

Abstract
BACKGROUND
The cause of most fetal anomalies is not determined prenatally. Exome sequencing has transformed genetic diagnosis after birth, but its usefulness for prenatal diagnosis is still emerging. Nonimmune hydrops fetalis (NIHF), a fetal abnormality that is often lethal, has numerous genetic causes; the extent to which exome sequencing can aid in its diagnosis is unclear.
METHODS
We evaluated a series of 127 consecutive unexplained cases of NIHF that were defined by the presence of fetal ascites, pleural or pericardial effusions, skin edema, cystic hygroma, increased nuchal translucency, or a combination of these conditions. The primary outcome was the diagnostic yield of exome sequencing for detecting genetic variants that were classified as either pathogenic or likely pathogenic according to the criteria of the American College of Medical Genetics and Genomics. Secondary outcomes were the percentage of cases associated with specific genetic disorders and the proportion of variants that were inherited.
RESULTS
In 37 of the 127 cases (29%), we identified diagnostic genetic variants, including those for disorders affecting the RAS–MAPK cell-signaling pathway (known as RASopathies) (30% of the genetic diagnoses); inborn errors of metabolism and musculoskeletal disorders (11% each); lymphatic, neurodevelopmental, cardiovascular, and hematologic disorders (8% each); and others. Prognoses ranged from a relatively mild outcome to death during the perinatal period. Overall, 68% of the cases (25 of 37) with diagnostic variants were autosomal dominant (of which 12% were inherited and 88% were de novo), 27% (10 of 37) were autosomal recessive (of which 95% were inherited and 5% were de novo), 1 was inherited X-linked recessive, and 1 was of uncertain inheritance. We identified potentially diagnostic variants in an additional 12 cases.
CONCLUSIONS
In this large case series of 127 fetuses with unexplained NIHF, we identified a diagnostic genetic variant in approximately one third of the cases. (Funded by the UCSF Center for Maternal-Fetal Precision Medicine and others; ClinicalTrials.gov number, NCT03412760.)
Prenatal diagnosis has historically been performed with the use of G-banded karyotyping to detect chromosomal abnormalities. This approach results in a diagnosis in 9 to 19% of fetal anomalies, and chromosomal microarray analysis provides an additional 6% yield.1–4 Therefore, the cause of the majority of fetal anomalies remains unknown.2,5 Identification of the cause of fetal anomalies is essential to determine prognosis, inform recurrence risk, and guide clinical management.
Recent studies in which exome sequencing was used to diagnose unexplained fetal anomalies showed diagnostic yields of 8.5% and 10%.6,7 These relatively low yields probably reflect the wide range of structural anomalies that were included, some of which were unlikely to be syndromic. In particular, limited data exist regarding the usefulness of exome sequencing for diagnosing specific, severe prenatal phenotypes.
In nonimmune hydrops fetalis (NIHF), a disorder that affects 1 in 1700 to 3000 pregnancies, fluid overload develops in the fetus and there is a high risk of stillbirth, preterm birth, and neonatal complications or death.8–11 Pregnant women with fetuses that have NIHF are also at risk for complications resulting from a form of preeclampsia called mirror syndrome.8,9 NIHF is a shared, severe presentation of many genetic disorders. Standard genetic testing with karyotyping or chromosomal microarray analysis identifies the cause of only 25% of NIHF cases and does not detect single-gene disorders.12–25 The contribution of single-gene disorders to NIHF is unknown but is potentially substantial. Some genetic disorders underlying NIHF portend mild long-term outcomes, whereas others are lethal despite treatment.8,12–24 An accurate diagnosis enables focused prenatal management and early, directed neonatal care to improve outcomes for this severe condition. The aims of this study were to establish the diagnostic yield of exome sequencing for single-gene disorders in unexplained NIHF and to describe the spectrum of underlying disorders.
METHODS
STUDY DESIGN AND PARTICIPANTS
We evaluated a series of consecutive NIHF cases with the use of exome sequencing. All five University of California (UC) Fetal–Maternal Consortium sites (UC, Davis; UC, Irvine; UC, Los Angeles; UC, San Diego; and UC, San Francisco [UCSF]) participated. Referrals were also accepted from providers across the United States. We aimed to enroll 100 participants on the basis of the prevalence of NIHF, but because recruitment was more rapid than anticipated, we exceeded our target. Approval for the study was obtained from the institutional review board at UCSF, and all the participants provided written informed consent.
We defined NIHF by the presence of one or more pathologic fetal fluid collections, including an increased thickness of nuchal translucency (≥3.5 mm), cystic hygroma, pleural effusion, pericardial effusion, ascites, skin edema, or a combination of these conditions. Although the current definition of NIHF (as defined by the Society for Maternal–Fetal Medicine) specifies at least two pathologic fluid collections, this definition is poorly supported; genetic disorders can be manifested by only one abnormal fluid collection, and the types of abnormal fluid collections may change during gestation.8,12–14,23,26,27 A nondiagnostic karyotype analysis or chromosomal microarray analysis was required for eligibility in the study. Cases in which concurrent fetal structural anomalies had been present in the index pregnancy with NIHF were eligible for inclusion, as were cases of ongoing pregnancy, stillbirth, termination, live birth, and infant death. Women could be enrolled either during the pregnancy with NIHF or after birth had occurred if NIHF had been documented prenatally but diagnostic testing had been deferred. We excluded cases in which there was established pathophysiological evidence of hydrops, including alloimmunization, congenital viral infection, or twin-to-twin transfusion syndrome. Additional exclusion criteria were an insufficient fetal or infant sample or the presence of a diagnostic result that had been obtained through gene-panel sequencing or other genetic testing.
