67 (dd, J = 2.1, 11.4 Hz, 1H), 6.33 (d, J = 9.0 Hz, 1H), 4.57–4.49 (m, 1H), 3.81 (t, J = 4.5 Hz, 4H), 3.52 (t, J = 5.1 Hz, 4H), 1.35 (d, J = 5.7 Hz, 6H). HRMS (m/z): [M]+ calculated for C38H36FN4O2, 599.2822; found, 599.2823.
4-(6-Fluoro-4-(5-isopropoxy-1H-indazol-3-yl)pyridin-2-yl)morpholine (FIPM)
A solution of 8 (100 mg, 0.17 mmol) in CH2Cl2 (980 μL) was added to trifluoroacetic acid (980 μL) and H2O (588 μL). The reaction mixture was stirred at room temperature for 6.5 h. After the removal of solvents, the residue was extracted with CH2Cl2 and the organic layer was washed with saturated aqueous NaHCO3, dried on Na2SO4 and concentrated under vacuum. The obtained crude product was purified using silica gel chromatography and recrystallized to generate FIPM (44 mg, 74%) as a white solid. mp: 152–153 °C. 1H NMR (300 MHz, CDCl3) δ 10.56 (br, 1H), 7.40 (d, J = 9.30 Hz, 1H), 7.35 (d, J = 2.10 Hz, 1H), 7.12 (dd, J = 2.4, 11.4 Hz, 1H), 7.01 (s, 1H), 6.77 (s, 1H), 4.65–4.55 (m, 1H), 3.88 (t, J = 4.2 Hz, 4H), 3.58 (t, J = 5.1 Hz, 4H), 1.39 (d, J = 6.3 Hz, 6H); 13C NMR (75.45 MHz, CDCl3) δ 163.44 (d, J = 233.82 Hz), 158.79 (d, J = 16.75 Hz), 153.63 (s), 147.32 (d, J = 8.68 Hz), 142.60 (d, J = 4.38 Hz), 137.46 (s), 121.42 (s), 120.69 (s), 111.17 (s), 103.97 (s), 100.66 (d, J = 4.38 Hz), 95.14 (d, J = 39.08 Hz), 71.23 (s), 66.57 (s), 45.40 (s), 22.00 (s); HRMS (m/z): [M]+ calculated for C19H22FN4O2, 357.1727; found, 357.1731.
3-(2,6-Dibromopyridin-4-yl)-5-isopropoxy-1-trityl-1H-indazole (10)
A mixture of 2,6-dibromo-4-(4,4,5,5-tetrameethyl-1,3,2-dioxaborolan-2-yl)pyridine (9, 435 mg, 1.2 mmol), 5 (544 mg, 1.0 mmol), K2CO3 (207 mg, 1.5 mmol), and Pd(PPh3)4 (116 mg, 0.10 mmol) in 1,4-dioxane/H2O (12 mL/4 mL) was heated at 100 °C for 24 h under N2. After cooling the reaction mixture was extracted with ethyl acetate and the organic layer was dried on Na2SO4 and concentrated under vacuum to leave a residue, which was purified by silica gel chromatography to produce 10 (252 mg, 38%) as a white solid. mp: 151–153 °C. 1H NMR (300 MHz, CDCl3) δ 7.90 (s, 2H), 7.32–7.17 (m, 16H), 6.70 (dd, J = 2.1, 11.7 Hz, 1H), 6.37 (d, J = 9.6 Hz, 1H), 4.60–4.52 (m,1H), 1.36 (d, J = 5.7 Hz, 6H), HRMS (m/z): [M]+ calculated for C34H28Br2N3O, 652.0599; found 652.0605.
