

Abstract
The contraction and relaxation of the heart is controlled by stimulation of the β1-adrenoreceptor (AR) signaling cascade, which leads to activation of cAMP-dependent protein kinase (PKA) and subsequent cardiac protein phosphorylation. Phosphorylation is counteracted by the main cardiac protein phosphatases, PP2A and PP1. Both kinase and phosphatases are sensitive to intramolecular disulfide formation in their catalytic subunits that inhibits their activity. Additionally, intermolecular disulfide formation between PKA type I regulatory subunits (PKA-RI) has been described to enhance PKA's affinity for protein kinase A anchoring proteins, which alters its subcellular distribution. Nitroxyl donors have been shown to affect contractility and relaxation, but the mechanistic basis for this effect is unclear. The present study investigates the impact of several nitroxyl donors and the thiol-oxidizing agent diamide on cardiac myocyte protein phosphorylation and oxidation. Although all tested compounds equally induced intermolecular disulfide formation in PKA-RI, only 1-nitrosocyclohexalycetate (NCA) and diamide induced reproducible protein phosphorylation. Phosphorylation occurred independently of β1-AR activation, but was abolished after pharmacological PKA inhibition and thus potentially attributable to increased PKA activity. NCA treatment of cardiac myocytes induced translocation of PKA and phosphatases to the myofilament compartment as shown by fractionation, immunofluorescence, and proximity ligation assays. Assessment of kinase and phosphatase activity within the myofilament fraction of cardiac myocytes after exposure to NCA revealed activation of PKA and inhibition of phosphatase activity thus explaining the increase in phosphorylation. The data suggest that the NCA-mediated effect on cardiac myocyte protein phosphorylation orchestrates alterations in the kinase/phosphatase balance.
Keywords: nitroxyl, oxidation, phosphorylation, kinase, phosphatase, cardiac myocyte, redox regulation, cardiomyocyte, cardiovascular disease, protein kinase A (PKA)
β1−Adrenoreceptor (AR)-mediated activation of cAMP-dependent protein kinase (PKA) is the main pathway that regulates cardiac contractility. Through phosphorylation of substrate proteins that mediate cardiac excitation-contraction coupling, PKA enhances cardiac myocyte inotropy and lusitropy (1). PKA is a heterotetrameric kinase comprising two regulatory and two catalytic subunits. Binding of two molecules of cAMP to each of the two regulatory subunits (PKA-R) induces the release of the active catalytic subunits (PKA-C). PKA kinase activity can be modulated by oxidation (2). Recently, oxidant-mediated formation of antiparallel interdisulfide bonds between Cys- 17 and -38 in neighboring PKA-RI subunits was described and shown to induce subcellular translocation of the cytosolic holoenzyme and a substrate-induced release of the catalytic subunits at the target site (3–6). Mice that cannot form the interdisulfide upon stimulation due to Cys-17 replacement by non-oxidizable serine were deficient in vascular endothelial growth factor-stimulated angiogenesis, supporting the physiological importance of this modification (5). Furthermore, S-glutathiolation at Cys-199 or intradisulfide formation between Cys-199 and Cys-343 within one catalytic subunit was shown to inhibit kinase activity (7, 8). Kinase-mediated phosphorylation is counteracted by phosphatases (9). Thereby, the serine/threonine phosphatase protein phosphatase 2A (PP2A) represents one of the most abundant and important phosphatases in cardiac myocytes. It is composed of a scaffold and a catalytic subunit (PP2A-C), which assemble to form the core enzyme. The combination with one of at least 18 regulatory subunits that confer cellular targeting and thus compartmentalized phosphatase action constitutes the active holoenzyme (10–12), which catalyzes the dephosphorylation of various PKA substrates such as cardiac myosin-binding protein C (cMyBP-C) (13), cardiac troponin I (cTnI) (14), and to a lesser extent phospholamban (PLN) (15). Equally to kinases, phosphatases are also susceptible to oxidation with a reported inhibition of phosphatase activity (16–18). Thereby, intradisulfide formation between Cys-266 and -269 in PP2A-C (19) as well as between Cys-39 and Cys-127 in protein phosphatase 1α (PP1α) (12, 16, 20) were reported as inhibitory redox switches in phosphatases.
Pharmacological compounds that release nitroxyl (HNO) have attracted attention for their beneficial effects on cardiac function (21–23). HNO donors combine vasorelaxing properties with positive cardiac inotropy and lusitropy and are currently tested clinically for the treatment of heart failure (24–28). Various chemically distinct HNO donor compounds with different release-kinetics have been developed in recent years (26). The cardiac effects of HNO-releasing compounds are mainly attributed to HNO-induced oxidative interdisulfide formation in cardiac myofilament and sarcoplasmic reticulum proteins (29) with positive effects on cardiac myocyte function (30, 31). Importantly, protein kinases and phosphatases that transduce extracellular signals into a contractile response via phosphorylation and dephosphorylation of substrate proteins have also been described to be oxidant-susceptible. Whether HNO donor compounds impact on contraction and relaxation parameters through alteration of protein kinase and phosphatase activity and thus cardiac myocyte protein phosphorylation, has not been reported before.
Results
Effect of HNO donors on cardiac myocyte contractility
We evaluated the impact of an experimental and a clinically relevant HNO donor, 1-nitrosocyclohexyl acetate (NCA) and CXL-1020, respectively, on cardiac myocyte contractile function. Contractility of single adult rat ventricular myocytes (ARVMs) was recorded in response to vehicle (DMSO; control; Fig. 1A), NCA (100 μmol/liter; Fig. 1B), or CXL-1020 (300 μmol/liter; Fig. 1C). Upon exposure to NCA, ARVMs displayed a significant but transient increase in sarcomere shortening and relaxation velocity, which reversed to baseline after ∼4.5 min, reaching a novel plateau state (Fig. 1B, Fig. S1B). The NCA-induced significant prolongation of the time to peak was visible after 1 min of treatment and this effect was maintained during the new plateau state (Fig. 1D). Importantly, a decrease in diastolic length and decreased response to the subsequent isoprenaline (ISO) application was observed. Significantly increased sarcomere shortening as well as maximal contraction and relaxation velocities were also elicited by CXL-1020, but, in contrast to NCA, persisted after a plateau was reached (Fig. 1C, Fig. S1C). Compared with baseline measurements (Fig. 1A, Fig. S1A), CXL-1020 additionally displayed significantly accelerated relaxation as reflected by a decreased time to baseline 50% (Fig. 1D). Exposure to CXL-1020 has been described previously to increase cellular cGMP levels via activation of soluble guanylate cyclase (sGC) with subsequent activation of cGMP-dependent protein kinase (PKG) (32). To rule out a contribution of CXL-1020–mediated PKG activity on the observed effect, experiments were repeated after 1H-(1,2,4)oxadiazolol(4,3-a)quinoxaline-1-one (ODQ) pretreatment to inhibit sGC (Fig. S2). There was no difference in any contractile parameter detectable between ARVMs treated with CXL-1020 alone or after pretreatment with ODQ (Fig. S2), excluding an effect of PKG on the CXL-1020–mediated positive inotropic effect.
Figure 1.
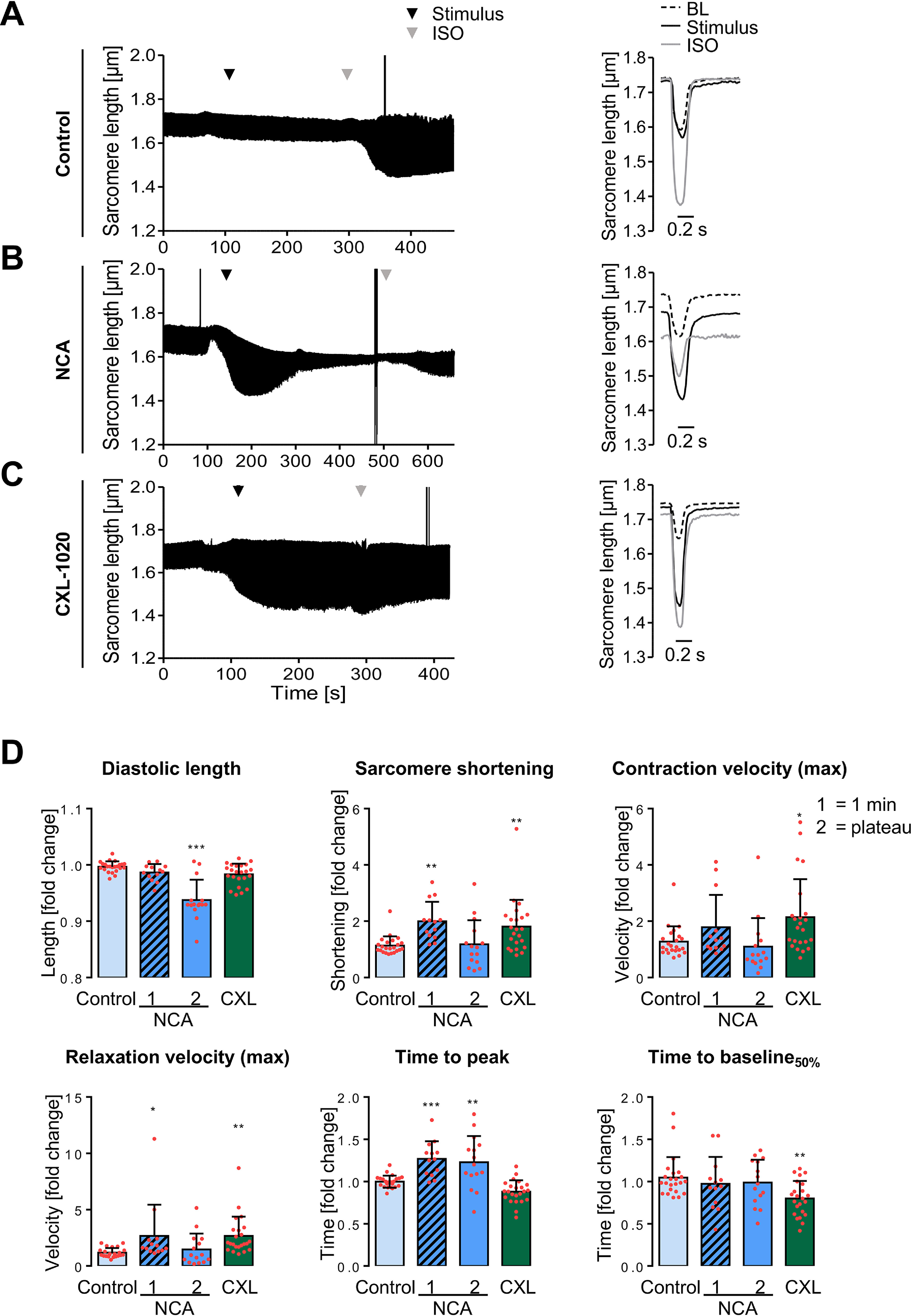
Impact of NCA and CXL-1020 on ARVM contractility. A–C, examples for contraction traces and corresponding single contraction peaks of single ARVMs paced at 1 Hz. Contraction characteristics were recorded under basal conditions (BL), upon exposure to different stimuli and in response to subsequent ISO application (10 nmol/liter). At baseline, ARVMs were supplemented with (A) vehicle (DMSO; control), (B) NCA (100 µmol/liter), or (C) CXL-1020 (300 µmol/liter). Time points of addition of the respective stimulus (black arrowhead) and ISO (gray arrowhead) are indicated. The single contraction peak shown for NCA represents the contractile response after 1 min of exposure. D, scatter plots compare effects of different stimuli after normalization to BL values from the respective cardiac myocyte and as fold-change of contractile parameters in response to vehicle. Scatter plots include NCA values after 1 min (1) of exposure and at the plateau state (2) of the transient effect (average 4.5 min of exposure). *, p < 0.05; **, p < 0.01; ***, p < 0.001 for comparison with the corresponding vehicle values by one-way ANOVA with Dunnett's post-test (F and p values): a, diastolic length: F = 25.80, p < 0.0001; b, sarcomere shortening: F = 6.08, p < 0.0010; c, contraction velocity (max): F = 4.10, p = 0.0097; d, relaxation velocity (max): F = 4.39, p = 0.0069; e, time to peak: F = 17.77, p < 0.0001; f, time to baseline 50%: F = 4.05, p = 0.0103).
Impact of oxidants on cardiac myocyte protein phosphorylation
To investigate whether NCA and CXL-1020 impact on the phosphorylation status of cardiac myocyte proteins at the concentration that was observed to induce increased contractility, ARVMs were exposed to NCA (100 μmol/liter) and CXL-1020 (300 μmol/liter) as well as to the HNO donor Angeli's salt (AS; 500 μmol/liter), hydrogen peroxide (H2O2; 100 μmol/liter), or the disulfide-inducing agent diamide (DIA; 500 μmol/liter). Samples were resolved by SDS-PAGE and phosphoproteins were stained by Pro-Q Diamond. In ARVMs exposed to the various oxidants, bands of increased intensity were observed at molecular masses of 150 and 25 kDa (Fig. 2A, arrows). Comparable signals were detected in response to the PKA-activating β1-AR agonist ISO, suggesting an impact of oxidant-mediated phosphorylation of PKA substrate proteins. Exposure of ARVMs to NCA led to enhanced phosphorylation of the 150-kDa PKA substrate protein cMyBP-C at Ser-273, -282, and -302, accounting for 28.1 ± 23.6, 59.5 ± 25.4, and 58.2 ± 39.9% of the response mediated by ISO, respectively (Fig. 2B). These phosphorylation sites in cMyBP-C, in particular Ser-282, have been shown to regulate actin-myosin interaction and contribute to positive inotropy and lusitropy in response to β1-AR stimulation (33–35). NCA exposure induced phosphorylation of PLN at Ser-16 that measured 23.3 ± 14.7% of the signal induced by ISO. This phosphorylation site in PLN regulates activity of the sarcoplasmic reticulum Ca2+-ATPase activity and contributes to positive inotropy and lusitropy in response to β1-AR stimulation (1). DIA mediated enhanced phosphorylation of cMyBP-C at Ser-282 (33.6 ± 18.1% of ISO response) and Ser-16 of PLN (11.0 ± 11.5% of the ISO response). Surprisingly, there was no significant impact on the phosphorylation of cTnI in response to oxidants detectable. The data suggest that NCA and DIA induce protein phosphorylation potentially by modulation of the PKA signaling pathway. Phosphorylation induced by exposure to AS or CXL-1020 was lower and the extent of the phosphorylation signal varied between experiments and batches, potentially attributed to the fast release kinetics of HNO by these donors. Exposure of ARVMs to increasing concentrations of the oxidants revealed concentration-dependent phosphorylation of cMyBP-C at Ser-282 and PLN at Ser-16 that was most pronounced for NCA and DIA. However, there was no impact on the phosphorylation of cTnI at Ser-22/23 detectable apart from a slight increase in response to the highest NCA concentration that remained statistically nonsignificant (Fig. 2, B and C). ISO was used to yield the maximal phosphorylation response for all cardiac proteins tested here.
Figure 2.
