

Abstract
Short tandemly repeated DNA sequences, termed microsatellites, are abundant in the human genome. These microsatellites exhibit length instability and susceptibility to DNA double-strand breaks (DSBs) due to their tendency to form stable non-B DNA structures. Replication-dependent microsatellite DSBs are linked to genome instability signatures in human developmental diseases and cancers. To probe the causes and consequences of microsatellite DSBs, we designed a dual-fluorescence reporter system to detect DSBs at expanded (CTG/CAG)n and polypurine/polypyrimidine (Pu/Py) mirror repeat structures alongside the c-myc replication origin integrated at a single ectopic chromosomal site. Restriction cleavage near the (CTG/CAG)100 microsatellite leads to homology-directed single-strand annealing between flanking AluY elements and reporter gene deletion that can be detected by flow cytometry. However, in the absence of restriction cleavage, endogenous and exogenous replication stressors induce DSBs at the (CTG/CAG)100 and Pu/Py microsatellites. DSBs map to a narrow region at the downstream edge of the (CTG)100 lagging-strand template. (CTG/CAG)n chromosome fragility is repeat length–dependent, whereas instability at the (Pu/Py) microsatellites depends on replication polarity. Strikingly, restriction-generated DSBs and replication-dependent DSBs are not repaired by the same mechanism. Knockdown of DNA damage response proteins increases (Rad18, polymerase (Pol) η, Pol κ) or decreases (Mus81) the sensitivity of the (CTG/CAG)100 microsatellites to replication stress. Replication stress and DSBs at the ectopic (CTG/CAG)100 microsatellite lead to break-induced replication and high-frequency mutagenesis at a flanking thymidine kinase gene. Our results show that non-B structure–prone microsatellites are susceptible to replication-dependent DSBs that cause genome instability.
Keywords: DNA replication, DNA repair, trinucleotide repeat disease, genomic instability, mutagenesis, myotonic dystrophy, cancer, break-induced replication, microsatellite instability, polycystic kidney disease
Approximately 3% of the human genome comprises microsatellites or short sequence repeats of 1–9 base pairs (1, 2). The structure of these ubiquitous microsatellites is dynamic and susceptible to expansions, contractions, and DNA double-strand breaks (DSBs) (2–5). The tendency of repetitive DNAs to form a variety of non-B DNA structures (hairpin, slipped strand, G quadruplex (G4), and triplex H-DNA) has been linked to interference with DNA replication and repair and to DSBs (6–10).
In humans, a growing cohort of neurodegenerative diseases has been attributed to DNA instability at microsatellite DNA noncanonical structures (3, 4, 7, 11–18). Thus, replication fork barriers are believed to provoke fork stalling and template switching (FoSTeS) and microhomology-mediated break-induced replication (MMBIR) (19–26). The induction of gross chromosomal rearrangements (GCRs) due to FoSTeS/MMBIR has been implicated in the etiology of several developmental disorders, including blepharophimosis syndrome (MIM no. 110100) (23), CHARGE syndrome (MIM no. 214800) (27), and Pelizaeus–Merzbacher disease (MIM no. 312080) (28).
In yeast, microsatellite DNAs promote chromosome breakage (29–35). In this model system, break-induced replication (BIR) has been shown to generate large-scale repeat expansions and mutations at a distance, as seen in human tumors (36–38). BIR is also a consequence of replication stress in human cells (39–44). Indeed in humans, replication-dependent single-ended DSBs (seDSBs) leading to BIR have been proposed to be responsible for copy number variation, several forms of GCR (19, 43, 45), oncogenesis (12–14, 42, 46–49), and multiple developmental disorders (27, 28, 47, 50–53).
Here, we focus on replication-dependent DSBs that occur at two types of microsatellite elements, an expanded (CTG/CAG)100 trinucleotide repeat from the 3′-UTR of the human DMPK locus and the 88-bp asymmetric polypurine/polypyrimidine (Pu/Py)88 mirror repeat from the PKD1 IVS21 locus. Much work has concentrated on (CTG/CAG) expansions in the DMPK gene, inasmuch as expansions of this microsatellite beyond ∼40 repeats promote further enlargement of the tract and genetic anticipation leading to myotonic dystrophy type 1 (DM1, chr19q13.32, MIM no. 160900) (for a recent review, see Ref. 54). Previous work showed that expanded (CTG/CAG) tracts can form hairpin structures in vivo (55, 56), and several reports have also shown that (CTG/CAG)n microsatellite repeats contribute to DNA DSBs in bacterial, yeast, and human model systems (29, 33, 57, 58).
The PKD1 (Pu/Py)88 asymmetric mirror repeats have the potential to form triplex H-DNA and G quadruplex DNA. In vitro, DNA triplex structures are visible in this sequence by atomic force microscopy (59). Mutations in the PKD1 gene are associated with at least 85% of the cases of autosomal dominant polycystic kidney disease (ADPKD) (chr16p13.3, MIM no. 173900). In more than 100 unrelated patients with ADPKD, mutations were at least twice as frequent in the exons flanking the PKD1 (Pu/Py)88 microsatellite as in 5′ exons 1–8 (60). Surprisingly, the (Pu/Py)88 microsatellite is not detectable as a hotspot for mutation in blood samples of ADPKD patients (61, 62).
We have shown that the PKD1 IVS21 mirror repeat also causes orientation-dependent fork stalling during replication in vitro and in vivo. When integrated alongside the c-myc replicator at an ectopic chromosomal site in the HeLa genome, the (Pu/Py)88 tract elicited a polar replication fork barrier. When the repeat was in the fork-stalling orientation, the binding of replication checkpoint proteins Rad9, RPA, and ATR near the repeat and the sensitivity of cells to Chk1 inhibition suggested that the DNA damage response is activated by replication fork stalling at this microsatellite (63).
In the present work, we describe a novel system to analyze replication-dependent DNA double-strand breaks in human cells, using fluorescent marker protein genes flanking the (CTG/CAG)100 or (Pu/Py)88 microsatellites. We find that the expanded (CTG/CAG)100 tract is sensitive to breakage following exposure to multiple forms of replication stress. Under nonperturbed conditions as well as after treatment with low-dose hydroxyurea (HU), DSBs occur in a narrow region near the downstream end of the (CTG/CAG) repeats. Moreover, these breaks are not repaired by the same mechanism as a restriction enzyme–generated DSB. Replication-dependent DSBs at the ectopic (CTG/CAG)100 microsatellite result in BIR and a greatly elevated frequency of mutagenesis of the neighboring thymidine kinase gene. The (Pu/Py)88 microsatellite is also sensitive to DSBs under unperturbed conditions and is highly vulnerable to DSBs when the purine-rich strand is the lagging-strand template for replication in cells treated with a G4-stabilizing drug.
Our results show that diverse forms of replication stress cause DSBs at microsatellite repeats prone to forming non-B DNA structures. The frequency of DSBs depends on the structure-selective Mus81 endonuclease and translesion polymerases. Invasion of the sister chromatid by the broken DNA results in complex rearrangements and a high rate of base substitutions during break-induced replication.
Results
A dual-fluorescence reporter system for analysis of DNA DSBs in vivo
Double-strand breaks are the most dangerous of DNA lesions because of the potential for error-prone repair, gross chromosomal rearrangement, and loss of heterozygosity. To identify factors affecting microsatellite DSBs and repair, we developed a system in which a DSB between two chromosomal reporter genes could be detected by microscopy or flow cytometry (Fig. 1). In this system, (CTG/CAG)100 or (Pu/Py)88 microsatellites were individually integrated at a single-copy FLP recombinase target (FRT) site at chromosome 18p11.22 in HeLa cells (64), bordered by the c-myc replication origin core (55, 65, 66), an I-Sce1 site, and two fluorescent protein marker genes (dTomato, eGFP) flanked by three identical AluYa5 elements (Fig. 1A) (67). Control cell lines were also constructed that contain the same starting construct except that the dual-fluorescence (DF)/myc cell line is missing the microsatellite sequences (Fig. 1B), and the DF cell line is additionally missing the c-myc origin core (Fig. 1C). The (CTG/CAG)23 and (CTG/CAG)100 sequences are pure (CTG/CAG) repeats (55). The sequence of the PKD1 (Pu/Py)88 microsatellite is shown in Fig. 1D. The cell lines are named to indicate the DNA sequence of the lagging-strand template when replicated from the c-myc origin (55, 63).
Figure 1.
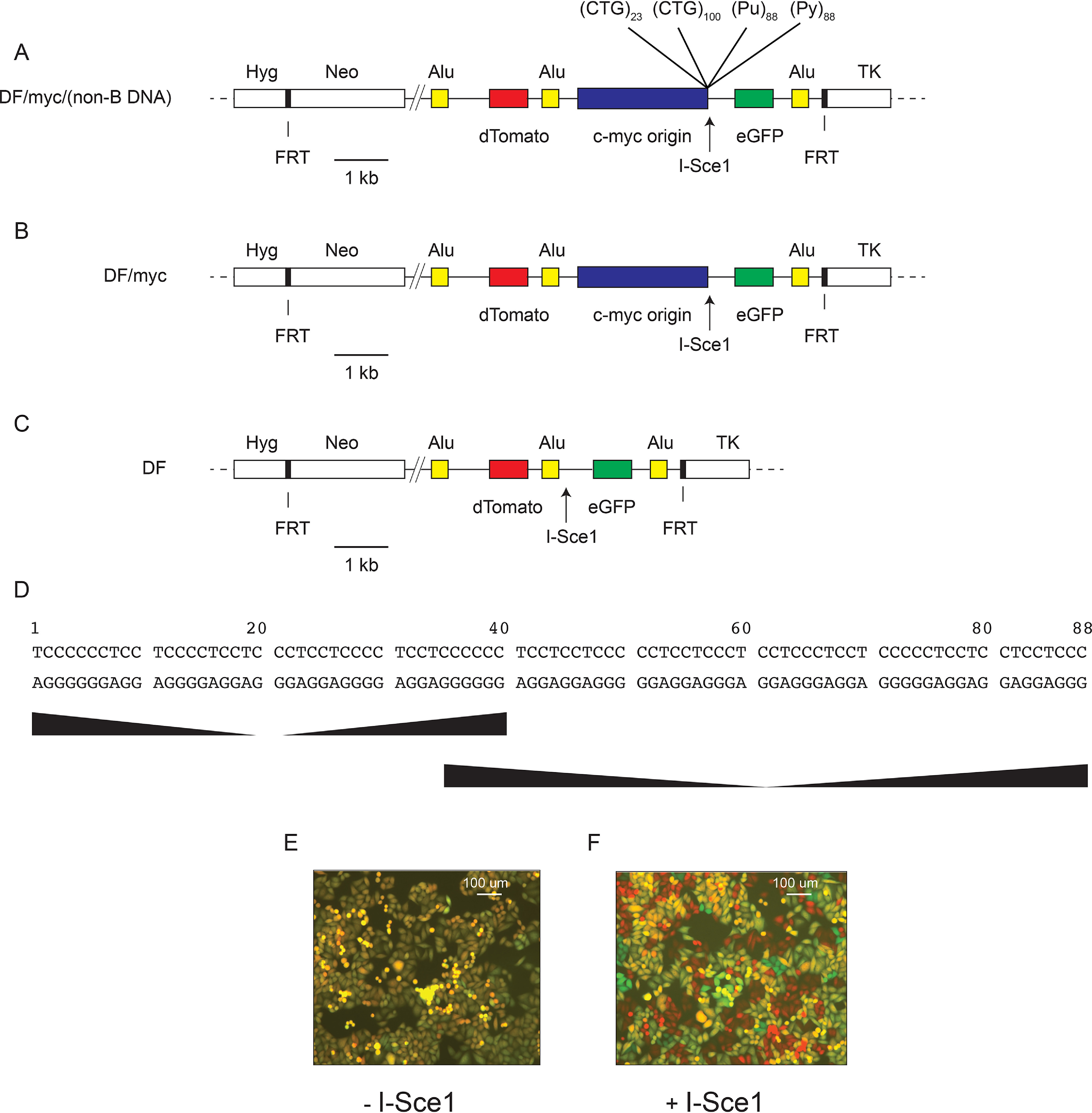
Maps of the ectopically integrated DF cell line constructs. A, DF/myc/(non-B DNA) cells contain the 2.4-kb c-myc core replication origin and individual microsatellites prone to forming non-B DNA. (CTG)23 and (CTG)100 are 23 repeats or 100 repeats of the CTG trinucleotide, respectively, in the lagging-strand template when replicated from the c-myc origin. (Pu)88 and (Py)88 refer to the 88-bp PKD1 microsatellite with the purine-rich strand or pyrimidine-rich strand, respectively, in the lagging-strand template when replicated from the c-myc origin. dTomato and eGFP genes are flanked by three identical AluYa5 repeats. B, DF/myc cells contain the same construct as in A but are missing the microsatellite sequences. C, DF cells contain the same construct as in A but are missing the ectopic c-myc origin and the microsatellite sequences. Hyg, hygromycin phosphotransferase (Hygr) gene; Neo, neomycin phosphotransferase (Neor) gene; TK, HSV thymidine kinase gene; FRT, S. cerevisiae FLP recombinase target, allowing site-directed integration. D, the PKD1 microsatellite sequence, showing two regions of mirror repeat symmetry; E, DF/myc(CTG)100 cells untreated; F, DF/myc(CTG)100 cells treated with I-Sce1.
The DF/myc(CTG)100 cell line (hereafter referred to as (CTG)100) expresses both the dTomato and eGFP reporter genes and fluoresces yellow (Fig. 1E). Transfection of an I-Sce1 expression vector results in double-strand DNA cleavage 25 bp downstream of the (CTG/CAG)100 microsatellite. This leads to intrachromosomal homology-directed recombination (single-strand annealing) between the second and third Alu elements (67), which eliminates the eGFP reporter. The half-lives of eGFP and dTomato reporter proteins are ∼24 h (68, 69); therefore, I-Sce1 digestion resulted in cells that appear red after allowing 4–8 days for turnover of the reporter proteins present before digestion (Fig. 1F).
Replication-dependent DSBs are not repaired in the same way as I-Sce1 DSBs
To quantitate these observations over the entire cell population, (CTG)100 cells were analyzed by flow cytometry. The (CTG)100 cells initially expressed both dTomato (red) and eGFP proteins and appeared in the upper right quadrant (yellow, double-positive) (Fig. 2, A and B). A small percentage of cells (<2%) had spontaneously lost either the green reporter (upper left quadrant, red cells), the red reporter (lower right quadrant, green cells), or both reporters (lower left quadrant, double-negative cells). When these cells were transfected with the I-Sce1 expression vector, more than 40% of the cells lost the green reporter (generating red cells) or both reporters (resulting in double-negative cells) by 4 days after treatment (Fig. 2C). The loss of the eGFP reporter gene is the result of intrachromosomal single-strand annealing between the second and third Alu elements, whereas the double-negative cells result from single-strand annealing between the first and third Alu elements (67).
Figure 2.