OUTCOMES
The primary outcome was the diagnostic yield of exome sequencing for detecting pathogenic or likely pathogenic variants in unexplained cases of NIHF. Secondary outcomes were the percentage of cases associated with specific genetic disorders and the proportion of variants that were inherited.
PROCEDURES
Participants provided informed consent either in person or by video call. Consent included their decision to receive or decline the results of secondary genomic findings (e.g., predisposition to cancer or cardiac disease), as recommended by the American College of Medical Genetics and Genomics (ACMG).28 Records were obtained and reviewed for medical and family history, previous genetic testing, detailed prenatal and postnatal phenotyping, and pregnancy outcomes. Cases were categorized according to the presence of NIHF features at the time of enrollment: increased nuchal translucency or cystic hygroma, a single abnormal fetal fluid collection (e.g., isolated ascites), and at least two abnormal fluid collections (pleural effusion, pericardial effusion, ascites, or skin edema).
For cases in which chorionic villus sampling, amniocentesis, or another prenatal procedure was performed, cultured cells or extracted DNA were used. For cases in which testing was carried out after pregnancy, umbilical-cord blood, a buccal-swab sample, or other tissue was obtained from the infant or stillborn fetus. Trio-exome sequencing, which requires a blood or saliva sample from each biologic parent and a sample from the fetus or infant, was performed whenever possible. In cases in which only the biologic mother was available, duo-exome sequencing was performed. In one case of a pregnancy resulting from a donor egg and donor sperm, a sample from only the fetus was sequenced. In cases in which an older sibling had been affected by NIHF, quad-exome sequencing, which included DNA from that sibling, was performed.
The UCSF Genomic Medicine Laboratory, which is certified by the Clinical Laboratory Improvement Amendments program, performed exome sequencing with the use of the Illumina HiSeq 2500 sequencer in rapid-run mode or with the Illumina NovaSeq 6000 sequencing system. Variant call format files were uploaded for variant filtering into Ingenuity Variant Analysis (Qiagen) before March 2020 and into Moon (Diploid) beginning in March 2020. Clinical informatics experts at the UCSF Genomic Medicine Laboratory manually curated the variants. In cases of ongoing pregnancies and live births, results of exome sequencing were prioritized to inform clinical management.
For both exome-sequencing analysis and genetic variant interpretation, detailed phenotypic data were incorporated as appropriate from prenatal laboratory and imaging findings, pathological findings in fetuses and infants, and examination, laboratory, and imaging findings in infants. Phenotypic data were described with the use of Human Phenotype Ontology terms to improve exome-sequencing findings.29 A multidisciplinary review of curated variants in the context of phenotypic features occurred for each case during weekly in-person board meetings at UCSF that included experts in clinical informatics, bioinformatics, clinical genetics, pathology, perinatology, pediatrics, and bioethics.
Genetic variants were classified according to ACMG and Association for Medical Pathology recommendations.30 Variants classified as pathogenic or likely pathogenic were considered to be diagnostic. Variants of uncertain clinical significance were considered to be potentially diagnostic and were reported if there was gene- or variant-level evidence to support strong potential for clinical significance but criteria for pathogenicity were not met. All reported genetic variants were confirmed by means of Sanger sequencing. Exome-sequencing results and a formal report were provided directly to participants and to referring providers.
STATISTICAL ANALYSIS
Percentages and proportions are reported for primary and secondary outcomes. For demographic variables, prenatal phenotypes, and pregnancy outcomes, categorical variables are summarized as percentages and proportions, and continuous variables are reported as median values with interquartile ranges. Data were analyzed with the use of Stata software, version 15.0 (StataCorp).
RESULTS
STUDY PARTICIPANTS
A total of 233 cases of NIHF were referred from October 2018 through May 2020. Overall, 106 women were not enrolled in the study because they were lost to follow-up, declined to participate, did not meet the inclusion criteria, had an inadequate fetal or infant sample available for exome sequencing, or had a diagnostic result from a karyotype analysis, chromosomal microarray analysis, or gene-panel sequencing (Fig. 1). Ultimately, 127 women were enrolled and underwent exome sequencing.
Figure 1. Study Enrollment and Testing.
