

Abstract
Febrile seizures, commonly in children between the ages of 3 months to 5 years, are a neurological abnormality characterized by neuronal hyper-excitability, that occur as a result of an increased core body temperature during a fever, which was caused by an underlying systemic infection. Such infections cause the immune system to elicit an inflammatory response resulting in the release of cytokines from macrophages. The cytokines such as interleukin (IL)- 1β, IL-6, and tumour necrosis factor-α (TNF-α) combat the infection in the localized area ultimately spilling over into circulation resulting in elevated cytokine levels. The cytokines, along with pathogen-associated molecular patterns (PAMPs) expressed on pathogens for example, lipopolysaccharide (LPS), interact with the blood brain barrier (BBB) causing a ‘leaky’ BBB which facilitates cytokines and LPS entry into the central nervous system. The cytokines activate the microglia which release their own cytokines, specifically IL1β. IL-β interacts with the brain endothelium resulting in the activation of cyclooxygenase 2 which catalyzes the production of prostaglandin 2 (PGE2). PGE2 enters the hypothalamic region and induces a fever. Abnormally increased IL-1β levels also progressively increases excitatory (glutamatergic) neurotransmission, and decreases inhibitory (GABAergic) neurotransmission, thus mediating the pathogenesis of convulsions. Current treatments for febrile seizures present with side effects that are detrimental to health, which fosters the need for an alternative, more affordable treatment with fewer adverse side effects, and 1 that is easily accessible, especially in low income areas that are also affected by other underlying socio-economic factors, in which febrile seizures are of growing concern.
Keywords: Febrile seizure, fever, IL-1β, neuroinflammation, treatments, convulsions
Introduction
A febrile seizure is a neurological abnormality that occurs as a result of a peripheral infection, to which the immune system reacts by producing an inflammatory response thereby, inducing a fever and subsequently increasing the core temperature of the body.1 The increase in temperature leads to increased neuronal excitability resulting in convulsions.2 Febrile seizures are categorized according to the duration and the number of times the convulsions occur.3 Simple febrile seizures have a life span of approximately 15 minutes and are caused by a distinct infection such as a gastrointestinal or respiratory infection.4 Complex febrile seizures have a life span of 15 to 30 minutes with more than 1 seizure occurring per episode of fever.5 Status epilepticus lasts longer than 30 minutes and occurs randomly as well as repeatedly in the brain during an infection.6 Simple febrile seizures are benign whilst complex febrile seizure and status epilepticus are more likely to develop into more critical conditions such as temporal lobe epilepsy or long term later in life.4–6 Currently, febrile seizures affect 3% to 5% of the world’s population of children aged between 3 months and 5 years.3 Febrile Infection Related Epilepsy Syndrome (FIRES) is a similar condition to febrile seizures which affects children between the aged between 3 years to 15 years.7 The seizures, however, are similar to complex febrile seizures in that they last between 15 to 30 minutes or status epilepticus lasting longer than 30 minutes in some cases.8 Although both febrile seizures and FIRES can occur during common childhood infections, the increased incidence of febrile seizures specifically in Africa can be attributed to limited medical resources, poor access to medical attention, as well as insufficient knowledge of the pathophysiology of febrile seizures.3 The high risk of malaria and high rate of malnutrition are some of the conditions that trigger febrile seizures in Africa.9
Pathophysiology of febrile seizures
Infection
Previous studies conducted on animal models using rats and mice, have reported that genetic and environmental factors are linked to the generation of febrile seizures4,10 Studies have shown that febrile seizures do have a genetic predisposition, where the risk for febrile seizure development with an affected sibling, is roughly 20%, that of which increases to 33% with affected parents.11 Genes mapped to chromosomes 19q, 19p13.3, 18p11.2, 8q13-21, 6q22-24, 5q14-15, 2q23-34 have been associated with increased risk for febrile seizure development, where a polygenic or multifactorial mode of inheritance is most probable.12,13 Studies have also associated febrile seizures with a number of mutations in the gene coding for the gamma-aminobutyric acid (GABA) A receptor γ2 subunit.12 GABA is an inhibitory neurotransmitter, which counteracts neuronal excitability, the receptor of which will be discussed later in this review. Environmental factors such as the exposure to peripheral infections, which include bacterial infections in the middle ear and throat and viral infections such as influenza, have been reported to stimulate an inflammatory response, thereby changing the set temperature of the body, resulting in an increase in the core body temperature and subsequently triggering febrile seizures.14,15 During a bacterial infection in a localized area, pathogen-associated molecular patterns (PAMPs) are expressed on the pathogen for example, lipopolysaccharide (LPS).16 Pathogen recognition receptors such as toll-like receptors (TLRs) found on the cells, which include macrophages and neutrophils, detect the PAMPs, and activate the innate (natural) immune response which is the second line of defense in the immune system.17,18 This line of defense elicits an inflammatory response upon infection.19 The inflammatory response is activated by releasing leukocytes such as macrophages and neutrophils to combat the infection, which in turn releases cytokines into the localized area of infection.19 Pro-inflammatory cytokines, which are released by the macrophages, include tumour necrosis factor (TNF-α), interleukin-6 (IL-6) and interleukin 1 β (IL-1 β).20 These pro-inflammatory cytokines are released concurrently with anti-inflammatory cytokines such as interleukin 1 receptor antagonist (IL-1Ra).21 However, if control of the localized inflammatory response is lost, the pro-inflammatory mediators spill into the circulation, resulting in systemic inflammation.22 This results in further production and release of cytokines in the circulation, elevating levels of peripheral cytokines which interact with the cells of the blood brain barrier (BBB), facilitating the entrance of these cytokines into the central nervous system (CNS).23,24 Cytokines in the CNS from LPS interaction on the BBB result in the recruitment of microglia, these are cells mainly responsible for combating infection in the CNS.15 Under normal conditions, as a form of negative feedback, the release of cytokines such as IL1β during inflammation activates the afferent vagus nerve signalling to the medulla oblongata, brainstem nuclei and the hypothalamus.25 Furthermore, the afferent signalling reaches the forebrain regions associated with integration of visceral sensory information and coordination of the autonomic function and behavioural responses.25 The activation of the afferent vagus nerve then activates the anti-inflammatory efferent vagus nerve cholinergic signalling in brain-to-immune communication as seen in the diagram in Figure 1.22,26 The activation of the signalling results in suppression of local and serum pro-inflammatory cytokine levels.22
Figure 1.
