

Abstract
Oxidative stress-induced damage to the retinal pigment epithelium (RPE) is considered to be a key factor in age-related macular degeneration (AMD) pathology. RPE cells are constantly exposed to oxidative stress that may lead to the accumulation of damaged cellular proteins, lipids, nucleic acids, and cellular organelles, including mitochondria. The ubiquitin-proteasome and the lysosomal/autophagy pathways are the two major proteolytic systems to remove damaged proteins and organelles. There is increasing evidence that proteostasis is disturbed in RPE as evidenced by lysosomal lipofuscin and extracellular drusen accumulation in AMD. Nuclear factor-erythroid 2-related factor-2 (NFE2L2) and peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) are master transcription factors in the regulation of antioxidant enzymes, clearance systems, and biogenesis of mitochondria. The precise cause of RPE degeneration and the onset and progression of AMD are not fully understood. However, mitochondria dysfunction, increased reactive oxygen species (ROS) production, and mitochondrial DNA (mtDNA) damage are observed together with increased protein aggregation and inflammation in AMD. In contrast, functional mitochondria prevent RPE cells damage and suppress inflammation. Here, we will discuss the role of mitochondria in RPE degeneration and AMD pathology focused on mtDNA damage and repair, autophagy/mitophagy signaling, and regulation of inflammation. Mitochondria are putative therapeutic targets to prevent or treat AMD.
Keywords: Age-related macular degeneration, Aggregation, Aging, Autophagy, Clearance, Degeneration, Mitochondria, Mitophagy, Retina, Retinal pigment epithelium
1. Introduction to RPE degeneration and development of AMD
AMD is associated with several environmental and genetic risk factors that are linked to increased oxidative stress (Fig. 1; Handa, 2012; Jarrett and Boulton, 2012; Fritsche et al., 2013; Kaarniranta et al., 2018a,b; Wang et al., 2018). Per definition, age is the main risk factor for AMD. The accumulation of oxidized proteins, lipids, and DNA in aged tissues provides evidence for an overall increase in oxidative stress with aging (Ames and Shigenaga, 1992; Moldogazieva et al., 2019). The main genetic factors in AMD are mutations in the genes of the complement factors H, B and I (CFH, CFB, CFI), complement components 3 and 2 (C3, C2), as well as Age-Related Maculopathy Susceptibility 2/High-Temperature Requirement protein A1 (ARMS2/HTRA1) gene regions (Fritsche et al., 2013). Evidence linking genetic backgrounds with elevated oxidative stress included the significantly higher RPE mtDNA damage reported in donors harboring the CFH high risk alleles (Ferrington et al., 2016a). Dysfunctional CFH reduces antioxidant capacity and energy metabolism of human RPE cells (Armento et al., 2020). Additionally, the presence of the ARMS2/HTRA1 risk alleles in induced pluripotent stem-RPE cells (iPSC-RPE) resulted in decreased antioxidant defenses, thereby making the RPE more susceptible to oxidative damage (Yang et al., 2014). Smoking, obesity, arteriosclerosis, hypertension, hypercholesterolemia, an unhealthy diet, and excessive light exposure are centralenvironmental or systemic risk factors in AMD (Fig. 1; Blasiak et al., 2019). Each of these environmental risk factors are known to increase oxidative stress and/or inflammation, characteristics that can precipitate retinal pathology (Datta et al., 2017; Rozing et al., 2019).
Fig. 1.

Mitochondrial DNA (mtDNA) in the neural retina and retinal pigment epithelial (RPE) cells can be damaged by many endogenous and exogenous factors, including some risk factors of age-related macular degeneration (AMD). Aging, the primary AMD risk factor, is also associated with induction of mtDNA damage. It is likely that mtDNA damage affects genes encoding the mitochondrial electron transport chain resulting in reactive oxygen species (ROS) overproduction, leading to more damage to mtDNA. These changes can be counterbalanced by efficient (+) antioxidant enzymes or DNA repair, but when these systems fail, accumulated damage to mtDNA can result in mitochondrial dysfunctions, energy crisis, cell degeneration and death observed in AMD.
The devastating effects of AMD are most apparent in the macula, a small, cone-rich area of the retina responsible for high acuity and color vision (Fig. 2A). The macula is under considerable oxidative stress due to its high metabolic rate, high oxygen tension, and the continuous production of reactive oxygen species (ROS) in the area (Stefánsson et al., 2011; Eells, 2019; Stefánsson et al., 2019). In AMD, pathological changes can be observed in the choriocapillaris, Bruch's membrane, RPE, and photoreceptors (Fig. 2B). However, it has been hypothesized that the RPE play a key role in the pathogenesis of AMD due to their central location between Bruch's membrane and photoreceptors where they serve as a conduit for oxygen, nutrients, and waste products between the outer retina and the choriocapillaris (Strauss, 2005; Kaarniranta et al., 2009). In clinical settings, a primary hallmark of AMD is the presence of drusen between the RPE and Bruch's membrane, with RPE degeneration occurring as the disease progresses (Fig. 2A and B). Since one of the main functions of the RPE is to maintain the health of the photoreceptors, RPE degeneration secondarily leads to dysfunction of the photoreceptors and visual disturbances associated with AMD (Fig. 2C).
Fig. 2.

Dry age-related macular degeneration: symptoms and signs. (A) Color fundus photograph from normal and dry AMD patient maculas. The central macula controls sharp and color vision (white ring). Drusen are the clinical hallmarks of AMD (dark ring). (B) Optical coherent tomography (OCT) from the healthy and AMD retinas. Arrows indicate different layers of the retina: 1) external limiting membrane, 2) ellipsoid layer, 3) RPE layer, 4) retinal nerve fiber layer (RNFL), 5) ganglion cell layer, 6) inner plexiform layer, 7) inner nuclear layer, 8) outer plexiform layer 9) outer nuclear layer. (C) Representative vision disturbances during the progression of AMD. (D) Light microscopic image of hematoxylin-eosin (HE) stained human retina from (D, left panel) healthy and (E, left panel) AMD donors. High power confocal images indicate extracellular ubiquitin (Ubq, middle panels) and intracellular p62 (right panels) stainings. Arrowheads show drusen accumulation.
Oxidative stress and damage have been suggested as key factors for triggering RPE degeneration. Oxidative stress ultimately involves an excess of ROS that are produced mainly in mitochondria (Handa, 2012; Blasiak et al., 2013). ROS oxidize many biological macromolecules, including proteins, lipids, and nucleic acids, and may evoke structural and functional changes in these molecules. In AMD pathology, excessive ROS and oxidized lipoproteins lead to protein misfolding, aggregation, and chronic activation of the innate immunity response (Ferrington et al., 2016b; Kauppinen et al., 2016; Kaarniranta et al., 2019). Various products of lipid peroxidation, such as advanced glycation-end products, carboxyethylpyrrole, malondialdehyde, and 4-hydroxynonenal, which are indicators of increased oxidative stress, have been detected in macula of AMD patients (Hyttinen et al., 2018). In addition to these products of oxidation, there is an accumulation of autofluorescent lipofuscin, cytoplasmic aggregates, and drusen in AMD that are indicators of disturbed proteostasis, which involves the coordinate regulation of protein synthesis, folding, and degradation. Evidence for defects in protein degradation in the RPE of donors with AMD includes the increased content of both ubiquitin (Ub) and sequestosome-1 (p62/SQSTM1), suggesting failure in the Ub-proteasome and lysosome/autophagy pathways, respectively (Fig. 2D and E). A deficiency in cellular proteostasis has been associated with multiple age-related degenerative disorders and has particularly devastating consequences in post-mitotic or quiescent cells, such as neurons and the RPE (Ferrington et al., 2016b).
Activation of protective pathways and upregulation of endogenous antioxidants are cellular responses against excessive oxidative stress. Antioxidative defense mechanisms can also improve autophagy, thereby increasing survival of RPE cells (Kaarniranta et al., 2017, 2018a). Among the protective pathways are the NFE2L2 and PGC-1α, which upregulate expression of antioxidants that protect against ROS-induced mitochondrial damage and cell death (Strom et al., 2016 https://onlinelibrary.wiley.com/doi/full/10.1111/acel.12650). PGC-1α also regulates the expression of many genes important for energy production and mitochondrial biogenesis (Kaarniranta et al., 2018a, b). Understanding how these pathways are involved in AMD and how they could be manipulated to protect RPE in vivo is critical to study.
We summarize data supporting the hypothesis that mitochondrial dysfunction in the RPE is a key event in AMD pathology. A significant increase in mtDNA damage is associated with normal aging, but it is more extensive with AMD. There is evidence that mitochondrial DNA damage repair and haplogroups are involved in the pathology of AMD. NFE2L2 and PGC-1α have a key role in mitochondrial quality control and in the regulation of protein aggregation and inflammation in AMD. We also discuss mitochondria as a putative therapy target to prevent or treat AMD.
2. RPE mitochondria damage and dysfunction during normal aging
Mitochondria are crucial organelles that produce adenosine triphosphate (ATP) via oxidative phosphorylation, which involves the transfer of electrons between multi-subunit complexes (I to IV) of the Electron Transport Chain (ETC) coupled with the reduction of molecular oxygen. Under normal circumstances, ~2–5% of the oxygen is incompletely reduced, leading to elevated ROS and superoxide production (Chen et al., 2003). Even the act of phagocytosis, one of the daily functions of the RPE, predisposes these cells to altered mitochondrial redox states and increased mtDNA damage (Jin et al., 2001; Liang and Godley, 2003). Several studies show a dual role for ROS in cell physiology: (i) Moderate amounts of ROS are essential for the maintenance of beneficial cellular processes, such as production of cellular energy and the regulation of mitochondria protective signaling pathways. Upregulation of these positive effect are capable of increasing lifespan in lower organisms (Giorgi et al., 2018), (ii) In contrast, excess ROS are pathogenic and may evoke accelerated cell degeneration or even cell death.
Mitochondrial ROS and oxidative damage are significantly increased with aging and aging-related diseases. For example, opening of the mitochondrial permeability transition pore, which is triggered by ROS and mitochondrial calcium overload, leads to apoptosis (Rottenberg and Hoek, 2017). The increased ROS levels promote oxidative damage to mitochondrial DNA, lipids, and proteins (Jarrett et al., 2008). During normal aging, a disruption of mitochondrial structure and an accumulation of mtDNA mutations have also been reported (Blasiak et al., 2013). These results are in line with earlier studies of Wang et al. who showed an accumulation of mtDNA damage with age in RPE and choroid of mice and rats (Wang et al., 2008). This effect was likely the result of the decrease in DNA repair capacity with age, which was supported by a decreased expression of genes encoding 8-oxo-guanine-DNA glycosylase 1 (OGG1), MutY DNA glycosylase, and thymine DNA glycosylase – enzymes critical for repairing oxidative damage to DNA. However, DNA repair is not the only way the cell may act against mtDNA damage as it can be prevented by antioxidant enzymes and small molecular weight antioxidants (Fig. 1; Tokarz et al., 2013). Furthermore, it seems that these studies suffer from the lack of evaluation of the DNA repair capacity in cells challenged by an endogenous DNA-damaging factor, which is important in the context of AMD pathogenesis, as aging is its main but not the only risk factor. In their subsequent work, Wang et al. showed that photoreceptors and retinal ganglion cells in rodents also displayed increased levels of mtDNA damage and again this effect was attributed to decreased DNA repair capacity (Wang et al., 2010).
Damage to the mitochondrial genome also involves a 4977 base pair deletion, known as the “Common Deletion”, which occurs with high frequency in patients with mitochondrial diseases, and also in aged post-mitotic and quiescent cells including the RPE and neural retina (Fig. 1; Barreau et al., 1996; Karunadharma et al., 2010; Nie et al., 2016; Huang et al., 2018). These results were confirmed in subsequent research, showing a biphasic increase in the ratio of this mutation in the retina. A slow increase occurred in donors between ages 14 and 60 years, followed by a rapid increase in damage for donors between 60 and 94 years of age (Barron et al., 2001). Preferentially, this mutation accumulated in the fovea and was linked with retinal dysfunctions observed in age-related maculopathies. Of note, while the Common Deletion increased in RPE with age for donors 34–88 years, it only increased in donors with advanced AMD, suggesting that this type of damage is not relevant to the early events in AMD pathogenesis (Karunadharma et al., 2010).
As AMD is per definition an age-dependent disease, the question whether it is associated with accelerated aging is often asked. Given the role of oxidative DNA damage in aging, the question about the role of mtDNA damage in AMD pathogenesis and its relations with aging also seems to be justified (Hyttinen et al., 2017). While the results obtained by Karunadharma et al. support the hypothesis that mtDNA damage increases with age, this damage was limited to the “Common Deletion” (Karunadharma et al., 2010). In contrast, the AMD-related mtDNA damage was distributed throughout the mitochondrial genome. These results suggest that AMD cannot be attributed only to accelerated aging, but it involves disease-specific changes that are superimposed on a background of typical age-related changes.
3. RPE mitochondria damage and dysfunction with AMD
With AMD, extensive changes to the mitochondrial genome have been reported. Comparison of mtDNA damage as measured by long-extension polymerase chain reaction (LX-PCR) in the macula of human donor RPE showed AMD donors had more mtDNA damage compared to the age-matched controls (Karunadharma et al., 2010; Terluk et al., 2015). Furthermore, the extent of RPE mtDNA damage correlated with AMD severity. Terluk and colleagues reported that the damage was localized in region of the mitochondrial genome that encode for the subunits of the ETC, thereby potentially leading to reduced ATP production (Terluk et al., 2015). Damage was also observed in the D-loop, the only non-coding region of the genome that is the site of initiation for mtDNA transcription and replication of one mtDNA strand. Of note, the background mutation rate in the D-loop is about an order higher than in the remaining, coding part of the mitochondrial genome (Allio et al., 2017). Damage to either the D-loop or region encoding the ETC proteins could lead to negative functional outcomes for the mitochondria.
Other clinically relevant information from this study included the finding that mtDNA damage was not limited to the macula as RPE harvested from the peripheral region also exhibited an AMD-related increase in damage (Terluk et al., 2015). It is also important to note that the AMD-related increase in mtDNA damage was not observed in the photoreceptors. A potential explanation may be related to the cell-specific difference in the pathways utilized to generate energy. In contrast with the RPE, which rely on ROS-producing mitochondria, photoreceptors utilize aerobic glycolysis, a pathway that does not produce excessive ROS (Ames et al., 1992; Medrano and Fox, 1995; Yu and Cringle, 2005; Hurley et al., 2015; Fisher and Ferrington, 2018).
Studies addressing the role of damage to mtDNA in AMD pathogenesis have also measured mtDNA damage in peripheral blood as a surrogate measure of the retina. However, there is evidence that mtDNA damage in peripheral blood shows different susceptibility to DNA-damaging factors than the retinal counterpart. This hypothesis is supported by results from Kenney et al., where they compared the extent of mtDNA deletions and rearrangements in retina, choroid, and blood of individuals with AMD and control subjects (Kenney et al., 2010). They noted higher levels of mtDNA rearrangements and deletions in the retina than blood for both normal and AMD samples. In addition, these observations reveal increased genetic variation in both the coding portion of mtDNA and the D-loop. The authors postulate that damage to these elements may lead to dysfunctional mitochondria and a reduced number of mtDNA copies (Udar et al., 2009).
There are several critical limitations that should be recognized when results from mtDNA damage analysis are interpreted. When the extent of mtDNA damage is evaluated with LX-PCR, the amount of PCR product depends on the amount of undamaged versus damaged template that is available for amplification. Damaged templates include DNA base modifications and single or double strand breaks that stop the DNA polymerase. However, the main oxidative modification to DNA bases is 8-oxo-guanine (8-oxo-G), which does not inhibit the DNA polymerase, so the LX-PCR assay would not recognize this modification. DNA fragmentation also limits or even eliminates the ability to quantify the amount of damage. Another limitation is that the type of mitochondrial damage cannot be determined. It is also important to note that there is great diversity in both the number of mitochondria per cell as well as the number of mtDNA copies per mitochondria. Since the assay is only measuring an average of many mitochondrial templates, the results cannot inform about the damage within a single cell or the distribution of damage among all the cells sampled. While acknowledging these limitations for LX-PCR, no other method for routine evaluation of mtDNA damage is currently available.