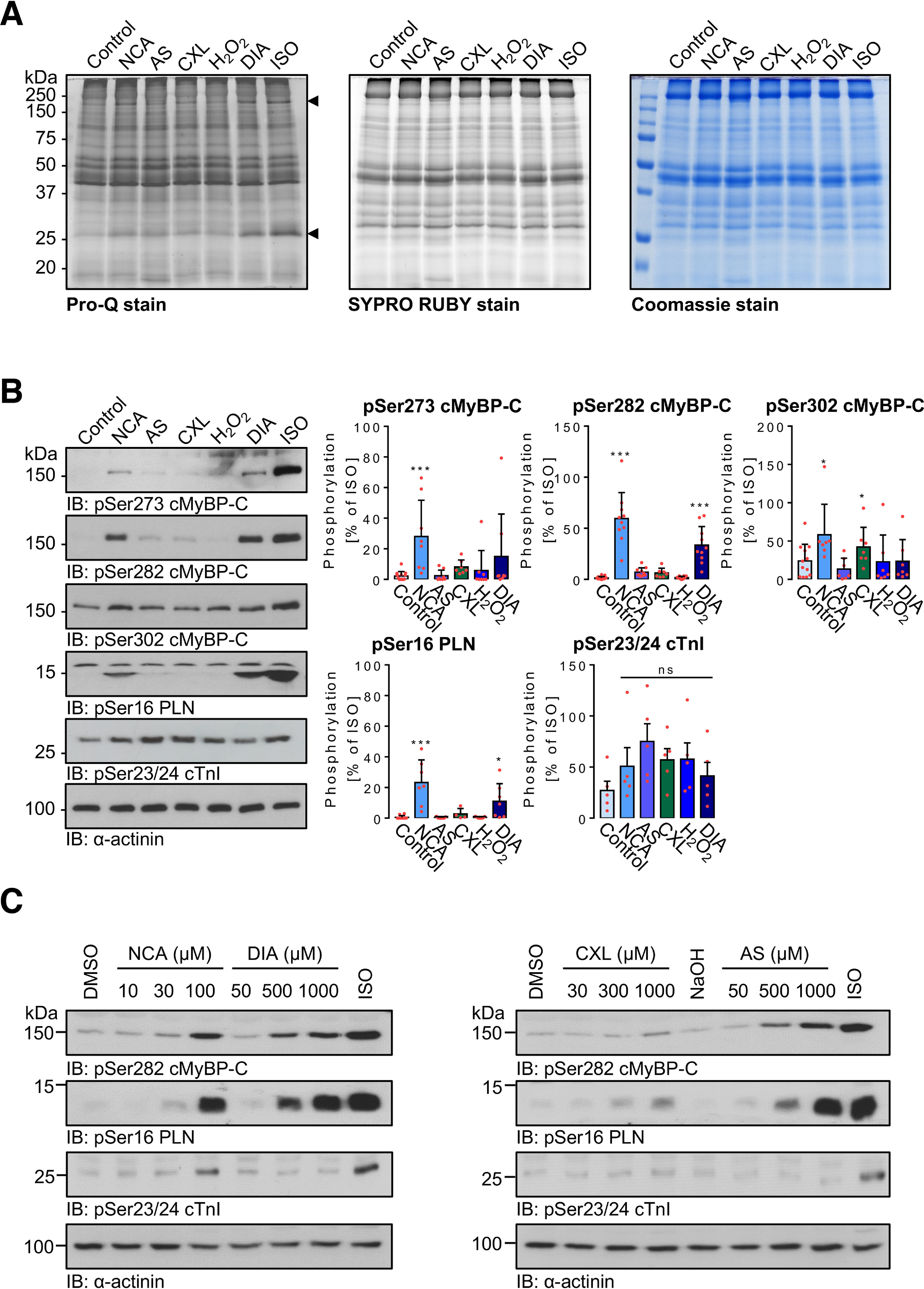
Impact of oxidants on protein phosphorylation in ARVMs. A, ARVMs were exposed to vehicle (control), NCA (100 µmol/liter, 30 min), AS (300 µmol/liter, 15 min), CXL-1020 (300 µmol/liter, 15 min), H2O2 (100 µmol/liter, 10 min), DIA (500 µmol/liter, 10 min), or ISO (10 nmol/liter, 10 min). Cell lysates were resolved by SDS-PAGE and gels were stained for phosphoproteins by ProQ-Diamond. Total protein was visualized by SYPRO Ruby and Coomassie staining. B, phosphorylation of cMyBP-C (pSer-273, pSer-282, and Ser-302), PLN (pSer-16), and cTnI (pSer-23/24) in response to oxidant exposure was investigated by immunoblotting (IB) and phospho-specific antibodies. Scatter plots summarize data of at least 7 (phospho-cMyBP-C), 4 (phospho-PLN), or 5 (phospho-cTnI) independent experiments normalized to α-actinin and expressed as % of signal induced by ISO. *, p < 0.05; ***, p < 0.001 for comparison with vehicle control by one-way ANOVA and Dunnett's post-test (F and p values): a, pSer-273 cMyBP-C: F = 79.70, p < 0.0001; b, pSer-282 cMyBP-C: F = 142.4, p < 0.0001; c, pSer-302 cMyBP-C: F = 15.27, p < 0.0001; d, pSer-16 PLB: F = 259.1, p < 0.0001; e, pcTnI: F = 1.246, p = 0.3192). C, ARVMs were treated with the indicated concentrations of NCA (for 30 min), DIA (for 10 min), CXL-1020 (for 15 min), and AS (for 15 min) or were exposed to vehicle (DMSO for 30 min or NaOH for 15 min) or ISO (10 nmol/liter, 10 min). Phosphorylation of cMyBP-C (pSer-282) and PLN (pSer-16) was analyzed by IB using the respective phospho-specific antibodies. Protein loading was assessed using an antibody against α-actinin. Blots are representative of three independent experiments. ns, not significant.
PKA activation in response to HNO donors in adult cardiac myocytes
Whether NCA and DIA exposure of ARVMs directly activates PKA was substantiated by FRET experiments in adenovirally transduced ARVMs expressing the protein kinase A (PKA) activity reporter AKAR3-NES. Application of NCA and DIA induced a change in biosensor fluorescence with 34.1 ± 29.7 and 31.5 ± 30.5% of the maximal response, respectively, which was elicited by subsequent application of forskolin/3-isobutyl-1-methylxanthine (FOR/IBMX) (Fig. 3A).
Figure 3.
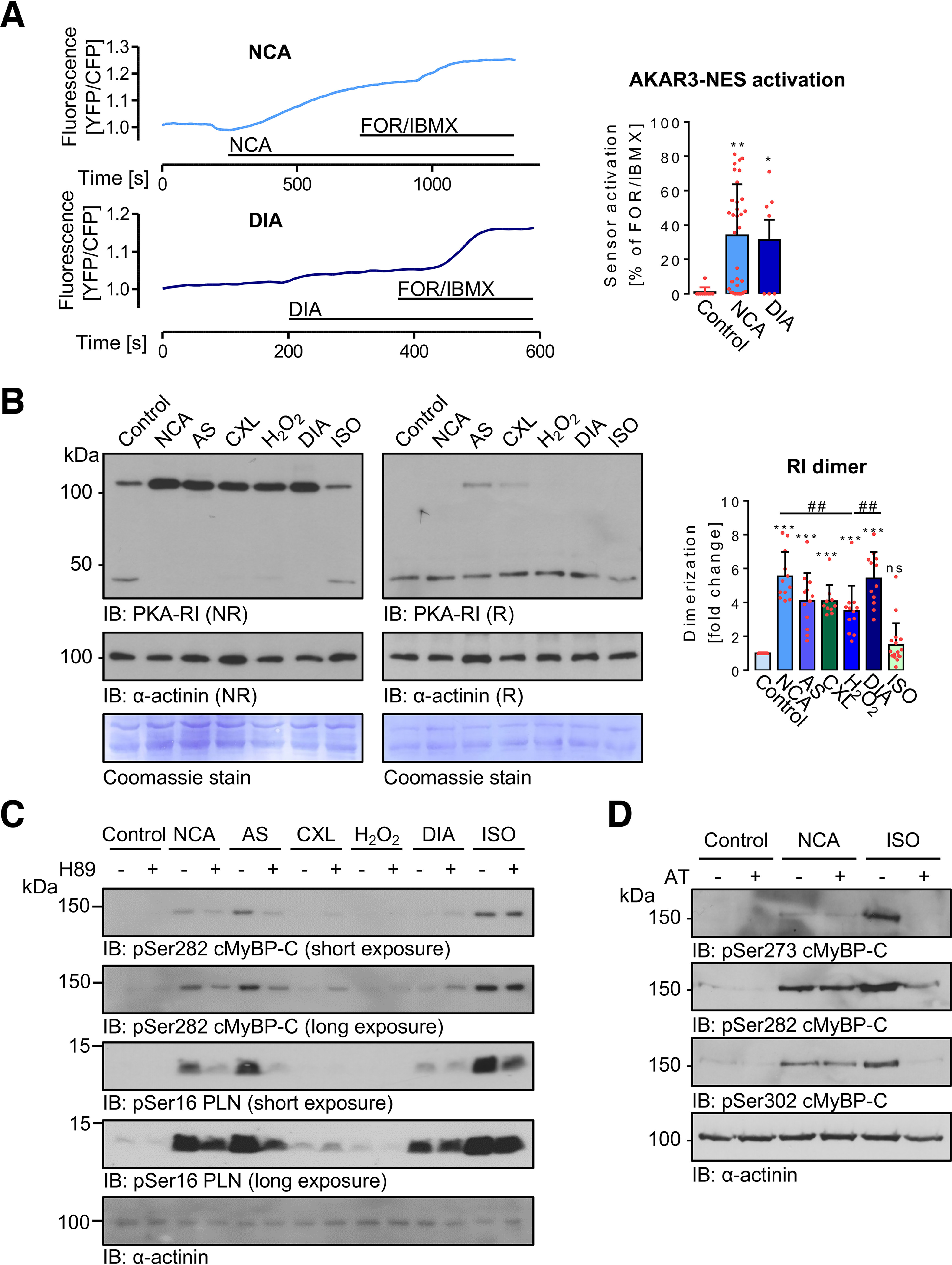
Impact of oxidants on PKA kinase activity and PKA-RI dimerization in ARVMs. A, ARVMs expressing the FRET PKA activity sensor AKAR3-NES were treated with vehicle (control, n = 10), NCA (100 µmol/liter, n = 32), or DIA (500 µmol/liter, n = 7) and sensor activation was recorded by fluorescence detection. Values are expressed as % of FOR/IBMX response, which was considered as the maximal signal. *, p < 0.05; **, p < 0.01 for comparison with vehicle control by one-way ANOVA and Dunnett's post-test (F = 5.96, p = 0.0050). B, ARVMs were exposed to vehicle (control), NCA (100 µmol/liter, 30 min), AS (300 µmol/liter, 15 min), CXL-1020 (300 µmol/liter, 15 min), H2O2 (100 µmol/liter, 10 min), DIA (500 µmol/liter, 10 min), or ISO (10 nmol/liter, 10 min). Myocyte lysates were then subjected to Western IB under NR conditions for analysis of the PKA-RI oxidation status. Samples were examined also under reducing conditions (R). RI dimer signals normalized to α-actinin (R) from 10 independent experiments are summarized in the scatter plots as fold-change of control signal. ***, p < 0.001 for comparison with control and ##, p < 0.01 for comparison between treatments by one-way ANOVA with Bonferroni post-test (F = 28.28, p < 0.0001). C, ARVMs were incubated for 30 min with H89 (10 µmol/liter) and then exposed to vehicle (control), NCA (100 µmol/liter, 30 min), AS (300 µmol/liter, 15 min), CXL-1020 (300 µmol/liter, 15 min), H2O2 (100 µmol/liter, 10 min), DIA (500 µmol/liter, 10 min), or ISO (10 nmol/liter, 10 min). Phosphorylation of cMyBP-C (Ser-282) and PLN (Ser-16) in lysates of stimulated ARVMs was investigated by IB using the respective phosphospecific antibodies. Chemiluminescence signals detected after a short and a long exposure are shown. Protein loading was examined using an anti-α-actinin antibody. Blots are representative of four independent experiments. D, following incubation with the β1-AR antagonist atenolol (AT, 1 µmol/liter), ARVMs were treated with vehicle (control), NCA (100 µmol/liter, 30 min) or ISO (10 nmol/liter, 10 min) and cMyBP-C phosphorylation at Ser-273, Ser-282, and Ser-302 analyzed by IB. Blots are representative of six independent experiments.
To investigate whether the mode of NCA-mediated PKA activation involves RI interdisulfide bond formation, the oxidation status of PKA-RI was assessed following oxidant treatment. The suitability of the PKA-RI antibody to unveil changes in the oxidation state of PKA-RIα was confirmed in isolated adult mouse ventricular myocytes from WT or knock-in mice that constitutively express a redox-deficient PKA-RIα C17S mutant exposed to H2O2 or ISO (Fig. S3). By Western immunoblot analyses under nonreducing conditions, enhanced formation of RI dimer was detected. Interdisulfide formation occurred to a similar extent following exposure to all tested oxidant stimuli (Fig. 3B). Notably, RI interdisulfide formation did not correlate with the previously shown phosphorylation of PKA substrate proteins (Fig. 2). Addition of a reducing agent revealed loss of PKA-RI dimer in favor of monomeric PKA-RI and confirmed the oxidative nature of dimer formation. Interestingly, only after exposure to AS or CXL-1020, a proportion of the PKA-RI dimer reproducibly remained under reducing conditions.
Pretreatment of ARVMs with the pharmacological PKA inhibitor H89 prior to oxidant exposure revealed attenuation of the phosphorylation of cMyBP-C at Ser-282 and PLN at Ser-16 in response to NCA, AS, and ISO, suggesting oxidant-mediated PKA activation involved in oxidant-mediated cardiac myocyte protein phosphorylation. In contrast, CXL-1020-mediated phosphorylation remained unaffected by H89 pretreatment suggesting a distinct mode of action (Fig. 3C). The involvement of a panel of other kinases (Ca2+-calmodulin-dependent kinase, protein kinase C, p90 ribosomal S6 kinase, and Rho-kinase) that have been described previously to phosphorylate cMyBP-C at Ser-282 (36) was excluded by the use of pharmacological inhibitors (Fig. S4). Importantly, ISO- but not NCA-mediated phosphorylation of cMyBP-C was abolished by pretreatment of ARVMs with the β-AR antagonist atenolol, confirming direct and receptor-independent PKA activation by NCA (Fig. 3D).
Oxidant-mediated PKA and phosphatase translocation to the cardiac myofilament compartment
Time-response experiments in ARVMs exposed to NCA revealed a quick onset of PKA-RI dimer formation, which was followed by phosphorylation of cMyBP-C at Ser-282 occurring with a timely delay. Phosphorylation was maintained for up to 100 min (Fig. 4A). To investigate whether oxidant exposure induces accumulation of PKA in the myofilament compartment, ARVMs were exposed to NCA or ISO, harvested under nonreducing conditions and the myofilament-containing Triton-insoluble fraction was isolated. Successful fractionation was confirmed by detection of the fraction-specific marker proteins GAPDH in the cytosolic, Na,K-ATPase (NKA) in the membrane, and cTnI in the myofilament fraction (Fig. S5). Exposure of ARVMs to NCA led to a significant accumulation of PKA-RI and PKA-C in the Triton-insoluble fraction (Fig. 4B). The catalytic and regulatory subunits of the PKA counteracting phosphatase PP2A-C and B56α were also found to accumulate after exposure to NCA in the Triton-insoluble fraction, whereas ISO treatment did not induce myofilament enrichment, neither of the kinase nor phosphatase subunits (Fig. 4B). Analysis of subcellular fractionation following time course treatment with NCA revealed a fast onset of myofilament translocation for PKA subunits within 10-30 min and PP2A-C and B56α within 3-10 min, culminating in cMyBP-C Ser-282 phosphorylation (Fig. 4B).
Figure 4.
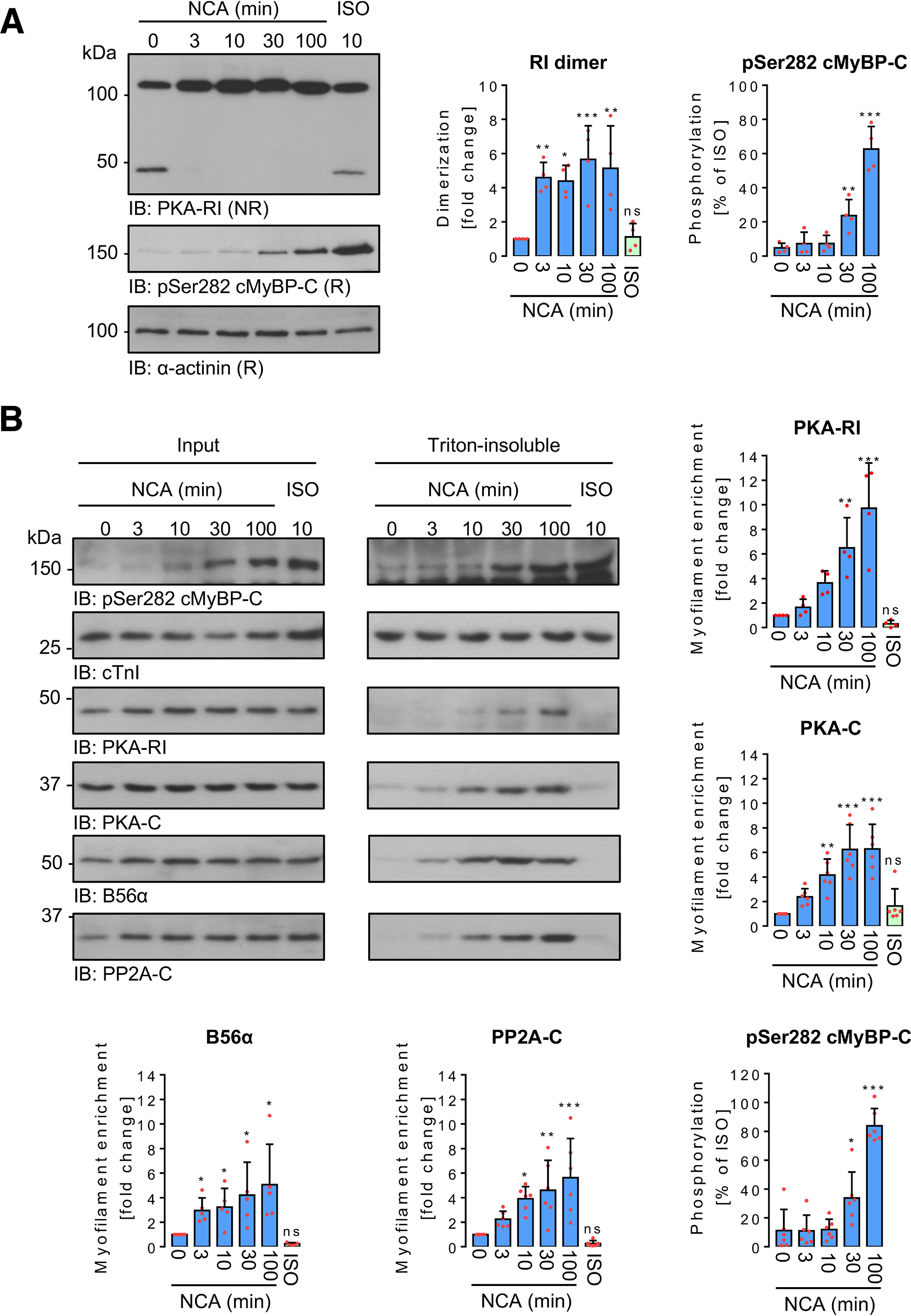
Time-dependent impact of NCA on PKA-RI dimerization and protein translocation in ARVMs. A, ARVMs were treated with vehicle for 100 min (0 min NCA), NCA (100 µmol/liter) for 3, 10, 30, and 100 min, or ISO (10 nmol/liter) for 10 min. PKA-RI and cMyBP-C phosphorylation at Ser-282 were detected in whole lysates by IB analysis under NR or R conditions, respectively. Scatter plots summarize RI dimer signal fold-change compared with control sample (0 min NCA) and cMyBP-C phosphorylation as % of ISO-response from 4 independent experiments. Band intensities were normalized to α-actinin. *, p < 0.05; **, p < 0.01; ***, p < 0.001 for comparison with vehicle control (0 min NCA) by one-way ANOVA with Dunnett's post-test (RI dimer: F = 8.23, p = 0.0003; pSer-282 cMyBP-C: F = 107.5, p < 0.0001). B, ARVMs were exposed to vehicle for 100 min (0 min NCA), NCA (100 µmol/liter) for 3, 10, 30, and 100 min or ISO (10 nmol/liter) for 10 min and subcellular fractionation was performed under NR conditions. Phosphorylation of cMyBP-C at Ser-282, as well as the presence of PKA-RI, PKA-C, B56α, and PP2A-C were detected in input lysates and Triton-insoluble fractions. Phosphorylation of cMyBP-C at Ser-282 (n = 6) normalized to Coomassie stain signals (not shown) is presented as % of the band intensity induced by ISO. PKA-RI (n = 4), PKA-C (n = 6), B56α (n = 5), and PP2A-C (n = 6) signals from Triton-insoluble fractions normalized to the corresponding inputs are shown as fold-change of the control signal (0 min NCA). *, p < 0.05; **, p < 0.01; ***, p < 0.001 for comparison with the corresponding vehicle control (sample 0 min) by one-way ANOVA with Dunnett's post-test (F and p values): a, PKA-RI, F = 15.18, p < 0.0001; b, PKA-C, F = 15.61, p < 0.0001; c, B56α, F = 4.78, p = 0.0036; d, PP2A-C, F = 9.16, p < 0.0001; e, pSer-282 cMyBP-C, F = 67.33, p < 0.0001). ns, not significant.