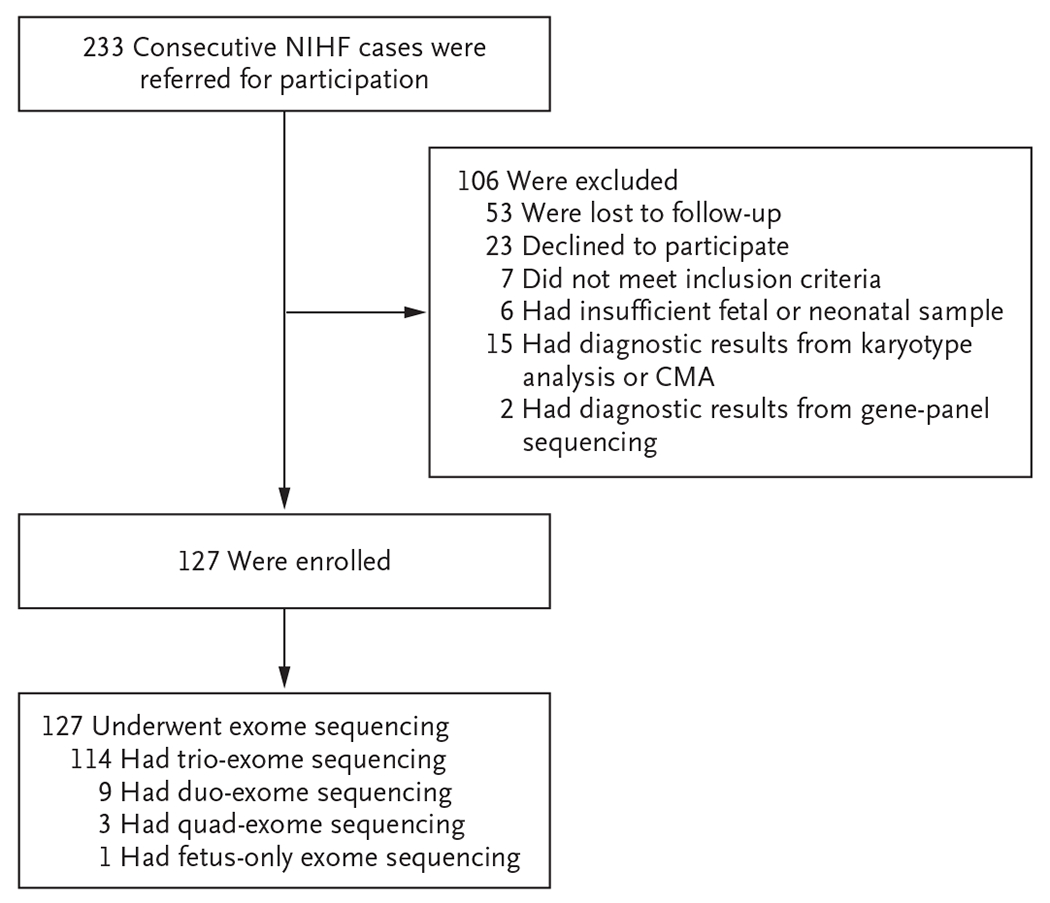
CMA denotes chromosomal microarray analysis, and NIHF nonimmune hydrops fetalis.
Before exome sequencing was performed, karyotype analysis only was performed in 4% (5 of the 127 cases), chromosomal microarray analysis only in 34% (43 cases), and both karyotype analysis and chromosomal microarray analysis in 62% (79 cases). Trio-exome sequencing was performed in 90% (114 cases), duo-exome sequencing in 7% (9 cases), quad-exome sequencing in 2% (3 cases), and proband-only-exome sequencing in 1% (1 case). Among the sources of fetal and infant DNA, 21% (in 27 cases) were from chorionic villus sampling, 57% (in 73 cases) from amniocentesis, 2% (in 2 cases) from fetal-blood sampling, 1% (in 1 case) from pleural fluid, 2% (in 3 cases) from umbilical-cord blood at delivery, 13% (in 17 cases) from placental tissue, and 3% (in 4 cases) from a buccal-swab sample. In total, 27 of the 127 samples (21%) were cultured (of which 85% were prenatal). In cases of ongoing pregnancies and live births, results were sent directly to participants and referring providers within 2 to 4 weeks after the receipt of samples at the laboratory. In cases of stillbirth, termination, and infant death, results were sent within 8 to 12 weeks.
Participants were enrolled in locations throughout the United States (Table 1), with 49% (62 of the 127 participants) within the University of California Fetal–Maternal Consortium and the remainder outside this system. A total of 58% (74 women) identified themselves as White, 15% (19 women) as Asian, 14% (18 women) as multiracial, 9% (12 women) as Hispanic or Latino, 2% (3 women) as Black, and 1% (1 woman) as unknown. Among the completed pregnancies, 31% (18 of 59) resulted in a live-born infant. Demographic characteristics, prenatal phenotypes, and pregnancy outcomes, according to exome-sequencing results, are provided in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Table 1.
Demographic Characteristics, Phenotypic Features, and Pregnancy Outcomes.*
| Characteristic | Value (N = 127) |
|---|---|
| Median maternal age (IQR) — yr | 32 (29–35) |
| Nulliparous — no. (%) | 57 (45) |
| Use of assisted reproductive technology — no. (%)† | 12 (9) |
| Median gestational age at diagnosis of NIHF (IQR) — wk | 20.0 (13.4–24.6) |
| Previous pregnancy with NIHF — no. (%) | 10 (8) |
| Biologic parents consanguineous — no. (%) | 4 (3) |
| Prenatal phenotype at enrollment — no. (%)‡ | |
| Increased nuchal translucency or cystic hygroma | 29 (23) |
| Isolated increased nuchal translucency or cystic hygroma | 15 (12) |
| Concurrent structural anomaly | 9 (7) |
| >1 additional abnormal fluid collection | 5 (4) |
| Single abnormal fetal fluid collection | 21 (17) |
| Isolated single abnormal fetal fluid collection | 4 (3) |
| Concurrent structural anomaly | 17 (13) |
| ≥2 abnormal fetal fluid collections | 77 (61) |
| Isolated abnormal fetal fluid collections | 39 (31) |
| Concurrent structural anomaly | 38 (30) |
| Fetal sex — no. (%) | |
| Female | 65 (51) |
| Male | 62 (49) |
| Maternal race or ethnic group — no. (%)§ | |
| White | 74 (58) |
| Asian | 19 (15) |
| Multiracial | 18 (14) |
| Hispanic or Latino | 12 (9) |
| Black | 3 (2) |
| Unknown | 1 (1) |
| Region of United States — no. (%) | |
| West | 86 (68) |
| Midwest | 13 (10) |
| South | 8 (6) |
| Northeast | 20 (16) |
| Pregnancy outcome — no. (%) | |
| Ongoing pregnancy | 23 (18) |
| Live-born infant | 18 (14) |
| Postnatal death | 26 (20) |
| Stillbirth¶ | 15 (12) |
| Pregnancy termination | 35 (28) |
| Selective reduction of affected twin | 3 (2) |
| Spontaneous loss of pregnancy before 20 wk of gestation | 7 (6) |
| Median gestational age at delivery (IQR) — wk∥ | 31.4 (27.6–34.1) |
Percentages may not total 100 because of rounding. IQR denotes interquartile range, and NIHF nonimmune hydrops fetalis.