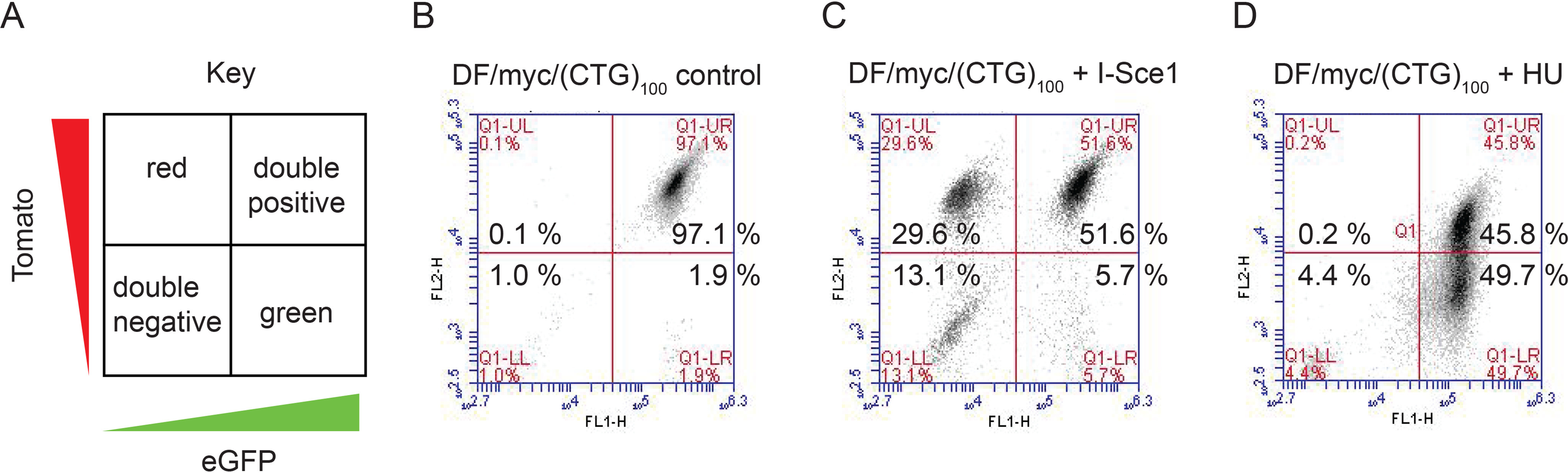
I-Sce1 DSBs are repaired by homologous recombination, but replication-dependent DSBs are refractory to homology-directed repair. A, key to flow cytometry results; B, untreated DF/myc(CTG)100 cells; C, DF/myc(CTG)100 cells transfected with an I-Sce1 expression plasmid; D, DF/myc(CTG)100 cells treated with low-dose hydroxyurea (0.2 mm HU). Similar results were observed after treatment of DF/myc(CTG)100 cells with aphidicolin (Fig. S2) or hydrogen peroxide (Fig. S3).
In striking contrast, approximately half of the (CTG)100 cells exposed to low-dose hydroxyurea (0.2 mm) for 96 h had lost the dTomato marker after 4 days of recovery (Fig. 2D). This HU treatment quickly arrests replication forks in S phase and induces a low level of the phosphorylated replication stress proteins γH2AX and pChk1345 (Fig. S1), consistent with previous reports that γH2AX marks stalled forks before DSBs are detectable (70).
DSBs were also induced in (CTG)100 cells by treatment with a low dose of the replication inhibitor aphidicolin (Fig. S2), or by using H2O2 as a source of ROS and replication stress (Fig. S3). In contrast, (CTG)23 cells did not exhibit these effects. We conclude that replication stress–dependent DSBs occur between the Tomato and eGFP marker genes near the ectopic (CTG)100 site. Based on the difference in flow cytometry patterns between cells treated with I-Sce1 and HU, we conclude that replication-dependent DSBs in the ectopic (CTG)100 locus are not repaired in the same way as “clean” restriction enzyme–generated DSBs and that replication-dependent DSBs caused by the (CTG/CAG)100 repeat are refractory to the most common repair pathways of homology-directed repair and nonhomologous end joining.
(CTG/CAG)n repeat length–dependent DSBs
To confirm that the HU-induced DSBs were dependent on the (CTG/CAG)100 repeat, several control cell clones were constructed and tested with HU, namely DF cells missing the c-myc origin and the (CTG/CAG)100 repeat (Fig. 3, A and B), DF/myc cells missing only the (CTG/CAG)100 repeat (Fig. 3, C and D), and DF/myc cells containing a shorter (CTG/CAG)23 repeat (Fig. 3, E and F). None of the control cell populations showed a significant difference in HU-induced DSBs (p > 0.999). Thus, the ectopic site displays replication-dependent DSBs contingent on the length of the (CTG/CAG)n microsatellite.
Figure 3.
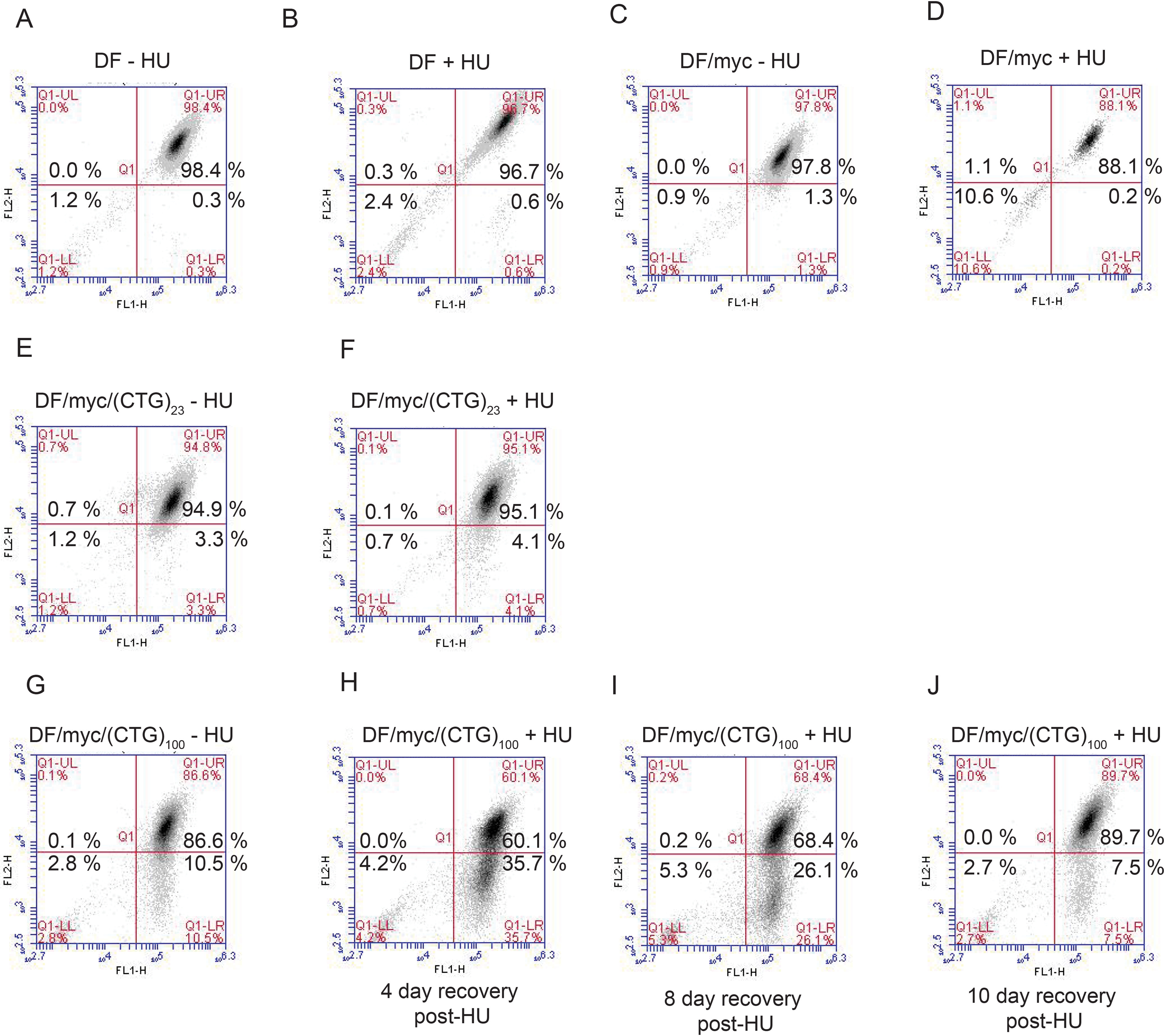
Chromosome fragility due to (CTG)100 repeats and replication stress. A, DF cells, untreated; B, DF cells treated with HU (0.2 mm hydroxyurea); C, DF/myc cells, untreated; D, DF/myc cells treated with HU; E, DF/myc(CTG)23 cells, untreated; F, DF/myc(CTG)23 cells treated with HU; G, DF/myc(CTG)100 cells treated with HU (48 h) and allowed to recover (H–J) for 4–10 days.
In previous work in which the position of the I-Sce1 site was changed, we showed that homology-directed repair that removes either the ectopic dTomato gene or the eGFP gene is not inherently deleterious to cells (67). Therefore, the loss of the dTomato gene raised the possibility that the replication-dependent DSBs that were resistant to recombination and deleted all or part of chromosome 18 containing the dTomato gene were also inimical to cell survival. To test this hypothesis, cells were allowed to recover for 4, 8, or 10 days following HU treatment (Fig. 3, G–I). The abundance of green cells in the culture suggests that an acentric fragment of chromosome 18 including the dTomato reporter gene may have been lost due to DSBs at the (CTG/CAG)100 microsatellite (see “Discussion”). The progressive loss of the green cells from the population (lower right quadrant) during the 4–10-day time course suggests that unrepaired replication-dependent DSBs had a lethal effect on these cells.
DNA DSBs are localized downstream of the (CTG/CAG)100 microsatellite
To determine the location of the replication-dependent DSBs, DNA was isolated from (CTG)100 cells treated with HU or I-Sce1 and subjected to linear amplification ligation–mediated PCR (lamPCR) (71, 72). The lamPCR primer was complementary to the single-copy eGFP gene (Fig. 4A) and designed to hybridize to the lagging-strand template DNA and leading-strand nascent DNA relative to the c-myc origin.
Figure 4.
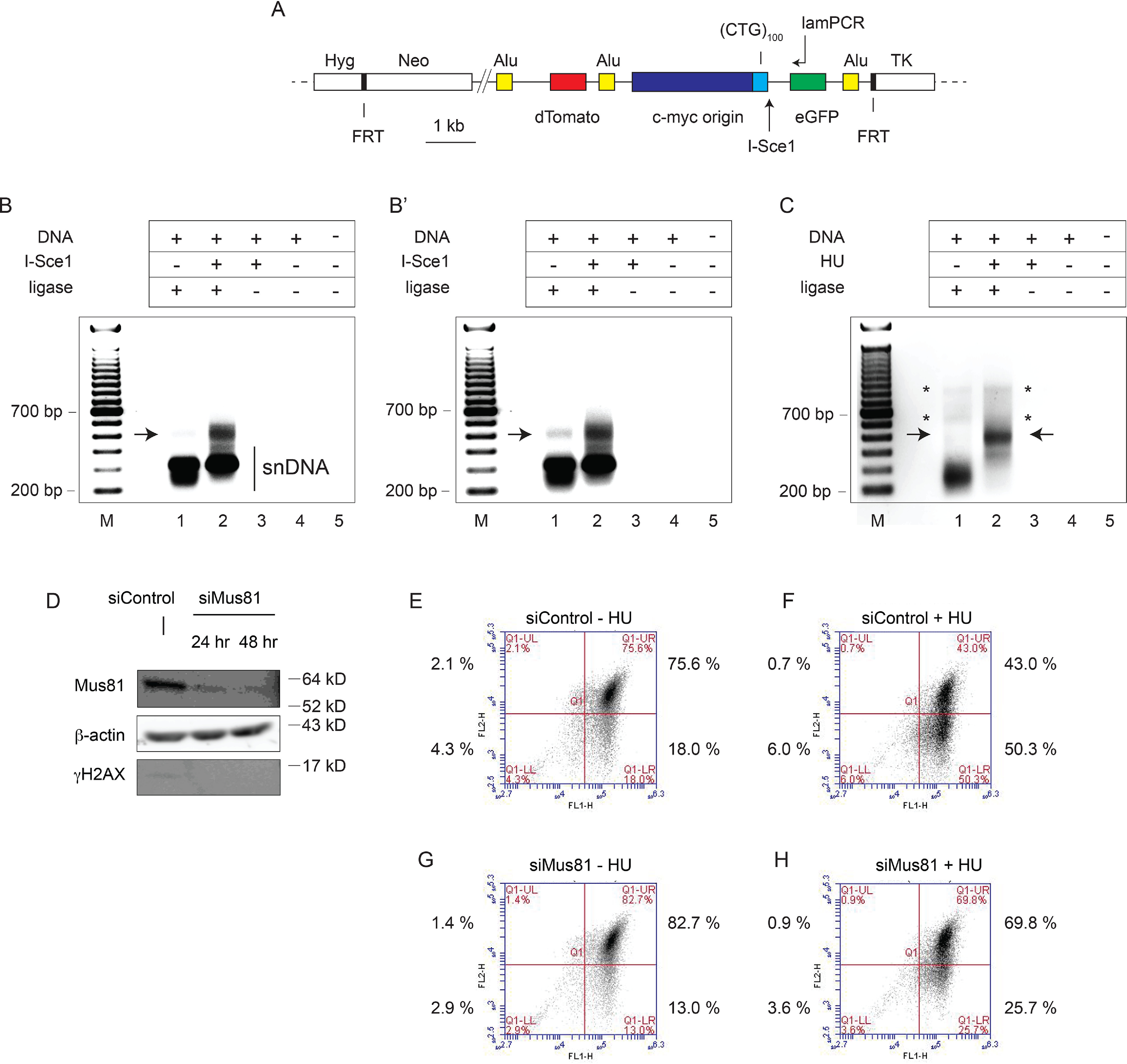
PCR mapping of I-Sce1 and replication-dependent DSBs in DF/myc(CTG)100 cells. A, diagram of the ectopic (CTG)100 insert showing the primer used for lamPCR (see “Experimental procedures”). Cells were transfected with an I-Sce1 expression plasmid or empty vector and incubated for 24 h before DNA isolation. Alternatively, cells were treated with 0.2 mm HU for 48 h. Genomic DNA was isolated and subjected to two rounds of lamPCR using the 5′-biotinylated lamPCR primer indicated. The biotinylated PCR products were captured on streptavidin-tagged magnetic beads and ligated to a 5′-phosphate, 3′-dideoxy adapter oligonucleotide (Circligase). Nested primers were used for exponential PCR amplification of the ligated template followed by gel electrophoresis. B, DF/myc(CTG)100 cells were treated with I-Sce1, and DSBs were mapped by lamPCR; B′, darker exposure of B showing endogenous DSB; C, DF/myc(CTG)100 cells were treated with HU, and DSBs were mapped by lamPCR. Arrows, bands indicating DSBs. Asterisks, putative extension products on unbroken leading-strand nascent DNA. These bands are not reproducible. D, DF/myc(CTG)100 cells were treated with nontargeting siControl siRNA or siRNA targeting Mus81 and analyzed by Western blotting. Flow cytometry was performed on cells treated with siControl (E), siControl plus HU (F), Mus81 siRNA (G), or Mus81 siRNA plus HU (H). Although the starting (CTG)100 culture had an increased percentage of green cells (cf. Fig. 3), the effects of HU treatment and the rescue by Mus81 knockdown were reproducible in three independent experiments (Fig. S4).
