

Abstract
Factor X activation by the intrinsic Xase complex, composed of factor IXa bound to factor VIIIa on membranes, is essential for the amplified blood coagulation response. The biological significance of this step is evident from bleeding arising from deficiencies in factors VIIIa or IXa in hemophilia. Here, we assess the mechanism(s) that enforce the distinctive specificity of intrinsic Xase for its biological substrate. Active-site function of IXa was assessed with a tripeptidyl substrate (PF-3688). The reversible S1 site binder, 4-aminobenzamidine (pAB), acted as a classical competitive inhibitor of PF-3688 cleavage by Xase. In contrast, pAB acted as a noncompetitive inhibitor of factor X activation. This disconnect between peptidyl substrate and protein substrate cleavage indicates a major role for interactions between factor X and extended sites on Xase in determining substrate affinity. Accordingly, an uncleavable factor X variant, not predicted to engage the active site of IXa within Xase, acted as a classical competitive inhibitor of factor X activation. Fluorescence studies confirmed the binding of factor X to Xase assembled with IXa with a covalently blocked active site. Our findings suggest that the recognition of factor X by the intrinsic Xase complex occurs through a multistep “dock-and-lock” pathway in which the initial interaction between factor X and intrinsic Xase occurs at exosites distant from the active site, followed by active-site docking and bond cleavage.
Keywords: factor X, intrinsic Xase, serine proteinase, exosites, serine protease, coagulation factor, factor VIII (FVIII), enzyme mechanism, enzyme kinetics, factor IXa, factor X activation
The serine proteinases of blood coagulation contain catalytic domains that are structurally similar and resemble trypsin, an ancestral member of the S1 peptidase family of which chymotrypsin is the archetype (1–3). Despite such homology, the trypsin-like proteinases of coagulation exhibit high specificity for their biological substrate(s) (4, 5). Narrow and distinctive specificity in this family of related proteinases was initially attributed to the recognition of sequences flanking cleavage sites in the cognate substrate akin to the preferential hydrolysis of one peptidyl substrate over the other by any one proteinase (6). However, these active site–centric ideas have gradually ceded to the burgeoning evidence that protein substrate affinity for several of the coagulation enzymes is determined by extended interactions between substrate sites removed from the cleavage site(s) and sites (exosites) on the enzyme or enzyme-cofactor complex distinct from the active site (7, 8). In these cases, the initial exosite-binding interaction not only drives affinity but also enforces specificity by both restricting and facilitating active-site engagement by the substrate followed by cleavage (7). Such a multistep pathway for substrate recognition is associated with signature disconnects between the kinetics of inhibition of active-site function relative to protein substrate cleavage (9, 10).
The activating cleavage of zymogen factor X to the proteinase, Xa, is key for both the initiation of coagulation and the subsequent amplification of flux through the coagulation cascade (11, 12). The latter is achieved by the intrinsic Xase complex, which assembles through reversible interactions between the proteinase IXa, its cofactor VIIIa, and membranes exposing negatively charged phospholipids (4). In a manner analogous to the other enzyme complexes of coagulation, the assembly of IXa into intrinsic Xase profoundly increases, by several orders of magnitude, its ability to cleave factor X (13). These properties of intrinsic Xase explain bleeding in hemophilia A or B associated with defects or deficiencies in either factor VIII or factor IX (14).
The prothrombinase complex of coagulation is the closest structural analog of the intrinsic Xase (4). The most comprehensive evidence for exosite-dependent substrate recognition and its ramifications for multiple aspects of function has been established in numerous studies of prothrombin activation by prothrombinase (7, 15). Given the similarities between intrinsic Xase and prothrombinase, it follows that some variation on analogous strategies is likely to apply to factor X activation by this enzyme complex as well. However, despite widespread interest in the basic and clinical science of the function of intrinsic Xase, details of the molecular basis for its substrate specificity remain incomplete. This partly reflects the limitations in the ability to compare active-site function of IXa within intrinsic Xase with factor X activation by the enzyme complex. Factor IXa is a notoriously poor enzyme for the cleavage of peptidyl substrates and typically requires the use of very high concentrations of alcohols for measurable rates of peptide hydrolysis (16, 17). This requirement limits interpretable kinetic studies comparing peptidyl substrate hydrolysis with factor X activation by intrinsic Xase to infer a role for exosite binding in protein substrate recognition by the enzyme complex.
Kinetic limitations aside, there is some evidence implicating a role for exosite binding in the recognition of factor X by intrinsic Xase. Fluorescence studies using intrinsic Xase assembled with IXa in which the active site has been occluded have provided evidence for factor X binding to the enzyme complex with affinity in line with the measured Km (18). This binding could be prevented by an inhibitory antibody targeting the A2 domain of VIIIa (18). The potential problem with these findings is that such implied exosite interactions could not be directly related to the productive pathway of factor X recognition by the intrinsic Xase. There is also evidence for the inhibition of X activation by intrinsic Xase by an aptamer targeting a surface of the substrate distant from the cleavage site (19). Whereas the data suggest that these substrate regions may be important in its binding to the enzyme complex, alternate kinetic interpretations have not been ruled out (19).
We have now exploited the finding that a fluorogenic peptidyl substrate, H-d-leucyl-phenyl-glycyl-arginine-7-amino-4-methylcoumarin (PF-3688), yields an experimentally accessible Km for IXa and intrinsic Xase in the absence of added alcohols. This has allowed interpretable kinetic studies to search for one aspect of the kinetic signature of exosite-dependent recognition of the protein substrate. Our kinetic strategy, combined with binding studies, now implicates an exosite-dependent dock-and-lock strategy for the recognition and cleavage of factor X by the intrinsic Xase.
Results
Peptidyl substrate cleavage
Factor IXa exhibits very weak amidolytic activity toward synthetic peptidyl substrates. Although measurable rates of peptidyl substrate cleavage have required the addition of high concentrations of ethylene glycol or other alcohols, the Km for most substrates remains indeterminate and limited by peptide solubility (16, 17, 20). The potential detrimental effect of alcohols on the assembly and stability of intrinsic Xase coupled with the high Km presents hurdles in the analysis of the active function of IXa within the enzyme complex.
Initial velocity measurements with PF-3688 revealed reasonable rates in the absence of added alcohols and yielded experimentally accessible Km values for IXa alone and IXa assembled within intrinsic Xase (Table 1). As has also been reported for the analogous prothrombinase complex, incorporation of IXa into the intrinsic Xase complex yielded a modest decrease in the rate and catalytic efficiency of peptidyl substrate cleavage. Given that the rate of cleavage of the cognate protein substrate is vastly increased following the assembly of prothrombinase or intrinsic Xase, the findings illustrate, once again, an obvious disconnect between the constraints affecting the kinetic constants for protein substrate cleavage relative to active-site function.
Table 1.