4-(6-Bromo-4-(5-isopropoxy-1-trityl-1H-indazol-3-yl)pyridin-2-yl)morpholine (1)
A mixture of 10 (206 mg, 0.32 mmol), xantphos (73 mg, 0.13 mmol), Pd(OAc)2 (7 mg, 0.032 mmol), Cs2CO3 (216 mg, 0.66 mmol) and morpholine (22 μL, 0.25 mmol) in 1,4-dioxane (2 mL) was heated at 100 °C for 4 h under N2. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated under vacuum to leave a residue, which was purified by silica gel chromatography to generate 1 (46 mg, 22%) as a white solid. mp: 201–203 °C. 1H NMR (300 MHz, CDCl3) δ 7.29–7.18 (m, 17H), 7.01 (s, 1H), 6.68 (dd, J = 2.4, 11.4 Hz, 1H), 6.33 (d, J = 9.0 Hz, 1H), 4.58–4.48 (m, 1H), 3.81 (t, J = 4.8 Hz, 4H), 3.52 (t, J = 4.8, 4H), 1.35 (d, J = 6.0 Hz, 6H); 13C NMR (75.45 MHz, CDCl3) δ 159.55, 153.22, 145.31, 142.40, 140.66, 138.69, 138.37, 130.32, 127.50, 127.48, 123.48, 118.23, 115.42, 114.76, 103.47, 102.49, 79.09, 70.74, 66.62, 45.40, 22.07; HRMS (m/z): [M]+ calculated for C38H36BrN4O2, 659.2022; found, 659.1985.
Radiosynthesis of [18F]FIPM
A solution of 1 (1.0 mg) and DABCO (12 mg) in DMSO (300 μL) was added to a reaction vial containing dry [18F]F– and K2CO3/Kryptofix222. The reaction mixture was heated at 150 °C for 20 min. After cooling, the reaction mixture was treated with 6 mol L–1 HCl (1 mL) at 110 °C for 20 min. After neutralization with 6 mol L–1 NaOH (1 mL) the reaction mixture was separated by HPLC under the following conditions: CAPCELL PAK C18 UG80 (10 mm i.d. × 250 mm), CH3CN/H2O (60/40, v/v) as the mobile phase, 5.0 mL min–1 flow rate, and 254 nm for UV detection. The radioactive fraction corresponding to [18F]FIPM (retention time: 11.5 min) was collected in a sterile flask in which Tween 80 (75 μL) in ethanol (300 μL) and 25% ascorbic acid (100 μL) were added before radiosynthesis, evaporated to dryness under vacuum, and re-dissolved in 2.5 mL of sterile normal saline to obtain [18F]FIPM. The radiochemical and chemical purity was measured under the following conditions: CAPCELL PAK C18 UG80 (4.6 mm i.d. × 250 mm), CH3CN/H2O (60/40, v/v) as the mobile phase, 1.0 mL min–1 flow rate, and 254 nm for UV detection. The retention time of [18F]FIPM was 10.0 min.
Measurement of lipophilicity (“shake flask method”)
The measure of log D value was carried out by mixing [18F]FIPM with n-octanol (3.0 g) and phosphate buffered saline (PBS, pH 7.4, 3.0 g, 0.1 M) in a test tube. The test tube was vortexed for 3 min at room temperature, followed by centrifugation at 3500g for 5 min. An aliquot of 0.6 mL PBS and 0.6 mL n-octanol was removed and weighed and the radioactivity in each component was measured with an autogamma counter (2480 Wizard, Perkin-Elmer, Waltham, MA). The log D value was calculated by comparing the ratio of counts per minute (cpm) per g of n-octanol to that of PBS and was expressed as log D = log[cpm per g (n-octanol)/cpm per g (PBS)].
Animal experiments
DdY mice (male, 8 weeks old, 37.1 ± 1.8 g) were purchased from Japan SLC (Shizuoka, Japan). Animals were housed under a 12 h dark-light cycle and were allowed free access to food pellets and water. All animal experiments were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of National Institute of Radiological Sciences (NIRS) and approved by the Animal Ethics Committee of the NIRS.