Subcellular compartmentalization of PKA and PP2A is regulated by their respective regulatory subunits. NCA-induced PKA-RI and B56α myofilament translocation was supported by immunofluorescence staining of ARVMs. Exposure to NCA resulted in a diffuse signal for PKA-RI with occasional appearance of transversal striations located nearby the characteristic doublet signals representing cMyBP-C (Fig. 5A, left panel). The signal obtained for B56α in response to NCA was more distinct and clearly located B56α at the M-band and to a lesser extent also at the Z-disc (Fig. 5A, right panel).
Figure 5.
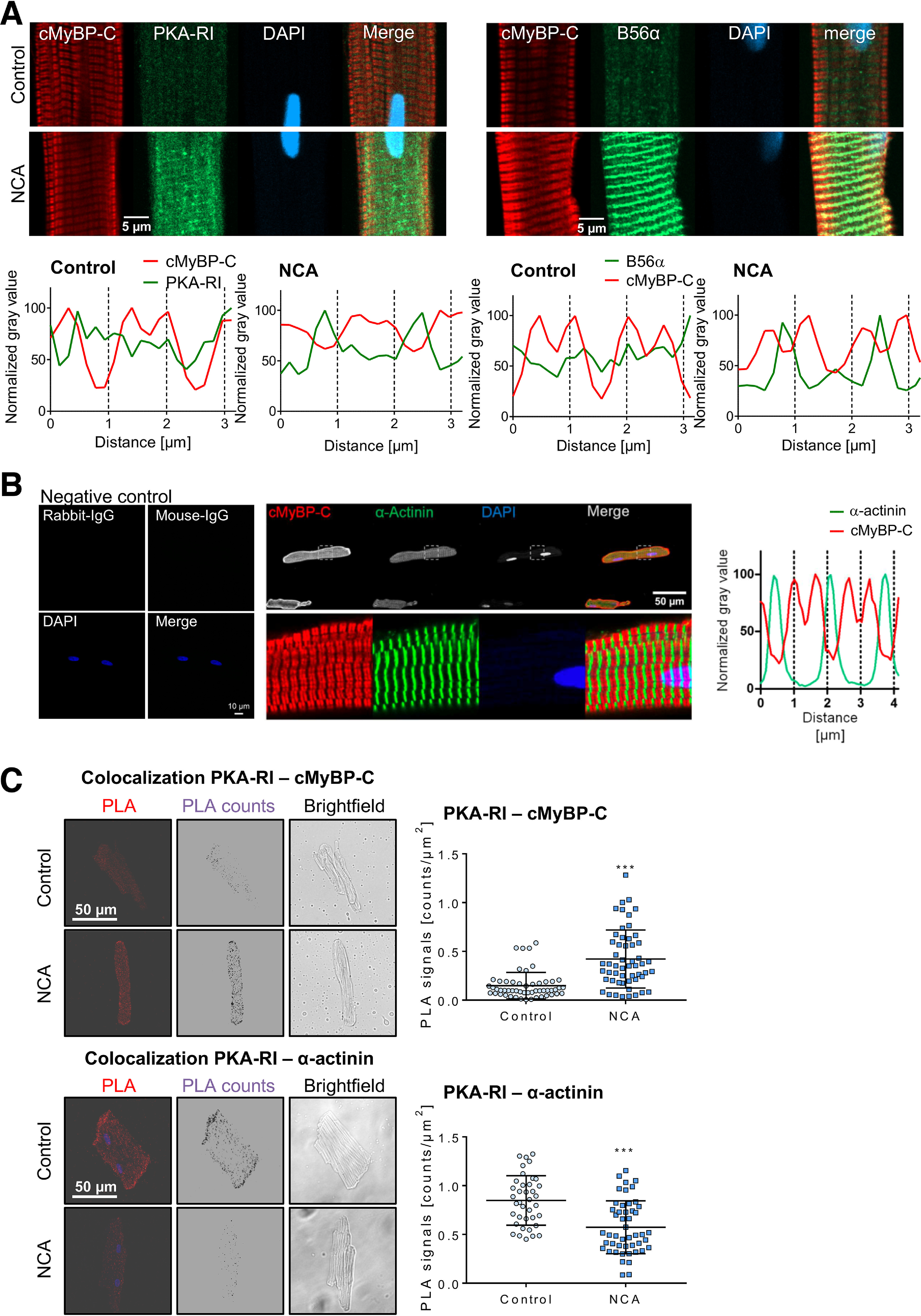
Fluorescence-based analysis of PKA-RI translocation. A, following incubation with vehicle (control) or NCA (100 µmol/liter, 30 min) ARVMs were subjected to immunofluorescence labeling of cMyBP-C and PKA-RI or B56α. DNA was visualized by DAPI. Displayed images are representative of at least 12 images from 2 to 3 individual sample sets. Longitudinal distribution of cMyBP-C (red) and PKA-RI or B56α (green) signals in the myofilament lattice are also shown. Intensity values are expressed as % of the highest intensity measured in the respective channel. Scale bar: 5 μm. B, ARVMs were stained with secondary antibodies (anti-mouse IgG; anti-rabbit IgG) alone as a negative control. Immunofluorescence for cMyBP-C (red) and α-actinin (green) confirmed their distinct sarcomeric location detected by the antibodies. DNA was visualized by DAPI. The merge displays the overlaid channels. Images are representative of at least 12 images from 2 to 3 individual sample sets. Longitudinal distribution of cMyBP-C (red) and α-actinin (green) signals in the myofilament lattice are also shown. Intensity values are expressed as % of the highest intensity measured in the respective channel. Scale bars: 10 μm (left) or 50 μm (right). C, ARVMs were treated with vehicle (control) or NCA (100 µmol/liter, 30 min) and the proximity between PKA-RI and cMyBP-C or α-actinin was investigated by PLA. PLA counts were obtained from microscopy Z-stack images. Confocal images show unprocessed PLA immunofluorescence signals (PLA), PLA counts after digital processing and a brightfield image of a representative cell. PLA counts/µm2 indicating the proximity between PKA-RI and cMyBP-C (control: n = 53; NCA: n = 54), PKA-RI and α-actinin (control: n = 38; NCA: n = 48) are visualized in the scatter plot. **, p < 0.001; ***, p < 0.001 for comparison between vehicle control and NCA-treated cells by two-tailed Student's t test. Scale bar: 50 μm.
A spatial rapprochement between PKA-RI within the myofilament lattice was further investigated by proximity ligation assay (PLA)-Duolink technology in ARVMs. The antibodies for cMyBP-C and α-actinin were tested prior to PLA experiments by immunofluorescence. This showed as expected the localization of cMyBP-C in characteristic doublets in the C-zone of sarcomeric A-bands at both sides of the M-line and α-actinin at the Z-discs. Respective IgG negative controls for the secondary antibodies did not show any signal (Fig. 5B). The number of PLA fluorescence signals suggested oxidant-mediated PKA-RI localization close to the C-zone of the A-band with a distinct colocalization of PKA-RI with cMyBP-C. In contrast, the presence of PKA-RI in the Z-disc was decreased as shown by a significantly reduced colocalization of PKA-RI with the Z-disc marker α-actinin (Fig. 5C). This observation was further corroborated by cAMP pulldown experiments. ARVMs were treated with vehicle, NCA, CXL-1020, or ISO and lysates were subjected to cAMP-agarose pulldown and analysis by LC–MS and Western immunoblot analysis (Fig. 6). As expected, cAMP-agarose captured PKA-RI with high affinity, which was present in all samples (Fig. 6). When the protein content within the pulldown fractions was analyzed by unbiased LC–MS analysis, cMyBP-C was among the proteins detected in the samples derived from ARVMs that were exposed to NCA (Fig. 6A, Table S1; ProteomeXchange PXD019808). This observation was subsequently confirmed by Western immunoblot analysis. Interestingly, cMyBP-C was only co-precipitated in response to HNO donor exposure when pulldown experiments were performed under nonreducing conditions (Fig. 6B). These data confirmed oxidant-mediated colocalization of PKA-RI and its substrate cMyBP-C.
Figure 6.
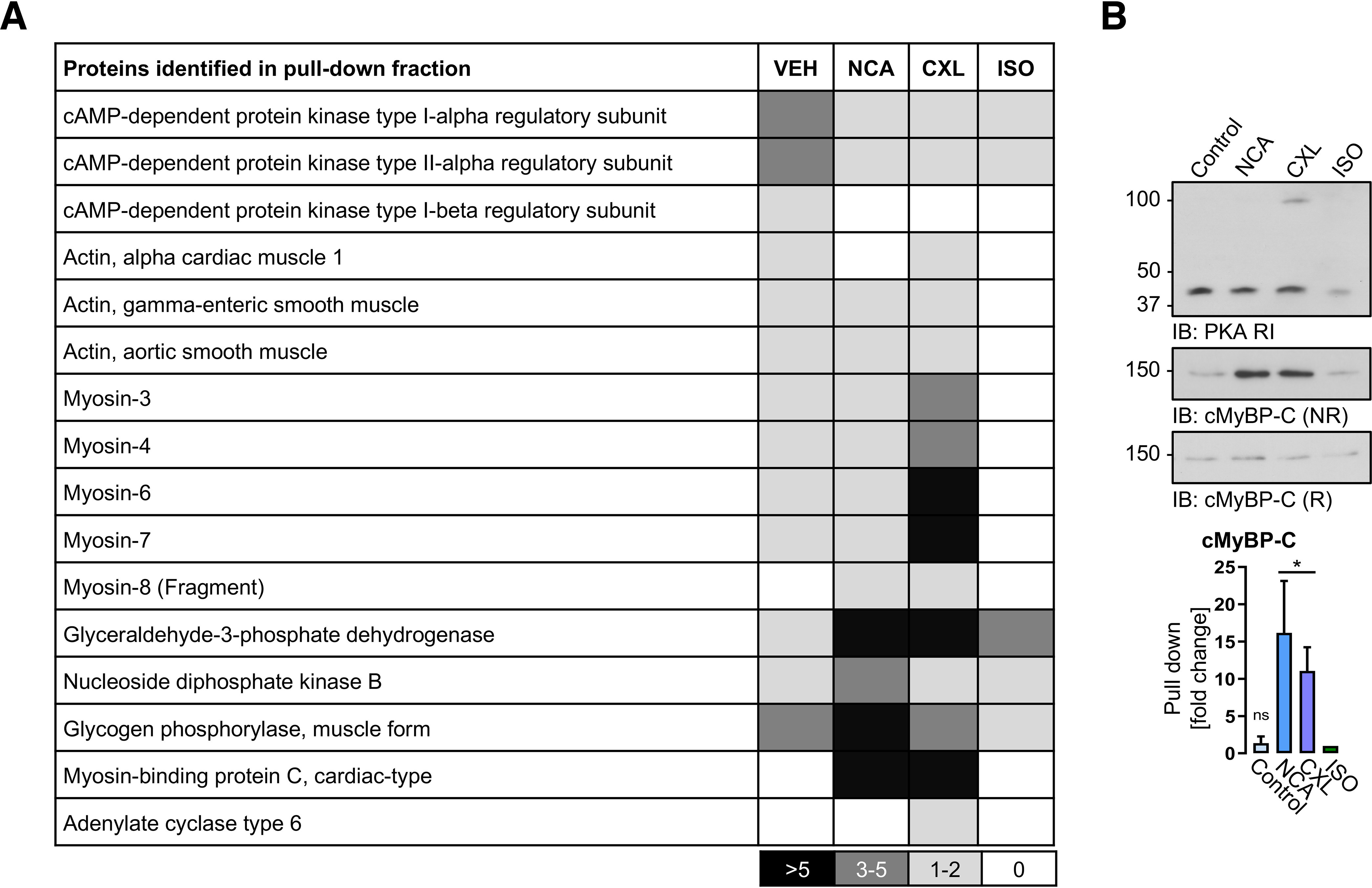
Identification and validation of proteins enriched by cAMP-agarose pulldown. ARVMs were treated with vehicle (DMSO), NCA (100 µmol/liter, 30 min), CXL-1020 (300 µmol/liter, 15 min), or ISO (10 nmol/liter, 10 min) and harvested under NR or R conditions. A, pulldown experiments using 8-AHA-cAMP-agarose were performed and analyzed by MS. Density chart reveals the number of identified peptides per protein hit. B, Western IB analysis of the pulldown samples for PKA-RI and cMyBP-C was performed under R or NR conditions. The scatter plots represent data of co-precipitated cMyBP-C in the pulldown fraction from 4 independent experiments. Results are expressed as fold-change over the ISO signal. *, p < 0.01 for comparison between the control (vehicle) and the HNO donor-treated group by one-way ANOVA with Dunnett's post-test (F = 8.38, p = 0.0028).
Furthermore, when cell fractionation was performed in the presence of a reducing agent, neither PKA nor PP2A were enriched in the Triton-insoluble fraction, suggesting indeed oxidation-dependent translocation and trapping of kinase and phosphatase in the myofilament compartment upon oxidant exposure (Fig. 7A). Importantly, oxidant-mediated myofilament enrichment also applied to PP1 and calcineurin (Fig. 7B).
Figure 7.