Kinetics of peptidyl substrate cleavage
Shown are kinetic constants obtained from initial velocity studies conducted with varying concentrations of PF-3688 either in the absence or presence of different fixed concentrations of 4-aminobenzamidine. Each data set was composed of 11 different concentrations of PF-3668 (0–400 μm). Inhibition studies used four different concentrations of pAB fixed at 0, 75, 150, and 300 μm.
| Enzymea | Inhibitor | Inhibition type | Vmaxb | Km | Ki |
|---|---|---|---|---|---|
| ΔF/min | µm | µm | |||
| IXa | None | NAc | 0.212 ± 0.006 | 267 ± 16 | NA |
| IXa | None | NA | 0.205 ± 0.008 | 213 ± 17 | NA |
| IXa | pAB | Competitive | 0.199 ± 0.008 | 200 ± 17 | 39 ± 2.2 |
| IXa-VIIIa-PCPS | None | NA | 0.369 ± 0.039 | 762 ± 111 | NA |
| IXa-VIIIa-PCPS | None | NA | 0.321 ± 0.031 | 652 ± 92 | NA |
| IXa-VIIIa-PCPS | pAB | Competitive | 0.343 ± 0.028 | 687 ± 79 | 111 ± 5 |
| IXa-VIIIa-PCPS | pAB | Competitive | 0.325 ± 0.021 | 669 ± 63 | 117 ± 5 |
aStudies done with 75 nm IXa or with 75 nm IXa, 90 nm VIIIa, and 100 μm PCPS.
bKinetic constants are reported ± 95% confidence limits.
cNA, not applicable.
Reversible inhibition of peptidyl substrate cleavage by active site–directed ligand
The arginine analog, 4-aminobenzamidine (pAB), is an established inhibitor of arginine-specific serine proteinases, including factor IXa (21, 22). Kinetic and structural studies have established that it binds in rapid equilibrium to the S1 pocket that confers arginine-specific recognition of substrates by this family of proteinases (23). As has been illustrated with several other coagulation proteinases, pAB is expected to act as a competitive inhibitor for substrates that interact in a limited way with the active site (21).
Initial velocity studies of PF-3688 cleavage by IXa alone (Fig. 1A) or in complex with VIIIa and PCPS (Fig. 1B) in the presence of different fixed concentrations of pAB yielded results consistent with classical competitive inhibition. The fitted kinetic constants illustrate that both the Km for PF-3688 and Ki for pAB are modestly increased by a factor of 2–3 when IXa is assembled in the intrinsic Xase (Table 1). The kinetic signature of classical competitive inhibition is that increasing concentrations of inhibitor increase Km without affecting Vmax because inhibition at any one inhibitor concentration can be completely overcome by saturating with substrate. This result implies that pAB and PF-3688 bind in a mutually exclusive way to the active site of IXa in solution or assembled into intrinsic Xase.
Figure 1.

Inhibition of peptidyl substrate cleavage by pAB. Initial velocities for the cleavage of PF-3688 were measured with 75 nm IXa (A) or 75 nm IXa, 90 nm VIIIa, and 100 μm PCPS (B) in the presence of 0 (open circles), 75 (filled circles), 150 (open triangles), or 300 μm (filled triangles) pAB. In both panels, the lines are drawn following analysis to the rate expression for competitive inhibition using the fitted constants Km = 200 ± 17 μm, Ki = 39 ± 2.2 μm, and V = 0.199 ± 0.008 ΔF/min (A) or Km = 687 ± 79 μm, Ki = 111 ± 5 μm, and V = 0.343 ± 0.028 ΔF/min (B).
Reversible inhibition of factor X activation
In contrast to the findings with the peptidyl substrate, initial velocity studies of factor X activation revealed a different inhibition profile by pAB (Fig. 2). Increasing concentrations of pAB markedly decrease Vmax without obviously increasing the Km for factor X. These data imply that pAB binding is not mutually exclusive with factor X binding to intrinsic Xase. Analysis distinguishing between different reversible inhibition types yielded best fits with the model for noncompetitive inhibition (Table 2). When taken with the kinetic pathway for noncompetitive inhibition, the findings illustrate that pAB and factor X can simultaneously bind to the enzyme (Scheme 1). It follows that the affinity of factor X for intrinsic Xase is substantially determined by interactions at sites distinct from the proteinase active site. Indeed, the fitted constants indicate that protein substrate affinity is modestly enhanced by bound pAB and pAB affinity is proportionately enhanced by bound factor X (i.e. α < 1, Scheme 1). Because active-site engagement by the substrate is required for catalysis, such a binding step to form E·S* must follow the initial formation of E·S before factor X can be cleaved to yield product (Scheme 1). Inhibition by pAB arises from a decreased Vmax because some fraction of the enzyme is tied up as E·S·I in the presence of inhibitor regardless of substrate concentration. This redistribution decreases the fraction of the E·S* species and thereby the rate of product formation even at saturating concentrations of substrate. This simple kinetic approach suggests a multistep dock-and-lock pathway for factor X recognition and cleavage by intrinsic Xase. Substrate affinity is enforced by an initial interaction between factor X and the enzyme complex at exosites, removed from the active site, followed by active-site engagement and cleavage. The second binding step affects Vmax and the perceived rate constant for catalysis. These points are evident from the rate expression for the two-step mechanism illustratively derived using the rapid equilibrium expression,
| (Eq. 1) |
where
| (Eq. 2) |
| (Eq. 3) |
Figure 2.

Inhibition of factor X activation by pAB. Initial velocities of Xa formation were measured using 0.1 nm IXa, 15 nm VIIIa, and 30 μm PCPS with 0 (open circles), 75 (filled circles), 150 (open triangles), or 300 μm (filled triangles) pAB. The lines are drawn following analysis according to noncompetitive inhibition using the fitted constants: V/[E] = 2.12 ± 0.04 s − 1, Km = 48.1 ± 3.2 nm, Ki = 161 ± 9 μm, and α = 0.34 ± 0.08 (see Scheme 1).
Table 2.
Inhibition of factor X activation by intrinsic Xase by active site–directed ligand
Kinetic constants obtained from initial velocity studies conducted with varying concentrations of factor X with different fixed concentrations of 4-aminobenzamidine. Studies were done using 14 different concentrations of factor X (0–0.3 μm), four different fixed concentrations of pAB (0, 75, 150, and 300 μm), and intrinsic Xase assembled using 0.1 nm IXa, 15 nm VIIIa, and 30 μm PCPS ([E] = 0.1 nm).
| Inhibition typea | V/Eb | Km | Ki | αc |
|---|---|---|---|---|
| s−1 | nm | µm | ||
| Noncompetitive | 2.07 ± 0.04 | 39.4 ± 2.6 | 163 ± 11 | 0.52 ± 0.09 |
| Noncompetitive | 1.95 ± 0.04 | 40.2 ± 2.4 | 131 ± 8 | 0.37 ± 0.07 |
| Noncompetitive | 2.12 ± 0.04 | 48.1 ± 3.2 | 161 ± 9 | 0.34 ± 0.08 |
aOther forms of reversible inhibition were eliminated by analysis according to alternate rate expressions as described “under Data Analysis”.
bKinetic constants are reported ± 95% confidence limits.
cMultiplier for Km at saturating pAB and for Ki at saturating factor X.
Scheme 1.