A total of 11 pregnancies resulted from in vitro fertilization and 1 from intrauterine insemination.
Shown are the phenotypic features that were observed on prenatal imaging at the time that exome sequencing was completed.
Race and ethnic group were reported by the participant.
Stillbirth was defined as intrauterine fetal death at 20 weeks of gestation or later.
The median and IQR were based on the ongoing pregnancies that did not result in spontaneous loss, termination, or stillbirth.
With regard to prenatal phenotype at enrollment, 23% of the cases (29 of 127) had increased nuchal translucency or cystic hygroma, 17% (21 cases) had a single abnormal fetal fluid collection, and 61% (77 cases) had at least two abnormal fetal fluid collections. In Table 1, these categories are further subdivided into isolated cases and cases with concurrent structural anomalies. Among the 15 cases of isolated increased nuchal translucency or cystic hygroma, the median thickness of nuchal translucency was 5.0 mm (interquartile range, 3.9 to 7.0).
PRIMARY AND SECONDARY OUTCOMES
No data were missing for the primary or secondary outcomes. We identified diagnostic variants in 37 of the 127 fetuses (29%); these variants caused a wide variety of genetic disorders (Table 2 and Fig. 2). Disorders affecting the RAS–MAPK cell-signaling pathway (known as RASopathies) composed the largest proportion (30%, 11 of 37 cases). Inborn errors of metabolism and musculoskeletal disorders each composed 11% (4 cases), and lymphatic, neurodevelopmental, cardiovascular, and hematologic disorders each composed 8% (3 cases). The least common disorders were immunologic disorders (5%, 2 cases), followed by renal disorders, ciliopathies, overgrowth syndromes, and others (3% each, 1 case). Among four consanguineous families, no diagnostic variants were identified.
Table 2.
Diagnostic Variants in Cases of NIHF.*
| Case No. | Gene | Genetic Disorder | Protein Alteration† | Inheritance | Recurrence Risk percent |
|---|---|---|---|---|---|
| RASopathies | |||||
| H025 | PTPN11 | Noonan syndrome | p.Ala72Pro | De novo (autosomal dominant) | 1–2 |
| H089 | PTPN11 | Noonan syndrome | p.Asn308Asp | De novo (autosomal dominant) | 1–2 |
| H086 | PTPN11 | Noonan syndrome | p.Phe285Ser | De novo (autosomal dominant) | 1–2 |
| H095 | PTPN11 | Noonan syndrome | p.Phe285Ser | De novo (autosomal dominant) | 1–2 |
| H036 | KRAS | Noonan syndrome | p.Thr74Pro | De novo (autosomal dominant) | 1–2 |
| H042 | RIT1 | Noonan syndrome | p.Phe82Leu | De novo (autosomal dominant) | 1–2 |
| H073 | SHOC2 | Noonan-like syndrome with loose anagen hair | p.Met173Ile | De novo (autosomal dominant) | 1–2 |
| H008 | HRAS | Costello syndrome | p.Gly12Ser | De novo (autosomal dominant) | 1–2 |
| H016 | HRAS | Costello syndrome | p.Gly13Arg | De novo (autosomal dominant) | 1–2 |
| H119 | HRAS | Costello syndrome | p.Gly12Asp | De novo (autosomal dominant) | 1–2 |
| H020 | BRAF | Cardiofaciocutaneous syndrome | p.Asn581His | De novo (autosomal dominant) | 1–2 |
| Inborn errors of metabolism | |||||
| H005 | NPC1 | Niemann–Pick disease type C | p.Ile1061Thr, p.Pro691Gln | Maternal and paternal (autosomal recessive) | 25 |
| H019 | GLB1 | GM1 gangliosidosis | p.Gly311Arg, c.75+1delG | Maternal and paternal (autosomal recessive) | 25 |
| H054 | GUSB | Mucopolysaccharidosis type VII | p.Leu12Pro, c.210+1G→A | Maternal and paternal (autosomal recessive) | 25 |