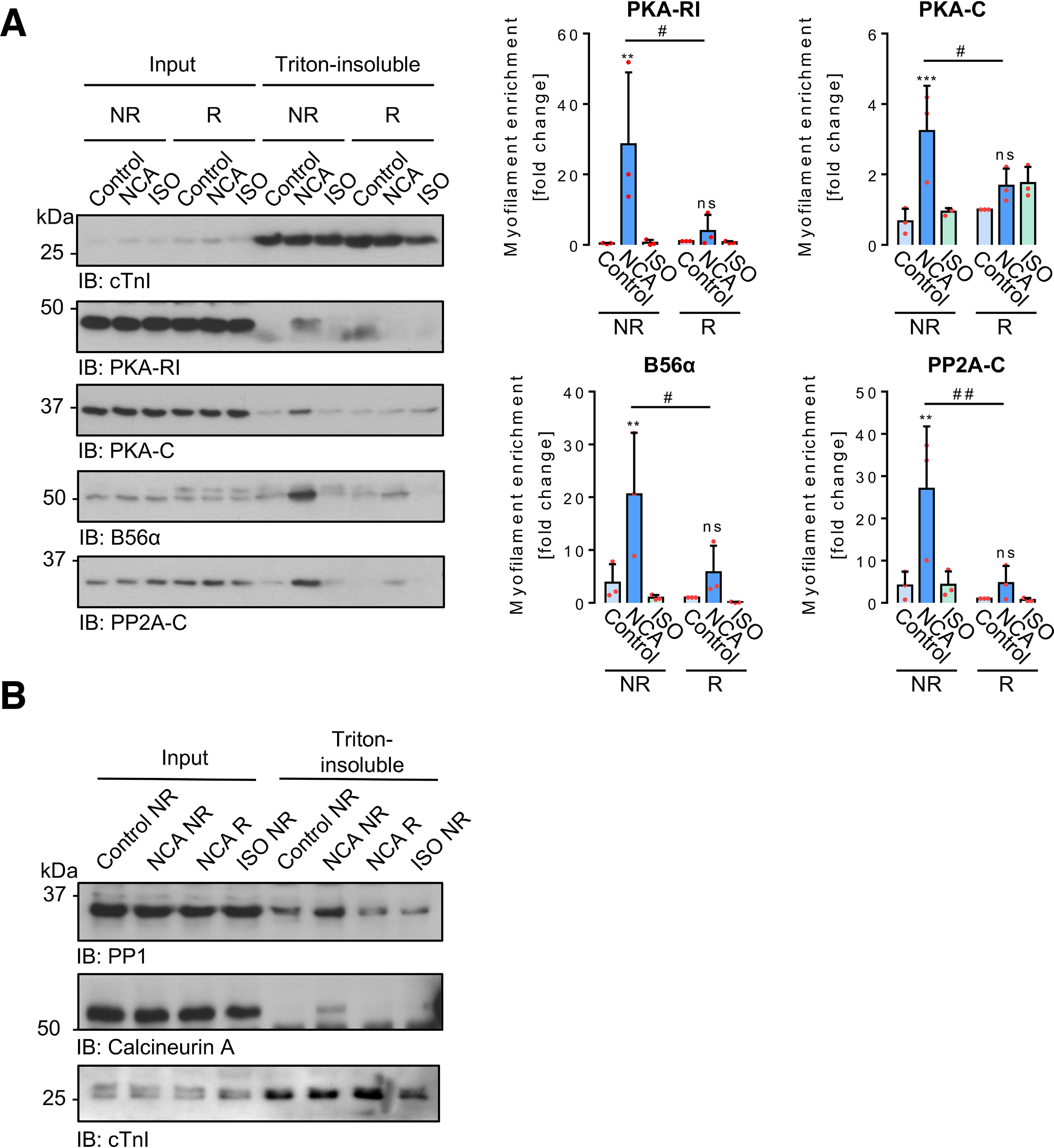
Oxidation-mediated effects of NCA on PKA and PP2A-C translocation. Following incubation of ARVMs with vehicle (control), NCA (100 µmol/liter, 30 min), or ISO (10 nmol/liter, 10 min), cells were harvested under NR or R conditions and separated into input Triton-soluble and Triton-insoluble fractions. cTnI was employed as the Triton-insoluble fraction marker protein. The content of PKA-RI, PKA-C, B56α, and PP2A-C in input lysates and in the Triton-insoluble fractions from both harvesting conditions was compared in 3 independent experiments. Signals obtained from Triton-insoluble samples were normalized to the related inputs and are presented as fold-change of vehicle control (R) in the scatter plots. **, p < 0.01; ***, p < 0.001 for comparison with the corresponding vehicle control; #, p < 0.05 for comparison between similar treatments from different harvesting conditions by two-way ANOVA with Bonferroni post-test (F and P values): a, PKA-RI, interaction, F = 4.24, p = 0.0403; treatment, F = 6.61, p = 0.0116; harvesting conditions, F = 3.91, p = 0.0714; b, PKA-C, interaction, F = 6.30, p = 0.0135; treatment, F = 11.03, p = 0.0019; harvesting conditions, F = 0.2196, p = 0.6477; c, B56α, interaction, F = 2.93, p = 0.0917; treatment,, F = 9.66, p = 0.0032; harvesting conditions, F = 5.89, p = 0.0320; d, PP2A-C, interaction, F = 4.23, p = 0.0406; treatment, F = 8.35, p = 0.0053; harvesting conditions, F = 9.86, p = 0.0085).
PKA and PP2A oxidation in response to oxidants in ARVMs
To investigate whether oxidant exposure alters the oxidation status of PKA and PP2A in intact cardiac myocytes, ARVMs were treated with vehicle (DMSO) or oxidants and subjected to biotin- or PEG-switch analyses (Fig. 8). For biotin-switch analysis, ARVMs were exposed to vehicle (DMSO) or NCA, oxidative posttranslational modifications on cysteines switched to biotin-labeling and biotinylated proteins were subsequently enriched by streptavidin pulldown. Equal protein content in the samples was assured by Coomassie and Sypro Ruby staining and enhanced oxidation of cellular proteins in response to NCA reflected by increased biotinylation was detected by Western immunoblotting using streptavidin-HRP (Fig. 8A). Immunoblot analysis of the eluted fractions revealed enrichment of PKA-RI, PKA-C, B56α, and PP2A-C in NCA-treated samples compared with vehicle controls, suggesting that NCA induced oxidation of PKA and PP2A subunits in intact ARVMs.
Figure 8.

Investigation of PKA and PP2A oxidation in intact ARVMs. A, biotin-switch analysis. Lysates from ARVMs exposed to vehicle (DMSO; 30 min) or NCA (100 µmol/liter; 30 min) were subjected to biotin labeling of oxidized thiol groups and subsequently biotinylated proteins were enriched by streptavidin pulldown. Total protein content before biotin labeling (Input; Lysate) or before streptavidin-pulldown (Input; Biotin) is demonstrated by Coomassie (left panel) or SYPRO Ruby staining (middle panel). Protein biotinylation levels correlated with protein oxidation in samples before (Input; Biotin) and after streptavidin-pulldown (eluate) was visualized by chemiluminescent detection with streptavidin-HRP (right panel). Blots are representative of three independent experiments. B, PKA-RI, PKA-C, B56α, and PP2A-C were detected in the streptavidin-pulldown samples by Western immunoblot analysis (IB). C, PEG-switch analysis. Lysates from ARVMs exposed to vehicle (control), NCA (100 µmol/liter, 30 min), AS (300 µmol/liter, 15 min), CXL-1020 (300 µmol/liter, 15 min), H2O2 (100 µmol/liter, 10 min), DIA (500 µmol/liter, 10 min), or ISO (10 nmol/liter, 10 min) were analyzed by nonreducing (left panel) or reducing (middle panel) Western immunoblot analysis for PKA-RI, PKA-C, B56α, and PP2A-C. Lysates were subjected to PEG tag labeling of oxidized thiol groups and subsequent detection of oxidized proteins via a mass shift was performed by western immunoblotting for PKA-RI, PKA-C, B56α, and PP2A-C (right panel). Blots are representative of three independent experiments.
An alternate methodology that requires lower protein input for analysis of protein oxidation, and thus allowed inclusion of all oxidant stimuli, is PEG-switch analysis that we have previously optimized for the detection of protein oxidation in cardiac myocytes (37). ARVMs were exposed to vehicle (DMSO), NCA, AS, CXL-1020, H2O2, DIA, or ISO and PEG-switch analysis was performed allowing detection of oxidized proteins due to a shift in molecular mass caused by linkage of the PEG-tag to a previously oxidized cysteine (Fig. 8C). Prior to PEG-switch, input samples were analyzed by Western immunoblotting under nonreducing or reducing conditions for PKA and PP2A subunits. Under nonreducing conditions, only the interdisulfide dimer of PKA-RI was detectable as previously observed. Interestingly, in samples treated with NCA and DIA, no band was detectable with the B56α antibody, suggesting that the loss in signal intensity could be attributable to oxidative modification of the protein. Under reducing conditions, PKA-RI was detectable as a monomer. Importantly, the loss in band intensity for B56α in response to NCA and DIA observed under nonreducing conditions revealed a signal, again suggesting the regaining of affinity of the antibody for its target under reducing conditions. PKA-C and PP2A-C were detectable in their monomeric state regardless of the experimental conditions. After PEG-switch, a shift to its dimer form was detectable for PKA-RI. Interestingly, a mass shift was also detectable for PKA-C and B56α in samples exposed to NCA, AS and DIA suggesting oxidation in intact ARVMs (Fig. 8C, black arrows), confirming the results obtained by biotin-switch. In contrast to the results from the biotin-switch analysis, no mass shift was detectable for PP2A-C, potentially reflecting the lack of access of the large PEG-tag to the cysteines in PP2A-C. Taken together, the data obtained by two different methods demonstrate that oxidation of PKA and PP2A subunits occurs in intact cardiac myocytes.
Effects of HNO donors on PKA and PP2A catalytic activity
To investigate whether direct exposure of purified PKA-C to NCA modulates kinase activity, in vitro kinase assays were performed using a recombinant His6-tagged C1-M-C2 domain of cMyBP-C. This substrate contains the phosphorylation sites Ser-273, Ser-282, and Ser-302 that are targeted by PKA. Under control conditions, addition of PKA-C induced robust phosphorylation of the substrate protein (Fig. 9A). Pretreatment with NCA completely abolished substrate phosphorylation, supporting previous reports that oxidation inhibits PKA-C activity (7). Pretreatment of PKA-C with ATP prior to oxidation rescued kinase activity as shown by significant substrate phosphorylation. As expected, addition of H89 to the reaction mixture inhibited PKA-C activity. Interestingly, when immunoblotting was performed under nonreducing conditions with an antibody recognizing PKA-C, a double band was detectable, revealing intradisulfide formation via Cys-199 and -343 as previously reported (38). Equal substrate presence in all samples was shown by immunoblotting with the anti-His antibody and Coomassie staining of the membrane.
Figure 9.
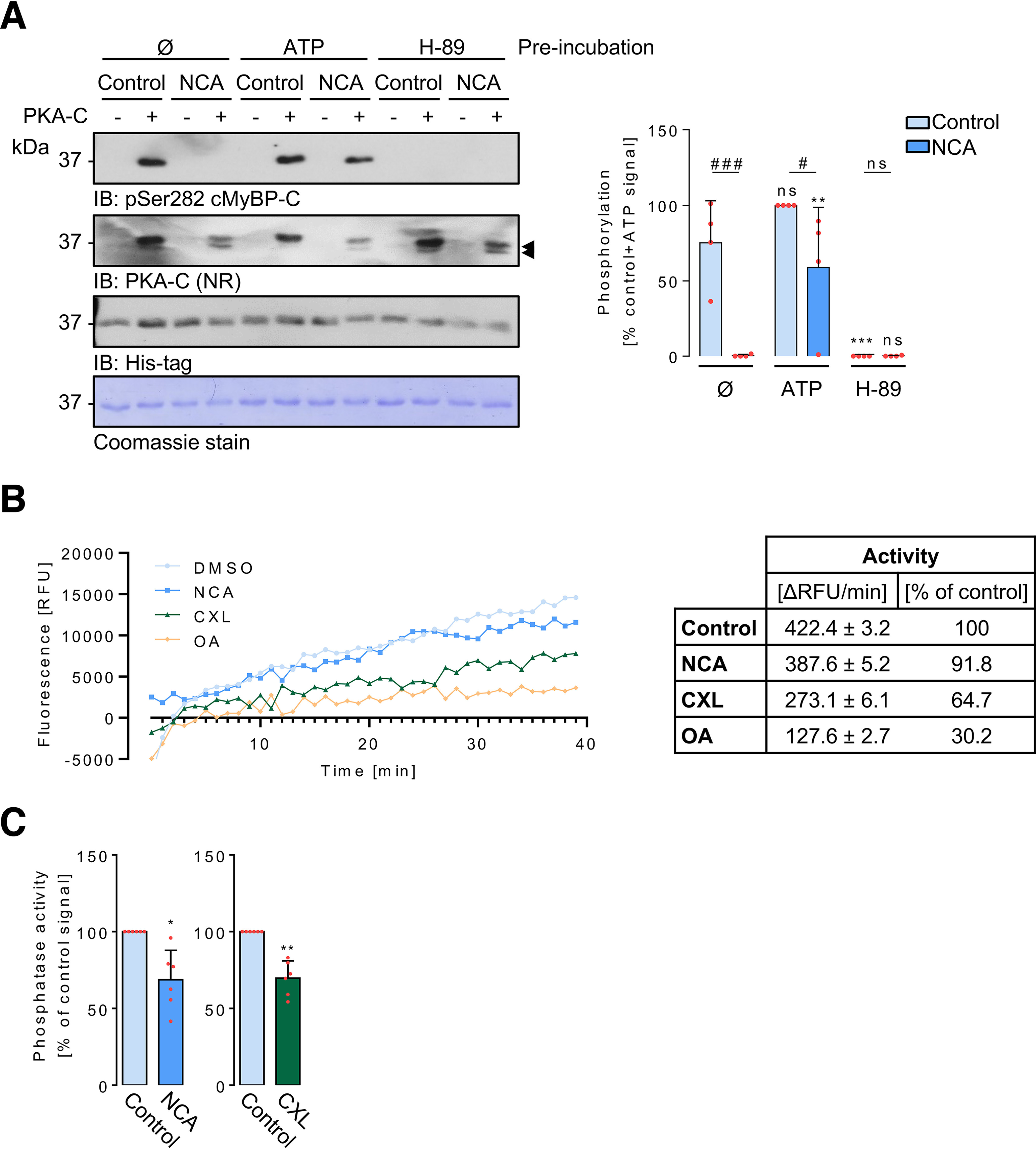
Oxidation-mediated effects on PKA and PP2A-C activity in vitro. A, active PKA catalytic subunit (PKA-C) was preincubated for 10 min with either ATP (100 µmol/liter), H89 (25 µmol/liter), or assay buffer (Ø) and then exposed to vehicle (control) or 100 µmol/liter NCA for 30 min. Then, the samples that initially did not contain ATP were supplemented with 100 µmol/liter of ATP and the in vitro kinase reaction was initiated by adding 100 pmol of recombinant His6-tagged C1-M-C2 cMyBP-C to each sample. Samples that did not contain kinase served as additional controls. Substrate phosphorylation and content was assessed under R conditions (sample reduction with 10% (v/v) β-mercaptoethanol) by IB using antibodies against pSer-282-cMyBP-C and the His6 tag. The presence and oxidation of PKA-C was monitored under NR conditions by IB using an anti-PKA-C antibody. Coomassie stain of the substrate blot is also shown as additional loading reference. The scatter plot summarizes results for C1-M-C2 phosphorylation from 4 independent experiments. Data are expressed as % of the signal of the vehicle control after ATP preincubation. **, p < 0.01; ***, p < 0.001 for comparison with the corresponding assay buffer (Ø)-pretreated sample; #, p < 0.05; ###, p < 0.001 for comparison with corresponding samples with and without NCA treatment by two-way ANOVA with Bonferroni post-test (interaction, F = 7.07, p = 0.0054; preincubation buffer, F = 31.64, p < 0.0001; treatment: F = 22.50, p = 0.0002). B, active recombinant human PP2A-C was incubated with vehicle (DMSO), NCA (100 μmol/liter), CXL-1020 (300 μmol/liter), or OA (10 nmol/liter), and the substrate DiFMUP (500 µmol/liter). Fluorescence reflecting phosphatase activity was monitored over time (representative traces). For each treatment, a phosphatase-free blank sample was included in the measurement. The table shows the activity of active recombinant PP2A-C at each condition, calculated from the slope of the linear regression of averaged RFU values and expressed as ΔRFU/min or % of the activity calculated under control conditions (n = 4-5 experiments). Measured fluorescence was normalized to blank values and expressed as % of the control signal. C, the data from B were reanalyzed to reflect the HNO donor incubation times in cell culture experiments. The scatter plots represent average phosphatase activity expressed as ΔRFU/min calculated from the slope of a linear curve fit after 15 min of CXL-1020 or 30 min of NCA treatment (n = 4-5 experiments) to reflect the cell culture conditions. *, p < 0.05; **, p < 0.01 two-tailed Student's t test for comparison of each time point with the corresponding vehicle control; ns, not significant.
Inhibition of PKA-C by NCA does not explain the increased phosphorylation that was observed. Therefore, the effect of HNO exposition on PP2A-C activity was investigated. Purified PP2A-C was subjected to NCA or CXL-1020 and dephosphorylation of the fluorogenic substrate 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) was assessed (Fig. 9B). The extent of phosphatase inhibition by HNO donors was compared with the effect observed in response to okadaic acid (OA), a prototypical serine/threonine phosphatase inhibitor that achieved ∼70% PP2A-C inhibition in our experiments. Exposure to CXL-1020 significantly reduced PP2A-C activity by ∼35% compared with the vehicle control. NCA reduced PP2A-C activity by ∼8% compared with the vehicle control. To match the exposure time of ARVMs to HNO donors in cell culture experiments, data were reanalyzed (Fig. 9C). The bar chart reflects the phosphatase activity following 15 or 30 min of exposure to CXL-1020 or NCA, respectively. In both cases, phosphatase activity was significantly inhibited by the HNO donors.