Kinetic pathway for factor X activation by intrinsic Xase. The scheme illustrates the conversion of substrate (S) to product (P) by the membrane-bound intrinsic Xase complex (E) in the presence of an active site–directed reversible inhibitor (I). KEXO denotes the equilibrium dissociation constant for the exosite-dependent interaction between S and E. Ks* is the equilibrium dissociation constant for the unimolecular step that leads to interactions between substrate and E at the active site followed by the catalytic step denoted by kcat. Ki denotes the equilibrium dissociation constant for the binding of I to E. The multiplier α corrects KEXO for the binding of S at saturating I and corrects Ki for I binding at saturating S. Exosite binding refers to interactions between S and E at sites distinct from the active site and elements flanking the scissile bond without specific knowledge of where these sites may lie on the interacting species.
Exosite-dependent inhibition of intrinsic Xase
Competitive inhibition of factor X activation by intrinsic Xase would require an approach that targets the initial exosite interaction between enzyme and protein substrate (Scheme 1). We assessed this possibility by using factor X variants, XS195A and XR15Q, that are expected to retain the ability to engage the intrinsic Xase complex in this way regardless of whether they can engage the active site. Mutation of the catalytic Ser195 to Ala in XS195A yields an alternate substrate but one that does not yield an active product in the initial velocity measurements of Xa formation. The variant XR15Q contains a Gln substitution at the P1 Arg and is expected to be uncleavable because it cannot engage the active site.
Initial velocity studies of factor X activation by intrinsic Xase in the presence of different fixed concentrations of XS195A (Fig. 3A) or XR15Q (Fig. 3B) indicated that both variants acted as classical competitive inhibitors of factor X activation. Fitted kinetic constants indicated that competitive inhibition by these variants was achieved with Ki values comparable with the Km for factor X (Table 3). Thus, in accordance with Scheme 1, competitive inhibition of factor X activation requires interference with exosite binding, and this can be accomplished even by the XR15Q variant that is not expected to effectively engage the active site of IXa within intrinsic Xase.
Figure 3.

Inhibition of factor X activation by factor X variants. Initial velocities of Xa formation were measured using 0.1 nm IXa, 15 nm VIIIa, and 30 μm PCPS in the presence of different fixed concentrations of XS195A (A) or XR15Q (B). In both panels, the concentration of factor X variant added as inhibitor was 0 (open circles), 0.1 (filled circles), 0.3 (open triangles), 0.5 (filled triangles), or 1 μm ( (open inverted triangles). The lines are drawn following analysis according to competitive inhibition using the fitted constants: V/[E] = 1.95 ± 0.04 s−1, Km = 60.4 ± 3.8 nm, and Ki = 118 ± 7 nm (A); V/[E] = 2.18 ± 0.04 s−1, Km = 60.4 ± 3 nm, and Ki = 38.1 ± 1.6 (B).
Table 3.
Inhibition of factor X activation by intrinsic Xase by factor X variants
Kinetic constants were obtained from initial velocity studies conducted with varying concentrations of factor X with different fixed concentrations of the indicated variant. Studies were done using 14 different concentrations of factor X (0–0.3 μm), five different fixed concentrations of the indicated variant (0, 0.1, 0.3, 0.5, and 1 μm), and intrinsic Xase assembled using 0.1 nm IXa, 15 nm VIIIa, and 30 μm PCPS ([E] = 0.1 nm).
| Inhibitor | Inhibition typea | V/Eb | Km | Ki |
|---|---|---|---|---|
| s−1 | nm | nm | ||
| XS195A | Competitive | 1.95 ± 0.04 | 60.4 ± 3.8 | 118 ± 7 |
| XS195A | Competitive | 2.98 ± 0.06 | 96.3 ± 5.2 | 347 ± 20 |
| XR15Q | Competitive | 2.18 ± 0.04 | 60.4 ± 3 | 38.1 ± 1.6 |
| XR15Q | Competitive | 2.80 ± 0.05 | 109.4 ± 5.2 | 114.9 ± 4.4 |
aOther forms of reversible inhibition were eliminated by analysis according to alternate rate expressions as described “under Data Analysis”.
bKinetic constants are reported ± 95% confidence limits.
The uncleavable nature of factor XR15Q
We verified the expected properties of XR15Q by examining its potential cleavage following prolonged incubation with a high concentration of intrinsic Xase followed by analysis using SDS-PAGE (Fig. 4). Whereas factor X from plasma was readily cleaved to Xa under these conditions, XR15Q yielded no evidence of Xa formation (Fig. 4). This verifies the inability of the Gln substitution at the P1 residue to allow for adequate active-site engagement and cleavage.
Figure 4.
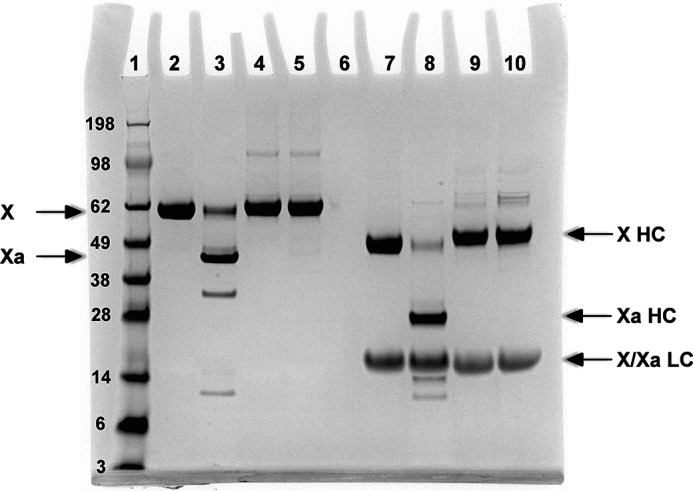
Cleavage of factor X variants by intrinsic Xase. Factor X variants (5 μm) were incubated with 50 nm IXa, 100 nm VIIIa, and 50 μm PCPS at 25 °C for 30 min. The products were separated by SDS-PAGE without (lanes 2–5) or after disulfide bond reduction (lanes 7–10), and bands were visualized by staining with Coomassie Brilliant Blue. Lane 1, molecular weight markers; lanes 2 and 7, uncleaved XWT; lanes 3 and 8, activated XWT; lanes 4 and 9, unactivated XR15Q; lanes 5 and 10, activated XR15Q. HC, heavy chain; LC, light chain. Molecular weights × 103 for the standards are listed in the left margin.
We also pursued the corollary approach of examining the ability of these substrate variants to displace pAB from the active site of the intrinsic Xase assembled using IXaS195A. However, fluorescence binding measurements revealed a very weak binding constant for pAB to IXaS195A in solution or assembled within intrinsic Xase, which rendered this approach impractical.
Binding of factor X variants to active site–blocked intrinsic Xase
Binding measurements were pursued to independently corroborate active site–independent interactions between factor X and intrinsic Xase. We employed a IXa derivative in which the active site was covalently occluded with a fluorescein derivative of a peptidyl chloromethyl ketone. The objective was to use the tethered fluorescent group as a reporter of interactions in the absence of catalysis and with a peptide irreversibly occluding the active site.