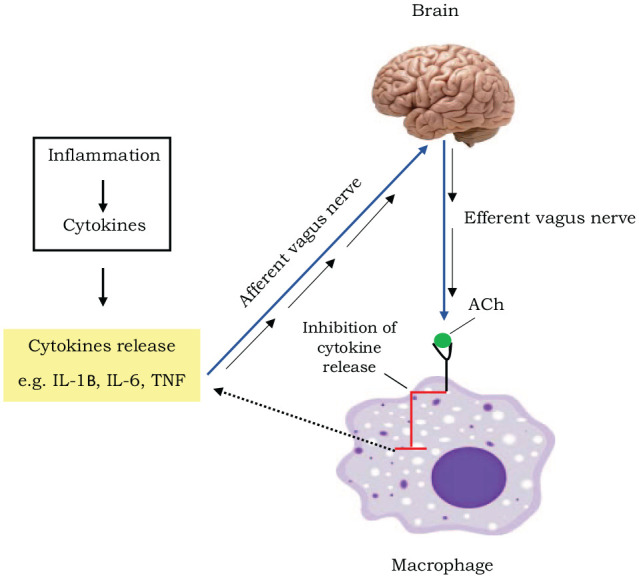
A diagram depicting the cholinergic anti-inflammatory reflex pathway: During an infection, an inflammatory response causes a release of cytokines such as Interleukin (IL) 1β, IL 6 and tumour necrosis factor α (TNF α) in the systemic circulation. This leads to the activation of the afferent vagus nerve signalling to the brain. This signalling subsequently results in the activation of the anti-inflammatory efferent vagus nerve cholinergic signalling with the release of neurotransmitter, acetylcholine (ACh). The ACh then inhibits the release of cytokines from the macrophages, decreasing systemic cytokine levels. Adapted from Pavlov, et al., 2012.26
It has been reported that acetylcholine, a neurotransmitter, inhibits the release of TNF-α, IL-1β and IL-18 from macrophages that were stimulated by LPS.22 However, dysregulation of this pathway during an immune challenge such as excessive LPS and excessive production of pro-inflammatory mediators can cause an imbalance of cytokines resulting in major alterations of the BBB resulting in a more permeable BBB.27,28
Blood-brain barrier integrity in inflammation
The key role of the BBB is to maintain homeostasis for optimal brain performance.29 The anatomy of the BBB is best described on 2 levels, histological and molecular.23 The barrier in its entirety restricts and regulates the movement of substances between the peripheral and cerebrospinal regions.30 The BBB at the histological level is formed by brain endothelial cells lining brain micro-vessels having tight junctions that restrict movement of substances between the cells.30 Furthermore, the brain endothelial cells have a basement membrane which contains a glycocalyx on the luminal surface as well as astrocytes.31,32 The molecular level of the BBB contains ectoenzymes that degrade or alter substances prior to entrance into CNS.23 The BBB is multi-layered with receptors and transporters in the multiple layers which are also responsible for movement of substances in and out of the cerebrospinal region.23 Systemic inflammation can change the integrity and responsiveness of the BBB causing what is known as ‘Blood-brain barrier change’.28 ‘Blood-brain barrier change’ can either be disruptive or non-disruptive, reflecting the presence or absence of physical alterations of the BBB.23 Disruptive BBB change involves alterations at the histological level such as endothelial cell damage or tight junction changes, while non-disruptive change occurs at molecular level.23 Other changes the BBB may go through include the alterations to the efflux transport system which are there to facilitate the entrance of various substance from the brain to the blood.33
LPS is a component of gram-negative bacterial cell walls known to disrupt the BBB and alter aspects of BBB function.34 TLRs, specifically TLR4, detect the LPS constituent resulting in the activation of the innate immune response.35 This results in transcription of pro-inflammatory and anti-inflammatory cytokines.36 The subsequent production and secretion of the cytokines by macrophages in the periphery triggers a cascade of downstream effects ultimately resulting in fever.37 The fever results in hyperthermia occurring in the body due to the increase in the set point temperature in the hypothalamus which has been shown to cause increased BBB permeability.38 LPS, along with the hyperthermia due to fever, induces disruptive BBB changes by altering tight junctions of the brain endothelial cells resulting in barrier dysfunction as well as functional and structural changes to astrocytes.23,38 LPS and the proinflammatory cytokines are also responsible for altering the expression of the efflux transporter, permeability-glycoprotein (P-gp) transporter which is found on the BBB.33,39 The disruptive BBB change results in increased permeability of BBB causing elevated levels of cytokine to be transported into the highly sterile cerebrospinal region recruiting immune cells of the CNS such as microglia which are involved in immune defence in the brain.35
Fever
There are 3 main types of glial cells in the CNS: astrocytes, oligodendrocytes and microglia, which perform different cellular functions in the CNS.40 Astrocytes and microglia act as immune cells in the CNS during inflammation, however, microglia are the main cells involved in overcoming infection in the CNS.40 Cytokines which have entered through the BBB activate the microglia.41 Microglia in the CNS when resting are characterized by small cell bodies with elaborate thin processes spread out in multiple directions.42 However, microglia have been shown to express TLRs such as TLR-3 and TLR-4 which recognise PAMPs including components of a virus infection or LPS resulting in the microglia becoming activated resulting in a change of their morphology.16,42,43 Activated microglia retract their processes which then become