| H068 | GUSB | Mucopolysaccharidosis type VII | Homozygous exon 9 deletion | Maternal and paternal (autosomal recessive) | 25 |
| Musculoskeletal disorders | |||||
| H026 | MYH3 | Multiple pterygium syndrome | p.Ile705Thr | De novo (autosomal dominant) | 1–2 |
| H034 | FGFR3 | Thanatophoric dysplasia type I | p.Arg248Cys | De novo (autosomal dominant) | 1–2 |
| H055 | KLHL40 | Nemaline myopathy | p.Thr506Pro | Maternal and paternal (autosomal recessive) | 25 |
| H085 | SF3B4 | Nager syndrome (also known as acrofacial dysostosis) | p.Met1? | De novo (autosomal dominant) | 1–2 |
| Lymphatic disorders | |||||
| H044 | FOXC2 | Lymphedema distichiasis syndrome | p.Glu343* | Maternal (autosomal dominant) | 50 |
| H050 | FLT4 | Milroy’s disease | p.Arg1041Trp | Paternal (autosomal dominant) | 50 |
| H072 | PIEZO1 | Generalized lymphatic dysplasia | p.Pro1906Lysfs*55, p.Ile2270Thr | Maternal and paternal (autosomal recessive) | 25 |
| Neurodevelopmental disorders | |||||
| H018 | WAC | DeSanto–Shinawi syndrome | p.Ser491fs*9 | De novo (autosomal dominant) | 1–2 |
| H063 | ZEB2 | Mowat–Wilson syndrome | p.Arg695* | De novo (autosomal dominant) | 1–2 |
| H083 | DHCR24 | Desmosterolosis | c.1218+1G→A, p.Gln402* | Maternal and paternal (autosomal recessive) | 25 |
| Cardiovascular disorders | |||||
| H007 | ACAD9 | Mitochondrial complex I deficiency | p.Pro370fs*13, p.Arg266Trp | De novo and maternal (autosomal recessive) | ≤25 |
| H058 | NEXN | Dilated and hypertrophic cardiomy-opathy | p.Arg216*, p.Lys536fs | De novo and maternal‡ | ≤50 |
| H080 | MYRF | Cardiac–urogenital syndrome | p.Ser264fs | De novo (autosomal dominant) | 1–2 |
| Hematologic disorders | |||||
| H027 | RPL11 | Diamond–Blackfan anemia | p.Phe105fs*15 | De novo (autosomal dominant) | 1–2 |
| H094 | PIEZO1 | Dehydrated hereditary stomatocytosis | p.Met870Ile | Suspected maternal mosaic (autosomal dominant) | ≤50 |
| H126 | PIEZO1 | Dehydrated hereditary stomatocytosis | p.Val598Met | De novo (autosomal dominant) | 1–2 |
| Immunologic disorders | |||||
| H056 | STAT3 | Hyper-IgE syndrome, multisystem infantile-onset autoimmune disease | p.Thr341Ile | De novo (autosomal dominant) | 1–2 |
| H113 | FOXP3 | IPEX syndrome | c.543–2→G | Maternal (X-linked recessive) | ≤50 if male fetus |
|
Renal disorder: H107 |
CEP55 | Multinucleated neurons, anhydramnios, renal dysplasia, cerebellar hypoplasia, and hydranencephaly | p.Arg64*, p.His458Arg | Maternal and paternal (autosomal recessive) | 25 |
|
Ciliopathy: H030 |
DNAH9 | Primary ciliary dyskinesia | p.Arg995fs*5 | Maternal and paternal (autosomal recessive) | 25 |
|
Overgrowth disorder: H109 |
SUZ12 | Imagawa–Matsumoto syndrome | p.Gly484fs | De novo (autosomal dominant) | 1–2 |
|
Other disorder: H124 |
CHD7 | CHARGE syndrome | p.Val1141fs | De novo (autosomal dominant) | 1–2 |
CHARGE denotes coloboma of the eye, heart anomaly, atresia of the choanae, retarded growth and development, and genital and ear anomalies; and IPEX immune dysregulation, polyendocrinopathy, enteropathy, X-linked.
Coding changes are shown for variants identified in intronic regions.
Two NEXN variants were identified: one of maternal inheritance and the other de novo, for which the cis or trans phase could not be definitively determined on the basis of exome sequencing.
Figure 2. Categories of Genetic Disorders Detected through Exome Sequencing in Cases of NIHF.
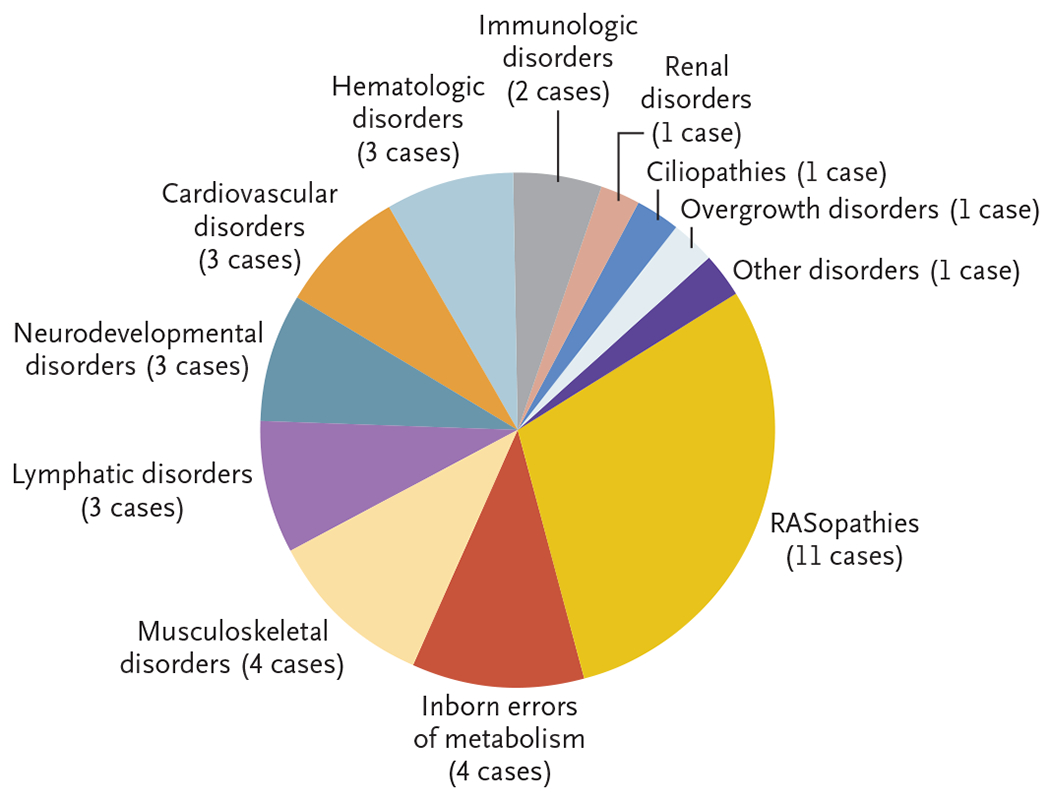
RASopathies were defined as disorders affecting the RAS–MAPK cell-signaling pathway.