In addition to mtDNA damage, other mitochondrial alterations directly related to AMD have been reported (Feher et al., 2006; Nordgaard et al., 2006; 2008). A transmission electron microscopy (TEM) analysis of human donor eyes affected by AMD showed a reduction in the number of mitochondria compared with age-matched controls with no AMD (Feher et al., 2006). In a proteomic analysis of the RPE, AMD-related changes were observed in the ATP synthase subunit that participates in oxidative phosphorylation (Nordgaard et al., 2008). The decrease in total ATP synthase subunits that coincides with the progression of AMD could lead to a malfunction of the ATP synthase complex. Since the ATP synthase complexes exist as oligomeric supercomplexes that maintain mitochondrial morphology and the mitochondrial membrane potential (Arselin et al., 2004; Bornhovd et al., 2006; Minauro-Sanmiguel et al., 2005), the decreased amount of the ATP synthase subunits with AMD could lead to defects in these critical processes. Another change in the mitochondrial proteome included the decline in the level of the mitochondrial heat shock protein (mtHsp70) observed in AMD RPE (Nordgaard et al., 2008). It functions as a molecular chaperone that regulates the ATP-dependent import of nuclear-encoded proteins into the mitochondrial matrix (Nordgaard et al., 2006, 2008; Kaarniranta et al., 2009). Therefore, decreased mtHsp70 levels could be detrimental to overall mitochondrial function and limit energy production in AMD.
Measurement of mitochondrial function in primary cultures of RPE provided direct evidence that energy production is significantly affected with AMD. Two different laboratories reported reduced mitochondrial function and ATP production in RPE cells isolated from human donors with AMD as compared with healthy subjects (Fig. 3, Ferrington et al., 2017; Golestaneh et al., 2017). Taken together, these results strengthen the hypothesis that mitochondrial damage and dysfunction may be involved in AMD pathology. Damage to RPE mitochondria associated with AMD not only decreased retinal bioenergetics, but could also disrupt intracellular calcium homeostasis and mitochondria-nuclear signaling, thereby having a more global impact on cell health (Rottenberg and Hoek, 2017; Fisher and Ferrington, 2018).
Fig. 3.
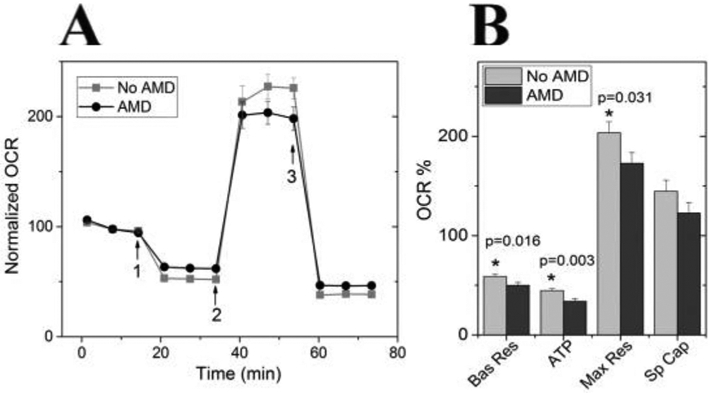
RPE from donors with AMD show reduced mitochondrial function. (A) Trace from an XF96 Extracellular Flux Analyzer shows the oxygen consumption rate (OCR) normalized to baseline for cells from non-diseased and AMD donors (No AMD n = 14; AMD n = 19). Arrows indicate injection of oligomycin (1), FCCP (2) and antimycin and rotenone (3) to perturb mitochondrial function. (B) Mitochondrial functional parameters were calculated from data shown in A. Probability values for significant differences, as determined by t-test comparing No AMD with AMD, is provided on the graphs. Bas Res = basal respiration; Max Res = maximal respiration; Sp Cap = spare capacity. All data are mean (±SEM). (* denotes p < 0.05). Data shown is similar to that in Ferrington et al. (2017), Redox Biol. publication.
This section summarized the studies to date showing a significant increase in mtDNA damage is associated with normal aging and is more extensive with AMD. The next section discusses how disturbed mtDNA damage repair (DDR) may play a central role in the regulation of mitochondria function in AMD (Chinnery et al., 2002; Blasiak et al., 2013).
4. Mitochondrial DNA damage and repair in AMD pathogenesis
Human mtDNA contains 13 intron-less polypeptide-encoding genes and several shorter genes coding for functional RNAs, including 12S and 16S ribosomal RNA and 22 tRNA. Therefore, it is very likely that damage induced in mtDNA will be localized in a coding sequence corresponding with either the polypeptide-encoding genes or the RNA, thereby limiting the production of fully functional proteins. Since the genes encode for subunits of ETC, the main source of energy in the cell, their damage can result in serious consequences for cell energetics.
As discussed previously, the mitochondria generate ROS as a byproduct of respiration. Therefore, mtDNA must be protected against damage or if damaged, must be repaired. Protection from ROS is provided by mitochondrial localized antioxidant enzymes, such as manganese superoxide dismutase (MnSOD), peroxiredoxins, catalase and glutathione peroxidases (GPXs): GPX1 and phospholipid-hydro peroxide GPX (PHGPX) (Holley et al., 2011). They neutralize ROS in a chain of reactions, in which MnSOD catalyzes the dismutation of superoxide radicals to hydrogen peroxide and molecular oxygen. Hydrogen peroxide is decomposed by peroxiredoxins that oxidize thioredoxin in this process. Hydrogen peroxide in mitochondria can be also removed by GPX1 and PHGPX. Previous research has reported a significant increase in both MnSOD and catalase in RPE from human AMD donors, suggesting the upregulation of these antioxidant enzymes was a compensatory response to the elevated oxidative stress induced by AMD (Decanini et al., 2007). Similar to nuclear DNA, which is protected from ROS damage by bound histones, nucleoid complexes of multiple proteins provide some physical protection to mtDNA from ROS-induced damage.
DNA damage elicits the response of a number of proteins that coordinate to repair the specific type of damage. This robust repair system in the nucleus includes: (i) base excision repair (BER) recovers damaged (oxidized, alkylated, hydrolyzed, deaminated) bases, (ii) nucleotide excision repair (NER) corrects lesions of double-stranded DNA (eg., pyrimidine dimers), (iii) mismatch repair (MMR) removes unpaired nucleotides, (iv) non-homologous end-joining (NHEJ) and homologous recombination both repair double-strand breaks in genomic DNA (Zinovkina, 2018). There are substantial differences in the DNA repair systems operating in the nucleus and mitochondria (Table 1).
Table 1.
A comparison of the DNA repair systems operating in the nucleus and mitochondria of the human nucleated cells
| Substrate | Repair system |
|
|---|---|---|
| Nucleus | Mitochondria | |
| Chemical modifications to DNA bases | BER1), DR | |
| UV photoproducts | NER | ? |
| Intrastrand crosslinks | NER | ? |
| Interstrand crosslinks | HRR + NER + FAP | ? |
| Mismatches, small loops | MMR | MMR (?) |
| Single-strand breaks | SSBR | ? |
| Double-strand breaks | NHEJ, HRR, SSA | HRR, MMEJ, ? |
Abbreviations used: BER, base excision repair; DR, direct repair; NER, nucleotide excision repair; HRR, homologous recombination repair; FAP, Fanconi anemia proteins; MMR, mismatch repair; SSBR, single-strand break repair; NHEJ, non-homologues end joining; SSA – single-strand annealing, MMEJ, microhomology-mediated end joining. The question mark indicates no evidence or uncertain occurrence.
BER proteins involve DNA glycosylases (UNG1, MYH, OGGI, NEIL1/2), endonucleases (APE1, FEN1, EXOG1), the helicase DNA2 and DNA polymerase γ. While these enzymes are found in both the mitochondria and nucleus, some have specific mitochondrial isoforms that are produced by alternative splicing. Other proteins that participate in BER include p53, which translocates into the mitochondria under conditions of oxidative stress where it interacts with DNA polymerase γ. Poly (ADP-ribose) polymerase (PARP1) is also involved in activation of the mitochondrial BER response to oxidative stress by interacting with DNA polymerase γ, transcription factor A mitochondrial (TFAM), and EXOG1.
Evidence for the presence of NHEJ, specifically the microhomology-mediated end joining (MMEJ), includes the mitochondrial localization of the main required enzymes, PARP1 and MRN (MRE11, Rad50-Nbs1)-CtlP complex (Zinovkina, 2018). Recently, biochemical evidence for the existence and activity of this pathway was provided (Tadi et al., 2016). Taken together, the presence of PARP1, active BER and MMEJ allows for repair of both single and double-strand breaks in mtDNA. While numerous enzymes from the other DDR pathways have been identified in the mitochondria, their exact function in this organelle is unknown or insufficiently studied. For example, the presence of some NER-related proteins in mitochondria suggests the possible action of NER, the most versatile pathway of DNA repair (Melis et al., 2013; Liu et al., 2015; Wu et al., 2018). Most but not all enzymes involved in homologous recombination are also present in the mitochondria, but to date, the existence of active recombination has not been definitively shown (Zinovkina, 2018). Thus, this area of research is being actively pursued due to the importance of DNA repair in health and disease.
Since accumulation of mtDNA damage is associated with normal and premature aging as well as serious human disorders, disturbed mtDDR may be involved in AMD (Fig. 1; Chinnery et al., 2002; Blasiak et al., 2013). While this possibility has not been extensively investigated, Lin et al. provides some evidence that mtDDR is reduced in aging and with AMD. Using cultured human RPE primary cells obtained from the macula and peripheral regions, they compared mtDNA damage and repair (Lin et al., 2011). While the extent of the damage in both regions increased with donor age, results show more damage to mtDNA in macular compared with RPE from the periphery. Moreover, efficacy of the repair of mtDNA damage decreased with age and was more reduced in macular compared with peripheral RPE. Heteroplasmic mtDNA mutations were more common in AMD than non-AMD eyes and were positively correlated with AMD severity. The authors concluded that defective mtDDR may be involved in AMD pathogenesis.
In summary, many reports point at mtDNA damage as an important contributing factor in AMD pathology (Fig. 1). Potential mechanisms responsible for the accumulation of mtDNA damage includes the decreased activity of antioxidant enzymes regulated by NFE2L2 and PGC-1α, reduced mtDNA repair proteins, or disturbance in clearance of damaged mitochondria. Several reports claim that mtDNA is more prone to damage than its nuclear counterpart and some suggest that mtDNA damage in the retina preferentially occurs in the macula than in the periphery (Lin et al., 2011). Another problem in the mutagenesis of mtDNA is the assessment of efficacy of DNA repair enzymes (Caston and Demple, 2017). They all are encoded by the nuclear genome, and after transcription and translation, they must be imported into the mitochondria. The conclusion of DNA repair deficiency on the basis of lowered overall expression of DNA repair enzymes may therefore be an underestimate of the content of active enzyme localized within the mitochondria. Therefore, mtDDR in AMD should be subjected to further studies as its many aspects have been unequivocally shown to be changed in this disease.
5. Mitochondrial haplogroups and AMD
The effect of mitochondrial sequence polymorphisms that are found in different populations, referred to as haplogroups, has also been studied. Haplogroups are populations of individuals who share a common maternal ancestor and are defined by their similarity in mtDNA sequence. The letter names of the haplogroups run from A to Z, being named alphabetically in the order of their discovery.
Several studies showed specific haplogroups demonstrated either a positive or negative association with AMD occurrence (Jones et al., 2007; Canter et al., 2008; SanGiovanni et al., 2009; Udar et al., 2009; Joachim et al., 2015). For example, the J haplogroup is linked with higher risk of AMD and the H haplogroup is linked with protection against this disease. A limitation of these reports is that they do not account for the influence of nDNA variants on the occurrence of AMD. To eliminate the impact of nDNA variants, Kenney et al. constructed cybrids using mitochondria-deprived ARPE-19 Rho0 cells merged with mitochondria from platelets of individuals belonging to either the H or J haplogroups (Kenney et al., 2013, 2014). In this experimental system, the nuclear genome was identical, but the cells contained the mitochondrial sequence that was specific for each haplogroup. They observed that J and H hybrids differentially expressed genes of the alternative complement pathway, inflammation, and apoptosis. J hybrids produced less ROS, had increased growth rate, and displayed a diminished expression of four genes involved in retinal degeneration as compared with their H counterparts. The authors also sequenced the entire mitochondrial genome to determine if rare mtDNA variants could be responsible for these results. They concluded that the changes they observed were not due to rare mtDNA variants, but rather to the mitochondrial genomic sequence associated with either the J or H haplogroup. These results show that the mitochondrial genome can significantly influence the expression of nuclear encoded genes, some clearly associated with increased risk for AMD.
6. Lipofuscin formation and monitoring in AMD
Lysosomes are key organelles that maintain the RPE-driven visual cycle (Sinha et al., 2016). They degrade phagocytosed photoreceptor outer segments (OS) in a circadian rhythm-controlled fashion via a process called heterophagy. The ROS formed due to constant light exposure, diurnal phagocytosis of lipid-rich OS, high retinal oxygen concentration and consumption, evoke malfunction of the lysosomes. Reduced capacity by lysosomes to degrade OS results in the accumulation of autofluorescent lipofuscin (Jarrett and Boulton, 2012). To date, it is largely accepted that lipofuscin is no longer only “normal age pigment”, but it is rather an indicator of RPE cell stress and a key biomarker in AMD (Kaarniranta et al., 2010).
Lipofuscin can have an effect on RPE through disruption of multiple pathways. Detection of lipid peroxidation end-products i.e., highly reactive electrophilic aldehydes like malondialdehyde (MDA) and 4-hydroxynonenal (HNE) in lipofuscin suggests a connection between its formation and increased oxidative stress (Schütt et al., 2002; Schütt et al., 2003; Ye et al., 2016). Lipofuscin also contains vitamin A-derived fluorophores, including A2-E, that decrease both heterophagy and the mitochondrial capacity to produce energy in RPE cells (Vives-Bauza et al., 2008; Kaarniranta et al., 2013; Marie et al., 2018; Alaimo et al., 2019). A2-E is also a photosensitizer that decreases lysosomal enzyme activity and disturbs cholesterol metabolism (Holz et al., 1999; Bermann et al., 2001; Lakkaraju et al., 2007; Guha et al., 2014; Choudhary et al., 2016; Ban et al., 2018; Lu et al., 2018; Ramachandra Rao et al., 2018).
Lipofuscin can be monitored using non-invasive, very sensitive, cost-effective, and rapid autofluorescence-based imaging techniques, which have become very important in ophthalmology clinics. Although there are multiple heterogenic fluorescent complexes in the RPE, the highest autofluorescence intensity comes from lipofuscin (Dysli et al., 2017). Every fluorophore is characterized by its own excitation and emission wavelength spectrum and exhibits an individual fluorescence lifetime. The fluorescence intensity depends on the molecular environment and the fluorophore's concentration. The clinical equipment measuring fundus autofluorescence (FAF) has an excitation wavelength of 488 nm and an emission bandwidth of 500–700 nm. Lipofuscin can be detected in that wavelength range, but it also covers the fluorescence of macular pigment, retinal carotenoids, different protein aggregates, and organelles. FAF shows an unspecific RPE cell emission, which is not only coming from lipofuscin. Therefore, the role of lipofuscin in AMD pathology may be difficult to determine based on FAF. However, FAF is very useful in clinics to do differential diagnosis for retinal diseases, since some of them have typical FAF characteristics, such as Stargard's disease, central serous chorioretinopathy and retinitis pigmentosa. FAF may provide data from the lesions associated with aging, stress conditions, or toxic effects in RPE cells.
Clinically advanced dry AMD, referred to as geographic atrophy (GA), possess elevated FAF around the atrophic lesion (Fig. 4). Currently, there are critical differences in opinions regarding the toxicity and influence of lipofuscin on the progression of AMD (Holz et al., 2007; Rudolf et al., 2013; Fleckenstein et al., 2018). Holz et al. showed that distinct phenotypic FAF patterns correlated with dry AMD progression and thus might be used as a prognostic factor (Holz et al., 2007). The amount of FAF in RPE adjacent to GA lesions correlated with the spread of the atrophic area during subsequent follow-up visits. Rudolf et al. documented that FAF around GA did not predict cells undergoing death. This group found that the increased FAF around the lesions was due to vertically superimposed cells and cellular fragments (Rudolf et al., 2013). The amount of autofluorescence, cell density, and packing geometric analysis revealed the uncoupling of lipofuscin content and RPE or photoreceptors cell numbers in AMD (Ach et al., 2014). Redistribution and loss of autofluorescent granules were present in the earliest AMD changes (Ach et al., 2015), and decreased autofluorescence was in line with the increased severity of RPE dysmorphia (Gambril et al., 2019).