Ultimately, the activity status of kinase and phosphatase within the myofilament compartment upon NCA-mediated translocation was explored (Fig. 10). ARVMs were pretreated with NCA and fractionation was performed under NR or R conditions. In vitro kinase assays were performed by addition of the PKA substrate C1-M-C2 of cMyBP-C. Only under NR conditions, substrate phosphorylation was detectable, which is in accordance with the previous results demonstrating oxidative modification as a prerequisite for translocation. Addition of DTT to the NR IVK reaction mixture significantly abolished substrate phosphorylation, suggesting reactivation of a phosphatase and subsequent dephosphorylation of the substrate in the sample. This was corroborated by addition of 100 nmol/liter of OA to the NR IVK reaction mixture to inhibit both, PP1α and PP2A. OA addition significantly increased substrate phosphorylation (Fig. 10A). The investigations on localized kinase activity were paralleled by experiments assaying myofilament phosphatase activity. ARVMs were subjected to NCA treatment and fractionation as described above under NR or R conditions. Phosphatase activity within the myofilament fraction was investigated by addition of DiFMUP to the myofilament samples. Increased fluorescence due to dephosphorylation of the fluorogenic substrate was detectable in samples harvested under both NR and R conditions (Fig. 10B). Addition of DTT to reduce the oxidative modification of the phosphatase and achieve reactivation revealed significantly increased phosphatase activity in the NR samples compared with the R samples. Taken together, substrate phosphorylation conveyed by HNO donors results from a combination of localized kinase activation and phosphatase inhibition.
Figure 10.
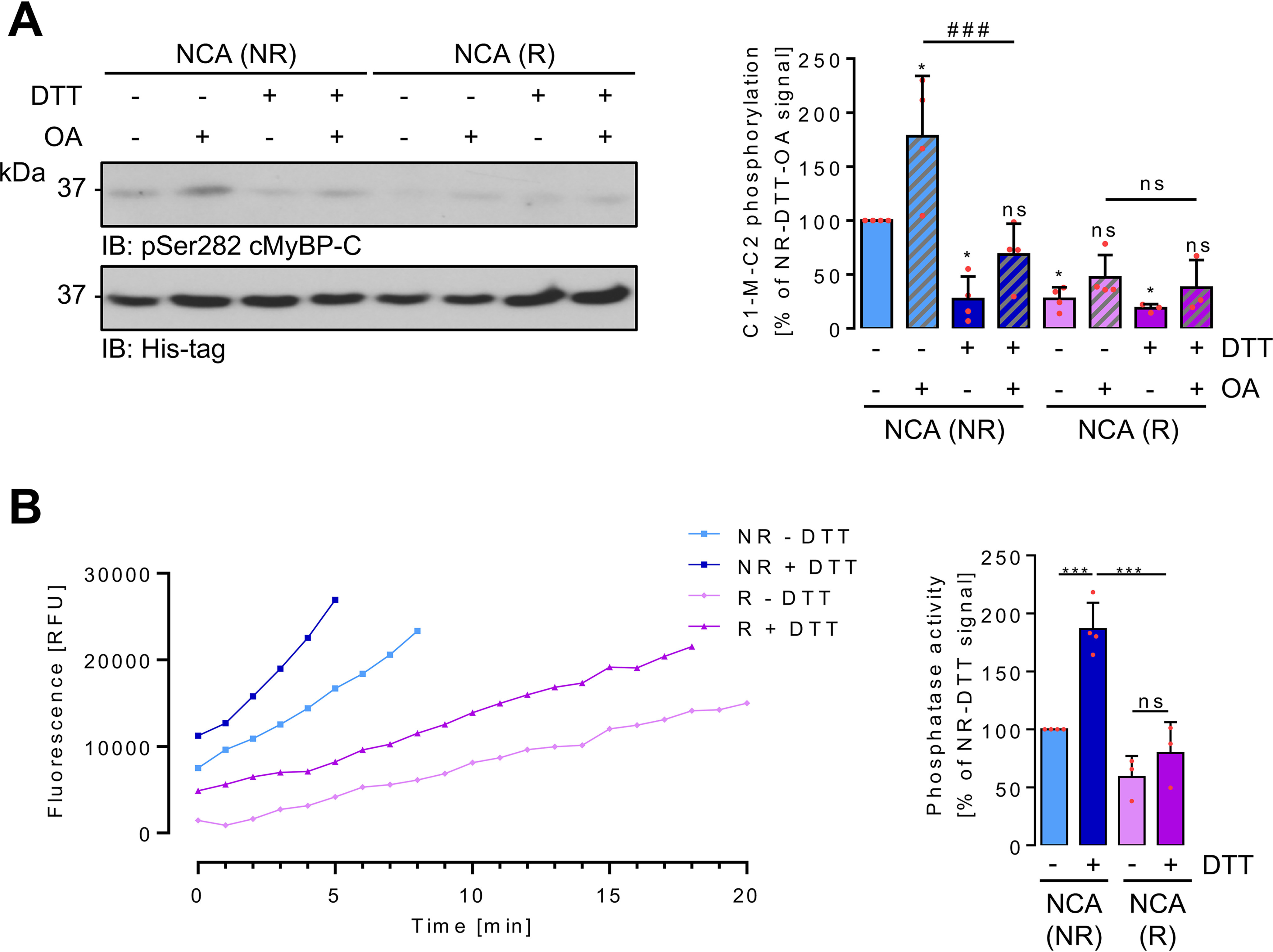
Oxidation-mediated effects on PKA and PP2A-C activity in myofilaments. A, Triton X-100–insoluble myofilament-containing fractions were prepared under NR or R conditions from ARVMs after exposure to NCA. Samples were split and in vitro phosphorylation of His6-tagged C1-M-C2 cMyBP-C at Ser-282 analyzed after addition of vehicle (DMSO), DTT to reduce oxidative modifications, or OA to inhibit PP1α and PP2A. Equal substrate protein content was demonstrated by immunoblotting with an anti-His antibody. The scatter plot summarizes results for C1-M-C2 phosphorylation from 3 to 4 independent experiments. Data are expressed as % of the signal of the NR signal without any additions. *, p < 0.05 for comparison with the NR nontreated sample; ####, p < 0.001 for comparison of the NR + OA sample in the presence or absence of DTT. Analysis by two-way ANOVA with Bonferroni post-test (interaction, F = 7.39, p = 0.0013; harvesting conditions, F = 36.99, p < 0.0001; IVK conditions, F = 14.22, p < 0.0001). B, Triton X-100–insoluble myofilament-containing fractions were prepared under NR or R conditions from ARVMs after exposure to NCA. Samples were split and phosphatase activity over time recorded after addition of vehicle (PBS) or DTT and DiFMUP (representative traces). The bar chart summarizes the results of 3 to 4 independent experiments expressed as % of the phosphatase activity measured under NR condition without any addition. ***, p < 0.001 for comparison with the NR DTT-treated sample. Analysis by two-way ANOVA with Bonferroni post-test (interaction, F = 10.14, p = 0.0097; harvesting conditions, F = 51,53, p < 0.0001; phosphatase assay conditions, F = 27,01, p < 0.0004). ns, nonsignificant.
Discussion
In the treatment of chronic heart failure, desensitization of β-ARs and poor prognosis in response to long-term treatment of patients with inotropic drugs are major obstacles that have to be addressed to achieve restoration of adequate cardiac contraction (39). Both, experimental and clinical data suggest therapeutic benefits of HNO donors in heart failure patients with reduced ejection fraction. This novel class of pharmacological agents has been shown to enhance cardiac contractility independently of β-AR stimulation (30), without development of tolerance or obvious adverse side effects, emphasizing their potential for clinical application. To date, cardiac inotropic and lusitropic effects of HNO donors have been entirely attributed to direct oxidative modification of protein components of the contractile machinery and those involved in Ca2+-cycling in cardiac myocytes (29). Here, we report a previously undescribed property of HNO donor compounds to indirectly impact on cardiac contractility by inducing oxidation of functionally important cysteines in PKA and PP2A thus modulating kinase and phosphatase activity and consequently prolonging basal substrate phosphorylation.
Information regarding the impact of phosphorylation on sarcomeric oxidation and vice versa is scarce. We showed previously that in human heart failure, increased S-glutathiolation of sarcomeric proteins was accompanied by reduced phosphorylation, whereas phosphorylation prevented subsequent oxidation. This suggested a negative cross-talk between different posttranslational modifications (37). Thus far, the induction of disulfide bonds between actin-tropomyosin and myosin light chain 1-myosin heavy chain accounted for the positive inotropic effects observed in response to HNO donors (29). In heart failure, hypophosphorylation of sarcomeric proteins is considered a major contributing factor for contractile dysfunction evoked by desensitization of the PKA signaling pathway in combination with increased expression and activity of protein phosphatases (37, 40, 41). This suggests that the contribution of HNO-mediated partial restoration of sarcomeric protein phosphorylation by an orchestrated modulation of receptor-independent kinase translocation and activation combined with phosphatase inhibition would assume greater significance under heart failure conditions with the potential for rescuing the positive inotropy by increasing cross-bridge cycling kinetics (33–35).
The main observation of our study was that HNO released by various donor compounds induced simultaneous and orchestrated spatiotemporal oxidation of multiple signaling components with a net result of increased cardiac protein phosphorylation. The order of events supported by our data are summarized schematically in Fig. 11 and suggests that HNO donors induce interdisulfide formation and translocation of PKA-RI and PKA-C to the myofilament compartment in the absence of receptor activation, which as we show occurred fast upon exposure to HNO. Thereby, a substrate-induced local release of the catalytic subunits might trigger phosphorylation of target proteins such as cMyBP-C. The lack of cTnI phosphorylation observed in the present study was surprising and suggests that either distinct spatiotemporal signaling upon oxidant exposure or more likely the inability of the antibody to detect cTnI phosphorylation under native pro-oxidative conditions might serve as an explanation. In line with previous reports (7, 8, 42), our in vitro data demonstrated oxidant-mediated inactivation of the catalytic subunit by intradisulfide formation, which cannot fully explain the increased phosphorylation of substrate proteins. However, a limitation of this in vitro experiment was the lack of the fully functional heterotetrameric PKA complex, but instead the catalytic PKA subunit was exposed to a pro-oxidant environment. This result provided the rationale to investigate the impact of HNO donor compounds on activity and subcellular localization of one of the main cardiac myocyte protein phosphatases PP2A. We reported previously that PP2A dynamically redistributed between myofilament and cytosolic compartment upon stimulation (43) and therefore hypothesized that HNO donors might affect this. Indeed, we found both, catalytic and targeting subunit of PP2A, coincidentally accumulating with the kinase subunits within the myofilament compartment upon oxidant exposure. These in vitro observations prompted us to monitor the actual activity of both, kinase and phosphatases in their native myofilament environment upon oxidant exposure. Although PKA activity increased moderately, pronounced inhibition of the myofilament-localized phosphatases was detectable, which could be reactivated by the reductant DTT. Combined with our in vitro observation that HNO donors inhibited PP2A activity, these findings strongly support the seminal observations made by the Taylor group (42) that the model-oxidant DIA impacted kinase and phosphatase activity, suggesting this as a general phenomenon. Thereby, basal or kinase-mediated substrate phosphorylation would accumulate upon HNO compound exposure and would be maintained over a longer time period, which the present study confirmed in time course experiments. Of note, also PP1 has been shown to form an inactivating intradisulfide between Cys-39 and Cys-127 upon exposure to oxidants (12, 16, 20). Importantly, the present study detects both main cardiac protein phosphatases accumulating in the myofilament fraction upon oxidant exposure. Overlap has been described regarding the spectrum of cardiac proteins impacting on cardiac myocyte contractility such as cMyBP-C (44) and cTnI (45) that are targeted by both, PP1α and PP2A. Therefore, despite the focus of the present manuscript on the contribution of PP2A in this experimental context, it is likely that HNO-mediated intradisulfide formation and subsequent inhibition of PP1α activity might also contribute to the increase in cardiac myocyte protein phosphorylation observed in response to HNO donors. This is also supported by the results obtained with OA that was on purpose applied at a concentration to inhibit both types of phosphatases.
Figure 11.

Summary scheme. Effect of HNO donor-mediated oxidation on kinase (PKA; left) and phosphatase (PP2A; right) signaling in adult rat ventricular myocytes. RI, regulatory type I PKA subunit; C, catalytic subunit of PKA (left) or PP2A (right); A, regulatory subunit; B, targeting subunit; P, protein phosphorylation.
When positive inotropy and lusitropy were described for HNO for the first time and also in studies that reported HNO-mediated oxidative modification of sarcoplasmic reticulum Ca2+-ATPase (46), PLN (31), and the ryanodine receptor (47), the prototypical donor compound AS was employed. Assuming the observed effects rely entirely on released HNO, results were translated to chemically distinct HNO donor compounds without further testing. In fact, AS, NCA, and CXL-1020 share the positive effects on vasorelaxation (32, 48, 49) and cardiac myocyte contractility (47, 50, 51). Nonetheless, there are considerable differences, as shown by deviating effects of NCA and AS on sarcomeric protein oxidation (29). In line with these latter observations, our data show a distinct potential of the experimental NCA and AS and the clinical CXL-1020 HNO donor compound to modulate cardiac myocyte protein phosphorylation. Interestingly, significant differences in ARVM contractility were observed in response to NCA and CXL-1020. With its short t½ value of 2.3 min, CXL-1020 is expected to produce higher concentrations of HNO than the slowly hydrolyzing donor NCA (t½: 800 min), rendering HNO release kinetics unlikely to explain the responses exclusively observed with NCA. The unexpectedly fast effect in RI dimerization, PKA translocation, and ARVM contractility suggested an additional direct interaction of NCA with protein thiols independent from HNO release to underlie these effects, as it had been previously suggested (52). The finding that NCA effects were shared by DIA, which acts by inducing disulfide bonds, further supported the potential of NCA to mediate protein oxidation by a direct mechanism in addition to HNO release.
Although PKA-RI dimer formation was induced by all tested oxidants, it did not correlate with the extent of substrate phosphorylation evoked by the different compounds. Thus, the role of oxidative RI dimerization as a direct readout for oxidant-mediated PKA activity remains ambiguous. Our study suggests that cardiac myocyte protein phosphorylation in response to oxidants depends on the direct oxidative modification of susceptible cysteines impacting on the activity and translocation of the catalytic subunits of both, PKA and PP2A. A similar observation was reported previously on thiol-disulfide inhibition of PKA and PP2A by DIA with impact on kinase and phosphatase activity (42). This combined inhibitory impact of oxidants may serve as a potential explanation of the NCA-mediated cardiac myocyte protein phosphorylation.
Conclusion
The present study demonstrates a role for HNO donors in the regulation of kinase and phosphatase signaling with impact on cardiac myocyte protein phosphorylation and function.
Experimental procedures
Materials
DMSO, H2O2, DIA, ISO, FOR, nonspecific goat serum (NGS), 4',6-diamidin-2-phenylindol (DAPI), atenolol, and H89 were from Sigma-Aldrich. KN-93, bisindolylmaleimide, and H1152 were from Tocris (Bristol, United Kingdom). BI-D1870 was from Enzo Life Sciences (Farmingdale, NY USA). NCA and CXL-1020 were synthesized by Axon Medchem (Groningen, Netherlands). Recombinant human PP2A-C (number 10011237; lot 0521439-1), AS and ODQ were purchased from Cayman Chemicals (Ann Arbor, MI USA). Pro-Q Diamond and SYPRO Ruby protein stain were from ThermoFisher Scientific (Waltham, MA USA). Rabbit polyclonal phospho-specific antibodies for pSer-273 and pSer-304 MyBP-C (dilution IB 1:1000) were gifts from S. Sadayappan (Cincinnati, OH USA). OA and the anti-pSer-282 cMyBP-C antibody (dilution IB 1:1000; rabbit polyclonal; number ALX-215-057-R050) was from Enzo Life Sciences. The anti-pSer-16 PLN antibody (dilution IB 1:2000; rabbit polyclonal; number A010-12AP; lot 642111) was from Badrilla (Leeds, UK). Antibodies against PKA-RI (dilution IB, 1:1000, and IF, 1:100; mouse monoclonal; number 610166; lot 5170502), PKA-C (dilution IB 1:1000; mouse monoclonal; number 610980, lot 5217836), and B56α (dilution IB 1:2000 and IF 1:100; mouse monoclonal; number 610615; lot 77324) were from BD Biosciences (San Jose, CA USA). Anti-α-actinin antibodies raised in mice (dilution IB, 1:1000, and IF, 1:200; number A7811; lot 127114807V) or rabbits (dilution IF, 1:800; number SAB2108642; lot QC12269) were from Sigma-Aldrich. The antibody against GAPDH (dilution IB, 1:2000; mouse monoclonal; number 5G4; lot 18105-G4-C5) was from HyTest (Turku, Finland). The antibodies against total cTnI (dilution IB, 1:1000; rabbit polyclonal; number 4002; lot 2) and pSer-22/23 cTnI (dilution IB, 1:500; rabbit polyclonal; number 4004; lot 4) were from Cell Signaling Technology (Danvers, MA USA). The anti-sodium potassium ATPase 1 (NKA1) antibody (dilution IB, 1:2000; mouse monoclonal; number 05-369; lot 2762285), digitonin, ATP, and PKA catalytic subunit from bovine heart (number 539576; lot D8MN079AK) were purchased from Merck Millipore (Billerica, MA USA). Antibodies against cMyBP-C (dilution IF, 1:100; rabbit polyclonal; number sc-67354; lot G1808), PP2A-C (dilution IB, 1:200; demethylated; mouse monoclonal; number sc-13601; lot L0716), PP1 (dilution IB, 1:300; mouse monoclonal; number sc-7482; lot J1117), calcineurin A (dilution IB, 1:300; number sc-17808; lot C1218) and His6 tag (dilution IB, 1:500; mouse monoclonal; sc-804; lot J0115) as well as IBMX were from Santa Cruz Biotechnology.