The I-Sce1 site 25 bp 3′ to the (CTG/CAG)100 microsatellite and ∼500 bp from the lamPCR primer was used as a landmark. As expected, when I-Sce1 was expressed in (CTG)100 cells, a major lamPCR band of ∼500 bp was observed (Fig. 4B). Lower-molecular weight bands (∼200–350 bp) were also observed in the undigested (Fig. 4B, lane 1) and I-Sce1–digested (Fig. 4B, lane 2) reactions, which we attribute to multiple leading-strand initiations near the c-myc origin (73, 74) that are not dependent on exogenous replication stress.
A discrete band of approximately the same size as the I-Sce1–generated lamPCR band could also be seen in a longer exposure of the PCR products from untreated cells (Fig. 4B′, lane 1). We propose that this band is due to DSBs close to the 3′ end of the (CTG/CAG)100 repeat and the I-Sce1 site that are generated during endogenous replication stress. The breadth of this band suggests that DSBs resulting during unperturbed growth are primarily the result of nuclease cleavage within a limited region near a stalled fork and not due to random torsional breakage throughout the (CTG/CAG)100 microsatellite.
When (CTG)100 cells were treated with HU, a band of ∼500 bp again appeared (Fig. 4C), suggesting that DSBs induced by HU treatment occur at or near the sites of DSBs due to endogenous replication stress and I-Sce1 cleavage. HU treatment also suppressed the 200–350 bp bands caused by nascent strand DNA annealing to the leftward-facing lamPCR primer. We conclude that endogenous replication stress, HU-induced fork stress, and I-Sce1 cleavage all produce DSBs at or near the 3′ end of the (CTG/CAG)100 microsatellite at the ectopic site in (CTG)100 cells.
Mus81 knockdown decreases (CTG/CAG)100 DSBs during replication stress
We have shown that (CTG/CAG)n repeats form hairpin structures in vivo that cause replication fork stalling (55, 56, 75). Inasmuch as the Mus81 nuclease has been strongly implicated in the cleavage of stalled replication forks (76–84), we wished to test whether knockdown of Mus81 (Fig. 4D) would affect the (CTG/CAG)100 DSBs. In this experiment, roughly 18% of (CTG)100 cells had suffered DSBs during unperturbed clonal growth (Fig. 4E). HU treatment of these cells significantly increased the percentage of green (DSB) cells in the population to greater than 40–50% (Fig. 4F and Fig. S4, p = 0.018). Consistent with the cleavage of stalled forks by Mus81, knockdown of the nuclease reproducibly resulted in a significant decrease in the percentage of cells with endogenous DSBs (cf. Fig. 4 (E and G) and Fig. S4, p = 0.049). Additionally, Mus81 knockdown dramatically decreased the percentage of DSBs induced by HU treatment from ∼50% to 25% of cells (cf. Fig. 4 (F and H), p = 0.003). Whereas these results are consistent with reports that Mus81 is a structure-selective nuclease that cleaves stalled replication forks (84–88), and our results show that Mus81 is involved in dTomato marker loss, we note that our experiments have not shown that it is specifically the nuclease activity of Mus81 that is responsible for the DSBs.
G quadruplex formation induces DSBs at the PKD1 polypurine/polypyrimidine microsatellite
The polycystic kidney disease type 1 (PKD1) locus harbors a polypurine-polypyrimidine (Pu/Py)88 tract of 88 base pairs in intron 21 (89), which is capable of forming intramolecular DNA triplex (H-DNA) and G quadruplex structures (90, 91). These structures have been strongly implicated in replication fork stalling and collapse (8, 92–94). Replication of the polypurine strand is blocked by non-B structure formation in vitro, and the PKD1 (Pu/Py)88 tract selectively inhibits replication when the polypurine strand is the lagging-strand template in vivo (63). To determine whether G quadruplex formation would sensitize this microsatellite to DSBs, we integrated the PKD1 (Pu/Py)88 repeat at the ectopic chromosomal site, in either the (Pu)88 or (Py)88 lagging-strand orientation when replicated from the c-myc origin (Fig. 5A).
Figure 5.
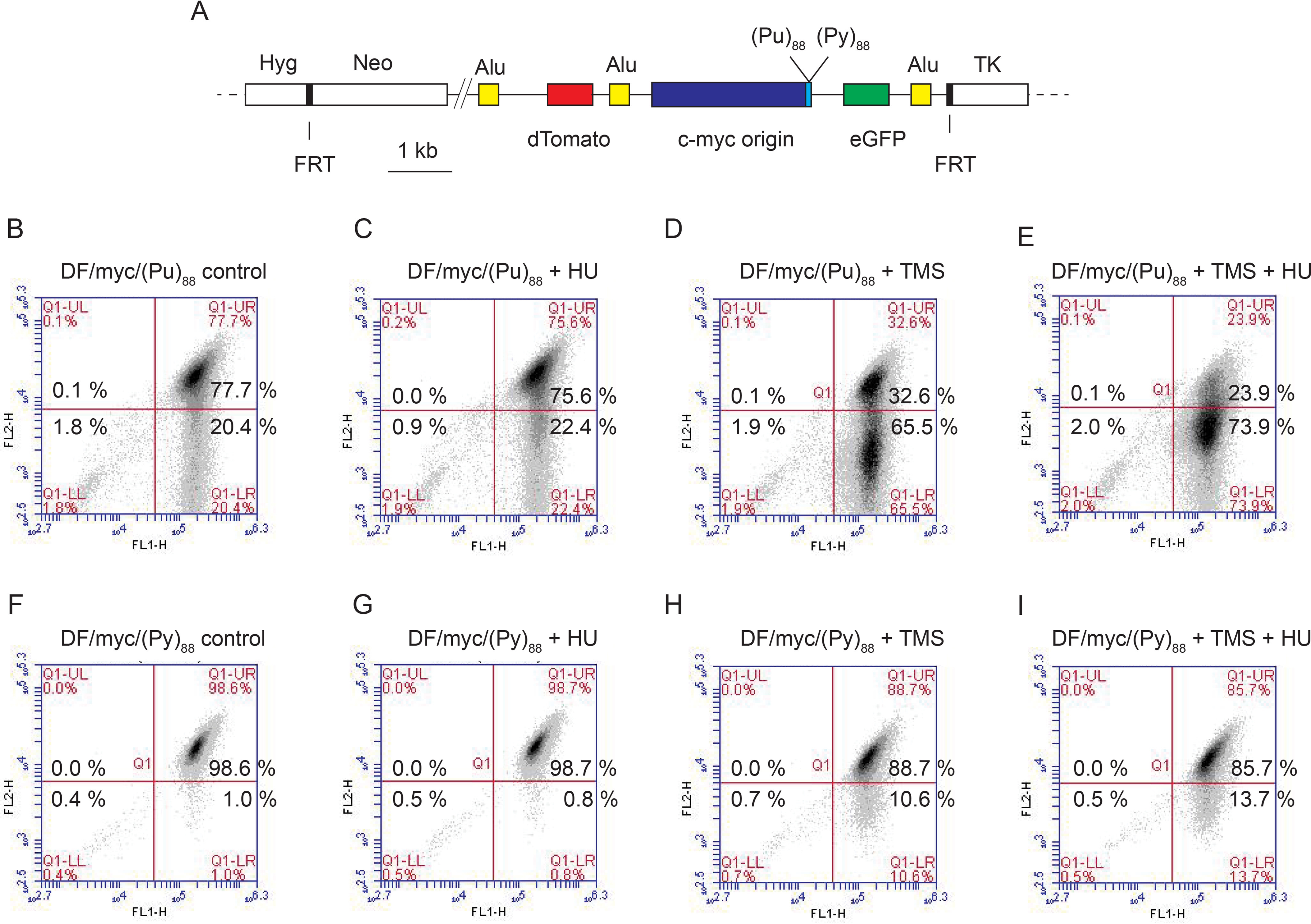
The PKD1 microsatellite is broken after replication stress and G-quadruplex formation, dependent on replication polarity. A, diagram of the ectopic (Pu/Py)88 inserts; B, DF/myc(Pu)88 control cells; C, DF/myc(Pu)88 cells treated with HU; D, DF/myc(Pu)88 cells treated with TMS; E, DF/myc(Pu)88 cells treated with TMS and HU; F, DF/myc(Py)88 control cells; G, DF/myc(Py)88 cells treated with HU; H, DF/myc(Py)88 cells treated with TMS; I, DF/myc(Py)88 cells treated with TMS and HU.
The starting culture of DF/myc/(Pu)88 cells (referred to as (Pu)88 cells) showed a significantly higher percentage of cells than the (Py)88 cells in the lower right (green) quadrant, resulting from DSBs occurring in the absence of exogenous stress (Fig. 5B and Fig. S6, p = 0.0001). Treatment of the (Pu)88 cells with HU did not significantly increase the percentage of green cells (Fig. 5C, p = 0.354); however, treatment of (Pu)88 cells with the G quadruplex–stabilizing drug telomestatin (TMS) (95, 96) markedly increased chromosome fragility at the ectopic site (Fig. 5D, p = 0.0045), and this effect was enhanced by co-administration of HU (Fig. 5E and Fig. S6, p = 0.024). These data suggest that replication fork slowing acts synergistically when G quadruplex formation is induced by the exogenous ligand. However, it remains to be seen whether endogenous levels of replication stress promote G4 versus H-DNA formation in the (Pu)88 repeat in vivo.
We showed previously that the PKD1 microsatellite in the (Pu)88 lagging-strand orientation blocked replication fork progress in vivo from the c-myc origin and elicited a constitutive DNA damage response, which was not observed when the ectopic site repeat was in the (Py)88 orientation (63). To test the effect of replication polarity on the stability of the PKD1 microsatellite, we also analyzed the stability of the ectopic site when (Py)88 was in the lagging-strand orientation. As shown in Fig. 5F, a significantly smaller percentage of DF/myc/(Py)88 cells (referred to as (Py)88 cells) than (Pu)88 cells were initially green in the absence of exogenous replication stress (cf. Fig. 5 (B and F) and Fig. S6, p = 0.0001). This result suggests that the ectopic (Pu/Py)88 tract is more sensitive to endogenous DSBs when the purine-rich strand is replicated as the lagging-strand template (i.e. in the fork-stalling orientation for replication). Nevertheless, compared with the effects of TMS on (Pu)88 cells, TMS had a reduced but statistically significant effect on the percentage of green (Py)88 cells in the absence (Fig. 5H, p = 0.006) or presence (Fig. 5I, p = 0.004) of HU, which is likely due to the presence of the NHE III1 G4 prone sequence in the c-myc replication origin core (97–99). In contrast, HU treatment did not have a significant effect on the flow cytometry profile of (Py)88 cells in the absence (Fig. 5G and Fig. S6, p = 0.355) or presence of TMS (Fig. 5I and Fig. S6, p = 0.483).
Indirect induction of (CTG/CAG)100 DSBs
TMS is a highly selective intramolecular G quadruplex ligand (95, 96, 100), which inhibits telomerase and causes telomere shortening in vivo (101). The results of TMS treatment of (Pu)88 and (Py)88 cells imply that DSBs occur preferentially when G4 prone sequences are present on lagging-strand templates and that G quadruplex formation contributes to fork stalling and replication-dependent DSBs.
Therefore, it was surprising that treatment of (CTG)100 cells with TMS resulted in DSBs between the dTomato and eGFP reporter genes (Fig. 6, A and B). The effect of HU on these cells was not additive to the effect of TMS (Fig. 6C), in contrast to the dramatic effect of HU on (CTG)100 cells in the absence of TMS (Fig. 2). These results suggest that the induction of DSBs by TMS or HU at the ectopic (CTG/CAG)100 microsatellite may both be related to replication fork stalling. To confirm that the TMS effect in (CTG)100 cells was due to the (CTG/CAG)100 repeat, we treated DF/myc cells with TMS and observed a significantly decreased appearance of green cells (Fig. 6 (D–F), p = 0.011).
Figure 6.

TMS indirectly induces (CTG/CAG)100 DSBs. A, DF/myc(CTG)100 cells, untreated; B, DF/myc(CTG)100 cells treated with TMS; C, DF/myc(CTG)100 cells treated with TMS and HU; D, DF/myc cells, untreated; E, DF/myc cells treated with TMS; F, DF/myc cells treated with TMS and HU; G, CD spectra of the indicated DNAs with or without TMS. Note the overlap of the (CTG)100 versus (CTG)100 + TMS spectra, (CAG)100 versus (CAG)100 + TMS spectra, and scrambled DNA versus scrambled DNA + TMS spectra.
The effect of TMS on DF/myc control cells was not statistically significantly different from its effect on (Py)88 cells (p = 0.149), suggesting that as in (Py)88 cells, the residual effect of TMS on the DF/myc control cells may be due to the NHE III1 G quadruplex–forming sequence in the 2.4 kb c-myc replication origin DNA (94, 98–100, 102, 103).
To test the possibility that TMS induces unexpected structural changes in the (CTG/CAG) microsatellite, we used CD to monitor the effect of TMS on CTG and CAG oligonucleotides (Fig. 6G). As anticipated, TMS caused a dramatic shift in the CD spectrum of a 22-mer oligonucleotide derived from the c-myc G quadruplex–forming promoter sequence (100, 102, 103). However, TMS had no discernible effect on the CD spectra of a scrambled DNA negative control, or (CTG)12 or (CAG)12 oligonucleotides, although the (CTG)12 and (CAG)12 sequences are known to form hairpins in vitro (104). This is consistent with the lack of stabilization of dsDNA by telomestatin or similar G4 ligands (105, 106). We conclude that TMS does not have a direct effect on (CTG/CAG) DNA structure in vitro and, therefore, that the effects of TMS on (CTG/CAG) stability in vivo are more likely to be an indirect result of activation of the DNA stress response (see “Discussion”), consistent with the observation that TMS leads to phosphorylation of the DNA damage response proteins Chk1, Chk2, and H2AX (107) (Fig. S5).
The ectopic (Pu)88 repeat is not sensitive to HU, whereas the ectopic (CTG)100 microsatellite is sensitive to multiple forms of replication stress, suggesting that the (Pu)88 G4 structure is responsible for DSBs. Nevertheless, the effects of TMS on (CTG)100 cells raise the possibility that TMS may contribute to DSBs at the ectopic G4 sequences in (Pu/Py)88 cells both in trans through the DNA damage response and directly by binding to G quadruplex–prone DNA.
Translesion DNA synthesis enzymes stabilize (CTG/CAG)100 against replication stress
Rad18 is important for translesion synthesis (TLS) in yeast and human cells (108, 109), where Rad18 monoubiquitination of proliferating cell nuclear antigen (PCNA) can recruit TLS DNA polymerases η, κ, ι, λ, ζ, and Rev1 to sites of replication fork stalling at non-B DNA structures (110–116). Because (CTG/CAG) sequences have been shown to form hairpin structures in vivo (55, 117), we wished to determine whether knockdown of Rad18 or the TLS polymerases Pol η or Pol κ would sensitize the (CTG/CAG)100 microsatellite to replication stress.