Biodistribution study in mice
A saline solution of [18F]FIPM (1.1 MBq, 2.0 pmol/100 μL) was administered intravenously via the tail vein. Three mice were sacrificed at each time point (5, 15, 30, 60 and 120 min after injection) by cervical dislocation. Blood samples were taken via cardiac puncture and tissues including the whole brain, heart, lung, liver, pancreas, spleen, kidney, adrenals, stomach, small intestine (including contents), large intestine (including contents), testes, muscle and bone samples were quickly harvested and weighed. Radioactivity in each of these tissues was counted on an autogamma counter (2480 Wizard2). Tissue uptake was calculated and expressed as a percentage of the injected dose per gram of organ tissue (% ID per g). All radioactivity measurements were corrected for decay.
Small-animal PET study in mice
The body temperature of an anesthetized (1.5% isoflurane) rat was maintained using a 40 °C water circulation system (T/Pump TP401, Gaymar Industries, Orchard Park, NY). A 24-gauge intravenous catheter (Terumo Medical Products, Tokyo) was placed into the tail vein of the mice for a bolus injection. A bolus of [18F]FIPM (5.3–6.7 MBq, 46–55 pmol) was injected at flow rate of 0.5 mL min–1. For blocking experiments, FIPM (1 mg kg–1, n = 2) was dissolved in saline containing 10% ethanol and 5% Tween 80, and was administered 1 min prior to the bolus injection of [18F]FIPM. The baseline mice (n = 2) were also pre-administrated with vehicle (saline containing 10% ethanol and 5% Tween 80). Dynamic emission scans in 3D list mode were performed for 60 min (10 s × 12 scans, 20 s × 3 scans, 30 s × 3 scans, 1 min × 3 scans, 2.5 min × 3 scans, and 5 min × 8 scans). Acquired PET dynamic images were reconstructed by filtered back projection using Hanning's filter with a Nyquist cutoff of 0.5 cycle per pixel. The TACs of [11C]1 were acquired from volumes of interest (VOIs) in the heart, lung, liver, kidney, brain, muscle, muscle and bone using the PMOD software (version 3.4; PMOD technology, Zurich, Switzerland). Radioactivity values were decay-corrected to the injection time and expressed as the standardized uptake value (SUV) normalized to the injected radioactivity and body weight of the rat. SUV was calculated according to the following formula: SUV = (radioactivity per milliliter tissue per injected radioactivity) × gram of body weight.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
We thank the staff of the National Institute of Radiological Sciences for their support with cyclotron operation and radionuclide production. We would like to thank Editage (www.editage.com) for English language editing. This study was supported in part by Grants-in-Aid for Scientific Research (Basic Research B: 17H04267) from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government.
References