thicker and fewer while also increasing the size of their cell bodies.42 In turn, activated microglia produce and release cytokines such as TNF-α, IL-1β and IL-6.41 Under normal conditions, cytokines play a vital role in various aspects of regular CNS function including regulation of sleep and neuronal development.44 However, elevated levels of cytokines during an infection have been shown to play a major role in the generation of fever in febrile seizure generation.45 This is not only seen in febrile seizures but in FIRES where elevated levels of cytokines, specifically IL-1β, activate brain endothelium in turn activating enzymes to produce major pro-inflammatory prostaglandins, including prostaglandin E2 (PGE2).24 The PGE2 is produced from arachidonic acid (AA), a lipid derived from membrane phospholipids which is catalyzed by phospholipase A2.46 Once AA is produced, IL-1β binds to the IL-1 receptor which mediates the activation cyclooxygenase-2 (COX-2), an enzyme expressed on the brain endothelial cells found in the preoptic region of the hypothalamus.47,48 COX-2 catalyzes the production of prostaglandins, specifically, oxidizing AA to produce PGE2.49 PGE2 then binds to EP3 prostaglandin receptors which are expressed by thermoregulatory neurons in the median preoptic nucleus within the hypothalamus to induce a fever.47,49 In non-febrile conditions, a negative feedback mechanism is activated by the body, releasing anti-inflammatory IL-1Ra which blocks and binds free IL-1β thus decreasing PGE2 production which subsequently decreases the generation of a fever.50 During febrile seizures, IL-1β and IL-1Ra are concurrently released resulting in IL-1β and IL-1Ra imbalance with IL-1β playing a major role in causing excitation and inhibition which triggers convulsions.21
Convulsions
There is overwhelming evidence that associates seizures with inflammation and elevated cytokine concentrations.45,51 A study showed that patients that were diagnosed with chronic seizure disorders had elevated levels of proinflammatory cytokines in the cerebral spinal fluid which may have contributed to the neuronal hyperexcitability underlying the seizures.45 Cytokines have a role in synaptic plasticity in areas of the brain such as the hippocampus, and synaptic effects on CNS neurons including those involved in central autonomic control (fever) and gastrointestinal control.52 IL-1β has been shown to have extensive effects in the generation of convulsions, especially febrile seizures.50 IL-1β and IL-1Ra are simultaneously released and compete for the same binding site, IL-1 type 1 receptor (Il-1RI).2 The binding favours IL-1β rather than IL1Ra leading to an imbalance between IL-1β and IL-1Ra.2 IL-1β acts on both the excitatory (glutamatergic) and inhibitory (GABAergic) circuits of the brain.21,44 Glutamate is the principle excitatory amino acid neurotransmitter released by the neurons in the CNS.53 It exerts its function by interacting with ionotropic receptors: αamino-3-hydroxy-5-methyl-isoxazolopropionic acid (AMPA) receptors, kainate receptors, as well as N-methyl-D-aspartate (NMDA) receptor.54,55 IL-1β and IL-1Ra stimulates ionotropic glutamate receptor interactions between glutamate and AMPA receptor.56 The binding of glutamate to AMPA receptor results in the influx of many sodium ions into the cell and a few potassium ions out of the cell resulting in membrane depolarization.56 Magnesium, found in the pore of the NMDA receptor ion channels, gets expelled into the cells during membrane depolarization.57 The binding of glutamate to NMDA receptors along with expulsion of magnesium results in the ion channels opening up causing an influx of calcium and sodium into the cell.51,54,57 The increase in calcium ions into the cell triggers a cascade of reactions and transcriptional changes which then results in convulsions.58 As a form of negative feedback, gamma-aminobutyric acid (GABA), an inhibitory neurotransmitter, counteracts neuronal excitability of glutamate by binding to either 1 of its 2 receptors, GABA A and GABA B.44 It has been reported that GABA A regulates chloride entry into the cell having a rapid inhibitory effect when the membrane is depolarized whilst GABAB regulates calcium entry and potassium efflux.59 The elevated levels of IL-1β have been shown to decrease calcium influx and increase potassium efflux.44 This is due the increased concentration of IL-1β decreasing levels of GABA to GABAA receptor interactions resulting in decrease in the GABAA receptor mediated currents in cultures.21,60 Several studies have also shown that decreased GABAA receptor mediated currents can be caused by hyperthermia which results from the infection.61,62 Therefore, the excitation and inhibition dysregulation along with the fever generated during inflammation results in the onset of seizure together known as febrile seizures as depicted in Figure 2.44
Figure 2.