Compared with cells transfected with control siRNA (Fig. 7, A, F, and K), combined control siRNA and HU treatment led to (CTG/CAG)100 DSBs in 35–50% of cells (Fig. 7 (B, G, and L) and Fig. S7, p = 5 × 10−6). Depletion of Rad18 (Fig. 7C) led to a reproducible increase in the percentage of cells with DSBs (green cells) (cf. Fig. 7 (A and D) and Fig. S7, p = 0.046), and Rad18 knockdown substantially increased the percentage of cells with DSBs when combined with HU treatment (cf. Fig. 7 (B and E) and Fig. S7, p = 0.037). Considered together, these results suggest that Rad18/TLS stabilizes the (CTG/CAG)100 microsatellite against the fork-slowing effects of endogenous and exogenous replication stress.
Figure 7.
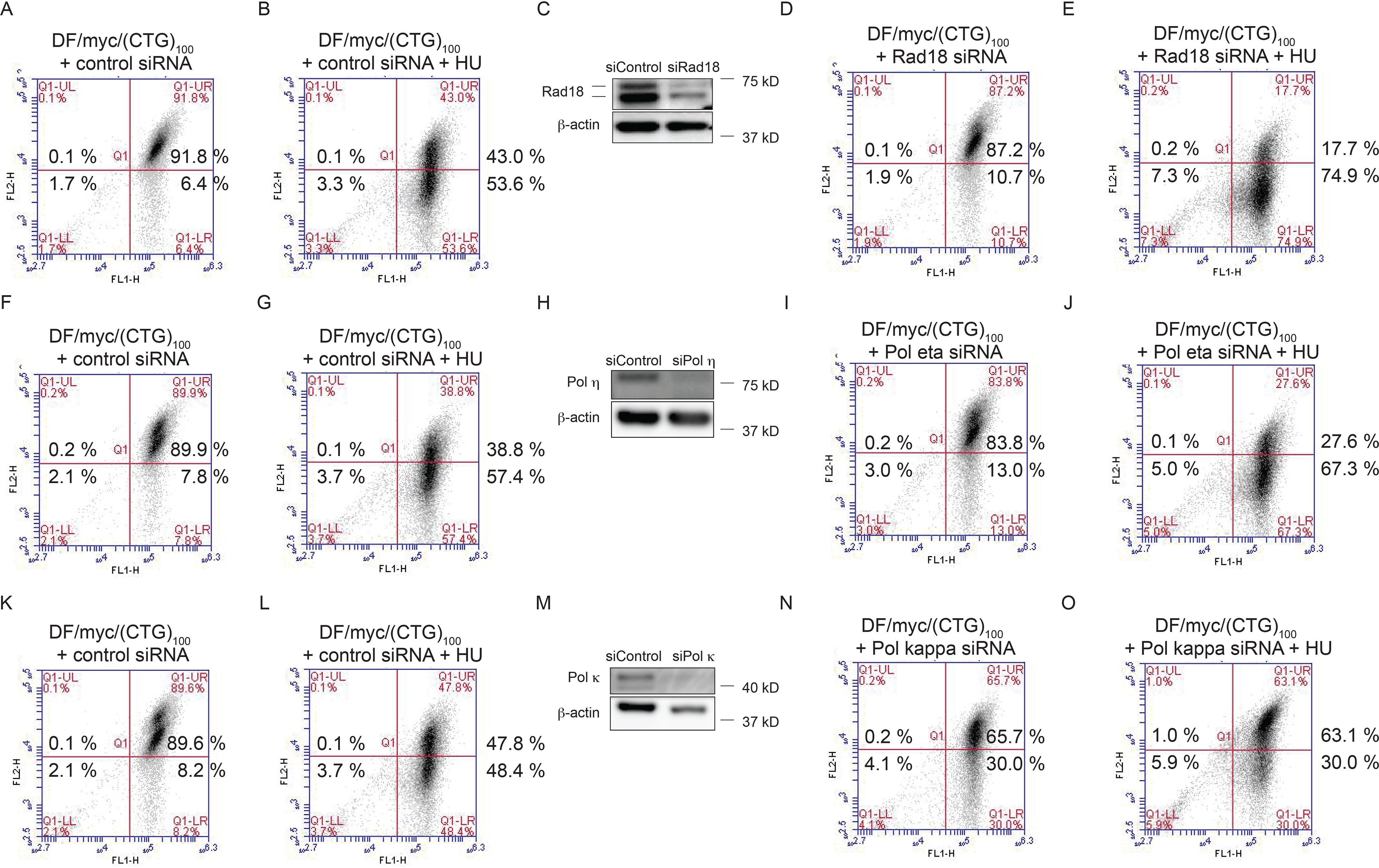
Translesion synthesis pathways affect (CTG/CAG)100 stability. DF/myc(CTG)100 cells were subjected to the following treatments: nontargeting control siRNA (A, F, and K), nontargeting siRNA and HU (B, G, and L); Rad18 siRNA (C and D); Rad18 siRNA plus HU (E); Pol η siRNA (H and I), Pol η siRNA plus HU (J), Pol κ siRNA (M and N), Pol κ siRNA plus HU (O), and Western blotting (C, H, and M).
TLS polymerases have been implicated in the bypass of DNA hairpin structures (e.g. by Escherichia coli Pol V synthesis across abasic DNA sites (118) and Saccharomyces cerevisiae Pol ζ/Rev1 primer extension by DNA template switching at hairpins (119)). In the current experiments, depletion of Pol η (cf. Fig. 7 (F and I) and Fig. S7) resulted in a statistically significant increase in green cells when compared with siControl (p = 0.041), whereas knockdown of Rev1 did not (not shown).
In addition, the effect of HU treatment was amplified by siRNA depletion of Rad18 (Fig. 7 (B and E) and Fig. S7, p = 0.022) or Pol η (Fig. 7 (G and J) and Fig. S7, p = 0.042), whereas Rev1 knockdown did not increase the effect of HU treatment (not shown). These results suggest that Pol η is one of the TLS polymerases involved in the restart of (CTG)100 stalled forks.
In contrast to the effects of knockdown of Rad18 or Pol η, depletion of Pol κ in the absence of HU treatment dramatically increased the fraction of cells containing DSBs (cf. Fig. 7 (K and N) and Fig. S7, p = 0.0007). Taken together, the modest effect of Rad18 knockdown versus the strong effect of Pol κ knockdown suggests that Pol κ may also have a fork restart function independent of Rad18 (120–122). Surprisingly, HU treatment did not augment the effect of Pol κ knockdown (Fig. 7O and Fig. S7, p = 0.185) These results indicate that Rad18, Pol η, and Pol κ are involved in resolving non-B DNA (123). In the presence of HU, Pol κ may interact with the stalled fork in a nonproductive manner; thus, when replication is inhibited by HU, fewer structures that lead to DSBs are formed when Pol κ is depleted.
Replication stress causes (CTG/CAG) BIR
Non-B DNA structure–prone repeats can induce mutagenesis at a distance in mammalian cells (9). These mutational events are thought to result from replication fork stalling at microsatellite repeats, fork breakage, and subsequent error-prone repair in a process termed repeat-induced mutagenesis (RIM) or BIR (9, 37, 124).
Our results have shown that the expanded (CTG/CAG) microsatellite stalls replication forks and induces replication-dependent DSBs. To test for RIM/BIR induced by the (CTG/CAG)100 microsatellite, we integrated a modified reporter plasmid, (CTG)100eGFP/TK, at the ectopic site such that the eGFP, FRT, and TK sequences become fused during FLP-mediated integration (Fig. 8A). We postulated that if BIR occurs following a hydroxyurea-induced, replication-dependent double-strand break at the (CTG/CAG)100 sequence, invasion of the broken end into the sister chromatid would result in mutagenesis of the neighboring TK gene ∼1 kb downstream (Fig. 8B).
Figure 8.
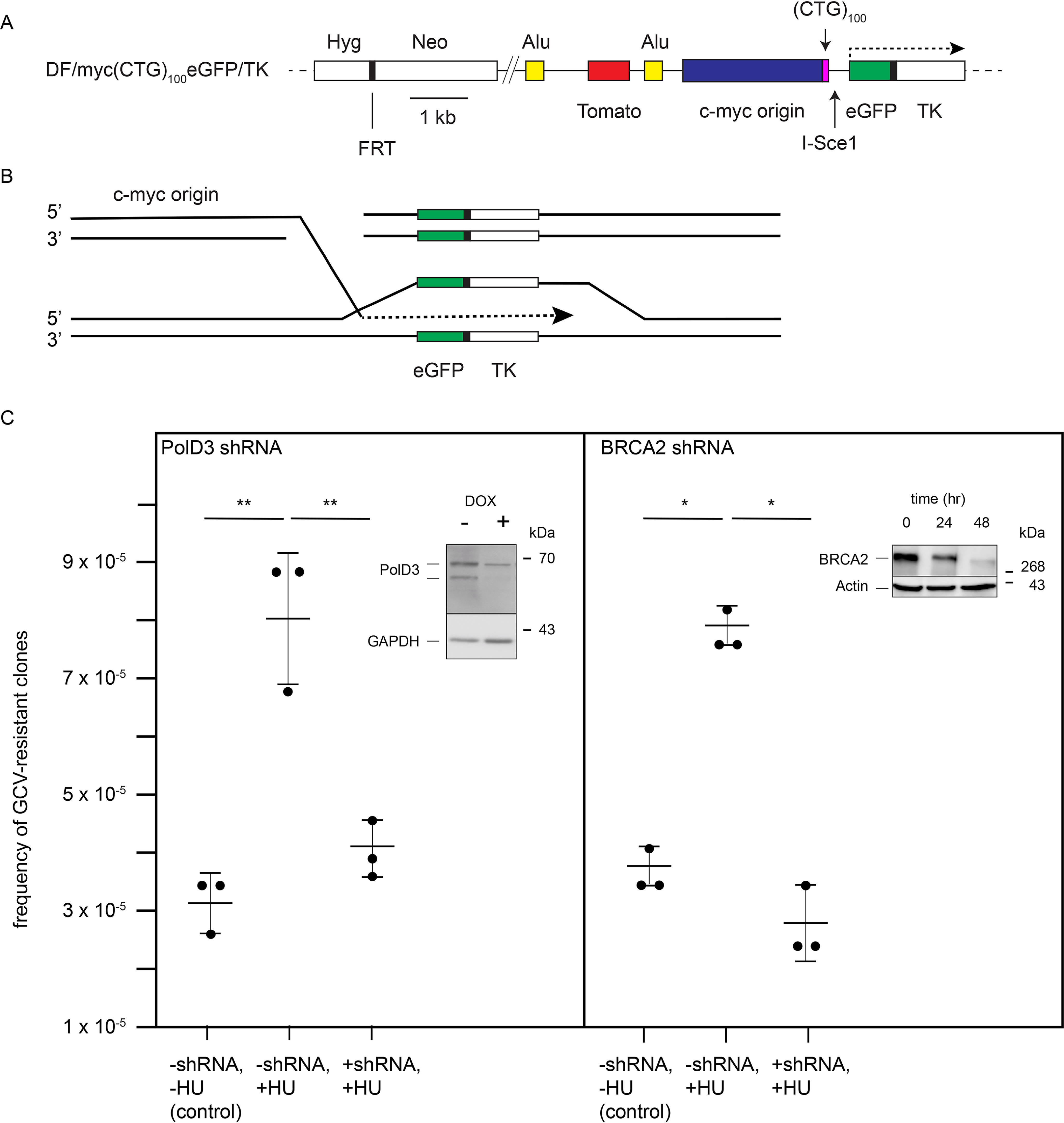
Repeat-induced mutagenesis. A, the DF/myc(CTG)100eGFP/TK ectopic site construct designed to detect BIR/RIM; B, model of repeat-induced mutagenesis of the thymidine kinase gene triggered by a replication-dependent DSB at the ectopic (CTG/CAG)100 repeat; C, GCVR colonies arising from BIR/RIM of the TK gene. Error bars, S.D. (n = 3 experiments).
Untreated DF/myc(CTG/CAG)100eGFP/TK cells produced ganciclovir (GCV)-resistant clones at a frequency of approximately three per 105 cells (Fig. 8C). These data are comparable with the frequency of GCV-resistant clones stemming from DSBs at an ectopic (CGG/CCG)153 repeat in a clonal population of murine erythroid leukemia cells (124). As in the case of the (CGG/CCG)153 murine erythroid leukemia cells, GCV resistance likely arose by endogenous replication stress and BIR during extended clonal outgrowth of the dual-fluorescence cell line.
When DF/myc(CTG/CAG)100eGFP/TK cells were treated with 0.2 mm HU followed by GCV selection, the frequency of GCV-resistant colonies rose to ∼7–8 colonies/105 cells (Fig. 8C, p = 4 × 10−5). Thus, acute treatment with HU produced a similar number of GCR-resistant cells as extended (>1-year) clonal outgrowth.
To confirm that the appearance of GCV-resistant cells is the result of BIR, we knocked down PolD3, which is necessary for BIR in yeast (40) and human cells (44, 124), or knocked down BRCA2, which mitigates DSBs under conditions of replication stress and promotes Rad51-dependent BIR in human cells (44, 125) (Fig. 8C). When DF/myc(CTG/CAG)100eGFP/TK cells were exposed to shRNA, knockdown of PolD3 (Fig. 8C, p = 0.015) or BRCA2 (Fig. 8C, p = 0.002) significantly decreased the frequency of GCV-resistant cells following HU treatment to the background levels of TK mutants accumulated during prolonged clonal outgrowth, supporting the view that DSBs at the (CTG/CAG) repeat lead to break-induced replication.
We previously used inverse PCR (iPCR) to show that replication stress leads to breakage at an ectopic (CTG/CAG)102 repeat in myc(CTG/CAG)102 cells (126), as well as DSBs at endogenous microsatellites across the genome (126). The (CTG/CAG)102 construct differed from the construct in DF/myc(CTG/CAG)100eGFP/TK cells in that there was no eGFP/TK fusion or GCV selection for cells undergoing BIR. DNA sequence analysis of iPCR products with nonallelic breakpoint junctions initiated within the ectopic site also showed that the broken site underwent nonrandom chromosomal translocations similar to genome rearrangements attributed to BIR in tumor cells (126).
Although there was no GCV selection for BIR in the myc(CTG/CAG)102 cells, we reanalyzed the iPCR DNA-sequencing data to focus on the distribution of mutations upstream and downstream of the ectopic repeat that had been repaired by homology-mediated templating of the sister chromatid (Fig. 9). The great majority (>95%) of mutations were single base substitutions. As predicted by BIR models, base substitutions were dramatically greater downstream of the (CTG/CAG)102 repeat than upstream (Fig. 9), consistent with the rightward replication of the repeat from the c-myc origin and with lamPCR mapping of the DSB at the downstream edge of the (CTG/CAG)100 repeat. Subtracting the frequency of nucleotide substitutions (PCR and sequencing errors, in vivo mutations) upstream of the repeat as background, the average frequency of nucleotide substitution downstream of the (CTG/CAG)102 repeat was ∼1.5 × 10−7 substitutions per bp. This value is at least 2–3 orders of magnitude greater than recent estimates of the natural mutation frequency in humans (40, 127, 128).