- Lees A. J., Hardy J., Revesz T. Lancet. 2009;373:2055. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Martinez-Martin P., Kurtis M. M. Ther. Adv. Neurol. Disord. 2012;5:105. doi: 10.1177/1756285611431974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. L., Zhang C. W. Front. Neurol. 2013;4:33. doi: 10.3389/fneur.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Del Tredici K., Braak H. Nat. Rev. Neurol. 2013;9:13. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- West A. B., Moore D. J., Biskup S., Bugayenko A., Smith W. W., Ross C. A., Dawson V. L., Dawson T. M. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16842. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachergus J., Mata I. F., Hulihan M., Taylor J. P., Lincoln S., Aasly J., Gibson J. M., Ross O. A., Lynch T., Wiley J., Payami H., Nutt J., Maraganore D. M., Czyzewski K., Styczynska M., Wszolek Z. K., Farrer M. J., Toft M. Am. J. Hum. Genet. 2005;76:672. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., de Silva R., Lees A., Martí-Massó J. F., Pérez-Tur J., Wood N. W., Singleton A. B. Neuron. 2004;44:595. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T. Neuron. 2004;44:601. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Dachsel J. C., Farrer M. J. Arch. Neurol. 2010;67:542. doi: 10.1001/archneurol.2010.79. [DOI] [PubMed] [Google Scholar]
- Deng X., Dzamko N., Prescott A., Davies P., Liu Q., Yang Q., Lee J. D., Patricelli M. P., Nomanbhoy T. K., Alessi D. R., Gray N. S. Nat. Chem. Biol. 2011;7:203. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith A. D., Bamborough P., Jandu K., Andreotti D., Mensah L., Dossang P., Choi H. G., Deng X., Zhang J., Alessi D. R., Gray N. S. Bioorg. Med. Chem. Lett. 2012;22:5625. doi: 10.1016/j.bmcl.2012.06.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada A. A., Liu X., Baker-Glenn C., Beresford A., Burdick D. J., Chambers M., Chan B. K., Chen H., Ding X., DiPasquale A. G., Dominguez S. L., Dotson J., Drummond J., Flagella M., Flynn S., Fuji R., Gill A., Gunzner-Toste J., Harris S. F., Heffron T. P., Kleinheinz T., Lee D. W., Le Pichon C. E., Lyssikatos J. P., Medhurst A. D., Moffat J. G., Mukund S., Nash K., Scearce-Levie K., Sheng Z., Shore D. G., Tran T., Trivedi N., Wang S., Zhang S., Zhang X., Zhao G., Zhu H., Sweeney Z. K. J. Med. Chem. 2012;55:9416. doi: 10.1021/jm301020q. [DOI] [PubMed] [Google Scholar]
- Kavanagh M. E., Doddareddy M. R., Kassiou M. Bioorg. Med. Chem. Lett. 2013;23:3690. doi: 10.1016/j.bmcl.2013.04.086. [DOI] [PubMed] [Google Scholar]
- Ding X., Dai X., Long K., Peng C., Andreotti D., Bamborough P., Eatherton A. J., Edge C., Jandu K. S., Nichols P. L., Philps O. J., Stasi L. P., Wan Z., Xiang J. N., Dong K., Dossang P., Ho M. H., Li Y., Mensah L., Guan X., Reith A. D., Ren F. Bioorg. Med. Chem. Lett. 2017;27:4034. doi: 10.1016/j.bmcl.2017.07.052. [DOI] [PubMed] [Google Scholar]
- Malik N., Gifford A. N., Sandell J., Tuchman D., Ding Y. S. Mol. Imaging Biol. 2017;19:837. doi: 10.1007/s11307-017-1070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan B. K., Estrada A., Marik J. and Sweeney Z. K., WO2013/079496A1, 2013.
- Wang M., Gao M., Xu Z., Zheng Q. H. Bioorg. Med. Chem. Lett. 2017;27:1351. doi: 10.1016/j.bmcl.2017.02.019. [DOI] [PubMed] [Google Scholar]
- Chen Z., Shao T., Gao W., Fu H., Collier T. L., Rong J., Deng X., Yu Q., Zhang X., Davenport A. T., Daunais J. B., Wey H. Y., Shao Y., Josephson L., Qiu W. W., Liang S. ChemMedChem. 2019;14:1580. doi: 10.1002/cmdc.201900321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N., Tuchman D., Sandell J., Gifford A., Ding Y. S. J. Nucl. Med. 2018;59:s1020. [Google Scholar]