Fig. 4.
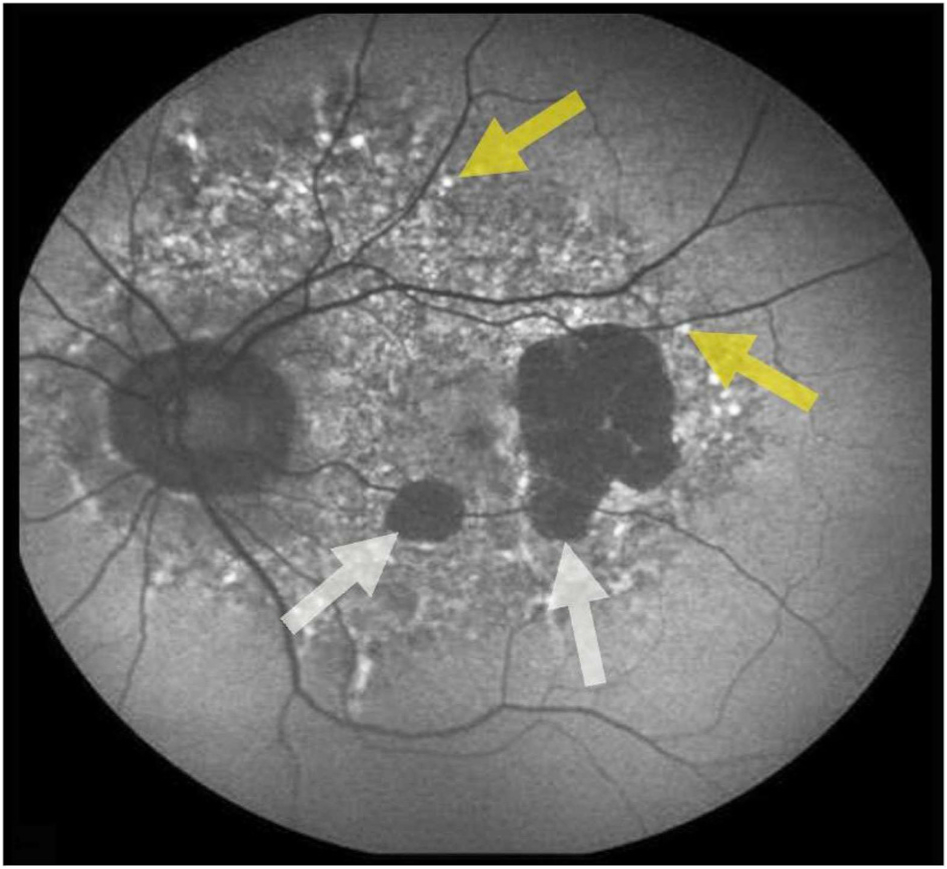
Fundus autofluorescence (FAF) image from a patient suffering geographic atrophy (GA; white arrows) in advanced AMD. Yellow arrows indicate increased autofluorescence due to lipofuscin accumulation and drusen. Photograph by Kai Kaarniranta.
Altogether, the accumulation of autofluorescent lipofuscin is a clinical hallmark of AMD. Lipofuscin reduces the lysosomal clearance capacity and increases cellular stress in RPE. FAF is used in clinics for diagnosis, differential diagnosis and following the progression of retinal diseases.
7. Mitochondria-lysosome axis in the regulation of protein clearance
Autophagy is a highly dynamic lysosomal catabolic process (Johansen and Lamark, 2020). It contributes to intracellular quality control and housekeeping that is extremely important in quiescent cells like RPE cells (Kaarniranta et al., 2013; Kim et al., 2013; Rodríguez-Muela et al., 2013). Autophagy may acts at basal levels, but is activated in response to environmental challenges. It is divided into non-selective and selective autophagy. Autophagy was initially characterized as non-selective bulk degradation that recycles amino acids and attempts to compensate for the lack of nutrients (Zaffagnini and Martens, 2016). Currently, we know that a selective autophagy is crucial by mediating the degradation of aggregated proteins, organelles, and invading pathogens (Johansen and Lamark, 2020). The molecular machinery for selective autophagy must ensure efficient recognition and sequestration of the cargo within autophagosomes. In selective autophagy particular substrate are targeted into the autophagosome by selective autophagy receptors.
Autophagy is divided into microautophagy, chaperone-mediated autophagy, and macroautophagy (Kaarniranta et al., 2013; Boya et al., 2016). Macroautophagy can be categorized into several selective types of autophagy according to their degradation targets, such as aggregates (aggrephagy), lipid droplets (lipophagy), exogenous pathogens (xenophagy), and organelles (for instance nucleophagy, ribophagy and mitophagy). Autophagy directs degradation of unnecessary or damaged cellular molecules and organelles by delivering them to lysosomes, in which they are then broken down by lysosomal hydrolytic enzymes (Fig. 5). Macroautophagy is mediated by the formation of a double membrane autophagosome. Cargo destined for degradation is sealed in autophagosomes and when combined with lysosomes, results in a structure called an autolysosome. Lysosomal enzymes degrade cargo.
Fig. 5.

The PINK1-Parkin-regulated mitophagy. Chronic oxidative stress and ROS evokes misfolding of proteins in RPE cells. Heat-shock proteins (Hsps) attempt to refold damaged proteins, but if not successful, the misfolded proteins are ubiquitinated (Ubq) and targeted for proteasomal clearance. Once proteasome activity is decreased or its capacity exceeded, proteins start to aggregate and are degraded via autophagy. Mitochondrial damage results in the accumulation of PINK1 and the activation of Parkin. Mitophagy receptors, such as OPTN (optineurin), TAXBP1 (Tax-1-binding protein 1), NDP52 (nuclear domain 10 protein 52), p62 and NBR1 (Neighbor of BRCA1 gene 1 protein), and contains an ubiquitin-binding site that allows their attachment to Parkin-ubiquitinated mitochondria. This process initiates the phagophore formation leading to sealed material degradation. Rab 5 and Rab7 control endosome maturation that finally leads to fusion of lysosomes and autophagosomes.
Decreased autophagy capacity has been strongly linked to the pathology of AMD (Fig. 5; Wang et al., 2009; Kaarniranta et al., 2013; Viiri et al., 2013; Mitter et al., 2014; Valapala et al., 2014; Ferrington et al., 2016b). The RPE-specific autophagy knockout leads to cellular degeneration resembling AMD (Kim et al., 2013). Since lysosomal enzyme activity is crucial to autophagy flux, lipofuscin components may ultimately decrease autophagy clearance capacity. This statement is supported by reports showing impaired autophagy coincides with lipofuscin granules and drusen deposits in RPE cells (Karlsson et al., 2013; Mitter et al., 2014; Felszeghy et al., 2019). Additionally, a decline in autophagic capacity accompanied by an increase in ROS production can elicit protein aggregation and activation of inflammasomes in RPE cells (see section 13; Piippo et al., 2014, 2018). Conversely, another study showed that autophagy induction alleviates A2E-induced RPE damage and reduced production of inflammatory cytokines and vascular endothelial growth factor A (VEGFA; Zhang et al., 2015).
To avoid the vicious cycle of extensive ROS production with further mitochondrial damage and ROS release, dysfunctional mitochondria must be removed from cells via mitophagy, a specialized form of selective macroautophagy, where the main cargo are mitochondria (Fig. 6; Lemasters, 2005; Gurubaran et al., 2020). In selective autophagy, specific cargo is targeted and ubiquitinated, later followed by recognition by/through autophagy adaptor molecules, such as autophagy adaptor protein p62/SQSTM1, followed by autolysosomal degradation (Viiri et al., 2013).
Fig. 6.
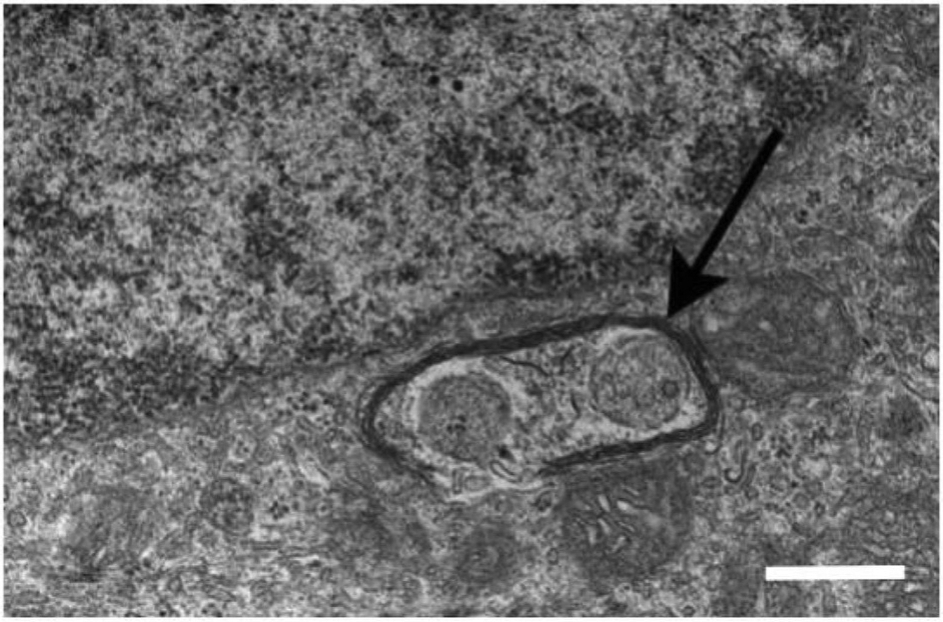
A transmission electron micrograph shows damaged mitochondria enclosed by autophagosome (arrow) in the RPE of a 20-month-old Cryba1 knockout mouse. The scale bar = 500 μm. The electron micgraph has been modified from the Sinha et al. (2016) Exp. Eye Res. publication.
Dysfunctional mitochondria and their exacerbated generation of ROS are among the earliest events in the progression of the numerous neurodegenerative diseases (Ratnayaka et al., 2007; Gureev and Popov, 2019). Since the maintenance of healthy mitochondria is essential to help RPE cells survival, dysfunction of this process in AMD can have harmful consequences. Under stress, both mitochondrial fission and fusion work together to maintain functional mitochondria (Kageyama et al., 2012). An increase in mitochondrial fission leads to mitochondrial fragmentation, whereas fusion results in mitochondrial elongation. Failure in any of the components of this machinery may lead to RPE degeneration (Brown et al., 2019). Once mitochondria are damaged, the inner membrane depolarizes and activates mitophagy, which does not always follow the canonical selective autophagy characteristics for clearance of protein aggregates (McWilliams et al., 2019). The PINK1-Parkin-derived pathway of mitophagy is commonly activated by ROS (Fig. 5; Hyttinen et al., 2018). First, the kinase PINK1 is stabilized in the outer mitochondrial membrane (OMM) after mitochondrial membrane potential disruption followed by PARKIN (a cytosolic E3-ubiquitin ligase) phosphorylation, and finally ubiquitination. These processes initiate the isolation membrane formation around the damaged mitochondrion by selective autophagy receptors. OMM attach to autophagosomes via their microtubule-associated protein 1 light chain 3 (LC3) interacting region (LIR) motifs. One group of mitophagy receptors contains an ubiquitin-binding domain, which allows its attachment to Parkin-ubiquitinated mitochondria, p62/SQSTM1, NBR1 (Neighbor of BRCA1 gene 1 protein), NDP52 (nuclear domain 10 protein 52), AMBRA1 (activating molecule in Beclin1-regulated autophagy), TAX1BP1 (Tax1 binding protein 1) and OPTN (optineurin) (Zaffagnini and Martens, 2016; Johansen and Lamark, 2020). During maturation, members of the Rab family (Ras-related guanosine tri-phophate-binding proteins) and SNAREs complex (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) control lysosome and autophagosome fusion process (Nakamura and Yoshimori, 2017; Gurubaran et al., 2020). Rab proteins are important regulators of vesicular transport. Rab7 binds to autophagosome membrane and regulates late-state fusion with the enzymatic lysosomes (Fig. 5; Hyttinen et al., 2013). Receptor-mediated mitophagy, also known as PINK1/Parkin-independent pathway, contain transmembrane domains and upon expression they constitutively localize to OMM. The LIR motif activity of mitophagy receptors is regulated by phosphorylation for binding to Bnip3 (BCL2 interacting protein 3), Nix/Bnip3L (BCL2-like 3), FUNDC1 (FUN14 domain containing 1) and Bcl2L13 (BCL2-like 13), FKBP8 (FK506-binding protein 8), PHB2 (prohibitin 2), NLRX1 (Nod-like receptor X1), AMBRA1, cardiolipin and ceramide that enhance mitophagy (Gurubaran et al., 2020; Johansen and Lamark, 2020).
The loss of mitochondrial membrane potential is one of the mitop0hagy-inducing signals (Azad et al., 2009; Twig and Shirihai, 2011). The induction of mitophagy triggered by rotenone, which blocks the complex I in ETC (Lee et al., 2014), is in line with the idea of the protective role of mitophagy in RPE cells and inversely, of destructive consequences upon dysfunctional intracellular clearance (Hyttinen et al., 2018). Interestingly, NFE2L2, a ROS sensor, can regulate the expression of PINK1 due to four AU-rich elements (ARE) in the promoter of the corresponding gene (Murata et al., 2015). Therefore, there is evidence that activation of NFE2L2 can directly regulate mitophagy.
Mouse models with genetic manipulation of gene expression for proteins involved in mitochondrial maintenance (PGC-1α), antioxidant response (PGC-1α, NFE2L2), or lysosomal acidification (Cryba1) have provided valuable insight into their effect on mitophagy (Sinha et al., 2016; Felszeghy et al., 2019; Gurubaran et al., 2020). Our data reveal that the deficiency in NFE2L2/PGC-1α and Cryba1 genes, relatively to increased oxidative stress, decrease mitophagy in mouse RPE cells. Recently, it was demonstrated that p62/SQSTM1 is able to bind directly to ubiquitinated mitochondrial OMM proteins (Lazarou et al., 2015). p62/SQSTM1 is recruited to mitochondrial clusters and is essential for the clearance of mitochondria in Parkin-dependent mitophagy (Geisler et al., 2010). p62/SQSTM1 expression is also controlled by NFE2L2 due to the presence of ARE in its promoter region (Jain et al., 2010). It seems that NFE2L2 and PGC-1α are central proteins in the regulation of selective autophagy in RPE cells.
8. NFE2L2 in mitochondrial quality control
NFE2L2/NRF2 is the key transcription factor in the sensing of oxidative stress (Lambros and Plafker, 2016). NFE2L2 is sometimes designated as NRF2, which can be missed with nuclear respiratory factor 2 (NRF2), which functions as a transcription factor activating the expression of key metabolic genes regulating cellular growth and nuclear genes required for mitochondrial respiration, DNA transcription, and replication (Baldelli et al., 2013). NFE2L2 was found to associate with the outer mitochondrial membrane and protect mitochondria from oxidant injury likely through direct interaction with mitochondria (Figs. 7 and 8; Strom et al., 2016). Upon activation, NFE2L2 is released from NFE2L2-KEAP1 (kelch-like ECH-associated protein 1) and the mitochondrial outer membrane serine/threonine protein phosphatase 5 (PGAM5) complex, allowing for the translocation of NFE2L2 from the cytosol into the nucleus. NFE2L2 can bind to an ARE to promote the transcription of more than 200 genes, including detoxification and antioxidant enzymes (heme oxygenase-1 (HO-1), NAD(P) H:quinone oxidoreductase-1 (NQO1), sulfiredoxin-1 (SRXN1), superoxide dismutase 2 (SOD2), PGAM5, per-oxiredoxins 3 and 5 (PRDX3 and PRDX5)), and enzymes involved in GSH metabolism (glutathione S-transferases, glutathione-synthesizing enzymes glutamate-cysteine ligase catalytic subunit (GCLC) and glutamate-cysteine ligase modifier subunit (GCLM)) (Sihvola and Levonen, 2017; Silva-Palacios et al., 2018; Di Malta et al., 2019).