Studies were pursued with IXa inactivated with a thioester derivative of Phe-Phe-Arg-chloromethyl ketone, which was then modified with fluorescein 5-maleimide to yield fluorescein-5-maleimide (F5M)-FFR-IXa (F-IXai). This derivative has previously proven useful to report interactions within intrinsic Xase (18). Incremental additions of thrombin cleaved VIII to a reaction mixture containing 50 nm F-IXai, and saturating PCPS produced a large and saturable increase in anisotropy (Δr ∼ 0.07) with fitted constants consistent with a tight interaction between F-IXai and VIIIa on the membrane surface (Fig. 5A). In a second series of studies, intrinsic Xase preformed with 50 nm F-IXai, 100 nm VIIIa, and saturating membranes was titrated with either XS195A or XR15Q (Fig. 5B). Equivalent saturable titration curves were obtained with either variant with a further increase in anisotropy (Δr ∼ 0.04). Combined analysis assuming 1 mol of factor X variant bound/mol of Xase revealed that either variant bound to the enzyme complex with an equilibrium dissociation constant of ∼15 nm. These findings illustrate that factor X can bind with high affinity to intrinsic Xase in a way that is independent of the active site of IXa or the complementary structures flanking the scissile bond in the substrate. The somewhat higher affinity obtained in these binding measurements relative to the affinity for the same variants inferred from the competitive inhibition studies may reflect the modest linkage observed between exosite binding and liganding at the active site within the enzyme complex.
Figure 5.

Binding studies using active site–blocked factor IXa. A, assembly of active site–blocked intrinsic Xase. A reaction mixture containing 50 nm F-IXai and 100 μm PCPS in assay buffer was titrated with incremental additions of factor VIIIa. B, binding of factor X variants to active site–blocked intrinsic Xase. Reaction mixtures containing 50 nm F-IXai, 100 nm VIIIa, and 100 μm PCPS in assay buffer were titrated with incremental additions of XS195A (open triangles), or XR15Q (filled triangles). The lines in both panels are drawn following global fitting as described under “Data analysis,” using the fitted parameters: KP,C= 3.1 ± 0.3 nm; n = 0.84 ± 0.09 mol of VIIIa bound/mol of F-IXai at saturation; KE,S = 14.7 ± 1.4 nm; rO = 0.209 ± 0.001; rE = 0.274 ± 0.001, and rES = 0.318 ± 0.001.
Discussion
The evidence presented in this study parallels the findings with extrinsic Xase (VIIa/tissue factor) and the more comprehensive studies with prothrombinase to illustrate that exosite-dependent protein substrate recognition is a prevalent strategy by which the enzyme complexes of coagulation recognize their cognate substrates (7, 8). The dock-and-lock approach to substrate binding (Scheme 1) delineated here and for the other two enzyme complexes as well likely underlies their narrow and distinctive specificities for their respective protein substrates (7). Thus, exosite binding allows these enzyme complexes to achieve high specificity despite the fact that the proteinases in each case bear catalytic domains that closely resemble each other and that of trypsin (7).
The kinetic approaches described here are necessary to address the question of whether exosite binding by factor X actually lies on the productive pathway for its recognition and cleavage by the intrinsic Xase. An essential aspect of this strategy is to address whether there is a clear-cut divergence in the inhibition kinetics of peptidyl substrates that target the active site in a limited way and the kinetics of cleavage of the protein substrate. The notably poor activity of IXa toward peptidyl substrates and the need for high concentrations of ethylene glycol or other alcohols to obtain reliable rates of peptidyl substrate cleavage or even measurable kinetic constants have prevented such an analysis (16). The finding that the use of PF-3688 circumvents these major hurdles was the significant advance that made the current studies possible.
Structural significance has been attributed to the ability of ethylene glycol to bind IXa and enhance the rate of cleavage of peptidyl substrates with the implication that it somehow replicates the effects of VIIIa and/or the assembly of intrinsic Xase on IXa active-site function (20). Such claims cannot be tested without directly comparing the kinetics of peptidyl substrate cleavage by IXa in solution and assembled into intrinsic Xase done in the absence of ethylene glycol. Our findings now show that the assembly of IXa into intrinsic Xase actually produces a modest decrease in the rate of peptidyl substrate cleavage through an increased Km. Thus, it is improbable that ethylene glycol binding to IXa produces changes in the proteinase that are relevant to those elicited by VIIIa.
The increased Km for the peptidyl substrate following assembly of IXa into intrinsic Xase parallels effects of comparable magnitude seen with the incorporation of Xa into prothrombinase (24, 25). A second parallel between prothrombinase and intrinsic Xase is the cofactor-dependent perturbation in fluorescence of a reporter group covalently bound at the active site through a peptidyl chloromethylketone tether (26–28). Such effects have been invoked to suggest that the incorporation of the proteinase into the enzyme complex alters active-site geometry to potentially explain the large increase in kcat for protein substrate cleavage (27, 28). Alternate approaches to test this possibility have failed to correlate cofactor-dependent perturbations in the active site with large enhancements in the function of prothrombinase (25). Instead, it appears that a major explanation lies in the exosite-dependent strategy used by prothrombinase for protein substrate cleavage, which is not likely to apply to Xa alone (7, 15). Although such questions are still premature to be addressed for intrinsic Xase, we speculate that concepts developed through more extensive work with prothrombinase will varyingly hold in this case as well.
The findings in previous studies have hinted at a role for exosite-dependent recognition of factor X by intrinsic Xase. Fluorescence studies, analogous to those described here, provided the first evidence that factor X could bind to Xase even when it was assembled with IXa blocked at the active site with a peptidyl chloromethylketone (18). At the time, there was uncertainty about how substrate binding inferred from such fluorescence changes might relate to the productive pathway for factor X binding and cleavage. Our work now provides the framework that links one to the other. It is also likely that VIIIa might play some role in binding factor X. This conclusion draws from the observations that IXa can bind (unactivated) factor VIII in the presence of membranes but the enhanced X activation associated with intrinsic Xase requires an activating cleavage of factor VIII between the A1 and A2 domains (18). It is possible that this cleavage is somehow associated with allowing for factor X binding, although that is not the only interpretation. In line with such reasoning, an inhibitory antibody targeting the A2 domain in factor VIII/VIIIa has been documented to inhibit the anisotropy change associated with factor X binding to intrinsic Xase in the fluorescence studies (18, 29).
A second line of evidence suggesting that binding of factor X to intrinsic Xase may involve substrate structures distinct from those flanking the scissile bond is provided by the inhibition of X activation by an RNA aptamer that binds factor X (19). The X-ray structure of factor Xa bound to the aptamer reveals that the aptamer occupies a surface on factor Xa that is distant from where a scissile bond would be expected in factor X (30). Interestingly, whereas the aptamer inhibits X activation by intrinsic Xase, it has no effect on Xa formation catalyzed by the extrinsic Xase (19). Given the current findings, the resulting implication is that the extrinsic and intrinsic Xase complexes utilize different surfaces on factor X to achieve exosite-dependent recognition (31).