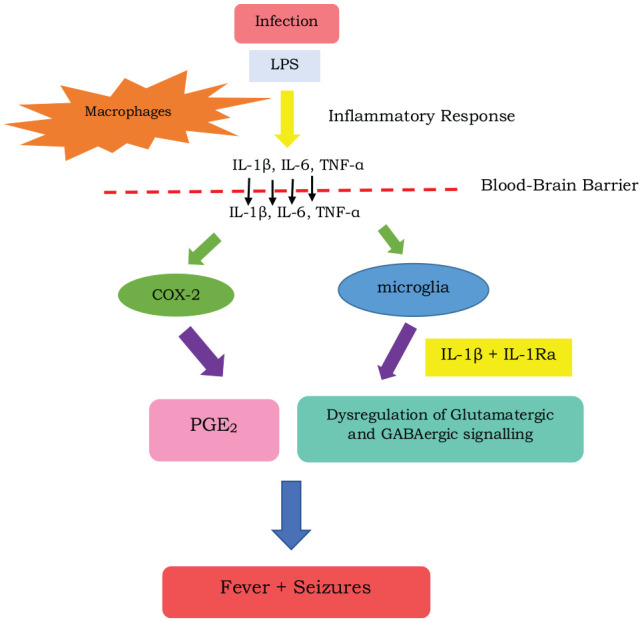
A diagram depicting the pathogenesis of febrile seizures: During an infection, lipopolysaccharide (LPS) is released, resulting in an inflammatory response. This causes macrophages to release cytokines such as interleukin (IL) 1β, IL 6 and tumour necrosis factor (TNF α) which, along with LPS, disrupts the blood-brain-barrier causing it to become leaky. Cytokines then enter through the blood-brain barrier and activate cyclooxygenase-2 (COX-2) and microglia. The COX-2 then catalyses the formation of prostaglandin- E2 (PGE2) which induces fever in the hypothalamus. In addition, activation of the microglia releases proinflammatory and anti-inflammatory cytokines which include Il-1β and interleukin 1 receptor antagonist (IL-1Ra) causing dysregulation of the glutamatergic and GABAergic circuits resulting in seizures. Adapted from Waruiru, et al., 2004.15
Treatment of febrile seizures
Several treatment regimens and drug combinations to treat febrile seizures have been proposed in recent years.63 Currently, febrile seizures are managed with antipyretic or antiepileptic drugs, however antipyretic drugs do not directly treat the febrile seizure and do not prevent further febrile seizures from occurring.64 Current antiepileptic treatment makes use of diazepam, which has been shown to be effective when administered in intermittent oral or rectal doses,15,65 or even intravenously66 or intranasally.67 Other prophylactic antiepileptic drugs used include phenobarbital, valproic acid, phenytoin, and carbamazepine.15,64 However, despite the documented use of these drugs in treating febrile seizures, they all present with side effects that could be detrimental to health.63,68 Diazepam causes side effects such as respiratory depression, bradycardia, dizziness, drowsiness, slurred speech, lethargy, ataxia, and hypotension.69 In addition, such side effects may make it difficult to detect more serious febrile illnesses such as meningitis or encephalitis.15 Phenobarbital, which is a GABA receptor agonist, causes side effects such as daytime drowsiness, transient sleep disturbances, impaired cognitive function, decreased memory, fussiness, attention deficit, and hyperactivity.53,65,70 Valproic acid increases GABA levels and has been used to treat febrile seizures, but its use is limited because of severe side effects such as pancreatitis, renal toxicity, fatal hepatotoxicity, and thrombocytopenia.65,71 In addition, there are also other underlying socio-economic factors regarding drug affordability and availability, as well as a social stigma associated with febrile seizures, especially in the African continent, which causes affected patients to avoid seeking proper medical care, all of which give rise to the need for an alternative, more affordable treatment with fewer adverse side effects.63,68,72
Searsia chirindensis has been shown to have therapeutically beneficial effects in treating various disorders, including neurological disorders, heart disease, and rheumatism.72 This plant contains triterpenoids and steroids, which have anti-inflammatory properties, as well as antioxidants (flavonoids) and tannin which scavenge reactive oxygen species.73–75 Qulu et al showed that treatment with plant extract of Searsia chirindensis reduced both seizure duration and IL1β levels in both non-stressed rats and prenatally stressed rats subjected to febrile seizures in the early postnatal period.68 The decrease in IL-1β levels could be attributed to the anti-inflammatory effects of Searsia, which possibly reduced the release of glutamate and subsequent seizure severity and duration.68 Therefore, Searsia may be a potentially important target for therapeutic interventions against prenatal stress and febrile seizures. Mkhize et al demonstrated a possible therapeutic effect of quercetin (a flavonoid known for its anti-inflammatory, anti-oxidant, and anti-convulsant properties) in a prenatally stressed febrile seizure rat model, in which there was a decrease in pro-inflammatory cytokines such as IL-1β when compared to untreated rats exposed to both prenatal stress and febrile seizures.76 A possible mechanism by which this occurs is that quercetin counteracts/restores the initially dysregulated hypothalamic- pituitary- adrenal (HPA) axis and higher basal glucocorticoid secretion caused by prenatal stress, which subsequently decreases IL-1β concentration.76,77 However, it was shown that quercetin is only therapeutically beneficial in the treatment of febrile seizures accompanied by prenatal stress, but not against febrile seizures alone.76 Therefore, further investigations need to be conducted to understand the mechanisms underlying the action of quercetin, which may also be a potentially important target for therapeutic interventions against prenatal stress and febrile seizures.