Fig. 7.

Schematic presentation of the oxidative stress influence on AMPK, NFE2L2, PGC-1 α, and autophagy pathway interactions in RPE. In response to oxidative stress, AMPK is phosphorylated, which subsequently phosphorylates NFEL2L and PGC-1α leading to their nuclear translocation and activation of genes that regulate mitochondrial biogenesis and antioxidant protein production. NFEL2L and PGC-1α also have a key role in the regulation of autophagy. Failure in AMPK –mediated signaling leads to disturbed proteostasis in RPE cells.
Fig. 8.
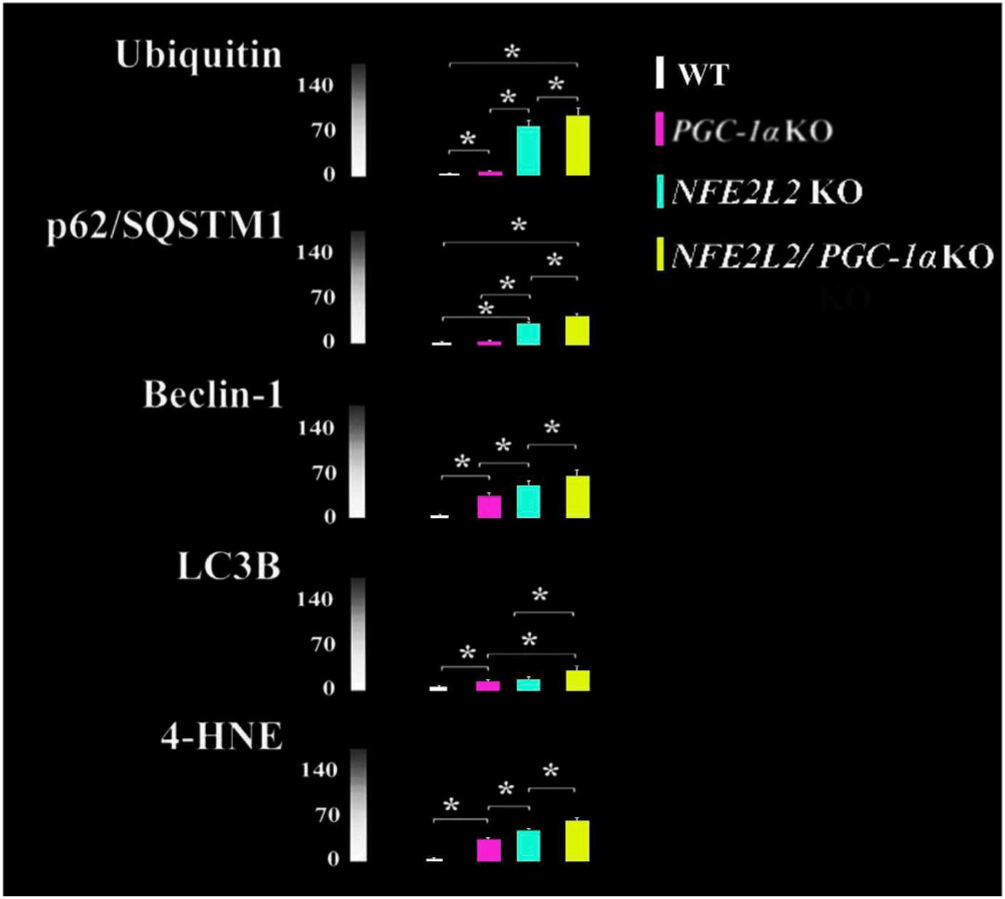
Increased protein aggregation (Ubiquitin), autophagy (p62/SQSTM1, Beclin-1, LC3B) and oxidative stress (4-HNE) markers in the RPE cells of PGC-1α KO, NFE2L2 KO and NFE2L2/PGC-1α dKO mice. Data represent average intensities from three different animals per genotype and n = 30. *p < 0.001 one-way ANOVA followed by Games-Howell post hoc test. Results are expressed as means ± SD. Colums represent similar results shown in the Felszeghy et al. (2019) Redox Biology publication.
Under basal conditions, cytosolic KEAP1 functions as an adapter for the E3 ubiquitin ligase and constitutively targets NFE2L2 for ubiquitylation and degradation via the ubiquitin proteasome system (UPS) (http://cancerres.aacrjournals.org/content/79/5/889.long; Kobayashi et al., 2004). However, in oxidative stress a conformational change in KEAP1 structure prevents the degradation of NFE2L2 by the proteasome. De novo synthesized NFE2L2 accumulates and translocates to the nucleus where it heterodimerizes and subsequently activates AREs. Since NFE2L2 also activates proteasomes and lysosomal autophagy, it regulates the quality control of proteins via the removal of misfolded and ubiquitinated proteins and damaged cellular organelles (Dick, 2017). It has been shown that the depletion of NFE2L2 disturbs mitochondrial motility, which is linked to degenerative diseases like AMD (Sachdeva et al., 2014; O'Mealey et al., 2017). Common NFE2L2-KEAP1 pathway induction and subsequent stimulation of the antioxidant response is estimated to prevent or slow the progression of degenerative and immunological processes and thus promote longevity (Murray et al., 2018).
High mitochondrial ROS production, combined with impaired antioxidant systems, results in detrimental protein aggregation (Scherz-Shouval et al., 2007; Korovila et al., 2017). Recently, impaired proteolysis in response to chronic oxidative stress has been suggested as a key contributor in AMD (Ferrington et al., 2016b). The proteasome and the lysosomal/autophagy degradation systems share the major responsibility of maintaining cellular proteostasis (Blasiak et al., 2019). Both systems recognize and actively select the material designated for degradation and recycling in cells. Degradation of damaged and misfolded proteins via the proteasome occurs in coordination with molecular chaperones (Ferrington et al., 2016b). Evidence of increased content of proteasome and heat shock proteins in the RPE from human donors with AMD is consistent with increased oxidative stress and extensive protein damage with this disease (Decanini et al., 2007). The proteasome degrades 80–90% of cellular proteins in eukaryotes, including short-lived, abnormal, denatured or damaged soluble proteins (Ciechanover, 2012). This proteolytic system also controls processes essential for cell viability, cell-cycle regulation, signal transduction, synaptic plasticity, pigmentation and circadian regulation (Juuti-Uusitalo et al., 2017; Gureev and Popov, 2019). Prior to degradation by the proteasome, the 76-amino acid ubiquitin polypeptide is covalently attached to the protein substrate via the ubiquitin-activating (E1), ubiquitin-conjugating (E2) and the ubiquitin ligase (E3) enzyme. Accumulated ubiquitin levels can be used as an surrogate indicator of the capacity of proteasomes to degrade damaged proteins. Data that suggests a decline in proteasome capacity with AMD, includes the increased ubiquitin levels in drusen localized under the macula from donors with AMD (Fig. 2E; Mullins et al., 2000; Viiri et al., 2013). In our NFE2L2/PGC-1α dKO mouse model of AMD, levels of protein ubiquitination were significantly increased in both RPE and photoreceptors (Fig. 8; Felszeghy et al., 2019).
In addition to basal homeostasis, autophagy increases in cellular stress conditions, which often coincides with the induction of the NFE2L2 pathway (Chen and Maltagliati, 2018). Beneficial effects of NFE2L2 pathway activation and autophagy induction have been recognized in cellular stress responses. For example, polyunsaturated fatty acids transiently increase ROS, and induce both NFE2L2 and autophagy (Johansson et al., 2015). These cellular changes coincide with decreased damage in RPE cells (Wang et al., 2014a, 2016a; Johansson et al., 2015; Kaarniranta et al., 2019). Other compounds with similar properties studied in RPE include isothiocyanate sulforaphane (O'Mealey et al., 2017), dimethyl fumarate (Lastres-Becker et al., 2016), triterpenoid derivatives RS9, RTA408 and glycyrrhizin (Liu et al., 2016; Saito et al., 2018; He et al., 2019), carotenoids fucoxanthin, lutein, lycopene, zeaxanthin and astaxanthin (Li et al., 2013; Zou et al., 2014; Yang et al., 2016; Frede et al., 2017), flavonol compounds fisetin and pinosylvin, (Hytti et al., 2015, 2017; Koskela et al., 2014), adenosine monophosphate kinase (AMPK) activators PF-06409577 and AICAR (5-aminoimidazole-4-carboxamide ribonucleoside) (Viiri et al., 2013, Li et al., 2018; Marchesi et al., 2018) are shown in Table 2. Isothiocyanate sulforaphane, lutein, zeaxanthin and AMPK activation effectively protects mitochondria under oxidative stress in RPE cells (Zou et al., 2014; O'Mealey et al., 2017; Su et al., 2014; Xu et al., 2018). Correspondingly, the deficiency of NFE2L2 in our mouse model results in reduced autophagic flux that may be due to impaired lysosomal enzyme activity (Figs. 7 and 8; Krohne et al., 2010; Dayalan Naidu et al., 2017; Felszeghy et al., 2019). It is important to note that we did not block autophagy flux with the specific autophagy inhibitors in our animal models (Klionsky et al., 2016). The use of autophagy flux inhibitors are more commonly used in cell culture conditions. Our interpreation for the decreased autophagy flux in vivo is based on the reduced number of autophagolysosomes in TEM and the increased autophagy markers in confocal micrographs (Felszeghy et al., 2019). Similar autophagy marker analysis has been used in ex vivo retina studies (Gómez-Sintes et al., 2017).
Table 2.
NFE2L2 activators.
| Compound | Effect | Model | Reference |
|---|---|---|---|
| Isothiocyanate sulforaphane | Activates KEAP-NFE2L2-ARE pathway Inhibits mitochondrial fission |
RPE-1 cells | O'Mealey et al. (2017) |
| Dimethyl fumarate | NFE2L2 activator Neuroprotection |
Primary microglias NFE2L2 gene modified mice |
Lastres-Becker et al. (2016) |
| RS9 | NFE2L2 activator Autophagy induction |
ARPE-19 cells | Saito et al. (2018) |
| RTA408 | Activates NFE2L2 Prevents apoptosis |
Primary human RPE cells |
Liu et al., 2016 |
| Glycyrrhizin | Increases expression of NFE2L2 and HO-1 | ARPE-19 cells C57BL/6 mice |
He et al. (2019) |
| Fucoxanthin | Suppress VEGF production Resists senescence Improves phagocytosis Reduces ROS production |
ARPE-19 cells Rabbit model |
Liu et al. (2016c) |
| Lutein | Activates NFE2L2 | ARPE-19 cells9 | Frede et al. (2017) |
| Lycopene | Inhibits nuclear factor-kappa B Activates NFE2L2 |
ARPE-19 cells | Yang et al. (2016) |
| Zeaxanthin | Improves anti-oxidant capacity Prevent cell death |
ARPE-19 cells | Zou et al. (2014) |
| Astaxanthin | Reduces ROS production Activates KEAP-NFE2L2-ARE pathway Prevents apoptosis |
ARPE-19 cells | Li et al. (2013) |
| Fisetin | Attenuates inflammation | ARPE-19 cells Primary human RPE cells |
Hytti et al. (2015) Hytti et al. (2017) |
| Pinosylvin | Protects against oxidative stress | ARPE-19 cells | Koskela et al., 2014 |
| PF06409577 | Activates AMPK Protects from UV radiation |
ARPE-19 cells | Li et al. (2018) |
| AICAR | Activates autophagy Decreases protein aggregation |
ARPE-19 cells |
Viiri et al. (2013) Marchesi et al. (2018) |
Proteasomal and autophagy clearance are connected via the scaffolding adaptor protein p62/SQSTM1 (Jain et al., 2010). p62/SQSTM1 is a multifunctional protein that undergoes phosphorylation prior to binding with ubiquitin and LC3 and subsequent degradation of protein aggregates, malfunctioning mitochondria, and pathogens in autophagosomes (Seibenhener et al., 2004; Matsumoto et al., 2011; Myeku and Figueiredo-Pereira, 2011). Proteasome inhibition induced ubiquitinated protein aggregates are degraded by autophagy in human RPE cells (Viiri et al., 2010, 2013). In line with accumulated ubiquitinated protein conjugates, p62/SQSTM1 accumulates in RPE cells of AMD donors having drusen maculopathy, suggesting suppressed proteasomal and autophagy activity (Viiri et al., 2013). In addition to autophagy regulation, p62/SQSTM1 stabilizes NFE2L2 through its binding with KEAP1, blocks the Keap1-NFE2L2 interaction, and activates the expression of genes having AREs (Lau et al., 2010; Komatsu et al., 2010). SQSTM1/p62 promotes autophagic degradation of KEAP1 and the activation of the NFE2L gene response. This forms a positive feedback loop for the activation of NFE2L2 via autophagy-related SQSTM1/p62 in response to oxidative stress (Ichimura and Komatsu, 2018; Shah et al., 2018). Moreover, NFE2L2 positively regulates p62/SQSTM1 gene expression, implying a positive feedback loop (Jain et al., 2010).
Various autophagy-related genes (Atg) have ARE sequences in their promoter regions, and several of these genes are known to be regulated by NFE2L2, including: Unc-51 like autophagy activating kinase (ULK1; autophagy initiation) p62/SQSTM1 (cargo recognition), Atg7 and gamma-aminobutyric acid receptor-associated protein-like 1 (GABAR-APL1; autophagosome formation), Atg2B and Atg5 (elongation), and Atg4D (autolysosome clearance) (Wang et al., 2007; Pajares et al., 2016). Our recent observations show an increase in NFE2L2 gene activity and subsequent upregulation of ROS-induced proteins that prevent damage has been found in RPE cultures from AMD donor eyes (Ferrington et al., 2017). Moreover, our data indicate that the marker of oxidative stress, 4-HNE, as well as autophagy markers p62/SQSTM1, Beclin-1 and LC and protein aggregation marker ubiquitin were significantly elevated in NFE2L2 KO compared to PGC-1α KO mice suggesting oxidative stress, insufficient proteasome function and autophagic clearance (Fig. 8). NFE2L2/PGC-1α double knockout (dKO) mice had the highest accumulation of all markers studied compared with single KO, suggesting the dKO mice had the most extensive oxidative stress and greater defects in the proteasome/autophagy clearance (Felszeghy et al., 2019). Ultrastructural analysis indicated an increased ratio of the damaged and healthy mitochondria in dKO RPE cells. We also observed that the dKO has larger autolysosomes, reduced basal infoldings and a moderate increase in lipofuscin level compared to intact normal cell structures in WT RPE cells (Fig. 9). Additionally, the number of melanosomes was higher in dKO mice. Moreover, Bruch's membrane elastic and collagenous layers were not often observed, and increased thickness of Bruch's membrane was detected along with an accumulation of electron dense amorphous material in dKO animals.
Fig. 9.
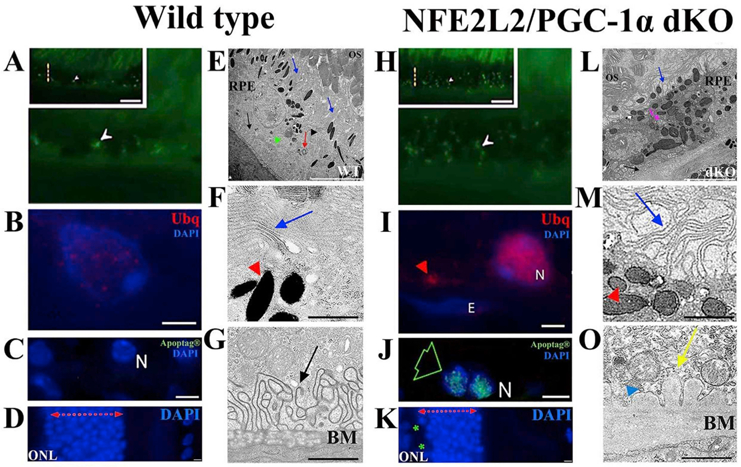
The pathological changes of RPE in NFE2L2/PGC-1α dKO mice. The representative confocal microscopic images of one-year old (A–D) and (H–K) dKO samples indicate various dry AMD resembling signs. The accumulation of intracellular lipofuscin-like material (white arrowhead; H), extracellular ubiquitin (Ubg) positivity (read arrowhead; I), restricted apoptosis (green arrow; J) and outer nuclear layer (ONL) atrophy (K) can be observed in the dKO samples. The TEM images of the WT (E–G) and dKO (L–O) samples. The basal folding of RPE is well-preserved in WT (E,G), while their disruption occurs in dKO (L,O). The thicker-disrupted Bruch's membrane (BM) of the dKO sample is shown in image L (black arrow), where electron-dense amorphous debris (yellow arrrow; O) and the membranous debris (blue arrowhead) are abundant compared to the WT sample (G). Alterations of microvilli structures (blue arrow; E,L), increased number of melanin pigments (red arrowheads; F,M), the damaged photoreceptor layer (E,L), enlarged vacuole-like structures (pink arrow; L and blue arrows; F,M) are more visible in dKO. N means nuclei and E endothelial cell nuclei. The scale bars for A, H = 5 μm, B, I and I–K = 2 μm, C, J = 5 μm, D, K = 2 μm, E,L = 5 μm, F, M and G, O Data represent similar results shown in the Felszeghy et al. (2019) Redox Biology publication.