Factor X binds PCPS membranes, and membrane binding by the substrate is considered to play an important role in the greatly increased rate of Xa formation that accompanies the assembly of the intrinsic Xase on the membrane surface (13). Drawing from the progress made in understanding the function of the vitamin K–dependent enzyme complex, two types of substrate-membrane interactions are likely important here (32). The first represents a general binding of factor X to the membrane surface, and the second is a membrane interaction in the vicinity of the enzyme complex, as illustrated in Scheme 1. If this second type of interaction plays a significant role in the oriented presentation of the substrate to the enzyme, then by definition it represents an exosite interaction relevant to the productive pathway for factor X binding and cleavage by intrinsic Xase. Studies with prothrombinase and extrinsic Xase have ruled out the possibility that this is the only exosite interaction involved by using cleaved forms of the substrate that cannot bind membranes or by incorporating tissue factor into pure phosphatidylcholine membranes to which factor X cannot bind (26, 31). Similar approaches have not been or are impractical to use in the present study. Thus, membrane binding by factor X in the vicinity of the membrane assembled intrinsic Xase could represent an important contributor to the exosite-binding strategy utilized by the enzyme complex.
In summary, our studies reveal that exosite-dependent substrate tethering is a central feature of the dock-and-lock strategy utilized by the intrinsic Xase for factor X recognition and cleavage. Exosite binding rather than active-site engagement is a major determinant of substrate affinity and binding specificity. Active-site engagement by structures flanking the scissile bond contributes to the observed rate constant for catalysis. Taken together with findings made with some of the other coagulation enzyme complexes, it follows that this multistep pathway for protein substrate recognition and cleavage is a prevalent strategy by which the structurally related proteinases of coagulation achieve specificity for their protein substrates.
Experimental procedures
Materials
The fluorogenic peptidyl substrate PF-3688 was custom-synthesized and provided purified as the TFA salt (Midwest Bio-Tech). The chromogenic peptidyl substrates for Xa (Spectrozyme Xa, Sekisui Diagnostics) and pAB (Alfa Aesar) were from the indicated suppliers. The concentration of pAB was determined using E293 = 15,000 m−1 cm−1 (21). Small unilamellar phospholipid vesicles composed of 75% (w/w) l-α-phosphatidylcholine and 25% (w/w) l-α-phosphatidylserine (Avanti Polar Lipids) were prepared by sonication followed by ultracentrifugation, and concentrations were determined by a colorimetric assay for phosphate following oxidation as described (33). Human plasma was obtained as a gift from the plasmapheresis unit of the hospital of the University of Pennsylvania. Hepes, Mes, Sephadex G-25, hydroxylamine, and N-hydroxysuccinimidyl-S-acetylthioacetate (SATA) were from Sigma. H-d-Phe-Phe-Arg-chloromethylketone (FFRck) was from Calbiochem. The acetothioacetyl adduct of FFRck (ATA-FFRck) was prepared by reacting FFRck with a 5-fold molar excess of SATA, purified, and quality-controlled as described previously (34). F5M was from Thermo Scientific. Matched-pair antibody sets for ELISA of human factors X and IX were from Affinity Biologicals. The chromatography resins Q-Sepharose Fast Flow, benzamidine-Sepharose, and Sephadex G-25 (GE Healthcare), Poros columns and resins (PerSeptive Biosystems), and Bio-Gel P6DG (Bio-Rad) were from the indicated sources. All kinetic and binding studies were carried out at 25 °C in 20 mm Hepes, 150 mm NaCl, 0.1% (w/v) PEG-8000, 5 mm CaCl2, pH 7.5 (assay buffer).
Proteins
Factors IX and X and prothrombin were purified from human plasma and freed of traces of cross-contaminating proteins as described (19, 35–37). Factor Xa and thrombin were prepared following preparative activation of the respective zymogens and purified as before (37, 38). IXaβ was produced by preparative activation of factor IX (0.5 mg/ml, 120 ml) in assay buffer with 100 nm bovine XIa (Enzyme Research Laboratories) for 2 h at 37 °C. Factor IXaβ was purified free of IX, IXα, and XIa by application to a column of Q-Sepharose (2.5 × 15 cm) equilibrated in buffer composed of 20 mm Hepes, 100 mm NaCl, 1 mm benzamidine, pH 7.4, and elution with a gradient of increasing NaCl (0.1–1 m, 150 ml) prepared in the same buffer. Fractions containing protein were pooled; dialyzed into 20 mm Mes, 1 mm benzamidine, pH 6.0; and applied to a column of immobilized heparin (Poros HE/M, 0.46 × 10 cm) equilibrated in the same buffer. Bound protein was eluted with a gradient of increasing NaCl (0–1.0 m, 65 min, 2 ml/min) in the same buffer. Fractions containing IXa were pooled; dialyzed into 20 mm Hepes, 0.15 m NaCl, pH 7.4; and applied to a column of benzamidine-Sepharose (1.6 × 15 cm) equilibrated in the same buffer. Bound IXa was eluted with buffer containing 1 mm benzamidine. Fractions containing IXa were precipitated by the addition of solid (NH4)2SO4 to 80% saturation. Precipitated protein was collected by centrifugation (56,000 × g, 20 min), dissolved in 50% (v/v) glycerol, and stored at −20 °C.
Recombinant human full-length factor VIII (Kogenate) was obtained as a generous gift from Bayer. The protein was freed from the pharmaceutical formulation by application to a Poros HQ/M (1 × 10-cm) column equilibrated in 20 mm Hepes, 0.15 m NaCl, 5 mm CaCl2, 0.05% (v/v) Tween 20, pH 7.4. Bound protein was eluted with a gradient of increasing NaCl (0.15–1 m, 2 ml/min, 15 min) in the same buffer. Fractions containing protein were pooled, treated with 4-amidinophenylmethanesulfonyl fluoride hydrochloride (25 μm), dialyzed into the column equilibration buffer, aliquoted, and stored frozen at −80 °C. When needed, factor VIII (42 nm) was activated to VIIIa in assay buffer at 25 °C by the addition of 5 nm thrombin. After 30 s of incubation, the activation reaction was terminated by the addition of 10 nm hirudin (CIBA-Geigy), and VIIIa was used immediately.
Recombinant factor X and variants were expressed by cloning an expression cassette encoding the human prothrombin signal sequence, the prothrombin propeptide sequence followed by the sequence for mature human factor X inserted into the phCMV1 expression vector. The 3′-UTR contained the sequence of the tripartite woodchuck hepatitis virus posttranscriptional regulatory element to enhance protein expression. Factor X expression constructs containing mutations to encode for XR15Q and XS195A were generated by QuikChange mutagenesis (Stratagene) and transfected into HEK293 cells as described (39). Stable clones expressing high levels of factor X variant (∼5 µg/ml) were identified by ELISA and expanded for large-scale expression in the presence of vitamin K as described (40). Factor X variants were purified from serum-free conditioned media (20 liters) using established procedures (40). Final yield of the putatively carboxylated version of each variant was ∼25 mg, and γ-carboxyglutamic acid content was determined by amino acid analysis following base hydrolysis (41). The variants XR15Q and XS195A contained 10.4 ± 0.4 and 10.7 ± 0.1 mol of γ-carboxyglutamate/mol of protein compared with 10.9 ± 0.8 for factor X isolated from human plasma and used as an internal control.
The purity of all protein preparations was evaluated by SDS-PAGE. Protein concentrations were calculated using the previously reported molecular weights and extinction coefficients (): IX, 55,000, 1.32 (42); IXaβ, 45,000, 1.43 (43); X and variants, 56,500, 1.16; Xa, 45,300, 1.16 (44); VIII, 300,000, 1.22 (45); thrombin, 37,500, 1.95 (46); hirudin, 6,956, 0.37 (calculated).