Figure 9.

High frequency of base substitutions due to BIR. A, base substitution analysis at the ectopic site following sister chromatid templated BIR. DNA was isolated from myc(CTG/CAG)102 cells treated with 0.2 μm aphidicolin, digested with MseI, and intramolecularly circularized. The circularized DNA was amplified by inverse PCR and analyzed by high-throughput sequencing. Nonallelic recombination junctions have been published previously (126). B, interpretative map of BIR at the ectopic site. C, quantitation of base substitutions at C, T, and G residues within the (CTG) repeat.
Within the (CTG)102 repeat, we observed expansions, contractions, inversions, and base substitutions. Interestingly, there was a strong third base periodicity of nucleotide substitutions at dG residues in the (CTG)102 template, which peaked dramatically near the center of the microsatellite (Fig. 9B). These data are consistent with the observation that dG:dC base pairs are preferential targets for single-base substitution mutations in tumor cells (129) and that a loop at the center of a single large (CTG)102 hairpin is a hotspot for mutagenesis during the process of BIR. Considered together, our data indicate that DSBs at (CTG/CAG) repeats lead to highly mutagenic break-induced replication.
Discussion
(CTG/CAG) microsatellite DSBs detected by flow cytometry
Microsatellite repeats prone to forming non-B DNA structures undergo expansion, contraction, and double-strand breakage in a variety of yeast and mammalian cell systems (7, 29, 36, 124, 130–135). Here, we used a dual-fluorescence reporter gene system to analyze DSBs in human cells. We show that DNA double-strand breaks occur at a relatively high frequency in an ectopic (CTG/CAG)100 microsatellite expanded beyond the WT range of repeats found in the human DMPK gene. These DSBs occur in unstressed cells and were dramatically increased in cells treated with four qualitatively different replication stressors (hydroxyurea (126, 136, 137), aphidicolin (56), hydrogen peroxide (138), and telomestatin (107, 111)), in agreement with the view that endogenous and exogenous replication stress leads to DNA DSBs. The low background level of DSBs at the ectopic site (CTG/CAG)23 repeat suggests that expanded (CTG/CAG)100 tracts promote replication-dependent breakage.
Replication-dependent DSBs at repeated sequences have been attributed to the propensity of these repeats to form noncanonical DNA structures (8, 35, 48, 139, 140). Consistent with this view, it has been shown that (CTG/CAG) repeats form hairpin structures in vivo (55, 56, 141). It is intriguing, therefore, that the cellular repair machinery treats a restriction enzyme–generated DSB differently than a replication-dependent DSB. We speculate that localized Mus81-dependent cleavage near the downstream edge of the (CTG/CAG)100 repeat is due to a noncanonical DNA structure that is refractory to replication and repair. Similar conclusions regarding the breakage and repair of structured ends have been obtained with an AT-rich repeat derived from the FRA16D common fragile site (30, 142).
The abundance of green cells (∼50%) after different forms of replication stress is consistent with models in which both ends of a replication-dependent DSB persist in the population (143); based on the abundance of dTomato− eGFP+ cells, we propose that the eGFP side of the DSB is replicated by a leftward moving replication fork (Figs. 8B and 10) (76) that produces two DSBs that are functionally single-ended.
Figure 10.

Proposed model for loss of the dTomato marker gene after replication stress. A, ideogram of chromosome 18 showing the FRT site at 18p11.22. B, integrated (CTG)100 construct. C, enlarged (not to scale) diagram of converging replication forks and the Mus81 cleavage substrate. D, proposed pathway for loss of the dTomato marker gene and generation of eGFP+ (green) cells.
Among other possibilities, the subsequent instability of the green cells may be due to the structure of the non-B DNA end per se, inhibition of the major pathways of repair (homologous recombination and nonhomologous end joining) with the downstream end, a nontelomeric structure of the downstream eGFP DSB end, and loss of DNA from the acentric upstream side of the DSB.
In contrast to the results presented here for the PKD1 microsatellite, Wenger et al. (144) reported the inability to detect fragile sites by cytogenetic G banding in blood cell cultures from congenital DM1 patients containing repeats as large as (CTG/CAG)1000 after treatment with replication stressors including 0.2 μm aphidicolin. Aside from differences in cell type, Wenger et al. (144) treated cells with bromodeoxyuridine, 5′-deoxy-5-fluorouridine, or aphidicolin for 24 h immediately before chromosome spreading, whereas the present experiments treated cells for 4 days prior to 4–10-day recovery in drug-free medium and flow cytometry. It is possible therefore that in the present experiments, DSBs occur during prolonged replication stress or during replication restart following drug treatment.
We observed as well that treatment of cells with telomestatin induced DSBs at the (CTG/CAG)100 ectopic site in vivo. Telomestatin is a known replication stressor of intramolecular G quadruplex–prone sequences, especially telomeres (101, 107, 145–147); however, no change in (CTG/CAG) oligonucleotide structure due to TMS could be detected in vitro by CD. These results suggest that telomestatin action at telomeres or other G quadruplex–prone sequences (107) can affect (CTG/CAG) stability in trans. We propose that G quadruplex formation elsewhere in the genome causes a diffusible state of replication stress in the cell that promotes fork slowing and non-B structure formation at the ectopic (CTG/CAG)100 repeat.
DSBs at the structure-prone (Pu/Py)88 microsatellite
During DNA replication, the lagging-strand template is expected to be more susceptible to structure formation than the leading-strand template, due to its relatively prolonged single-strandedness (6, 35). Consistent with this model, replication of the PKD1 intron 21 (Pu)88 sequence as the lagging-strand template leads to replisome stalling; recruitment of RPA, Rad9, and ATR to the stalled fork; and induction of a DNA damage response (63).
In the current work, the (Pu/Py)88 repeat displayed replication polarity–dependent instability in the absence of exogenous stress. Administration of the G quadruplex–binding ligand telomestatin strongly enhanced the sensitivity of the (Pu/Py)88 tract to DSBs in the same replication polarity–dependent manner, which we attribute to the stabilization of G quadruplex DNA structures in the lagging-strand template. However, the (Pu/Py)88 tract also has the ability to form triplex H-DNA structures in the absence of telomestatin (63, 89, 90, 148). Therefore, whereas the present results indicate that induced G quadruplex formation can stall replication forks and cause DSBs, the sensitivity of the lagging-strand (Pu)88 tract to DSBs in unperturbed cells could also be the result of H-DNA formation. This possibility is currently under investigation.
In contrast to the DSB sensitivity of the ectopic PKD1 IVS21 (Pu/Py)88 tract, PCR analysis of 57 patients with autosomal dominant polycystic kidney disease (ADPKD1) showed no hotspot for mutation in the PKD1 gene, although mutations were 2–3 times more frequent in the exons surrounding IVS21 than in exons 1–8 (60). Similarly, in samples from 15 tuberous sclerosis (TSC) patients in which deletions in the upstream TSC2 gene extended into PKD1, multiplex ligation-dependent probe amplification did not show clustering of breakpoints near the IVS21 (Pu/Py)88 tract (62). One explanation for these observations may be a strong selection against PKD1 DSBs, which includes origin choice (149–152) to avoid lagging-strand replication of the PKD1 (Pu)88 sequence.
Translesion polymerases mediate (CTG/CAG) stability
The TLS polymerases comprise a group of functionally divergent enzymes that can bypass non-B DNA and template lesions that stall replicative polymerases (123). Rad18-dependent ubiquitination of the PCNA scaffold allows TLS polymerase exchange and access to the unreplicated lesion (111, 115, 153–155). Multiple TLS polymerases can bind to ubiquitinated PCNA (123); thus, in the absence of one TLS polymerase, alternative postreplication repair pathways can be employed (115, 123). In the dual-fluorescence assay, siRNA knockdown of Rad18 or Pol η caused a marked increase in ectopic instability when combined with HU treatment. In contrast, depletion of Pol κ led to a large increase in DSBs in otherwise unstressed cells but minimized the effect of HU treatment. We suggest that Rad18 and Pol η assist in the replication of the (CTG/CAG)100 repeat, particularly during HU-induced fork slowing, whereas depletion of Pol κ in the presence of HU decreases the formation of a subset of difficult-to-replicate structures. Intriguingly, translesion synthesis has also been implicated in break-induced replication (156, 157). Our results are consistent with reports that Pol η and Pol κ are involved in the replication of common fragile sites (158) and that knockdown of these polymerases enhances DSBs in HeLa cells transfected with plasmids containing c-myc G4-prone DNA (111).
(CTG/CAG)100 break-induced replication
Early replicating fragile sites (ERFSs) are detected as DNA breaks in the absence of exogenous replication stress but are increased on release from hydroxyurea treatment or ATR inhibition (159). Tubbs et al. (143) recently showed that a subset of ERFSs close to replication origins containing poly(dA:dT) tracts are highly sensitive to DSBs in lymphocyte cultures treated with HU. The 2:1 ratio of DNA ends on opposite sides of the DNA breaks suggested that both arms of the replication fork are broken into DSBs, in contrast to the single-ended DSB model of RIM and BIR (40, 160–163).
The ectopic (CTG/CAG)100 repeat resembles an ERFS in its proximity to an origin (143), its early replication (164), and its sensitivity to several forms of replication stress (159). However, breakage at the (CTG)100 may differ from the model for ERFS DSBs on both arms of a fork based on RIM/BIR mutagenesis at the flanking TK sequence upon HU treatment (i.e. the 3′ end of a replication-dependent DSB generated by (CTG)100 fork collapse can invade and mutagenize the TK gene of an intact sister chromatid).
In the dual-fluorescence system, the frequency of GCVR colonies is comparable with recently reported values for BIR initiated by (CGG/CCG)153 repeats in murine cells (124) and may reflect the rate of error-prone DNA synthesis during break-induced replication. However, additional factors (efficiency of mismatch repair, incidence of sister chromatid (versus nonallelic) invasion, efficiency of synthesis on the sister chromatid template, frequency of template switching) may also affect the observed frequency of mutagenesis.
A variety of replication stressors, including HU, induced breaks between the dTomato and eGFP reporter genes at the ectopic site. HU also induced a (CTG/CAG)100 DSB localized to the edge of the repeat downstream of the c-myc replication origin. The induction of GCV-resistant clones by HU treatment and the decrease in TK mutant clones following knockdown of PolD3 or BRCA2 suggest that HU causes BIR in this system. Considered with the strong preferential occurrence of mutations downstream of the ectopic repeat in (CTG/CAG)102 cells, these data are consistent with a model in which replication stress at the (CTG/CAG) microsatellite leads to DSBs in forks originating at the c-myc origin, resulting in BIR and mutagenesis of the downstream TK gene.
We propose that Mus81 cleavage of the stalled rightward moving fork results in a covalently open or closed hairpin end (165) of the dTomato chromosome fragment (Fig. 10). Alternatively, Mus81 cleavage may occur as the leftward moving fork stalls at a hairpin structure or template-switched/chicken-foot reversed fork structure. The abundance of green cells after HU treatment implies that the eGFP gene downstream of the (CTG/CAG)100 repeat DSB is replicated by a leftward moving replication fork and is preserved in cells that have lost the dTomato gene (76).
We speculate that the non-B DNA structure of the dTomato end blocks repair and yields two DSBs that are functionally single-ended. The unligated DSB also leads to loss of the acentric dTomato chromosome fragment.
A small fraction of eGFP ends may undergo BIR and generate GCVS cells that are green (mutant dTomato) or yellow, whereas the majority of green cells die, inasmuch as a single unresolved DSB can cause apoptosis (166, 167). BIR of the dTomato end gives cells that are GCVR (TK mutant) and red (eGFP mutant) or yellow.
Recently, Mayle et al. (76) showed that knockout of Mus81 in yeast could increase BIR mutagenesis. In this system, a mutant form of the FLP recombinase was used to generate a long-lived DNA nick that could be converted to a seDSB when traversed by a replication fork. The authors concluded that Mus81 cleavage of the BIR D-loop normally reduced BIR mutagenesis. In contrast, the present results suggest that earlier cleavage by Mus81 at stalled forks may also increase DSBs. Further experiments are under way to analyze the structures of the right and left DSB ends and test the effects of enzymes involved in BIR in processing DSBs in this system.
Experimental procedures
Cell culture
HeLa/406 acceptor cells contain a single FRT site (65). Cell lines were derived by co-transfecting HeLa/406 cells with dual-fluorescence donor plasmids and the FLP recombinase expression vector pOG44 (168). Cells were maintained on Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 5% CO2 at 37 °C. GCV-resistant cell growth in control (no HU)- or HU-treated cells was assayed in 96-well plates using resazurin (Biotium, catalog no. 30025) according to the manufacturer's directions (169) after 14 days of GCV selection, as described (124).
Hydroxyurea, aphidicolin, and telomestatin treatment assays
Cells were treated at a final hydroxyurea concentration of 0.2 mm, aphidicolin at a final concentration of 0.2 μm, and TMS at a final concentration of 0.5 μm. The reagents were added to the medium 24 h after plating the cells and maintained until the start of recovery (2–4 days). Cells were treated with 200 μm H2O2 for 15 min. Treatment and recovery of cells was in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 5% CO2 at 37 °C.
I-Sce1 transfection
DF2/myc/(CTG/CAG)100 cells were transfected with Lipofectamine 2000 (Invitrogen, catalog no. 11668-019) and 2 μg of I-Sce1 plasmid (Addgene no. 26477) in a 6-well plate, 24 h postplating. During transfection, medium without antibiotic was used. 8 days after transfection, cells were harvested for analysis by flow cytometry.
siRNA/shRNA treatment
The siRNAs used to knock down translesion synthesis polymerases were generously provided by Yanzhe Gao (University of North Carolina). Cells were transfected with Lipofectamine 2000 (Invitrogen 11668-019) and 100 nm (final concentration) of siRNA in a 6-well plate, 24 h postplating. Control experiments were performed using AllStars negative control siRNA (Qiagen, catalog no. 1027281). Cells were allowed to recover 48 h post-transfection for 4 days and then analyzed by flow cytometry. PolD3 was knocked down in the presence of doxycycline (1.25 μg/ml) in cells stably transfected with a SMARTvector Inducible Lentiviral PolD3 shRNA (V3SH11252-226758280, Dharmacon/Horizon).
Western blotting
Whole cell lysates from treated or untreated cells were prepared. After SDS-PAGE, membranes were probed using a 1:1000 dilution of antibodies against pChk1 (Cell Signaling, catalog no. 2341), BRCA2 antibody (Calbiochem, catalog no. 3146957; 1:1000), or PolD3 (Invitrogen, catalog no. PA536951; 1:500). Primary antibodies against Pol, Pol κ (1:2000 dilution), Rad18, or Rev1 (1:5000 dilution) were generously provided by Yanzhe Gao (University of North Carolina).