The horseradish peroxidase (HRP)-linked sheep anti-mouse IgG (dilution 1:2000; number NA931V; lot 16953154), the HRP-linked donkey anti-rabbit IgG (dilution 1:2000; number NA9340V; lot 16997954), the enhanced chemiluminescence (ECL) kit, Hyperfilm, polyvinylidene fluoride and nitrocellulose membrane were from GE Healthcare (Buckinghamshire, UK). The AKAR3-NES FRET sensor was a kind gift of G. Vandecasteele (Paris). Triton X-100 was purchased from Roth (Arlesheim, Germany). 8-Aminohexylamino (8-AHA)-cAMP-agarose was from Biolog Life Science Institute GmbH (catalog number A028-06; Bremen, Germany). DiFMUP was purchased from Endotherm (Saarbrücken, Germany). Complete™ protease inhibitors were from Roche Diagnostics (Risch, Switzerland). Homogenates from adult mouse ventricular myocytes isolated from WT or C17S PKA knock-in mice exposed to hydrogen peroxide (100 μmol/liter, 10 min) or isoprenaline (10 nmol/liter, 10 min) were kindly provided by Prof. Philip Eaton from Queen Mary University London, UK.
Isolation of ARVMs
The isolation of primary ARVMs from male Wistar rats was performed as described previously (37) and executed in compliance with the German law for the protection of animals and the Guide for the Care and Use of Laboratory Animals issued by the National Research Council (USA) Committee (2011). ARVMs were cultured for 24 h in laminin-coated 6-well–plates containing Medium 199 supplemented with 5 mmol/liter of taurine, 2 mmol/liter of carnitine, 2 mmol/liter of creatine, and 100 units/ml of penicillin-streptomycin.
Oxidant treatment of ARVMs and sample analysis
ARVMs were exposed to vehicle DMSO (0.1% (v/v), 30 min), NCA (100 μmol/liter, 30 min), AS (500 μmol/liter, 15 min), CXL-1020 (300 μmol/liter, 15 min), H2O2 (100 μmol/liter, 10 min), DIA (500 μmol/liter, 10 min) or ISO (10 nmol/liter, 10 min) and harvested in nonreducing (NR) (3 times in mmol/liter: Tris-HCl 187.5, pH 6.8, maleimide 100, 6% (w/v) SDS, 0.03% (w/v) bromphenol blue, 30% (v/v) glycerol) or reducing (R) Laemmli sample buffer (3 times in mmol/liter: Tris-HCl 187.5, pH 6.8, 6% (w/v) SDS, 0.03% (w/v) bromphenol blue, 30% (v/v) glycerol, 9% (v/v) β-mercaptoethanol). Lysates were subjected to SDS-PAGE and phosphoproteins were visualized with Pro-Q Diamond using a Typhoon 9400 imager (GE Healthcare). Total protein content was visualized by re-staining with SYPRO Ruby and Coomassie Brilliant Blue. Alternatively, samples were analyzed by Western immunoblotting. For time course treatment, ARVMs were exposed to vehicle DMSO (0.1% (v/v)), NCA (100 μmol/liter; 3, 10, 30, and 100 min), or ISO (10 nmol/liter; 10 min) and analyzed by immunoblot analysis.
Pharmacological treatment of ARVMs
ARVMs were exposed to the respective solvent vehicle (DMSO, 0.1% (v/v) or PBS), atenolol (1 μmol/liter, 5 min), to inhibit β1-AR; H89 (10 μmol/liter, 30 min) to inhibit PKA, KN-93 (20 μmol/liter, 30 min) to inhibit Ca2+-calmodulin–dependent kinase, H1152 (300 nmol/liter, 4 h) to inhibit Rho-kinase, bisindolylmaleimide (2 μmol/liter, 15 min) to inhibit protein kinase C, and BI-D1870 (10 μmol/liter, 30 min) to inhibit p90 ribosomal S6 kinase prior to oxidant treatment as described above.
FRET measurements in ARVMs
Isolated ARVMs were seeded onto glass slides and transduced at multiplicity of infection of 300 with an adenovirus encoding the AKAR3-NES biosensor that was provided for the present study and had been extensively characterized by Vandecasteele and colleagues (53). The ratio of light emitted by CFP and YFP of this biosensor changes upon phosphorylation by PKA. After 48 h of cultivation, glass slides were transferred into a microscopy cell chamber and baseline fluorescence recorded in FRET buffer (in mmol/liter: NaCl 144, HEPES 10, KCl 5.4, MgCl2 1, pH 7.3), before NCA (100 μmol/liter) or DIA (500 μmol/liter) were applied. When a plateau was reached, maximal sensor activation was achieved by combined addition of FOR (10 μmol/liter)/IBMX (100 μmol/liter). Measurements were performed using a DMI3000b inverted microscope with a ×40 objective (Leica, Wetzlar, Germany). Signals from CFP and YFP were simultaneously recorded using a DV2 DualView emission splitting system (Photometrics, Tucson, AR USA) and an OptiMOS™ Camera (QImaging, Surrey, Canada). CFP excitation was induced by a Light source pE-100 (440 nm; CoolLED, Andover, UK). Data processing was performed with ImageJ μManager with a customized plugin and Microsoft Excel. FRET changes in response to the applied stimuli were normalized to baseline values and expressed as % of the effect induced by FOR/IBMX.
Subcellular fractionation
Following the treatment as described above, ARVMs were scraped into 180 μl/well of NR fractionation buffer (in mmol/liter: Tris-HCl 50, pH 7.5, NaF 100, EGTA 5, EDTA 2, 0.05% (w/v) digitonin, protease inhibitor) or R fractionation buffer supplemented with DTT (5 mmol/liter) and snap frozen in liquid N2. After thawing, ARVMs were harvested and subjected to subcellular fractionation as previously described (43). In brief, lysates were centrifuged at 10,000 × g for 2 min at 4 °C and the supernatant representing the cytosolic fraction was collected. The pellet was resuspended in fractionation buffer containing 1% Triton X-100 and after centrifugation, the supernatant comprising membrane-associated proteins was collected. The remaining myofilament containing detergent-insoluble pellet was resolved in the sample buffer. Cytosolic and membrane fractions were supplemented with sample buffer before analysis by Western immunoblotting.
cAMP-agarose pulldown
After treatment of ARVMs, cells were harvested in 200 μl of lysis buffer (with or without DTT)/well of a 6-well–plate (in mmol/liter: HEPES 30 pH 7.4, EDTA 2, EGTA 2, NaCl 150, 1% (v/v) Triton X-100, NaF 2, protease inhibitors). Lysates were centrifuged at 10,000 × g for 10 min at 4 °C and the supernatant was collected, supplemented with 30 μl of 8-AHA-cAMP-agarose, and incubated under rotation at 4 °C for 4 h. Beads were washed twice with lysis buffer supplemented with detergent and protease inhibitors and twice without.
Tryptic in-gel digestion
In-gel digestion was done following Shevchenko et al. (54). Shrinking and swelling was performed with 100% ACN and 100 mmol/liter of NH4HCO3. In-gel reduction was achieved with 10 mmol/liter of DTT (dissolved in 100 mmol/liter of NH4HCO3). Alkylation was performed with 55 mmol/liter of iodacetamide (dissolved in 100 mmol/liter of NH4HCO3). Proteins in the gel pieces were digested by covering them with a trypsin solution (8 ng/μl of sequencing-grade trypsin, dissolved in 50 mmol/liter of NH4HCO3 containing 10% ACN) and incubating the mixture at 37 °C for overnight. Tryptic peptides were yielded by extraction with 2% FA, 80% ACN. The extract was evaporated. For LC–MS/MS analysis, samples were dissolved in 20 μl of 0.1% FA.
LC–MS/MS analysis
Protein identification via analysis of the tryptic peptides by LC–MS/MS was achieved by injection of the samples into a nano-liquid chromatography system (Dionex UltiMate 3000 RSLCnano, Thermo Scientific, Bremen, Germany) coupled via electrospray ionization to a mass spectrometer (MS) equipped with a quadrupole, a linear trap and an Orbitrap (Orbitrap Fusion, Thermo Scientific, Bremen, Germany) or with nano liquid chromatography system (nanoACQUITYy, Waters, Manchester, UK) coupled via electrospray ionization to a MS consisting of a quadrupole and an Orbitrap mass analyzer (Orbitrap QExcactive, Thermo Scientific, Bremen, Germany). The samples were injected (5 μl/min) to a trapping column (Acclaim PepMap μ-precolumn, C18, 300 μm × 5 mm, 5 μm, 100 Å, Thermo Scientific, Bremen, Germany; nanoACQUITY UPLC Symmetry C18 trap column, 180 μm × 20 mm, 5 μm, 100 Å; buffer A, 0.1% FA in HPLC-H2O; buffer B, 0.1% FA in ACN) with 2% buffer B. After sample injection the trapping column was washed for 5 min with 2% buffer B (5 μl/min). Peptides were moved to (200 nl/min) the separation column and separated on this column (Acclaim PepMap 100, C18, 75 μm × 250 mm, 2 μm, 100 Å, Thermo Scientific, Bremen, Germany; nanoAcquity UPLC column, BEH 130 C18, Waters; 75 μm × 250 mm, 1.7 μm, 100 Å; 200 nl/min, gradient: 2−30% B in 30 min). The spray was formed by a fused-silica emitter (I.D. 10 μm, New Objective, Woburn, MA USA) at a capillary voltage of 1650 V. Mass spectra were measured in the positive ion mode. LC–MS/MS analysis with the Orbitrap Fusion were carried out in data-dependent acquisition mode applying top speed mode. An HCD collision energy of 28%, an intensity threshold of 2e5, and an isolation width of 1.6 m/z was used. A MS scan was performed every second over a m/z range from 400 to 1500 (resolution of 120,000 FWHM at m/z 200; transient length = 256 ms; maximum injection time = 50 ms; AGC target = 2e5). MS/MS spectra were obtained in the ion trap (scan rate = 66 kDa/s; maximum injection time = 200 ms; AGC target = 1e4). LC–MS/MS runs with the Orbitrap QExcactive were done on the MS level over a m/z range from 400 to 1500 (resolution of 70,000 FWHM at m/z 200; transient length = 256 ms; injection time = 100 ms, AGC target = 3e6). MS/MS measurements were carried out applying the data-dependent acquisition mode mode (Top5; HCD collision energy of 30%; resolution of 17,000 FWHM at m/z 200; transient length = 64 ms; injection time = 100 ms; AGC target = 3e6; underfill ratio of 10%; isolation width of 2 m/z).
LC–MS/MS data analysis
With Proteome Discoverer 2.0 (Thermo Scientific, Bremen, Germany) the LC–MS/MS data were processed. Identification of the proteins from the MS/MS spectra were performed with the search engine Sequest HT using the Rattus norvegicus SwissProt database (release June 2016, 7961 entries; RRID:SCR_002380) and a contaminant database. For the searches, the following parameters were applied: precursor mass tolerance: 10 ppm; fragment mass tolerance: 0.2 Da; fully tryptic digestion. Two missed cleavages were allowed. Carbamidomethylation on cysteine residues as a fixed modification and oxidation of methionine residues as a variable modification was used for the search. Peptides with a false discovery rate of 1% using Percolator were identified. At least two unique peptides per protein were used as a condition for a reliable identification.
In vitro kinase assay
For the in vitro kinase (IVK) assays, 1250 IU/sample active PKA catalytic subunit were diluted in 5 μl of assay buffer (30 mmol/liter of Tris, pH 7.4, 15 mmol/liter MgCl2) and preincubated for 10 min at 37 °C with either 100 μmol/liter of ATP, 25 μmol/liter of H89 or the equivalent volume of assay buffer (Ø). This was followed by a 30-min treatment with vehicle DMSO (control) or NCA (100 μmol/liter) at 37 °C. Subsequently, ATP (100 μmol/liter) was added to the H89- or Ø-pretreated samples and the appropriate volume of assay buffer was added so that the final reaction volume would be 50 μl/sample. The kinase reaction was initiated by adding 100 pmol/sample of recombinant His6-tagged C1-M-C2 domain (amino acid residues 153-450) of human cMyBP-C as substrate (prepared as described in Ref. 37), proceeded for 30 min at 30 °C and terminated by addition of 25 μl/sample 3 times with nonreducing sample buffer. For each condition, a blank sample devoid of kinase was also prepared.
In vitro PP2A-C activity assay
The PP2A-C activity was analyzed in vitro by a fluorescence assay in which fluorogenic DiFMUP was used as phosphatase substrate. The substrate was initially prepared as a 10 mmol/liter stock solution in 50 mmol/liter of Tris-HCl buffer (pH 7.0) and stored at −20 °C. Before each experiment, this stock was further diluted to 1 mmol/liter with phosphatase assay buffer (40 mmol/liter of Tris-HCl, pH 7.5, 34 mmol/liter of MgCl2, 4 mmol/liter of EDTA, 0.005% (w/v) BSA). The initial stock solutions of NCA (100 mmol/liter), CXL-1020 (300 mmol/liter), and the serine/threonine protein phosphatase inhibitor OA (10 μmol/liter) were prepared in DMSO. These were then diluted in phosphatase assay buffer to acquire solutions of 250 μmol/liter of NCA, 750 μmol/liter of CXL-1020, and 25 nmol/liter of OA. For each reaction, 40 μl of compound solution were added to 50 μl phosphatase assay buffer containing DiFMUP in a black-walled 96-well–plate. The reaction was started by adding 10 μl/well of 12.1 microunits/μl of recombinant human PP2A-C solution prepared in phosphatase assay buffer (final concentration, 121 μm/well). This resulted in a final volume of 100 μl/sample and final concentrations of 500 μmol/liter of DiFMUP, 100 μmol/liter of NCA, 300 μmol/liters of CXL-1020, and 10 nmol/liter of OA. As a blank for each sample, a phosphatase-free control was prepared and measured in parallel. The dephosphorylation of the DiFMUP substrate to fluorescent DiFMU was measured in a Tecan Safire II microplate reader (Männerdorf, Switzerland) at excitation/emission wavelengths of λ358/455 for 40 min (1 read/min) at 30 °C. Average activities expressed as ΔRFU/min were calculated from the slope of a linear curve fit through the origin of the coordinate system from averaged normalized values of 4-5 independent assays. For the reanalysis of the data to reflect cell culture conditions, phosphatase activity in response to CXL-1020 exposure was calculated involving data points from 15 to 40 min and for NCA from 30 to 40 min.
Myofilament kinase and phosphatase assay
ARVMs were cultured in 6-well–plates and 4 wells were combined per intervention. After pharmacological treatment, ARVMs were lysed in 200 μl of fractionation buffer/well (in mmol/liter: Tris-HCl 50, pH 7.4, EGTA 5, EDTA 2, NaF 100, supplemented with 1% (v/v) Triton X-100 and Complete protease inhibitors). Harvesting under reducing conditions occurred with fractionation buffer containing 5 mmol/liter of DTT. The crude lysates were then aliquoted in two sets of tubes (labeled A and B; 350 μl lysate/tube) and centrifuged for 5 min, at 3000 × g and 4 °C. After removal of the supernatant, the pellets that contained the Triton X-100–insoluble myofilament fraction were washed with assay buffer (in mmol/liter: Tris-HCl 40, pH 7.5, MgCl2 34, EDTA 4, supplemented with 0.005% (w/v) BSA) and centrifuged again as described above. Each myofilament pellet was then resuspended in 120 μl of assay buffer without DTT (set A) or with 5 mmol/liter of DTT (set B).
For the kinase assays, each reaction was prepared in a final volume of 60 μl and contained 50 μl of myofilament suspension, 100 μmol/liter of ATP, 100 pmol of substrate (recombinantly expressed His6-tagged C1-M-C2 domain) and either vehicle (DMSO) or 100 nmol/liter of the pan-phosphatase inhibitor OA. The reactions were carried out at 30 °C for 30 min and terminated by addition of 30 μl of 3× Laemmli sample buffer.