Preparation of fluorescent, active site–blocked IXa
Active site–blocked IXa was prepared by incubating IXa with 10-fold molar excess ATA-FFRck in assay buffer at 4 °C for 48 h, and free ATA-FFRck was removed by gel filtration using Sephadex G-25 equilibrated in 0.1 m Hepes, 0.15 m NaCl, pH 7.5. Purified ATA-FFR-IXa was treated with 10 eq of F5M, 0.1 m hydroxylamine and incubated overnight at 4 °C. Free probe was removed by gel filtration using Bio-Gel P-6DG equilibrated in assay buffer. Aliquots of F5M-FFR-IXa were stored at −80 °C.
Kinetics of peptidyl substrate cleavage
Initial velocities of PF-3688 hydrolysis by IXa alone or in complex with VIIIa and PCPS were measured in assay buffer. Reaction mixtures (100 µl) containing increasing concentrations of PF-3688 (0–800 μm) in assay buffer were prepared in a Corning black polystyrene 96-well assay plate. Reactions were initiated by the addition of an equal volume of IXa or mixture of IXa, VIIIa, and PCPS in assay buffer to achieve a final concentration of 75 nm IXa or of 75 nm IXa, 90 nm VIIIa, and 100 μm PCPS. PF-3688 cleavage was measured by continuously monitoring fluorescence for 45 min with λex = 360 nm, λem = 460 nm using a SpectraMax Gemini kinetic plate reader (Molecular Devices). Initial rates were determined from the linear increase in fluorescence over a substantial period of time. The initial rate was linearly dependent on the concentration of enzyme. For inhibition studies, the initial 2× substrate solution also contained different fixed concentrations of pAB (0, 150, 300, and 600 μm).
Kinetics of factor X activation
Initial velocities of factor X activation were obtained from discontinuous measurements of Xa formation. Reaction mixtures (100 µl) containing increasing concentrations of factor X (0–0.6 μm) in assay buffer with different fixed concentrations of pAB (0, 150, 300, and 600 μm) or the variants, XS195A and XR15Q (0, 0.2, 0.6, 1, and 2 μm) were prepared in 1.2-ml microdilution tubes (USA Scientific). Factor X activation was initiated by the addition of an equal volume of enzyme solution in assay buffer to achieve final concentrations of 0.1 nm IXa, 15 nm VIIIa, and 30 μm PCPS. Following mixing, the reactions were allowed to proceed at 25 °C in a thermostatted water bath. Aliquots (10 µl) were withdrawn at various times (0, 30, 60, 90, 120, and 180 s) and quenched by mixing with 140 µl of 20 mm Hepes, 0.15 m NaCl, 0.1% (w/v) PEG-8000, 50 mm EDTA, pH 7.5, in a clear polystyrene 96-well assay plate (Corning). Factor Xa formed was assessed by the addition of 50 µl of 500 μm Spectrozyme Xa prepared in the quenching buffer, and initial velocities of substrate hydrolysis were determined by the increase in absorbance at 405 nm using a SpectraMax 250 kinetic plate reader (Molecular Devices). The concentration of Xa formed as a function of time was determined as described previously (31).
Cleavage of factor X variants by intrinsic Xase
Factor X cleavage was assessed following prolonged incubation of 5 μm factor X isolated from plasma (XWT) or recombinant XR15Q with 50 nm IXa, 100 nm VIIIa, and 50 μm PCPS at 25 °C. After 30 min, samples containing ∼4 μg of factor X were analyzed by SDS-PAGE without or with disulfide bond reduction and visualized with by staining with Coomassie Brilliant Blue R-250.
Steady-state fluorescence measurements
Fluorescence anisotropy was measured at 25 °C using a PTI QuantaMaster fluorescence spectrophotometer (Photon Technology Inc.). Measurements were made in T-format using λex = 490 nm, λem = 520 nm with KV-500 long-pass filters (Schott Glass) in the emission beams. Reaction mixtures (2.5 ml) in 1 × 1-cm stirred quartz cuvettes initially contained 50 nm F-IXai and 100 μm PCPS in assay buffer and were titrated by incremental additions of factor VIIIa. Anisotropy was measured following each addition and corrected for scattering contributions measured with a parallel reaction mixture containing 100 μm PCPS. To avoid systematic error from the time-dependent loss of factor VIIIa function, factor VIII was freshly activated with thrombin before each successive addition.
For studies of factor X binding, the initial reaction mixture contained 50 nm F-IXai, 100 nm VIIIa, and 100 μm PCPS in assay buffer. Anisotropy was measured following incremental additions of either XS195A or XR15Q (0–300 nm).
Data analysis
The data illustrated are representative of at least two separate experiments done at an equivalent level of detail. Initial velocity and inhibition studies were analyzed according to the appropriate rate expressions by nonlinear least squares analysis using custom software that employs the Levenberg–Marquardt algorithm (47). For kinetic studies with more than one variable, all data were simultaneously fit to the stated rate expression. For inhibition studies, alternate models were ruled out by fits to different rate expressions and selection of the most reliable fitting model using the criteria of systematic deviation of residuals, root mean squared deviation, uncertainty in fitted parameters, and the reduced χ2 statistic as described previously (48). The fitted constants are reported ± 95% confidence limits.
For the binding studies, experimental design and interpretation were according to the criteria described extensively for the analogous prothrombinase complex (49). The titrations were done at saturating PCPS so that all free and bound species were bound to the membrane surface and binding inferred from the anisotropy change would correspond to the interaction of the titrant with the fixed species on the membrane surface (49). The fluorescence titration data were analyzed according to the following equilibria,
where n moles of cofactor (C) bind per mole of proteinase (P) with equilibrium dissociation constant KP,C to form the enzyme complex (E). Although the stoichiometry of cofactor binding to proteinase is expected to be 1 (50), the term n is included to account for systematic errors in the active concentration of VIIIa and in determining the concentration of IXa modified with the fluorophore. One mole of substrate (S) binds per mole of E in an active site–independent way to form E·S with equilibrium dissociation constant KE,S. P, E, and E·S differ with respect to measured anisotropy denoted by rO, rE, and rES.
In the first phase of fitting, the factor VIIIa titration data were locally analyzed by nonlinear least squares using the equations described (49), to account for the first equilibrium step and obtain fitted constants for KP,C, n, rO, and rE. The term n from this analysis was used to scale the fixed concentration of P. Subsequently, all anisotropy data obtained with varying concentrations of VIIIa and X variants were globally analyzed to both sets of equilibria using KinTek Explorer (version 6.3) and the rapid equilibrium assumption (51). Global analysis yielded fitted values for KP,C, KE,S, rO, rE, and rES. The fitted value for n from the first analysis is also reported, although it was not obtained from the global analysis. This approach was used to circumvent the inability of KinTek Explorer to fit stoichiometries in the global analyses.
Data availability
Data for all figures are contained within the article. Additional data backing the kinetic analyses in the tables are available from S. K. (skrishna@pennmedicine.upenn.edu).