Flow cytometry
Cell were trypsinized and centrifuged at 300 × g for 3 min. Medium was aspirated, and cells were washed with cold PBS. After a final wash with PBS, cells were analyzed using a C-Flow® Plus Accuri cytometer. All of the results that compare the effect of treatments on a single cell line within a figure were obtained contemporaneously from sister subcultures split from the same cell population.
Statistical analysis
Student's two-tailed t test was used to analyze the statistical significance of the experimental results versus the corresponding paired controls using the “percent green cells” shown in Figs. 4 and 10 and Figs. S1–S7 and generate p values (GraphPad Prism 8). A value of p < 0.05 was taken to indicate statistical significance.
Unpaired Student's two-tailed t tests were used to compare (Pu)88 versus (Py)88 cells (Fig. 5) and (CTG)100 versus DF/myc cells treated with TMS with or without HU (Fig. 6), as these cells were derived from separate clonal outgrowths using different integrant constructs.
DRAQ-7 flow cytometry
After treatment of cells with replication stress–inducing agent followed by recovery, cells were centrifuged at 500 × g for 3 min. Medium was aspirated, and cells were washed with cold PBS and spun down at 500 × g for 3 min. Cells were permeabilized with 70% ethanol at −20 °C for 20 min or overnight. Cells were centrifuged and washed and resuspended in 1 ml of PBS with RNase A (0.75 μg/ml final concentration) and incubated at 37 °C for 20 min. Finally, DRAQ7 dye (170) (Abcam, catalog no. ab109202) was added at a final concentration of 7.5 μm. Cells were incubated in the dark for 25 min and analyzed using a CFlow® Plus Accuri cytometer.
CD
CD spectra were collected using a Jasco J-815 CD spectropolarimeter (Jasco Inc., Easton, MD). Spectra were recorded from 320 to 220 nm with a bandwidth of 1.0 nm, scan rate of 50 nm/min, and time constant of 1 s. All DNA samples were dissolved in 10 mm Tris, 1 mm EDTA, pH 7.4, and diluted in water to a working concentration of 10 μm. Telomestatin was added to DNA samples at a final concentration of 50 μm. The CD spectra represent the average of four scans taken at 25 °C and baseline-corrected for buffer. The oligonucleotides used for CD were as follows: c-myc G4, 5′-TGA GGG TGG GGA GGG TGG GTA A, (CTG)12, and (CAG)12.
Ligation-mediated lamPCR
LamPCR was performed based on previously described conditions with the following modifications (71, 72). Genomic DNA was isolated from untransfected DF2/myc/(CTG/CAG)100 cells, 24 h after transfection with the I-Sce1 expression plasmid, or after 4 days of 0.2 mm HU treatment. Linear amplification was performed using 5 μg of DNA and the downstream biotinylated primer 3′ F0-biot ((biotin)5′-GTCAGCTTGCCGTAGGTGG-3′) for 50 cycles. A second aliquot (0.5 μl) of HotStarTaq was added, and amplification was performed for an additional 50 cycles.
The linear amplification products were captured on streptavidin beads, washed, and ligated overnight to the adapter oligonucleotide (5′-pATCGACAACAACTCTCCTCCTCCGTGCGddC-3′) (71). The beads were washed, and the ligated products were amplified with the adapter reverse complement primer (5′-CGCACGGAGGAGGAGAGTTGTTGTCGAT-3′) and the nested downstream primer 3′F1 (5′-GCTGAACTTGTGGCCGTTTA-3′). Products were electrophoresed on 1.5% agarose gels.
Data availability
All data are contained in the article and supporting information.
Supplementary Material
Acknowledgments
We thank Yanzhe Gao (University of North Carolina) for reagents, David Hitch (Wright State University) for intellectual and technical contributions, and Eric Brown (University of Pennsylvania) for comments on this work.
This article contains supporting information.
Author contributions—R. Y. G., E. J. R., C. C. G., S. D. R., F. J. D., J. R. B., K. S., H. H., and M. L. conceptualization; R. Y. G., J. R. B., K. S., and M. L. data curation; R. Y. G., E. J. R., C. C. G., S. D. R., F. J. D., J. R. B., and K. S. formal analysis; R. Y. G., S. D. R., and F. J. D. validation; R. Y. G., E. J. R., S. D. R., F. J. D., J. R. B., K. S., and M. L. investigation; R. Y. G., J. R. B., K. S., and M. L. visualization; R. Y. G., E. J. R., C. C. G., S. D. R., F. J. D., J. R. B., K. S., H. H., and M. L. methodology; R. Y. G., E. J. R., C. C. G., S. D. R., F. J. D., J. R. B., K. S., H. H., and M. L. writing-review and editing; S. D. R. and F. J. D. software; K. S., H. H., and M. L. resources; M. L. supervision; M. L. funding acquisition; M. L. writing-original draft; M. L. project administration.
Funding and additional information—This work was supported by the Wright State University Biomedical Sciences Ph.D. Program (to J. B.) and National Institutes of Health Grant GM122976 (to M. L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- DSB
- double-strand break
- FoSTeS
- fork stalling and template switching
- MMBIR
- microhomology-mediated break-induced replication
- GCR
- gross chromosomal rearrangement
- BIR
- break-induced replication
- seDSB
- single-ended DSB
- Pu/Py
- polypurine/polypyrimidine
- ADPKD
- autosomal dominant polycystic kidney disease
- HU
- hydroxyurea
- FRT
- FLP recombinase target
- lamPCR
- linear amplification ligation–mediated PCR
- TMS
- telomestatin
- TLS
- translesion synthesis
- PCNA
- proliferating cell nuclear antigen
- Pol
- polymerase
- iPCR
- inverse PCR
- TSC
- tuberous sclerosis
- ERFS
- early replicating fragile site
- GCV
- ganciclovir
- DF
- dual-fluorescence
- G4
- G quadruplex.
References
- 1. Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- 2. Subramanian S., Mishra R. K., and Singh L. (2003) Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biol. 4, R13 10.1186/gb-2003-4-2-r13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neil A. J., Kim J. C., and Mirkin S. M. (2017) Precarious maintenance of simple DNA repeats in eukaryotes. Bioessays 39, 1700077 10.1002/bies.201700077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim J. C., and Mirkin S. M. (2013) The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 23, 280–288 10.1016/j.gde.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. López Castel A., Cleary J. D., and Pearson C. E. (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 11, 165–170 10.1038/nrm2854 [DOI] [PubMed] [Google Scholar]
- 6. Mirkin S. M. (2006) DNA structures, repeat expansions and human hereditary disorders. Curr. Opin. Struct. Biol. 16, 351–358 10.1016/j.sbi.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 7. Mirkin S. M. (2007) Expandable DNA repeats and human disease. Nature 447, 932–940 10.1038/nature05977 [DOI] [PubMed] [Google Scholar]
- 8. Voineagu I., Freudenreich C. H., and Mirkin S. M. (2009) Checkpoint responses to unusual structures formed by DNA repeats. Mol. Carcinog. 48, 309–318 10.1002/mc.20512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang G., and Vasquez K. M. (2014) Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair (Amst.) 19, 143–151 10.1016/j.dnarep.2014.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shastri N., Tsai Y. C., Hile S., Jordan D., Powell B., Chen J., Maloney D., Dose M., Lo Y., Anastassiadis T., Rivera O., Kim T., Shah S., Borole P., Asija K., et al. (2018) Genome-wide identification of structure-forming repeats as principal sites of fork collapse upon ATR inhibition. Mol. Cell 72, 222–238.e11 10.1016/j.molcel.2018.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidt M. H., and Pearson C. E. (2016) Disease-associated repeat instability and mismatch repair. DNA Repair (Amst.) 38, 117–126 10.1016/j.dnarep.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 12. Kumari D., Hayward B., Nakamura A. J., Bonner W. M., and Usdin K. (2015) Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat. Res. 781, 14–21 10.1016/j.mrfmmm.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Usdin K. (2006) Bending the rules: unusual nucleic acid structures and disease pathology in the repeat expansion diseases. in Genetic Instabilities and Neurological Diseases (Wells R. D., and Ashizawa T., eds) pp. 617–635, Elsevier, Amsterdam [Google Scholar]
- 14. Usdin K., and Grabczyk E. (2000) DNA repeat expansions and human disease. Cell. Mol. Life Sci. 57, 914–931 10.1007/PL00000734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium (2019) CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell 178, 887–900.e14 10.1016/j.cell.2019.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao X. N., Lokanga R., Allette K., Gazy I., Wu D., and Usdin K. (2016) A MutSβ-dependent contribution of MutSα to repeat expansions in fragile X premutation mice? PLoS Genet. 12, e1006190 10.1371/journal.pgen.1006190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumari D., and Usdin K. (2016) Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 25, 3689–3698 10.1093/hmg/ddw215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerhardt J., Bhalla A. D., Butler J. S., Puckett J. W., Dervan P. B., Rosenwaks Z., and Napierala M. (2016) Stalled DNA replication forks at the endogenous GAA repeats drive repeat expansion in Friedreich's ataxia cells. Cell Rep. 16, 1218–1227 10.1016/j.celrep.2016.06.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J. A., Carvalho C. M., and Lupski J. R. (2007) A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247 10.1016/j.cell.2007.11.037 [DOI] [PubMed] [Google Scholar]
- 20. Cocquempot O., Brault V., Babinet C., and Herault Y. (2009) Fork stalling and template switching as a mechanism for polyalanine tract expansion affecting the DYC mutant of HOXD13, a new murine model of synpolydactyly. Genetics 183, 23–30 10.1534/genetics.109.104695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang F., Khajavi M., Connolly A. M., Towne C. F., Batish S. D., and Lupski J. R. (2009) The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 41, 849–853 10.1038/ng.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koumbaris G., Hatzisevastou-Loukidou H., Alexandrou A., Ioannides M., Christodoulou C., Fitzgerald T., Rajan D., Clayton S., Kitsiou-Tzeli S., Vermeesch J. R., Skordis N., Antoniou P., Kurg A., Georgiou I., Carter N. P., et al. (2011) FoSTeS, MMBIR and NAHR at the human proximal Xp region and the mechanisms of human Xq isochromosome formation. Hum. Mol. Genet. 20, 1925–1936 10.1093/hmg/ddr074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verdin H., D'Haene B., Beysen D., Novikova Y., Menten B., Sante T., Lapunzina P., Nevado J., Carvalho C. M., Lupski J. R., and De Baere E. (2013) Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet. 9, e1003358 10.1371/journal.pgen.1003358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hsiao M. C., Piotrowski A., Callens T., Fu C., Wimmer K., Claes K. B., and Messiaen L. (2015) Decoding NF1 intragenic copy-number variations. Am. J. Hum. Genet. 97, 238–249 10.1016/j.ajhg.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vogt J., Wernstedt A., Ripperger T., Pabst B., Zschocke J., Kratz C., and Wimmer K. (2016) PMS2 inactivation by a complex rearrangement involving an HERV retroelement and the inverted 100-kb duplicon on 7p22.1. Eur. J. Hum. Genet. 24, 1598–1604 10.1038/ejhg.2016.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deshpande M., and Gerhardt J. (2018) Break-induced replication sparks CGG-repeat instability. Nat. Struct. Mol. Biol. 25, 643–644 10.1038/s41594-018-0103-z [DOI] [PubMed] [Google Scholar]
- 27. Vatta M., Niu Z., Lupski J. R., Putnam P., Spoonamore K. G., Fang P., Eng C. M., and Willis A. S. (2013) Evidence for replicative mechanism in a CHD7 rearrangement in a patient with CHARGE syndrome. Am. J. Med. Genet. A 161A, 3182–3186 10.1002/ajmg.a.36178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beck C. R., Carvalho C. M., Banser L., Gambin T., Stubbolo D., Yuan B., Sperle K., McCahan S. M., Henneke M., Seeman P., Garbern J. Y., Hobson G. M., and Lupski J. R. (2015) Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genet. 11, e1005050 10.1371/journal.pgen.1005050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sundararajan R., Gellon L., Zunder R. M., and Freudenreich C. H. (2010) Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 184, 65–77 10.1534/genetics.109.111039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang H., and Freudenreich C. H. (2007) An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 27, 367–379 10.1016/j.molcel.2007.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Freudenreich C. H., and Lahiri M. (2004) Structure-forming CAG/CTG repeat sequences are sensitive to breakage in the absence of Mrc1 checkpoint function and S-phase checkpoint signaling: implications for trinucleotide repeat expansion diseases. Cell Cycle 3, 1370–1374 10.4161/cc.3.11.1246 [DOI] [PubMed] [Google Scholar]
- 32. Balakumaran B. S., Freudenreich C. H., and Zakian V. A. (2000) CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum. Mol. Genet. 9, 93–100 10.1093/hmg/9.1.93 [DOI] [PubMed] [Google Scholar]
- 33. Freudenreich C. H., Kantrow S. M., and Zakian V. A. (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856 10.1126/science.279.5352.853 [DOI] [PubMed] [Google Scholar]
- 34. Kim H. M., Narayanan V., Mieczkowski P. A., Petes T. D., Krasilnikova M. M., Mirkin S. M., and Lobachev K. S. (2008) Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 27, 2896–2906 10.1038/emboj.2008.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voineagu I., Narayanan V., Lobachev K. S., and Mirkin S. M. (2008) Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. U. S. A. 105, 9936–9941 10.1073/pnas.0804510105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim J. C., Harris S. T., Dinter T., Shah K. A., and Mirkin S. M. (2017) The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat. Struct. Mol. Biol. 24, 55–60 10.1038/nsmb.3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah K. A., and Mirkin S. M. (2015) The hidden side of unstable DNA repeats: mutagenesis at a distance. DNA Repair (Amst.) 32, 106–112 10.1016/j.dnarep.2015.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anand R. P., Tsaponina O., Greenwell P. W., Lee C. S., Du W., Petes T. D., and Haber J. E. (2014) Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev. 28, 2394–2406 10.1101/gad.250258.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sotiriou S. K., Kamileri I., Lugli N., Evangelou K., Da-Re C., Huber F., Padayachy L., Tardy S., Nicati N. L., Barriot S., Ochs F., Lukas C., Lukas J., Gorgoulis V. G., Scapozza L., et al. (2016) Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell 64, 1127–1134 10.1016/j.molcel.2016.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sakofsky C. J., and Malkova A. (2017) Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 52, 395–413 10.1080/10409238.2017.1314444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leffak M. (2017) Break-induced replication links microsatellite expansion to complex genome rearrangements. Bioessays 39, 1700025 10.1002/bies.201700025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carvalho C. M. B., Pfundt R., King D. A., Lindsay S. J., Zuccherato L. W., Macville M. V. E., Liu P., Johnson D., Stankiewicz P., Brown C. W., Shaw C. A., Hurles M. E., Ira G., Hastings P. J., Brunner H. G., et al. (2015) Absence of heterozygosity due to template switching during replicative rearrangements. Am. J. Hum. Genet. 96, 555–564 10.1016/j.ajhg.2015.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakofsky C. J., Roberts S. A., Malc E., Mieczkowski P. A., Resnick M. A., Gordenin D. A., and Malkova A. (2014) Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 7, 1640–1648 10.1016/j.celrep.2014.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Costantino L., Sotiriou S. K., Rantala J. K., Magin S., Mladenov E., Helleday T., Haber J. E., Iliakis G., Kallioniemi O. P., and Halazonetis T. D. (2014) Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91 10.1126/science.1243211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kramara J., Osia B., and Malkova A. (2017) Break-induced replication: an unhealthy choice for stress relief? Nat. Struct. Mol. Biol. 24, 11–12 10.1038/nsmb.3361 [DOI] [PubMed] [Google Scholar]