The 11 cases with RASopathies included the Noonan syndrome (caused by mutations in PTPN11, KRAS, and RIT1), Noonan-like syndrome with loose anagen hair (SHOC2), cardiofaciocutaneous syndrome (BRAF), and the Costello syndrome (HRAS). All the RASopathy variants were de novo and were autosomal dominant. There were 4 cases with inborn errors of metabolism, including Niemann–Pick disease type C (NPC1), GM1 gangliosidosis (GLB1), and mucopolysaccharidosis type VII (GUSB); all were autosomal recessive and inherited from parents who were heterozygous carriers. Four cases with musculoskeletal disorders were seen, including the Nager syndrome (SF3B4), thanatophoric dysplasia type I (FGFR3), nemaline myopathy (KLHL40), and multiple pterygium syndrome (MYH3). The SF3B4, FGFR3, and MYH3 variants were autosomal dominant and de novo, whereas both KLHL40 variants for autosomal recessive nemaline myopathy were inherited from carrier parents. Among the 3 cases with lymphatic disorders (Milroy’s disease, lymphedema distichiasis syndrome, and generalized lymphatic dysplasia), all the variants (FLT4, FOXC2, and PIEZO1) were inherited. The FLT4 and FOXC2 variants were inherited from a parent with previously unexplained mild swelling in the legs and feet. Three cases with neurodevelopmental disorders were seen, including the DeSanto–Shinawi syndrome (WAC), the Mowat–Wilson syndrome (ZEB2), and desmosterolosis (DHCR24). The autosomal dominant WAC and ZEB2 variants were de novo, whereas both variants for autosomal recessive desmosterolosis were inherited. There were 3 cases with hematologic disorders, including 1 case of Diamond–Blackfan anemia (RPL11) and 2 cases of dehydrated hereditary stomatocytosis (PIEZO1). All were autosomal dominant; the RPL11 variant and one PIEZO1 variant were de novo, and the other PIEZO1 variant resulted from suspected maternal mosaicism. Table 2 and Figure 2 show all the diagnoses, and Table S2 shows full genomic details, the prenatal phenotype, and the pregnancy outcome for each diagnostic variant.
Exome-sequencing results informed the risk of recurrence on the basis of Mendelian recurrence estimates, which ranged from 1 to 2% with de novo variants31 to 50% with inherited autosomal dominant variants (Table 2). Overall, 68% of the cases of diagnostic variants (25 of 37) were autosomal dominant, 27% (10 of 37) were autosomal recessive, 1 was X-linked recessive (FOXP3), and 1 had uncertain inheritance. In the case with uncertain inheritance, 1 maternally inherited NEXN variant and 1 de novo NEXN variant were seen, but the phase remained unclear on the basis of exome sequencing; these variants were associated with autosomal dominant dilated and hypertrophic cardiomyopathy. The majority of autosomal dominant variants were de novo (88%, 22 of 25 variants) and 36% (9 of 25) were novel, whereas the majority of autosomal recessive variants were inherited (95%, 19 of 20 variants) and 80% (16 of 20) were novel.
Among the 29 cases with increased nuchal translucency or cystic hygroma (either isolated or concurrent with other anomalies), 31% (9 of 29) had a diagnostic variant (Table S1). However, among the cases with isolated increased nuchal translucency or cystic hygroma, the diagnostic yield was 7% (1 of 15); nuchal translucency measured 4.5 mm thick for the 1 diagnostic case (CHARGE syndrome [coloboma of the eye, heart anomaly, atresia of the choanae, retarded growth and development, and genital and ear anomalies]). Among the 77 cases with at least two abnormal fluid collections, 26 (34%) had a diagnostic variant. Further details of diagnostic yield according to phenotype are provided in Table S1.
VARIANTS OF POTENTIAL CLINICAL SIGNIFICANCE
We identified a variant with gene-level or variant-level evidence of potential clinical significance in 12 of the 127 affected fetuses (9%), but these variants did not meet the criteria for pathogenicity or likely pathogenicity (Table S3). Potentially implicated disorders included a RASopathy, generalized lymphatic dysplasia, several neurodevelopmental disorders, and others. In some cases, such as with POU3F3-associated emerging developmental delay disorder, gene-level evidence was insufficient. In other cases, such as with ERCC5-associated cerebrooculofacioskeletal syndrome, the gene–disease fit was strong, but variant-level data were insufficient.
In an additional 2% of the fetuses (2 of 127), one genetic variant was detected for an autosomal recessive disorder consistent with the phenotype. However, a second variant in the same gene was not identified. These genes were CNTN1 (Compton–North congenital myopathy) and RYR1 (lethal multiple pterygium syndrome).
SECONDARY FINDINGS
In total, 91% of the families of the participants (115 of 127) chose to receive secondary findings; 3% (4 of 115) had a pathogenic or likely pathogenic variant in each of APOB (familial hypercholesterolemia), MYH7 (familial hypertrophic cardiomyopathy), PTEN (PTEN hamartoma tumor syndrome), and BRCA1 (hereditary breast and ovarian cancer syndrome).