Animal models of febrile seizures
Studies on the pathophysiology of febrile seizures are difficult to conduct in humans due to difficulties in recruitment, individual variability, and compliance etc., as well as ethical issues that may arise regarding human trials.37 Therefore, animal models are used to mimic and understand the mechanisms underlying the human pathophysiology of febrile seizures, which allows for the of study seizures in a controlled laboratory environment, and for the production of more reliable results.78 As previously shown, such studies on febrile seizures are commonly conducted on rodent models, particularly Sprague Dawley or Wistar rats.1,2,24,37,79 There are various animal models of febrile seizures that have been previously used. The methodologies used to develop these models vary, and include the use of chemoconvulsant drugs (pentylenetetrazole, kainic acid, pilocarpine) to chemically induce seizures,1,20,37,80 electrical stimulation,80 heated water bath79,81 or heated stream of air.2 Yagoubi et al induced seizures in rat pups on postnatal day 11, by inducing hyperthermia by means of a heated water bath.79 The rats were placed in glass bottles, which were then placed in a water bath heated to 40°C to 45°C, for approximately half an hour until the body temperature was higher than 39.5°C (which is characteristic of fever).79 The rats were transferred to room temperature following the observation of myoclonic jerks, which then progressed into a tonic-clonic generalised seizure.79 This method of febrile seizure induction was successful since the seizures were observed in most animals.79 Dubé et al induced hyperthermia in rat pups on postnatal day 14 and 15, whereby the rats were placed in glass containers and subjected to a regulated stream of air until the core body temperature reached approximately 41°C (which is characteristic of fever).2 This subsequently resulted in the development of febrile seizures.2 Dai et al induced seizures in adult rats on postnatal day 60, using both the maximal electroshock method and pentylenetetrazole administration.80 The maximal electroshock involved the use of a rodent shocker, which delivered a constant current through a pair of ear-clip electrodes.80 The pentylenetetrazole (PTZ) administration involved a single intraperitoneal injection of 60 mg/kg PTZ to 60-day old rats, to produce clonic seizures.80 As can be seen, there are various febrile seizure models, with each model testing different aspects of the pathophysiology underlying the different types of febrile seizures. Of interest is the model employed by Heida et al which involves the generation of immune generated fever to lower seizure threshold.37 This was achieved by the administration of LPS, which as aforementioned, is a component of the gram-negative bacterial cell wall, used to induce fever and mimic infection.37 This fever induction can occur via different mechanisms, which encompass signalling through circumventricular organs, perivascular endothelial cells, and vagal afferents, all of which occur either by direct action of LPS in these brain areas, or by LPS -induced peripheral cytokines.37 Since not all animals develop seizures while febrile, it was deduced that those animals susceptible to febrile seizures have some sort of abnormal excitability, therefore the LPS administration was accompanied by a sub-threshold dose of the convulsant drug, kainic acid (ionotropic glutamate receptor agonist).37 Particularly, this model was developed by an intraperitoneal injection of 200 µg/kg LPS, followed by an intraperitoneal injection of 1.75 mg/kg kainic acid 2.5 hours later, and was successful in inducing febrile seizures in more than 50% of treated animals.37 Therefore, this model is considered a favourable model in many labs, since physiological fever was induced to lower seizure threshold which closely represents the natural occurrence of febrile seizures in children with underlying febrile infections, and also since febrile seizures occurred at clinically relevant temperatures, which lasted approximately 60 minutes (which represents prolonged complex febrile seizures or febrile status epilepticus).1,36,37,68,82 Furthermore, a prenatally stressed febrile seizure rat model, as shown by Qulu et al, was developed by subjecting pregnant female rats to restraint stress for 1 hour daily, for a total of 7 days, from gestational day 14 to 20.1 Following the birth of the rat pups, febrile seizures were induced on postnatal day 14 with an intraperitoneal injection of 200 µg/kg LPS, followed by an intraperitoneal injection of 1.75 mg/kg kainic acid 2.5 hours later.1 The development of this particular model provides a means to conduct further investigations on prenatal stress and febrile seizures.