- 46. Zhang F., Carvalho C. M., and Lupski J. R. (2009) Complex human chromosomal and genomic rearrangements. Trends Genet. 25, 298–307 10.1016/j.tig.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holland A. J., and Cleveland D. W. (2012) Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 18, 1630–1638 10.1038/nm.2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nambiar M., Goldsmith G., Moorthy B. T., Lieber M. R., Joshi M. V., Choudhary B., Hosur R. V., and Raghavan S. C. (2011) Formation of a G-quadruplex at the BCL2 major breakpoint region of the t(14;18) translocation in follicular lymphoma. Nucleic Acids Res. 39, 936–948 10.1093/nar/gkq824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nambiar M., Kari V., and Raghavan S. C. (2008) Chromosomal translocations in cancer. Biochim. Biophys. Acta 1786, 139–152 10.1016/j.bbcan.2008.07.005 [DOI] [PubMed] [Google Scholar]
- 50. Nagamani S. C., Zhang F., Shchelochkov O. A., Bi W., Ou Z., Scaglia F., Probst F. J., Shinawi M., Eng C., Hunter J. V., Sparagana S., Lagoe E., Fong C. T., Pearson M., Doco-Fenzy M., et al. (2009) Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J. Med. Genet. 46, 825–833 10.1136/jmg.2009.067637 [DOI] [PubMed] [Google Scholar]
- 51. Pehlivan D., Hullings M., Carvalho C. M., Gonzaga-Jauregui C. G., Loy E., Jackson L. G., Krantz I. D., Deardorff M. A., and Lupski J. R. (2012) NIPBL rearrangements in Cornelia de Lange syndrome: evidence for replicative mechanism and genotype-phenotype correlation. Genet. Med. 14, 313–322 10.1038/gim.2011.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lupski J. R. (2015) Structural variation mutagenesis of the human genome: Impact on disease and evolution. Environ. Mol. Mutagen. 56, 419–436 10.1002/em.21943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Carvalho C. M., and Lupski J. R. (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 17, 224–238 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pettersson O. J., Aagaard L., Jensen T. G., and Damgaard C. K. (2015) Molecular mechanisms in DM1—a focus on foci. Nucleic Acids Res. 43, 2433–2441 10.1093/nar/gkv029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu G., Chen X., Bissler J. J., Sinden R. R., and Leffak M. (2010) Replication-dependent instability at (CTG)·(CAG) repeat hairpins in human cells. Nat. Chem. Biol. 6, 652–659 10.1038/nchembio.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu G., Chen X., and Leffak M. (2013) Oligodeoxynucleotide Binding to (CTG)·(CAG) microsatellite repeats inhibits replication fork stalling, hairpin formation, and genome instability. Mol. Cell Biol. 33, 571–581 10.1128/MCB.01265-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blackwood J. K., Okely E. A., Zahra R., Eykelenboom J. K., and Leach D. R. (2010) DNA tandem repeat instability in the Escherichia coli chromosome is stimulated by mismatch repair at an adjacent CAG·CTG trinucleotide repeat. Proc. Natl. Acad. Sci. U. S. A. 107, 22582–22586 10.1073/pnas.1012906108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Frizzell A., Nguyen J. H., Petalcorin M. I., Turner K. D., Boulton S. J., Freudenreich C. H., and Lahue R. S. (2014) RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep. 6, 827–835 10.1016/j.celrep.2014.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tiner W. J. Sr., Potaman V. N., Sinden R. R., and Lyubchenko Y. L. (2001) The structure of intramolecular triplex DNA: atomic force microscopy study. J. Mol. Biol. 314, 353–357 10.1006/jmbi.2001.5174 [DOI] [PubMed] [Google Scholar]
- 60. Rossetti S., Strmecki L., Gamble V., Burton S., Sneddon V., Peral B., Roy S., Bakkaloglu A., Komel R., Winearls C. G., and Harris P. C. (2001) Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am. J. Hum. Genet. 68, 46–63 10.1086/316939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kozlowski P., Roberts P., Dabora S., Franz D., Bissler J., Northrup H., Au K. S., Lazarus R., Domanska-Pakiela D., Kotulska K., Jozwiak S., and Kwiatkowski D. J. (2007) Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum. Genet. 121, 389–400 10.1007/s00439-006-0308-9 [DOI] [PubMed] [Google Scholar]
- 62. Kozlowski P., Bissler J., Pei Y., and Kwiatkowski D. J. (2008) Analysis of PKD1 for genomic deletion by multiplex ligation-dependent probe assay: absence of hot spots. Genomics 91, 203–208 10.1016/j.ygeno.2007.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu G., Myers S., Chen X., Bissler J. J., Sinden R. R., and Leffak M. (2012) Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine-polypyrimidine (Pu-Py) tract. J. Biol. Chem. 287, 33412–33423 10.1074/jbc.M112.402503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu G., Bissler J. J., Sinden R. R., and Leffak M. (2007) Unstable spinocerebellar ataxia type 10 (ATTCT)*(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell Biol. 27, 7828–7838 10.1128/MCB.01276-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu G., Malott M., and Leffak M. (2003) Multiple functional elements comprise a mammalian chromosomal replicator. Mol. Cell Biol. 23, 1832–1842 10.1128/mcb.23.5.1832-1842.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen X., Liu G., and Leffak M. (2013) Activation of a human chromosomal replication origin by protein tethering. Nucleic Acids Res. 41, 6460–6474 10.1093/nar/gkt368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lewis T. W., Barthelemy J. R., Virts E. L., Kennedy F. M., Gadgil R. Y., Wiek C., Linka R. M., Zhang F., Andreassen P. R., Hanenberg H., and Leffak M. (2019) Deficiency of the Fanconi anemia E2 ubiqitin conjugase UBE2T only partially abrogates Alu-mediated recombination in a new model of homology dependent recombination. Nucleic Acids Res. 47, 3503–3520 10.1093/nar/gkz026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li X., Zhao X., Fang Y., Jiang X., Duong T., Fan C., Huang C. C., and Kain S. R. (1998) Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–34975 10.1074/jbc.273.52.34970 [DOI] [PubMed] [Google Scholar]
- 69. Snapp E. L. (2009) Fluorescent proteins: a cell biologist's user guide. Trends Cell Biol. 19, 649–655 10.1016/j.tcb.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Petermann E., Orta M. L., Issaeva N., Schultz N., and Helleday T. (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37, 492–502 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li T. W., and Weeks K. M. (2006) Structure-independent and quantitative ligation of single-stranded DNA. Anal. Biochem. 349, 242–246 10.1016/j.ab.2005.11.002 [DOI] [PubMed] [Google Scholar]
- 72. Paruzynski A., Arens A., Gabriel R., Bartholomae C. C., Scholz S., Wang W., Wolf S., Glimm H., Schmidt M., and von Kalle C. (2010) Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat. Protoc. 5, 1379–1395 10.1038/nprot.2010.87 [DOI] [PubMed] [Google Scholar]
- 73. Trivedi A., Waltz S. E., Kamath S., and Leffak M. (1998) Multiple initiations in the c-myc replication origin independent of chromosomal location. DNA Cell Biol. 17, 885–896 10.1089/dna.1998.17.885 [DOI] [PubMed] [Google Scholar]
- 74. Waltz S. E., Trivedi A. A., and Leffak M. (1996) DNA replication initiates non-randomly at multiple sites near the c-myc gene in HeLa cells. Nucleic Acids Res. 24, 1887–1894 10.1093/nar/24.10.1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu G., Chen X., Gao Y., Lewis T., Barthelemy J., and Leffak M. (2012) Altered replication in human cells promotes DMPK (CTG)n·(CAG)n repeat instability. Mol. Cell Biol. 32, 1618–1632 10.1128/MCB.06727-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mayle R., Campbell I. M., Beck C. R., Yu Y., Wilson M., Shaw C. A., Bjergbaek L., Lupski J. R., and Ira G. (2015) DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349, 742–747 10.1126/science.aaa8391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bétous R., Glick G. G., Zhao R., and Cortez D. (2013) Identification and characterization of SMARCAL1 protein complexes. PLoS ONE 8, e63149 10.1371/journal.pone.0063149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dehé P. M., and Gaillard P. H. L. (2017) Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 18, 315–330 10.1038/nrm.2016.177 [DOI] [PubMed] [Google Scholar]
- 79. Duda H., Arter M., Gloggnitzer J., Teloni F., Wild P., Blanco M. G., Altmeyer M., and Matos J. (2016) A mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev. Cell 39, 740–755 10.1016/j.devcel.2016.11.017 [DOI] [PubMed] [Google Scholar]
- 80. Franchitto A., Pirzio L. M., Prosperi E., Sapora O., Bignami M., and Pichierri P. (2008) Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 183, 241–252 10.1083/jcb.200803173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fugger K., Chu W. K., Haahr P., Kousholt A. N., Beck H., Payne M. J., Hanada K., Hickson I. D., and Sørensen C. S. (2013) FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 4, 1423 10.1038/ncomms2395 [DOI] [PubMed] [Google Scholar]
- 82. Xing M., Wang X., Palmai-Pallag T., Shen H., Helleday T., Hickson I. D., and Ying S. (2015) Acute MUS81 depletion leads to replication fork slowing and a constitutive DNA damage response. Oncotarget 6, 37638–37646 10.18632/oncotarget.5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fu H., Martin M. M., Regairaz M., Huang L., You Y., Lin C. M., Ryan M., Kim R., Shimura T., Pommier Y., and Aladjem M. I. (2015) The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nat. Commun. 6, 6746 10.1038/ncomms7746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pepe A., and West S. C. (2014) MUS81-EME2 promotes replication fork restart. Cell Rep. 7, 1048–1055 10.1016/j.celrep.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Regairaz M., Zhang Y. W., Fu H., Agama K. K., Tata N., Agrawal S., Aladjem M. I., and Pommier Y. (2011) Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 195, 739–749 10.1083/jcb.201104003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pepe A., and West S. C. (2014) Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 42, 3833–3845 10.1093/nar/gkt1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lemaçon D., Jackson J., Quinet A., Brickner J. R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N., and Vindigni A. (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 10.1038/s41467-017-01180-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Quinet A., Lemaçon D., and Vindigni A. (2017) Replication fork reversal: players and guardians. Mol. Cell 68, 830–833 10.1016/j.molcel.2017.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Blaszak R. T., Potaman V., Sinden R. R., and Bissler J. J. (1999) DNA structural transitions within the PKD1 gene. Nucleic Acids Res 27, 2610–2617 10.1093/nar/27.13.2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Patel H. P., Lu L., Blaszak R. T., and Bissler J. J. (2004) PKD1 intron 21: triplex DNA formation and effect on replication. Nucleic Acids Res. 32, 1460–1468 10.1093/nar/gkh312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Guédin A., Gros J., Alberti P., and Mergny J. L. (2010) How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 38, 7858–7868 10.1093/nar/gkq639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bacolla A., Tainer J. A., Vasquez K. M., and Cooper D. N. (2016) Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 44, 5673–5688 10.1093/nar/gkw261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhao J., Bacolla A., Wang G., and Vasquez K. M. (2010) Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 67, 43–62 10.1007/s00018-009-0131-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Belotserkovskii B. P., De Silva E., Tornaletti S., Wang G., Vasquez K. M., and Hanawalt P. C. (2007) A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem. 282, 32433–32441 10.1074/jbc.M704618200 [DOI] [PubMed] [Google Scholar]
- 95. De Cian A., Guittat L., Shin-Ya K., Riou J. F., and Mergny J. L. (2005) Affinity and selectivity of G4 ligands measured by FRET. Nucleic Acids Symp. Ser. (Oxf.) 49, 235–236 10.1093/nass/49.1.235 [DOI] [PubMed] [Google Scholar]
- 96. Gomez D., Paterski R., Lemarteleur T., Shin-Ya K., Mergny J. L., and Riou J. F. (2004) Interaction of telomestatin with the telomeric single-strand overhang. J. Biol. Chem. 279, 41487–41494 10.1074/jbc.M406123200 [DOI] [PubMed] [Google Scholar]
- 97. Del Mundo I. M. A., Zewail-Foote M., Kerwin S. M., and Vasquez K. M. (2017) Alternative DNA structure formation in the mutagenic human c-MYC promoter. Nucleic Acids Res. 45, 4929–4943 10.1093/nar/gkx100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mathad R. I., Hatzakis E., Dai J., and Yang D. (2011) c-MYC promoter G-quadruplex formed at the 5'-end of NHE III1 element: insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 39, 9023–9033 10.1093/nar/gkr612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yang D., and Hurley L. H. (2006) Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids 25, 951–968 10.1080/15257770600809913 [DOI] [PubMed] [Google Scholar]
- 100. Kim M. Y., Gleason-Guzman M., Izbicka E., Nishioka D., and Hurley L. H. (2003) The different biological effects of telomestatin and TMPyP4 can be attributed to their selectivity for interaction with intramolecular or intermolecular G-quadruplex structures. Cancer Res. 63, 3247–3256 [PubMed] [Google Scholar]
- 101. Tauchi T., Shin-Ya K., Sashida G., Sumi M., Nakajima A., Shimamoto T., Ohyashiki J. H., and Ohyashiki K. (2003) Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: involvement of ATM-dependent DNA damage response pathways. Oncogene 22, 5338–5347 10.1038/sj.onc.1206833 [DOI] [PubMed] [Google Scholar]
- 102. Siddiqui-Jain A., Grand C. L., Bearss D. J., and Hurley L. H. (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U. S. A. 99, 11593–11598 10.1073/pnas.182256799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bedrat A., Lacroix L., and Mergny J. L. (2016) Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 44, 1746–1759 10.1093/nar/gkw006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Figueroa A. A., Cattie D., and Delaney S. (2011) Structure of even/odd trinucleotide repeat sequences modulates persistence of non-B conformations and conversion to duplex. Biochemistry 50, 4441–4450 10.1021/bi200397b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pilch D. S., Barbieri C. M., Rzuczek S. G., Lavoie E. J., and Rice J. E. (2008) Targeting human telomeric G-quadruplex DNA with oxazole-containing macrocyclic compounds. Biochimie 90, 1233–1249 10.1016/j.biochi.2008.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nakamura T., Okabe S., Yoshida H., Iida K., Ma Y., Sasaki S., Yamori T., Shin-Ya K., Nakano I., Nagasawa K., and Seimiya H. (2017) Targeting glioma stem cells in vivo by a G-quadruplex-stabilizing synthetic macrocyclic hexaoxazole. Sci. Rep. 7, 3605 10.1038/s41598-017-03785-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hasegawa D., Okabe S., Okamoto K., Nakano I., Shin-Ya K., and Seimiya H. (2016) G-quadruplex ligand-induced DNA damage response coupled with telomere dysfunction and replication stress in glioma stem cells. Biochem. Biophys. Res. Commun. 471, 75–81 10.1016/j.bbrc.2016.01.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bailly V., Lauder S., Prakash S., and Prakash L. (1997) Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J. Biol. Chem. 272, 23360–23365 10.1074/jbc.272.37.23360 [DOI] [PubMed] [Google Scholar]