DISCUSSION
In this large series of NIHF cases unexplained by standard genetic testing, exome sequencing was used to identify diagnostic variants in 29% of the cases. A variant of potential clinical significance was detected in an additional 9% of the cases, many of which were probably associated with the phenotype but were novel variants and emerging genes. The yield in our series is substantially higher than the 8.5% and 10% yields that were reported in studies of unselected fetal anomalies,6,7 findings that reflect the burden of single-gene disorders underlying NIHF. The postnatal prognoses for the diseases we identified ranged from relatively mild to severely affected with limited life expectancy, and diagnoses affected both counseling and direct clinical care.
RASopathies were common in our series. The Noonan syndrome has been well established in its association with NIHF,12–15,23 but in utero manifestations of RASopathies beyond the Noonan syndrome are less well characterized. In contrast to approximately half of RASopathy variants being inherited in postnatal series,32 the de novo nature of all the RASopathy variants in our series highlights those capable of severe in utero presentations. Also common were cases of inborn errors of metabolism, as well as musculoskeletal, lymphatic, neurodevelopmental, cardiovascular, and hematologic disorders. Despite having similar prenatal phenotypes, these disorders are associated with a wide range of outcomes, from relatively mild lymphedema to probable perinatal death, and their clinical management differs greatly.
Establishing a diagnosis allows precise determination of the risk of recurrence and can guide perinatal care. Two thirds of the diagnostic variants were autosomal dominant; 12% were inherited with a 50% recurrence risk, as compared with a 1 to 2% recurrence risk for the many de novo cases. In contrast, 27% of the diagnostic variants were autosomal recessive, and nearly all were associated with a 25% recurrence risk. Identifying these diagnoses improves the accuracy of counseling, allows the option of preimplantation genetic diagnosis in future pregnancies, and improves clinical care in the affected pregnancy. Examples from our series include screening for fetal anemia in pregnancies with dehydrated hereditary stomatocytosis and Diamond–Blackfan anemia to determine whether intrauterine transfusions are indicated, as well as prenatal magnetic resonance imaging and pediatric neurology consults in a case of the Imagawa–Matsumoto syndrome to plan for postnatal needs. Furthermore, only 31% of the completed pregnancies resulted in a live-born infant, highlighting the critical need for accurate diagnosis to guide perinatal care and improve outcomes.
Our study has several important strengths. Our series represents a large, national population. It highlights the importance of accurate prenatal diagnosis for NIHF, contributes data about the scope of underlying genetic disorders, and identifies novel variants that can portend a poor prognosis. Exome-sequencing analysis and the multidisciplinary UCSF board reviews incorporated thorough details of evolving prenatal phenotypic data, pathological findings in fetuses and infants, and postnatal phenotypic data, which are critical for accurate identification and interpretation of genetic variants.
However, this study is not without limitations. Although the participants were geographically diverse, more than half identified themselves as White. Among cases with increased nuchal translucency or cystic hygroma, many later showed additional fluid collections or concurrent anomalies, and the diagnostic yield for isolated increased nuchal translucency or cystic hygroma cases was low. Further studies are warranted to determine the usefulness of exome sequencing for isolated increased nuchal translucency or cystic hygroma, since the risk of subsequent pathological conditions is unknown. There are limitations of prenatal phenotyping, especially in early gestation. Because accurate genetic variant classification relies in part on phenotypic fit, it is possible that disease-causing variants were missed or incorrectly classified. Although some copy-number variants and intronic variants were detected, exome sequencing is not designed to routinely detect these genomic changes. Future studies in which whole-genome sequencing or functional assays are used to evaluate additional genomic changes when exome sequencing shows normal results are warranted. Finally, it is possible that providers and patients motivated by the desire for genomic information were more likely to participate, potentially affecting the generalization of our results.
Exome sequencing identified a diagnostic variant in 29% of NIHF cases unexplained by standard genetic testing. These data support the use of exome sequencing for NIHF cases with nondiagnostic results of chromosomal microarray analysis or karyotype analysis in order to inform prognosis, establish recurrence risk, and direct prenatal and postnatal clinical care.
Supplementary Material
Acknowledgments
The views expressed in this article are those of the authors and do not necessarily reflect the views of the National Institutes of Health, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, or the National Human Genome Research Institute.
Supported by the University of California, San Francisco (UCSF), Center for Maternal–Fetal Precision Medicine, the Fetal Health Foundation, the Brianna Marie Foundation, Ultragenyx (for studies conducted through the UCSF Center for Maternal–Fetal Precision Medicine), and grants (5K12HD001262-18, supporting Dr. Sparks, and U01HG009599, to Drs. Slavotinek and Norton) from the National Institutes of Health.