Conclusion
In conclusion, inflammation, though a defence response against invaders and pathogens during an infection in children, has been shown to have adverse effects in the central nervous system. The pro-inflammatory cytokines, especially IL-1β, are the main factors that induce fever. The increase of IL-1β in the CNS also results in an imbalance in glutamate and GABA causing an imbalance in excitation and inhibition transmission in the brain resulting in convulsions which accompany the fever, giving rise to the onset of febrile seizures. The aim of this review was to highlight the increased susceptibility of young offspring to the development of febrile seizures, or other infections; as well as to raise awareness of the dire need for further investigations into producing an affordable, easily accessible treatment for febrile seizures, especially in low income areas that are also affected by other underlying socio-economic factors, in which febrile seizures are of growing concern.
Acknowledgments
The authors wish to thank the University of KwaZulu-Natal Biomedical Research Unit (BRU) and Professor Vivienne Russell for their writing assistance and proof reading of this review article.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: “a University of KwaZulu-Natal (UKZN) Developing Research Innovation, Localisation and Leadership in South Africa (DRILL) fellow. DRILL, is a NIH D43 grant (D43TW010131) awarded to UKZN in 2015 to support a research training and induction programme for early career academics. The content is solely the responsibility of the authors and does not necessarily represent the official views of DRILL and the National Institutes of Health.”
Declaration of conflicting interests:The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Please note that there is shared authorship for the first author, which is between Shreyal Maikoo and Palesa Mosili
Author Contributions: All the authors contributed equally to writing and editing the paper. Lihle Qulu designed and funded the manuscript.
ORCID iD: Lihle Qulu  https://orcid.org/0000-0003-2800-5711
https://orcid.org/0000-0003-2800-5711
References
- 1. Qulu L, Daniels WM, Mabandla MV. Exposure to prenatal stress enhances the development of seizures in young rats. Metab Brain Dis. 2012;27:399-404. [DOI] [PubMed] [Google Scholar]
- 2. Dubé C, Vezzani A, Behrens M, Bartfai T, Baram TZ. Interleukin-1β contributes to the generation of experimental febrile seizures. Ann Neurol. 2005;57:152-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mung’ala-Odera V, White S, Meehan R, et al. Prevalence, incidence and risk factors of epilepsy in older children in rural Kenya. Seizure. 2008;17:396-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dubé CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev. 2009;31:366-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia. 1996;37:126-133. [DOI] [PubMed] [Google Scholar]
- 6. Cherian A, Thomas SV. Status epilepticus. Ann Indian Acad Neurol. 2009;12:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fox K, Wells ME, Tennison M, Vaughn B. Febrile infection-related epilepsy syndrome (FIRES): a literature review and case study. Neurodiagn J. 2017;57:224-233. [DOI] [PubMed] [Google Scholar]
- 8. Appenzeller S, Helbig I, Stephani U, et al. Febrile infection-related epilepsy syndrome (FIRES) is not caused by SCN1A, POLG, PCDH19 mutations or rare copy number variations. Dev Med Child Neurol. 2012;54:1144-1148. [DOI] [PubMed] [Google Scholar]
- 9. Kariuki SM, Abubakar A, Kombe M, et al. Prevalence, risk factors and behavioural and emotional comorbidity of acute seizures in young Kenyan children: a population-based study. BMC Med. 2018;16:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pociot F, Mølvig J, Wogensen L, Worsaae H, Nerup J. A Taql polymorphism in the human interleukin-1β (IL-1β) gene correlates with IL-1β secretion in vitro. Eur J Clin Invest. 1992;22:396-402. [DOI] [PubMed] [Google Scholar]
- 11. Van Zeijl J, Mullaart R, Galama J. The pathogenesis of febrile seizures: is there a role for specific infections? Rev Med Virol. 2002;12(2):93-106. [DOI] [PubMed] [Google Scholar]
- 12. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci. 2006;26:2590-2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vestergaard M, Basso O, Henriksen TB, Østergaard JR, Olsen J. Risk factors for febrile convulsions. Epidemiology. 2002;13:282-287. [DOI] [PubMed] [Google Scholar]
- 14. Bakken IJ, Aaberg KM, Ghaderi S, et al. Febrile seizures after 2009 influenza A (H1N1) vaccination and infection: a nationwide registry-based study. BMC Infect Dis. 2015;15:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waruiru C, Appleton R. Febrile seizures: an update. Arch Dis Child. 2004;89:751-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawabe T, Suda T, Fukui M, Imamura R, Umemura M. Pathogen-associated molecular patterns. J Immunol. 2003;171:1868-1874. [DOI] [PubMed] [Google Scholar]
- 17. Delves PJ, Roitt IM. The immune system. N Engl J Med. 2000;343:37-49. [DOI] [PubMed] [Google Scholar]