- Chen Z., Shao T., Fu H., Wang L., Shao Y., Wey H. Y., Collier L., Josephson L., Liang S. J. Nucl. Med. 2019;60:s1102. [Google Scholar]
- Ding Y. S., Clancy D., Carlucci G., Ramos N., Jackson R., Boada F., Bruno S., Ciaccio J. J. Nucl. Med. 2019;60:s1105. [Google Scholar]
- Chen Z., Wang L., Ishii H., Yamasaki T., Shao T., Shao Y., Josephson L., Zhang M. R., Liang S. J. Nucl. Med. 2019;60:s1609. [Google Scholar]
- Chen X., Mou Z., Zhang Y., Yang H., Li Z., Xie F. J. Labelled Compd. Radiopharm. 2019;62(Suppl. 1):S174. [Google Scholar]
- Henderson J. L., Kormos B. L., Hayward M. M., Coffman K. J., Jasti J., Kurumbail R. G., Wager T. T., Verhoest P. R., Noell G. S., Chen Y., Needle E., Berger Z., Steyn S. J. C., Houle C., Hirst W. D., Galatsis P. J. Med. Chem. 2015;58:419. doi: 10.1021/jm5014055. [DOI] [PubMed] [Google Scholar]
- Scott J. D., DeMong D. E., Greshock T. J., Basu K., Dai X., Harris J., Li A. Hruza A. S. W., Lin S. I., Liu H., Macala M. K., Hu Z., Mei H., Zhang H., Walsh P., Poirier M., Shi Z. C., Xiao L., Agnihotri G., Baptista M. A., Columbus J., Fell M. J., Hyde L. A., Kuvelkar R., Lin Y., Mirescu C., Morrow J. A., Yin Z., Zhang X., Zhou X., Chang R. K., Embrey M. W., Sanders J. M., Tiscia H. E., Drolet R. E., Kern J. T., Sur S. M., Renger J. J., Bilodeau M. T., Kennedy M. E., Parker E. M., Stamford A. W., Nargund R., McCauley J. A., Miller M. W. J. Med. Chem. 2017;60:2983. doi: 10.1021/acs.jmedchem.7b00045. [DOI] [PubMed] [Google Scholar]
- Fell M. J., Mirescu C., Basu K., Cheewatrakoolpong B., DeMong D. E., Ellis J. M., Hyde L. A., Lin Y., Markgraf C. G., Mei H., Miller M., Poulet F. M., Scott J. D., Smith M. D., Yin Z., Zhou X., Parker E. M., Kennedy M. E., Morrow J. A. J. Pharmacol. Exp. Ther. 2015;355:397. doi: 10.1124/jpet.115.227587. [DOI] [PubMed] [Google Scholar]
- Zhang M. R., Maeda J., Furutsuka K., Yoshida Y., Ogawa M., Suhara T., Suzuki K. Bioorg. Med. Chem. Lett. 2003;1:201. doi: 10.1016/s0960-894x(02)00886-7. [DOI] [PubMed] [Google Scholar]
- Zhang M. R., Maeda J., Ogawa M., Noguchi J., Ito T., Yoshida Y., Okauchi T., Obayashi S., Suhara T., Suzuki K. J. Med. Chem. 2004;47:2228. doi: 10.1021/jm0304919. [DOI] [PubMed] [Google Scholar]
- Fujinaga M., Kumata K., Zhang Y., Hatori A., Yamasaki T., Mori W., Ohkubo T., Xie L., Nengaki N., Zhang M. R. Org. Biomol. Chem. 2018;16:8325. doi: 10.1039/c8ob01700j. [DOI] [PubMed] [Google Scholar]
- Kumata K., Zhang Y., Fujinaga M., Ohkubo T., Mori W., Yamasaki T., Hanyu M., Xie L., Hatori A., Zhang M. R. Bioorg. Med. Chem. 2018;26:4817. doi: 10.1016/j.bmc.2018.08.017. [DOI] [PubMed] [Google Scholar]
- Akula M. R., Blevins D. W., Kabalka G. W., Osborne D. J. Labelled Compd. Radiopharm. 2015;58(Suppl. 1):S198. [Google Scholar]
- Naumiec G. R., Cai L., Lu S., Pike V. W. Eur. J. Org. Chem. 2017;45:6593. doi: 10.1002/ejoc.201700970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura T., Suzuki H., Mukai K., Zhang M. R., Yoshida Y., Nemoto K., Suzuki K. J. Labelled Compd. Radiopharm. 2007;50(Suppl. 1):S202. [Google Scholar]
- Miklossy J., Arai T., Guo J. P., Klegeris A., Yu S., McGeer E. G., McGeer P. L. J. Neuropathol. Exp. Neurol. 2006;65:953. doi: 10.1097/01.jnen.0000235121.98052.54. [DOI] [PMC free article] [PubMed] [Google Scholar]