We thank personnel at the University of California Fetal–Maternal Consortium and the members of the UCSF Center for Maternal–Fetal Precision Medicine, especially Barbara Koenig, Ph.D., for input with regard to this study, personnel at the UCSF Genomic Medicine Laboratory and the UCSF Institute for Human Genetics for genetic testing, and each of the families who participated in this study and the institutions and providers who referred or supported the participants.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Staebler M, Donner C, Van Regemorter N, et al. Should determination of the karyotype be systematic for all malformations detected by obstetrical ultrasound? Prenat Diagn 2005;25:567–73. [DOI] [PubMed] [Google Scholar]
- 2.Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril 2018;109:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American College of Obstetricians and Gynecologists Committee on Genetics. Committee opinion no. 581: the use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol 2013;122:1374–7. [DOI] [PubMed] [Google Scholar]
- 5.Feldkamp ML, Carey JC, Byrne JLB, Krikov S, Botto LD. Etiology and clinical presentation of birth defects: population based study. BMJ 2017;357:j2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 2019;393:758–67. [DOI] [PubMed] [Google Scholar]
- 7.Lord J, McMullan DJ, Eberhardt RY, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 2019;393:747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Society for Maternal-Fetal Medicine (SMFM). Society for Maternal-Fetal Medicine (SMFM) clinical guideline #7: non-immune hydrops fetalis. Am J Obstet Gynecol 2015;212:127–39. [DOI] [PubMed] [Google Scholar]
- 9.Gedikbasi A, Oztarhan K, Gunenc Z, et al. Preeclampsia due to fetal non-immune hydrops: mirror syndrome and review of literature. Hypertens Pregnancy 2011;30:322–30. [DOI] [PubMed] [Google Scholar]
- 10.Fukushima K, Morokuma S, Fujita Y, et al. Short-term and long-term outcomes of 214 cases of non-immune hydrops fetalis. Early Hum Dev 2011;87:571–5. [DOI] [PubMed] [Google Scholar]
- 11.Steurer MA, Peyvandi S, Baer RJ, et al. Epidemiology of live born infants with nonimmune hydrops fetalis — insights from a population-based dataset. J Pediatr 2017;187:182–188.e3. [DOI] [PubMed] [Google Scholar]
- 12.Mardy AH, Chetty SP, Norton ME, Sparks TN. A system-based approach to the genetic etiologies of non-immune hydrops fetalis. Prenat Diagn 2019;39:732–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croonen EA, Nillesen WM, Stuurman KE, et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with abnormal ultrasound findings. Eur J Hum Genet 2013;21:936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sparks TN, Thao K, Lianoglou BR, et al. Nonimmune hydrops fetalis: identifying the underlying genetic etiology. Genet Med 2019;21:1339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellini C, Donarini G, Paladini D, et al. Etiology of non-immune hydrops fetalis: an update. Am J Med Genet A 2015;167A:1082–8. [DOI] [PubMed] [Google Scholar]
- 16.Daniel-Spiegel E, Ghalamkarpour A, Spiegel R, et al. Hydrops fetalis: an unusual prenatal presentation of hereditary congenital lymphedema. Prenat Diagn 2005;25:1015–8. [DOI] [PubMed] [Google Scholar]
- 17.Overcash RT, Gibu CK, Jones MC, Ramos GA, Andreasen TS. Maternal and fetal capillary malformation-arteriovenous malformation (CM-AVM) due to a novel RASA1 mutation presenting with prenatal non-immune hydrops fetalis. Am J Med Genet A 2015;167A:2440–3. [DOI] [PubMed] [Google Scholar]
- 18.Burin MG, Scholz AP, Gus R, et al. Investigation of lysosomal storage diseases in nonimmune hydrops fetalis. Prenat Diagn 2004;24:653–7. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Y, Verp MS, Knutel T, Hibbard JU. Mucopolysaccharidosis type VII as a cause of recurrent non-immune hydrops fetalis. J Perinat Med 2003;31:535–7. [DOI] [PubMed] [Google Scholar]
- 20.Gimovsky AC, Luzi P, Berghella V. Lysosomal storage disease as an etiology of nonimmune hydrops. Am J Obstet Gynecol 2015;212:281–90. [DOI] [PubMed] [Google Scholar]
- 21.Surmeli-Onay O, Yakarisik S, Korkmaz A, et al. Prenatal-onset Niemann-Pick type C disease with nonimmune hydrops fetalis. Pediatr Neonatol 2013;54:344–7. [DOI] [PubMed] [Google Scholar]
- 22.Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R. Infantile lethal variant of Simpson-Golabi-Behmel syndrome associated with hydrops fetalis. Am J Med Genet 1995;59:329–33. [DOI] [PubMed] [Google Scholar]
- 23.Gedikbasi A, Gul A, Sargin A, Ceylan Y. Cystic hygroma and lymphangioma: associated findings, perinatal outcome and prognostic factors in live-born infants. Arch Gynecol Obstet 2007;276:491–8. [DOI] [PubMed] [Google Scholar]
- 24.Derderian SC, Trivedi S, Farrell J, et al. Outcomes of fetal intervention for primary hydrothorax. J Pediatr Surg 2014;49:900–3. [DOI] [PubMed] [Google Scholar]
- 25.Mardy AH, Rangwala N, Hernandez-Cruz Y, et al. Utility of chromosomal microarray for diagnosis in cases of nonimmune hydrops fetalis. Prenat Diagn 2020;40:492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baer RJ, Norton ME, Shaw GM, et al. Risk of selected structural abnormalities in infants after increased nuchal translucency measurement. Am J Obstet Gynecol 2014;211(6):675.e1–675.e19. [DOI] [PubMed] [Google Scholar]
- 27.Maya I, Yacobson S, Kahana S, et al. Cut-off value of nuchal translucency as indication for chromosomal microarray analysis. Ultrasound Obstet Gynecol 2017;50:332–5. [DOI] [PubMed] [Google Scholar]
- 28.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249–55. [DOI] [PubMed] [Google Scholar]
- 29.Masino AJ, Dechene ET, Dulik MC, et al. Clinical phenotype-based gene prioritization: an initial study using semantic similarity and the Human Phenotype Ontology. BMC Bioinformatics 2014;15:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell IM, Stewart JR, James RA, et al. Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet 2014;95:345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelb BD, Cavé H, Dillon MW, et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med 2018;20:1334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.