- 18. Janeway CA, Jr, Medzhitov R. Innate immune recognition. Ann Rev Immunol. 2002;20:197-216. [DOI] [PubMed] [Google Scholar]
- 19. Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol. 2010;125:S24-S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heida JG, Teskey GC, Pittman QJ. Febrile convulsions induced by the combination of lipopolysaccharide and low-dose kainic acid enhance seizure susceptibility, not epileptogenesis, in rats. Epilepsia. 2005;46:1898-1905. [DOI] [PubMed] [Google Scholar]
- 21. Heida JG, Pittman QJ. Causal links between brain cytokines and experimental febrile convulsions in the rat. Epilepsia. 2005;46:1906-1913. [DOI] [PubMed] [Google Scholar]
- 22. Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med. 2003;9:125-134. [PMC free article] [PubMed] [Google Scholar]
- 23. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1-12. [DOI] [PubMed] [Google Scholar]
- 24. Zotova N, Chereshnev V, Gusev EY. Systemic inflammation: methodological approaches to identification of the common pathological process. PLoS One. 2016;11:e0155138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci. 2000;85:49-59. [DOI] [PubMed] [Google Scholar]
- 26. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex—linking immunity and metabolism. Nat Rev Endocrinol. 2012;8:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung S. Febrile seizures. Korean J Pediatr. 2014;57:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elwood E, Lim Z, Naveed H, Galea I. The effect of systemic inflammation on human brain barrier function. Brain Behav Immun. 2017;62:35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campos-Bedolla P, Walter FR, Veszelka S, Deli MA. Role of the blood–brain barrier in the nutrition of the central nervous system. Arch Med Res. 2014;45:610-638. [DOI] [PubMed] [Google Scholar]
- 30. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13-25. [DOI] [PubMed] [Google Scholar]
- 31. Helms HC, Abbott NJ, Burek M, et al. In vitro models of the blood–brain barrier: an overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. 2016;36:862-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vaccaro KK, Liang BT, Sheard BE, Perlman RL. Monensin inhibits catecholamine synthesis in pheochromocytoma cells. J Pharmacol Exp Ther. 1982;221:536-540. [PubMed] [Google Scholar]
- 33. Erickson MA, Banks WA. Neuroimmune axes of the blood–brain barriers and blood–brain interfaces: bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol Rev. 2018;70:278-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schumann RR, Flaggs G, Gray P, et al. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429-1431. [DOI] [PubMed] [Google Scholar]
- 35. Banks WA, Gray AM, Erickson MA, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. 2015;12:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cassim S, Qulu L, Mabandla MV. Prenatal stress and early life febrile convulsions compromise hippocampal genes MeCP2/REST function in mid-adolescent life of Sprague-Dawley rats. Neurobiol Learn Mem. 2015;125:195-201. [DOI] [PubMed] [Google Scholar]
- 37. Heida JG, Moshé SL, Pittman QJ. The role of interleukin-1β in febrile seizures. Brain Dev. 2009;31:388-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kiyatkin EA, Sharma HS. Permeability of the blood–brain barrier depends on brain temperature. Neuroscience. 2009;161:926-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Erickson MA, Hansen K, Banks WA. Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood–brain barrier: protection by the antioxidant N-acetylcysteine. Brain Behav Immun. 2012;26:1085-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jäkel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front Cell Neurosci. 2017;11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nayak D, Roth TL, McGavern DB. Microglia development and function. Ann Rev Immunol. 2014;32:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013;36:209-217. [DOI] [PubMed] [Google Scholar]
- 43. Vontell R, Supramaniam V, Thornton C, et al. Toll-like receptor 3 expression in glia and neurons alters in response to white matter injury in preterm infants. Dev Neurosci. 2013;35:130-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33:116-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia. 2011;52:26-32. [DOI] [PubMed] [Google Scholar]
- 46. Rosenberger TA, Villacreses NE, Contreras MA, Bonventre JV, Rapoport SI. Brain lipid metabolism in the cPLA2 knockout mouse. J Lipid Res. 2003;44:109-117. [DOI] [PubMed] [Google Scholar]
- 47. Choi S-H, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and-2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci. 2009;30:174-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Conti B. Prostaglandin E2 that triggers fever is synthesized through an endocannabinoid-dependent pathway. Temperature. 2016;3:25-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Evans SS, Repasky EA, Fisher DT. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol. 2015;15:335-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232. [DOI] [PubMed] [Google Scholar]
- 51. Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64:93-109. [DOI] [PubMed] [Google Scholar]