Author contributions—M. G. B. and S. K. conceptualization; M. G. B. data curation; M. G. B. and S. K. formal analysis; M. G. B. investigation; M. G. B. and S. K. methodology; M. G. B. writing-original draft; S. K. supervision; S. K. funding acquisition; S. K. project administration; S. K. writing-review and editing.
Funding and additional information—This work was supported by National Institutes of Health Grants HL-125422 and HL-139420 (to S. K). M. G. B. was additionally supported by an Early Career Investigator award from Bayer and a Martin Villar Basic Research award from Grifols. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PF-3688
- H-d-leucyl-phenyl-glycyl-arginine-7-amino-4-methylcoumarin
- FFRck
- H-d-Phe-Phe-Arg-chloromethylketone
- ATA-FFRck
- acetylthioacetyl-FFRck
- IXai
- factor IXa inactivated with ATA-FFRck
- F5M
- fluorescein-5-maleimide
- F-IXai
- IXai modified with F5M following thioester hydrolysis of IXai
- pAB
- 4-aminobenzamidine
- PCPS
- small unilamellar phospholipid vesicles composed of 75% (w/w) L-α-phosphatidylcholine and 25% (w/w) L-α-phosphatidylserine
- SATA
- N-hydroxysuccinimidyl-S-acetylthioacetate
- XR15Q
- factor X with a Gln substitution at Arg15
- XS195A
- factor X with an Ala substitution at Ser195.
References
- 1. Barrett A. J., Rawlings N. D., and Woessner J. F. (2004) Handbook of Proteolytic Enzymes, Vol. 2, pp. 1417–1447, Elsevier, Amsterdam [Google Scholar]
- 2. Patthy L. (1985) Evolution of the proteases of blood coagulation and fibrinolysis by assembly from modules. Cell 41, 657–663 10.1016/S0092-8674(85)80046-5 [DOI] [PubMed] [Google Scholar]
- 3. Neurath H. (1984) Evolution of proteolytic enzymes. Science 224, 350–357 10.1126/science.6369538 [DOI] [PubMed] [Google Scholar]
- 4. Mann K. G., Jenny R. J., and Krishnaswamy S. (1988) Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Annu. Rev. Biochem. 57, 915–956 10.1146/annurev.bi.57.070188.004411 [DOI] [PubMed] [Google Scholar]
- 5. Mann K. G., Nesheim M. E., Church W. R., Haley P., and Krishnaswamy S. (1990) Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 76, 1–16 10.1182/blood.V76.1.1.bloodjournal7611 [DOI] [PubMed] [Google Scholar]
- 6. Perona J. J., and Craik C. S. (1997) Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J. Biol. Chem. 272, 29987–29990 10.1074/jbc.272.48.29987 [DOI] [PubMed] [Google Scholar]
- 7. Krishnaswamy S. (2005) Exosite-driven substrate specificity and function in coagulation. J. Thromb. Hemost. 3, 54–67 10.1111/j.1538-7836.2004.01021.x [DOI] [PubMed] [Google Scholar]
- 8. Bock P. E., Panizzi P., and Verhamme I. M. (2007) Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 5, 81–94 10.1111/j.1538-7836.2007.02496.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krishnaswamy S., and Betz A. (1997) Exosites determine macromolecular substrate recognition by prothrombinase. Biochemistry 36, 12080–12086 10.1021/bi970979+ [DOI] [PubMed] [Google Scholar]
- 10. Blat Y. (2010) Non-competitive inhibition by active site binders. Chem. Biol. Drug Des. 75, 535–540 10.1111/j.1747-0285.2010.00972.x [DOI] [PubMed] [Google Scholar]
- 11. Smith S. A., Travers R. J., and Morrissey J. H. (2015) How it all starts: initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 50, 326–336 10.3109/10409238.2015.1050550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mann K. G. (2003) Thrombin formation. Chest 124, 4S–10S 10.1378/chest.124.3_suppl.4s [DOI] [PubMed] [Google Scholar]
- 13. van Dieijen G., Tans G., Rosing J., and Hemker H. C. (1981) The role of phospholipid and factor VIIIa in the activation of bovine factor X. J. Biol. Chem. 256, 3433–3442 [PubMed] [Google Scholar]
- 14. Davie E. W., Fujikawa K., and Kisiel W. (1991) The coagulation cascade: initiation, maintenance, and regulation. Biochemistry 30, 10363–10370 10.1021/bi00107a001 [DOI] [PubMed] [Google Scholar]
- 15. Krishnaswamy S. (2013) The transition of prothrombin to thrombin. J. Thromb. Hemost. 11, 265–276 10.1111/jth.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stürzebecher J., Kopetzki E., Bode W., and Hopfner K. P. (1997) Dramatic enhancement of the catalytic activity of coagulation factor IXa by alcohols. FEBS Lett. 412, 295–300 10.1016/S0014-5793(97)00811-9 [DOI] [PubMed] [Google Scholar]
- 17. Neuenschwander P. F., Deadmond K. J., Zepeda K., and Rutland J. (2012) Correlation of factor IXa subsite modulations with effects on substrate discrimination. J. Thromb. Haemost. 10, 382–389 10.1111/j.1538-7836.2011.04605.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lollar P., Parker E. T., Curtis J. E., Helgerson S. L., Hoyer L. W., Scott M. E., and Scandella D. (1994) Inhibition of human factor VIIIa by anti-A2 subunit antibodies. J. Clin. Invest. 93, 2497–2504 10.1172/JCI117259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buddai S. K., Layzer J. M., Lu G., Rusconi C. P., Sullenger B. A., Monroe D. M., and Krishnaswamy S. (2010) An anticoagulant RNA aptamer that inhibits proteinase-cofactor interactions within prothrombinase. J. Biol. Chem. 285, 5212–5223 10.1074/jbc.M109.049833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zögg T., and Brandstetter H. (2009) Structural basis of the cofactor- and substrate-assisted activation of human coagulation factor IXa. Structure 17, 1669–1678 10.1016/j.str.2009.10.011 [DOI] [PubMed] [Google Scholar]
- 21. Evans S. A., Olson S. T., and Shore J. D. (1982) Aminobenzamidine as a fluorescent probe for the active site of serine proteases. J. Biol. Chem. 257, 3014–3017 [PubMed] [Google Scholar]
- 22. Schmidt A. E., Stewart J. E., Mathur A., Krishnaswamy S., and Bajaj S. P. (2005) Na+ site in blood coagulation factor IXa: effect on catalysis and factor VIIIa binding. J. Mol. Biol. 350, 78–91 10.1016/j.jmb.2005.04.052 [DOI] [PubMed] [Google Scholar]
- 23. Brandstetter H., Turk D., Hoeffken H. W., Grosse D., Stürzebecher J., Martin P. D., Edwards B. F., and Bode W. (1992) Refined 2.3 Å X-ray crystal structure of bovine thrombin complexes formed with the benzamidine and arginine-based thrombin inhibitors NAPAP, 4-TAPAP and MQPA: a starting point for improving antithrombotics. J. Mol. Biol. 226, 1085–1099 10.1016/0022-2836(92)91054-S [DOI] [PubMed] [Google Scholar]