Data from human donor eyes reveal that during AMD development, many cytoprotective mechanisms are first upregulated, including antioxidant production, molecular chaperone production, and improved clearance. Once the disease shifts to the chronic state, cytoprotection is followed by a decreased capacity to respond to stress. Our recent studies in mouse models suggest that the most promising target for pharmacological intervention on mitochondria is the NFE2L2 signaling cascade (Figs. 7 and 8). The rationale for this statement includes that NFE2L2 participates in the maintenance of mitochondrial homeostasis, which is provided by an optimal ratio in the processes of mitochondrial biogenesis and mitophagy, as well as the optimal ratio of ROS production and ROS scavenging. As shown previously, NFE2L2 activators are capable of modulating these processes and maintaining mitochondrial homeostasis in neurons (Gureev and Popov, 2019). Since, the RPE is composed of quiescent cells, they could be a viable option for treating AMD patients early in the disease.
9. PGC-1α in mitochondrial quality control
The peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family consists of PGC-1α, PGC-Ιβ and PRC (PGC-1-related coactivator). These proteins control mitochondrial biogenesis, respiratory function, and the mitochondrial antioxidant defense system. PGC-1α enhances RPE metabolic functions and resistance to oxidative stress through the induction of oxidative metabolic genes, antioxidant enzymes, and regulating autophagy (Fig. 8; Egger et al., 2012; Roggia and Ueta, 2015; Iacovelli et al., 2016; Felszeghy et al., 2019; Rosales et al., 2019). Deficiency of PGC-1α increases mitochondrial damage, as well as the oxidative stress markers 3′-nitrotyrosine and 4-HNE (Lu et al., 2010; Felszeghy et al., 2019.). The activation of the energy sensor AMPK induces autophagy and regulates PGC-1α activities (Jäger et al., 2007; Viiri et 2013; Marchesi et al., 2018). Revealing the importance of PGC-1α in the pathogenesis of AMD, iPSC-derived RPE cells isolated from dry AMD patients showed decreased PGC-1α expression (Golestaneh et al., 2016).
Our global KO mouse models for PGC-1α exhibited dry AMD-like degenerative signs but were not as extensive as that observed with NFE2L2 or NFE2L2/PGC-1α deficiency (Figs. 8 and 10; Felszeghy et al., 2019). However, data from Ferrington's lab indicate that primary RPE cells isolated from AMD donors expressed higher levels of PGC-1α and were more resistant to oxidative stress (Fig. 3, Ferrington et al., 2017). This reveals the capacity of RPE cells to respond to increased oxidative stress by different defense systems as we have previously documented (Decanini et al., 2007; Viiri et al., 2013). Individual genetic variations and severity of AMD may affect RPE stress responses (Kaarniranta et al., 2018a,b). Recent papers indicate that PGC-1α is required to maintain the functional and phenotypic status of RPE by supporting the cells' oxidative metabolism and autophagy-mediated repression of epithelial-mesenchymal transition (Zhang et al., 2018; Rosales et al., 2019). PGC-1α cooperates with p53 to attenuate the effects of oxidative stress and its consequences for mitochondria (Kaarniranta et al., 2018a).
Fig. 10.
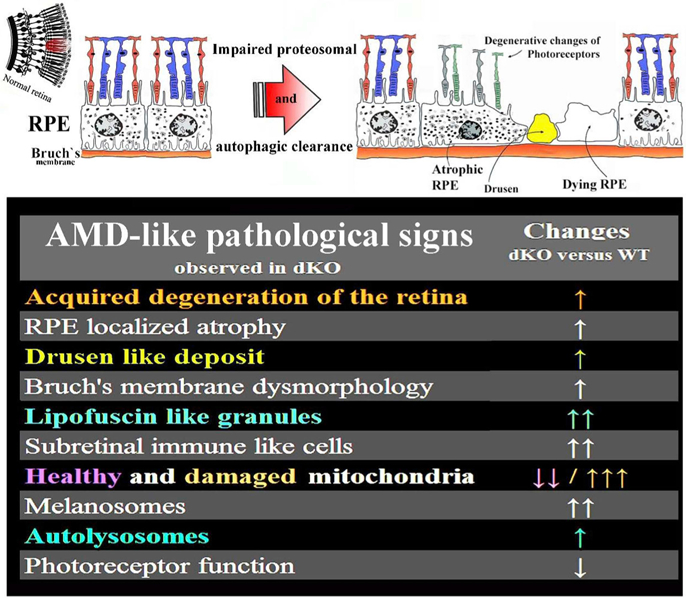
Graphical summary of the AMD hallmarks detected in one-year-old NFE2L2/PGC-1α gene modified mice. Summary modified from the Felszeghy et al. (2019) Redox Biology publication.
The PGC-1α promoter contains consensus sequences recognized by NFE2L2 (St Pierre et al., 2006). PGC-1α and NFE2L2 have overlapping functions in promoting the mitochondria protective response (Fig. 8; Baldelli et al., 2013). Similar to NFE2L2, PGC-1α induces expression of several genes coding for antioxidant enzymes, such as SOD2, SOD1, Gpx1and uncoupling proteins (UCP1/2) thus giving mechanistic evidence for their overlapping functions in the regulation of the antioxidant system (Baldelli et al., 2013; Ježek et al., 2018). Notably, NFE2L2 expression is controlled by PGC-1α suggesting a feed-forward loop promoting expression of protective genes, thereby amplifying the effect. The activation of the PGC-1α/NFE2L2 loop increases the activity of the proteasome and autophagy leading to decreased protein aggregation, a characteristic of aging and neurodegenerative diseases. Our PGC-1α and NFE2L2 KO mice support this evidence since they both show increased ubiquitinated protein conjugates, lipofuscin- and drusen-like aggregate formation, relatively impaired autophagy flux, and recruitment of inflammatory cells (Felszeghy et al., 2019). All these effects suggest altered protein-organelle quality control in RPE cells, which has been suggested to contribute to AMD (Ferrington et al., 2016b). Mitochondrial quality control may be improved by AMPK, which is a master regulator of autophagy and mitophagy. Sirtuin 1 (SIRT1) phosphorylation by AMPK leads to deacetylation and activation of the PGC-1α. AMPK is a putative pharmacological target in the prevention of neurodegenerative inflammatory disease (Kaarniranta et al., 2018a; Salminen et al., 2012).
10. Mitochondrial dysfunction in the induction of inflammation
Increasing evidence reveals that protein damage and aggregation induce inflammation in AMD (Anderson et al., 2010; Ferrington et al., 2016b; Chen et al., 2019). Para-inflammation is a beneficial host defense that can be shifted to chronic inflammation, which is associated with the pathological processes of advanced AMD. Inflammation is a rapid response to factors endangering cellular homeostasis, such as damaged macromolecules, dysfunctional mitochondria, as well as prolonged oxidative stress (Shadrach et al., 2013; Mao et al., 2014; Kauppinen et al., 2016). Currently, it is well known that mitochondrial damage and ROS production induce inflammation by activating the NOD-like receptor family, pyrin domain containing 3 (NLRP3) signaling pathway in RPE cells (Kauppinen et al., 2012; Shi et al., 2015; Wang et al., 2016b, 2017). Due to aging and AMD pathology, autophagy and antioxidant systems decline, resulting in the accumulation of damaged mitochondria and its subsequent harmful effects, such as ROS production and inflammasome activation (Zhou et al., 2011; Lee et al., 2012; Datta et al., 2017). Degenerating RPE cells with dysfunctional autophagy and mitochondria are recognized as dangerous material in the tissue since they can induce inflammasome activation in surrounding macrophages (Liu et al., 2016a; Wang et al., 2019). On the other hand, extensive autophagy evokes cell death in RPE, subsequently triggering cytokine production in macrophages (Szatmári-Tóth et al., 2016; Szatmári-Tóth et al., 2019). Additionally, specific haplogroups arising from inherited variation in mitochondrial DNA can participate in the pathogenesis of AMD, e.g., through altered complement functions and their predisposition to promote inflammasome signaling (Kenney et al., 2013, 2014; Malik et al., 2014; Atilano et al., 2015, Gao et al., 2015). Finally, challenges to mitochondrial function have been shown to result in changes in nuclear inflammatory genes expression and the development of the “inflamm-aging” RPE cell phenotype (Miceli and Jazwinski, 2005; Cannizzo et al., 2011; Salminen et al., 2012).
Mitochondria can contribute to inflammation in multiple ways. Inflammasome activation is stimulated by ATP, oxidized mtDNA, or cardiolipin (Kauppinen et al., 2016). Furthermore, mtDNA-induced toll-like receptor 9 (TLR9) activation promotes interferon regulatory factor 3 (IRF3)-mediated type I interferon (IFN) release, the production of inflammatory cytokines through nuclear factor-kappa B (NF-κB) or the mitogen mitogen-activated protein kinase (MAPK)-mediated signaling following mtDNA-activated cyclic guanosine monophosphate-adenosine monophopshate synthase/stimulator of interferon genes (cGAS/STING) pathway, and formyl peptide-driven chemotaxis (Bader and Winklhofer, 2019). Of those, especially inflammasome signaling and the mtDNA-mediated cGAS/STING pathway, activation have been implicated in RPE cells as mitochondria-related mediators of inflammation. mtDNA-driven NF-κB or MAPK activation, as well as ATP-regulated P2Y1 receptor signaling are also able to implement the priming signal for the inflammasome activation in RPE cells (Prager et al., 2016; Doktor et al., 2018; Fann et al., 2018). NF-κB p65 activity may be a critical upstream initiator of p62/SQSTM1 expression in RPE cells under oxidative stress that links inflammatory signaling to autophagy and NFE2L2 cascades (Song et al., 2017). Furthermore, Advanced Glycation End (AGE) product activates NF-κB and renders it more responsive to pro-inflammatory stimuli and extracellular matrix modifications in RPE cells (Kay et al., 2014; Sharif et al., 2019).
Recent reviews discuss the role of NOD-Like, Toll-Like, AGE product receptors, pentraxins, and complement system in AMD pathology (Kauppinen et al., 2016; Kaarniranta et al., 2019). Accelerated recycling of the membrane-bound complement regulator CD59 to the RPE cell surface inhibits the late activation of complement cascade. Once complement attack occurs, fusion of lysosomes with the RPE plasma membrane suppresses elevations in intracellular calcium and prevents mitochondrial injury (Tan et al., 2016). Disturbances in CD59 recycling and lysosome exocytosis may lead to mitochondrial fragmentation and increased ROS production in RPE. Complement factor B deficiency and smoking extract induced oxidative stress evoked increase in a RPE size that can be interpreted as a prognosis factor of cell death (Woodell et al., 2013; Chen et al., 2016). Evidence of inflammation was also present in mouse models exhibiting an AMD-like phenotype. Deficient NFE2L2 induces a pro-inflammatory environment by generating C3a and C3b in response to smoking (Wang et al., 2014b). NFE2L2/PGC-1α dKO mice exhibited significantly increased ionized calcium binding adaptor molecule 1 (Iba-1) mononuclear phagocyte marker and enlarged RPE size (Felszeghy et al., 2019).
The accumulation of Alu-retroelement RNA or DICER1 deficiency are known to drive mtDNA release from RPE cells through mitochondrial permeability transition (MPT) pores (Kerur et al., 2018). By interacting with cGAS, mtDNA promotes noncanonical NLRP3 inflammasome activation by caspase-4 in human and caspase-11 in mouse (Tarallo et al., 2012; Kerur et al., 2018). cGAS, IFN-β and caspase-4 are among factors that have been found in increased amounts in the eyes of dry AMD patients (Kerur et al., 2018). The cGAS pathway is important in viral resistance, but it has recently been associated also with the pathogenesis of chronic diseases, including AMD (West et al., 2015; Kerur et al., 2018). In addition to MPT and BAX/BAK pores, mtDNA can escape from mitochondria also within mitochondrial-derived vesicles. It has been hypothesized that mtDNA may be released from the endo-lysosomal pathway or from autophagosomes due to incomplete destruction of engulfed mitochondria and thereby end up in the cytosol where it can activate the NLRP3 inflammasome (Bader and Winklhofer, 2019).
Taken together, insufficient autophagy elevates the amount of damaged mitochondria and ROS production in AMD. Chronic oxidative stress coincides with the detrimental signaling effects of Toll-Like, AGE product receptors, pentraxins, complement system and inflammasomes.
11. Future directions
Mitochondria are organelles essential for maintaining RPE cell function. However, over the course of aging, mitochondria exhibit decreased rates of oxidative phosphorylation, increased ROS generation, and increased amounts of mtDNA damage and mutations (Murphy, 2009; Kageyama et al., 2012). Mitochondria in RPE are particularly susceptible to oxidative damage with aging and especially in pathological processes like those occurring in AMD (Núñez-Álvarez et al., 2019). While inflammation has been a therapeutic target in various clinical trials to treat AMD, outcomes have been relatively poor. Since mitochondria are connected to the regulation of cytoprotection, proteolysis, and inflammation, they might be a promising therapeutic target in early stages of AMD development.
Humanin (HN), a member of a family of mitochondrial-derived peptides, is expressed from an open reading frame of mitochondrial 16S ribosomal RNA (rRNA) (Matsunaga et al., 2016; Sreekumar et al., 2016). Humanin exhibits cytoprotective properties against oxidative stressors (e.g. by activating autophagy pathway, inhibiting ROS generation, upregulating mitochondrial GSH and activation of caspases 3 and; Minasyan et al., 2017; Gong et al., 2018). The mechanism behind humanin's cytoprotective properties likely involves activation of AMPK (Yong and Tang, 2018).
We have previously shown that AMPK activator AICAR also exhibits cytoprotective properties by accelerating protein clearance in RPE via autophagy (Viiri et al., 2013; Marchesi et al., 2018). AMPK has a pleiotrophic role in the protection of mitochondria. It can stimulate mitochondria biogenesis through increasing the gene transcription regulated by PGC-1α and can acutely trigger the destruction of existing defective mitochondria through ULK1-dependent mitophagy. Moreover, it is the key player in activating NFE2L2 signaling cascades and anti-oxidant production. Thus, AMPK has the net effect of replacing existing defective mitochondria with new functional mitochondria. The agonist of AMPK has cytoprotective effects by reducing detrimental protein aggregation in RPE cells (Salminen et al., 2012).
Our enhanced understanding of mitochondrial dysfunction in dry AMD pathogenesis may lead to the development of targeted therapies. However, defects in multiple biological pathways leading to different AMD phenotypes suggest that one type of treatment for all AMD patients may not be universally effective in the preventing or reversing the disease. One can predict that personalized medicine will increase in clinics not only in follow-ups and treatments, but also for selecting patients and planning clinical trials to find optimal endpoints (Barot et al., 2011). Targeting to correct mitochondrial dysfunction in the RPE, an essential factor in AMD pathology, is a viable option. Therefore, the aim in AMD therapy is to find drugs that better control mitochondria fusion/fission balance and/or activates autophagy to remove damaged mitochondria (Osborne et al., 2014; Kaarniranta et al., 2018c). One can estimate that this strategy could include the suppression of chronic oxidative stress and inflammation in the RPE (Kaarniranta et al., 2018c). In addition to new drug candidates and therapeutic targets, there are demands to develop longer-lasting agents or new drug delivery systems (Del Amo et al., 2017; Nashine et al., 2018; Schottler et al., 2018; Sharma et al., 2018; Popov, 2019). Possible breakthroughs in AMD treatments may open new therapeutic options in other retinal diseases.