- 53. Lasoń W, Chlebicka M, Rejdak K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol Rep. 2013;65:787-801. [DOI] [PubMed] [Google Scholar]
- 54. Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31:3-16. [DOI] [PubMed] [Google Scholar]
- 55. Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm. 2014;121:799-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126-134. [DOI] [PubMed] [Google Scholar]
- 57. Lüscher C, Malenka RC. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol. 2012;4:a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barker-Haliski M, White HS. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med. 2015;5:a022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42:8-12. [DOI] [PubMed] [Google Scholar]
- 60. Schäfers M, Sorkin L. Effect of cytokines on neuronal excitability. Neurosci Lett. 2008;437:188-193. [DOI] [PubMed] [Google Scholar]
- 61. Qu L, Leung LS. Effects of temperature elevation on neuronal inhibition in hippocampal neurons of immature and mature rats. J Neurosci Res. 2009;87:2773-2785. [DOI] [PubMed] [Google Scholar]
- 62. Qu L, Liu X, Wu C, Leung LS. Hyperthermia decreases GABAergic synaptic transmission in hippocampal neurons of immature rats. Neurobiol Dis. 2007;27:320-327. [DOI] [PubMed] [Google Scholar]
- 63. Chen X, Wang J, Su X, et al. Prophylactic treatment of the recurrence of febrile convulsion by different drugs: a meta-analysis. Int J Clin Exp Med. 2017;10:6453-6460. [Google Scholar]
- 64. Leung AK, Robson WLM. Febrile seizures. J Pediatr Health Care. 2007;21:250-255. [DOI] [PubMed] [Google Scholar]
- 65. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. Pediatr Neurol. 2000;23:11-17. [DOI] [PubMed] [Google Scholar]
- 66. Mukherjee A, Mukherjee A. Febrile convulsion–an overview. J Indian Med Assoc. 2002;100:317. [PubMed] [Google Scholar]
- 67. Lahat E, Goldman M, Barr J, Bistritzer T, Berkovitch M. Comparison of intranasal midazolam with intravenous diazepam for treating febrile seizures in children: prospective randomised study. BMJ. 2000;321:83-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qulu L, Daniels WM, Russell V, Mabandla MV. Searsia chirindensis reverses the potentiating effect of prenatal stress on the development of febrile seizures and decreased plasma interleukin-1β levels. Neurosci Res. 2016;103:54-58. [DOI] [PubMed] [Google Scholar]
- 69. Sagraves R. Febrile seizures—treatment and prevention or not? J Pediatr Health Care. 1999;13:79-83. [DOI] [PubMed] [Google Scholar]
- 70. Farwell JR, Lee YJ, Hirtz DG, Sulzbacher SI, Ellenberg JH, Nelson KB. Phenobarbital for febrile seizures—effects on intelligence and on seizure recurrence. N Engl J Med. 1990;322:364-369. [DOI] [PubMed] [Google Scholar]
- 71. Millar JS. Evaluation and treatment of the child with febrile seizure. Am Fam Physician. 2006;73:1761-1764. [PubMed] [Google Scholar]
- 72. Ojewole JA. Anticonvulsant effect of Rhus chirindensis (Baker F.)(Anacardiaceae) stem-bark aqueous extract in mice. J Ethnopharmacol. 2008;117:130-135. [DOI] [PubMed] [Google Scholar]
- 73. Dehmlow EV, Van Ree T, Jakupovic J, Take E, Kuensting H., Jr Notes on activity tests and constituents of two supposed medicinal plants from South Africa, Englerophytum magalismontanum and Diospyros lycioides desf. subsp. sericea. J Chem Res Synop. 1998;5:252-253. [Google Scholar]
- 74. Greaves M. Anti-inflammatory action of corticosteroids. Postgrad Med J. 1976;52:631-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kohen R, Nyska A. Invited review: oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol. 2002;30:620-650. [DOI] [PubMed] [Google Scholar]
- 76. Mkhize NVP, Qulu L, Mabandla MV. The effect of quercetin on pro-and anti-inflammatory cytokines in a prenatally stressed rat model of febrile seizures. J Exp Neurosci. 2017;11:1179069517704668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Charil A, Laplante DP, Vaillancourt C, King S. Prenatal stress and brain development. Brain Res Rev. 2010;65:56-79. [DOI] [PubMed] [Google Scholar]
- 78. Stables JP, Bertram EH, White HS, et al. Models for epilepsy and epileptogenesis: report from the NIH workshop, Bethesda, Maryland. Epilepsia. 2002;43:1410-1420. [DOI] [PubMed] [Google Scholar]
- 79. Yagoubi N, Jomni Y, Sakly M. Hyperthermia-induced febrile seizures have moderate and transient effects on spatial learning in immature rats. Behav Neurol. 2015;2015:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dai Y-J, Xu Z-H, Feng B, et al. Gender difference in acquired seizure susceptibility in adult rats after early complex febrile seizures. Neurosci Bull. 2014;30:913-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhou J, Wang F, Zhang J, Gao H, Yang Y, Fu R. Repeated febrile convulsions impair hippocampal neurons and cause synaptic damage in immature rats: neuroprotective effect of fructose-1, 6-diphosphate. Neural Regen Res. 2014;9:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Qulu L, Daniels WM, Mabandla MV. Exposure to prenatal stress has deleterious effects on hippocampal function in a febrile seizure rat model. Brain Res. 2015;1624:506-514. [DOI] [PubMed] [Google Scholar]