- 24. Nesheim M. E., Eid S., and Mann K. G. (1981) Assembly of the prothrombinase complex in the absence of prothrombin. J. Biol. Chem. 256, 9874–9882 [PubMed] [Google Scholar]
- 25. Walker R. K., and Krishnaswamy S. (1993) The influence of factor Va on the active site of factor Xa. J. Biol. Chem. 268, 13920–13929 [PubMed] [Google Scholar]
- 26. Betz A., and Krishnaswamy S. (1998) Regions remote from the site of cleavage determine macromolecular substrate recognition by the prothrombinase complex. J. Biol. Chem. 273, 10709–10718 10.1074/jbc.273.17.10709 [DOI] [PubMed] [Google Scholar]
- 27. Husten E. J., Esmon C. T., and Johnson A. E. (1987) The active site of blood coagulation factor Xa: its distance from the phospholipid surface and its conformational sensitivity to components of the prothrombinase complex. J. Biol. Chem. 262, 12953–12961 [PubMed] [Google Scholar]
- 28. Nesheim M. E., Kettner C., Shaw E., and Mann K. G. (1981) Cofactor dependence of factor Xa incorporation into the prothrombinase complex. J. Biol. Chem. 256, 6537–6540 [PubMed] [Google Scholar]
- 29. Pittman D. D., and Kaufman R. J. (1988) Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII). Proc. Natl. Acad. Sci. U. S. A. 85, 2429–2433 10.1073/pnas.85.8.2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gunaratne R., Kumar S., Frederiksen J. W., Stayrook S., Lohrmann J. L., Perry K., Bompiani K. M., Chabata C. V., Thalji N. K., Ho M. D., Arepally G., Camire R. M., Krishnaswamy S., and Sullenger B. A. (2018) Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass. Nat. Biotechnol. 36, 606–613 10.1038/nbt.4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baugh R. J., Dickinson C. D., Ruf W., and Krishnaswamy S. (2000) Exosite interactions determine the affinity of factor X for the extrinsic Xase complex. J. Biol. Chem. 275, 28826–28833 10.1074/jbc.M005266200 [DOI] [PubMed] [Google Scholar]
- 32. Krishnaswamy S., Field K. A., Edgington T. S., Morrissey J. H., and Mann K. G. (1992) Role of the membrane surface in the activation of human factor X. J. Biol. Chem. 267, 26110–26120 [PubMed] [Google Scholar]
- 33. Walker R. K., and Krishnaswamy S. (1994) The activation of prothrombin by the prothrombinase complex: the contribution of the substrate-membrane interaction to catalysis. J. Biol. Chem. 269, 27441–27450 [PubMed] [Google Scholar]
- 34. Bock P. E. (1993) Thioester peptide chloromethyl ketones: reagents for active site-selective labeling of serine proteases with spectroscopic probes. Methods Enzymol. 222, 478–503 10.1016/0076-6879(93)22030-j [DOI] [PubMed] [Google Scholar]
- 35. Baugh R. J., and Krishnaswamy S. (1996) Role of the activation peptide domain in human factor X activation by the extrinsic Xase complex. J. Biol. Chem. 271, 16126–16134 10.1074/jbc.271.27.16126 [DOI] [PubMed] [Google Scholar]
- 36. Orcutt S. J., Pietropaolo C., and Krishnaswamy S. (2002) Extended interactions with prothrombinase enforce affinity and specificity for its macromolecular substrate. J. Biol. Chem. 277, 46191–46196 10.1074/jbc.M208677200 [DOI] [PubMed] [Google Scholar]
- 37. Lu G., Broze G. J. Jr., and Krishnaswamy S. (2004) Formation of factors IXa and Xa by the extrinsic pathway: differential regulation by tissue factor pathway inhibitor and antithrombin III. J. Biol. Chem. 279, 17241–17249 10.1074/jbc.M312827200 [DOI] [PubMed] [Google Scholar]
- 38. Mann K. G., Elion J., Butkowski R. J., Downing M., and Nesheim M. E. (1981) Prothrombin. Methods Enzymol. 80, 286–302 10.1016/S0076-6879(81)80025-0 [DOI] [PubMed] [Google Scholar]
- 39. Orcutt S. J., and Krishnaswamy S. (2004) Binding of substrate in two conformations to human prothrombinase drives consecutive cleavage at two sites in prothrombin. J. Biol. Chem. 279, 54927–54936 10.1074/jbc.M410866200 [DOI] [PubMed] [Google Scholar]
- 40. Camire R. M., Larson P. J., Stafford D. W., and High K. A. (2000) Enhanced 4-carboxylation of recombinant factor X using a chimeric construct containing the prothrombin propeptide. Biochemistry 39, 14322–14329 10.1021/bi001074q [DOI] [PubMed] [Google Scholar]
- 41. Price P. A. (1983) Analysis for γ-carboxyglutamic acid. Methods Enzymol. 91, 13–17 10.1016/S0076-6879(83)91005-4 [DOI] [PubMed] [Google Scholar]
- 42. Di Scipio R. G., Kurachi K., and Davie E. W. (1978) Activation of human factor IX (Christmas factor). J. Clin. Invest. 61, 1528–1538 10.1172/JCI109073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheehan J. P., Kobbervig C. E., and Kirkpatrick H. M. (2003) Heparin inhibits the intrinsic tenase complex by interacting with an exosite on factor IXa. Biochemistry 42, 11316–11325 10.1021/bi0342923 [DOI] [PubMed] [Google Scholar]
- 44. Di Scipio R. G., Hermodson M. A., and Davie E. W. (1977) Activation of human factor X (Stuart factor) by a protease from Russell's viper venom. Biochemistry 16, 5253–5260 10.1021/bi00643a015 [DOI] [PubMed] [Google Scholar]
- 45. Cao W., Krishnaswamy S., Camire R. M., Lenting P. J., and Zheng X. L. (2008) Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc. Natl. Acad. Sci. U. S. A. 105, 7416–7421 10.1073/pnas.0801735105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lundblad R. L., Kingdon H. S., and Mann K. G. (1976) Thrombin. Methods Enzymol. 45, 156–176 10.1016/S0076-6879(76)45017-6 [DOI] [PubMed] [Google Scholar]
- 47. Bevington P. R., and Robinson K. D. (1992) Data Reduction and Error Analysis for the Physical Sciences, pp. 141–167, McGraw-Hill, New York [Google Scholar]
- 48. Straume M., and Johnson M. L. (1992) Analysis of residuals: criteria for determining the goodness-of-fit. Methods Enzymol. 210, 87–105 10.1016/0076-6879(92)10007-Z [DOI] [PubMed] [Google Scholar]
- 49. Krishnaswamy S. (1990) Prothrombinase complex assembly: contributions of protein-protein and protein-membrane interactions toward complex formation. J. Biol. Chem. 265, 3708–3718 [PubMed] [Google Scholar]
- 50. Duffy E. J., Parker E. T., Mutucumarana V. P., Johnson A. E., and Lollar P. (1992) Binding of factor VIIIa and factor VIII to factor IXa on phospholipid vesicles. J. Biol. Chem. 267, 17006–17011 [PubMed] [Google Scholar]
- 51. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data for all figures are contained within the article. Additional data backing the kinetic analyses in the tables are available from S. K. (skrishna@pennmedicine.upenn.edu).