Acknowledgements
This work was supported by the Kuopio University Hospital (KK), the Finnish Eye Foundation (KK), The Sigrid Juselius Foundation (KK), the Health Research Council of the Academy of Finland (AK 297267, 307341, 328443, KK 296840), the Päivikki and Sakari Sohlberg Foundation (AK, KK), the National Institutes of Health/National Eye Institute (R01 EY028554 and EY026012), the Lindsay Family Foundation and an anonymous benefactor for AMD research (DAF), National Science Centre, Poland (JB, Grant number 2017/27/B/NZ3/00872). RBF/IRRF Catalyst Award for Innovative Research Approaches for AMD (DS) and the Jennifer Salvitti Davis Chair in Ophthalmology (DS). We thank Iswariyaraja Sridevi Gurubaran, Ali Koskela and Johanna Viiri for technical assistance in preparing figures.
References
- Ach T, Huisingh C, McGwin G Jr., Messinger JD, Zhang T, Bentley MJ, Gutierrez DB, Ablonczy Z, Smith RT, Sloan KR, Curcio CA, 2014. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci 55, 4832–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ach T, Tolstik E, Messinger JD, Zarubina AV, Heintzmann R, Curcio CA, 2015. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 56, 3242–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaimo A, Liñares GG, Bujjamer JM, Gorojod RM, Alcon SP, Martínez JH, Baldessari A, Grecco HE, Kotler ML, 2019. Toxicity of blue led light and A2E is associated to mitochondrial dynamics impairment in ARPE-19 cells: implications for age-related macular degeneration. Arch. Toxicol 93, 1401–1415. [DOI] [PubMed] [Google Scholar]
- Allio R, Donega S, Galtier N, Nabholz B, 2017. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Mol. Biol. Evol 34, 2762–2772. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, 1992. Oxidants are a major contributor to aging. Ann. N. Y. Acad. Sci 21, 85–96. [DOI] [PubMed] [Google Scholar]
- Ames A 3rd, Li YY, Heher EC, Kimble CR, 1992. Energy metabolism of rabbit retina as related to function: high cost of sodium transport. J. Neurosci 12, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV, 2010. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog. Retin. Eye Res 29, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armento A, Honisch S, Panagiotakopoulou V, Sonntag I, Jacob A, Kilger E, Deleidi M, Clark S, Ueffing M, 2020. Loss of Complement Factor H impairs antioxidant capacity and energy metabolism of human RPE cells. bioRxiv. 10.1101/2020.01.08.898551. [DOI] [PMC free article] [PubMed]
- Arselin G, Vaillier J, Salin B, Schaeffer J, Giraud MF, Dautant A, Brèthes D, Velours J, 2004. The modulation in subunits e and g amounts of yeast ATP synthase modifies mitochondrial cristae morphology. J. Biol. Chem 279, 40392–40399. [DOI] [PubMed] [Google Scholar]
- Atilano SR, Malik D, Chwa M, Cáceres-Del-Carpio J, Nesburn AB, Boyer DS, Kuppermann BD, Jazwinski SM, Miceli MV, Wallace DC, Udar N, Kenney MC, 2015. Mitochondrial DNA variants can mediate methylation status of inflammation, angiogenesis and signaling genes. Hum. Mol. Genet 24, 4491–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad MB, Chen Y, Gibson SB, 2009. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progressionand treatment. Antioxidants Redox Signal. 11, 777–790. [DOI] [PubMed] [Google Scholar]
- Bader V, Winklhofer KF, 2019. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol S1084–9521 (18), 30191–30195 2019. [DOI] [PubMed] [Google Scholar]
- Baldelli S, Aquilano K, Ciriolo MR, 2013. Punctum on two different transcription factors regulated by PGC-1α: nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 1830, 4137–4146. [DOI] [PubMed] [Google Scholar]
- Ban N, Lee TJ, Sene A, Choudhary M, Lekwuwa M, Dong Z, Santeford A, Lin JB, Malek G, Ory DS, Apte RS, 2018. Impaired monocyte cholesterol clearance initiates age-related retinal degeneration and vision loss. JCI Insight 3, PAR120824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barot M, Gokulgandhi MR, Mitra AK, 2011. Mitochondrial dysfunction in retinal diseases. Curr. Eye Res 36, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau E, Brossas JY, Courtois Y, Treton JA, 1996. Accumulation of mitochondrial DNA deletions in human retina during aging. Invest. Ophthalmol. Vis. Sci 37, 384–391. [PubMed] [Google Scholar]
- Barron MJ, Johnson MA, Andrews RM, Clarke MP, Griffiths PG, Bristow E, He LP, Durham S, Turnbull DM, 2001. Mitochondrial abnormalities in ageing macular photoreceptors. Invest. Ophthalmol. Vis. Sci 42, 3016–3022. [PubMed] [Google Scholar]
- Bermann M, Schütt F, Holz FG, Kopitz J, 2001. Does A2E, a retinoid component of lipofuscin and inhibitor of lysosomal degradative functions, directly affect the activity of lysosomal hydrolases? Exp. Eye Res 72, 191–195. [DOI] [PubMed] [Google Scholar]
- Blasiak J, Glowacki S, Kauppinen A, Kaarniranta K, 2013. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci 14, 2996–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasiak J, Pawlowska E, Szczepanska J, Kaarniranta K, 2019. Interplay between autophagy and the ubiquitin-proteasome system and its role in the pathogenesis of age-related macular degeneration. Int. J. Mol. Sci 20, E210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhovd C, Vogel F, Neupert W, Reichert AS, 2006. Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J. Biol. Chem 281, 13990–13998. [DOI] [PubMed] [Google Scholar]
- Boya P, Esteban-Martínez L, Serrano-Puebla A, Gómez-Sintes R, Villarejo-Zori B, 2016. Autophagy in the eye: development, degeneration, and aging. Prog. Retin. Eye Res 55, 206–245. [DOI] [PubMed] [Google Scholar]
- Brown EE, DeWeerd AJ, Ildefonso CJ, Lewin AS, Ash JD, 2019. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol 101201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L, 2011. Oxidative stress, inflamm-aging and immunosenescence. J. Proteomics 7, 2313–2323. [DOI] [PubMed] [Google Scholar]
- Canter JA, Olson LM, Spencer K, Schnetz-Boutaud N, Anderson B, Hauser MA, Schmidt S, Postel EA, Agarwal A, Pericak-Vance MA, Sternberg P Jr., Haines JL, 2008. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PloS One 3, e2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caston RA, Demple B, 2017. Risky repair: DNA-protein crosslinks formed by mitochondrial base excision DNA repair enzymes acting on free radical lesions. Free Radic. Biol. Med 107, 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QM, Maltagliati AJ, 2018. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom 50, 77–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ, 2003. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem 278, 36027–36031. [DOI] [PubMed] [Google Scholar]
- Chen M, Rajapakse D, Fraczek M, Luo C, Forrester JV, Xu H, 2016. Retinal pigment epithelial cell multinucleation in the aging eye - a mechanism to repair damage and maintain homoeostasis. Aging Cell 15, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Luo C, Zhao J, Devarajan G, Xu H, 2019. Immune regulation in the aging retina. Prog. Retin. Eye Res 69, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Samuels DC, Elson J, Turnbull DM, 2002. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet 360, 1323–1325. [DOI] [PubMed] [Google Scholar]
- Choudhary M, Ding JD, Qi X, Boulton ME, Yao PL, Peters JM, Malek G, 2016. PPARβ/δ selectively regulates phenotypic features of age-related macular degeneration. Aging (Albany NY) 8, 1952–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, 2012. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim. Biophys. Acta 1824, 3–13. [DOI] [PubMed] [Google Scholar]
- Datta S, Cano M, Ebrhimi K, Wang L, Handa JT, 2017. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res 60, 201–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayalan Naidu S, Dikovskaya D, Gaurilcikaite E, Knatko EV, Healy ZR, Mohan H, Koh G, Laurell A, Ball G, Olagnier D-, de la Vega L, Ganley IG, Talalay P, Dinkova-Kostova AT, 2017. Transcription factors NRF2 and HSF1 have opposing functions in autophagy. Sci. Rep 8, 11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW, 2007. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol 143, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Amo EM, Rimpelä AK, Heikkinen E, Kari OK, Ramsay E, Lajunen T, Schmitt M, Pelkonen L, Bhattacharya M, Richardson D, Subriz i A., Turunen T, Reinisalo M, Itkonen J, Toropainen E, Casteleijn M, Kidron H, Antopolsky M, Vellonen KS, Ruponen M, Urtti A, 2017. Pharmacokinetic aspects of retinal drug delivery. Prog. Retin. Eye Res 57, 134–185. [DOI] [PubMed] [Google Scholar]
- Di Malta C, Cinque L, Settembre C, 2019. Transcriptional regulation of autophagy: mechanisms and diseases. Front. Cell Dev. Biol 7, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick I, 2017. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem 86, 193–224. [DOI] [PubMed] [Google Scholar]
- Doktor F, Prager P, Wiedemann P, Kohen L, Bringmann A, Hollborn M, 2018. Hypoxic expression of NLRP3 and VEGF in cultured retinal pigment epithelial cells: contribution of P2Y2 receptor signaling. Purinergic Signal. 14, 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dysli C, Wolf S, Berezin MY, Sauer L, Hammer M, Zinkernagel MS, 2017. Fluorescence lifetime imaging ophthalmoscopy. Prog. Retin. Eye Res 60, 120–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eells JT, 2019. Mitochondrial dysfunction in the aging retina. Biology 8, E31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger AM, Samardzija V, Sothilingam N, Tanimoto C, Lange S, Salatino L, Fang M, Garcia-Garrido S, Beck MJ, Okoniewski A, Neutzner MW, Seeliger C, Grimm C, Handschin C, 2012. PGC-1alpha determines light damage susceptibility of the murine retina. PloS One 7, e31272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann DY, Lim YA, Cheng YL, Lok KZ, Chunduri P, Baik SH, Drummond GR, Dheen ST, Sobey CG, Jo DG, Chen CL, Arumugam TV, 2018. Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol 55, 1082–1096. [DOI] [PubMed] [Google Scholar]
- Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C, 2006. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 27, 983–993. [DOI] [PubMed] [Google Scholar]
- Felszeghy S, Viiri J, Paterno JJ, Hyttinen JMT, Koskela A, Chen M, Leinonen H, Tanila H, Kivinen N, Koistinen A, Toropainen E, Amadio M, Smedowski A, Reinisalo M, Winiarczyk M, Mackiewicz J, Mutikainen M, Ruotsalainen AK, Kettunen M, Jokivarsi K, Sinha D, Kinnunen K, Petrovski G, Blasiak J, Bjørkøy G, Koskelainen A, Skottman H, Urtti A, Salminen A, Kannan R, Ferrington DA, Xu H, Levonen AL, Tavi P, Kauppinen A, Kaarniranta K, 2019. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 20, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Kapphahn RJ, Leary MM, Atilano SR, Terluk MR, Karunadharma P, Chen GK, Ratnapriya R, Swaroop A, Montezuma SR, Kenney MC, 2016a. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res 145, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Sinha D, Kaarniranta K, 2016b. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res 51, 69–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Ebeling MC, Kapphahn RJ, Terluk MR, Fisher CR, Polanco JR, Roehrich H, Leary MM, Geng Z, Dutton JR, Montezuma SR, 2017. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol 13, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher CR, Ferrington DA, 2018. Perspective on AMD pathobiology: a bioenergetic crisis in the RPE. Invest. Ophthalmol. Vis. Sci 59, AMD41–AMD47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein M, Mitchell P, Freund KB, Sadda S, Holz FG, Brittain C, Henry EC, Ferrara D, 2018. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology 125, 369–390. [DOI] [PubMed] [Google Scholar]
- Frede K, Ebert F, Kipp AP, Schwerdtle T, Baldermann S, 2017. Lutein activates the transcription factor Nrf2 in human retinal pigment epithelial cells. J. Agric. Food Chem 65, 5944–5952. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP Jr., Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis NN, Keilhauer C, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Saïd S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel J, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Léveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR, Gene Consortium AMD, 2013. Seven new loci associated with age-related macular degeneration. Nat. Genet 45 433–439, 439e1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambril JA, Sloan KR, Swain TA, Huisingh C, Zarubina AV, Messinger JD, Ach T, Curcio CA, 2019. Quantifying retinal pigment epithelium dysmorphia and loss of histologic autofluorescence in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 60, 2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Liu RT, Cao S, Cui JZ, Wang A, To E, Matsubara JA, 2015. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediat. Inflamm 2015, 690243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W, 2010. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol 12, 119–131. [DOI] [PubMed] [Google Scholar]
- Giorgi C, Marchi S, Simoes ICM, Ren Z, Morciano G, Perrone M, Patalas-Krawczyk P, Borchard S, Jędrak P, Pierzynowska K, Szymański J, Wang DQ, Portincasa P, Węgrzyn G, Zischka H, Dobrzyn P, Bonora M, Duszynski J, Rimessi A, Karkucinska-Wieckowska A, Dobrzyn A, Szabadkai G, Zavan B, Oliveira PJ, Sardao VA, Pinton P, Wieckowski MR, 2018. Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell. Mol. Biol 340, 209–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golestaneh N, Chu Y, Cheng SK, Cao H, Poliakov E, Berinstein DM, 2016. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med 14, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golestaneh N, Chu Y, Xiao YY, Stoleru GL, Theos AC, 2017. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 8, e2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sintes R, Villarejo-Zori B, Serrano-Puebla A, Esteban-Martínez L, Sierra-Filardi E, Ramírez-Pardo I, Rodríguez-Muela N, Boya P, 2017. Standard assays for the study of autophagy in the ex vivo retina. Cells 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, Kuliawat R, Liu H, Kühn B, Cuervo AM, Muzumdar R, 2018. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol 217, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha S, Coffey EE, Lu W, Lim JC, Beckel JM, Laties AM, Boesze-Battaglia K, Mitchell CH, 2014. Approaches for detecting lysosomal alkalinization and impaired degradation in fresh and cultured RPE cells: evidence for a role in retinal degenerations. Exp. Eye Res 126, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureev AP, Popov VN, 2019. Nrf2/ARE pathway as a therapeutic target for the treatment of Parkinson diseases. Neurochem. Res 44, 2273–2279. [DOI] [PubMed] [Google Scholar]
- Gurubaran RS, Viiri J, Koskela A, Hyttinen JMT, Paterno J, Kis G, Antal M, Urtti A, Kauppinen A, Felszeghy S, Kaarniranta K, 2020. Mitophagy in the retinal pigment epithelium of dry age-related macular degeneration investigated in the NFE2L2/PGC-1α−/− Mouse Model. Int. J. Mol. Sci 21, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa JT, 2012. How does the macula protect itself from oxidative stress? Mol. Aspect. Med 33, 418–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Wei D, Liu H, Zhu C, Lu Y, Ke Z, Jiang S, Huang J, 2019. Glycyrrhizin protects against sodium iodate-induced RPE and retinal injury though activation of AKT and Nrf2/HO-1 pathway. J. Cell Mol. Med 23, 3495–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK, 2011. Manganese superoxide dismutase: guardian of the powerhouse. Int. J. Mol. Sci 12, 7114–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz FG, Schütt F, Kopitz J, Eldred GE, Kruse FE, Völcke HE, Cantz M, 1999. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest. Ophthalmol. Vis. Sci 40, 737–743. [PubMed] [Google Scholar]
- Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HP, Schmitz-Valckenberg S, FAM-Study Group, 2007. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am. J. Ophthalmol 143, 463–472. [DOI] [PubMed] [Google Scholar]
- Huang YH, Chen CM, Lee YS, Chang KH, Chen HW, Chen YC, 2018. Detection of mitochondrial DNA with 4977 bp deletion in leukocytes of patients with ischemic stroke. PloS One 13, e0193175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JB, Lindsay KJ, Du J, 2015. Glucose, lactate and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res 93, 1079–1092 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hytti M, Piippo N, Korhonen E, Honkakoski P, Kaarniranta K, Kauppinen A, 2015. Fisetin and luteolin protect human retinal pigment epithelial cells from oxidative stress-induced cell death and regulate inflammation. Sci. Rep 5, 17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hytti M, Szabó D, Piippo N, Korhonen E, Honkakoski P, Kaarniranta K, Petrovski G, Kauppinen A, 2017. Two dietary polyphenols, fisetin and luteolin, reduce inflammation but augment DNA damage-induced toxicity in human RPE cells. J. Nutr. Biochem 42, 37–42. [DOI] [PubMed] [Google Scholar]
- Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K, 2013. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim. Biophys. Acta 1833, 503–510. [DOI] [PubMed] [Google Scholar]
- Hyttinen JMT, Błasiak J, Niittykoski M, Kinnunen K, Kauppinen A, Salminen A, Kaarniranta K, 2017. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res. Rev 36, 64–77. [DOI] [PubMed] [Google Scholar]
- Hyttinen JMT, Viiri J, Kaarniranta K, Błasiak J, 2018. Mitochondrial quality control in AMD: does mitophagy play a pivotal role? Cell. Mol. Life Sci 75, 2991–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovelli J, Rowe GC, Khadka A, Diaz-Aguilar D, Spencer C, Arany Z, Saint-Geniez M, 2016. PGC-1α induces human RPE oxidative metabolism and antioxidant capacity. Invest. Ophthalmol. Vis. Sci 57, 1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Komatsu M, 2018. Activation of p62/SQSTM1-keap1-nuclear factor erythroid 2-related factor 2 pathway in cancer. Front. Oncol 8, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM, 2007. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U. S. A 104, 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, McMahon M, Hayes JD, Johansen T, 2010. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem 85, 22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Boulton ME, 2012. Consequences of oxidative stress in age-related macular degeneration. Mol. Aspect. Med 33, 399–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Lin H, Godley BF, Boulton ME, 2008. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res 27, 596–607. [DOI] [PubMed] [Google Scholar]
- Ježek P, Holendová B, Garlid KD, Jabůrek M, 2018. Mitochondrial uncoupling proteins: subtle regulators of cellular redox signaling. Antioxidants Redox Signal. 29, 667–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin GF, Hurst JS, Godley BF, 2001. Rod outer segments mediate mitochondrial DNA damage and apoptosis in human retinal pigment epithelium. Curr. Eye Res 23, 11–19. [DOI] [PubMed] [Google Scholar]
- Joachim N, Mitchell P, Burlutsky G, Kifley A, Wang JJ, 2015. The incidence and progression of age-related macular degeneration over 15 Years: the blue mountains eye study. Ophthalmology 122, 2482–2489. [DOI] [PubMed] [Google Scholar]
- Johansen T, Lamark T, 2020. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol 432, 80–103. [DOI] [PubMed] [Google Scholar]
- Johansson I, Monsen VT, Pettersen K, Mildenberger J, Misund K, Kaarniranta K, Schønberg S, Bjørkøy G, 2015. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy 11, 1636–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MM, Manwaring N, Wang JJ, Rochtchina E, Mitchell P, Sue CM, 2007. Mitochondrial DNA haplogroups and age-related maculopathy. Arch. Ophthalmol 125, 1235–1240. [DOI] [PubMed] [Google Scholar]
- Juuti-Uusitalo K, Koskela A, Kivinen N, Viiri J, Hyttinen JMT, Reinisalo M, Koistinen A, Uusitalo H, Sinha D, Skottman H, Kaarniranta K, 2017. Autophagy regulates proteasome inhibitor-induced pigmentation in human embryonic stem cell-derived retinal pigment epithelial cells. Int. J. Mol. Sci 18, E1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Salminen A, Eskelinen EL, Kopitz J, 2009. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD). Ageing Res. Rev 8, 128–139. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Hyttinen J, Ryhanen T, Viiri J, Paimela T, Toropainen E, Sorri I, Salminen A, 2010. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front. Biosci. (Elite Ed) 2, 1374–1384. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Veréb Z, Salminen A, Boulton ME, Petrovski G, 2013. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 9, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Tokarz P, Koskela A, Paterno J, Blasiak J, 2017. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol 33, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Kajdanek J, Morawiec J, Pawlowska E, Blasiak J, 2018a. PGC-1α protects RPE cells of the aging retina against oxidative stress-induced degeneration through the regulation of senescence and mitochondrial quality control. The significance for AMD pathogenesis. Int. J. Mol. Sci 19, E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Pawlowska E, Szczepanska J, Jablkowska A, Blasiak J, 2018b. Role of mitochondrial DNA damage in ROS-mediated pathogenesis of age-related macular degeneration (AMD). Int. J. Mol. Sci 20, E2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Xu H, Kauppinen A, 2018c. Mechanistical retinal drug targets and challenges. Adv. Drug Deliv. Rev 126, 177–184. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Koskela A, Felszeghy S, Kivinen N, Salminen A, Kauppinen A, 2019. Fatty acids and oxidized lipoproteins contribute to autophagy and innate immunity responses upon the degeneration of retinal pigment epithelium and development of age-related macular degeneration. Biochimie 159, 49–54. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H, 2012. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol 197, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson M, Frennesson C, Gustafsson T, Brunk UT, Nilsson SE, Kurz T, 2013. Autophagy of iron-binding proteins may contribute to the oxidative stress resistance of ARPE-19 cells. Exp. Eye Res 116, 359–365. [PubMed] [Google Scholar]
- Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA, 2010. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 51, 5470–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K, 2012. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells–implications for age-related macular degeneration (AMD). Immunol. Lett 147, 29–33. [DOI] [PubMed] [Google Scholar]
- Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K, 2016. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci 73, 1765–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay P, Yang YC, Hiscott P, Gray D, Maminishkis A, Paraoan L, 2014. Age-related changes of cystatin C expression and polarized secretion by retinal pigment epithelium: potential age-related macular degeneration links. Invest. Ophthalmol. Vis. Sci 55, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MC, Atilano SR, Boyer D, Chwa M, Chak G, Chinichian S, Coskun P, Wallace DC, Nesburn AB, Udar NS, 2010. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest. Ophthalmol. Vis. Sci 51, 4289–4297. [DOI] [PubMed] [Google Scholar]
- Kenney MC, Chwa M, Atilano SR, Pavlis JM, Falatoonzadeh P, Ramirez C, Malik D, Hsu T, Woo G, Soe K, Nesburn AB, Boyer DS, Kuppermann BD, Jazwinski SM, Miceli MV, Wallace DC, Udar N, 2013. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PloS One 8, e54339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, Caceres-del-Carpio J, Nesburn AB, Boyer DS, Kuppermann BD, Vawter M, Jazwinski SM, Miceli M, Wallace DC, Udar N, 2014. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial-nuclear interactions. Hum. Mol. Genet 23, 3537–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, Varshney A, Yasuma R, Fowler BJ, Baghdasaryan E, Marion KM, Huang X, Yasuma T, Hirano Y, Serbulea V, Ambati M, Ambati VL, Kajiwara Y, Ambati K, Hirahara S, Bastos-Carvalho A, Ogura Y, Terasaki H, Oshika T, Kim KB, Hinton DR, Leitinger N, Cambier JC, Buxbaum JD, Kenney MC, Jazwinski SM, Nagai H, Hara I, West AP, Fitzgerald KA, Sadda SR, Gelfand BD, Ambati J, 2018. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med 24, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-Y, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy T, Chan C-C, Zhou Z, Green DR, Ferguson TA, 2013. Noncanonical autophagy promotes the visual cycle. Cell 155, 725–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. , 2016. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M, 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M, 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol 12, 213–223. [DOI] [PubMed] [Google Scholar]
- Korovila I, Hugo M, Castro JP, Weber D, Höhn A, Grune T, Jung T, 2017. Proteostasis, oxidative stress and aging. Redox Biol 13, 550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskela A, Reinisalo M, Hyttinen JM, Kaarniranta K, Karjalainen RO, 2014. Pinosylvin-mediated protection against oxidative stress in human retinal pigment epithelial cells. Mol. Vis 20, 760–769. [PMC free article] [PubMed] [Google Scholar]
- Krohne TU, Stratmann NK, Kopitz J, Holz FG, 2010. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp. Eye Res 90, 465–471. [DOI] [PubMed] [Google Scholar]
- Lakkaraju A, Finnemann SC, Rodriguez-Boulan E, 2007. The lipofuscin fluorophore A2E perturbs cholesterol metabolism in retinal pigment epithelial cells. Proc. Natl. Acad. Sci. U.S.A 104, 11026–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambros ML, Plafker SM, 2016. Oxidative stress and the Nrf2 anti-oxidant transcription factor in age-related macular degeneration. Adv. Exp. Med. Biol 854, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastres-Becker I, García-Yagüe AJ, Scannevin RH, Casarejos MJ, Kügler S, Rábano A, Cuadrado A, 2016. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson's disease. Antioxidants Redox Signal. 25, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD, 2010. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell Biol 30, 3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ, 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Giordano S, Zhang J, 2012. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J 441, 523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Oh JS, Rho JH, Jeong NY, Kwon YH, Jeong WJ, Ryu WY, Ahn HB, Park WC, Rho SH, Yoon YG, Jeong SY, Choi YH, Kim HY, Yoo YH, 2014. Retinal pigment epithelial cells undergoing mitotic catastrophe are vulnerable to autophagy inhibition. Cell Death Dis. 5, e1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, 2005. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. [DOI] [PubMed] [Google Scholar]
- Li Z, Dong X, Liu H, Chen X, Shi H, Fan Y, Hou D, Zhang X, 2013. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis 19, 1656–1666. [PMC free article] [PubMed] [Google Scholar]
- Li XF, Liu XM, Huang DR, Cao HJ, Wang JY, 2018. PF-06409577 activates AMPK signaling to protect retinal pigment epithelium cells from UV radiation. Biochem. Biophys. Res. Commun 501, 293–299. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Godley BF, 2003. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res 76, 397–403. [DOI] [PubMed] [Google Scholar]
- Lin H, Xu H, Liang FQ, Liang H, Gupta P, Havey AN, Boulton ME, Godley BF, 2011. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 52, 3521–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Fang H, Chi Z, Wu Z, Wei D, Mo D, Niu K, Balajee AS, Hei TK, Nie L, Zhao Y, 2015. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res. 43, 5476–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Copland DA, Theodoropoulou S, Chiu HA, Barba MD, Mak KW, Mack M, Nicholson LB, Dick AD, 2016a. Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis. Sci. Rep 6, 20639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu M, Zhang X, Chen Q, Chen H, Sun L, Liu G, 2016c. Protective effect of fucoxanthin isolated from laminaria japonica against visible light-induced retinal damage both in vitro and in vivo. J. Agric. Food Chem 64, 416–424. [DOI] [PubMed] [Google Scholar]
- Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, Chen Y, 2010. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxidants Redox Signal. 13, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ward K, Xavier C, Jann J, Clark AF, Pang IH, Wu H, 2016. The novel triterpenoid RTA 408 protects human retinal pigment epithelial cells against H2O2-induced cell injury via NF-E2-related factor 2 (Nrf2) activation. Redox Biol. 8, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Gómez NM, Lim JC, Guha S, O'Brien-Jenkins A, Coffey EE, Campagno KE, McCaughey SA, Laties AM, Carlsson LG, Mitchell CH, 2018. The P2Y(12) receptor antagonist ticagrelor reduces lysosomal pH and autofluorescence in retinal pigmented epithelial cells from the ABCA4(−/−) mouse model of retinal degeneration. Front. Pharmacol 19, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik D, Hsu T, Falatoonzadeh P, Cáceres-del-Carpio J, Tarek M, Chwa M, Atilano SR, Ramirez C, Nesburn AB, Boyer DS, Kuppermann BD, Jazwinski SM, Miceli MV, Wallace DC, Udar N, Kenney MC, 2014. Human retinal transmitochondrial cybrids with J or H mtDNA haplogroups respond differently to ultraviolet radiation: implications for retinal diseases. PloS One 9, e99003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Seo SJ, Biswal MR, Li H, Conners M, Nandyala A, Jones K, Le YZ, Lewin AS, 2014. Mitochondrial oxidative stress in the retinal pigment epithelium leads to localized retinaldegeneration. Invest. Ophthalmol. Vis. Sci 55, 4613–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi N, Thongon N, Pascale A, Provenzani A, Koskela A, Korhonen E, Smedowski A, Govoni S, Kauppinen A, Kaarniranta K, Amadio M, 2018. Autophagy stimulus promotes early HuR protein activation and p62/SQSTM1 protein synthesis in ARPE-19 cells by triggering erk1/2, p38(MAPK), and JNK kinase pathways. Oxid. Med. Cell. Longev 2018, 4956080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie M, Bigot K, Angebault C, Barrau C, Gondouin P, Pagan D, Fouquet S, Villette T, Sahel JA, Lenaers G, Picaud S, 2018. Light action spectrum on oxidative stress and mitochondrial damage in A2E-loaded retinal pigment epithelium cells. Cell Death Dis. 9, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N, 2011. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 44, 279–289. [DOI] [PubMed] [Google Scholar]
- Matsunaga D, Sreekumar PG, Ishikawa K, Terasaki H, Barron E, Cohen P, Kannan R, Hinton DR, 2016. Humanin protects RPE cells from endoplasmic reticulum stress-induced apoptosis by upregulation of mitochondrial glutathione. PloS One 11, e0165150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Villarejo-Zori B, Ball G, Boya P, Ganley IG, 2019. A comparative map of macroautophagy and mitophagy in the vertebrate eye. Autophagy 15, 1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano CJ, Fox DA, 1995. Oxygen consumption in the rat outer and inner retina: light- and pharmacologically-induced inhibition. Exp. Eye Res 61, 273–284. [DOI] [PubMed] [Google Scholar]
- Melis JP, van Steeg H, Luijten M, 2013. Oxidative DNA damage and nucleotide excision repair. Antioxidants Redox Signal. 18, 2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli MV, Jazwinski SM, 2005. Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 46, 1765–1773. [DOI] [PubMed] [Google Scholar]
- Minasyan L, Sreekumar PG, Hinton DR, Kannan R, 2017. Protective mechanisms of the mitochondrial – derived peptide humanin in oxidative and endoplasmic reticulum stress in RPE cells. Oxid. Med. Cell Longev 1675230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minauro-Sanmiguel F, Wilkens S, Garcia JJ, 2005. Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc. Natl. Acad. Sci. U.S.A 102, 12356–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W Jr., Ding J, Bowes Rickman C, Boulton M, 2014. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 10, 1989–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldogazieva NT, Mokhosoev IM, Mel'nikova TI, Porozov YB, Terentiev AA, 2019. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid. Med. Cell. Longevity 3085756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Russell SR, Anderson DH, Hageman GS, 2000. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 14, 835–846. [PubMed] [Google Scholar]
- Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M, 2015. NRF2 regulates PINK1 expression under oxidative stress conditions. PloS One 10, e0142438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochem. J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray D, Mirzayans R, McBride WH, 2018. Defenses against pro-oxidant forces - maintenance of cellular and genomic integrity and longevity. Radiat. Res 190, 331–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N, Figueiredo-Pereira ME, 2011. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J. Biol. Chem 286, 22426–22440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Yoshimori T, 2017. New insights into autophagosome-lysosome fusion. J. Cell Sci 130, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Nashine S, Cohen P, Nesburn AB, Kuppermann BD, Kenney MC, 2018. Characterizing the protective effects of SHLP2, a mitochondrial-derived peptide, in maculardegeneration. Sci. Rep 8, 15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Chen G, He J, Zhang F, Li M, Wang Q, Zhou H, Lyu J, Bai Y, 2016. Mitochondrial common deletion is elevated in blood of breast cancer patients mediated by oxidative stress. Mitochondrion 26, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgaard CL, Berg KM, Kapphahn RJ, Reilly C, Feng X, Olsen TW, Ferrington DA, 2006. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 47, 815–822. [DOI] [PubMed] [Google Scholar]
- Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA, 2008. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 49, 2848–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüñez-Álvarez C, Suárez-Barrio C, Del Olmo Aguado S, Osborne NN, 2019. Blue light negatively affects the survival of ARPE19 cells through an action on their mitochondria and blunted by red light. Acta Ophthalmol. 97, e103–e115. [DOI] [PubMed] [Google Scholar]
- O'Mealey GB, Berry WL, Plafker SM, 2017. Sulforaphane is a Nrf2-independent inhibitor of mitochondrial fission. Redox Biol 11, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Álvarez CN, del Olmo Aguado S, 2014. Targeting mitochondrial dysfunction as in aging and glaucoma. Drug Discov. Today 19, 1613–1622. [DOI] [PubMed] [Google Scholar]
- Pajares M, Jiménez-Moreno N, García-Yagüe ÁJ, Escoll M, de Ceballos ML, Van Leuven F, Rábano A, Yamamoto M, Rojo AI, Cuadrado A, 2016. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12, 1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piippo N, Korkmaz A, Hytti M, Kinnunen K, Salminen A, Atalay M, Kaarniranta K, Kauppinen A, 2014. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochim. Biophys. Acta 1843, 3038–3046. [DOI] [PubMed] [Google Scholar]
- Piippo N, Korhonen E, Hytti M, Kinnunen K, Kaarniranta K, Kauppinen A, 2018. Oxidative stress is the principal contributor to inflammasome activation in retinal pigment epithelium cells with defunct proteasomes and autophagy. Cell. Physiol. Biochem 49, 359–367. [DOI] [PubMed] [Google Scholar]
- Popov LD, 2019. Mitochondrial peptides-appropriate options for therapeutic exploitation. Cell Tissue Res. 377, 161–165. [DOI] [PubMed] [Google Scholar]
- Prager P, Hollborn M, Steffen A, Wiedemann P, Kohen L, Bringmann A, 2016. P2Y1 receptor signaling contributes to high salt-induced priming of the NLRP3 inflammasome in retinal pigment epithelial cells. PloS One 11, e0165653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandra Rao S, Pfeffer BA, Más Gómez N, Skelton LA, Keiko U, Sparrow JR, Rowsam AM, Mitchell CH, Fliesler SJ, 2018. Compromised phagosome maturation underlies RPE pathology in cell culture and whole animal models of Smith-Lemli-Opitz Syndrome. Autophagy 14, 1796–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnayaka A, Paraoan L, Spiller DG, Hiscott P, Nelson G, White MR, Grierson I, 2007. A dual Golgi- and mitochondria-localised Ala25Ser precursor cystatin C: an additional tool for characterising intracellular mis-localisation leading to increased AMD susceptibility. Exp. Eye Res 84, 1135–1139. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muela N, Koga H, García-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P, 2013. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggia MF, Ueta T, 2015. alphavbeta5 integrin/FAK/PGC-1alpha pathway confers protective effects on retinal pigment epithelium. PloS One 10, e0134870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales MAB, u DY, Iacovelli J, Saint-Geniez M, 2019. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci Alliance 2 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, Hoek JB, 2017. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozing MP, Durhuus JA, Krogh Nielsen M, Subhi Y, Kirkwood TB, Westendorp RG, Sørensen TL, 2019. Age-related macular degeneration: a two-level model hypothesis. Prog. Retin. Eye Res 100825. 10.1016/j.preteyeres.2019.100825. ([Epub ahead of print]). [DOI] [PubMed] [Google Scholar]
- Rudolf M, Vogt SD, Curcio CA, Huisingh C, McGwin G Jr., Wagner A, Grisanti S, Read RW, 2013. Histologic basis of variations in retinal pigment epithelium autofluorescence in eyes with geographic atrophy. Ophthalmology 120, 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva MM, Cano M, Handa JT, 2014. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res 119, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Kuse Y, Inoue Y, Nakamura S, Hara H, Shimazawa M, 2018. Transient acceleration of autophagic degradation by pharmacological Nrf2 activation is important for retinal pigment epithelium cell survival. Redox Biol 19, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Kauppinen A, 2012. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 4, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanGiovanni JP, Arking DE, Iyengar SK, Elashoff M, Clemons TE, Reed GF, Henning AK, Sivakumaran TA, Xu X, DeWan A, Agron E, Rochtchina E, Sue CM, Wang JJ, Mitchell P, Hoh J, Francis PJ, Klein ML, Chew EY, Chakravarti A, 2009. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PloS One 4, e5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z, 2007. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottler J, Randoll N, Lucius R, Caliebe A, Roider J, Klettner A, 2018. Long-term treatment with anti-VEGF does not induce cell aging in primary retinal pigment epithelium. Exp. Eye Res 171, 1–11. [DOI] [PubMed] [Google Scholar]
- Schütt F, Bergmann M, Holz FG, Kopitz J, 2002. Isolation of intact lysosomes from human RPE cells and effects of A2-E on the integrity of the lysosomal and other cellular membranes. Graefes Arch. Clin. Exp. Ophthalmol 240, 983–988. [DOI] [PubMed] [Google Scholar]
- Schütt F, Bergmann M, Holz FG, Kopitz J, 2003. Proteins modified by malondialdehyde,4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci 44, 3663–3668. [DOI] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW, 2004. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol 24, 8055–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadrach KG, Rayborn ME, Hollyfield JG, Bonilha VL, 2013. DJ-1-dependent regulation of oxidative stress in the retinal pigment epithelium (RPE). PloS One 8, e67983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SZA, Zhao D, Hussain T, Sabir N, Mangi MH, Yang L, 2018. p62-Keap1-NRF2-ARE pathway: a contentious player for selective targeting of autophagy, oxidative stress and mitochondrial dysfunction in prion diseases. Front. Mol. Neurosci 11, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif U, Mahmud NM, Kay P, Yang YC, Harding SP, Grierson I, Kamalden TA, Jackson MJ, Paraoan L, 2019. Advanced glycation end products-related modulation of cathepsin L and NF-κB signalling effectors in retinal pigment epithelium lead to augmented response to TNFα J. Cell Mol. Med 23, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Liaw K, Sharma R, Zhang Z, Kannan S, Kannan RM, 2018. Targeting mitochondrial dysfunction and oxidative stress in activated microglia using dendrimer-based therapeutics. Theranostics 8, 5529–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Zhang Z, Wang X, Li R, Hou W, Bi W, Zhang X, 2015. Inhibition of autophagy induces IL-1β release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem. Biophys. Res. Commun 463, 1071–1076. [DOI] [PubMed] [Google Scholar]
- Sihvola V, Levonen AL, 2017. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys 617, 94–100. [DOI] [PubMed] [Google Scholar]
- Silva-Palacios A, Ostolga-Chavarría M, Zazueta C, Königsberg M, 2018. Nrf2: molecular and epigenetic regulation during aging. Ageing Res. Rev 47, 31–40. [DOI] [PubMed] [Google Scholar]
- Sinha D, Valapala M, Shang P, Hose S, Grebe R, Lutty GA, Zigler JS Jr., Kaarniranta K, Handa JT, 2016. Lysosomes: regulators of autophagy in the retinal pigmented epithelium. Exp. Eye Res 144, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Mitter SK, Qi X, Beli E, Rao HV, Ding J, Ip CS, Gu H, Akin D, Dunn WA Jr., Bowes Rickman, C., Lewin AS, Grant MB, Boulton ME, 2017. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PloS One 12, e0171940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, Cohen P, Kannan R, Hinton DR, 2016. The mitochondrial-derived peptide Humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest. Ophthalmol. Vis. Sci 57, 1238–1253 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM, 2006. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408. [DOI] [PubMed] [Google Scholar]
- Stefánsson E, Geirsdóttir A, Sigurdsson H, 2011. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res 30, 72–80. [DOI] [PubMed] [Google Scholar]
- Stefánsson E, Olafsdottir OB, Eliasdottir TS, Vehmeijer W, Einarsdottir AB, Bek T, Torp TL, Grauslund J, Eysteinsson T, Karlsson RA, Van Keer K, Stalmans I, Vandewalle E, Todorova MG, Hammer M, Garhöfer G, Schmetterer L, Šín M, Hardarson SH, 2019. Retinal oximetry: metabolic imaging for diseases of the retina and brain. Prog. Retin. Eye Res 70, 1–22. [DOI] [PubMed] [Google Scholar]
- Strauss O, 2005. The retinal pigment epithelium in visual function. Physiol. Rev 85, 845–881. [DOI] [PubMed] [Google Scholar]
- Strom J, Xu B, Tian X, Chen QM, 2016. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 30, 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Chan CM, Chen HM, Wu CC, Hsiao CY, Lee PL, Lin VC, Hung CF, 2014. Lutein inhibits the migration of retinal pigment epithelial cells via cytosolic and mitochondrial Akt pathways (lutein inhibits RPE cells migration). Int. J. Mol. Sci 15, 13755–13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmári-Tóth M, Kristóf E, Veréb Z, Akhtar S, Facskó A, Fésüs L, Kauppinen A, Kaarniranta K, Petrovski G, 2016. Clearance of autophagy-associated dying retinal pigment epithelial cells - a possible source for inflammation in age-related macular degeneration. Cell Death Dis. 7, e2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmári-Tóth M, Ilmarinen T, Mikhailova A, Skottman H, Kauppinen A, Kaarniranta K, Kristóf E, Lytvynchuk L, Veréb Z, Fésüs L, Petrovski G, 2019. Human embryonic stem cell-derived retinal pigment epithelium-role in dead cell clearance and inflammation. Int. J. Mol. Sci 20, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadi SK, Sebastian R, Dahal S, Babu RK, Choudhary B, Raghavan SC, 2016. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell 27, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan LX, Toops KA, Lakkaraju A, 2016. Protective responses to sublytic complement in the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A 113, 8789–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Nüñez G, Baffi JZ, Kleinman ME, Ambati J, 2012. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terluk MR, Kapphahn RJ, Soukup LM, Gong H, Gallardo C, Montezuma SR, Ferrington DA, 2015. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci 35, 7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz P, Kaarniranta K, Blasiak J, 2013. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 14, 461–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Shirihai OS, 2011. The interplay between mitochondrial dynamics and mitophagy. Antioxidants Redox Signal. 14, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udar N, Atilano SR, Memarzadeh M, Boyer DS, Chwa M, Lu S, Maguen B, Langberg J, Coskun P, Wallace DC, Nesburn AB, Khatibi N, Hertzog D, Le K, Hwang D, Kenney MC, 2009. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 50, 2966–2974. [DOI] [PubMed] [Google Scholar]
- Valapala M, Wilson C, Hose S, Bhutto IA, Grebe R, Dong A, Greenbaum S, Gu L, Sengupta S, Cano M, Hackett S, Xu G, Lutty GA, Dong L, Sergeev Y, Handa JT, Campochiaro P, Wawrousek E, Zigler JS Jr., Sinha D, 2014. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 10, 480–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viiri J, Hyttinen JM, Ryhänen T, Rilla K, Paimela T, Kuusisto E, Siitonen A, Urtti A, Salminen A, Kaarniranta K, 2010. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol. Vis 16, 1399–1414. [PMC free article] [PubMed] [Google Scholar]
- Viiri J, Amadio M, Marchesi N, Hyttinen JM, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D'Agostino VG, Govoni S, Pascale A, Agostini H, Petrovski G, Salminen A, Kaarniranta K, 2013. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PloS One 8, e69563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Anand M, Shiraz AK, Magrane J, Gao J, Vollmer-Snarr HR, Manfredi G, Finnemann SC, 2008. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem 283, 24770–24780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tomso DJ, Chorley BN, Cho HY, Cheung VG, Kleeberger SR, Bell DA, 2007. Identification of polymorphic antioxidant response elements in the human genome. Hum. Mol. Genet 1188, 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Neufeld AH, 2008. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol. Vis 14, 644–651. [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH, 2009. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PloS One 4, e4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Neufeld AH, 2010. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol. Aging 31, 2002–2010. [DOI] [PubMed] [Google Scholar]
- Wang L, Cano M, Handa JT, 2014a. p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochim. Biophys. Acta 1843, 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kondo N, Cano M, Ebrahimi K, Yoshida T, Barnett BP, Biswal S, Handa JT, 2014b. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic. Biol. Med 70, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ebrahimi KB, Chyn M, Cano M, Handa JT, 2016a. Biology of p62/sequestosome-1 in age-related macular degeneration (AMD). Adv. Exp. Med. Biol 854, 17–22. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hanus JW, Abu-Asab MS, Shen D, Ogilvy A, Ou J, Chu XK, Shi G, Li W, Wang S, Chan CC, 2016b. NLRP3 upregulation in retinal pigment epithelium in age-related macular degeneration. Int. J. Mol. Sci 17, E73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yao Y, Zhu X, Zhang K, Zhou F, Zhu L, 2017. Amyloid β induces NLRP3 inflammasome activation in retinal pigment epithelial cells via NADPH oxidase- and mitochondria-dependent ROS production. J. Biochem. Mol. Toxicol 31, 6. [DOI] [PubMed] [Google Scholar]
- Wang J, Zibetti C, Shang P, Sripathi SR, Zhang P, Cano M, Hoang T, Xia S, Ji H, Merbs SL, Zack DJ, Handa JT, Sinha D, Blackshaw S, Qian J, 2018. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun 9, 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang X, Cheng Y, Ouyang W, Sang X, Liu J, Su Y, Liu Y, Li C, Yang L, Jin L, Wang Z, 2019. Autophagy dysfunction, cellular senescence, and abnormal immune-inflammatory responses in AMD: from mechanisms to therapeutic potential. Oxid. Med. Cell Longev 3632169 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS, 2015. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodell A, Coughlin B, Kunchithapautham K, Casey S, Williamson T, Ferrell WD, Atkinson C, Jones BW, Rohrer B, 2013. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PloS One 8, e67894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Tan Q, Niu K, Zhu Y, Wei D, Zhao Y, Fang H, 2018. MMS19 localizes to mitochondria and protects the mitochondrial genome from oxidative damage. Biochem. Cell. Biol 96, 4. [DOI] [PubMed] [Google Scholar]
- Xu L, Kong L, Wang J, Ash JD, 2018. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A 115, 10475–10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Li Y, Chan L, Tsai YT, Wu WH, Nguyen HV, Hsu CW, Li X, Brown LM, Egli D, Sparrow JR, Tsang SH, 2014. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum. Mol. Genet 23, 3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PM, Wu ZZ, Zhang YQ, Wung BS, 2016. Lycopene inhibits ICAM-1 expression and NF-κB activation by Nrf2-regulated cell redox state in human retinal pigment epithelial cells. Life Sci. 155, 94–101. [DOI] [PubMed] [Google Scholar]
- Ye F, Kaneko H, Hayashi Y, Takayama K, Hwang SJ, Nishizawa Y, Kimoto R, Nagasaka Y, Tsunekawa T, Matsuura T, Yasukawa T, Kondo T, Terasaki H, 2016. Malondialdehyde induces autophagy dysfunction and VEGF secretion in the retinal pigment epithelium in age-related macular degeneration. Free Radic. Biol. Med 94, 121–134. [DOI] [PubMed] [Google Scholar]
- Yong CQY, Tang BL, 2018. A mitochondrial encoded messenger at the nucleus. Cells 7, E105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, 2005. Retinal degeneration and local oxygen metabolism. Exp. Eye Res 80, 745–751. [DOI] [PubMed] [Google Scholar]
- Zaffagnini G, Martens S, 2016. Mechanisms of selective autophagy. J. Mol. Biol 428, 1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bai Y, Huang L, Qi Y, Zhang Q, Li S, Wu Y, Li X, 2015. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: implications for age-related macular degeneration. Cell Death Dis. 6, e1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chu Y, Mowery J, Konkel B, Galli S, Theos AC, Golestaneh N, 2018. Pgc-1α repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model Mech 211, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J, 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]
- Zinovkina LA, 2018. Mechanisms of mitochondrial DNA repair in mammals. Biochemistry (Mosc.) 83, 233–249. [DOI] [PubMed] [Google Scholar]
- Zou X, Gao J, Zheng Y, Wang X, Chen C, Cao K, Xu J, Li Y, Lu W, Liu J, Feng Z, 2014. Zeaxanthin induces Nrf2-mediated phase II enzymes in protection of cell death. Cell Death Dis. 5, e1218. [DOI] [PMC free article] [PubMed] [Google Scholar]