For the phosphatase assays, 25 μl of myofilament suspension were mixed with DiFMUP (500 μmol/liter, final concentration) in assay buffer without (set A) or with 5 mmol/liter of DTT (set B) at a final volume of 100 μl/reaction. Measurements were performed as described under the “In vitro PP2A-C activity assay” section. For each condition measured, a linear regression was used to calculate the slope from the available number of measurements.
SDS-PAGE and Western immunoblot analysis
Protein samples in reducing or nonreducing sample buffer were resolved by SDS-PAGE and analyzed by Western immunoblotting as described previously (55). In brief, gels were prepared using acrylamide/bisacrylamide (Bio-Rad, Feldkirchen, Germany; number 1610156). Running gels contained a buffer composed of (in mmol/liter) 375 Tris base (pH 8.0), 3.5 SDS, 4.38 ammonium persulfate (APS), 0.1% (v/v) N,N,N',N'-tetramethylethylenediamine (TEMED). Thereby, 7.5% (v/v) polyacrylamide gels were used for the analysis of cMyBP-C phosphorylation, α-actinin, and NKA1; 10% (v/v) for the analysis of PKA-RI, PKA-C, PP2A-C, B56α, PP1, calcineurin, GAPDH, and His6 tag; 15% (v/v) for pSer-16 PLN and pSer-23/24 cTnI. Stacking gels were composed of 3.5% (v/v) polyacrylamide in a buffer comprising (in mmol/liter): 125 Tris base (pH 6.8), 3.5 SDS, 4.38 APS, and 0.1% (v/v) TEMED. As secondary antibodies, HRP-linked sheep anti-mouse IgG or the HRP-linked donkey anti-rabbit IgG at a dilution of 1:2000 was used. Band density quantification was performed using the GelQuant.NET software provided by BiochemLabSolutions (RRID:SCR_015703). Validation of the respective phosphospecific antibodies recognizing cMyBP-C, cTnI, and PLN phosphorylation has been described previously (35, 55–61). Full blots with respective molecular mass markers are shown in Fig. S6.
For analysis of protein phosphorylation in ARVMs, all signals were normalized to the respective loading control (α-actinin) from the same blot and data were presented as % fold-change over the isoprenaline-positive control. For the analysis of the interdisulfide-induced PKA-RI dimer formation, after normalization to the loading control, the ratio of dimer to monomer signal density of the same lane was calculated and expressed as fold-change over the control value for each experiment. For analysis of protein translocation, signals in the insoluble fraction were normalized to the respective input signals and data were expressed as fold-change over the control value for each experiment. In the IVK assays, the phosphorylation signals were normalized to the total substrate loading and expressed as % of the vehicle control after ATP pre-incubation.
Immunofluorescence labeling
After pharmacological treatment, the plasma membrane of ARVMs was removed by incubation for 15 min with 1% Triton X-100 in PBS, cells were fixed in 4% paraformaldehyde (PFA) in PBS and blocking was performed with 5% NGS in BSA/Gold buffer (in mmol/liter: NaCl 155, Tris 10, EGTA 2, MgCl2 2, pH 7.2, and 1% (w/v) BSA). Immunofluorescence staining of cMyBP-C was performed with the primary anti-cMyBP-C antibody and secondary sheep anti-rabbit IgG: DyLight®459 (dilution 1:100; number STAR36D549GA; lot 0203; Bio-Rad, Feldkirchen, Germany). PKA-RI and B56α were labeled by a combination of anti-PKA-RI or anti-B56α antibodies, respectively, biotinylated goat anti-mouse antibody (dilution 1:500; number A10519; lot 1871954; ThermoFisher Scientific, Waltham, MA USA) and Alexa Fluor®546 streptavidin (dilution 1:2000; number S11225; lot 1584283; ThermoFisher Scientific) to enhance signal intensity. Cell nuclei were stained with DAPI (dilution 1:100). Specimens were analyzed by confocal imaging with the LSM 800 laser scanning microscope and Plan-NEOFLUAR ×40 oil immersion objective. ImageJ (62) was used for image processing and the determination of longitudinal signal intensity distributions in immunolabeled ARVMs.
Proximity ligation assay
The proximity between PKA-RI and cMyBP-C or α-actinin was assessed by PLA using the Duolink® In Situ Orange Starter Kit Mouse/Rabbit (Merck Millipore). ARVMs were exposed to DMSO (0.1% (v/v), 30 min) or NCA (100 μmol/liter, 30 min) and the plasma membrane was removed with 1% Triton X-100 in PBS before fixing in 4% PFA in PBS. Unspecific sites were blocked with 5% NGS in BSA/Gold buffer and PLA was performed according to the manufacturer's instructions. Fluorescence signals indicating a distance of maximally 40 nm between PKA-RI and cMyBP-C or α-actinin were detected by Z-stack imaging at 2-μm intervals with a confocal laser scanning microscope LSM 800 with a Plan-NEOFLUAR ×40 oil immersion objective and a Cy3 filter (Carl Zeiss, Oberkochen, Germany). PLA counts were obtained from the maximum intensity projection of each Z-stack experiment and thresholding was performed by the “Intermodes” function of ImageJ.
To allow better image visualization, digital image processing was performed using GIMP version 2.10.14. Specifically, brightness was adjusted in all confocal images in an identical manner. For the images obtained after measuring “PLA counts,” the color mode was changed to grayscale, brightness was then adjusted, and color was finally inverted. All PLA count images were processed in the same way to allow comparison between images. The brightfield images were adjusted regarding brightness and contrast in an independent fashion, which does not affect the results but allows better visible cells. Adjustments were applied on the entire field of each image.
Biotin-switch analysis
The biotin-switch protocol was adapted from the PEG-switch protocol that was previously described (63). In brief, cultured ARVMs were treated with vehicle (DMSO; 30 min) or NCA (100 μmol/liter, 30 min). After a washing step with 1 ml of ice-cold Tris-HCl buffer (100 mmol/liter, pH 7.4), 70 μl of ice-cold biotin-switch harvesting buffer (in mmol/liter: NaCl 150, N-ethylmaleimide 100, Tris-HCl 100, pH 7.4, supplemented with 1% (v/v) Triton X-100 and protease inhibitors) was pipetted on the cells and the cell culture plate frozen in liquid N2. The ARVMs were thawed, harvested, and incubated at room temperature for 10 min. SDS (1%; applied as 20% solution) was added to the lysates and samples were incubated at 50 °C and 300 rpm for 25 min to block free thiol groups by alkylation with N-ethylmaleimide in the harvesting buffer. Subsequently, a sample volume of 130 μl was desalted by a Zeba™ Spin desalting column (ThermoFisher Scientific) to remove N-ethylmaleimide. The residual volume was kept as input. Oxidized protein thiol groups of desalted samples were reduced by the addition of 50 mmol/liter of DTT. Following 20 min of incubation at room temperature, DTT was removed by another desalting step. The biotinylation reagent EZ-Link® Maleimide-PEG2-Biotin (1 mmol/liter; number 21901BID; ThermoFisher Scientific) and 0.5% SDS (applied as 20% solution) were added sequentially and samples incubated on a laboratory agitator at room temperature for 2 h in the dark. To remove unbound maleimide-PEG2-Biotin, 4 volumes of acetone (−20 °C) were added to the samples and proteins were precipitated at −20 °C for 1 h. Proteins were pelleted by centrifugation at 16,000 × g and 4 °C for 10 min. The supernatant was removed, and the pellets washed twice with 731 μl of 80% acetone (−20 °C) by detaching the pellet from the tube, centrifugation and discarding the supernatant. Residual acetone was left to evaporate for 2 min, then 400 μl of Tris-HCl buffer (100 mmol/liter, pH 7.4) were added to the protein pellets. Resuspension was supported by vortexing and ultrasonic homogenization. After a quick centrifugation step, 40 μl of the supernatant were kept as input (biotinylated). The residual volume was added to 100-μl streptavidin-agarose beads (50% slurry) and incubated overnight on a laboratory agitator at 4 °C to bind biotinylated proteins. The beads were washed three times with 1 ml of RIPA buffer (in mmol/liter: NaCl 150, Tris-HCl 25, pH 7.4, SDS 0.1% (w/v), sodium deoxycholate 0.1% (w/v), Triton X-100 1% (v/v)) for 5 min at room temperature on the agitator, followed by two washes with Tris-HCl buffer (100 mmol/liter, pH 7.4). After each washing step, the beads were sedimented by centrifugation (1 min, 3000 × g). Eventually, 50 μl of elution buffer was added to the beads and the samples incubated at room temperature and at 95 °C for 15 min each. The beads were pelleted by centrifugation at 16,100 × g for 10 min. Subsequently, the supernatant was decanted and supplemented with 6× reducing Laemmli sample buffer. The precipitation of proteins, which indicated the biotinylation of cysteine residues as a consequence of protein oxidation in response to the initial treatment, was analyzed by SDS-PAGE and Western immunoblotting.
PEG-switch analysis
The PEG-switch protocol was performed as described before (37, 63). This method has the advantage over the previously described Biotin-switch method that less starting material is needed. In brief, cultured ARVMs were treated with vehicle DMSO (30 min), NCA (100 μmol/liter, 30 min), AS (500 μmol/liter, 15 min), CXL-1020 (300 μmol/liter, 15 min), H2O2 (100 μmol/liter, 10 min), DIA (500 μmol/liter, 10 min), or ISO (10 nmol/liter, 10 min). After a washing step with 2 ml of ice-cold Tris-HCl buffer (100 mmol/liter, pH 7.4), 100 μl of ice-cold PEG-switch harvesting buffer (in mmol/liter: maleimide 100, Tris-HCl 100, pH 7.4, SDS 34.7, supplemented with protease inhibitors) was pipetted on the cells and the cell culture plate frozen in liquid N2. The ARVMs were thawed, collected, and 130 μl of each lysate was incubated at 50 °C and 300 rpm for 25 min to block free thiol groups by alkylation with maleimide contained in the harvesting buffer. The residual sample volume was kept as input and one-half of each supplemented with 3× nonreducing or reducing Laemmli sample buffer. Maleimide from the harvesting buffer was removed by desalting with Zeba™ Spin desalting columns (ThermoFisher Scientific). Desalted samples were supplemented with 50 mmol/liter of DTT, causing the reduction of oxidized protein thiols. Followed by 20 min incubation at room temperature, another desalting step was performed to remove DTT. After sequential addition of the thiol-labeling reagent PEG-maleimide (2 mmol/liter, 5 kDa; number 63187; Sigma-Aldrich) and 0.5% SDS (applied as 20% solution), samples were incubated on a laboratory agitator at room temperature for 2 h in the dark. Finally, each sample was supplemented with 3× nonreducing Laemmli sample buffer and subjected to Western immunoblot analysis. The detection of a shift in molecular weight of a protein indicated PEG labeling and is considered as a surrogate for the oxidation of cysteine residues in response to the initial treatment.
Single cell contractility measurements
After isolation, ARVMs were maintained in IonOptix buffer (in mmol/liter: NaCl 135, glucose 20, HEPES 10, pH 7.46, KCl 4.7, CaCl2 1.5, MgSO4 1.2, KH2PO4 0.6, Na2HPO4 0.6) for at least 1 h. During pacing at 1 Hz (15 V, pulse duration: 4 ms), diastolic sarcomere length (in μm), sarcomere shortening (as % of diastolic sarcomere length), maximal contraction and relaxation velocities were reached (maximal change in sarcomere length over time; dL/dt max), as well as the time needed to reach maximal contraction (time to peak, in s) and the state of 50% maximal relaxation (time to baseline 50%; in s) were assessed under basal conditions or after pretreatment with ODQ (0.3 μmol/liter, 10 min) during exposure to DMSO (0.001% (v/v)), NCA (100 μmol/liter), or CXL-1020 (300 μmol/liter). ISO (10 nmol/liter) was added at the end of the treatment protocol to ensure cell viability. Contractile parameters were recorded using MyoPacer EP, MyoCam-S, and a Fluorescence System Interface unit (IonOptix, Westwood, MA USA). The IonWizard software was used for data recording and analysis.
Statistical analysis
All data were analyzed by GraphPad PrismSoftware Inc., San Diego, CA, USA) apart from the data concerning FRET measurements, which were analyzed by Origin (OriginLab Corporation, Northampton, MA, USA). Data from the contractility measurements, immunoblot analyses, and activity assays were statistically compared by one-way or two-way ANOVA followed by Dunnett's or Bonferroni post hoc test, as indicated in the respective figure legends. Two-tailed Student's t test was used to compare data obtained from PLA assays, from the cAMP-agarose pulldown experiments, and the PP2A in vitro assay. For each experiment, n numbers reflect biological replicates and are mentioned in the respective figure legends. Quantitative data are given as mean ± S.D. and p < 0.05 was considered as significant.
Data availability
All data are located within this manuscript. The MS proteomics data set has been deposited to the ProteomeXchange Consortium via the PRIDE (64) partner repository with the data set identifier PXD019808. The protein identifications are presented in Table S1.
Supplementary Material
Acknowledgments
We are grateful to the expert technical support provided by the Core Facility Mass Spectrometric Proteomics-UKE.
This article contains supporting information.
Author contributions—S. Diering, K. S., M. G., L. R., S. H., A. P., J. R., S. Schulz, M. B., J. P., U. F., S. Donzelli, V. O. N., E. E., and F. C. data curation; S. Diering, K. S., S. H., F. F., E. E., and F. C. formal analysis; S. Diering and F. C. investigation; S. Diering and F. C. methodology; S. Diering and F. C. writing-original draft; K. S. and FC supervision; F. C. conceptualization; F. C. funding acquisition; F. C. project administration; S. Sadayappan provided an antibody.
Funding and additional information—This work was supported by Deutsche Forschungsgemeinschaft Grant DFG CU 53/5-1, the Werner Otto Stiftung (8/89 and 7/92), the Deutsche Stiftung für Herzforschung (F/19/19), and the Deutsches Zentrum für Herz-Kreislauf-Forschung (DZHK) (to F. C.).
Conflict of interest—The authors declare that they have no conflict of interest with the content of this article.
- AR
- adrenoceptor
- ARVMs
- adult rat ventricular myocytes
- AS
- Angeli's salt
- APS
- ammonium persulfate
- cMyBP-C
- cardiac myosin-binding protein
- cTnI
- cardiac troponin I
- DAPI
- 4',6-diamidin-2-phenylindol
- DIA
- diamide
- DiFMUP
- 6,8-difluoro-4-methylumbelliferyl phosphate
- FOR
- forskolin
- HNO
- nitroxyl
- IBMX
- 3-isobutyl-1-methylxanthine
- ISO
- isoprenaline
- IVK
- in vitro kinase
- NCA
- 1-nitrosocyclohexyl acetate
- NGS
- nonspecific goat serum
- NKA
- sodium potassium ATPase
- NR
- nonreducing
- OA
- okadaic acid
- ODQ
- 1H-(1,2,4)oxadiazolol(4,3-a)quinoxaline-1-one
- PEG
- polyethylene glycol
- PFA
- paraformaldehyde
- PLA
- proximity ligation assay
- PLN
- phospholamban
- PKA
- cAMP-dependent protein kinase
- PKA-C
- PKA catalytic subunit
- PKA-R
- PKA regulatory subunit
- PKG
- cGMP-dependent protein kinase
- PP1
- protein phosphatase 1
- PP2A-C
- protein phosphatase 2A catalytic subunit
- R
- reducing
- sGC
- soluble guanylate cyclase
- TEMED
- N,N,N',N'-tetramethylethylenediamine
- IB
- immunoblotting
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- ACN
- acetonitrile
- ANOVA
- analysis of variance
- HRP
- horseradish peroxidase
- 8-AHA
- 8-aminohexylamino
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IB
- immunoblot
- IF
- immunofluorescence
- RFU
- relative fluorescence unit.