- 109. Yoon J. H., Prakash S., and Prakash L. (2012) Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc. Natl. Acad. Sci. U. S. A. 109, 7799–7804 10.1073/pnas.1204105109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yang W., and Gao Y. (2018) Translesion and repair DNA polymerases: diverse structure and mechanism. Annu. Rev. Biochem. 87, 239–261 10.1146/annurev-biochem-062917-012405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bétous R., Rey L., Wang G., Pillaire M. J., Puget N., Selves J., Biard D. S., Shin-Ya K., Vasquez K. M., Cazaux C., and Hoffmann J. S. (2009) Role of TLS DNA polymerases η and κ in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 48, 369–378 10.1002/mc.20509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bergoglio V., Boyer A. S., Walsh E., Naim V., Legube G., Lee M. Y., Rey L., Rosselli F., Cazaux C., Eckert K. A., and Hoffmann J. S. (2013) DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 201, 395–408 10.1083/jcb.201207066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Day T. A., Palle K., Barkley L. R., Kakusho N., Zou Y., Tateishi S., Verreault A., Masai H., and Vaziri C. (2010) Phosphorylated Rad18 directs DNA polymerase η to sites of stalled replication. J. Cell Biol. 191, 953–966 10.1083/jcb.201006043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Garg P., and Burgers P. M. (2005) Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1. Proc. Natl. Acad. Sci. U. S. A. 102, 18361–18366 10.1073/pnas.0505949102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Prakash S., Johnson R. E., and Prakash L. (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74, 317–353 10.1146/annurev.biochem.74.082803.133250 [DOI] [PubMed] [Google Scholar]
- 116. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., and Yamaizumi M. (2004) Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23, 3886–3896 10.1038/sj.emboj.7600383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Axford M. M., Wang Y. H., Nakamori M., Zannis-Hadjopoulos M., Thornton C. A., and Pearson C. E. (2013) Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLoS Genet. 9, e1003866 10.1371/journal.pgen.1003866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Maor-Shoshani A., Ben-Ari V., and Livneh Z. (2003) Lesion bypass DNA polymerases replicate across non-DNA segments. Proc. Natl. Acad. Sci. U. S. A. 100, 14760–14765 10.1073/pnas.2433503100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Northam M. R., Moore E. A., Mertz T. M., Binz S. K., Stith C. M., Stepchenkova E. I., Wendt K. L., Burgers P. M., and Shcherbakova P. V. (2014) DNA polymerases ζ and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res. 42, 290–306 10.1093/nar/gkt830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lewis J. S., Spenkelink L. M., Schauer G. D., Yurieva O., Mueller S. H., Natarajan V., Kaur G., Maher C., Kay C., O'Donnell M. E., and van Oijen A. M. (2020) Tunability of DNA polymerase stability during eukaryotic DNA replication. Mol Cell 77, 17–25.e15 10.1016/j.molcel.2019.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schmutz V., Janel-Bintz R., Wagner J., Biard D., Shiomi N., Fuchs R. P., and Cordonnier A. M. (2010) Role of the ubiquitin-binding domain of Polη in Rad18-independent translesion DNA synthesis in human cell extracts. Nucleic Acids Res. 38, 6456–6465 10.1093/nar/gkq403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Okada T., Sonoda E., Yamashita Y. M., Koyoshi S., Tateishi S., Yamaizumi M., Takata M., Ogawa O., and Takeda S. (2002) Involvement of vertebrate polκ in Rad18-independent postreplication repair of UV damage. J. Biol. Chem. 277, 48690–48695 10.1074/jbc.M207957200 [DOI] [PubMed] [Google Scholar]
- 123. Gao Y., Mutter-Rottmayer E., Zlatanou A., Vaziri C., and Yang Y. (2017) Mechanisms of post-replication DNA repair. Genes (Basel) 8, 64 10.3390/genes8020064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kononenko A. V., Ebersole T., Vasquez K. M., and Mirkin S. M. (2018) Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 25, 669–676 10.1038/s41594-018-0094-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bhowmick R., Minocherhomji S., and Hickson I. D. (2016) RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 64, 1117–1126 10.1016/j.molcel.2016.10.037 [DOI] [PubMed] [Google Scholar]
- 126. Barthelemy J., Hanenberg H., and Leffak M. (2016) FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 44, 6803–6816 10.1093/nar/gkw433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nachman M. W., and Crowell S. L. (2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lynch M. (2010) Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. U. S. A. 107, 961–968 10.1073/pnas.0912629107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bacolla A., Temiz N. A., Yi M., Ivanic J., Cer R. Z., Donohue D. E., Ball E. V., Mudunuri U. S., Wang G., Jain A., Volfovsky N., Luke B. T., Stephens R. M., Cooper D. N., Collins J. R., et al. (2013) Guanine holes are prominent targets for mutation in cancer and inherited disease. PLoS Genet. 9, e1003816 10.1371/journal.pgen.1003816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gomes-Pereira M., Foiry L., Nicole A., Huguet A., Junien C., Munnich A., and Gourdon G. (2007) CTG trinucleotide repeat “big jumps”: large expansions, small mice. PLoS Genet. 3, e52 10.1371/journal.pgen.0030052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shishkin A. A., Voineagu I., Matera R., Cherng N., Chernet B. T., Krasilnikova M. M., Narayanan V., Lobachev K. S., and Mirkin S. M. (2009) Large-scale expansions of Friedreich's ataxia GAA repeats in yeast. Mol. Cell 35, 82–92 10.1016/j.molcel.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Saini N., Zhang Y., Nishida Y., Sheng Z., Choudhury S., Mieczkowski P., and Lobachev K. S. (2013) Fragile DNA motifs trigger mutagenesis at distant chromosomal loci in Saccharomyces cerevisiae. PLoS Genet. 9, e1003551 10.1371/journal.pgen.1003551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhao J., Wang G., Del Mundo I. M., McKinney J. A., Lu X., Bacolla A., Boulware S. B., Zhang C., Zhang H., Ren P., Freudenreich C. H., and Vasquez K. M. (2018) Distinct mechanisms of nuclease-directed DNA-structure-induced genetic instability in cancer genomes. Cell Rep. 22, 1200–1210 10.1016/j.celrep.2018.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Usdin K., House N. C., and Freudenreich C. H. (2015) Repeat instability during DNA repair: insights from model systems. Crit. Rev. Biochem. Mol. Biol. 50, 142–167 10.3109/10409238.2014.999192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Cherng N., Shishkin A. A., Schlager L. I., Tuck R. H., Sloan L., Matera R., Sarkar P. S., Ashizawa T., Freudenreich C. H., and Mirkin S. M. (2011) Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc. Natl. Acad. Sci. U. S. A. 108, 2843–2848 10.1073/pnas.1009409108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Singh A., and Xu Y. J. (2016) The Cell Killing Mechanisms of Hydroxyurea. Genes (Basel) 7, 99 10.3390/genes7110099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Feng W., Di Rienzi S. C., Raghuraman M. K., and Brewer B. J. (2011) Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 1, 327–335 10.1534/g3.111.000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Venkatachalam G., Surana U., and Clément M. V. (2017) Replication stress-induced endogenous DNA damage drives cellular senescence induced by a sub-lethal oxidative stress. Nucleic Acids Res. 45, 10564–10582 10.1093/nar/gkx684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lukusa T., and Fryns J. P. (2008) Human chromosome fragility. Biochim. Biophys. Acta 1779, 3–16 10.1016/j.bbagrm.2007.10.005 [DOI] [PubMed] [Google Scholar]
- 140. Wang G., and Vasquez K. M. (2006) Non-B DNA structure-induced genetic instability. Mutat. Res. 598, 103–119 10.1016/j.mrfmmm.2006.01.019 [DOI] [PubMed] [Google Scholar]
- 141. Avila Figueroa A., and Delaney S. (2010) Mechanistic studies of hairpin to duplex conversion for trinucleotide repeat sequences. J. Biol. Chem. 285, 14648–14657 10.1074/jbc.M109.061853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wang H., Li Y., Truong L. N., Shi L. Z., Hwang P. Y., He J., Do J., Cho M. J., Li H., Negrete A., Shiloach J., Berns M. W., Shen B., Chen L., and Wu X. (2014) CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 54, 1012–1021 10.1016/j.molcel.2014.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tubbs A., Sridharan S., van Wietmarschen N., Maman Y., Callen E., Stanlie A., Wu W., Wu X., Day A., Wong N., Yin M., Canela A., Fu H., Redon C., Pruitt S. C., et al. (2018) Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142.e19 10.1016/j.cell.2018.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wenger S. L., Giangreco C. A., Tarleton J., and Wessel H. B. (1996) Inability to induce fragile sites at CTG repeats in congenital myotonic dystrophy. Am. J. Med. Genet. 66, 60–63 10.1002/(SICI)1096-8628(19961202)66:1<60::AID-AJMG13>3.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- 145. Kim M. Y., Vankayalapati H., Shin-Ya K., Wierzba K., and Hurley L. H. (2002) Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular g-quadruplex. J. Am. Chem. Soc. 124, 2098–2099 10.1021/ja017308q [DOI] [PubMed] [Google Scholar]
- 146. Sun D., Guo K., Rusche J. J., and Hurley L. H. (2005) Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 33, 6070–6080 10.1093/nar/gki917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Temime-Smaali N., Guittat L., Sidibe A., Shin-Ya K., Trentesaux C., and Riou J. F. (2009) The G-quadruplex ligand telomestatin impairs binding of topoisomerase IIIα to G-quadruplex-forming oligonucleotides and uncaps telomeres in ALT cells. PLoS ONE 4, e6919 10.1371/journal.pone.0006919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Van Raay T. J., Burn T. C., Connors T. D., Petry L. R., Germino G. G., Klinger K. W., and Landes G. M. (1996) A 2.5 kb polypyrimidine tract in the PKD1 gene contains at least 23 H-DNA-forming sequences. Microb. Comp. Genomics 1, 317–327 10.1089/mcg.1996.1.317 [DOI] [PubMed] [Google Scholar]
- 149. Mirkin E. V., and Mirkin S. M. (2014) To switch or not to switch: at the origin of repeat expansion disease. Mol. Cell 53, 1–3 10.1016/j.molcel.2013.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Gerhardt J., Tomishima M. J., Zaninovic N., Colak D., Yan Z., Zhan Q., Rosenwaks Z., Jaffrey S. R., and Schildkraut C. L. (2014) The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 53, 19–31 10.1016/j.molcel.2013.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Prioleau M. N., and MacAlpine D. M. (2016) DNA replication origins-where do we begin? Genes Dev. 30, 1683–1697 10.1101/gad.285114.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Stevanoni M., Palumbo E., and Russo A. (2016) The replication of frataxin gene is assured by activation of dormant origins in the presence of a GAA-repeat expansion. PLoS Genet. 12, e1006201 10.1371/journal.pgen.1006201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhao L., and Washington M. T. (2017) Translesion synthesis: insights into the selection and switching of DNA polymerases. Genes (Basel) 8, 24 10.3390/genes8010024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Vujanovic M., Krietsch J., Raso M. C., Terraneo N., Zellweger R., Schmid J. A., Taglialatela A., Huang J. W., Holland C. L., Zwicky K., Herrador R., Jacobs H., Cortez D., Ciccia A., Penengo L., et al. (2017) Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell 67, 882–890.e5 10.1016/j.molcel.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Vaisman A., and Woodgate R. (2017) Translesion DNA polymerases in eukaryotes: what makes them tick? Crit. Rev. Biochem. Mol. Biol. 52, 274–303 10.1080/10409238.2017.1291576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wong R. P., Garcia-Rodriguez N., Zilio N., Hanulova M., and Ulrich H. D. (2020) Processing of DNA polymerase-blocking lesions during genome replication is spatially and temporally segregated from replication forks. Mol. Cell 77, 3–16.e14 10.1016/j.molcel.2019.09.015 [DOI] [PubMed] [Google Scholar]
- 157. Sakofsky C. J., Ayyar S., Deem A. K., Chung W. H., Ira G., and Malkova A. (2015) Translesion polymerases drive microhomology-mediated break-induced replication leading to complex chromosomal rearrangements. Mol. Cell 60, 860–872 10.1016/j.molcel.2015.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Barnes R. P., Hile S. E., Lee M. Y., and Eckert K. A. (2017) DNA polymerases η and κ exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst.) 57, 1–11 10.1016/j.dnarep.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Barlow J. H., Faryabi R. B., Callén E., Wong N., Malhowski A., Chen H. T., Gutierrez-Cruz G., Sun H. W., McKinnon P., Wright G., Casellas R., Robbiani D. F., Staudt L., Fernandez-Capetillo O., and Nussenzweig A. (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632 10.1016/j.cell.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kramara J., Osia B., and Malkova A. (2018) Break-induced replication: the where, the why, and the how. Trends Genet. 34, 518–531 10.1016/j.tig.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Donnianni R. A., and Symington L. S. (2013) Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 110, 13475–13480 10.1073/pnas.1309800110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ruff P., Donnianni R. A., Glancy E., Oh J., and Symington L. S. (2016) RPA stabilization of single-stranded DNA is critical for break-induced replication. Cell Rep. 17, 3359–3368 10.1016/j.celrep.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J. E., Lobachev K. S., and Malkova A. (2013) Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392 10.1038/nature12584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kemp M. G., Ghosh M., Liu G., and Leffak M. (2005) The histone deacetylase inhibitor trichostatin A alters the pattern of DNA replication origin activity in human cells. Nucleic Acids Res. 33, 325–336 10.1093/nar/gki177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zhang Y., Saini N., Sheng Z., and Lobachev K. S. (2013) Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet. 9, e1003979 10.1371/journal.pgen.1003979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Huang L. C., Clarkin K. C., and Wahl G. M. (1996) Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc. Natl. Acad. Sci. U. S. A. 93, 4827–4832 10.1073/pnas.93.10.4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Rich T., Allen R. L., and Wyllie A. H. (2000) Defying death after DNA damage. Nature 407, 777–783 10.1038/35037717 [DOI] [PubMed] [Google Scholar]
- 168. O'Gorman S., Fox D. T., and Wahl G. M. (1991) Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 251, 1351–1355 10.1126/science.1900642 [DOI] [PubMed] [Google Scholar]
- 169. Kanagaraj R., Huehn D., MacKellar A., Menigatti M., Zheng L., Urban V., Shevelev I., Greenleaf A. L., and Janscak P. (2010) RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 38, 8131–8140 10.1093/nar/gkq697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wlodkowic D., Akagi J., Dobrucki J., Errington R., Smith P. J., Takeda K., and Darzynkiewicz Z. (2013) Kinetic viability assays using DRAQ7 probe. Curr. Protoc. Cytom Chapter 9, 9.41 10.1002/0471142956.cy0941s65 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained in the article and supporting information.