References
- 1. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 2. Cuello F., and Eaton P. (2019) Cysteine-based redox sensing and its role in signaling by cyclic nucleotide-dependent kinases in the cardiovascular system. Annu. Rev. Physiol. 81, 63–87 10.1146/annurev-physiol-020518-114417 [DOI] [PubMed] [Google Scholar]
- 3. Banky P., Huang L. J., and Taylor S. S. (1998) Dimerization/docking domain of the type Iα regulatory subunit of cAMP-dependent protein kinase: requirements for dimerization and docking are distinct but overlapping. J. Biol. Chem. 273, 35048–35055 10.1074/jbc.273.52.35048 [DOI] [PubMed] [Google Scholar]
- 4. Brennan J. P., Bardswell S. C., Burgoyne J. R., Fuller W., Schröder E., Wait R., Begum S., Kentish J. C., and Eaton P. (2006) Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 281, 21827–21836 10.1074/jbc.M603952200 [DOI] [PubMed] [Google Scholar]
- 5. Burgoyne J. R., Rudyk O., Cho H. J., Prysyazhna O., Hathaway N., Weeks A., Evans R., Ng T., Schröder K., Brandes R. P., Shah A. M., and Eaton P. (2015) Deficient angiogenesis in redox-dead Cys17Ser PKARIα knock-in mice. Nat. Commun. 6, 7920 10.1038/ncomms8920 [DOI] [PubMed] [Google Scholar]
- 6. Taylor S. S., Kim C., Cheng C. Y., Brown S. H., Wu J., and Kannan N. (2008) Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim. Biophys. Acta 1784, 16–26 10.1016/j.bbapap.2007.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Humphries K. M., Juliano C., and Taylor S. S. (2002) Regulation of cAMP-dependent protein kinase activity by glutathionylation. J. Biol. Chem. 277, 43505–43511 10.1074/jbc.M207088200 [DOI] [PubMed] [Google Scholar]
- 8. Humphries K. M., Deal M. S., and Taylor S. S. (2005) Enhanced dephosphorylation of cAMP-dependent protein kinase by oxidation and thiol modification. J. Biol. Chem. 280, 2750–2758 10.1074/jbc.M410242200 [DOI] [PubMed] [Google Scholar]
- 9. Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., and Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711 10.1016/j.cell.2004.05.018 [DOI] [PubMed] [Google Scholar]
- 10. Janssens V., and Goris J. (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353, 417–439 10.1042/0264-6021:3530417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 10.1016/j.cell.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 12. Lubbers E. R., and Mohler P. J. (2016) Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J. Mol. Cell. Cardiol. 101, 127–133 10.1016/j.yjmcc.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlender K. K., Hegazy M. G., and Thysseril T. J. (1987) Dephosphorylation of cardiac myofibril C-protein by protein phosphatase 1 and protein phosphatase 2A. Biochim. Biophys. Acta 928, 312–319 10.1016/0167-4889(87)90191-1 [DOI] [PubMed] [Google Scholar]
- 14. Jideama N. M., Crawford B. H., Hussain A. K., and Raynor R. L. (2006) Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int. J. Biol. Sci. 2, 1–9 10.7150/ijbs.2.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. MacDougall L. K., Jones L. R., and Cohen P. (1991) Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. 196, 725–734 10.1111/j.1432-1033.1991.tb15871.x [DOI] [PubMed] [Google Scholar]
- 16. Singh S., Lämmle S., Giese H., Kammerer S., Meyer-Roxlau S., Alfar E. A., Dihazi H., Guan K., El-Armouche A., and Richter F. (2018) The reduced activity of PP-1α under redox stress condition is a consequence of GSH-mediated transient disulfide formation. Sci. Rep. 8, 17711 10.1038/s41598-018-36267-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rao R. K., and Clayton L. W. (2002) Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem. Biophys. Res. Commun. 293, 610–616 10.1016/S0006-291X(02)00268-1 [DOI] [PubMed] [Google Scholar]
- 18. Foley T. D., and Kintner M. E. (2005) Brain PP2A is modified by thiol-disulfide exchange and intermolecular disulfide formation. Biochem. Biophys. Res. Commun. 330, 1224–1229 10.1016/j.bbrc.2005.03.108 [DOI] [PubMed] [Google Scholar]
- 19. Foley T. D., Petro L. A., Stredny C. M., and Coppa T. M. (2007) Oxidative inhibition of protein phosphatase 2A activity: role of catalytic subunit disulfides. Neurochem. Res. 32, 1957–1964 10.1007/s11064-007-9394-x [DOI] [PubMed] [Google Scholar]
- 20. O'Loghlen A., Pérez-Morgado M. I., Salinas M., and Martín M. E. (2003) Reversible inhibition of the protein phosphatase 1 by hydrogen peroxide: potential regulation of eIF2a phosphorylation in differentiated PC12 cells. Arch. Biochem. Biophys 417, 194–202 10.1016/S0003-9861(03)00368-0 [DOI] [PubMed] [Google Scholar]
- 21. Felker G. M., Borentain M., Cleland J. G., DeSouza M. M., Kessler P. D., O'Connor C. M., Seiffert D., Teerlink J. R., Voors A. A., and McMurray J. J. V. (2019) Rationale and design for the development of a novel nitroxyl donor in patients with acute heart failure. Eur. J. Heart Fail. 21, 1022–1031 10.1002/ejhf.1504 [DOI] [PubMed] [Google Scholar]
- 22. Hartman J. C., Del Rio C. L., Reardon J. E., Zhang K., and Sabbah H. N. (2018) Intravenous infusion of the novel HNO donor BMS-986231 is associated with beneficial inotropic, lusitropic, and vasodilatory properties in 2 canine models of heart failure. JACC Basic Transl. Sci. 3, 625–638 10.1016/j.jacbts.2018.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo Y., Xu J., Wu L., Deng Y., Wang J., and An J. (2019) Advances in research on treatment of heart failure with nitrosyl hydrogen. Heart Fail. Rev. 24, 941–948 10.1007/s10741-019-09800-6 [DOI] [PubMed] [Google Scholar]
- 24. Parissis J., Bistola V., Ikonomidis I., and Triposkiadis F. (2017) Nitroxyl donors for acute heart failure: promising newcomers. Eur. J. Heart Fail. 19, 1333–1334 10.1002/ejhf.793 [DOI] [PubMed] [Google Scholar]
- 25. Tita C., Gilbert E. M., Van Bakel A. B., Grzybowski J., Haas G. J., Jarrah M., Dunlap S. H., Gottlieb S. S., Klapholz M., Patel P. C., Pfister R., Seidler T., Shah K. B., Zieliński T., Venuti R. P., et al. (2017) A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur. J. Heart Fail. 19, 1321–1332 10.1002/ejhf.897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kemp-Harper B. K., Horowitz J. D., and Ritchie R. H. (2016) Therapeutic potential of nitroxyl (HNO) donors in the management of acute decompensated heart failure. Drugs 76, 1337–1348 10.1007/s40265-016-0631-y [DOI] [PubMed] [Google Scholar]
- 27. Cook J. C., Tran R. H., Patterson J. H., and Rodgers J. E. (2016) Evolving therapies for the management of chronic and acute decompensated heart failure. Am. J. Health Syst. Pharm. 73, 1745–1754 10.2146/ajhp150635 [DOI] [PubMed] [Google Scholar]
- 28. Singh A., Laribi S., Teerlink J. R., and Mebazaa A. (2017) Agents with vasodilator properties in acute heart failure. Eur. Heart J. 38, 317–325 10.1093/eurheartj/ehv755 [DOI] [PubMed] [Google Scholar]
- 29. Gao W. D., Murray C. I., Tian Y., Zhong X., DuMond J. F., Shen X., Stanley B. A., Foster D. B., Wink D. A., King S. B., Van Eyk J. E., and Paolocci N. (2012) Nitroxyl-mediated disulfide bond formation between cardiac myofilament cysteines enhances contractile function. Circ. Res. 111, 1002–1011 10.1161/CIRCRESAHA.112.270827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paolocci N., Katori T., Champion H. C., St John M. E., Miranda K. M., Fukuto J. M., Wink D. A., and Kass D. A. (2003) Positive inotropic and lusitropic effects of HNO/NO− in failing hearts: independence from β-adrenergic signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 5537–5542 10.1073/pnas.0937302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sivakumaran V., Stanley B. A., Tocchetti C. G., Ballin J. D., Caceres V., Zhou L., Keceli G., Rainer P. P., Lee D. I., Huke S., Ziolo M. T., Kranias E. G., Toscano J. P., Wilson G. M., O'Rourke B., et al. (2013) HNO enhances SERCA2a activity and cardiomyocyte function by promoting redox-dependent phospholamban oligomerization. Antioxid. Redox Signal. 19, 1185–1197 10.1089/ars.2012.5057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu G., Groneberg D., Sikka G., Hori D., Ranek M. J., Nakamura T., Takimoto E., Paolocci N., Berkowitz D. E., Friebe A., and Kass D. A. (2015) Soluble guanylate cyclase is required for systemic vasodilation but not positive inotropy induced by nitroxyl in the mouse. Hypertension 65, 385–392 10.1161/HYPERTENSIONAHA.114.04285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gautel M., Zuffardi O., Freiburg A., and Labeit S. (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 14, 1952–1960 10.1002/j.1460-2075.1995.tb07187.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gruen M., and Gautel M. (1999) Mutations in β-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 286, 933–949 10.1006/jmbi.1998.2522 [DOI] [PubMed] [Google Scholar]
- 35. Sadayappan S., Gulick J., Osinska H., Barefield D., Cuello F., Avkiran M., Lasko V. M., Lorenz J. N., Maillet M., Martin J. L., Brown J. H., Bers D. M., Molkentin J. D., James J., and Robbins J. (2011) A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 109, 141–150 10.1161/CIRCRESAHA.111.242560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carrier L., Mearini G., Stathopoulou K., and Cuello F. (2015) Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573, 188–197 10.1016/j.gene.2015.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stathopoulou K., Wittig I., Heidler J., Piasecki A., Richter F., Diering S., van der Velden J., Buck F., Donzelli S., Schröder E., Wijnker P. J., Voigt N., Dobrev D., Sadayappan S., Eschenhagen T., et al. (2016) S-Glutathiolation impairs phosphoregulation and function of cardiac myosin-binding protein C in human heart failure. FASEB J. 30, 1849–1864 10.1096/fj.201500048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. First E. A., and Taylor S. S. (1989) Selective modification of the catalytic subunit of cAMP-dependent protein kinase with sulfhydryl-specific fluorescent probes. Biochemistry 28, 3598–3605 10.1021/bi00434a067 [DOI] [PubMed] [Google Scholar]
- 39. Maack C., Eschenhagen T., Hamdani N., Heinzel F. R., Lyon A. R., Manstein D. J., Metzger J., Papp Z., Tocchetti C. G., Yilmaz M. B., Anker S. D., Balligand J. L., Bauersachs J., Brutsaert D., Carrier L., et al. (2019) Treatments targeting inotropy. Eur. Heart J. 40, 3626–3644 10.1093/eurheartj/ehy600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. El-Armouche A., Pohlmann L., Schlossarek S., Starbatty J., Yeh Y. H., Nattel S., Dobrev D., Eschenhagen T., and Carrier L. (2007) Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J. Mol. Cell Cardiol. 43, 223–229 10.1016/j.yjmcc.2007.05.003 [DOI] [PubMed] [Google Scholar]
- 41. Bardswell S. C., Cuello F., Kentish J. C., and Avkiran M. (2012) cMyBP-C as a promiscuous substrate: phosphorylation by non-PKA kinases and its potential significance. J. Muscle Res. Cell Motil. 33, 53–60 10.1007/s10974-011-9276-3 [DOI] [PubMed] [Google Scholar]
- 42. Humphries K. M., Pennypacker J. K., and Taylor S. S. (2007) Redox regulation of cAMP-dependent protein kinase signaling: kinase versus phosphatase inactivation. J. Biol. Chem. 282, 22072–22079 10.1074/jbc.M702582200 [DOI] [PubMed] [Google Scholar]
- 43. Yin X., Cuello F., Mayr U., Hao Z., Hornshaw M., Ehler E., Avkiran M., and Mayr M. (2010) Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol. Cell. Proteomics 9, 497–509 10.1074/mcp.M900275-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuster D. W., Bawazeer A. C., Zaremba R., Goebel M., Boontje N. M., and van der Velden J. (2012) Cardiac myosin binding protein C phosphorylation in cardiac disease. J. Muscle Res. Cell Motil. 33, 43–52 10.1007/s10974-011-9280-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Solaro R. J., and Kobayashi T. (2011) Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 286, 9935–9940 10.1074/jbc.R110.197731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lancel S., Zhang J., Evangelista A., Trucillo M. P., Tong X., Siwik D. A., Cohen R. A., and Colucci W. S. (2009) Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ. Res. 104, 720–723 10.1161/CIRCRESAHA.108.188441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tocchetti C. G., Wang W., Froehlich J. P., Huke S., Aon M. A., Wilson G. M., Di Benedetto G., O'Rourke B., Gao W. D., Wink D. A., Toscano J. P., Zaccolo M., Bers D. M., Valdivia H. H., Cheng H., et al. (2007) Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ. Res. 100, 96–104 10.1161/01.RES.0000253904.53601.c9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fukuto J. M., Chiang K., Hszieh R., Wong P., and Chaudhuri G. (1992) The pharmacological activity of nitroxyl: a potent vasodilator with activity similar to nitric oxide and/or endothelium-derived relaxing factor. J. Pharmacol. Exp. Ther. 263, 546–551 [PubMed] [Google Scholar]
- 49. Donzelli S., Fischer G., King B. S., Niemann C., DuMond J. F., Heeren J., Wieboldt H., Baldus S., Gerloff C., Eschenhagen T., Carrier L., Boger R. H., and Espey M. G. (2013) Pharmacological characterization of 1-nitrosocyclohexyl acetate, a long-acting nitroxyl donor that shows vasorelaxant and antiaggregatory effects. J. Pharmacol. Exp. Ther. 344, 339–347 10.1124/jpet.112.199836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. El-Armouche A., Wahab A., Wittkopper K., Schulze T., Bottcher F., Pohlmann L., King S. B., DuMond J. F., Gerloff C., Boger R. H., Eschenhagen T., Carrier L., and Donzelli S. (2010) The new HNO donor, 1-nitrosocyclohexyl acetate, increases contractile force in normal and β-adrenergically desensitized ventricular myocytes. Biochem. Biophys. Res. Commun. 402, 340–344 10.1016/j.bbrc.2010.10.030 [DOI] [PubMed] [Google Scholar]
- 51. Sabbah H. N., Tocchetti C. G., Wang M., Daya S., Gupta R. C., Tunin R. S., Mazhari R., Takimoto E., Paolocci N., Cowart D., Colucci W. S., and Kass D. A. (2013) Nitroxyl (HNO): a novel approach for the acute treatment of heart failure. Circ. Heart Fail. 6, 1250–1258 10.1161/CIRCHEARTFAILURE.113.000632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shoman M. E., DuMond J. F., Isbell T. S., Crawford J. H., Brandon A., Honovar J., Vitturi D. A., White C. R., Patel R. P., and King S. B. (2011) Acyloxy nitroso compounds as nitroxyl (HNO) donors: kinetics, reactions with thiols, and vasodilation properties. J. Med. Chem. 54, 1059–1070 10.1021/jm101432z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cazabat L., Ragazzon B., Varin A., Potier-Cartereau M., Vandier C., Vezzosi D., Risk-Rabin M., Guellich A., Schittl J., Lechene P., Richter W., Nikolaev V. O., Zhang J., Bertherat J., and Vandecasteele G. (2014) Inactivation of the Carney complex gene 1 (PRKAR1A) alters spatiotemporal regulation of cAMP and cAMP-dependent protein kinase: a study using genetically encoded FRET-based reporters. Hum. Mol. Genet. 23, 1163–1174 10.1093/hmg/ddt510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- 55. Stathopoulou K., Schobesberger S., Bork N. I., Sprenger J. U., Perera R. K., Sotoud H., Geertz B., David J. P., Christ T., Nikolaev V. O., and Cuello F. (2019) Divergent off-target effects of RSK N-terminal and C-terminal kinase inhibitors in cardiac myocytes. Cell Signal. 63, 109362 10.1016/j.cellsig.2019.109362 [DOI] [PubMed] [Google Scholar]
- 56. Bardswell S. C., Cuello F., Rowland A. J., Sadayappan S., Robbins J., Gautel M., Walker J. W., Kentish J. C., and Avkiran M. (2010) Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J. Biol. Chem. 285, 5674–5682 10.1074/jbc.M109.066456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cuello F., Bardswell S. C., Haworth R. S., Ehler E., Sadayappan S., Kentish J. C., and Avkiran M. (2011) Novel role for p90 ribosomal S6 kinase in the regulation of cardiac myofilament phosphorylation. J. Biol. Chem. 286, 5300–5310 10.1074/jbc.M110.202713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cuello F., Bardswell S. C., Haworth R. S., Yin X., Lutz S., Wieland T., Mayr M., Kentish J. C., and Avkiran M. (2007) Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ. Res. 100, 864–873 10.1161/01.RES.0000260809.15393.fa [DOI] [PubMed] [Google Scholar]
- 59. Haworth R. S., Cuello F., Herron T. J., Franzen G., Kentish J. C., Gautel M., and Avkiran M. (2004) Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ. Res. 95, 1091–1099 10.1161/01.RES.0000149299.34793.3c [DOI] [PubMed] [Google Scholar]
- 60. Luo W., Chu G., Sato Y., Zhou Z., Kadambi V. J., and Kranias E. G. (1998) Transgenic approaches to define the functional role of dual site phospholamban phosphorylation. J. Biol. Chem. 273, 4734–4739 10.1074/jbc.273.8.4734 [DOI] [PubMed] [Google Scholar]
- 61. Chu G., Lester J. W., Young K. B., Luo W., Zhai J., and Kranias E. G. (2000) A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to β-agonists. J. Biol. Chem. 275, 38938–38943 10.1074/jbc.M004079200 [DOI] [PubMed] [Google Scholar]
- 62. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Burgoyne J. R., Oviosu O., and Eaton P. (2013) The PEG-switch assay: a fast semi-quantitative method to determine protein reversible cysteine oxidation. J. Pharmacol. Toxicol. Methods 68, 297–301 10.1016/j.vascn.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 64. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are located within this manuscript. The MS proteomics data set has been deposited to the ProteomeXchange Consortium via the PRIDE (64) partner repository with the data set identifier PXD019808. The protein identifications are presented in Table S1.